
Manipulating and quantifying temperature-triggered coalescence with microcentrifugation†
Huanhuan
Feng
ab,
Dmitry
Ershov
a,
Thomas
Krebs
cd,
Karin
Schroen
c,
Martien A.
Cohen Stuart
a,
Jasper
van der Gucht
a and
Joris
Sprakel
*a
aLaboratory of Physical Chemistry and Colloid Science, Wageningen University, Dreijenplein 6, 6703 HB Wageningen, The Netherlands. E-mail: joris.sprakel@wur.nl
bDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX Eindhoven, The Netherlands
cFood Process Engineering Group, Wageningen University, Bomenweg 2, 6703 HD Wageningen, The Netherlands
dFMC Separation Systems, Delta 101, 6825MN Arnhem, The Netherlands
First published on 6th October 2014
Abstract
In this paper we describe a new approach to quantify the stability and coalescence kinetics of thermally switchable emulsions using an imaging-based microcentrifugation method. We first show that combining synchronized high-speed imaging with microfluidic centrifugation allows the direct measurement of the thermodynamic stability of emulsions, as expressed by the critical disjoining pressure. We apply this to a thermoresponsive emulsion, allowing us to measure the critical disjoining pressure as a function of temperature. The same method, combined with quantitative image analysis, also gives access to droplet-scale details of the coalescence process. We illustrate this by measuring temperature-dependent coalescence rates and by analysing the temperature-induced switching between two distinct microscopic mechanisms by which dense emulsions can destabilise to form a homogeneous oil phase.
Introduction
The formation of a homogeneous film from a previously dispersed phase, for example during the drying of emulsions or dispersions, is an essential stage in the surface application of dispersed coating systems. Especially for waterborne paints, which by definition are dispersions of liquid or solid particles in a continuous water phase, the way in which the final film forms is crucial in determining its properties. With the increasing political and societal pressure to phase out the emission of hazardous organic compounds from both consumer and industrial coatings and adhesives, this has become a key issue in the future of a sustainable coatings industry. A thorough understanding of how film formation takes place at all length scales, and how it can be manipulated, is required to improve the quality of waterborne paints to the standards of their solvent-based counterparts. Nonetheless, despite the direct implications, a microscopic understanding of how and where coalescence occurs in a dense packing of emulsion droplets, and how it can be effectively manipulated from the outside, is lacking. This is exacerbated by the absence of model materials and measurement techniques that allow the quantitative analysis of coalescence at the scale of individual particles.Recently, we reported that coalescence in dense suspensions subjected to a unilateral pressure, as occurs in a drying film, may occur in two physically distinct modes; occurring either localized to the drying front, where single particles preferentially coalesce with a homogeneous oil phase, or in a random manner throughout the bulk of the film, where droplet–droplet coalescence is equally likely as droplet–front coalescence.1 Theoretical analysis of the experimental data suggested that the type of coalescence process that occurs depends mainly on the initial stability of the emulsion, as expressed by the so-called critical disjoining pressure, which gives the minimum pressure with which two droplets need to be pushed together before film rupture and coalescence can occur. In these experiments however, drying was used to develop a pressure gradient to induce de-emulsification; this introduces many complications, such as the development of solute, e.g. surfactant, gradients, throughout the film and solute concentrations, which evolve in time. To further advance our fundamental understanding of emulsion coalescence and its implications for film formation, new methods and materials are required in which these phenomena can be systematically studied and manipulated under well-defined conditions.
Microfluidic methods promise to offer the required experimental prerequisites of control of fluid flows, internal and environmental control parameters, applied pressures and allowing facile high-speed and high-resolution imaging at the scale of individual droplets. These advantages have been used previously to study, for example the effects of applied pressure on the coalescence of individual droplets,2 electric field-induced coalescence,3 flow-induced coalescence in dilute emulsions4 and the effects of packing geometry on coalescence in dense emulsion layers.5
In this paper, we extend the microfluidic toolbox to study coalescence in dense systems by applying a microcentrifugation method developed by Krebs et al.4 combined with a temperature-triggerable emulsion system. Centrifugation of an emulsion placed in a microchip allows us to apply a well-defined unilateral driving force for compaction and coalescence of an emulsion; using a temperature-trigger, the initial stability of the emulsion can be directly manipulated. By combining this approach with synchronized high-speed imaging, we can monitor, manipulate and quantify, in situ and at the level of individual droplets, both the thermodynamic and kinetic aspects of the coalescence process. This opens a wide variety of possibilities to gain new insight into film formation in dense dispersed systems.
