
A novel device to concurrently assess leukocyte extravasation and interstitial migration within a defined 3D environment†
Raffaella
Molteni‡
a,
Elena
Bianchi‡
b,
Paolo
Patete‡
c,
Monica
Fabbri‡
a,
Guido
Baroni‡
c,
Gabriele
Dubini‡
b and
Ruggero
Pardi‡
*ad
aDivision of Immunology, Transplantation and Infectious Diseases, Leukocyte Biology Unit, San Raffaele Scientific Institute, Milan, Italy. E-mail: pardi.ruggero@hsr.it; Fax: +39 02 2643 4723; Tel: +39 02 2643 4731
bDepartment of Chemistry, Materials and Chemical Engineering, Laboratory of Biological Structure Mechanics, Politecnico di Milano, Milan, Italy
cDepartment of Electronics, Information and Bioengineering, Politecnico di Milano, Milan, Italy
dVita-Salute San Raffaele University School of Medicine, Milan, Italy
First published on 29th September 2014
Abstract
Leukocyte extravasation and interstitial migration are key events during inflammation. Traditional in vitro techniques address only specific steps of cell recruitment to tissues and fail to recapitulate the whole process in an appropriate three-dimensional (3D) microenvironment. Herein, we describe a device that enables us to qualitatively and quantitatively assess in 4D the interdependent steps underlying leukocyte trafficking in a close-to-physiology in vitro context. Real-time tracking of cells, from initial adhesion to the endothelium and subsequent diapedesis to interstitial migration towards the source of the chemoattractant within the 3D collagen matrix, is enabled by the use of optically transparent porous membranes laid over the matrix. Unique features of the device, such as the use of non-planar surfaces and the contribution of physiological flow to the establishment of a persistent chemoattractant gradient, were assessed by numerical simulations and validated by proof-of-concept, simultaneous testing of differentially treated primary mouse neutrophils. This microfluidic platform offers new and versatile tools to thoroughly investigate the stepwise process of circulating cell recruitment to target tissues in vitro and to test novel therapeutics targeting various steps of the process.
Introduction
Leukocyte extravasation and interstitial migration are interdependent events that play crucial roles in the inflammatory response to injury or infection.1,2 Leukocyte extravasation is a higher-order process initiated with capture of free-flowing leukocytes and subsequent rolling onto the vascular endothelium, primarily mediated by selectins.3 These events precede and facilitate leukocyte recognition of chemokines immobilized on the endothelial apical surface leading to integrin-dependent arrest and adhesion strengthening.4–6 Shear-resistant leukocytes can crawl along and eventually migrate across the endothelial cells (diapedesis or transendothelial migration).7 After leaving the vascular region, leukocytes migrate through the interstitial tissue to approach the site of inflammation.8 In line with evidence suggesting that each of the above steps prepares the leukocytes for the subsequent one,2,9,10 emerging data support a role for transendothelial migration in regulating the behavior and cellular functions of leukocytes in the extravascular tissue.11 There is evidence that transmigrating neutrophils exert a mechanical force that may disrupt endothelial cell–cell contacts and influence penetration of the basement membrane.12 Moreover, transendothelial migration mediates changes in leukocyte integrin and protease expression/activation, which might increase the ability of leukocytes to interact with and penetrate the basement membrane. In turn, interaction with the basement membrane could facilitate leukocyte motility in the extracellular matrix and enhance cellular responses crucial for an effective inflammatory response.2,13Spatiotemporal analysis of leukocyte trafficking towards inflammatory sites has been limited by difficulties in recreating such a complex sequence of concurrent steps in vitro. Indeed, although a number of in vitro assays have increased knowledge about the mechanisms of leukocyte recruitment, conventional devices address only specific steps and fail to recapitulate the whole process in an in vivo-like milieu.14 A variety of established microfluidic-based technologies allow to monitor the first key sequence of events occurring in the vascular compartment, the multistep extravasation process, but are unable to concurrently address the migratory cell responses in the extravascular tissues. Indeed, the parallel-plate flow chamber15 and the capillary flow chamber with a cylindrical configuration16 are engineered to specifically investigate leukocyte behavior on adhesive substrates and, if possible, diapedesis under controlled fluid dynamic conditions. Moreover, the Boyden chamber,17 a traditional device developed for shear-free cell chemotaxis/transmigration assays, has been implemented in a transwell flow chamber for monitoring leukocyte transendothelial migration under flow.18 Further development of such technologies led to an advanced transwell flow chamber where endothelial and stromal cells are co-cultured onto two opposite sides of a porous filter to better mimic the in vivo vascular conditions.19 Conversely, other conventional devices address the specific ability of cells to migrate towards a gradient of chemokines within two-dimensional environments, typically occurring during leukocyte crawling along the vascular endothelium. They include the Dunn chamber20 and the Zigmond chamber,21 where cells migrate on a glass coverslip guided by a diffusive gradient across a bridge between two wells; the under-agarose assay,22 measuring cells migrating on a glass coverslip underneath an agarose gel across which a gradient is formed; and the micropipette set-up,23 releasing chemoattractants to create gradients and microfabricated devices able to generate stable linear chemokine gradients.24,25 Finally, more recent solutions allow the assessment of interstitial migration within a three-dimensional (3D) microenvironment, as it occurs during extravascular leukocyte migration, but fail to recapitulate the upstream vascular events. Basic collagen-based models have been established to examine leukocyte motility in a tissue-like context.26–28 More recent microfluidic devices have introduced the possibility of exposing cells embedded in a 3D extracellular matrix to more controlled and/or stable chemotactic gradients.29–32 Additionally, an advanced microfluidic tool has been engineered to model, in a shear-free microenvironment, leukocyte diapedesis across an endothelial layer and subsequent interstitial cell migration.33
Herein, we describe a microfluidic device that overcomes the limitations of the aforementioned in vitro assays by providing a comprehensive method for in vitro modeling of the whole process of leukocyte recruitment to target tissues. The platform allows us i) to reproducibly recapitulate in an in vivo-like microenvironment the interdependent steps underlying leukocyte extravasation and interstitial migration; ii) to perform real-time high-resolution 2D and 3D imaging of leukocyte extravasation and interstitial migration, respectively; and iii) to concurrently compare the behavior of multiple sets of differentially labeled leukocytes. Image processing and data analysis are performed using an in-house developed program together with a commercially available software tool. The possibility of modulating microenvironment-related parameters, including shear flow, extracellular matrix composition and chemoattractant gradients, makes this microfluidic chamber highly versatile. As such, it provides a method for deeply and reliably understanding the mechanisms of leukocyte recruitment and might help identify and validate novel anti-inflammatory drug targets. We assessed the potential of the above tool by investigating neutrophil trafficking in response to the inflammatory keratinocyte-derived chemokine (KC).
