
Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer's disease†
JiSoo
Park‡
a,
Bo Kyeong
Lee‡
b,
Gi Seok
Jeong
b,
Jung Keun
Hyun
cd,
C. Justin
Lee
ae and
Sang-Hoon
Lee
*ab
aKU-KIST Graduate School of Converging Science and Technology, Korea University, Seoul 136-701, Republic of Korea
bSchool of Biomedical Engineering, College of Health Science, Korea University, Seoul 136-703, Republic of Korea. E-mail: dbiomed@korea.ac.kr; Fax: +82 2 921 6818; Tel: +82 2 940 2881
cDepartment of Nanobiomedical Science and BK21 PLUS NBM Global Research Center for Regenerative Medicine, Institute of Tissue Regeneration Engineering (ITREN), Republic of Korea
dDepartment of Nanobiomedical Science, Dankook University, Cheonan 330-714, Republic of Korea
eCenter for Neuroscience, Brain Science Institute, Korea Institute of Science and Technology (KIST), Seoul, Republic of Korea
First published on 29th September 2014
Abstract
There has been a growing need for in vitro models of neurodegenerative diseases such as Alzheimer's disease that would enable a better understanding of etiology and faster development of treatment strategies. However, meeting this demand has been held back by the limited ability to mimic the in vivo microenvironment in an in vitro system. Here, we developed a microfluidic chip based on three-dimensional (3D) neurospheroids that more closely mimics the in vivo brain microenvironment by providing a constant flow of fluid that is readily observed in the interstitial space of the brain. Uniform neurospheroids, with cell–cell interactions and contacts in all directions, were formed in concave microwell arrays, and a slow interstitial level of flow was maintained using an osmotic micropump system. Using this platform, we investigated the effect of flow on neurospheroid size, neural network, and neural differentiation. Neurospheroids cultured with flow were larger and formed more robust and complex neural networks than those cultured under static conditions, suggesting an effect of the interstitial level of slow and diffusion-dominant flow on continuous nutrient, oxygen, and cytokine transport and removal of metabolic wastes. We also tested the toxic effects of amyloid-β, which is generally considered to be the major contributor in Alzheimer's disease. Amyloid-β treatment via an osmotic micropump significantly reduced the viability of neurospheroids and caused a significantly more destruction of neural networks, compared to the amyloid-β treatment under static conditions. By adding in vivo-like microenvironments, we propose this 3D culture-based microfluidic chip as an in vitro brain model for neurodegenerative disease and high-throughput drug screening.
1. Introduction
Alzheimer's disease (AD), classified as a serious brain disorder, is one of the most costly diseases to society and imposes an enormous burden on caregivers. Because AD is an age-related neurodegenerative disorder, it is a growing threat in developed countries where the elderly segment of the population is dramatically increasing. Because of its severity, AD has been the subject of extensive studies of disease pathogenesis and clinical trials of various compounds. However, the main cause of AD has not been clearly established, and treatments that effectively stop or reverse the progression of the disease have not yet been developed. Although several competing hypotheses for the cause of AD have been suggested, including genetics, amyloid-β and tau proteins, confirming the major cause of AD remains a challenge. Among the hypotheses proposed, the accumulation of amyloid-β is widely recognized as a major disease contributor, and diverse studies of an amyloid-related mechanism involving pruning of neuronal connections in the brain have been carried out using in vivo animal models and in vitro model systems. Most research on neurologic diseases has been performed in animal models. However, because of the high costs of animal models, uncertain translation of their results to humans and ethical concerns associated with human studies, there is increasing demand for an in vitro brain model that more faithfully mimics the in vivo microenvironment of the brain. Despite the growing appreciation of the need for such an in vitro brain model for neurologic disease studies, no satisfactory model exists.To date, most in vitro studies of neurologic diseases are based on two-dimensional (2D) culture methods, which lack cell–cell contacts and interactions that are essential features of three-dimensional (3D) brain tissue. Cell–cell contacts and interactions are important not only for morphogenesis but also for cell signaling. Many studies have demonstrated differences between 2D and 3D cultures, highlighting the importance of 3D culture effects on various neural cell phenotypes.1–6 Since cells in a 3D culture maintain cell–cell contacts and interactions in all directions that mimic the in vivo cytoarchitecture, the extracellular environment of these cells provides the possibility of spatiotemporal cell-extrinsic stimuli, a situation quite different from that of cells cultured using 2D culture methods. Mechanical cell signaling properties, which recent studies have shown are key factors in determining morphology and differentiation,7–11 also differ between cells in 3D- and 2D-cultured cells. Another factor that is overlooked in current in vitro brain models is interstitial fluid flow. In brain tissue in vivo, the interstitial fluid serves the critical function of delivering nutrients through the brain tissue and clearing away metabolic wastes.12,13 Interstitial flow in the brain is also known to affect cell–cell communication between non-synaptic neurons.14 For these reasons, in vitro brain models designed to better represent the in vivo environment of the brain should not neglect the 3D cytoarchitecture and interstitial flow.