Experimental section
All reagents were purchased from Sigma-Aldrich and used as received.Thermoresponsive surfactants
We synthesize a thermally-switchable surfactant by means of Atom Transfer Radical Polymerization (ATRP) using an alkyl-functional initiator; details of the synthesis have been reported elsewhere.6 The resulting surfactant, composed of a hydrophobic C18-alkyl tail, and a thermoresponsive block of poly(di(ethyleneglycol)methacrylate-co-poly(ethylene glycol)methacrylate) displays a lower critical solution temperature (LCST) around 34 °C in aqueous solution, as measured by light scattering and calorimetry.6 At temperatures below the LCST, the hydrophilic block is well dissolved in water; when the temperature is raised above the LCST the hydrophilic block collapsed and becomes insoluble.Emulsions
Monodisperse emulsions of silicone oil-in-water are prepared in a standard flow-focusing microfluidic device (Micronit Microfluidics BV); silicone oil, with viscosity 0.1 Pa s, is hydrodynamically focused at a tapered nozzle by the continuous phase, consisting of a 5 g L−1 solution of the thermoresponsive surfactant. The size of the droplets can be tuned by varying the flow rates of both continuous and dispersed phases;7,8 we produce droplets with a final diameter of 200 μm, giving a single layer of droplets in the coalescence chambers. Upon loading the emulsions in the sample chambers, some coalescence occurs due to the hydrophobic walls of the microliter pipette, introducing modest polydispersity; however, this does not influence the quantitative analysis of our experiments, which is equally suited for the analysis of even highly polydisperse emulsions.Microcentrifugation & data analysis
The emulsions are compressed and subjected to a unilateral pressure by applying a constant centrifugal force to a microfluidic chamber (Micronit Microfluidics BV, the Netherlands) that holds a monolayer of emulsion droplets (see Fig. 1).4 The sample is mounted on a sample holder, which can be rotated at variable speed, for the experiments presented here to a total acceleration of 450g where g is the gravitational acceleration, with an externally controlled motor. We heat the sample through a continuous stream of warm air and monitor the temperature of the measurement cell using an infrared thermometer. We vary the temperature between 25 and 45 °C, representing temperatures both above and below the LCST of the surfactant. Images of the sample are captured using a high-speed camera, synchronized with the rotating sample chamber, mounted on a bright field microscope. All experiments were repeated 5 times to ensure reproducibility of our results. To extract quantitative information from the images we developed an image processing routine in Matlab. First, we spatially filter all images to reduce high-frequency noise, e.g. resulting from dust or camera pixel noise. We then discard images in which the synchronization between the moving sample and the camera was not ideal, resulting in only partial capture of the sample cell. This is done by