Materials and method
Additional methods about technical validation of the device, use of inhibitors, cell staining protocols, flow cytometry, culture and characterization of endothelial cells, development of cell tracking software, and statistical analysis are available in the ESI.†Device assembly
The microfluidic device described here (Fig. 1a) consists of two main steel components (top and bottom, Fig. 1b). Between them, a number of layers are assembled (Fig. 1b) in order to model the in vivo-like 3D microenvironment where leukocytes traffic (Fig. 1c). A functional multilayer chamber is obtained by assembling its components as follows (Fig. 1a, b):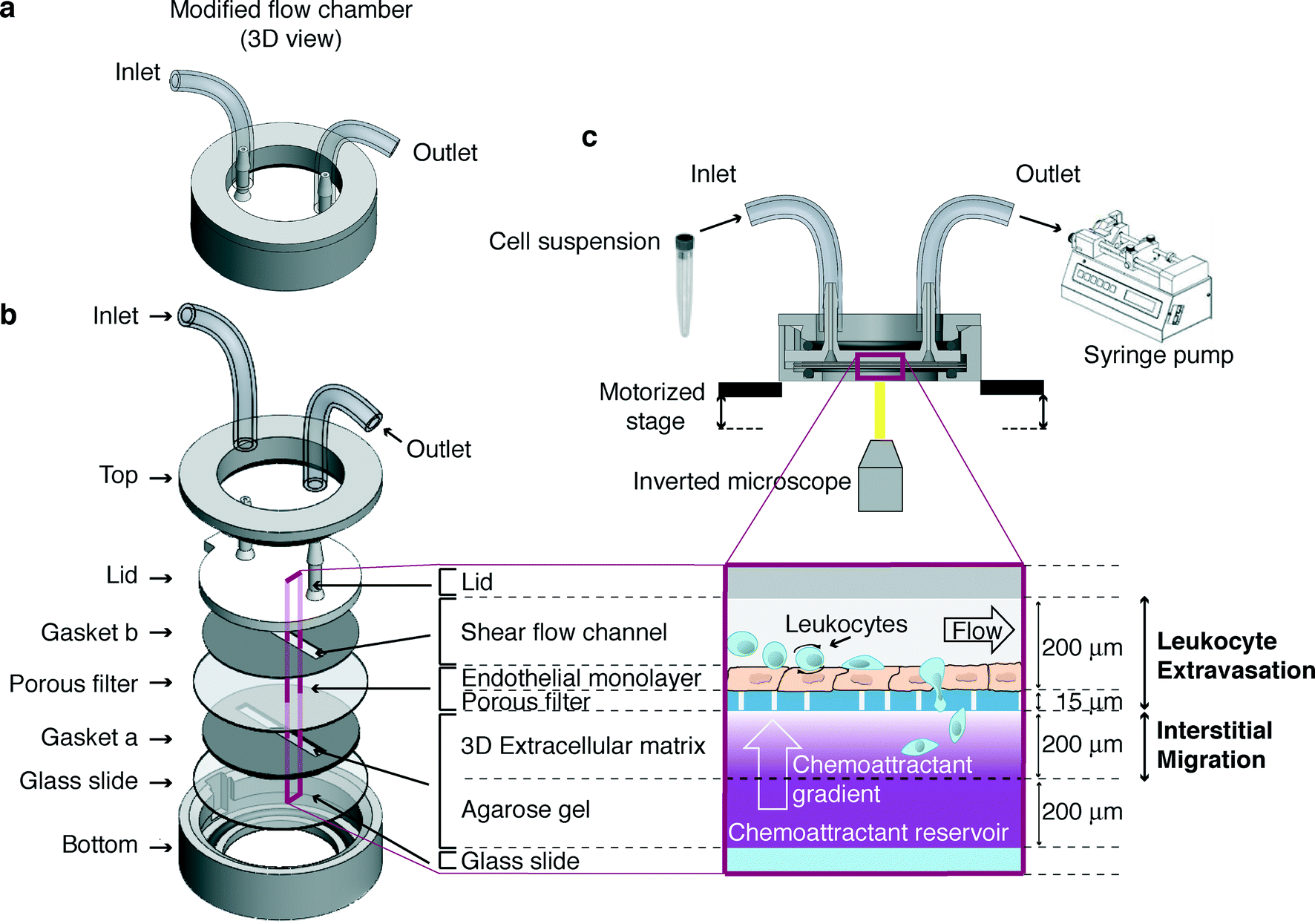 | ||
| Fig. 1 Overview of the microfluidic device. (a) Three-dimensional (3D) representation of the device. Tubing connected to the inlet and outlet ports allows cells to flow into the chamber and recapitulate leukocyte trafficking in vitro. (b) Schematics of the layers that are assembled during the device set-up. (c) Experimental set-up (not drawn to scale). Leukocytes enter through an inlet port and flow onto an endothelium-coated filter (inset corresponding to the observation area) under a controlled shear stress operated by a syringe pump. The filter lays over a 3D extracellular matrix where chemoattractants diffusing from an underlying reservoir can form a gradient. The multistep leukocyte extravasation process and interstitial migration are sequentially imaged by using an inverted microscope equipped with a motorized stage for 3D acquisition. | ||
1) a silicone gapped gasket (gasket “a” in Fig. 1b, 400 μm thick) is laid over a glass slide (25 mm diameter, VWR International);
2) the slide and the overlapped gasket are placed in the steel bottom component;
3) 1.2% agarose solution (UltraPure™ Agarose 1000; Life Technologies-Invitrogen) is prepared in Hank's balanced salt solution (HBSS; Life Technologies-Invitrogen) without phenol red, supplemented with 1 mM Ca2+/Mg2+, 10 mM HEPES (Gibco-Invitrogen) and 0.1% bovine serum albumin (BSA; Sigma-Aldrich). Upon cooling the solution at 37 °C, the keratinocyte-derived chemokine (KC, 2 μg mL−1 final concentration; R&D Systems) is either added or not;
4) the chemoattractant containing agarose solution is pipetted into the gasket “a” gap (50% of the gasket gap volume) and is allowed to gel for a few minutes;
5) a collagen I solution (1.6 mg mL−1 final concentration; BD Biosciences) is then pipetted over the agarose gel (50% of the gasket “a” gap volume) and is allowed to gel for 30 minutes at 37 °C. A slight positive meniscus should form when collagen is added in order to prevent air bubbles from being trapped when the filter is applied;
6) the endothelium-coated filter (Millipore®, 8 μm pore size), pre-coated with 2 μg mL−1 of KC, is placed over the gasket “a” with the endothelium-coated side facing up;
7) another silicone gapped gasket (gasket “b” in Fig. 1b, 200 μm thick) is allowed to adhere to the plastic lid, aligning the far ends of the channel-shaped gap with the inlet/outlet ports;
8) the lid and the adhered gapped gasket “b” are placed over the underlying layers, aligning the gasket “b” gap with the underlying one containing gelled matrices; the layers are aligned with one another and kept in place by coupling the lid feature to the cavity of the bottom piece;
9) the steel top component is then screwed to the bottom one;
10) finally, tubing is connected to the lid fitting, enabling cell suspension to enter the channel sucked by a programmable withdrawal syringe pump.
In such a platform, cells are allowed to i) flow onto the endothelial cells under a defined shear stress; ii) establish adhesive interactions; iii) transmigrate across the endothelial layer; and iv) interstitially migrate through the extracellular matrix where a chemoattractant gradient can be formed under an appropriate flow within the channel.
All components of the device are sterilized using 70% ethanol.
Computational modelling of wall shear stress distribution on the porous filter
The morphology of the porous filter surface, after settling on the collagen matrix, was reconstructed in a computer-aided design (CAD) in silico model to estimate the wall shear stress on the endothelial monolayer by means of computational fluid dynamic (CFD) techniques. Three completely assembled devices filled with working medium were observed with an inverted microscope (UltraVIEW ERS spinning disk confocal microscope, objective 20×; PerkinElmer) equipped with a motorized stage for 3D acquisition. A regular grid of about 150 points was used in the x–y plane (7 points along the x-direction and about 12 along the y-direction), while the origin for the z-coordinate axis was set at the lateral edges of the filter, where it is confined between the two silicone gaskets. A non-planar and non-porous surface (w = 2.5 mm in width and y = 10.5 mm in length, approximately) was then reconstructed by spline interpolation of the points and inserted in the 3D model of a portion of the flow chamber. The height of the channel at the lateral walls is 200 μm. The tolerance of the z-coordinate measurements was estimated as ±2 μm.Three independent CFD simulations (ANSYS.14; ANSYS, Inc., Canonsburg, PA, USA – Fluent) of fluid flowing in the flow channel were performed to test three selected conditions of wall shear stress stimuli with a flow of 160, 320, and 1060 μL min−1. Two simulations were set for each reconstructed flow channel, alternatively evaluating both the flow directions in the case of a flow rate of 1060 μL min−1. The working fluid was characterized by the density ρ = 1060 kg m−3 and the viscosity μ = 0.00076 Pa s, which are typical properties of the working fluid at 37 °C. The obtained wall shear stress was then normalized with reference to the value of the wall shear stress τwp in a planar parallel-plate flow chamber for a given flow rate (eqn (1)).
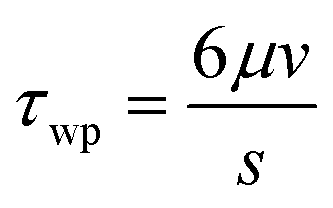 | (1) |
Computational modelling of chemokine gradient evolution in the multilayer chamber
The generation and persistence over time of a chemokine gradient across the collagen matrix were estimated by using a dedicated in silico model (ANSYS, Inc., Canonsburg, PA, USA – Fluent) representing a 3D portion of the multilayer chamber. Advection–diffusion of the chemokine (with a molecular weight of 8 kDa and a diffusion coefficient Dk = 1.42 × 10−6 cm2 s−1 (ref. 34)) across the four compartments (agarose gel, extracellular matrix, porous filter and flow channel) was modeled in a slice portion of the device representing the total height of the multilayer chamber. According to the nominal superficial density of pores (105 cm−2), Ø = 8 μm cylindrical pores were modeled through the thickness of the filter, connecting the upper flow channel to the matrix volume under the filter. Darcy's law (eqn (2)) was used to describe the flow in agarose (k = 9.26 × 10−16 m2 (ref. 35)) and extracellular matrix (k = 1.49 × 10−14 m2 (ref. 36)) compartments, which were modeled as porous media (porosity, 95%):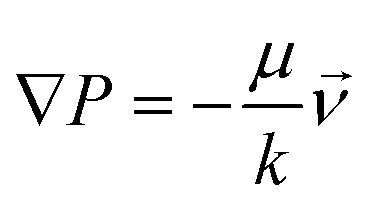 | (2) |
The simulations were conducted in the presence (320 μL min−1) and absence of flow in the upper flow channel. The contribution of the endothelial monolayer, thin (5–7 μm) and porous, was not included in the simulation (see details in the ESI†).
Cell culture
Murine neutrophils were differentiated from myeloid progenitors immortalized with estrogen-regulated Hoxb8, as previously described.37 Mouse progenitors were maintained undifferentiated in Opti-MEM® (Life Technologies-Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Euroclone), 1% penicillin/streptomycin (Life Technologies-Invitrogen), 30 μM beta-mercaptoethanol (Life Technologies-Invitrogen), 20 ng mL−1 stem cell factor (PeproTech) and 1 μM beta-estradiol (Sigma-Aldrich). Differentiation of myeloid progenitors into polymorphonuclear granulocytes was obtained by adding 20 ng mL−1 recombinant human granulocyte colony-stimulating factor (Myelostim – Italfarmaco) to the medium in the absence of estradiol.Brain endothelioma cells (bEnd.3; ATCC® catalog no. CRL-2299) were cultured in Dulbecco's modified Eagle's medium (Life Technologies-Invitrogen) supplemented with 15% FBS and 1% penicillin/streptomycin.
Murine neutrophils used in in vitro functional assays were suspended in HBSS without phenol red, supplemented with 1 mM Ca2+/Mg2+, 10 mM HEPES and 0.1% BSA.
In vitro assay to sequentially monitor leukocyte extravasation and interstitial migration
Neutrophils labeled with either Green or Orange CellTracker™ dyes were suspended in a working medium (3 × 106 cells per milliliter) and perfused through the flow chamber at a flow rate of 160 μL min−1 (0.12 Pa of expected shear stress) for 3 minutes. The flow rate was then increased to 320 μL min−1 (0.24 Pa of expected shear stress) till the end of the acquisition. The multistep leukocyte extravasation process was imaged bi-dimensionally for 4 minutes by focusing on the endothelial cell plane where leukocyte–endothelium interactions occurred. All cellular interactions with the substrate were determined by automated analysis of the first 3 minute acquisition with the in-house developed software DedICATE (DynamIc Cell Adhesion Tracking software). i) “Rolling” cells were defined as cells that roll for at least three cell diameters; ii) “transient” tethers were defined as cells attached briefly (<3 s) to the substrate; and iii) “arrest” (firm) tethers were defined as tethered cells that immediately stopped for at least 3 s.38 “Arrest” interactions of different time duration (10 s and 20 s) were analyzed. Frequencies of adhesive categories were determined as the percentage of total cells interacting with the endothelium.For analysis of adhesion strengthening, the number of cells arrested upon perfusion at 160 μL min−1 that detached under increased flow rate (320 μL min−1) was determined. The number of detaching cells was expressed relative to the number of cells that originally settled on the endothelial monolayer.
Once neutrophils have adhered onto and migrated across the endothelial cells, interstitial migration within the collagen matrix, occurring in the presence of flow within the channel (with a flow rate of 320 μL min−1), was imaged three-dimensionally for 30 minutes. Time-lapse imaging of 25 z planes spanning 100 μm across the collagen gel was performed. Z stacks were acquired between the top plane, proximal to the endothelial monolayer, and the bottom one, corresponding to the z plane which is 100 μm away from the top layer. Migrating cells were tracked manually with Volocity 3D Image Analysis Software (Perkin-Elmer) in order to quantify cell motility parameters including i) track length (cell path), defined as accumulated distance; ii) track vector (displacement or Euclidean distance), defined as the length of the straight line between the cell start point and end point; iii) speed, defined as velocity along the accumulated distance; and iv) directionality, defined as the ratio between Euclidean distance and accumulated distance (indicative of cell motility directionality).
For quantitative analysis of cell distribution throughout the z planes in the presence or absence of KC, cells in each z plane of a z stack were counted at the beginning (0 min) and at the end (30 min) of the 3D acquisition in the same observation field. The number of cells in each z plane was expressed relative to the total number of cells in the z stack at a selected time point.
Both leukocyte extravasation and interstitial migration under flow were performed at 37 °C.
Video acquisition
Real-time imaging of leukocyte trafficking was performed by using an UltraVIEW ERS spinning disk confocal microscope (set-up for fluorescence imaging, objective 20×; PerkinElmer) equipped with a stage incubator and a motorized stage for 3D acquisition.Movie analysis software
To analyze cell behavior during the extravasation process, an ad hoc software tool (DedICATE – DynamIc Cell Adhesion Tracking software) was developed. DedICATE is able to recognize each cell in the acquired movies and track its movement in the acquisition window. Such an analysis tool was implemented in C++ by means of third-party libraries such as Qt© (Digia Oyj, Helsinki, Finland) and OpenCV© (Itseez, Nizhny Novgorod, Russia). The cell movement analysis is made up of three steps: I) cell recognition, II) track reconstruction and III) track clustering (see details in the ESI†).Results
Microfluidic device design
The microfluidic device described here (Fig. 1a) consists of two main steel components (top and bottom, Fig. 1b). Between them, a number of layers are assembled (Fig. 1b) in order to model the in vivo-like 3D microenvironment where leukocytes traffic (Fig. 1c). Cells are allowed to enter through an inlet port and to flow along a channel created by a plastic lid and a silicone gapped gasket over an endothelium-coated filter. The channel design and the flow create a controlled wall shear stress on the endothelial cell monolayer, which can be modified during the assay by operating a syringe pump. This portion of the chamber mimics the vascular milieu where the multistep leukocyte extravasation process occurs. The polycarbonate filter (optically transparent, deformable and porous) lies over a gelled 3D collagen I matrix where a chemoattractant, if present, can diffuse from an underlying agarose gel serving as a reservoir of the chemoattractant. In the presence of an appropriate flow, this results in the formation of a concentration gradient by chemoattractant diffusion along the z-axis of the collagen matrix, which is perpendicular to the endothelial layer on top. Such collagen I-based and agarose gel layers are created sequentially into a silicone gapped gasket underlying the filter laid over a glass slide located in the bottom part of the device. This second portion of the chamber recapitulates the extravascular microenvironment where leukocyte 3D interstitial migration takes place.The operating set-up encompasses the above-described device suitably integrated in a system comprising a withdrawal syringe pump operated to generate the flow, a spinning disk confocal microscope (set-up for fluorescence imaging) equipped with a stage incubator and a motorized stage for 3D imaging, a software for real-time acquisition and analysis of interstitial migration, and an in-house developed software tool (DynamIc Cell Adhesion Tracking software – DedICATE) for image processing and analysis of leukocyte–endothelial cell interactions (Fig. 1c).
In summary, in our platform, leukocytes are allowed to flow onto the endothelial cells and establish a sequence of adhesive events (tethering, rolling, and arrest) culminating with transmigration across the endothelium and interstitial migration within a 3D extracellular matrix where a chemoattractant gradient perpendicular to the endothelial layer is formed.
Technical validation of the platform
The herein described microdevice aims at reconstituting a microenvironment characterized by the presence of a chemical gradient under a shear flow generating a controlled mechanical stimulus on cells.In order to validate the presence of a controlled wall shear stress on the filter surface, its geometrical configuration was reproduced in silico after measuring the z coordinates of a number of points on the filter surface. Fluid dynamic numerical simulations were performed in a flow chamber model incorporating the reconstructed surfaces. The contribution of the non-planar features, explained by the non-regular surface of the collagen matrix where the filter is overlaid, was evaluated. A color contour map of the wall shear stress on a reconstructed portion of the filter surface (in a representative non-planar configuration), normalized to the expected shear stress value in a planar parallel-plate flow chamber (τwp), is shown in Fig. 2a. Wall shear stress distribution, evaluated in a defined test area unaffected by boundary effects, was found to cover a range of values around the expected one with a maximum variability of +33%. The entity of the shear stress on the filter relates not only to the local non-planar conformation but also to the conformation of the whole filter: the local flow rate is actually affected by the distribution of the total flow rate along the cross section of the flow chamber. These findings confirmed that a suitable setting of the flow rate allows for the generation of a physiological wall shear stress in spite of the presence of non-planar surfaces within the chamber.
 | ||
| Fig. 2 Technical validation of the platform. (a) Contour map, from a 3D numerical model, of wall shear stress distribution on the reconstructed portion of the filter surface in a representative configuration. Shear stress values are normalized to the expected stress value τwp in a rectangular cross section channel. The outlined test area, which includes the biological observation field, corresponds to the region where flow is not affected by the presence of lateral walls and the inlet/outlet (~13.9 mm2, 0.35 mm from the lateral gasket and 1.4 mm from the inlet/outlet sections). For a flow rate equal to 160 μL min−1, the average shear stress imposed on the filter surface is 0.12 Pa. This results, in the defined test area, in a maximum shear stress on the surface of 0.16 Pa (red) and a minimum of about 0.12 Pa. For a flow rate equal to 320 μL min−1, the average shear stress imposed on the filter surface is 0.24 Pa. This results, in the defined test area, in a maximum shear stress on the surface of 0.32 Pa (red) and a minimum of about 0.24 Pa. (b–e) Chemokine distribution over time through the multilayer chamber resulting from 3D numerical simulations in the presence or absence of flow (320 μL min−1). A chemokine concentration gradient (ΔC/ΔZ) across the filter and collagen layers as a function of the distance from the filter along the z-axis (ΔZ) in the presence (b) or absence (d) of flow is depicted. Curve lines (enlarged in insets c and e) represent the chemokine concentration gradient distribution at different time intervals upon channel filling with fluid. (f) Difference of chemokine concentration across the filter and the collagen layers over time in the absence (blue) or presence (red) of flow. | ||
The generation and persistence over time of a chemokine gradient across the collagen matrix (Fig. 2b–e) were estimated by a dedicated time-dependent numerical simulation representing a 3D portion of the microchamber in the presence (Fig. 2b, c) or absence (Fig. 2d, e) of flow. All conditions were pre-set in order to reproduce the most critical experimental scenario corresponding to the fastest depletion of the chemokine gradient. Under both conditions, at t = 0 min, collagen and agarose were demonstrated to have the same chemokine concentration due to the diffusion phenomena occurring during gel polymerization and device set-up. Indeed, the time to reach this condition computed by an ad hoc simulation (the average concentration of chemokine in the collagen layer equal to 0.999 × 10−6 g mL−1) is about 14 min. At t > 0 min, the channel was filled by fluid. Simulation showed that the chemokine diffused in the fluid through the adluminal filter pores, giving rise to a chemotactic gradient in the collagen layer (Fig. 2b–e). The concentration of chemokine in the collagen matrix decreased in time (data not shown) as the chemotactic gradient did (Fig. 2b–e). Notably, under flow conditions (Fig. 2b, c), the chemokine gradient was higher and more persistent than in the absence of flow (Fig. 2d, e). As a consequence, the difference of chemokine concentration across the filter and the collagen increased over time in the presence of flow compared to the absence of flow (Fig. 2f). Interestingly, these findings demonstrate that our microdevice configuration enabled the establishment and maintenance of a chemotactic gradient, which was maximal in the presence of flow, to guide the cells for the entire duration of the assay.
In vitro modeling of concurrent steps in the extravasation process
The enabling potential of the device was demonstrated through a real-time in vitro reconstitution of leukocyte trafficking within vascular and extravascular in vivo-like microenvironments. We chose as a cellular model primary mouse neutrophils differentiated from reversibly immortalized myeloid precursors (Fig. 3a)37 as they play a key role in vascular inflammation.9,39 Such neutrophils exhibited morphological differentiation, upregulated typical surface differentiation markers including the neutrophil antigen Gr-1, the myeloid integrin Mac-1, and the KC receptor CXCR-2 and, consistently, downregulated the general myeloid transmembrane receptor CD177 and the macrophage marker F4/80 (Fig. S1†).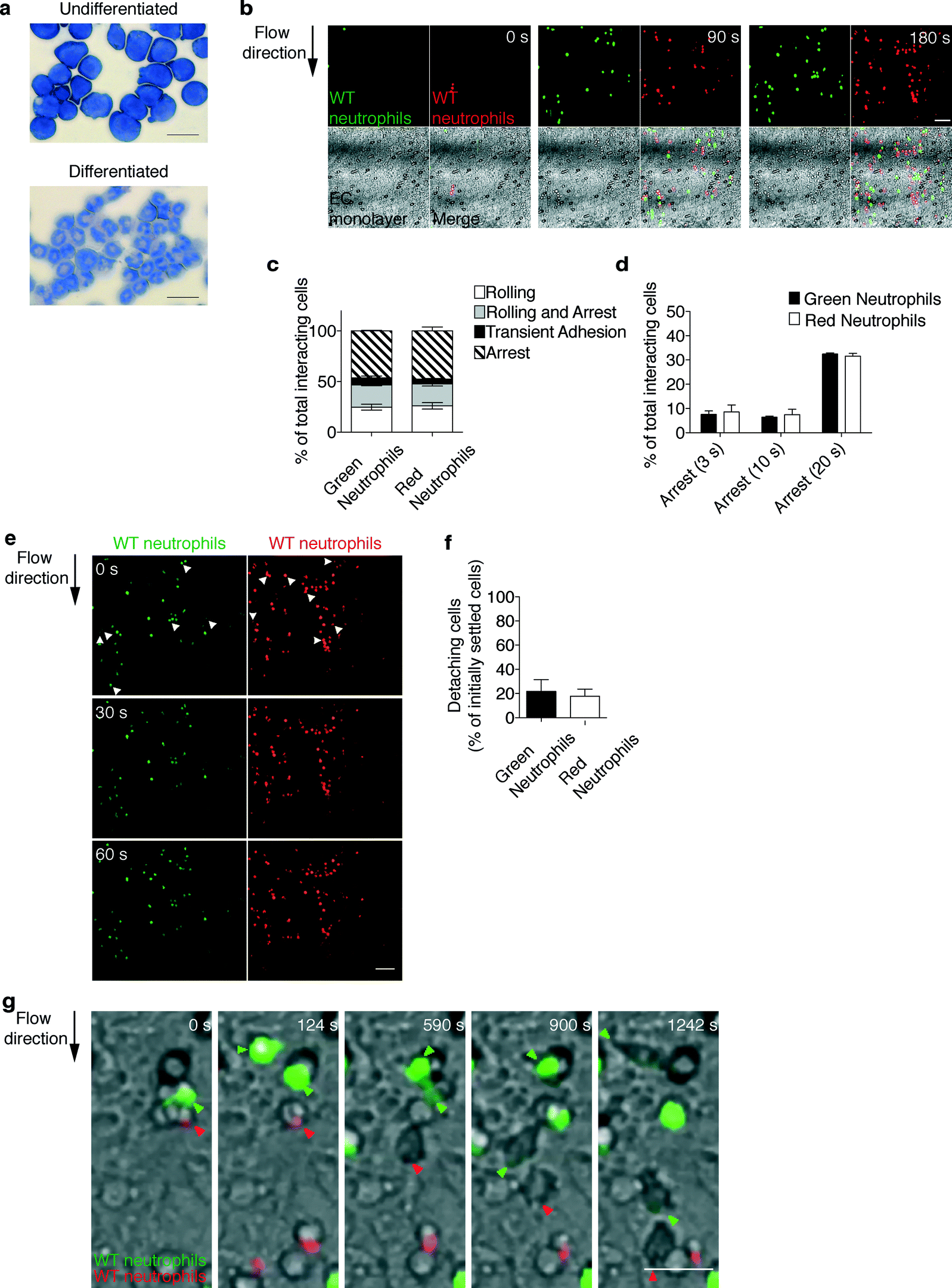 | ||