With the development of microtechnologies, a myriad of microsystems that provide in vivo-like microenvironments, such as organ-on-a-chip and cell chips, have been suggested, and several studies have presented 3D culture methods using microfabrication technology.15–20 Among these systems, concave microwell arrays offer the advantages of homogeneous spheroidal micro-tissue formation and size control.15,16 Using this system, we reported the effect of 3D culture on neural tissue.15 Recent studies have reported the development of microfluidic-based systems that provide an interstitial level of flow on cells in vitro and described its effects on cultured cells.21–25 Interstitial flow in the brain is very slow, with speeds ranging from about 0.1 to 0.3 μL min−1;14 most pumps capable of maintaining this range of flow rate are complicated and expensive. However, an osmotic pump developed by Park et al. provides such slow flow without using any complicated devices. This relatively simple device can easily be used to control the flow rate and can be operated in a cell culture incubator for several weeks without an external power source.15,21–23,25
Here, we developed an in vivo-mimicking microfluidic 3D brain model with an interstitial level of flow by combining concave microwell arrays with an osmotic micropump system. Using this brain model, we investigated the effect of flow on 3D micro-spheroidal neural tissue (neurospheroids). The effect of flow on 2D neural cultures has been previously reported,21 but there are no reports on the effect of interstitial flow on in vitro-cultured 3D neurospheroids. The flow provided by the osmotic micropump system was about 0.15 μL min−1, comparable to the level of interstitial flow. To investigate flow effects, we prepared two types of brain model: a static model (neurospheroids cultured without flow) and a dynamic model (neurospheroids cultured with flow). Changes in neurospheroid size and neural network formation between neurospheroids were investigated in both static and dynamic models. To demonstrate the potential of this system as an in vitro brain model for neurologic disease studies, we performed the first test of the effects of amyloid-β on 3D neurospheroids cultured with interstitial flow in vitro. By culturing neurospheroids in parallel with and without amyloid-β, we were able to mimic the normal and Alzheimer's disease brain simultaneously on a single platform. The proposed 3D brain-on-a-chip provides an interstitial level of flow that mimics the in vivo microenvironment and enables long-term in vitro observation without the need for peripheral devices. It could be a valuable in vitro brain model for studies designed to achieve a better understanding of neurologic disease pathology or develop strategies for treating diseases such as Alzheimer's disease. This chip may help to better understand or monitor specific pathways in neurodegenerative diseases because of its simplicity with limited variables in contrast to complex animal models. Moreover, with further development, it could be a substitute for animal models in drug development applications, surpassing the limits of animal models for drug screening.
2. Materials and methods
2.1 Design and operation
The design and operation of the microfluidic chip were inspired by the previously reported artificial liver chip (ALC).23Fig. 1 illustrates the chip design concept. The chip contains concave microwells for the formation of homogeneous 3D neurospheroids with a uniform size. The osmotic micropump system is connected to the outlet to provide a continuous flow of medium at a rate of 0.15 μL min−1. Osmosis is driven by the concentration difference between pure distilled water and 0.05 M poly(ethylene glycol) (PEG) solution separated by a semipermeable cellophane film. A tube coil of 3 cm diameter, wound with flexible polytetrafluoroethylene (PTFE) tubing (inner diameter, 1.0 mm; outer diameter, 1.5 mm), is connected at the outlet of the chip to act as a reservoir of pure distilled water to sustain the flow over time; ~4 mL of 0.05 M PEG solution in a 35 mm Petri dish provided the osmosis effect (Fig. S1†). By providing a 3D cytoarchitecture and interstitial flow, this chip approximates the microenvironment of normal and AD brains, facilitating the investigation of amyloid-β effects on 3D neural tissue (Fig. 2). | ||