detecting the edges of the sample chamber, and requiring that each image used for further processing has two black edges that demarcate the glass sample walls (see ESI†). We then use an edge detection algorithm to locate areas of continuous intensity in our thresholded images to identify the position and size of all droplets and the sizes of the continuous water and oil phases (see Fig. 2). These data are then used to extract quantitative parameters regarding the coalescence process.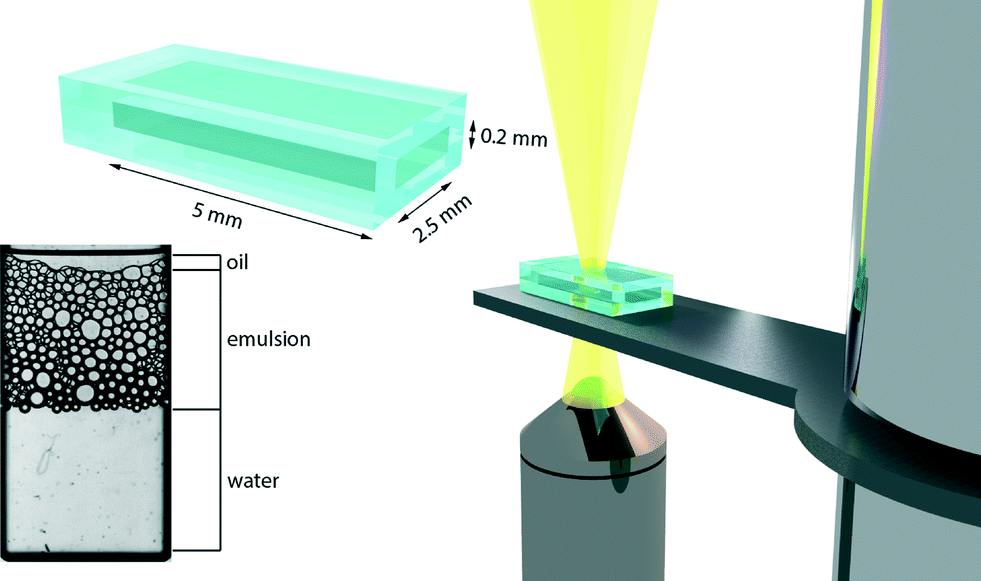 | ||
| Fig. 1 Schematic illustration of the microcentrifugation set-up, including the glass microchip (dimensions as shown), sample holder & motor drive, which is mounted on an inverted microscope connected to a high-speed camera. Bottom left corner shows an example of a raw image from the synchronized high-speed camera, indicating the continuous oil and water phases and the emulsion layer. | ||
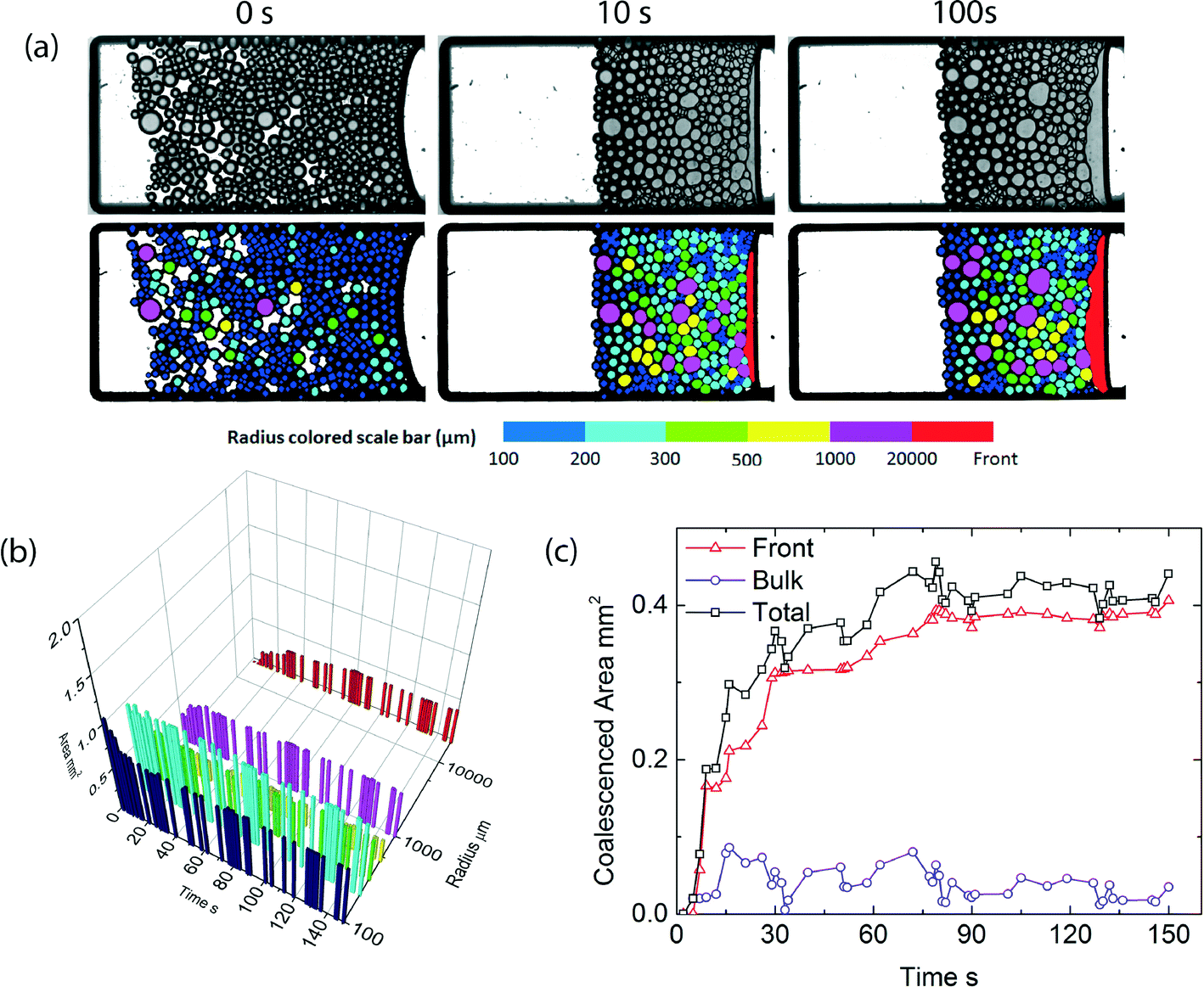 | ||
| Fig. 2 a) Images of a microfluidic centrifugation experiment, at T = 25 °C < TLCST, α = 450g, before (top) and after (bottom) image processing, at three different time intervals, b) volume of oil stored in various droplets size classes, and the continuous oil front, c) amount of coalesced oil as a function of time, separated into the amount coalesced at the macroscopic oil front and within the packed film of droplets. | ||
Results & discussion
Emulsions of silicone oil-in-water, stabilized by our thermoresponsive surfactant,6 are prepared by microfluidics and then collected in small sample chambers designed for the microcentrifugation experiments. The droplets are prepared with diameters of approximately 200 microns, which thus form a single layer in the sample chambers with smallest dimension, orthogonal to the imaging plane, of also 200 micrometers. Initially, droplets are present throughout the sample, but upon applying a constant centrifugal stress of 450g, in which g is the centrifugal constant, the droplets, which have a lower density then the continuous phase, move towards the emulsion–air interface moving to the right side of the images. Within a few seconds, they become jammed and deform due to the centrifugal stress, as shown in the image sequence in Fig. 2; subsequently, the droplets begin to coalesce. This leads to an increase in the volume of the continuous oil phase and a reduction in the volume of the packed emulsion. To extract quantitative information from the large number of images collected during an experiment, we apply an image processing routine described above. Example raw images and images after passing through this algorithm are shown in the top and bottom row of Fig. 2, respectively. All droplets, as well as the oil and water bulk phases, are recognized and colored according to their area.Critical disjoining pressure
