
Multiplexed fluidic plunger mechanism for the measurement of red blood cell deformability†
Marie-Eve
Myrand-Lapierre
a,
Xiaoyan
Deng
a,
Richard R.
Ang
a,
Kerryn
Matthews
a,
Aline T.
Santoso
a and
Hongshen
Ma
*abc
aDepartment of Mechanical Engineering, University of British Columbia, 2054-6250 Applied Science Lane, Vancouver, BC, Canada V6T 1Z4. E-mail: hongma@mech.ubc.ca
bDepartment of Urologic Science, University of British Columbia, Vancouver, BC, Canada
cVancouver Prostate Centre, Vancouver General Hospital, Vancouver, BC, Canada
First published on 17th October 2014
Abstract
The extraordinary deformability of red blood cells gives them the ability to repeatedly transit through the microvasculature of the human body. The loss of this capability is part of the pathology of a wide range of diseases including malaria, hemoglobinopathies, and micronutrient deficiencies. We report on a technique for multiplexed measurements of the pressure required to deform individual red blood cell through micrometer-scale constrictions. This measurement is performed by first infusing single red blood cells into a parallel array of ~1.7 μm funnel-shaped constrictions. Next, a saw-tooth pressure waveform is applied across the constrictions to squeeze each cell through its constriction. The threshold deformation pressure is then determined by relating the pressure–time data with the video of the deformation process. Our key innovation is a self-compensating fluidic network that ensures identical pressures are applied to each cell regardless of its position, as well as the presence of cells in neighboring constrictions. These characteristics ensure the consistency of the measurement process and robustness against blockages of the constrictions by rigid cells and debris. We evaluate this technique using in vitro cultures of RBCs infected with P. falciparum, the parasite that causes malaria, to demonstrate the ability to profile the deformability signature of a heterogeneous sample.
Introduction
Red blood cells (RBCs) perform the critical function of transporting oxygen and carbon dioxide between tissues in the human body. This capability is enabled in part by their extraordinary mechanical deformability where discoid-shaped RBCs, 8 μm in diameter and 2 μm in thickness, can repeatedly deform through microcapillaries less than 2.5 μm in diameter, as well as inter-endothelial clefts in the spleen ranging from 0.5–1 μm.1 A loss of this extraordinary deformability can result in microvascular occlusion and impairment of blood flow, leading to tissue necrosis and ultimately, organ failure.2 Not surprisingly, the loss of RBC deformability is associated with the pathology of many diseases including malaria,3–6 hemoglobinopathies,4–8 and micronutrient deficiencies.9,10 Therefore, the analysis of RBC deformability presents a potential means to develop a biophysical signature for rapidly analyzing disease status and severity. A key limitation in the development of such biophysical signatures is that pathological cells often comprises of only a small subset of the overall cell population. Therefore, a large number of cells need to be tested in order to ensure sufficient sampling of the pathological cells.Traditional technologies for characterizing RBC deformability can be divided into bulk flow methods and single cell methods. Bulk flow methods, such as ektacytometry11,12 and micropore filtration,13,14 provide a measure of the average deformability of thousands of cells, but obscure information on subpopulations of diseased cells.15 Single cell techniques, such as micropipette aspiration,16,17 optical tweezers,18–20 and atomic force microscopy,21,22 measure single cells individually. However, these methods typically require complex experiments performed by trained personnel using expensive equipment,23 and therefore cannot provide sufficient throughput to measure large populations of cells in which a subset are diseased cells.
Recent advances in microfluidic mechanisms for measuring RBC deformability include approaches based on hydrodynamic stretching,24 wedging in tapered constrictions,25,26 transit time through constrictions,27,28 and transit pressure through constrictions (or the measurement of pressure required to deform single cells through constrictions).29 Hydrodynamic stretching requires precise lateral cell alignment in a flow stream, which is difficult to achieve for RBCs.24 Furthermore, cell deformability is quantified by observing the stretched cells using a high-speed camera and specialized microscopy equipment, and as a result, exclude this technique from many point-of-care applications. Wedging in tapered constrictions relies on optical measurements of the position of compressed RBCs with micrometer accuracy and therefore has limited sensitivity.25 Transit time through constrictions measures the relaxation of the RBC membrane in response to bending.27 Transit pressure through constrictions mimics the physiological transport of RBCs through the microvasculature, as well as the mechanism of splenic clearance, and is therefore potentially highly sensitive to disease pathologies.29 Both transit time and transit pressure techniques, however, rely on pushing multiple RBCs through a single micro-meter scale constriction and are therefore limited by rigid cells obstructing the constriction. This problem is especially pronounced in the analysis of RBCs infected with malaria, where increased rigidity and cytoadherence of the parasitized RBCs greatly increase the potential for obstructing the constriction. Furthermore, a key issue for all three constriction-based methods (wedging, transit time, and transit pressure) is the need to multiplex the measurement process in order to achieve sufficient throughput to profile heterogeneous RBC samples where pathological cells comprise of a small subpopulation. However, previous multiplexing mechanisms have not been able to ensure that consistent deformation pressures are applied uniformly to each constriction, and therefore limiting their ability to distinguish healthy and pathological red cells.30
To address the need for populational single-cell profiling of RBC deformability, we developed the Multiplexed Fluidic Plunger (MFP) mechanism, which deforms multiple single RBCs simultaneously through a linear array of micrometer scale funnel-shaped constrictions using a saw-tooth pressure waveform. Our key innovation is the ability to ensure each cell is deformed using an identical pressure, which is achieved through a self-compensating fluidic network that delivers a consistent pressure simultaneously to an array of constrictions irrespective of position in the array and the presence of cells in the constrictions. We apply this mechanism to determine the deformability profile of in vitro samples of RBCs infected with Plasmodium falciparum, the parasite that causes malaria, to demonstrate the potential to detect a pathological subpopulation in a heterogeneous cell sample.