| Fig. 1 Schematic diagram of a three-dimensional brain-on-a-chip with an interstitial level of flow. The chip contains a concave microwell array for the formation of homogeneous neurospheroids with a uniform size with 3D cytoarchitecture. The osmotic micropump system is connected to the outlet to provide a continuous flow of medium at the level of interstitial flow. By providing a 3D cytoarchitecture and interstitial flow, this chip approximates the microenvironment of normal and AD brains, facilitating the investigation of amyloid-β effects on 3D neural tissue. | ||
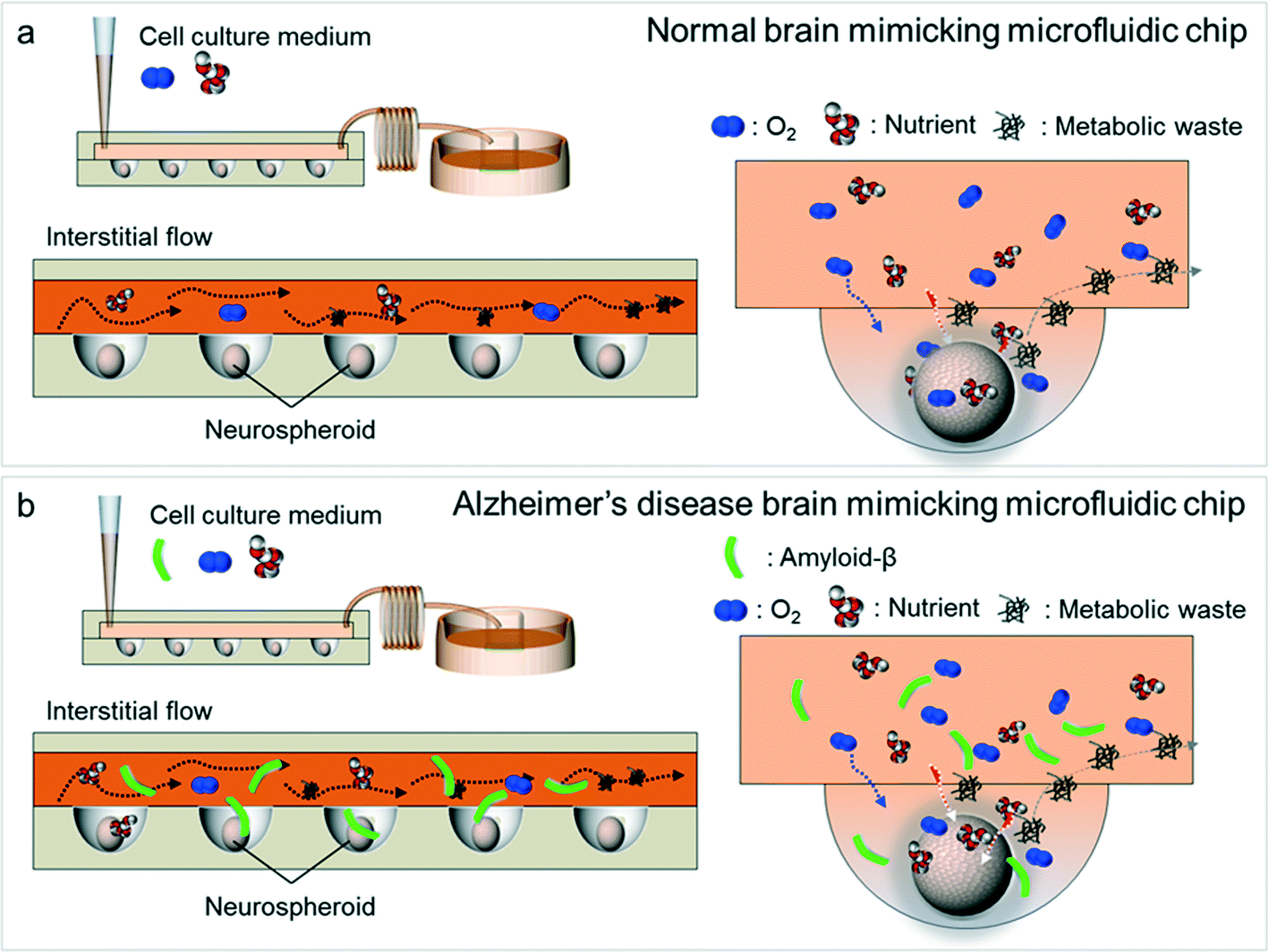 | ||
| Fig. 2 Schematic diagrams of normal brain mimicking microfluidic chip (a) and Alzheimer's disease brain mimicking microfluidic chip (b). (a) Neurospheroids on normal brain mimicking microfluidic chip are cultured under dynamic conditions with a flow of normal medium containing oxygen and nutrient for 10 days. (b) Neurospheroids on Alzheimer's disease brain mimicking microfluidic chip are cultured under dynamic conditions with a flow of normal medium containing oxygen and nutrient for 7 days and then incubated with a medium containing 5 μM synthetic amyloid-β (1–42) for 3 days. | ||
2.2 Chip fabrication
The chip fabrication process also followed that of the ALC.23 First, the top chamber (height, 200 μm) and bottom layer, which contains an array structure containing 50 cylindrical wells (diameter, 600 μm; height, 400 μm), were fabricated using a standard soft lithography procedure. To fabricate the concave microwell array, we used the surface tension of the PDMS prepolymer as described in previous studies.17,26 A polydimethylsiloxane (PDMS) prepolymer consisting of a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of PDMS precursor (Sylgard 184) and curing agent was poured into the cylindrical microwells and allowed to completely fill all microwells. Thereafter, the prepolymer was racked out with a glass slide, with application of slight pressure to the soft PDMS microwell plate. The remaining PDMS prepolymer in each microwell then formed a concave meniscus through surface tension. The final concave structure was formed by thermal curing of the prepolymer in the oven (80 °C for 2 h), after which inlet and outlet holes in the top chamber were punched with a sharp needle. Finally, the top and the bottom layer were bonded by treating with oxygen plasma for 20 s. For the osmotic micropump, cubic PDMS chambers (1 × 1 × 1 cm) with a cellulose membrane window (5 × 5 mm) were fabricated; the cellulose membrane was bonded to the PDMS chamber using the PDMS prepolymer as an adhesive, following a previously reported process.22
:1 mixture of PDMS precursor (Sylgard 184) and curing agent was poured into the cylindrical microwells and allowed to completely fill all microwells. Thereafter, the prepolymer was racked out with a glass slide, with application of slight pressure to the soft PDMS microwell plate. The remaining PDMS prepolymer in each microwell then formed a concave meniscus through surface tension. The final concave structure was formed by thermal curing of the prepolymer in the oven (80 °C for 2 h), after which inlet and outlet holes in the top chamber were punched with a sharp needle. Finally, the top and the bottom layer were bonded by treating with oxygen plasma for 20 s. For the osmotic micropump, cubic PDMS chambers (1 × 1 × 1 cm) with a cellulose membrane window (5 × 5 mm) were fabricated; the cellulose membrane was bonded to the PDMS chamber using the PDMS prepolymer as an adhesive, following a previously reported process.22
2.3 Isolation of prenatal rat cortical neurons
Primary cortical neurons were isolated from cerebral cortical regions of embryonic day 16 rat embryos (DBL, Incheon, South Korea) using a surgical procedure authorized by the Institutional Animal Care and Use Committee (IACUC) of Texas Southern University. Neural progenitor cells were maintained in Neurobasal medium (Gibco) supplemented with B-27 Supplement (Gibco), 0.5 mM L-glutamine, and 1% antibiotic solution containing 10000 units penicillin (Gibco) and streptomycin. We call it ‘normal medium’ in this paper to discriminate from the medium containing amyloid-β that was treated in groups IA and IIA.