Upon bringing two emulsion drops together, the water film that separates the droplets becomes thinner. This leads to a restoring pressure that acts to separate the droplets, which is known as the disjoining pressure. For two drops to coalesce, the water film that separates them needs to rupture; this can only occur if the pressure applied to the droplets exceeds a critical disjoining pressure Π*. The critical disjoining pressure is a thermodynamic measure for the stability of an emulsion, and therefore an important parameter to classify emulsions. Various methods are established to measure critical disjoining pressures; either on free-standing thin films, for example using interferometry9 or atomic force microscopy,10 or between emulsion droplets using the film trapping technique11 or by centrifugation.12 In our microcentrifugation experiments we apply the latter method to directly measure Π*. In steady-state, when the emulsion is left to equilibrate sufficiently long at fixed centrifugal acceleration, a well-defined gradient in pressure builds-up in the concentrated emulsion. The pressure within this packed layer is highest at the oil-front and lowest at the water-end of the emulsion and is determined by the centrifugal pressure that emulsion droplets exert on each other. Coalescence will occur, until the total volume of the emulsion, that pushes on the front, is reduced to the point where the pressure exerted on the droplets at the front drops below the critical disjoining pressure.Thus, from the total height h (see inset Fig. 3b) of the packed emulsion, and knowing the density difference Δρ, between oil and water, the disjoining pressure can be precisely measured as Π = Δραh, in which h is the height of the cream layer and the α = 450g is the centrifugal acceleration, with g = 9.81 m2 s−1 the normal gravitational acceleration in absence of centrifugal forces. At room temperature, below the LCST of the thermoresponsive block of our surfactant, we find a critical disjoining pressure, for droplets of approximately 200 micrometers, of around 300 Pa (Fig. 3b). As the critical disjoining pressure is a function of the droplet size, making a direct comparison with values reported for other surfactants in literature difficult, this disjoining pressure is approximately two orders of magnitude lower than that for the common, and effective, surfactant sodium dodecyl sulfate.1,13 Nevertheless, this is sufficient to stabilize these emulsions against coalescence for several months in absence of centrifugal forces, as reported previously.6 Upon increasing the temperature, to above the LCST of the surfactant, we see a gradual but significant decrease of the critical disjoining pressure; at 45 °C, it has decreased by a factor of 6 to only Π* ≈ 50 Pa (Fig. 3b). Note that the decrease in disjoining pressure is not abrupt at the LCST: Π* gradually decreases when increasing the temperature above the LCST; a similar effect was seen for the adhesive forces between two surfaces coated with thermoresponsive surfactants.14 As the critical disjoining pressure is a function of droplet size, we note that the gradually decrease in Π*, as opposed to the sudden decrease in interfacial tension reported previously6 may be caused by the polydispersity in the droplets during these experiments.