| Fig. 3 Real-time imaging and automated tracking of neutrophil extravasation. (a) Cellular model: murine neutrophils differentiated from immortalized myeloid precursors. Diff-Quik staining of myeloid precursors and differentiated neutrophils. Scale bars, 20 μm. (b–d) The multistep adhesion cascade: comparative analysis of green- and red-labeled wild-type (WT) neutrophils to determine the ability to roll and adhere onto the endothelial cells (EC) under flow. Representative time sequence (b) (see also movie S3†) of neutrophils flowing onto an endothelium-coated filter. Scale bar, 50 μm. Quantitative analysis of leukocyte–endothelium interactions (c) and cells arresting onto the endothelium for increasing times (d). Frequencies of adhesive categories are determined as the percentage of total interacting cells. Data are expressed as mean ± SEM of three independent experiments. (e, f) Comparative analysis of differentially labeled wild-type neutrophils to determine the ability to resist shear-induced detachment from endothelial cells. Representative fluorescent images (e) of neutrophils settled onto the endothelial cells under a defined shear stress (0.12 Pa of expected shear stress, 0 s), which are subjected to increased shear stress (0.24 Pa of expected shear stress, 30 s and 60 s). Arrows indicate settled cells detaching under increased shear stress (0.24 Pa). Scale bar, 50 μm. Quantitative analysis of cells detaching under increased flow rate (320 μL min−1, 0.24 Pa of expected shear stress) for 1 min (f). Data are expressed as mean ± SEM of three independent experiments. (g) Leukocyte transendothelial migration: representative time sequence of green- and red-labeled neutrophils (as indicated by arrows) migrating across the endothelial cells under shear flow (see also movie S4†). Fluorescence of cells moves out of focus due to their movement underneath the endothelium. Differential interference contrast imaging of the same microscopic field confirms that the fading fluorescence signal by individual arrested cells is a feature of transmigrating leukocytes. Scale bar, 20 μm. | ||
To perform a biological validation of the device, we first examined the multistep extravasation process of neutrophils flowing in the presence of physiological shear stress onto the keratinocyte-derived chemokine (KC)-coated endothelial cells (bEnd.3) treated with the inflammatory cytokine TNF alpha. bEnd.3 endothelial cells constitutively expressed the platelet endothelial cell adhesion molecule-1 (PECAM-1), which is involved in leukocyte transmigration, and showed TNF alpha-inducible expression of pro-adhesive molecules, including E-selectin and vascular cell adhesion molecule-1 (VCAM-1) (Fig. S2a†). Endothelial cells at confluency showed a similar distribution on the plate and filter, as demonstrated by the green-labeled endothelial cell pattern over such distinct surfaces (Fig. S2b†). The physiological relevance of the extravasation process recapitulated by our device was further confirmed by results showing that application of flow did not alter the density as well as the morphology of the endothelial layer (movies S1 and S2 and Fig. S2c†). To rule out the possibility that cell labeling with different cytosolic dyes could affect leukocyte behavior, we compared wild-type (WT) neutrophils, labeled with either Green or Orange CellTracker™ dyes, to determine the ability to i) tether, roll, or arrest onto the endothelial cells (EC) (Fig. 3b–d and movie S3†), ii) develop shear-resistant adhesion (Fig. 3e, f) and iii) migrate across the endothelium under flow (Fig. 3g and movie S4†). Data from time-lapse acquisition of neutrophil adhesion under flow conditions (Fig. 3b and movie S3†) were processed by the in-house developed software (DedICATE) for automated tracking and analysis of leukocyte–endothelium interactions. The custom-designed software is able to quickly track the cells and output their adhesive category (transient adhesion, rolling, rolling and arrest, or arrest for a selected time) based on cell velocity. As expected, green- and red-labeled neutrophils behaved similarly, with the majority of them establishing chemokine-induced arrest to the endothelium (Fig. 3c). Most of the arrested cells exhibited long-lasting adhesive interactions (>20 s, Fig. 3d) and resisted shear-induced detachment (Fig. 3e, f), which is a prerequisite for efficient diapedesis. Transendothelial migration (Fig. 3g and movie S4†) occurred comparably as well, as determined by manual scoring (data not shown) of cells whose fluorescence noticeably moved out of focus due to their movement underneath the endothelium. Differential interference contrast imaging of the same microscopic field confirmed that a fading fluorescence signal by individual arrested cells was a feature of transmigrating leukocytes.
In vitro modeling of interstitial migration
The main advancement of the device described here lies in the possibility of performing sequential imaging of leukocyte extravasation and the subsequent interstitial migration step. Indeed, once neutrophils in our platform have adhered onto and migrated across the endothelial layer (proximal to the top layer in Fig. 4a and movies S5 and S6†), their interstitial migration within the 3D collagen matrix (between the top and bottom layers) can be imaged three-dimensionally (Fig. 4a and movies S5 and S6†) and quantified (Fig. 4b–f). In order to validate this potential, WT green- and red-labeled neutrophils were compared to determine the ability to migrate in the x-, y-, and z-axes. To dissect the mechanisms underlying 3D interstitial migration that have only recently begun to emerge, we compared the leukocyte migratory behavior in the presence or absence of the inflammatory chemokine KC gradient along the z-axis of the collagen matrix (Fig. 4a and movies S5 and S6† from top to bottom). As expected, the migratory behavior of green and red cells was comparable (Fig. 4d). With respect to the absence of KC, in the presence of a KC gradient, neutrophils migrated farther (in the lower z planes) towards the source of the chemoattractant, as shown in Fig. 4a where the end time point (30 min) of the migration process under both conditions is depicted. Notably, real-time 3D imaging in our set-up allowed for a higher-content and more sophisticated analysis of cell motility than those in conventional assays. Indeed, 3D manual cell tracking revealed that in the presence of a chemotactic gradient, neutrophils were more motile (Fig. 4b) and showed higher displacements along the x-, y-, and z-axes (Fig. 4c). As expected, neutrophils showed significantly increased speed, displacement and directionality when exposed to the chemokine (Fig. 4e). Interestingly, while the cell distribution throughout the z planes at the beginning of the migration process was comparable under the two conditions (Fig. 4f, 0 min), the number of neutrophils migrated farther from the origin (the middle to bottom z planes) significantly increased in the presence of a KC gradient (Fig. 4f, 30 min). Altogether, these findings suggest that in the presence of a chemokine gradient within the collagen matrix, neutrophils are highly motile and display increased directional and persistent motility. Finally, the versatility of our device enables morphological studies and functional analyses. Indeed, high-resolution imaging revealed amoeboid features of neutrophil interstitial movement resulting from cycles of morphological expansion and contraction over time (Fig. 4g and movies S7 and S8†).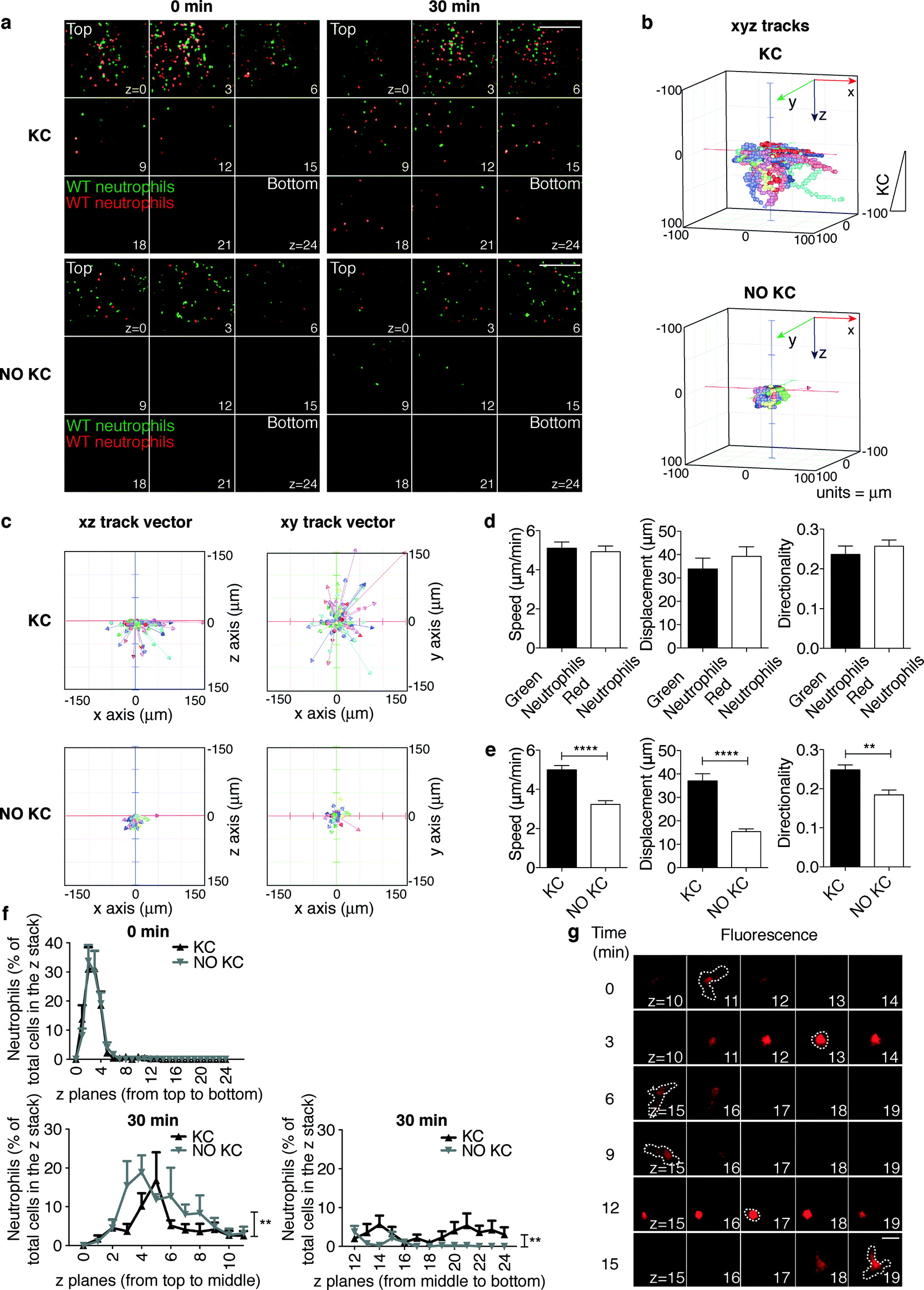 | ||
| Fig. 4 Real-time imaging and tracking of neutrophil interstitial migration in the presence or absence of a KC gradient. (a) Still images from representative movies (movies S5 and S6†) of neutrophil distribution throughout the z planes at the beginning (0 min) and at the end (30 min) of the migration process. 9 planes out of 25 (from z = 0, proximal to the endothelium, to z = 24) acquired along the z-axis through the collagen matrix are shown. Z step = 4.17 μm. Scale bar, 200 μm. (b–f) Quantification of neutrophil interstitial migration: 3D cell migration tracks (b); xz and xy track vectors (c); KC-dependent speed, displacement and directionality of differentially labeled neutrophils (d); neutrophil speed, displacement and directionality in the presence or absence of a KC gradient (e). **P = 0.001 and ****P < 0.0001 (unpaired Student t test); quantitation of cell distribution throughout the z planes (f) at time points 0 min and 30 min of the same observation field. **P < 0.01 for the two curves in the presence or absence of KC (two-way ANOVA). Data are representative of at least three independent experiments (mean and SEM in d and e; mean and SEM of six observation fields in f). At least 50 single cells/experiment/sample were tracked. (g) Amoeboid-like morphology of migrating neutrophils across the z planes. A time sequence of a representative neutrophil (see also movies S7 and S8†) is shown. Dotted lines trace cell morphology at each time point in the z plane where the cell is in focus, as indicated by its highest fluorescence. Scale bars, 10 μm. | ||
Simultaneous assessment of control versus pharmacologically inhibited neutrophils
To further validate the platform and gain insight into neutrophil trafficking during inflammation, we assessed the effect of pertussis toxin (PTX), an irreversible inhibitor of Gαi-coupled receptors, on interdependent steps of the extravasation process reconstructed in our in vitro system (Fig. 5 and S3 and movies S9 and S10†). Pretreatment with PTX (red cells) affected the ability of neutrophils to interact with the endothelium under flow (movie S9†) leading to an impairment in the firm adhesion step (Fig. 5a). PTX-treated neutrophils were less stably adherent, as indicated by a lower number of cells establishing long-duration adhesion (Fig. 5b), and were strikingly impaired in developing shear-resistant adhesion (Fig. S3a, b†). Notably, transendothelial migration as well was hindered by pretreatment with PTX (movie S10† and data not shown). In line with previous results in lymphocytes,40,41 these findings indicate that Gi protein signals are required for neutrophil firm arrest, adhesion strengthening and diapedesis.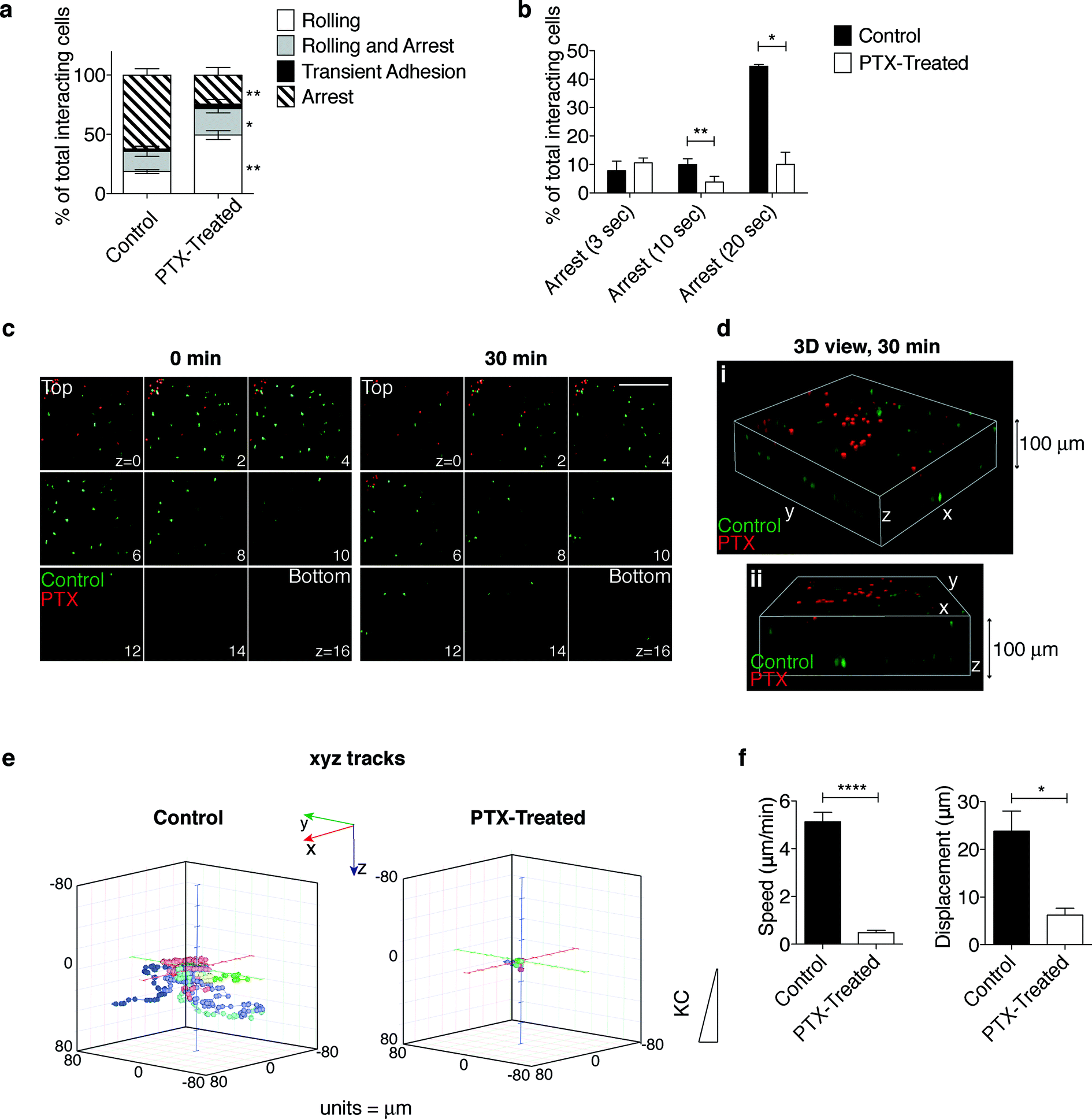 | ||
| Fig. 5 Effect of pertussis toxin (PTX)-mediated inactivation of Gαi protein on neutrophil extravasation and interstitial migration. (a, b) The multistep adhesion cascade: comparative analysis of control and PTX-treated neutrophils to determine the ability to roll and adhere onto the endothelial cells under flow. Quantitative analysis of leukocyte–endothelium interactions (a) and cells arresting onto the endothelium for increasing times (b) (see the representative time sequence of neutrophils flowing onto the endothelium-coated filter in movie S9†). Frequencies of adhesive categories are determined as the percentage of total interacting cells. *P < 0.05 and **P < 0.01 (paired Student t test). Data are expressed as mean ± SEM of three independent experiments. (c) Still images from a representative movie (movie S11†) of neutrophil distribution throughout the z planes at the beginning (0 min) and at the end (30 min) of the migration process in the presence of KC. 9 planes (from z = 0, proximal to the endothelial monolayer, to z = 16) out of 25 (from z = 0 to z = 24) acquired along the z-axis through the collagen matrix are shown. Z step = 4.17 μm. Scale bar, 200 μm. (d) 3D reconstruction of a representative observation field (two 3D views, i and ii, of the same field) at the end (30 min) of the migration process. (e, f) Quantification of neutrophil interstitial migration: 3D cell migration tracks (e); neutrophil speed and displacement (f). *P < 0.05 and ****P < 0.0001 (unpaired Student t test); data are representative of three independent experiments (mean and SEM). | ||
Time-lapse analysis of cells migrating towards KC through the collagen matrix after diapedesis (between the top and bottom layers, Fig. 5c, d and movie S11†) showed that PTX-treated cells, which displayed a severe impairment in transendothelial migration, were confined on the upper layers (Fig. 5c, 30 min, and 5d). Interestingly, 3D manual cell tracking demonstrated that while control neutrophils were highly motile, 3D migration and displacements along the x-, y-, and z-axes of PTX-treated cells were strikingly impaired (Fig. 5e and S3c†). Consistently, both cell speed and displacement were negatively affected by treatment with PTX (Fig. 5f).
Discussion
Modeling the complexity of leukocyte extravasation at sites of inflammation or injury is highly desirable, as each step in the process involves potentially “druggable” molecular targets. Prior work using both in vitro and in vivo models has established that the individual steps in the trans-endothelial migration process are tightly interconnected, i.e. each step is profoundly affected by the preceding one and in turn dictates the outcome of the next step in the process. As such, the overall process should be assessed at the systems level. The new device we have engineered aims at fulfilling the above requirements as it includes i) close-to-physiology, defined and adjustable 3D microenvironmental parameters such as shear stress over a non-planar vascular-like surface; ii) endothelial cells lining the shear flow-exposed surface and overlying the appropriate 3D extracellular matrix; iii) a flow-dependent establishment of a long-term chemoattractant gradient across the extracellular matrix; iv) sequential time-lapse high-resolution 2D and 3D imaging; v) a user-adjustable in-house developed software tool for quantitative analysis; vi) the possibility of concurrently comparing the behavior of multiple sets of differentially treated/labeled leukocytes.A prominent feature of our device is the combined usage of shear stress-exposed endothelium laid over a non-planar porous filter, which bears closer resemblance to the non-planar in vivo features of the vascular environment compared to solid phase supports. Flow numerical simulations demonstrate that physiological shear stress can be generated in spite of the presence of non-planar surfaces within the chamber. Furthermore, we have established (data not shown) that pre-coating of the porous membrane with cytokine-treated endothelial cells is a requirement for the entire process to occur, as the porous filter alone does not support transmigration even in the presence of an underlying chemokine gradient. Combined simulation and experimental validation of the device have revealed that shear flow along the endothelium-coated filter is actually required to establish and maintain a long-term chemoattractant gradient within the collagen matrix. Although the use of flow to generate stable chemoattractant gradients in microfluidic systems has already been established,24,42 the usage of shear flow within the vascular-like channel to create a gradient across the extracellular matrix below is distinct of our device geometry. Computational modeling demonstrates that flow along the channel, a key feature of the in vivo vascular environment, contributes to the establishment of a chemoattractant gradient across the matrix, which diffuses from the agarose gel (serving as a chemoattractant reservoir) to the channel fluid through the adluminal filter pores. Additionally, both the extracellular matrix composition and the microvascular-like structure (in vivo composed of endothelial cells, pericytes and a basement membrane) can be finely adjusted to suit different physiopathological contexts. The above features potentially allow to extend the range of cell types and pathophysiological conditions that could be modeled using our device. Extravasation of circulating tumor cells43 or homing of stem cells44 are but a few examples of relevant biological processes that could be assessed using this novel device.
By combining real-time high-resolution 2D (x, y) and 3D (x, y, z) imaging with ad hoc-developed analysis software, we can compare side-by-side multiple sets of differentially treated and labeled leukocytes. Leukocyte extravasation and interstitial migration can be thoroughly characterized both qualitatively and quantitatively in 2D and 3D, respectively. The in-house developed software DedICATE allows automatic fast tracking and quantitation of cell interactions with the endothelium during leukocyte extravasation. Moreover, commercially available software tools applied to our platform allow to measure three-dimensionally traditional migration-related parameters that in conventional assays are often acquired and quantified only two-dimensionally. Interestingly, high-resolution imaging of cell morphology enables additional analysis of cell migration patterns (e.g. the amoeboid pattern).
As a proof of concept for the biological validation of the tool, we compared differentially labeled/treated leukocytes to determine their ability to establish adhesion with the endothelium, transmigrate and migrate interstitially. Quantitative analysis of leukocyte behavior reveals that i) firm adhesion and transendothelial migration of murine neutrophils are strictly dependent on Gi protein signals and, ii) in the presence of a chemotactic gradient, neutrophils exhibit a persistent chemotactic amoeboid-like behavior within the extracellular matrix.
In summary, we have developed and validated both technically and biologically a microfluidic chamber that provides a versatile method for reliably and reproducibly investigating in vitro the mechanisms underlying leukocyte trafficking. The key features of this device are as follows: i) reconstruction of a physiological 3D microenvironment, ii) versatility, iii) easy handling and iv) capability of reproducing the whole process of leukocyte recruitment into tissues using a single assay, making it suitable for broad applications, from basic leukocyte biology studies to high-content functional readouts aimed at the identification and pre-clinical validation of novel anti-inflammatory drugs targeting inflammatory cell migration.
Acknowledgements
We wish to thank Mauro Biffi, Alembic – Advanced Light and Electron Microscopy BioImaging Center – Cesare Covino, for technical help, Carolina Lage Crespo for support in image analysis, Prof. Ronen Alon for critical discussion, Dr. Mark Kamps for providing the Hoxb8 construct, Gian Luigi Frigerio and Katia Laganà for helping with the device fabrication and Italfarmaco for providing Myelostim. Support: EMDM project (Regione Lombardia), AIRC-IG, PRIN 2010–2011 and RF-2011-02351229 project to R. P.References
- R. Molteni, M. Fabbri, J. R. Bender and R. Pardi, Curr. Opin. Cell Biol., 2006, 18, 491–498 CrossRef CAS PubMed.
- K. Ley, C. Laudanna, M. I. Cybulsky and S. Nourshargh, Nat. Rev. Immunol., 2007, 7, 678–689 CrossRef CAS PubMed.
- R. P. McEver, Curr. Opin. Cell Biol., 2002, 14, 581–586 CrossRef CAS PubMed.
- C. Laudanna, J. Y. Kim, G. Constantin and E. Butcher, Immunol. Rev., 2002, 186, 37–46 CrossRef CAS PubMed.
- R. Molteni, C. L. Crespo, S. Feigelson, C. Moser, M. Fabbri, V. Grabovsky, F. Krombach, C. Laudanna, R. Alon and R. Pardi, Blood, 2009, 114, 1073–1082 CrossRef CAS PubMed.
- R. Alon and K. Ley, Curr. Opin. Cell Biol., 2008, 20, 525–532 CrossRef CAS PubMed.
- W. A. Muller, Circ. Res., 2009, 105, 223–230 CrossRef CAS PubMed.
- P. Y. Lam and A. Huttenlocher, Curr. Opin. Cell Biol., 2013, 25, 650–658 CrossRef CAS PubMed.
- E. Kolaczkowska and P. Kubes, Nat. Rev. Immunol., 2013, 13, 159–175 CrossRef CAS PubMed.
- B. Yap and R. D. Kamm, J. Appl. Physiol., 2005, 98, 1930–1939 CrossRef PubMed.
- S. Nourshargh, P. L. Hordijk and M. Sixt, Nat. Rev. Mol. Cell Biol., 2010, 11, 366–378 CrossRef CAS PubMed.
- A. Rabodzey, P. Alcaide, F. W. Luscinskas and B. Ladoux, Biophys. J., 2008, 95, 1428–1438 CrossRef CAS PubMed.
- S. Nourshargh and F. M. Marelli-Berg, Trends Immunol., 2005, 26, 157–165 CrossRef CAS PubMed.
- E. Bianchi, R. Molteni, R. Pardi and G. Dubini, J. Biomech., 2013, 46, 276–283 CrossRef PubMed.
- C. Lawson, M. Rose and S. Wolf, Methods Mol. Biol., 2010, 616, 31–47 CAS.
- B. M. Cooke, S. Usami, I. Perry and G. B. Nash, Microvasc. Res., 1993, 45, 33–45 CrossRef CAS PubMed.
- S. Boyden, J. Exp. Med., 1962, 115, 453–466 CrossRef CAS PubMed.
- T. H. Schreiber, V. Shinder, D. W. Cain, R. Alon and R. Sackstein, Blood, 2007, 109, 1381–1386 CrossRef CAS PubMed.
- H. M. McGettrick, A. Filer, G. E. Rainger, C. D. Buckley and G. B. Nash, Biochem. Soc. Trans., 2007, 35, 1161–1162 CrossRef CAS PubMed.
- D. Zicha, G. A. Dunn and A. F. Brown, J. Cell Sci., 1991, 99(Pt 4), 769–775 Search PubMed.
- S. H. Zigmond, J. Cell Biol., 1977, 75, 606–616 CrossRef CAS PubMed.
- R. D. Nelson, P. G. Quie and R. L. Simmons, J. Immunol., 1975, 115, 1650–1656 CAS.
- G. Gerisch and H. U. Keller, J. Cell Sci., 1981, 52, 1–10 CAS.
- N. Li Jeon, H. Baskaran, S. K. Dertinger, G. M. Whitesides, L. Van de Water and M. Toner, Nat. Biotechnol., 2002, 20, 826–830 CrossRef PubMed.
- D. Irimia, S. Y. Liu, W. G. Tharp, A. Samadani, M. Toner and M. C. Poznansky, Lab Chip, 2006, 6, 191–198 RSC.
- M. Sixt and T. Lammermann, Methods Mol. Biol., 2011, 769, 149–165 CAS.
- K. Wolf, S. Alexander, V. Schacht, L. M. Coussens, U. H. von Andrian, J. van Rheenen, E. Deryugina and P. Friedl, Semin. Cell Dev. Biol., 2009, 20, 931–941 CrossRef CAS PubMed.
- M. Gunzer, A. Schafer, S. Borgmann, S. Grabbe, K. S. Zanker, E. B. Brocker, E. Kampgen and P. Friedl, Immunity, 2000, 13, 323–332 CrossRef CAS PubMed.
- V. V. Abhyankar, M. W. Toepke, C. L. Cortesio, M. A. Lokuta, A. Huttenlocher and D. J. Beebe, Lab Chip, 2008, 8, 1507–1515 RSC.
- W. Saadi, S. W. Rhee, F. Lin, B. Vahidi, B. G. Chung and N. L. Jeon, Biomed. Microdevices, 2007, 9, 627–635 CrossRef PubMed.
- U. Haessler, Y. Kalinin, M. A. Swartz and M. Wu, Biomed. Microdevices, 2009, 11, 827–835 CrossRef CAS PubMed.
- E. K. Sackmann, E. Berthier, E. W. Young, M. A. Shelef, S. A. Wernimont, A. Huttenlocher and D. J. Beebe, Blood, 2012, 120, e45–53 CrossRef CAS PubMed.
- S. Han, J. J. Yan, Y. Shin, J. J. Jeon, J. Won, H. E. Jeong, R. D. Kamm, Y. J. Kim and S. Chung, Lab Chip, 2012, 12, 3861–3865 RSC.
- X. Zhao, S. Jain, H. Benjamin Larman, S. Gonzalez and D. J. Irvine, Biomaterials, 2005, 26, 5048–5063 CrossRef CAS PubMed.
- E. M. Johnson and W. M. Deen, AIChE J., 1996, 42, 1220–1224 CrossRef CAS.
- S. Ramanujan, A. Pluen, T. D. McKee, E. B. Brown, Y. Boucher and R. K. Jain, Biophys. J., 2002, 83, 1650–1660 CrossRef CAS PubMed.
- G. G. Wang, K. R. Calvo, M. P. Pasillas, D. B. Sykes, H. Hacker and M. P. Kamps, Nat. Methods, 2006, 3, 287–293 CrossRef CAS PubMed.
- R. Pasvolsky, S. W. Feigelson, S. S. Kilic, A. J. Simon, G. Tal-Lapidot, V. Grabovsky, J. R. Crittenden, N. Amariglio, M. Safran, A. M. Graybiel, G. Rechavi, S. Ben-Dor, A. Etzioni and R. Alon, J. Exp. Med., 2007, 204, 1571–1582 CAS.
- M. Phillipson and P. Kubes, Nat. Med., 2011, 17, 1381–1390 CrossRef CAS PubMed.
- Z. Shulman, S. J. Cohen, B. Roediger, V. Kalchenko, R. Jain, V. Grabovsky, E. Klein, V. Shinder, L. Stoler-Barak, S. W. Feigelson, T. Meshel, S. M. Nurmi, I. Goldstein, O. Hartley, C. G. Gahmberg, A. Etzioni, W. Weninger, A. Ben-Baruch and R. Alon, Nat. Immunol., 2012, 13, 67–76 CrossRef CAS PubMed.
- R. A. Warnock, S. Askari, E. C. Butcher and U. H. von Andrian, J. Exp. Med., 1998, 187, 205–216 CrossRef CAS PubMed.
- G. M. Walker, J. Sai, A. Richmond, M. Stremler, C. Y. Chung and J. P. Wikswo, Lab Chip, 2005, 5, 611–618 RSC.
- N. Reymond, B. B. d'Agua and A. J. Ridley, Nat. Rev. Cancer, 2013, 13, 858–870 CrossRef CAS PubMed.
- D. J. Laird, U. H. von Andrian and A. J. Wagers, Cell, 2008, 132, 612–630 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Including supplementary materials and methods, three supplementary figures, supplementary movies and supplementary movie legends. See DOI: 10.1039/c4lc00741g |
| ‡ Author contributions: R. M., E. B., G. D., and R. P. invented and developed the device. R. M. coordinated the study, designed and performed the biological validation of the device, including the refinement of the image analysis software, and wrote and revised the manuscript and the figures. E. B. performed the technical validation and wrote the related part of the manuscript. P. P. developed the software “DynamIc Cell Adhesion Tracking software” (DedICATE) and wrote its respective method. M. F. generated the mouse myeloid progenitor clone for neutrophil differentiation. G. B. supervised the development of the software “DynamIc Cell Adhesion Tracking software” (DedICATE). G. D. supervised the technical validation of the device and the related manuscript write-up. R. P. designed the study, contributed to writing the manuscript, and supervised the whole project and the manuscript write-up. |
| This journal is © The Royal Society of Chemistry 2015 |