2.4 Cell seeding and culture of prenatal rat cortical neurons
A neural progenitor cell suspension (100 μL of 4 × 107 cells mL−1) was loaded into the inlet of the chip using a micropipette. The cells in the suspension medium sank into and were trapped in concave wells by gravity and then were allowed to stabilize by leaving undisturbed for about 5 min. This procedure was repeated two times, after which a constant flow of the medium from the inlet to the outlet was applied to wash out untrapped cells. Finally, chips with concave microwells containing nearly identical numbers of neural progenitor cells were cultured for 10 days to allow neurospheroid formation. The following four groups of neurospheroids, providing different culture conditions, were employed: group I, neurospheroids cultured without flow (static conditions); group IA, static conditions with added amyloid-β; group II, neurospheroids cultured with flow (dynamic conditions); group IIA, dynamic conditions with added amyloid-β. The medium was changed every other day in the static condition groups, group I and group IA. For treatment with amyloid-β, neurospheroids were cultured in normal medium for 7 days and then incubated with a medium containing 5 μM synthetic amyloid-β (1–42) for 3 days.2.5 Cell viability assay
The viability of neural cells cultured in the chip was assessed using a Live/Dead assay kit (Invitrogen) as described by the manufacturer. Briefly, cells were incubated in a solution of Neurobasal medium containing 2 μL of calcein AM solution and 5 μL of ethidium homodimer-1 solution at 37 °C for 40 min. Stained cells were observed using a confocal laser-scanning microscope (Olympus, Japan). Also, a cell viability test using a Cell Counting Kit-8 (CCK-8; Dojindo, Tabaru, Japan) was conducted. Neurospheroids were harvested from the chip and plated into a 96-well plate (100 μL medium per microwell). Then, 10 μL of CCK-8 solution was added to each well and the plate was incubated at 37 °C in a humidified 5% CO2 atmosphere for 3 h. The absorbance of each well was read at 450 nm using a microplate reader (PerkinElmer, Akron, OH, USA).2.6 Fixation and immunocytochemistry
Cultured neurospheroids in the four groups were fixed with 4% paraformaldehyde (PFA) for 20 min at 4 °C and then washed with 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). The cells were incubated in 0.1% Triton X-100 in PBS for 20 min at room temperature and then washed with PBS containing 0.1% BSA. The cells were blocked for 30 min at 4 °C to reduce non-specific protein adsorption and then incubated overnight at 4 °C with primary antibodies against the following proteins: β-III tubulin (Santa Cruz Biotechnology), synapsin IIa (Santa Cruz Biotechnology), and nestin (Santa Cruz Biotechnology). Cells were rinsed with PBS containing 0.1% BSA and incubated with Alexa Fluor 488- or 594-conjugated secondary antibody (Invitrogen) for 90 min at 4 °C. Amyloid plaque staining was done after fixation with 4% PFA. After the fixation, the neurospheroids were washed with PBS three times and were incubated in 1 mM thioflavin S (Sigma Aldrich) which had been dissolved in 50% ethanol for 10 min. Then they were rinsed with 80% ethanol twice and washed with PBS three times. Fluorescence images were collected using a confocal laser-scanning microscope (Olympus, Japan) after counterstaining the cell nuclei with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Invitrogen).2.7 Scanning electron microscopy (SEM)
For SEM imaging, the culture medium was removed and the neurospheroids formed in the concave microwells were washed three times with 1 mL of PBS. Neurospheroids were then fixed by incubating with 1 mL of 2.5% glutaraldehyde solution in PBS for 10 min at room temperature. The fixed spheroids were subsequently dehydrated with a series of graded ethanol (75% and 95%, 1 time each; 100%, 3 times) for 25 min each. After dehydration, the spheroids were immersed in hexamethyldisilazane (Sigma-Aldrich) for 10 min. The spheroids were mounted on top of a sample holder using carbon tape, coated with palladium alloy, and observed under a scanning electron microscope (JEOL Ltd, Tokyo, Japan).2.8 Statistical analysis
All quantitative data were expressed as means ± standard errors of the mean. All collected data were analyzed using unpaired t tests using the statistical software SPSS, Version 10.0 (SPSS, Chicago, IL, USA). P values less than 0.05 were considered statistically significant.3. Results
3.1 Time course of the size distribution of neurospheroids cultured with or without flow
To determine the effects of an interstitial level of flow on neurospheroids, we cultured neurospheroids in two different model systems: a static, no-flow model (group I) and a dynamic, slow-flow model (group II). The neurospheroid size was measured every day from day 0 (1 h after cell seeding) to day 10. Fig. 3(a) and (b) show the size distribution of neurospheroids over time. On day 0, the average size of neurospheroids in group I and group II was 451.68 and 447.87 μm, respectively, indicating that similar numbers of neural progenitor cells were seeded in both groups. During the first 3 days after seeding, the sizes of neurospheroids in both groups decreased due to aggregation, as previously reported.15 However, thereafter (days 4 to 10), the changes in spheroid size differed depending on the presence of flow. In group II (with flow), the size of neurospheroids slightly increased, whereas the size of neurospheroids in group I (without flow) was essentially unchanged from day 4 to day 10. The average size of neurospheroids on day 10 in group I and group II was 280.95 and 357.15 μm, respectively. Also, the average size of neurospheroids over 10 days in culture was greater in group II than that in group I. | ||