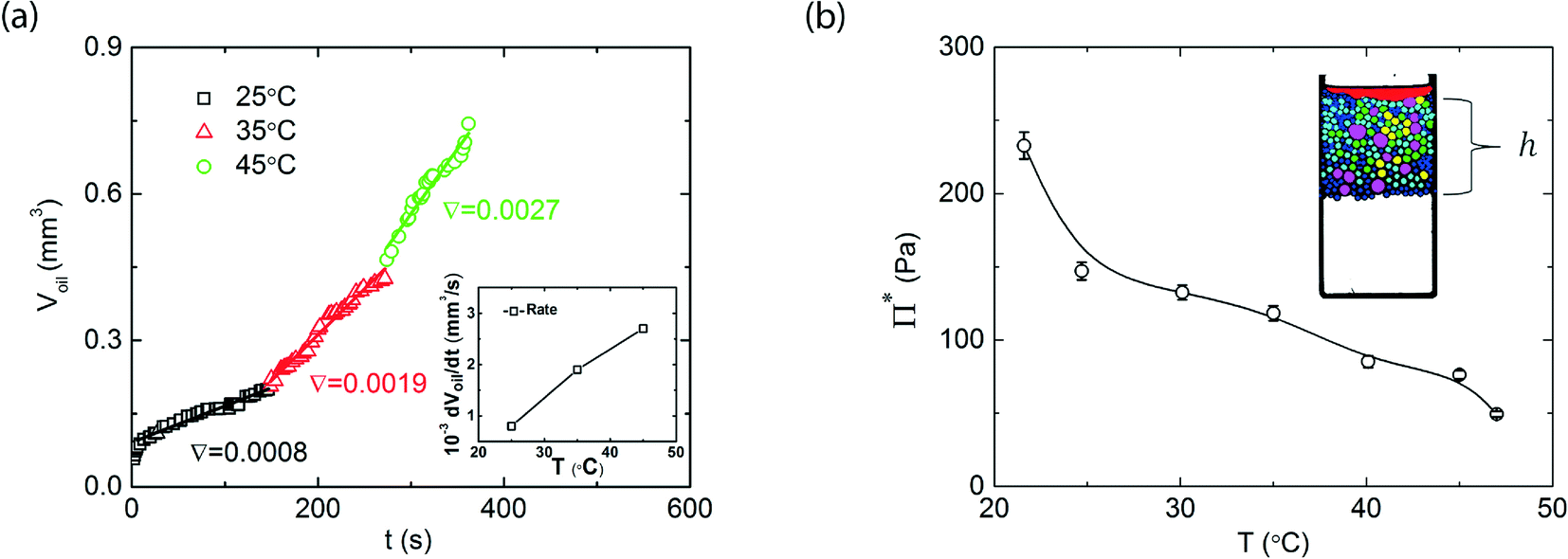 | ||
| Fig. 3 a) Coalesced volume as a function of time for three different temperatures, at a constant acceleration of 450g, inset shows the measured coalescence rate as a function of temperature. b) Critical disjoining pressures as a function of the temperature. | ||
The significant decrease in critical disjoining pressure with increasing temperature above the LCST also sheds new light on our previous observation that an emulsion, stabilized by these surfactants, could be triggered from stable to unstable with increasing the temperature;6 when an emulsion is prepared at osmotic pressures between the critical disjoining pressure at room temperature and that above the LCST, it will be stable at room temperature. Once the temperature is raised, the osmotic pressure will remain the same yet exceed the decreased critical disjoining pressure, leading to rapid destabilization.
Coalescence dynamics
The ratio of the actual osmotic pressure in the emulsion and the critical disjoining pressure determines whether coalescence can occur; however, this consideration is based on thermodynamic arguments and thus only gives insight into the equilibrium stability of the system. Once the critical disjoining pressure is (locally) exceeded, coalescence can occur. The kinetics of this process are governed by a complex interplay of the time scale for film rupture, typically assumed to be an activated process, film drainage, governed by hydrodynamic factors such as the film thickness and viscosity of the continuous phase, and in some cases, when the dispersed phase itself is highly viscous, the viscous flows associated with droplet merging.In our experimental approach, the kinetics of coalescence can be ideally studied; we record images at a frame rate of 5000 fps to capture still frames of the fast spinning sample cell. As the coalescence we observe occurs over relatively long time scales, we choose to analyse only 5 images s−1; more images are available and can be used for systems in which these processes occur much faster. Our image analysis gives access to the area of the bulk oil phase at the front of the sample, indicated in red, at the right hand side of the images in Fig. 2. As we know the height of the lithographically produced sample chamber precisely, we can track the change in volume of the bulk oil phase Voil, as shown in Fig. 3. Also here we can clearly see the effect of our switchable surfactant. While coalescence occurs relatively slowly, with a rate dVoil/dt = 8 × 10−4 mm3 s−1 at 25 °C, below the LCST of the surfactant, increasing the temperature to around the LCST, 35 °C, or above the LCST, at 45 °C, greatly enhances the coalescence rate. These results are summarized in the inset in Fig. 3a, which show the coalescence rates as a function of temperature. As a control experiment, to rule out that convective flows or other experimental artefacts influence our measurements, we repeat these experiments on an emulsion, stabilised by 10 mM SDS, which is not thermoresponsive and should be fully stable at these pressures. Indeed, we observe no coalescence at all temperatures or during rapid temperature changes (see ESI†).