Results and discussion
Mechanism principles
The principle of transit pressure measurements can be understood by considering the infusion of a single cell into a microchannel containing a constriction with a cross-section smaller than the diameter of the cell. Before the cell reaches the constriction, the applied pressure is distributed across the microchannel. Once the cell flows into the constriction, it forms a temporary seal with the constriction to block the flow of liquid. Consequently, the applied pressure focuses across the cell, effectively acting as a fluidic plunger to remotely push on the cell (Fig. 1). Varying the applied pressure while observing the position of the cell enables the measurement of the pressure required to push the cell through the constriction. The cross-section of the constriction is selected to allow the cell to establish a temporary seal against the constriction. For RBCs, a 1.5–2.0 μm wide constriction with a thickness of 3.0–3.7 μm was found to be appropriate.1,29,31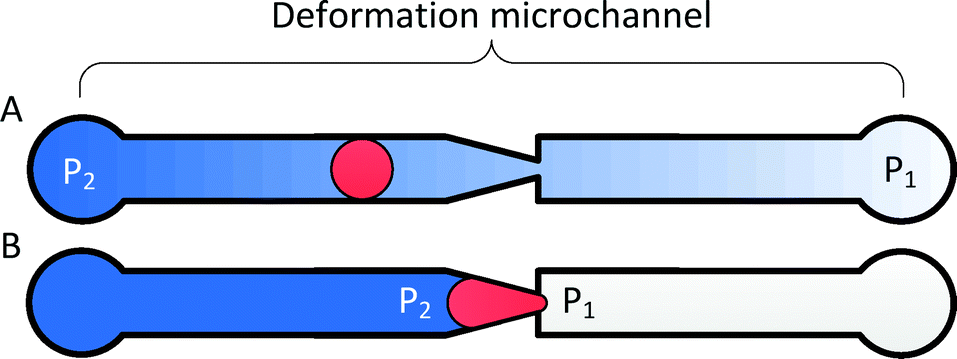 | ||
| Fig. 1 The fluidic plunger mechanism. (A) When a cell is not trapped in a constriction, the applied pressure (P2–P1) is distributed across the deformation microchannel; (B) when a cell is trapped in a constriction, the applied pressure focuses across the cell. | ||
To multiplex and automate this process, RBCs are deformed in a parallel array of deformation microchannels using a saw-tooth pressure waveform (Fig. 2A–B). The deformation microchannels each contains a funnel-shaped constriction at its entrance and is collectively fed by a loading microchannel. At the start of the measurement process, single RBCs are loaded into the mouth of each constriction at a modest pressure that is insufficient for them to transit. The presence of this cell blocks fluid flow into its residing deformation microchannel and prevents other cells from loading into the constriction. In rare instances, two RBCs are simultaneously loaded into the same deformation microchannel and are excluded from measurement during the data analysis. Once the majority of the constrictions are loaded with cells, a saw-tooth pressure waveform is applied while simultaneously recording a video of the deformation process (ESI† Video S1). The threshold transit pressure is then determined by relating the position of the cells with the pressure–time data of the saw-tooth waveform.
 | ||
| Fig. 2 Cell loading and pressure measurement process. (A) To measure the threshold deformation pressure, single cells are first loaded into the funnel constrictions at a modest pressure; (B) a saw-tooth pressure waveform is then applied and the threshold deformation pressure is determined by relating the position of the cell with the pressure–time waveform; (C–D) a key challenge is the multiplexing error caused by variation in the streamlines of the loading microchannel with constriction occupancy, which results in an inconsistency in PD. | ||
A key challenge to obtain consistent threshold pressure measurements is the application of a consistent pressure across multiple deformation microchannels when different numbers of funnel constrictions are occupied with cells. This phenomenon can be understood by considering fluid flow in the following two situations: 1) when the constrictions contain no cells, streamlines in the loading microchannels are evenly distributed across the deformation microchannels (Fig. 2C). 2) When one or more of the funnel constrictions are occupied with cells that block fluid flow in that channel, streamlines in the loading microchannel are skewed to feed fluid into the remaining unblocked deformation microchannels (Fig. 2D). The difference in the combined loading and deformation microchannel hydrodynamic resistances between these two situations causes an inconsistency in the resulting pressure across deformation microchannels (PD).
To estimate the potential error in the magnitude of PD for a device with N deformation microchannels, we consider the worst-case pressure error, which occurs between when the deformation microchannels are occupied with only a single cell and when the deformation microchannels are completely filled with cells. The pressures measured across the deformation microchannels in these two situations can be estimated as follows:
1) Deformation microchannels occupied with a single cell:
 | (1) |
2) Deformation microchannels completely occupied:
| PD,N = PCD | (2) |
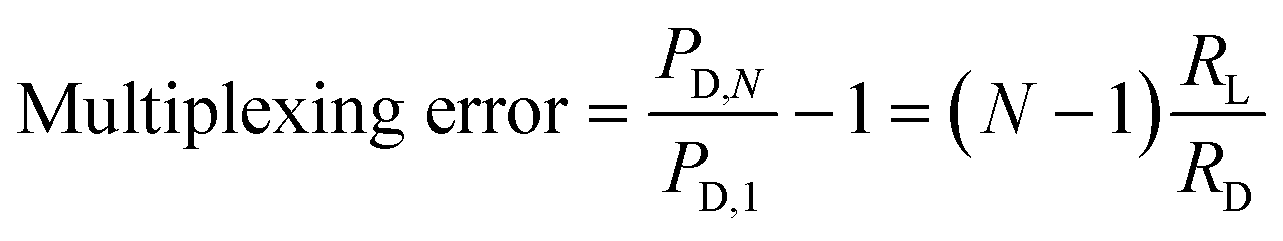 | (3) |
Therefore, maximizing RD/RL based on a desired number of parallel deformation microchannels minimizes the multiplexing error. Since significant natural variability exists for most biological systems, a multiplexing error of less than 3% is considered to be sufficient to observe most pathological effects.
Device design
The multiplexed fluidic plunger device is a single layer PDMS microfluidic device consisting of parallel deformation microchannels, bypass microchannels, loading microchannels, and inlet microchannels, where the geometries of these microchannels are designed to ensure that a consistent and precisely controlled pressure is simultaneously applied to all deformation microchannels (Fig. 3 and 4).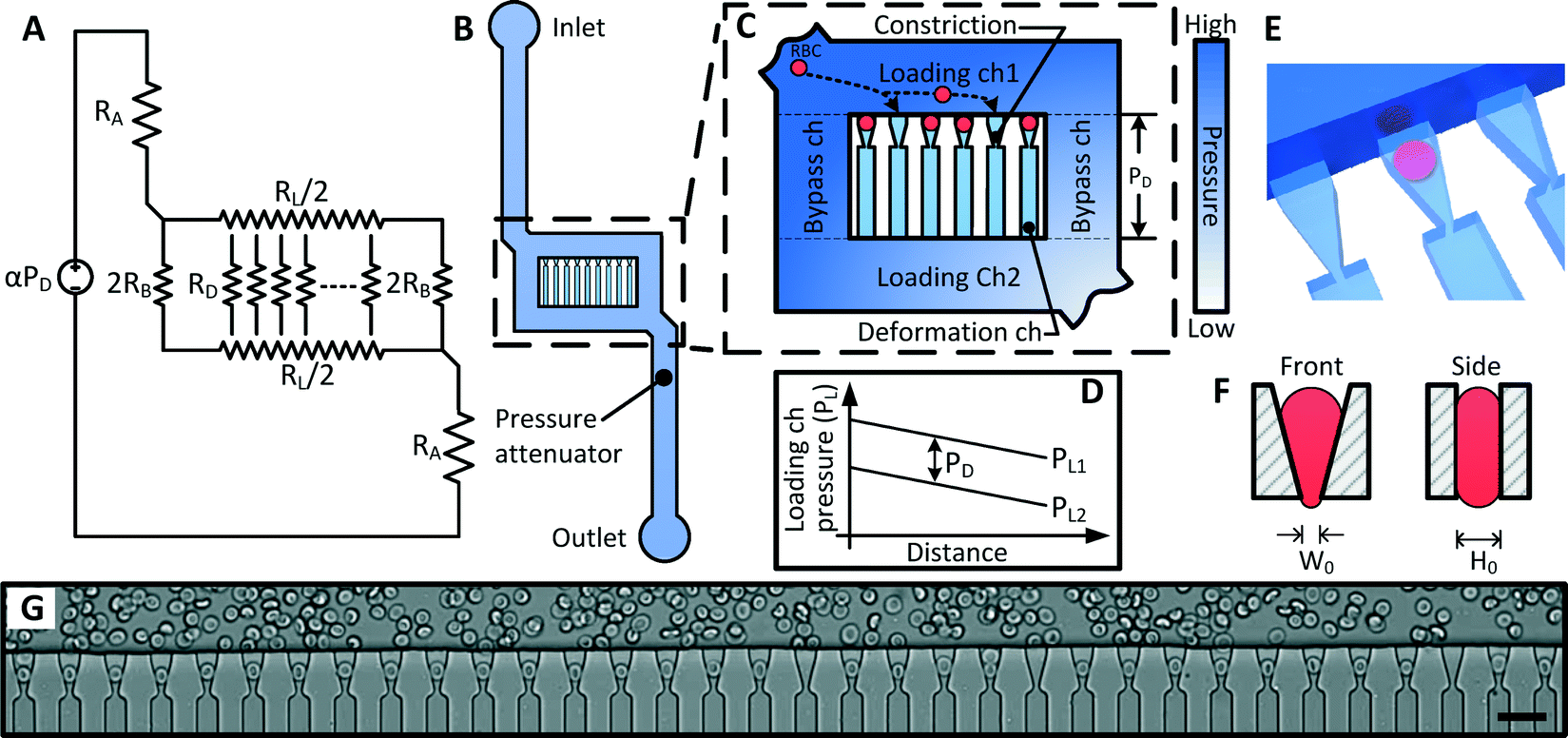 | ||
| Fig. 3 Design of the Multiplex Fluidic Plunger (MFP) device. (A) Equivalent hydrodynamic circuit of the MFP device, where α is the pressure divider ration, PD is the deformation pressure, RA, RB, RL and RD are the hydrodynamic resistance of the pressure attenuator, bypass, loading and deformation microchannels respectively; (B) structure of the MFP device; (C) magnified view of the microchannel array showing the deformation, loading and bypass microchannels; (D) pressure in the two Loading microchannels (PL) as a function of position. The difference between these pressure profiles is the pressure across the deformation microchannel (PD), which remains constant; (E) 3D model of the loading and deformation microchannels showing the RBC in the planar position inside the deformation microchannel; (F) schematic of the front and side view of a loaded constriction; (G) Micrograph of deformation microchannels at the opening of the constrictions (scale bar = 20 μm). | ||
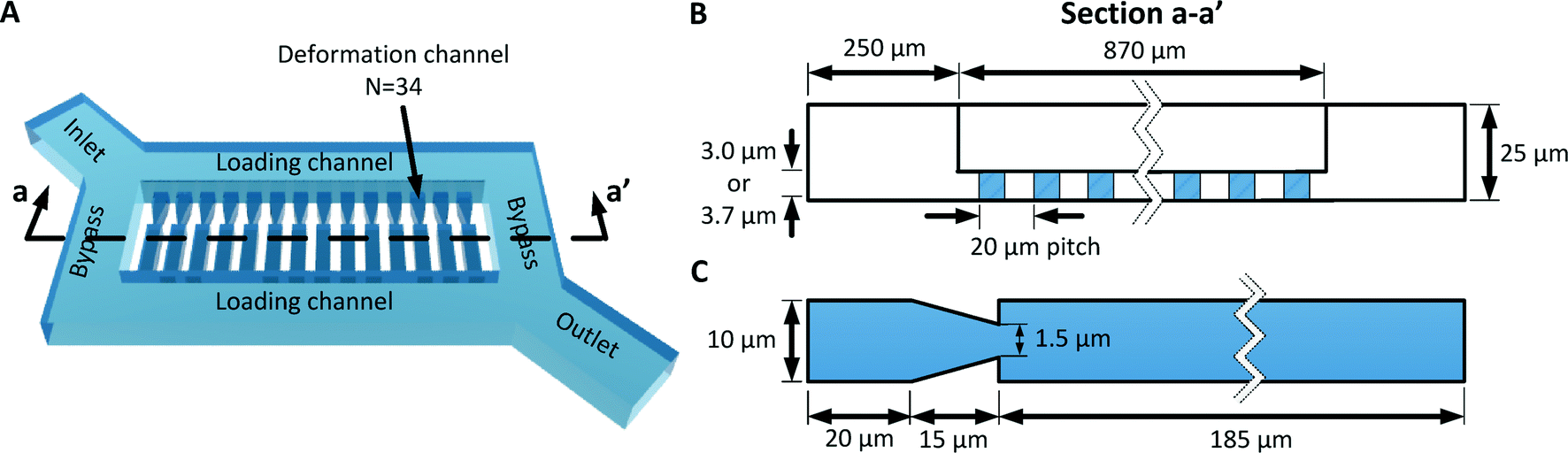 | ||
| Fig. 4 (A) 3D model of the MFP device. (B) Cross section of the device showing the geometry of the bypass and deformation microchannels. (C) Detailed design of the deformation microchannel. | ||
As discussed in the previous section, the deformation microchannel consists of a single constriction in a much longer microchannel. The length of the microchannel is selected to maximize its hydrodynamic resistance relative to the loading microchannel in order to minimize multiplexing error according to eqn (3). The geometry of the deformation microchannel is also selected to match the intended cell sample. For RBCs, the thickness of the deformation microchannels is selected to be similar to the thickness of the RBCs to orientate the cells into the planar configuration as they deform through the constrictions (Fig. 3E, F). Indeed, if the deformation microchannel is too thick, the RBCs would rotate to a perpendicular orientation to the plane of the microchannel and could transit through the funnel constriction without creating a temporary seal required for the fluidic plunger effect. In our studies, normal human RBCs are tested using a microchannel thickness of 3.0 μm. While a deformation microchannel thickness of 3.7 μm was used for RBCs parasitized by P. falciparum, which may contain altered membranes and irregular bulges.32 The maximum number of deformation microchannels is limited by the field of view of the microscopy equipment since all the deformation microchannels must be simultaneously visualized in order to identify the threshold pressure of each individual RBC. For the purposes of the experimental validation, a prototype containing 34 channels was developed.