| Fig. 3 Time course of the size distribution of neurospheroids cultured with (dynamic conditions, group II) or without flow (static conditions, group I). (a) Daily optical images of neurospheroids of group I and group II from day 0 to day 10. (b) Size distribution analysis of neurospheroids of group I and group II over time. Data represent means ± standard deviations of 40 independent experiments (**p < 0.01, two-tailed). (c) Cell viability of group I and group II was quantified by CCK-8 assays on days 1, 4, and 10. Data represent means ± standard deviations of 6 independent experiments (*p < 0.05, two-tailed). | ||
3.2 Interstitial flow enhances synapse formation leading to formation of a robust neural network
We next examined the formation of neural networks, which are important for communication between neurons, and thus brain function, in group I and group II. The pattern of network formation on the chip was different between the two groups. SEM images of neurospheroids in group I and group II (Fig. 4a and c, respectively) revealed greater neurite extension in neurospheroids in group II than that in group I, leading to more robust neural network formation. Optical images of the chip (Fig. 4b and d) show more active neural network formation in group II as well. For quantitative analysis of these optical images, the chip was divided into ten sections by column (each containing five microwells) from inlet to outlet, and each section was analyzed by determining the total number of neurites that extended from microwells and the average size of neurospheroids. In group II, the number of neurites extending from microwells was high near the inlet and decreased towards the outlet of the chip. In contrast, no neurites were detected extending from group I microwells (Fig. 4e). Moreover, neurospheroids near the inlet in group II were larger and become smaller toward the outlet, whereas the sizes of neurospheroids are almost the same throughout the chip in group I (Fig. 4f). To further examine the formation of neural networks in neurospheroids, we immunostained neurospheroids for the synaptic marker synapsin IIa (Fig. 5). The intensity of synapsin IIa immunostaining was higher in the dynamic model than in the static model, showing that interstitial flow enhances synapse formation and leads to the development of a complex neural network by providing continuous nutrient, cytokine, and oxygen transport. | ||
| Fig. 4 Neural network formation in group I and group II. (a and c) SEM images and (b and d) optical images of group I and group II. White arrows indicate neurites. For quantitative analysis of their optical images (b and d), the chip was divided into ten sections by column (each containing five concave microwells) from inlet to outlet. (e) Average number of neurites extending from microwells of each section from inlet to outlet of group I and group II. (f) Average size of neurospheroids of each section from inlet to outlet of group I and group II. | ||
 | ||
| Fig. 5 (a) Immunofluorescence images of neurospheroids of group I (static, normal medium), group IA (static, medium + amyloid-β (Aβ)), group II (dynamic, normal medium), and group IIA (dynamic, medium + Aβ) immunostained against synaptic marker synapsin IIa (red) and neural progenitor/stem cell marker nestin (green). (b and c) Quantified results of the immunofluorescence images by the intensity. Data represent means ± standard deviations of 8 independent experiments (*p < 0.05, **p < 0.01, two-tailed). | ||
3.3 Interstitial flow enhances differentiation of neural progenitor cells into neurons
We immunostained neurospheroids in the two groups for the neural progenitor/stem cell marker nestin and the neuronal marker β-III tubulin (Fig. 5 and 6). Nestin levels in neurospheroids were higher in group I, whereas β-III tubulin levels were higher in group II. These results indicate that under static conditions, the stemness of neural progenitor cells was maintained; in contrast, under dynamic conditions, the differentiation of neural progenitor cells into neurons was enhanced. | ||
| Fig. 6 (a) Immunofluorescence images of neurospheroids of group I (static, normal medium), group IA (static, medium + amyloid-β (Aβ)), group II (dynamic, normal medium), and group IIA (dynamic, medium + Aβ) stained Aβ with thioflavin S (green) and immunostained against neuronal marker β-III tubulin (red). (b and c) Quantified results of the immunofluorescence images by the intensity. Data represent means ± standard deviations of 8 independent experiments (**p < 0.01, two-tailed). | ||
3.4 Neurotoxic effects of amyloid-β