As mentioned above, multiple factors influence the coalescence kinetics; one important contribution is the time required for the breaking of the thin film between two droplets. We can assume that film breaking is an activated process, as it occurs through an intermediate stage in which the total surface of the drops connected through a small liquid bridge15 is larger than that of the two individual drops; this intermediate state thus possesses a higher free energy, which provides an activation barrier against coalescence. The rate of transitions over this barrier, required for droplet coalescence, will depend on the absolute height of this energy barrier, but also on the difference ΔΠ = Π − Π*between actual pressure Π and critical disjoining pressure Π*. If the difference is negative, the film is stable and coalescence will not occur; if the difference is positive, but small, transitions over the energy barrier will occur but infrequently, leading to low coalescence rates, and when the difference becomes larger, individual coalescence events become more frequent, thus increasing the macroscopic coalescence rate. As an increase in temperature decreases the critical disjoining pressure for our thermoresponsive surfactants, as shown in Fig. 3a, but the centrifugal pressure remains constant, increases in temperature in effect increase ΔΠ, and thus increase the coalescence rate.
Modes of coalescence
While the analysis above, where we only consider the increase in volume of the bulk oil phase, gives direct access to the overall coalescence rate as a function of temperature, it does not describe how the coalescence process occurs spatially. Recently, we showed that coalescence in concentrated emulsions subjected to a unidirectional pressure can occur in two distinct modes.1 When the critical disjoining pressure is relatively low, coalescence occurs throughout the bulk of the sample in which several nuclei of coalesced drops form, which subsequently rapidly grow until the entire sample has been destabilized. By contrast, when the critical disjoining pressure of the system is high, the pressure is only high enough to induce coalescence at the front end of the sample; in this scenario, coalescence only occurs at the front where the emulsion meets the bulk oil phase. These observations suggest that the thermoresponsive emulsions we use here, in which we can gradually tune the disjoining pressure through small changes in temperature, could be suitable to induce a change from front to bulk coalescence on demand.At temperatures below the LCST of the surfactant, where the critical disjoining pressure is high, we indeed observe visually that coalescence only occurs at the front end of the sample (Fig. 2a). As expected for front coalescence, there should be little coarsening in the packed emulsion layer; histograms of the total amount of oil stored in droplets of various sizes, indeed show little changes, while the total amount of oil in the bulk oil layer steadily increases (Fig. 2b). From this we can extract the amount of coalescence occurring in the sample; hardly any coalescence occurs in the bulk (packed emulsion layer), and coalescence is completely dominated by droplets merging with the oil front (Fig. 2c). The local pressure in the packed emulsion layer subjected to centrifugation is lowest at the emulsion–water interface and reaches a maximum at the emulsion–oil front. When the initial critical disjoining pressure is high, at low temperatures, apparently the local pressure only exceeds the critical value at this front, leading to front coalescence. Note that the volume of coalesced droplets as shown in Fig. 2c, occasionally shows a small decrease; this is the noise on the data caused by the minor inaccuracy of the edge finding algorithm we apply to our images to identify the individual droplets and the homogeneous oil phase.