The purpose of the bypass microchannel is to provide a microchannel parallel to the deformation microchannels with significantly smaller hydrodynamic resistance in order to dictate the pressure applied across the deformation microchannels (Fig. 3A). Specifically, we selected the hydrodynamic resistance of the bypass microchannels to be ~0.002 times the combined hydrodynamic resistance of the deformation microchannels (Table 1). Additionally, the bypass microchannel combines with the inlet microchannels to attenuate pressure applied from an external source. Typical pressures required to deform single RBCs through a 1.5 to 2 μm funnel-shaped constriction range between 1 to 25 Pa.31 Such small pressures are extremely difficult to generate reliably using external instrumentation and therefore require additional fluidic circuitry to produce the necessary pressure on-chip. The pressure divider fluidic circuit, used previously by others,33,34 produces an attenuated pressure from an external source using a segment of a long microchannel. For this device, the long microchannel is the inlet microchannel while the segment is the bypass microchannel (Fig. 3A). Therefore, the attenuation factor (α) is the ratio of the hydrodynamic resistances of the bypass microchannel (RB) and inlet microchannels (RA) as
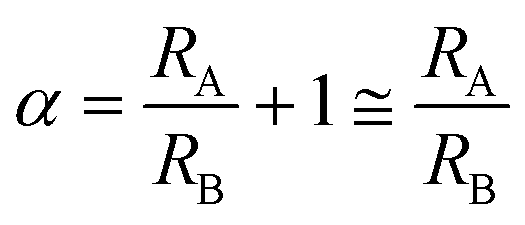 | (4) |
| Microchannel | Parameter | Value | Unit |
|---|---|---|---|
| Loading | R L | 5.70E + 12 | Pa s m−3 |
| Bypass | R B | 3.60E + 11 | Pa s m−3 |
| Deformation | R D | 7.80E + 15 | Pa s m−3 |
| N | 34 | ||
| N R B/RD | 0.20% | ||
| R L/RD | 0.07% | ||
| Multiplexing error | 2.42% |
The purpose of the loading microchannel is to infuse RBCs into the mouth of the deformation microchannels (Fig. 3G). As discussed in the previous section, the magnitude of the multiplexing error is determined by the ratio of the hydrodynamic resistance of the loading microchannel and the deformation microchannels. Therefore, it is desirable to decrease the resistance of the loading microchannels as much as practically feasible. However, if the resistance of the loading microchannel is too small, then the relative fluid flow into the deformation microchannel will be too small and the time to load the deformation microchannels with RBCs will be unreasonably long. In practice, for a device with 34 parallel deformation channels, we found that a RL/RD ratio of 0.0007 reduced the multiplexing error to <3% (eqn (3), Table 1) and allowed for RBCs to be loaded into the deformation microchannel in a reasonable amount of time.
The bifurcation of the sample flow in the loading and bypass microchannels around the deformation microchannels also performs the important function of ensuring that an identical pressure is applied simultaneously to all deformation microchannels. Specifically, since the inlet of the deformation microchannels are spatially separated along the loading microchannels, the pressure at these inlet points will vary along the loading microchannel as shown in Fig. 3D. However, since the outlet of the deformation microchannel is also distributed along another loading microchannel with matched geometry, the pressure difference across all of the deformation microchannels are kept at a constant value of PD. The pressure distribution in the loading and bypass microchannels has been modeled using a finite element model, which confirmed the consistency of the pressure difference across the deformation microchannels (ESI† Fig. S1 and S2).
In summary, the deformation microchannels are designed to constrain the RBCs and deform them through a constriction. The bypass and the inlet microchannels are designed to attenuate an external pressure and apply it across the deformation microchannels. The loading microchannels are designed to minimize multiplexing error and allow RBCs to be loaded into the entrance of the deformation microchannels. Finally, bifurcation flow around the deformation microchannels ensures that a consistent pressure is applied across the deformation microchannels irrespective of the position of the deformation microchannel. The detailed geometries of these microchannels are shown in Fig. 4 while key design parameters are summarized in Table 1.
Measurement process and data processing
Threshold deformation pressure measurements involve initially filling the device with buffer fluid by pressurizing the outlet reservoir. Once the device is filled, the sample is pipetted into the inlet reservoir. Next, a small pressure, insufficient for the RBCs to transit the constrictions, is applied to load the cells into the entrance of the constriction. Once most of the constrictions are occupied by RBCs, the applied pressure is incrementally increased while recording a video of the deformation process. The experimental setup requires <10 minutes, while the process of infusing RBCs in to the 34 deformation channels and then applying the deformation pressure requires ~1 minute.The threshold deformation pressure is determined from the recorded video and pressure–time data. Video analysis software was developed to perform threshold pressure measurements in a semi-automated fashion by converting the recorded videos of the deformation process into a rapidly human readable format. To reliably detect the deformation of single cells through the constrictions, the position of the constrictions must be first detected to accommodate small variations in the position and angle within the camera's field of view. To register the position of the funnel constrictions, the boundaries of the device are detected using the distinct lines of the device to create a smaller area for refined device position registration. To achieve acceptable alignment with the expected cell transit region, small alignment markers on either side of the deformation microchannels are detected. In the event that these side markers are not visible due to poor focus the user can also manually align the device. To generate the human readable view, the critical points in each funnel are converted into their respective intensity values and graphed on a color graded chart representing the intensity over time (ESI† Fig. S1). Because cells transiting the constriction create an abnormality in the intensity of the constriction, the transit of a cell is very apparent. Additionally, this process helps to identify when cells become too rigid to deform and are stuck at the funnel constriction. Coupled with the displayed graph is a cursor-driven live-updating video viewer, which dynamically focuses a near zoomed field of view and allows the user to quickly look through the video in search of the point where each RBC transits through the constriction to record the corresponding applied pressure.
Mechanism evaluation
To experimentally validate the ability of the MFP mechanism to eliminate the multiplexing error, we measured the threshold deformation pressures from nearly empty (defined as ≤10% funnels occupied) and nearly full (defined as ≥80% funnels occupied) funnel arrays using identical fresh RBC samples. The distributions of the threshold pressures from these two cases are statistically identical (p = 0.45, Fig. 5A), which confirms the elimination of the multiplexing error.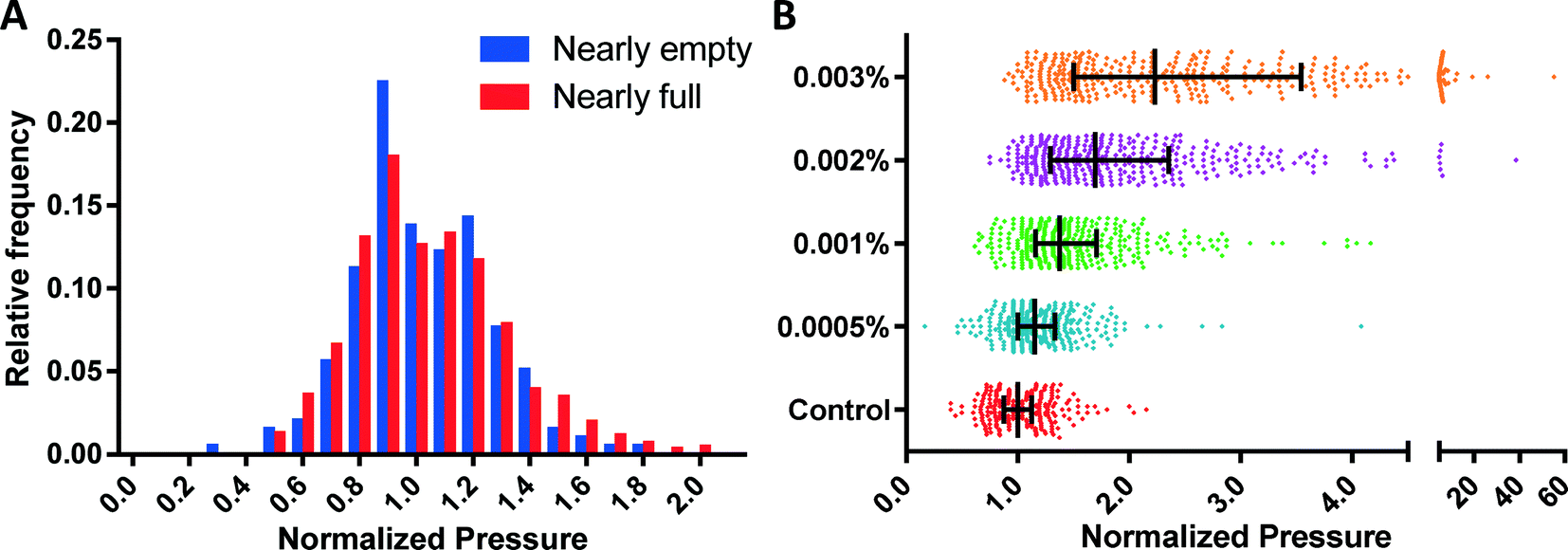 | ||
| Fig. 5 MFP mechanism validation. (A) Distribution of measured deformation pressures with the funnel array nearly empty (<10% occupancy, N = 196) and nearly full (>80% occupancy, N = 864), which show no distinction (P = 0.45); (B) sensitivity of MFP device tested using glutaraldehyde treatment of RBCs. Measured values are normalized to the median of the control with N ≥ 335 at each test condition. | ||
The sensitivity of the MFP mechanism was established by measuring the deformability profiles of RBC samples treated with small amounts of glutaraldehyde (GTA). GTA is a common fixative agent, which induces cross-linking and stabilization of proteins in the red blood cell membrane and thus artificially reduces their deformability in a concentration dependent manner.35,36 Control and GTA treated-RBCs were measured using the same device. The RBC deformability patterns obtained (Fig. 5B) using the MFP device can reliably differentiate between control and 0.0005% GTA-treated RBCs (p < 0.005), which is similar to or better than the sensitivities of ektacytometry and other microfluidic methods.12,27,31
Deformability profiling of RBCs parasitized by P. falciparum from in vitro cultures
The key strength of the MFP mechanism is its ability to measure multiple single cells simultaneously to perform a robust, high-throughput profiling of a RBC population. This capability enables the detection and analysis of subsets of pathologic cells, which is precisely the situation in malaria, where parasitized RBCs typically account for a fraction of the overall population. We initially verified the reduced deformability of infected RBCs (iRBCs) grown in vitro by separately testing the uninfected and infected RBC sample. Purified iRBCs were obtained using magnetic separation, which preferentially selects for the advanced stages (trophozoite and schizont) of infection. Expectedly, these iRBCs were significantly less deformable than control RBCs (p < 0.0001) obtained from the same donor (Fig. 6A).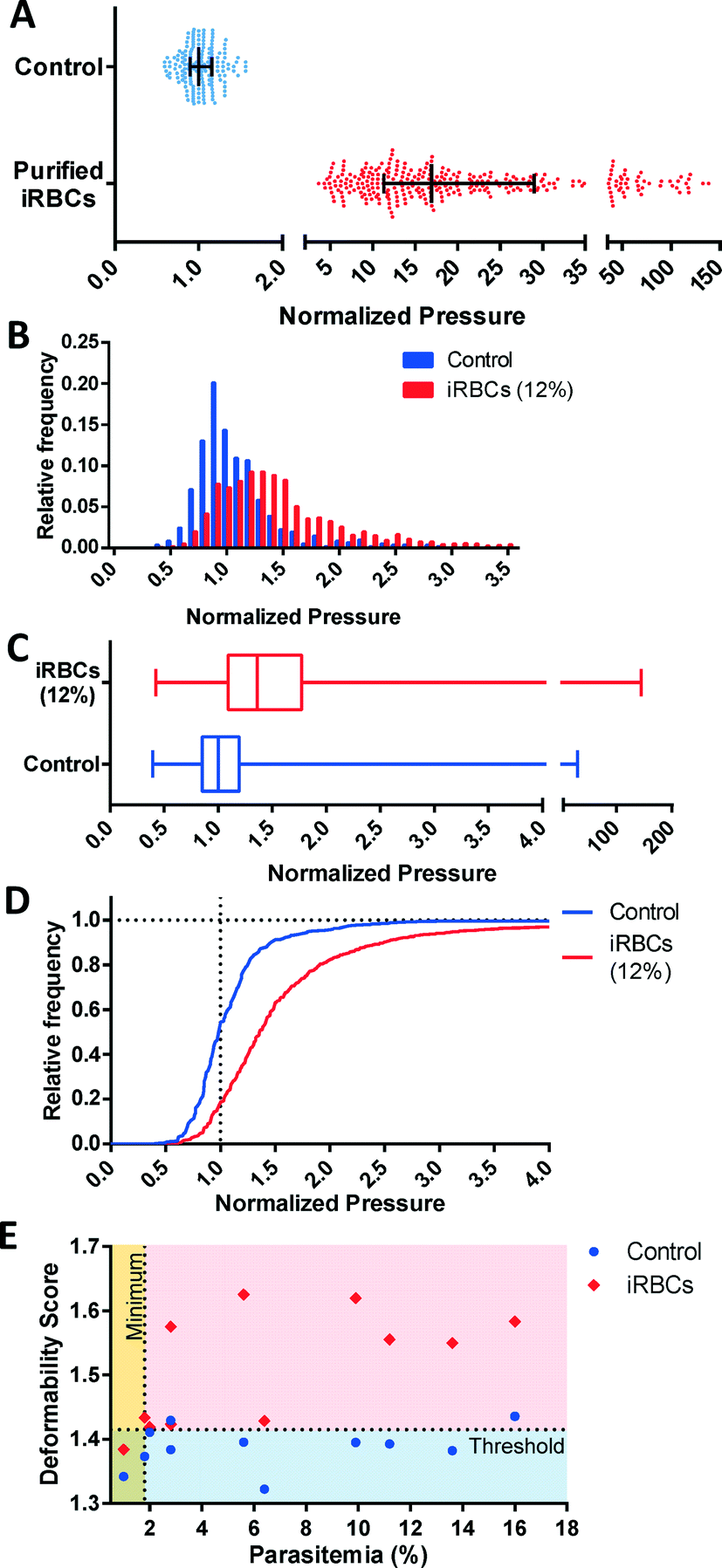 | ||
| Fig. 6 Deformability profiles of RBCs parasitized by P. falciparum from in vitro cultures. (A) Bar graph showing a decrease of deformability of purified iRBCs relative to fresh RBCs from healthy donors (N = 177 for control, N = 300 for purified iRBC, p < 0.0001). (B) Histogram, (C) Box plot and (D) Cumulative histogram for iRBCs (12% parasitemia) and control red blood cells used for the parasite culture (N = 1609 for 12% iRBCs, N = 622 for control, p < 0.0001). In each case, the measured pressures are normalized to the median of the control. (E) Deformability score as a function of parasitemia for iRBCs (N ≥ 527) and control samples (N ≥ 301). Control samples are uninfected red blood cells used to culture each iRBC sample. These results indicate a DS = 1.415 detects malaria infections with 82% specificity at 1.8% minimum detectable parasitemia. | ||
To investigate the potential to use multiplexed single-cell deformability profiling to detect malaria infection in vitro, the deformability profiles of iRBC samples were tested at various parasitemia levels. The deformability profile for a typical iRBC sample at 12 ± 1% parasitemia is shown relative to control in Fig. 6B–D. The control sample is the unexposed red cells used to feed the parasite culture. The iRBC distribution is clearly distinguishable from control (p < 0.0001) with a greater median pressure and wider distribution. The control deformability profile appears to be a balanced normal distribution while the iRBC profile appears to also be approximately normal with a rightward skew because of the presence of rigidified RBCs from infection. Subtracting the iRBC profile from the control profile shows the rigidified RBCs comprises of greater than the fraction of cells expected from the 12% parasitemia sample, which suggest that uninfected RBCs have also been rigidified by the iRBCs. This biophysical modification has been previously observed by others and likely arises from the release of free heme into the culture as schizonts rupture, which induces oxidative stress on the RBC membrane.37–40
At <10% parasitemia, the deformability profile between iRBC and control becomes more similar and the rigidification of uninfected RBCs becomes an increasingly larger confounding factor. Consequently, simple statistical parameters, such as mean and median of the overall population are less likely to be affected by the presence of the subpopulation of iRBCs. To detect the presence of iRBCs in these situations, the Deformability Score (DS) parameter is created to evaluate the more rigid segment of the measured cells. Specifically, DS is defined as follows,
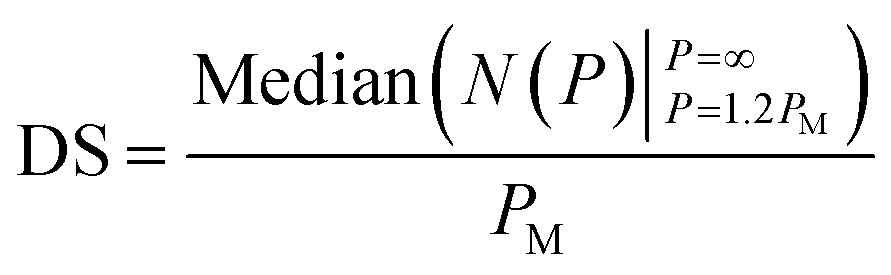 | (5) |
| PM = Median(N(P)|P=∞P=0) | (6) |
The relationship between DS and parasitemia for infected and control samples are shown in Fig. 6E. The control samples are unexposed red cells used in each iRBC culture and tested on the same day. These results show a reasonable separation between infected and uninfected samples with DS = 1.415 corresponding to a detection specificity of 82% and a minimum detectable parasitemia at 1.8%. While this detection threshold is at a higher parasitemia than clinical malaria cases, which often have parasitemia levels less than 1%, the ability to profile iRBC samples at this parasitemia is nonetheless useful for assessing the properties of in vitro malaria samples, as well as the their response to antimalarial drugs. Future improvements to our measurement methodology and device design aim to further increase measurement throughput in order to reach clinically relevant parasitemia levels.
Conclusions
We described the multiplexed fluidic plunger mechanism for measuring the mechanical deformability of individual red blood cells. The key innovation of this work is the ability to apply a precisely controlled pressure to multiple single red blood cells simultaneously in order to squeeze them through micrometer-scale constrictions to measure their deformability. This capability enables the profiling of a heterogeneous cell sample where pathological cells comprise of a small subpopulation of the overall sample, and thus provide a promising approach for establishing the biophysical signature for diseases that affect the deformability of RBCs and other cells.Methods
Microfabrication
Molds for the microfluidic devices were fabricated on silicon wafer substrates using photolithography of two different thicknesses of SU8 photoresist. The deformation microchannels were fabricated using SU-8 3005 photoresist (MicroChem) thinned with cyclopentanone at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume. The remaining microstructures were fabricated using SU-8 3025 with alignment marks first created using SU-8 2015. The patterns for the microstructures were drawn using DraftSight. After fabrication, the thicknesses of the microstructures are confirmed using a profilometer (Alpha Step 200).
:1 by volume. The remaining microstructures were fabricated using SU-8 3025 with alignment marks first created using SU-8 2015. The patterns for the microstructures were drawn using DraftSight. After fabrication, the thicknesses of the microstructures are confirmed using a profilometer (Alpha Step 200).
Soft-lithography
Microfluidic devices were made using replica molding of Polydimethylsiloxane (PDMS) silicon. Replicas of the microfabricated silicon wafers were first made using a polyurethane-based plastic (Smooth-Cast ONYX, Smooth-On) as described by Desai et al.41 Holes were punched for the fluidic reservoirs into the PDMS master using a 6 mm hole punch (Technical Innovations). Sylgard-184 PDMS (Ellsworth Adhesives), mixed at a ratio of 10:1 (w/w) base to hardener, was poured into the mold to fabricate the microfluidic device. The pre-cured PDMS was degassed in a vacuum desiccator for 15 minutes prior to baking for 2 hours at 65 °C.
To prevent RBCs from sticking to the glass slide, the device was bonded onto a thin PDMS surface, made by spin-coating RTV615 PDMS (Momentive Performance Material) at a ratio of 10:1 (w/w) base to hardener, onto a blank wafer. The layer was baked at 65 °C for 1 hour. The device and the PDMS coated wafer were then exposed to oxygen plasma (Model PDC-001, Harrick Plasma) for 75 s and then joined to create a permanent covalent bond between them. To strengthen the bond, the device was further baked for 15 minutes at 65 °C, after which, the resulting device was peeled off and bonded onto a standard microscope slide (Fisher Scientific) using the same process.
Cell sample preparation
Whole blood was collected into 6 ml BD EDTA vacutainer tubes from healthy donors with informed consent. In some cases, a droplet of whole blood was collected using a finger-prick lancet (Unistik 3, Owen Mumford, Fisher). Blood was diluted to 30% (vol/vol) in Phosphate Buffered Saline (PBS, Gibco) with 0.2% (wt/vol) Pluronic F127 solution (Sigma).For device sensitivity experiments, whole blood from a single donor was first diluted to 5% (vol/vol) in PBS. Glutaraldehyde was added at concentrations of 0.0005, 0.001, 0.002 and 0.003% (vol/vol) and incubated for 30 minutes.
In vitro cultures of Plasmodium falciparum were prepared as described by Radfar, et al.42 Briefly, RBCs were washed, infected with P. falciparum (3D7 strain) and incubated in a hypoxic incubator (5% O2 and 6% CO2) at 37 °C. The culture was maintained by adding RBC and RPMI-1640 culture media (Invitrogen) implemented with 25 mM HEPES (Sigma), 0.5% (wt/vol) AlbuMAX I (Life Technologies), 100 μM hypoxanthine (Sigma), 12.5 μg ml−1 gentamicin (Sigma) and 1.77 mM sodium bicarbonate (Sigma) on alternating days. RBCs for culturing were obtained from donors (8 in total) with informed consent by the Canadian Blood Services’s Networked Centre for Applied Development and stored in a standard blood bag. To create samples with very low parasitemia, infected blood samples were diluted using uninfected blood.
Parasitemia was measured using Giemsa staining (Sigma-Aldrich).43 Briefly, blood samples were spread onto a microscope slide, fixed using methanol, and washed using DI water. Giemsa staining and PBS were mixed in a 1:5 volume ratio and applied on the RBCs for 20 minutes. The stain was removed and the slide was washed with DI water. The parasitemia was determined by counting ~1000 cells using a 100× oil immersion objective (Nikon).
To obtain purified infected RBCs (iRBCs), P. falciparum cultures were first washed using culture media and then added to a LS column (Miltenyl Biotec) surrounded by Neodymium Super Magnets (Applied Magnets).44 The late-stage iRBCs, i.e. the late-trophozoites and schizonts, were held in the column due to the presence of hemozoin (iron-containing by-product of the hemoglobin produced by the parasite).45 Next, the magnets were removed and the remaining cells are extracted from the column using a syringe and added to buffer prior to deformability measurements.
Experimental apparatus and protocol
Inlet and outlet areas of the microfluidic device are punched with 6 mm diameter holes to serve as sample and buffer reservoirs. Female luer-lock connectors are inserted into these reservoirs to form a water and airtight seal. The reservoirs are pressured using the MFCS-2C (Fluigent SA) pneumatic pressure control system through 0.5 mm ID flexible Tygon tubing (Cole-Parmer). This pressure control system is capable of generating precise pressure with a resolution of 0.25 mbar (25 Pa) and a range of 1000 mbar. Pressure measurements are performed with the microfluidic device mounted on an inverted microscope (Nikon Ti-U) while observed using a 20× objective and a 1.45 megapixel Digital CCD camera (QIClick-F-M-12, QImaging). An in-house data acquisition software was developed in which the field of view of the microscope and the applied pressures of the different ports are simultaneously visualized.Acknowledgements
This work was made possible by funding from the Canadian Institutes of Health Research, Canadian Blood Services, Health Canada, and the Natural Sciences and Engineering Research Council of Canada. The authors would like to acknowledge Dr. Petra Rohrbach and Sarah Reiling for help with the malaria culture, Dr. Mark Scott and Dr. Boris Stoeber for helpful discussions, and Hantao Yuan for helping to prepare some of the figures.References
- J. M. Kwan, Q. Guo, D. L. Kyluik-Price, H. Ma and M. D. Scott, Am. J. Hematol., 2013, 88, 682–689 CrossRef CAS PubMed.
- S. Yedgar, A. Koshkaryev and G. Alexander, Pathophysiol. Haemostasis Thromb., 2002, 32, 263–268 CrossRef CAS PubMed.
- B. M. Cooke, N. Mohandas and R. L. Coppel, Semin. Hematol., 2004, 41, 173–188 CrossRef PubMed.
- F. C. Mokken, M. Kedaria, C. P. Henny, M. R. Hardeman and A. W. Gelb, Ann. Hematol., 1992, 64, 113–122 CrossRef CAS.
- G. Y. H. Lee and C. T. Lim, Trends Biotechnol., 2007, 25, 111–118 CrossRef CAS PubMed.
- J. Stuart and G. B. Nash, Blood Rev., 1990, 4, 141–147 CrossRef CAS.
- M. W. Kenny, M. Meakin, D. J. Worthington and J. Stuart, Br. J. Haematol., 1981, 49, 103–109 CrossRef CAS PubMed.
- D. K. Wood, A. Soriano, L. Mahadevan, J. M. Higgins and S. N. Bhatia, Sci. Transl. Med., 2012, 4, 123ra26 Search PubMed.
- R. Yip, M. R. Clark, S. Jain, S. B. Shohet and P. R. Dallman, Blood, 1983, 62, 99–106 CAS.
- A. Vayá, M. Simó, M. Santaolaria, J. Todolí and J. Aznar, Clin. Hemorheol. Microcirc., 2005, 33, 75–80 Search PubMed.
- Z. Wen, Z. Yan, L. Song, H. Dou, D. Sun, Z. Lü, Y. Shi and H. Xiao, Sci. China, Ser. C: Life Sci., 1998, 41, 195–202 CrossRef CAS PubMed.
- O. K. Baskurt, M. R. Hardeman, M. Uyuklu, P. Ulker, M. Cengiz, N. Nemeth, S. Shin, T. Alexy and H. J. Meiselman, Biorheology, 2009, 46, 251–264 Search PubMed.
- P. F. Leblond and L. Coulombe, J. Lab. Clin. Med., 1979, 94, 133–143 CAS.
- T. L. Berezina, S. B. Zaets, C. Morgan, C. R. Spillert, M. Kamiyama, Z. Spolarics, E. A. Deitch and G. W. Machiedo, J. Surg. Res., 2002, 102, 6–12 CrossRef CAS PubMed.
- J. Stuart, J. Clin. Pathol., 1985, 38, 965–977 CrossRef CAS.
- G. B. Nash, E. O’Brien, E. C. Gordon-Smith and J. A. Dormandy, Blood, 1989, 74, 855–861 CAS.
- R. P. Hebbel, A. Leung and N. Mohandas, Blood, 1990, 76, 1015–1020 CAS.
- J. P. Mills, L. Qie, M. Dao, C. T. Lim and S. Suresh, Mech. Chem. Biosyst., 2004, 1, 169–180 CAS.
- M. Dao, C. T. Lim and S. Suresh, J. Mech. Phys. Solids, 2003, 51, 2259–2280 CrossRef PubMed.
- S. Hénon, G. Lenormand, A. Richert and F. Gallet, Biophys. J., 1999, 76, 1145–1151 CrossRef.
- I. Dulińska, M. Targosz, W. Strojny, M. Lekka, P. Czuba, W. Balwierz and M. Szymoński, J. Biochem. Biophys. Methods, 2006, 66, 1–11 CrossRef PubMed.
- M. Lekka, M. Fornal, G. Pyka-Fościak, K. Lebed, B. Wizner, T. Grodzicki and J. Styczeń, Biorheology, 2005, 42, 307–317 Search PubMed.
- M. Musielak, Clin. Hemorheol. Microcirc., 2009, 42, 47–64 CAS.
- D. R. Gossett, H. T. K. Tse, S. A. Lee, Y. Ying, A. G. Lindgren, O. O. Yang, J. Rao, A. T. Clark and D. Di Carlo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7630–7635 CrossRef CAS PubMed.
- S. C. Gifford, M. G. Frank, J. Derganc, C. Gabel, R. H. Austin, T. Yoshida and M. W. Bitensky, Biophys. J., 2003, 84, 623–633 CrossRef CAS.
- X. Sun, W. D. Weinlandt, H. Patel, M. Wu and C. J. Hernandez, Lab Chip, 2014, 14, 2491–2498 RSC.
- H. Bow, I. V. Pivkin, M. Diez-Silva, S. J. Goldfless, M. Dao, J. C. Niles, S. Suresh and J. Han, Lab Chip, 2011, 11, 1065 RSC.
- M. J. Rosenbluth, W. A. Lam and D. A. Fletcher, Lab Chip, 2008, 8, 1062–1070 RSC.
- Q. Guo, S. J. Reiling, P. Rohrbach and H. Ma, Lab Chip, 2012, 12, 1143–1150 RSC.
- T. Herricks, M. Antia and P. K. Rathod, Cell. Microbiol., 2009, 11, 1340–1353 CrossRef CAS PubMed.
- Q. Guo, S. P. Duffy, K. Matthews, A. T. Santoso, M. D. Scott and H. Ma, J. Biomech., 2014, 47, 1767–1776 CrossRef PubMed.
- A. Esposito, J.-B. Choimet, J. N. Skepper, J. M. A. Mauritz, V. L. Lew, C. F. Kaminski and T. Tiffert, Biophys. J., 2010, 99, 953–960 CrossRef CAS PubMed.
- Q. Guo, S. Park and H. Ma, Lab Chip, 2012, 12, 2687–2695 RSC.
- Q. Guo, S. M. McFaul and H. Ma, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 051910 CrossRef.
- M. Komorowska, M. Koter, G. Bartosz and J. Gomułkiewicz, Biochim. Biophys. Acta, 1982, 686, 94–98 CAS.
- T. L. Steck, J. Mol. Biol., 1972, 66, 295–305 CrossRef CAS.
- J. P. Mills, L. Qie, M. Dao, K. S. W. Tan, C. T. Lim and S. Suresh, MRS Online Proc. Libr., 2004, 844, 179–184 CAS.
- A. M. Dondorp, P. A. Kager, J. Vreeken and N. J. White, Parasitol. Today, 2000, 16, 228–232 CrossRef CAS.
- F. Nuchsongsin, K. Chotivanich, P. Charunwatthana, O. S. Fausta, D. Taramelli, N. P. Day, N. J. White and A. M. Dondorp, Am. J. Trop. Med. Hyg., 2007, 77, 617–622 Search PubMed.
- F. Omodeo-Salè, A. Motti, N. Basilico, S. Parapini, P. Olliaro and D. Taramelli, Blood, 2003, 102, 705–711 CrossRef PubMed.
- S. P. Desai, D. M. Freeman and J. Voldman, Lab Chip, 2009, 9, 1631–1637 RSC.
- A. Radfar, D. Méndez, C. Moneriz, M. Linares, P. Marín-García, A. Puyet, A. Diez and J. M. Bautista, Nat. Protoc., 2009, 4, 1899–1915 CAS.
- K. Moll, I. Ljungström, H. Perlmann, A. Scherf and M. Wahlgren, Methods in Malaria Research, MR4/ATCC, Manassas, 5th edn, 2008 Search PubMed.
- C. C. Kim, E. B. Wilson and J. L. DeRisi, Malar. J., 2010, 9, 17–21 CrossRef PubMed.
- S. Hackett, J. Hamzah, T. M. E. Davis and T. G. St Pierre, Biochim. Biophys. Acta, Mol. Basis Dis., 2009, 1792, 93–99 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc01100g |
| This journal is © The Royal Society of Chemistry 2015 |