To study how amyloid-β affects neurospheroids, we cultured spheroids in the presence of amyloid-β under static conditions (group IA) and dynamic conditions (group IIA); neurospheroids cultured in the absence of amyloid-β (group I and group II) served as controls. To assess the presence and distribution of amyloid-β in neurospheroids, we stained neurospheroids with thioflavin S, which binds β-sheet-rich structures and thus stains amyloid-β. As shown in Fig. 6, the fluorescence intensity increased after treatment of neurospheroids with amyloid-β. We also observed that the amount of amyloid-β remaining in the neurospheroids in group IIA was greater than that in group IA. We next performed a Live/Dead assay to assess the viability of cells in neurospheroids in each group (Fig. 7). The intensity of red fluorescence corresponding to dead cells was higher in groups treated with amyloid-β regardless of the presence of flow. Quantification of Live/Dead assay results showed that the percentage of viable cells in group IA and group IIA was lower than that in group I and group II, respectively, consistent with previous reports of the apoptotic and neurotoxic effects of amyloid-β.15,21,27,28 Neurospheroids were also immunostained for synapsin IIa, nestin, and β-III tubulin. Groups IA and IIA showed lower levels of synapsin IIa, indicating greater destruction of neural networks in groups IA and IIA than in groups I and II (Fig. 5). This result was reinforced by β-III tubulin immunostaining results, which showed lower levels of β-III tubulin in groups IA and IIA (Fig. 6). Nestin immunostaining in groups IA and IIA was also less intense than that in groups I and II (Fig. 5), consistent with previous reports.29–31 Collectively, these data indicate that the neurotoxic effects of amyloid-β decreased cell viability, destroyed neural networks, which would be predicted to cause synaptic dysfunction, and reduced the level of nestin in neurospheroids. | ||
| Fig. 7 Cell viability of neurospheroids of group I (static, normal medium), group IA (static, medium + amyloid-β (Aβ)), group II (dynamic, normal medium), and group IIA (dynamic, medium + Aβ) as a result of the live/dead assay on day 10. Calcein AM stains live cells (green) and ethidium homodimer stains dead cells (red). | ||
4. Discussion
In this study, we simultaneously realized two in vivo brain conditions—3D cytoarchitecture and interstitial fluid flow—and investigated the effects of an interstitial level of flow and amyloid-β protein toxicity on neurospheroids, demonstrating the potential of this in vitro brain model as a drug-screening and cytotoxicity testing tool.We first found that an interstitial level of flow influenced the size distribution of neurospheroids. During the first 3 days after seeding, the size of neurospheroids cultured under both static (group I) and dynamic (group II) conditions decreased as a result of aggregation through cell–cell interactions, as previously reported.15 However, there were considerable differences in the size distributions of the two groups. Neurospheroids in group II were larger at all points during culture than neurospheroids in group I and visibly increased in size between day 4 and day 10, whereas the size of neurospheroids in group I remained the same from day 3 to day 10. This difference suggests that an interstitial level of flow is involved in accelerating differentiation of neural progenitor cells into mature neurons, a process accompanied by neuritogenesis, neurite outgrowth, and synaptogenesis,32 and thus increases the volume of neurospheroids. Also, as Fig. 3(c) shows, the absorbance at 450 nm increased gradually from day 1 to day 10 in group II, while group I increased for the first 4 days a bit and decreased again. Since the intensity result of the CCK-8 assay is proportional to the number of cells, this data showed that the number of cells in neurospheroids in group II gradually increased from day 1 to day 10 and this might explain the size distribution of neurospheroids. We confirmed this possibility by immunostaining neurospheroids with nestin, a marker for neural progenitor cells, and β-III tubulin, a marker for neurons. The results of these immunohistochemistry analyses revealed greater expression of nestin in neurospheroids in group I, indicating the presence of more early-stage neural progenitor cells under these static culture conditions. In contrast, β-III tubulin levels were higher in neurospheroids cultured with flow. Since β-III tubulin is a component of neurites, a higher level of β-III tubulin translates into more extended neurites—a characteristic of mature neurons. This enhanced differentiation likely indicates the effect of continuous nutrient, oxygen, and cytokine transport and removal of metabolic wastes caused by interstitial flow. These differences also show that interstitial flow has the propensity to stimulate proliferation of neural progenitor cells. We further found that interstitial flow influenced neural network formation in neurospheroids by providing a continuous medium in diffusion-dominant laminar flow. Both qualitative and quantitative assessments revealed more robust neural network formation in neurospheroids cultured with flow, possibly because of the continuous supply of fresh medium rich in oxygen and nutrients and clearance of metabolic wastes by flow.33,34 One interesting aspect of neural network formation in our system was the observation that the total number of neurites extending from each microwell decreased from inlet to outlet (Fig. 4). This phenomenon could be due to the effect of the dilution of cytokines and the removal of metabolic wastes and their interactions from the inlet to the outlet. Although further investigations are required, the reason seems to be from the insufficient supply of nutrients and oxygen to the outlet. Generally, it is well known that the flow rate should be higher or equal to the critical perfusion rate to guarantee that cells receive enough nutrients and oxygen in dynamic culture.35 The interstitial level of slow flow that we demonstrated on this chip is extremely slow flow, that is, diffusion-dominant laminar flow, that provides continuous transport of nutrient, oxygen, and cytokine and removal of metabolic wastes, which seemed to have affected size distribution, neural network formation, and synapse formation.