By contrast, when we repeat this experiment at higher temperatures, leading to a significant reduction in the initial emulsion stability, expressed by the lower critical disjoining pressure, we observe a completely different coalescence process (Fig. 4a). Droplets coalesce both with the homogeneous oil front and their droplet neighbors, leading to rapid coarsening of the packed emulsion bed (Fig. 4b). In this scenario, the local pressure exceeds the critical pressure in a much larger area of the emulsion layer, as a result coalescence occurs in the bulk of the sample. For this case, the total amount of coalescence is initially dominated by droplet–droplet coalescence events, until the packed emulsion layer becomes so coarse that these droplets also merge with the oil front (Fig. 4c). Emulsions which are initially marginally stable show a clear preference from droplet–droplet coalescence throughout the packed emulsion layer, even in areas where the total centrifugal pressure is relatively low. Note, full droplet size distributions for both scenarios are available in the ESI.†
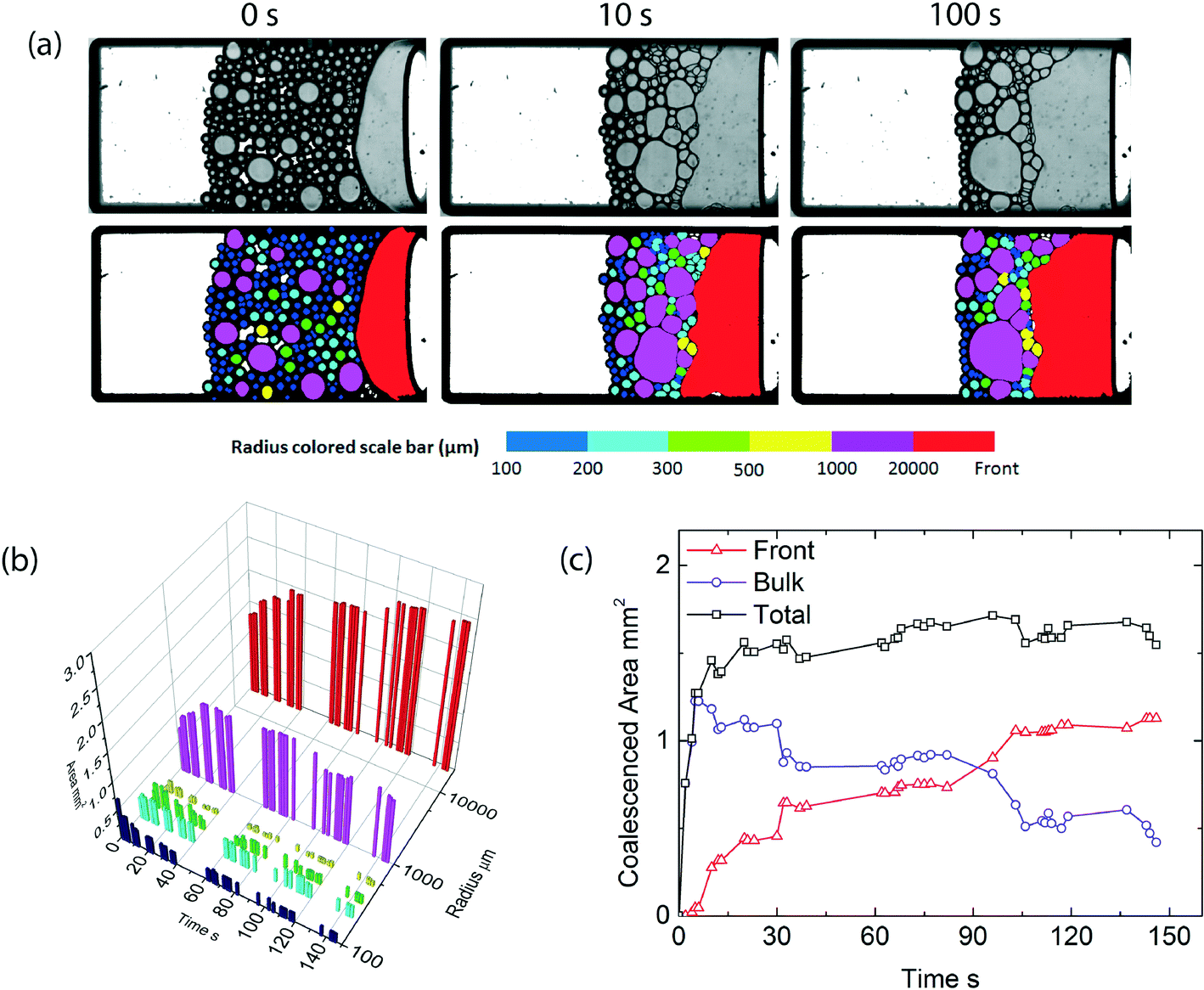 | ||
| Fig. 4 a) Images of a microfluidic centrifugation experiment, at T = 45 °C > TLCST, α = 450g, before (top) and after (bottom) image processing, at three different time intervals, b) volume of oil stored in various droplets size classes, and the continuous oil front, c) amount of coalesced oil as a function of time, separated into the amount coalesced at the macroscopic oil front and within the packed film of droplets. | ||
This highlights that the mode of coalescence, either occurring throughout the bulk of the sample, or being restricted to the front, is indeed determined by the disjoining pressure. Moreover, the introduction of thermoresponsivity to the emulsions allows us to trigger these two distinctly different modes on demand and study them at the scale of individual particles using the microcentrifugation method. For the system we study here, the transition from bulk to front coalescence occurs at a critical disjoining pressure of approximately 50 Pa. In a previous paper, dealing with emulsions stabilized by ionic surfactants, the transition from front-to-bulk coalescence occurred at much larger values for the critical disjoining pressure.1 We therefore speculate that these two modes we observe are a universal feature of any concentrated emulsion system, the critical value of the maximum disjoining pressure at which the transition occurs depends on a variety of system details such as the droplet size and size distribution, the strength of the capillary pressure gradient in the dense emulsions, which in turn depends on the nature of the stress (gravitational versus drying), and the specific nature of the surfactants or stabilisers used at the oil–water interface. Future work should elucidate which parameters influence the transition to arrive at a universal view of coalescence in dense emulsions.
Conclusion
Despite a wealth of research on the coalescence kinetics of two individual droplets dispersed in a continuous phase, the destabilisation and film formation in dense emulsions remain ill understood at the level of individual droplets. This is partially due to the complex nature of the problem in which equilibrium thermodynamics (critical disjoining pressures), activated processes (film rupture) and fluid dynamics (film drainage, fluid transport through the packed emulsion) interplay. Moreover, suitable methods to study coalescence in dense emulsions remain scarce; especially when control over the applied forces, thermodynamic properties and onset of coalescence are required. Here we reported on a new approach to study coalescence in dense thermoresponsive emulsions using a microfluidic-based microcentrifugation method. We have shown that both thermodynamic and kinetic properties can be measured through automated image analysis, and that the temperature-responsivity of the surfactants can be used to trigger different modes of coalescence on demand. These developments form a stepping stone for further investigations into the governing mechanisms that dominate emulsion (in)stability and film formation.Acknowledgements
The work of HF is part of the research programme of the Dutch Polymer Institute (DPI), project #675. JS & JvdG acknowledge the Netherlands Organization for Scientific Research (NWO) for financial support.Notes and references
- H. Feng, J. Sprakel, D. Ershov, T. Krebs, M. A. C. Stuart and J. van der Gucht, Soft Matter, 2013, 9, 2810–2815 RSC.
- N. Bremond, A. R. Thiam and J. Bibette, Phys. Rev. Lett., 2008, 100, 024501 CrossRef.
- C. Priest, S. Herminghaus and R. Seemann, Appl. Phys. Lett., 2006, 89, 134101 CrossRef PubMed.
- T. Krebs, D. Ershov, C. G. P. H. Schroen and R. M. Boom, Soft Matter, 2013, 9, 4026–4035 RSC.
- N. Bremond, H. Doméjean and J. Bibette, Phys. Rev. Lett., 2011, 106, 214502 CrossRef.
- H. Feng, N. A. L. Verstappen, A. J. C. Kuehne and J. Sprakel, Polym. Chem., 2013, 4, 1842–1847 RSC.
- W. L. Ong, J. S. Hua, B. L. Zhang, T. Y. Teo, J. L. Zhuo, N. T. Nguyen, N. Ranganathan and L. Yobas, Sens. Actuators, A, 2007, 138, 203–212 CrossRef CAS PubMed.
- T. T. Fu, Y. N. Wu, Y. G. Ma and H. Z. Li, Chem. Eng. Sci., 2012, 84, 207–217 CrossRef CAS PubMed.
- D. Exerowa, T. Kolarov and K. Khristov, Colloids Surf., 1987, 22, 161–169 CrossRef.
- S. Basu and M. M. Sharma, J. Colloid Interface Sci., 1996, 181, 443–455 CrossRef CAS.
- A. Hadjiiski, S. Tcholakova, I. B. Ivanov, T. D. Gurkov and E. F. Leonard, Langmuir, 2002, 18, 127–138 CrossRef CAS.
- G. Narsimhan, Colloids Surf., 1992, 62, 41–55 CrossRef CAS.
- J. Bibette, D. C. Morse, T. A. Witten and D. A. Weitz, Phys. Rev. Lett., 1992, 69, 2439–2442 CrossRef CAS.
- T. E. Kodger and J. Sprakel, Adv. Funct. Mater., 2013, 23, 475–482 CrossRef CAS.
- J. Lyklema, in Fundamentals of Interface and Colloid Science, 2005, ch. 6, vol. V Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00773e |
| This journal is © The Royal Society of Chemistry 2015 |