To confirm the applicability of this in vitro brain model for drug screening or toxicity testing, we treated neurospheroids with amyloid-β, which is generally considered a cause of AD,36 and tested neurotoxicity by fluorescence imaging and SEM. We also assessed neural network formation by immunostaining for β-III tubulin and synapsin IIa, a pre-synaptic nerve terminal-specific phosphoprotein that plays a key role in modulating the release of neurotransmitters in the nervous system.37 The reduction in synapsin IIa levels observed after treatment suggests that amyloid-β induces neural network destruction. Consistent with this interpretation, β-III tubulin levels were also reduced by amyloid-β treatment, as were the total numbers and lengths of neurites extending from microwells.
We also noted the presence of more amyloid-β and dead cells in neurospheroids cultured under flow conditions. Under static conditions, the access of amyloid-β to the interior of neurospheroids, and thus its ability to cause neurospheroid degradation, is limited by simple diffusion. However, with an interstitial level of flow, soluble amyloid-β can infiltrate more deeply into neurospheroids causing more neural cells to undergo apoptosis. This finding is seemingly at odds with previous reports that interstitial fluid movement contributes to the removal of interstitial solutes through exchange with cerebrospinal fluid.33,34 However, in our experiments, a continuous supply of amyloid-β was provided to neurospheroids during the last 3 days of culture; thus, without a supply of fresh medium to serve the function of cerebrospinal fluid, amyloid-β clearance was not observed. Accordingly, rather than conflicting with these previous studies, our results reinforce the importance of the exchange of interstitial fluid with cerebrospinal fluid. Additional studies, in which fresh medium is supplied after amyloid-β treatment, will be needed to confirm that amyloid-β is cleared by interstitial flow.
5. Conclusion
Using a biomimetic approach, we developed a brain-on-a-chip that creates a 3D cytoarchitecture and interstitial flow. To the best of our knowledge, there are no previous reports describing a system that combines both of these important features of the in vivo brain microenvironment. 3D culture is essential for establishing normal cell–cell contacts and interactions, and interstitial flow plays an important role not only in nutrient delivery and metabolic waste clearance but also in neural differentiation and morphogenesis by providing a continuous supplement of medium containing nutrient and oxygen. Consistent with this, neurospheroids cultured under dynamic conditions were larger and showed a more robust neural network than neurospheroids cultured under static conditions. We also used this system to investigate the neurotoxic effects of amyloid-β, demonstrating decreased cell viability and increased neural destruction and synaptic dysfunction, which are pathophysiological features of Alzheimer's disease in vivo. The in vivo-like microenvironment provided by this 3D culture-based microfluidic chip has great potential as an in vitro brain model. As such, this platform could fill the gap between traditional in vitro neural cell culture models and in vivo brain studies, serving as a more reliable tool for studying neurologic disease pathology and treatment strategies as well as drug screening applications.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (no. NRF-2012R1A2A1A05026340) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number HI14C0522).References
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CrossRef CAS PubMed.
- D. K. Cullen, J. A. Wolf, V. N. Vernekar, J. Vukasinovic and M. C. LaPlaca, Neural Tissue Engineering and Biohybridized Microsystems for Neurobiological Investigation In Vitro (Part 1), 2011, vol. 39, pp. 201–240 Search PubMed.
- K. M. Smalley, M. Lioni and M. Herlyn, In Vitro Cell. Dev. Biol.: Anim., 2006, 42, 242–247 CrossRef CAS PubMed.
- J. P. Frampton, M. R. Hynd, M. L. Shuler and W. Shain, Biomed. Mater., 2011, 6, 015002 CrossRef CAS PubMed.
- H. R. Irons, D. K. Cullen, N. P. Shapiro, N. A. Lambert, R. H. Lee and M. C. LaPlaca, J. Neural Eng., 2008, 5, 333 CrossRef PubMed.
- M. Schindler, A. Nur-E-Kamal, I. Ahmed, J. Kamal, H.-Y. Liu, N. Amor, A. Ponery, D. Crockett, T. Grafe, H. Y. Chung, T. Weik, E. Jones and S. Meiners, Cell Biochem. Biophys., 2006, 45, 215–227 CrossRef CAS.
- D. K. Cullen and M. C. LaPlaca, Neuronal response to high rate shear deformation depends on heterogeneity of the local strain field, Liebert, Larchmont, NY, ETATS-UNIS, 2006 Search PubMed.
- S. M. Willerth, K. J. Arendas, D. I. Gottlieb and S. E. Sakiyama-Elbert, Biomaterials, 2006, 27, 5990–6003 CrossRef CAS PubMed.
- T.-H. Chun, K. B. Hotary, F. Sabeh, A. R. Saltiel, E. D. Allen and S. J. Weiss, Cell, 2006, 125, 577–591 CrossRef CAS PubMed.
- D. E. Ingber, Annu. Rev. Physiol., 1997, 59, 575–599 CrossRef CAS PubMed.
- E. Cukierman, R. Pankov, D. R. Stevens and K. M. Yamada, Science, 2001, 294, 1708–1712 CrossRef CAS PubMed.
- J. M. Rutkowski and M. A. Swartz, Trends Cell Biol., 2007, 17, 44–50 CrossRef CAS PubMed.
- M. A. Swartz and M. E. Fleury, Annu. Rev. Biomed. Eng., 2007, 9, 229–256 CrossRef CAS PubMed.
- N. J. Abbott, Neurochem. Int., 2004, 45, 545–552 CrossRef CAS PubMed.
- Y. J. Choi, J. Park and S.-H. Lee, Biomaterials, 2013, 34, 2938–2946 CrossRef CAS PubMed.
- Y. Y. Choi, B. G. Chung, D. H. Lee, A. Khademhosseini, J.-H. Kim and S.-H. Lee, Biomaterials, 2010, 31, 4296–4303 CrossRef CAS PubMed.
- G. S. Jeong, J. H. Song, A. R. Kang, Y. Jun, J. H. Kim, J. Y. Chang and S.-H. Lee, Adv. Healthcare Mater., 2013, 2, 119–125 CrossRef CAS PubMed.
- A. Kunze, M. Giugliano, A. Valero and P. Renaud, Biomaterials, 2011, 32, 2088–2098 CrossRef CAS PubMed.
- M. Kato-Negishi, Y. Tsuda, H. Onoe and S. Takeuchi, Biomaterials, 2010, 31, 8939–8945 CrossRef CAS PubMed.
- W. Wang, K. Itaka, S. Ohba, N. Nishiyama, U.-I. Chung, Y. Yamasaki and K. Kataoka, Biomaterials, 2009, 30, 2705–2715 CrossRef CAS PubMed.
- Y. J. Choi, S. Chae, J. H. Kim, K. F. Barald, J. Y. Park and S.-H. Lee, Sci. Rep., 2013, 3 Search PubMed.
- J. Y. Park, C. M. Hwang, S. H. Lee and S.-H. Lee, Lab Chip, 2007, 7, 1673–1680 RSC.
- S.-A. Lee, D. Y. No, E. Kang, J. Ju, D.-S. Kim and S.-H. Lee, Lab Chip, 2013, 13, 3529–3537 RSC.
- R. Gómez-Sjöberg, A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Anal. Chem., 2007, 79, 8557–8563 CrossRef PubMed.
- J. Y. Park, S.-K. Kim, D.-H. Woo, E.-J. Lee, J.-H. Kim and S.-H. Lee, Stem Cells, 2009, 27, 2646–2654 CrossRef CAS PubMed.
- G. S. Jeong, Y. Jun, J. H. Song, S. H. Shin and S.-H. Lee, Lab Chip, 2012, 12, 159–166 RSC.
- D. T. Loo, A. Copani, C. J. Pike, E. R. Whittemore, A. J. Walencewicz and C. W. Cotman, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7951–7955 CrossRef CAS.
- C. Pike, D. Burdick, A. Walencewicz, C. Glabe and C. Cotman, J. Neurosci., 1993, 13, 1676–1687 CAS.
- N. J. Haughey, A. Nath, S. L. Chan, A. C. Borchard, M. S. Rao and M. P. Mattson, J. Neurochem., 2002, 83, 1509–1524 CrossRef CAS.
- B. A. Yanker, Mechanisms of Neuronal Degeneration in Alzheimer's Disease, Cell Press, Cambridge, MA, ETATS-UNIS, 1996 Search PubMed.
- I.-S. Lee, K. Jung, I.-S. Kim and K. I. Park, Exp. Mol. Med., 2013, 45, e60 CrossRef PubMed.
- T. Tojima and E. Ito, Prog. Neurobiol., 2004, 72, 183–193 CrossRef CAS PubMed.
- J. J. Iliff, H. Lee, M. Yu, T. Feng, J. Logan, M. Nedergaard and H. Benveniste, J. Clin. Invest., 2013, 123, 1299–1309 CAS.
- J. J. Iliff, M. Wang, Y. Liao, B. A. Plogg, W. Peng, G. A. Gundersen, H. Benveniste, G. E. Vates, R. Deane, S. A. Goldman, E. A. Nagelhus and M. Nedergaard, Sci. Transl. Med., 2012, 4, 147ra111 Search PubMed.
- E. W. K. Young and D. J. Beebe, Chem. Soc. Rev., 2010, 39, 1036–1048 RSC.
- S. S. Kar, P. M. Stephen, D. Westaway and H. T. J. Mount, Interactions between β-amyloid and central cholinergic neurons: implications for Alzheimer's disease, Canadian Medical Association, Ottawa, ON, CANADA, 2004 Search PubMed.
- M. Bähler, F. Benfenati, F. Valtorta and P. Greengard, BioEssays, 1990, 12, 259–263 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00962b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |