DOI:
10.1039/C4GC02052A
(Paper)
Green Chem., 2015,
17, 1572-1580
Enhanced catalytic activity of palladium nanoparticles confined inside porous carbon in methanol electro-oxidation†
Received
21st October 2014
, Accepted 4th December 2014
First published on 5th December 2014
Abstract
The effects of the confinement of Pd nanoparticles (NPs) inside hollow carbon matrices are evaluated based on their performance in methanol (MeOH) electro-oxidation (EOx). The performance of three catalyst systems were compared: (1) Pd NPs exclusively confined inside the hollow carbon NPs, (2) Pd NPs distributed between the pores and the surface of porous carbon and (3) Pd NPs bound to the surface of activated charcoal. When NPs are exclusively confined, the electrocatalytic activity improves due to the higher proximity of the NPs to the reactants inside the cavity and minimal leaching of the NPs over 300 cycles. NP confinement yields a high anodic peak current density (AP-CD) of 142 mA cm−2 (626 cm2 mgPd−1) at a lower anodic potential of −0.3 V and an excellent re-usability, as compared to the partially confined and exposed NPs. A minimal loading of 1.2–2.5 wt% Pd NPs inside the mesoporous carbon resulted in a high electrochemically active surface area (ECSA), which clearly underlines the positive effect of the confinement of metal NPs.
Introduction
The high energy conversion efficiency and power density of direct MeOH fuel cells (DMFCs) makes them attractive to address the demand for clean and renewable energy.1 DMFCs utilize the MeOH EOx reaction and provide easy storability and transport of MeOH, cost-effectiveness of the cell design and eco-friendly applications.2–4 Different heterogeneous nanocatalysts with various morphologies are being explored for MeOH EOx in order to enhance the activity of DMFC.5,6 In a confined geometry, embedding the catalyst NPs inside nanochannels or pores has the potential to increase the catalytic performance of nanocatalysts.7,8 The confinement effect on the catalyst NPs implies an enhancement of the catalytic activity as the confined space provides the ideal conditions to entrap the nanocatalyst particles, however providing the leeway for catalysis. In the last decade, metal NPs encapsulated within hollow nanostructures have generated a great deal of attention in heterogeneous catalysis.9–11
Among the confining supports, carbon nanomaterials are most popular because of their chemical stability, regenerability, mechanical strength, high porosity and ease of surface functionalization.12 One-dimensional carbon nanostructures such as tubes, fibers and whiskers,13,14 along with nanospheres and cages,15 are desirable host materials for anchoring and confining the catalyst NPs in heterogeneous catalysis reactions. However, the most commonly used support is mesoporous carbon because of its large surface area,16 where the NPs can be embedded inside the pores or immobilized on the surface.17–24 Among the catalyst NP systems, Pd NPs demonstrate excellent catalytic activity towards various heterogeneous catalysis reactions.25–28 The Pd nanocatalysts are the best balance between cost-effectiveness and decent activity as compared to other metal NPs. The Pd NPs have a versatile applicability and are the best alternative to the expensive Pt in fuel cells.4,29 Although Pd NPs confined inside the carrier support were tested in organic molecular transformations,30 so far there is no report on the confinement effect of Pd NPs in the electro-oxidation of alcohols.
In this context, encapsulating the Pd NPs within well-defined carbon nanostructures could enhance the activity of the nanocatalyst. Pd NPs are encapsulated inside the pores of carbon NPs and the performance of these confined electrocatalysts is enhanced in MeOH EOx. Two confined systems were synthesized. A 150–200 nm Pd@C1 system was synthesized from Pd@SiO2 by successive C-coating of Pd@SiO2@C and the removal of the SiO2 template. The second partially confined Pd@C2 system was synthesized by encapsulating Pd NPs within carbonized sucrose, followed by annealing. The carbon confined Pd NPs exhibited an excellent catalytic activity and reusability in comparison with a conventional Pd/C system comprising Pd NPs chemically immobilized on activated charcoal. The simple strategy of NP confinement may be generalized to other metal NPs confined inside various nanostructured supports for an improved performance in fuel cells and other applications.22,23
Results and discussion
Structural and morphological studies
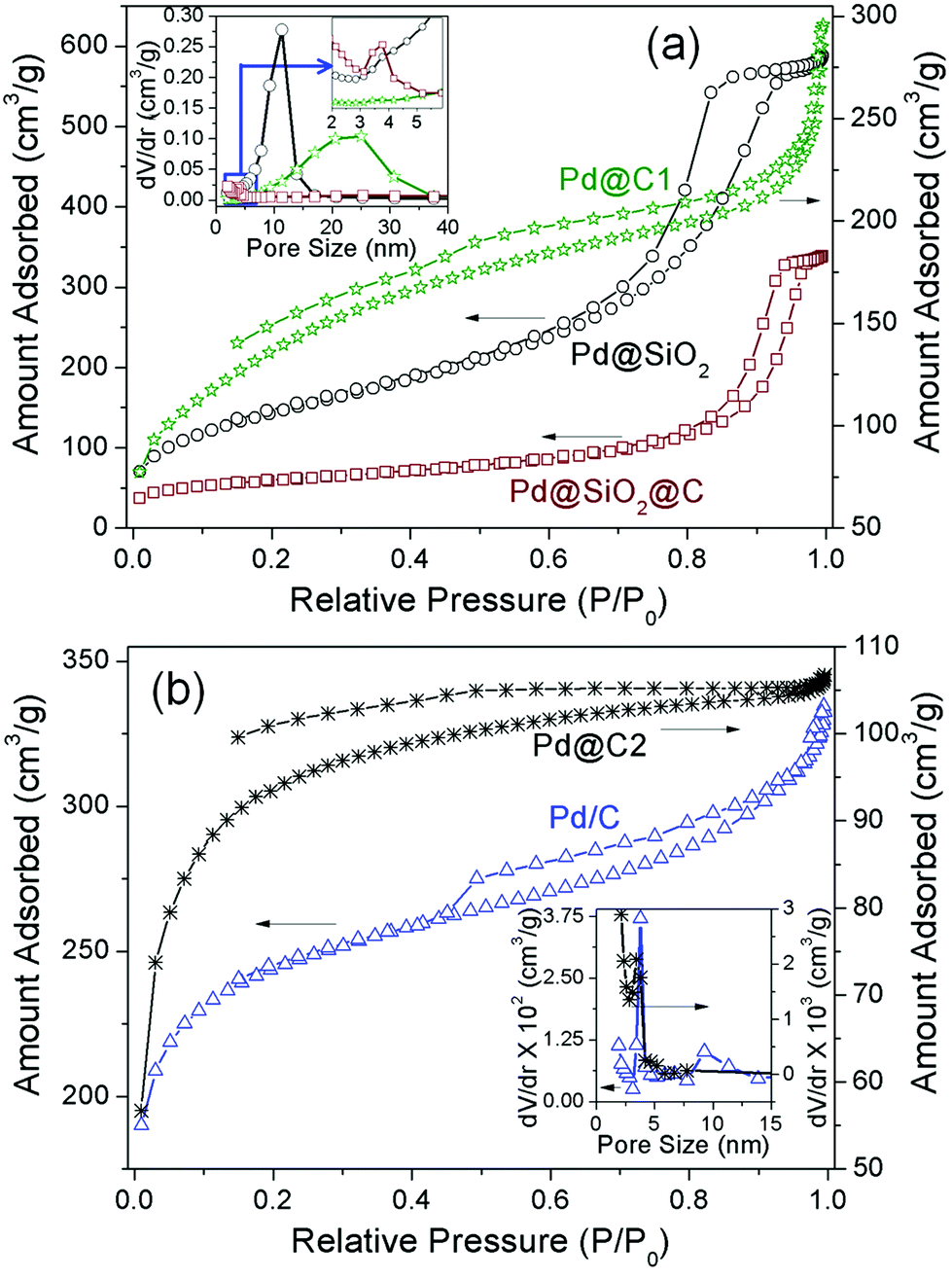
The surface area and porosity measurement studies provide an insight into the structural evolution and confinement effects of the Pd@C1 catalyst. Fig. 1a shows the N2 adsorption–desorption curves and the pore characteristics, the parameters of which are tabulated in Table 1. The N2 sorption curve of Pd@SiO2 NPs exhibits a type IV isotherm with an H2 hysteresis loop showing the limited uptake of the absorbate at a high relative pressure (P/P0) which indicates capillary condensation inside the 5–15 nm interconnected open mesopores in the SiO2 shell.31,32 After carbonization and annealing, the surface area reduces by 62% in Pd@SiO2@C due to the blocking of the SiO2 pores by the carbon layer. Consequently the hysteresis loop changes to H1 type due to the constriction of the pores, evident from a much lower pore volume and very small pore size distribution. HF etching of SiO2 leads to a 3.3 times increase in surface area along with an increase in pore volume and broad pore size distribution in Pd@C1. The non-reversible Type II isotherm is indicative of both monolayer and multilayer adsorption. The hollow cavities created by HF treatment initially adsorb the N2 molecules, followed by the appearance of a linear middle region in the sorption curve which shows the completion of monolayer adsorption and the beginning of a multilayer adsorption. In spite of the huge increase in surface area for Pd@C1, the slight increase in the total micropore volume can be attributed to graphitization and hence desensitization of the outermost carbon layer to N2 absorption after annealing at 900 °C.33 In fact the low pressure hysteresis is a signature of sub-nm wide pores associated with the irreversible uptake of adsorbate (N2) molecules.25 These Å-wide pores in the C-shell provide the percolation pathway for the KOH and methanol molecules to reach the confined Pd NPs for the latter's electro-oxidation. The N2 sorption characteristics of Pd@C2 and Pd/C are presented in Fig. 1b and Table 1. The commercial activated charcoal in Pd/C possesses a high surface area of 779 ± 5 m2 g−1, with pores of diameter 3.8 and 9.2 nm, whereas Pd@C2 has a comparatively lesser surface area of 302 ± 1 m2 g−1. The Pd NPs confined inside the bulk of Pd@C2 have access to the reactant molecules through the open 3.4 nm pores as well as the sub-nm pores.
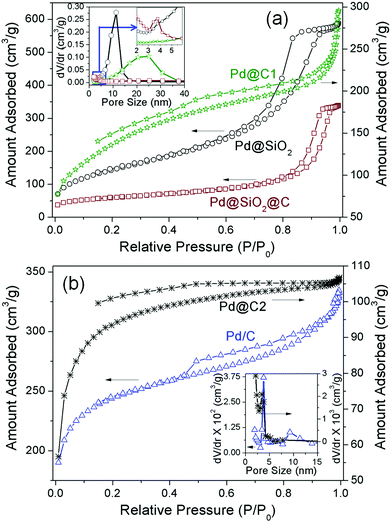 |
| | Fig. 1 N2 sorption isotherms and (inset) pore size distributions. | |
Table 1 BET surface area (SBET), total pore volume (Vtotal) and pore diameter (D) of the samples
| Sample |
S
BET (m2 g−1) |
V
total (cm3 g−1) |
D (nm) |
| Pd@SiO2 |
520 ± 3 |
0.86 ± 0.01 |
9.5 (sharp) |
| Pd@SiO2@C |
198 ± 3 |
0.480 ± 0.005 |
2.5 (weak) |
| Pd@C1 |
653 ± 3 |
0.6 ± 0.03 |
20.0 (broad) |
| Pd@C2 |
302 ± 1 |
0.02 |
3.4 |
| Pd/C |
779 ± 2 |
0.2 ± 0.03 |
3.8 (sharp), 9.2 (broad) |
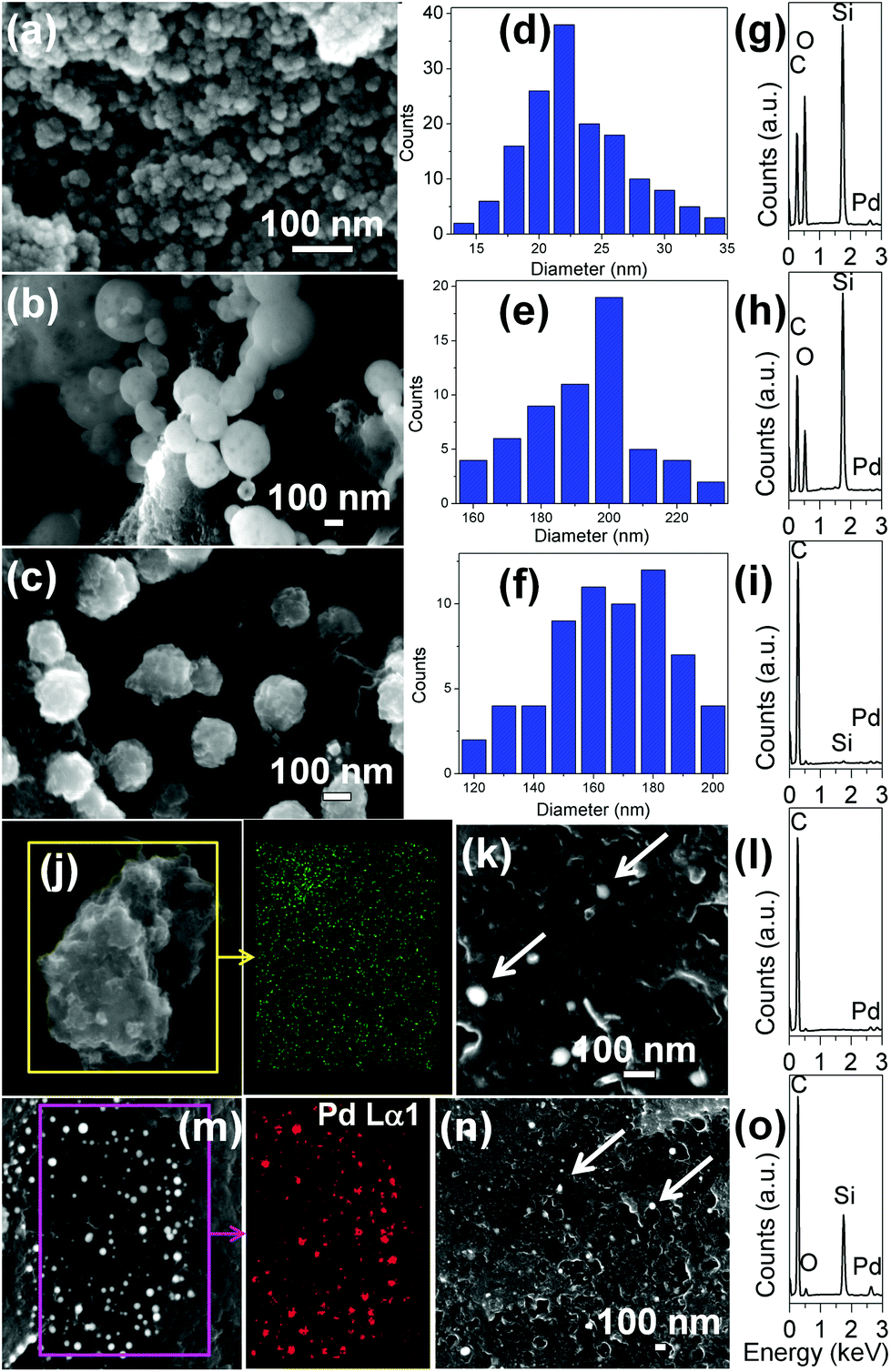
The field emission scanning electron microscopy (FESEM) images and elemental analyses (Fig. 2a–j) reveal the structural properties of Pd@C1. After carbonization and annealing of ∼22 nm Pd@SiO2 NPs, 180–200 nm Pd@SiO2@C particles were obtained. HF treatment of the Pd@SiO2@C particles breaks the continuous C-layer of Pd@SiO2@C resulting in well-defined 150–180 nm Pd@C1 particles with an uneven surface and 0.8 wt% leftover SiO2 (Fig. 2g–i). Pd Lα1 mapping in Fig. 2j shows the uniform distribution of Pd NPs. In the absence of the SiO2 template in Pd@C2, the Pd NPs are not perfectly confined since a large fraction of the NPs (arrows in Fig. 2k) reside on the carbon surface. Fig. 2m indicates the distribution of Pd NPs both inside (weaker dots) and outside (brighter dots) the mesoporous matrix. The partial NP confinement in Pd@C2 emphasizes the importance of the SiO2 template to generate catalyst NPs exclusively confined inside C-matrices of well-defined morphologies. The third Pd/C sample has Pd NPs uniformly dispersed over the surface of activated charcoal as observed in Fig. 2n. The average Pd wt% in Pd@C1, Pd@C2 and Pd/C was found to be 1.2, 2.4 and 2.5, respectively by energy dispersive X-ray spectral (EDS) analyses.
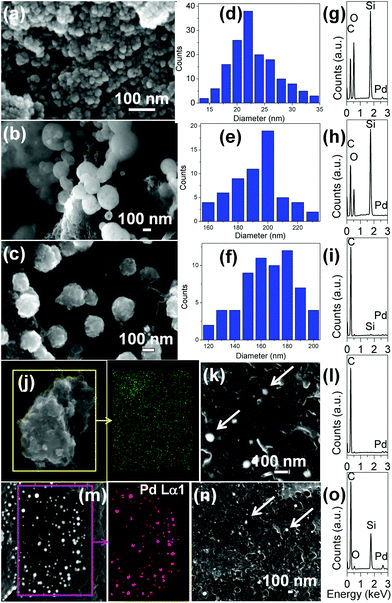 |
| | Fig. 2 FESEM images of (a) Pd@SiO2, (b) Pd@SiO2@C, (c) Pd@C1, diameter histograms of (d) Pd@SiO2, (e) Pd@SiO2@C, (f) Pd@C1 and EDS spectra of (g) Pd@SiO2, (h) Pd@SiO2@C, (i) Pd@C1. (j) Elemental mapping of Pd Lα1 over a selected region of Pd@C1. (k) FESEM image of Pd@C2 and (l) its EDS spectrum. (m) Elemental mapping of Pd Lα1 over a selected region of Pd@C2. (n) FESEM image of Pd/C and (o) its EDS spectrum. | |
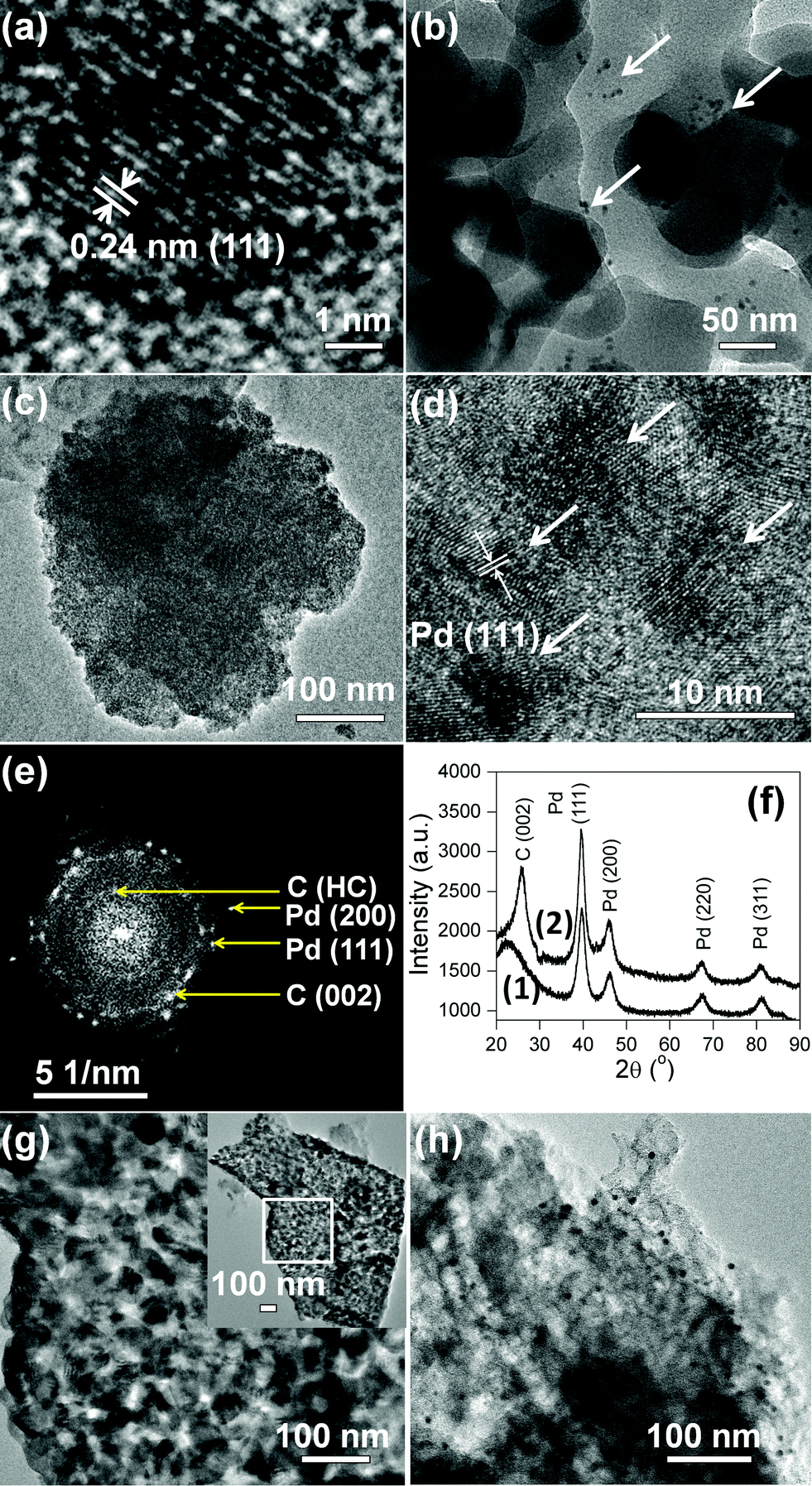
Transmission electron microscopy (TEM) images corroborate the morphologies observed in FESEM. Fig. 3a shows the transmission electron micrograph (TEM) of a single 5.3 ± 0.2 nm Pd NP inside the SiO2 shell. The inter-linked Pd@SiO2 NPs appear ∼50 nm in diameter (Fig. 3b). The arrows in Fig. 3b indicate the dispersed Pd NPs inside the SiO2 matrix. Individual mesoporous ∼165 nm Pd@C1 particles with colonized ∼8.5 nm Pd NPs inside the mesopores are observed in Fig. 3c and d. The increase in the size of the Pd NPs by ∼3 nm was due to annealing at 900 °C. The selected area electron diffraction (SAED) pattern in Fig. 3e shows the Pd (111) and (200) reflections. At 900 °C, carbon becomes partially graphitic. The d-spacing of 0.5 nm can result from the regions of high curvature of the immediate carbon layers surrounding each Pd NP.34 The SAED pattern is duly supported by the powder X-ray diffraction (PXRD) patterns of Pd@SiO2 and Pd@C1 in Fig. 3f. The peak at 22.5° in Fig. 3f(1) represents the amorphous SiO2 shell. The crystallite size of Pd NPs obtained from the PXRD peak broadening using Debye Scherrer's equation was 8.2 nm, which correlates well with the size of Pd NPs in Pd@C1. Although PXRD patterns do not provide evidence of any Pd2+, the presence of a minor fraction of amorphous PdO is most likely.25 Peak fitting of the X-ray photoelectron spectrum (XPS) of the Pd 3d level of Pd@C1 (Fig. S1, ESI†) shows the presence of 35% Pd2+ species. The metallic nature of the Pd NPs could be retained due to annealing under N2. The 12–16 nm pores of Pd@C2 are clearly observed in Fig. 3g. The dispersed Pd NPs on activated charcoal in Pd/C can be visualized from the dark spots on the carbon matrix in Fig. 3h.
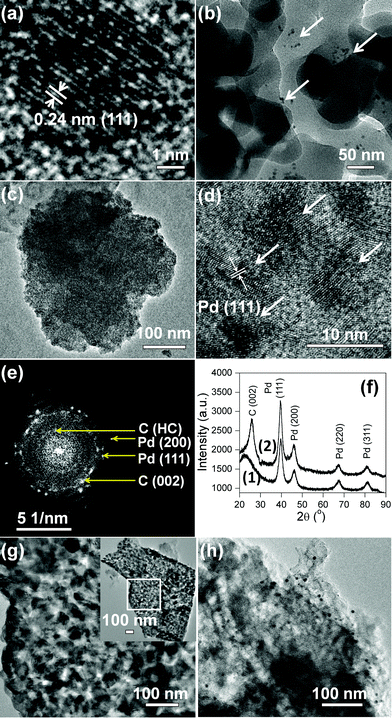 |
| | Fig. 3 TEM images of (a) Pd NP, (b) Pd@SiO2, (c, d) Pd@C1. (e) SAED pattern of Pd@C1. HC denotes “high curvature”. (f) XRD patterns of (1) Pd@SiO2 and (2) Pd@C1. TEM images of (g) Pd@C2, inset shows the entire matrix, and (h) Pd/C. | |
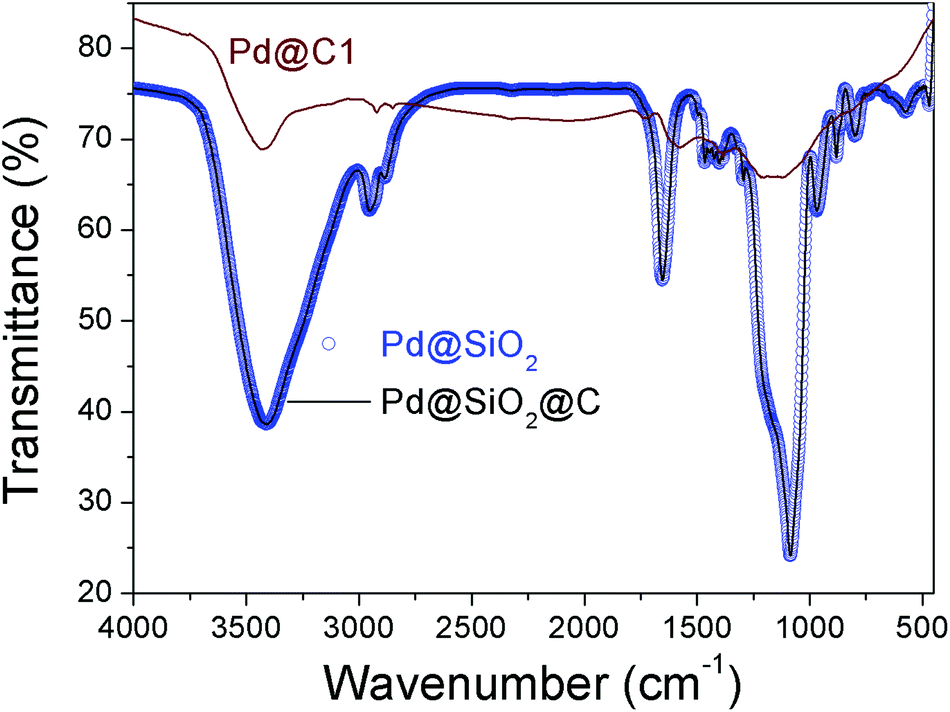
The chemical bonding characteristics of Pd@C1 at different stages of synthesis were studied by Fourier transform infrared (FTIR) spectroscopy and the spectra are presented in Fig. 4. For Pd@SiO2 and Pd@SiO2@C, the wavenumber and intensity of the peaks are similar indicating no major change in the bonding after carbonization. The 3420 cm−1 peak is due to the O–H stretching from adsorbed moisture and the peak at 2945 cm−1 arises from the Si–CH2 stretching due to bonding interactions between cetrimonium bromide (CTAB) surfactant molecules on the surface of Pd NPs and the overlying SiO2. The peaks at 1654 and 1440 cm−1 are the N–CH3 stretching vibrations of CTAB.35 The 1085, 950 and 882 cm−1 peaks account for the Si–O–Si stretching in TO3, TO2 and TO1 modes respectively.36 The absence of all the above peaks except a weaker O–H stretching at 3420 cm−1 in Pd@C1 confirms the complete removal of CTAB after annealing at 900 °C and SiO2 after the HF treatment.
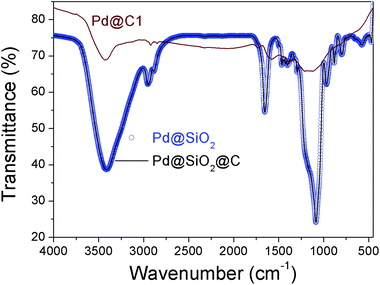 |
| | Fig. 4 FTIR spectra of Pd@SiO2, Pd@SiO2@C and Pd@C1. | |
Electrocatalytic activities
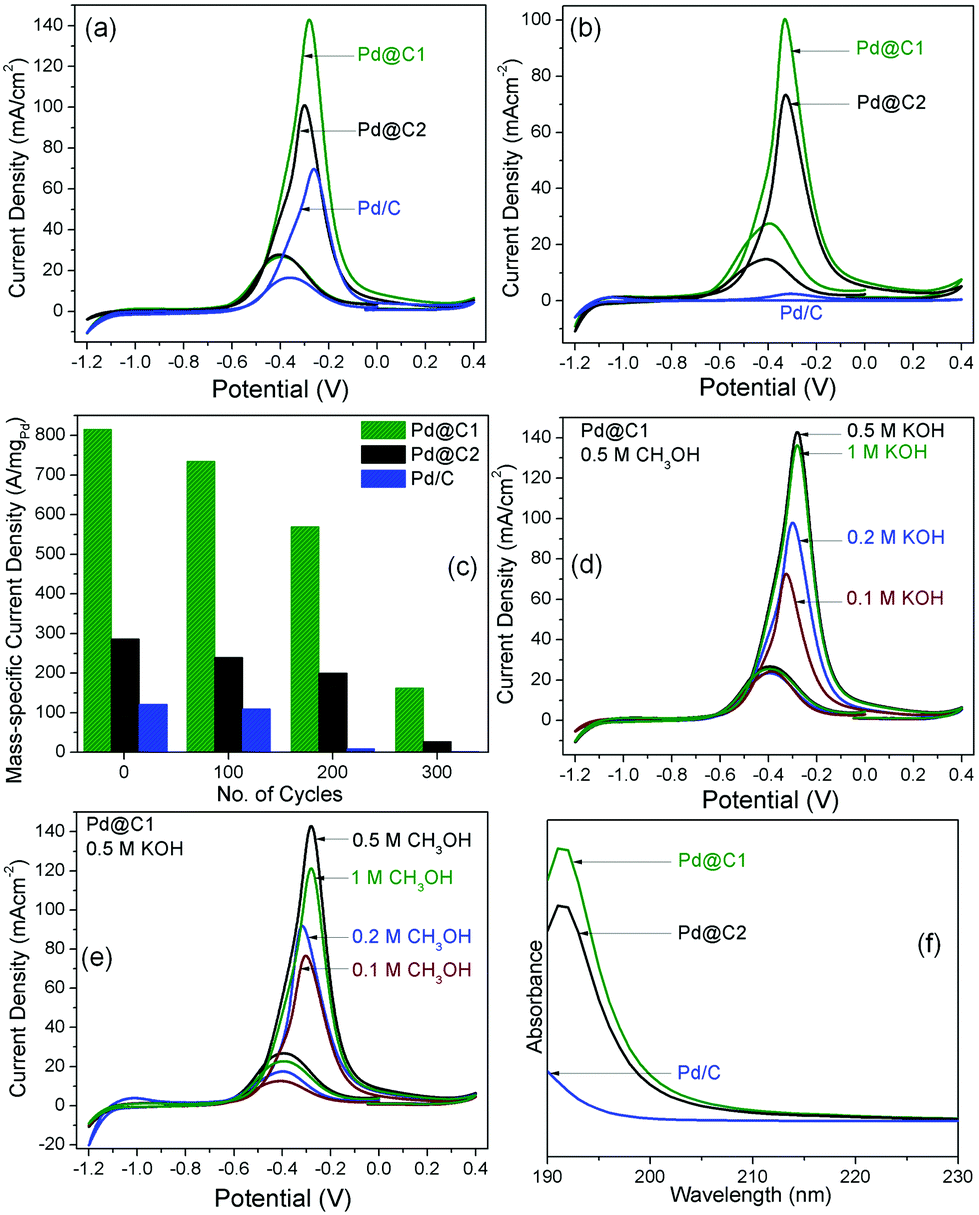
Mesoporous carbon without Pd NP loading showed a peak current density 0.9 mA cm−2 (Fig. S2†). The catalytic activity of mesoporous carbon probably arises from a large fraction of defect sites and the ECSA was observed to be 1.71 cm2 mgCarbon−1.37Fig. 5 presents the cyclic voltammograms (CVs) for MeOH EOx using the heterogeneous catalysts coated on glassy carbon electrodes and the parameters are tabulated in Table 2. The ECSA of the catalyst systems are compared with other reports.2,4,21,38–43 The solutions were stirred at 250 rpm to evenly distribute MeOH on the electrode surface, remove the concentration gradient of OH− ions and to prevent the accumulation of the oxidation products on the electrode surface resulting in a fast decrease in the anodic potential and the high catalytic activity (Table S3†). It is clearly observed from Fig. 5a that the peak of the anodic oxidation remains at −0.30 to −0.25 V versus the saturated calomel electrode (SCE). The corresponding AP-CD of Pd@C1 is 1.4 and 2 times higher than that for Pd@C2 and Pd/C catalysts, respectively. The utility of NP confinement and the superior activity of Pd@C1 were also prominent from the performance of the electrocatalysts in the recyclability tests up to the 300th cycle (Fig. 5b and c). After the 200th electro-oxidation cycle, the confined catalyst Pd@C1 shows a decent activity with an AP-CD of 100 mA cm−2 (176 cm2 mgPd−1). The re-usability of Pd@C2 was comparatively lower. In contrast, the activity of Pd/C was lost after the 200th cycle. Interestingly, the AP-CD of Pd@C1 and Pd@C2 was still maintained after the 300th cycle. The performance of Pd@C1 is comparatively better than the recently reported catalyst nanosystems used for MeOH EOx (Table 2).3,4,38–41,44,45 The anodic peak potential was observed at one of the lowest reported values of ∼−0.3 V,45 whereas the AP-CD of 142 mA cm−2 (626 cm2 mgPd−1) is the highest among the reported Pd NP catalysts. Fig. 5d shows the change in the AP-CD of Pd@C1 for MeOH EOx with an increasing pH of the solution. It was observed that with 0.5 M MeOH, the AP-CD increased from 70/80 (297/322 cm2 mgPd−1) to 142/141 mA cm−2 (626/613 cm2 mgPd−1) with/without stirring for an increasing pH from 0.1 to 0.5 M KOH, respectively (Table S3†). The MeOH EOx occurs simultaneously with the oxidation of the Pd NPs and the competition of adsorption of MeOH and OH− on the NP surface. A higher pH increases the concentration of both chemically and physically adsorbed active OH− on the NP surface which enhances the AP-CD.46 However, with 1 M KOH, the AP-CD slightly decreased to 135 mA cm−2 (607 cm2 mgPd−1). Again, by altering the MeOH concentration keeping 0.5 M KOH, the AP-CD of Pd@C1 increases from 76/103 mA cm−2 (357/378 cm2 mgPd−1) for 0.1 M to 142/141 mA cm−2 (626/613 cm2 mgPd−1) for 0.5 M MeOH and decreases to 120/133 mA cm−2 (591/603 cm2 mgPd−1) for 1 M MeOH with/without stirring (Fig. 5e and Table S4†). Increasing the MeOH concentration beyond 0.5 M results in the chances of MeOH crossover across the permeable Nafion layer posing multiple mass transport issues.47,48 Optimizing the MeOH concentration to reduce its crossover is always a challenge. In this study, 0.5 M KOH and 0.5 M MeOH were found to be optimum.
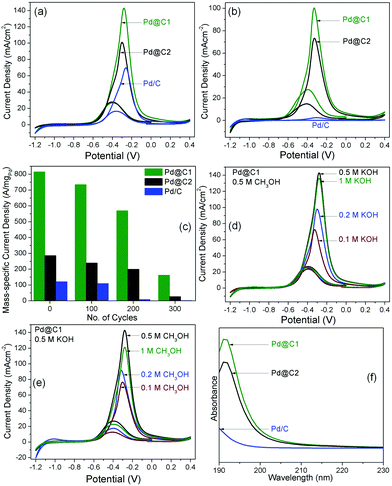 |
| | Fig. 5 CVs for MeOH EOx with the catalyst systems after (a) 1st and (b) 200th cycle with 0.5 M MeOH and 0.5 M KOH. (c) Bar diagrams showing the changes in AP-CD of the samples up to the 300th cycle of electro-oxidation. Performance of Pd@C1 with (d) 0.1 M MeOH and varying concentrations of KOH and (e) 0.1 M KOH and varying concentrations of MeOH. All the plots were obtained after stirring the solutions. (f) Solid state UV-vis absorption spectra of MeOH in the recovered catalysts after the 200th cycle. | |
Table 2 Peak voltage in the 1st cycle (V1(peak)), AP-CD in the 1st cycle (i1), after increased cycles for MeOH EOx with stirring the solution (icycles), ECSA in the 1st cycle and ECSAcycles after increased cycles for MeOH EOx. The reactant concentrations in molarity are mentioned for n (NaOH) and h (MeOH)
| Sample |
n + h (M) |
V
1(peak) (V) |
i
1 (mA cm−2) |
ECSA (cm2 mgPd−1) |
i
cycles (mA cm−2) |
ECSAcycles (cm2 mgPd−1) |
|
Without stirring.
|
| Pd@C1 |
0.5n + 0.5h |
−0.29 |
142 |
626 |
128100 |
516100 |
| 141a |
613a |
100200 |
404200 |
| 29300 |
139300 |
| Pd@C2 |
0.5n + 0.5h |
−0.30 |
100 |
234 |
85100 |
202100 |
| 120a |
191a |
72200 |
176200 |
| 10300 |
22300 |
| Pd/C |
0.5n + 0.5h |
−0.25 |
70 |
162 |
41100 |
95100 |
| 108a |
189a |
3200 |
7.6200 |
| 1300 |
2.2300 |
| Pt–Pd NPs/reduced graphene oxide2 |
0.5n + 0.5h |
0.7 |
120 |
197 |
— |
— |
| Pd–Ni–P NPs/C4 |
0.5n + 1.0h |
0.7 |
∼0.6 |
629 |
∼0.7400 |
— |
| Pd NPs on ordered mesoporous carbon21 |
0.5n + 1.0h |
−0.25 |
180/mgPd |
520 |
— |
— |
| Pd NP/low-defect graphene38 |
0.5n + 1.0h |
−0.1 |
26.7 |
830 |
— |
— |
| Pd34Pt66 NPs39 |
0.1n + 0.1h |
0.68 |
55 |
700 |
— |
— |
| Pd NP/WC40 |
1.0n + 2.0h |
−0.1 |
1.4 |
227 |
— |
— |
| Porous Pd NPs41 |
1.0n + 0.5h |
−0.1 |
120 |
230 |
— |
— |
| Pd NP based nano-membrane42 |
0.5n + 1.0h |
−0.15 |
60/mgPd |
573 |
— |
— |
| Pt–Pd nanowire43 |
0.5 M H2SO4 +0.5h |
0.7 |
50 |
907 |
— |
— |
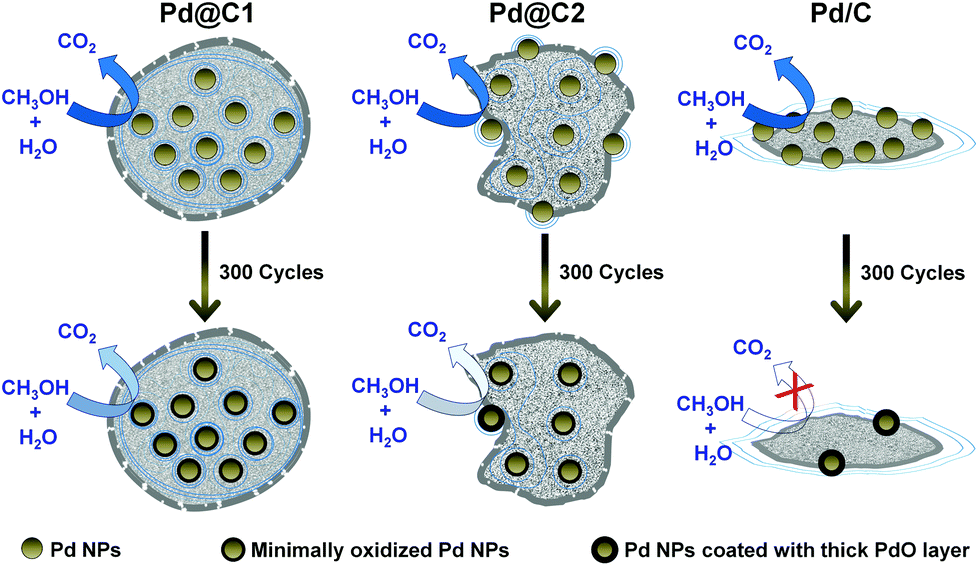
The morphology of the catalyst systems after the electro-oxidation cycles is shown in Fig. 6. After 200 cycles of MeOH EOx, the core–shell structure of Pd@C1 disintegrates, however the Pd NPs were observed to be trapped within the carbon matrix. The Pd NPs aggregated after continued cycles and ∼40 nm diameter NPs are observed after 200 cycles in Fig. 6b. The elemental line scan over a selected area of the used Pd@C1 shows a prominent Pd signal indicating decent anchoring of the NPs. After 300 cycles, the core–shell structure of Pd@C1 completely breaks, however Pd NPs are still observed correlating the ECSA (139 cm2 mgPd−1) after 300 cycles. The morphology of Pd@C2 after 200 cycles consists of a scattered mass of Pd and C along with carbon shells. These shells rupture after 300 cycles. The elemental line scan (Fig. 6h) shows the presence of Pd NPs, although the relative content of Pd is much lower than that of Pd@C1 after 300 cycles (Fig. 6e). The morphologies of Pd/C after 200 and 300 cycles are similar and the elemental analysis in Fig. 6k shows the presence of negligible Pd after 300 cycles of MeOH EOx. Fig. 7 shows a cartoon view of the performance of the three catalysts. Although, the confinement of NPs in the mesopores of the carbon support of Pd@C1 indeed can lead to an enhancement of the catalytic activity and stability by providing excellent anchoring possibilities, there is also a trade off in this effect. The confinement can hinder decent fuel diffusion and lead to kinetic problems in the MeOH EOx process. Also the diffusion of the surrounding carbon can cause the deactivation of the catalyst. However, the 20 nm and Å-wide pores come to the rescue and allow the intake of the fluid inside the cavity containing the NPs, and thus a better contact could be established between the trapped reactant molecules and the confined catalyst NPs. In Pd@C1, MeOH, KOH and H2O come in contact with the Pd NPs after percolating through the open pores at the wall of the hollow cavity. It is noteworthy that Pd@C1 with Pd NPs half the wt% of Pd@C2 and Pd/C could perform a better MeOH EOx. Actually, the normal Pd NP loading used in scientific studies and commercial catalysts is at least ∼20 wt%. In Pd@C1, since the Pd NPs are exclusively confined inside the mesopores, the Pd loading is expectedly low. In spite of doubling the Pd NP loading in Pd@C2 and Pd/C, the electrocatalytic activities were lower as compared to Pd@C1, which clearly underlines the positive influence of the confinement of the metal NP catalyst. The confinement of NPs has several advantages such as minimal surface oxidation after prolonged electrocatalytic cycles, zero leaching from inside the hollow cavity and increased redox properties of the confined NPs due to greater electron density.49,50 The above factors ensure better mass transport around the NPs and smaller electrical resistance, which provides a better AP-CD. The drop in AP-CD after repeated cycles could be due to the limited surface oxidation of the NPs after prolonged use and the OH− ions from abundant H2O adsorbed on the surface of Pd NPs which compete with the OH− in solution.47 When the Pd NPs are partially confined and the rest exposed to the surface of mesoporous carbon as in Pd@C2, the Pd NPs at the surface become vulnerable to oxidation and leaching. With continued cycles, even if the confined NPs experience lesser surface oxidation, the exposed NPs become increasingly oxidized and the AP-CD also decreases. In Pd/C, the chances of surface oxidation and leaching are the highest, decreasing its activity. The moisture content of Pd@C1, Pd@C2 and Pd/C after the 200th cycle was estimated to be 14.7, 10.3 and 7.9 wt%, which after the 300th cycle reduced to 12.9, 7.6 and 2.1 wt%, respectively. The excess moisture in Pd@C1 indicates that both MeOH and KOH are trapped inside the hollow cavity, justifying its superior activity. Solid state UV-visible spectra in Fig. 5f show the absorbance of MeOH present in the three catalysts and the shift of the peaks signifies modifications in the ground and excited states due to molecular interactions such as hydrogen bonding.51 The leftover MeOH in Pd@C1 and Pd@C2 is mostly from those trapped under confinement, whereas in Pd/C, the leftover MeOH is solely from inside the pores of activated charcoal. Inside the hollow cavity, the trapped MeOH molecules will have free OH− sites, whereas inside the nm- and Å-sized pores, the molecules will have a larger tendency to self-organize via hydrogen bonding. Thus from Pd@C1 to Pd@C2 and Pd/C, an intensity decrease in the absorption due to self-aggregation and a blue-shift due to the loss of terminal free OH− groups is observed.
 |
| | Fig. 6 (a) FESEM image, (b) TEM and (c) elemental line scan of Pd@C1 after 200 cycles of MeOH EOx. (d) FESEM image and (e) elemental line scan of Pd@C1 after 300 cycles. FESEM images of Pd@C2 after (f) 200 and (g) 300 cycles. (h) Elemental line scan of Pd@C2 after 300 cycles. FESEM images of Pd/C after (i) 200 and (j) 300 cycles. (k) Elemental line scan of Pd/C after 300 cycles. Arrows indicate the agglomerated Pd NPs. All scale bars correspond to 200 nm. | |
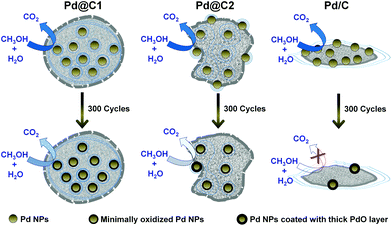 |
| | Fig. 7 Schematic cartoon showing the performance of the catalysts during MeOH EOx in their 1st (top panel) and 300th (bottom panel) cycle. The greater color intensity of the curved arrows indicates better catalytic activity and vice versa. | |
Conclusions
In summary, we have shown that confining the NP catalysts inside the well-defined hollow carbon structures can significantly enhance the activity of the catalyst in MeOH EOx as compared to the partially confined and exposed NP catalysts. According to our literature survey, this is the first report on the confinement effect of NP catalysts in MeOH EOx, which also provides a significantly high AP-CD of 142 mA cm−2 (626 cm2 mgPd−1) as compared to other Pd NP catalyst systems. The adapted and highly efficient process of Pd@C1 may be applicable to prepare various metal NPs confined within supports.
Experimental section
Materials
Tetraethyl orthosilicate (TEOS, Si(OC2H5)4, Merck India, synthesis grade), CTAB (Sigma Aldrich, 98%), potassium tetrachloropalladate (K2PdCl4, Arora Matthey Limited), polyvinylpyrrolidone (PVP, Loba Chemie, Mol. Wt. 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), ethanol (absolute for analysis, Merck India), ammonium hydroxide (25%, EMPLURA, Merck India), hydrofluoric acid (HF, Merck, 40%), sucrose (pure grade, Merck India), hydrochloric acid (HCl, 35% Merck India), MeOH (EMPLURA, Merck India), potassium hydroxide pellets (EMPLURA, Merck India) and activated charcoal (Merck India) were used without further purification.
000), ethanol (absolute for analysis, Merck India), ammonium hydroxide (25%, EMPLURA, Merck India), hydrofluoric acid (HF, Merck, 40%), sucrose (pure grade, Merck India), hydrochloric acid (HCl, 35% Merck India), MeOH (EMPLURA, Merck India), potassium hydroxide pellets (EMPLURA, Merck India) and activated charcoal (Merck India) were used without further purification.
Characterization
The surface area and porosity measurements were carried out with a Micromeritics Gemini VII surface area analyzer. The nitrogen adsorption/desorption isotherms were reported by BJH (Barrett–Joyner–Halenda) surface/volume mesopore analysis. The micropore volume was calculated using the Frenkel–Halsey–Hill isotherm equation. Each sample was degassed at 300 °C for 2 h. FESEM images were obtained using Carl Zeiss SUPRA 55VP FESEM. EDAX studies were performed with the Oxford Instruments X-Max with INCA software coupled to the FESEM. TEM images were recorded by UHR-FEG-TEM, JEOL, JEM 2100 F model using a 200 kV electron source. The XRD measurements were performed with a Rigaku (mini flex II, Japan) powder X-ray diffractometer having Cu Kα = 1.54059 Å radiation. XPS measurements were performed on the samples mounted on copper stubs with a silver paste, using Al Kα radiation (1486.6 eV) in a commercial photoelectron spectrometer from VSW Scientific Instruments. The base pressure of the chamber was maintained around 5 × 10−10 mbar during the experiments. The Fourier transform infrared (FTIR) studies were performed with a Perkin Elmer spectrum RX1 with KBr pellets. Ultraviolet-visible absorption spectra were collected using a JASCO V-670 spectrophotometer. Moisture % in the samples was measured using a Sartorius MA-150 analyzer by heating the samples up to 200 °C.
Materials synthesis
Synthesis of Pd NPs.
Pd NPs were synthesized as reported by Narayanan et al.52 with slight modifications. The palladium precursor solution was prepared by adding 0.16 g anhydrous K2PdCl4 with 6 mL of 0.2 M HCl and diluting it to 250 mL with de-ionized water. 15 mL of this precursor solution, 21 mL of de-ionized water, 0.0067 g of PVP and 2 drops of 1 M HCl were mixed together and heated. 15 mL ethanol was added to the mixture at the initial stages of refluxing, continued for 3 h. Upon completion of the reaction, a dark brown colloid of Pd NPs was collected.
Preparation of Pd@C1
Synthesis of Pd@SiO2 (Step 1).
The colloidal solution of Pd NPs was re-dispersed in a solution of 5 vol% NH4OH in ethanol. Soon after that 15 mL 10 vol% TEOS in ethanol and 0.05 g CTAB were mixed with 10 mL of re-dispersed Pd NP solution. The mixture was kept under constant stirring at 600 rpm for 4 h. Upon completion of the reaction, the product was filtered and washed thrice with distilled water and ethanol followed by drying at 70 °C for 3 h.
Carbonization of Pd@SiO2 (Step 2).
0.3 g of dried as-prepared Pd@SiO2 sample was dispersed in 10 mL ethanol and 20 mL of 0.5 M aqueous solution of sucrose was added to it. After adding 5 mL of 2 M HCl to this mixture, it was refluxed for 6 h and cooled to room temperature. A dark brown colloid was obtained as the product, which was collected using a rotary evaporator by evaporating the solvent, washed with ethanol and dried, followed by annealing at 900 °C for 2 h under N2. Upon completion of the annealing and cooling, the grey powder of Pd@SiO2@C was collected.
Removal of SiO2 (Step 3).
The powdered sample of Pd@SiO2@C was added to 10 mL of 5 vol% HF in water and sonicated for 2 h. After SiO2 was etched out, the solid was filtered and washed with ethanol and distilled water thrice followed by drying at 70 °C for 3 h.
Preparation of Pd@C2
30 mL of the colloidal Pd NP solution was mixed with 20 mL of 0.5 M aqueous solution of sucrose. After adding 5 mL of 2 M HCl to this mixture, it was refluxed for 6 h and cooled to 25 °C. A dark brown colloid was obtained as the product, which was collected by evaporating the solvent, washed with ethanol and dried followed by annealing at 900 °C for 2 h under an inert atmosphere. Upon completion of the annealing and cooling, the black powder of Pd@C2 was collected.
Preparation of Pd/C catalyst
The conventional catalyst where Pd NPs are anchored on the surface of activated charcoal was prepared as follows: 0.1 g of activated charcoal, 0.0067 g of PVP, 15 mL of a previously prepared K2PdCl4 solution and 21 mL de-ionized water were mixed together and the mixture was stirred for 0.5 h at room temperature. Afterwards the mixture was heated with constant stirring at 600 rpm. When the solution began to reflux, 15 mL of ethanol was added to it and kept under refluxing conditions for 3 h. After the reaction was complete, the solid product was filtered out and washed thrice with ethanol and distilled water followed by drying at 70 °C for 3 h.
Electrochemical measurements
Electrocatalytic activities of the catalyst samples were measured with a conventional three electrode cell in a CHI604D electrochemical workstation. The three electrode cell was assembled by using a Pt wire as the counter electrode, a saturated calomel electrode (SCE) as the reference electrode and a glassy carbon (GC) disk (3 mm diameter) coated with the catalyst as the working electrode. The working electrode was designed as follows: 1 mg of the powdered catalyst was dispersed in a mixed solution containing 250 μL distilled water, 250 μL of ethanol and 25 μL of 5% aqueous Nafion solution. 5 μL of the resulting suspension was carefully drop casted on to the surface of GC disk followed by drying at 25 °C for 0.5 h. All electrochemical measurements were performed without and with mechanical stirring at 250 rpm. The scanning rate was maintained constant at 50 mV s−1.
Acknowledgements
The Department of Science and Technology (DST), Government of India is duly acknowledged for the financial support under grant no. SR/S1/PC-28/2011. The authors thank Prof. S. M. Shivaprasad, JNCASR India for the XPS measurement. AD and SK thank the University Grants Commission (UGC) and Council of Scientific and Industrial Research (CSIR), New Delhi, for their fellowships, respectively.
Notes and references
- S. K. Kamarudin, F. Achmad and W. R. W. Daud, Int. J. Hydrogen Energy, 2009, 34, 6902–6916 CrossRef CAS PubMed.
- Y. Lu, Y. Jiang, H. Wu and W. Chen, J. Phys. Chem. C, 2013, 117, 2926–2938 CAS.
- L. Zhao, J. P. Thomas, N. F. Heinig, M. Abd-Ellah, X. Wang and K. T. Leung, J. Mater. Chem. C, 2014, 2, 2707–2714 RSC.
- M. Zhao, K. Abe, S. Yamaura and Y. Yamamoto, Chem. Mater., 2014, 26, 1056–1061 CrossRef CAS.
- S. Guo, S. Dong and E. Wang, Chem. Commun., 2010, 46, 1869–1871 RSC.
- G. Lu and A. Wieckowski, Curr. Opin. Colloid Interface Sci., 2000, 5, 95–100 CrossRef CAS.
- J. Zhang, S. Guo, J. Wei, Q. Xu, W. Yan, J. Fu, S. Wang, M. Cao and Z. Chen, Chem. – Eur. J., 2013, 19, 16087–16092 CrossRef CAS PubMed.
- N. A. M. Barakat, M. A. Abdelkareem, A. Yousef, S. S. Al-Deyab, M. El-Newehy and H. Y. Kim, Int. J. Hydrogen Energy, 2013, 38, 3387–3394 CrossRef CAS PubMed.
- D. Wang, G. Yang, Q. Ma, M. Wu, Y. Tan, Y. Yoneyama and N. Tsubaki, ACS Catal., 2012, 2, 1958–1966 CrossRef CAS.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, P. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- Y. Liu, W. Zhang, S. Li, C. Cui, J. Wu, H. Chen and F. Huo, Chem. Mater., 2014, 26, 1119–1125 CrossRef CAS.
- F. Rodríguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- Z. Chen, Z. Guan, M. Li, Q. Yang and C. Li, Angew. Chem., Int. Ed., 2011, 50, 4913–4917 CrossRef CAS PubMed.
- A. Datta, P. Dutta, A. Sadhu, S. Maiti and S. Bhattacharyya, J. Nanopart. Res., 2013, 15, 1808 CrossRef.
- G. Wang, J. Hilgert, F. H. Richter, F. Wang, H. Bongard, B. Spliethoff, C. Weidenthaler and F. Schüth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed.
- K. Ha, G. Kwak, K. Jun, J. Hwang and J. Lee, Chem. Commun., 2013, 49, 5141–5143 RSC.
- S. Bhattacharyya and A. Gedanken, J. Phys. Chem. C, 2008, 112, 659–665 CAS.
- S. Bhattacharyya, A. Gabashvili, N. Perkas and A. Gedanken, J. Phys. Chem. C, 2007, 111, 11161–11167 CAS.
- B. Fıçıcılar, A. Bayrakçeken and I. Eroğlu, J. Power Sources, 2009, 193, 17–23 CrossRef PubMed.
- H. Wang, X. Bo, J. Bai, L. Wang and L. Guo, J. Electroanal. Chem., 2011, 662, 281–287 CrossRef CAS PubMed.
- G. Z. Hu, F. Nitze, X. Jia, T. Sharifi, H. R. Barzegar, E. Gracia-Espino and T. Wagberg, RSC Adv., 2014, 4, 676–682 RSC.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- O. David, W. J. N. Meester, H. Bieräugel, H. E. Schoemaker, H. Hiemstra and J. H. Van Maarseveen, Angew. Chem., Int. Ed., 2003, 42, 4373–4375 CrossRef CAS PubMed.
- G. Hu, F. Nitze, E. Gracia-Espino, J. Ma, H. R. Barzegar, T. Sharifi, X. Jia, A. Shchukarev, L. Lu, C. Ma, G. Yang and T. Wågberg, Nat. Commun., 2014, 5, 5253 CrossRef CAS PubMed.
- A. Datta, A. Sadhu, S. Santra, S. M. Shivaprasad, S. K. Mandal and S. Bhattacharyya, Chem. Commun., 2014, 50, 10510–10512 RSC.
- A. Balouch, A. A. Umar, A. A. Shah, M. M. Salleh and M. Oyama, ACS Appl. Mater. Interfaces, 2013, 5, 9843–9849 CAS.
- Y. Huang, Z. Lin and R. Cao, Chem. – Eur. J., 2011, 17, 12706–12712 CrossRef CAS PubMed.
- M. S. Kwon, N. Kim, C. M. Park, J. S. Lee, K. Y. Kang and J. Park, Org. Lett., 2005, 7, 1077–1079 CrossRef CAS PubMed.
- E. Antolini, Energy Environ. Sci., 2009, 2, 915–931 CAS.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- S. Kubo, R. J. White, N. Yoshizawa, M. Antonietti and M.-M. Titirici, Chem. Mater., 2011, 23, 4882–4885 CrossRef CAS.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591–594 CrossRef CAS PubMed.
-
Y. Gogotsi, Carbon Nanomaterials, CRC Press & Francis Group, Boca Raton, 2006, p. 164 Search PubMed.
- R. B. Viana, A. B. F. da Silva and A. S. Pimentel, Adv. Phys. Chem., 2012, 2012, 903272 Search PubMed.
- J. Osswald and K. T. Fehr, J. Mater. Sci., 2006, 41, 1335–1339 CrossRef CAS.
- G. Hu, F. Nitze, T. Sharifi, H. R. Barzegar and T. Wågberg, J. Mater. Chem., 2012, 22, 8541–8548 RSC.
- H. Huang and X. Wang, J. Mater. Chem., 2012, 22, 22533–22541 RSC.
- Y. Liu, M. Chi, V. Mazumder, K. L. More, S. Soled, J. D. Henao and S. Sun, Chem. Mater., 2011, 23, 4199–4203 CrossRef CAS.
- Y.-W. Lee, A.-R. Ko, S.-B. Han, H.-S. Kim, D.-Y. Kim, S.-J. Kim and K.-W. Park, Chem. Commun., 2010, 46, 9241–9243 RSC.
- X. Wang, W. Wang, Z. Qi, C. Zhao, H. Ji and Z. Zhang, Electrochem. Commun., 2009, 11, 1896–1899 CrossRef CAS PubMed.
- H. Wu, H. Li, Y. Zhai, X. Zu and Y. Jin, Adv. Mater., 2012, 24, 1594–1597 CrossRef CAS PubMed.
- S. Guo, S. Dong and E. Wang, Chem. Commun., 2010, 46, 1869–1871 RSC.
- Z. J. Mellinger, T. G. Kelly and J. G. Chen, ACS Catal., 2012, 2, 751–758 CrossRef CAS.
- P. K. Shen, Z. Yan, H. Meng, M. Wu, G. Cui, R. Wang, L. wang, K. Si and H. Fu, RSC Adv., 2011, 1, 191–198 RSC.
- B. Ballarin, A. Mignani, E. Scavetta, M. Giorgetti, D. Tonelli, E. Boanini, C. Mousty and V. Prevot, Langmuir, 2012, 28, 15065–15074 CrossRef CAS PubMed.
- S. Yan, S. Zhang, Y. Lin and G. Liu, J. Phys. Chem. C, 2011, 115, 6986–6993 CAS.
- T. S. Zhao, C. Xu, R. Chen and W. W. Yang, Prog. Energy Combust. Sci., 2009, 35, 275–292 CrossRef CAS PubMed.
- W. Chen, Z. Fan, X. Pan and X. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS PubMed.
- S. Guo, X. Pan, H. Gao, Z. Yang, J. Zhao and X. Bao, Chem. – Eur. J., 2010, 16, 5379–5384 CrossRef CAS PubMed.
- Y. Morisawa, A. Ikehata, N. Higashi and Y. Ozaki, Chem. Phys. Lett., 2009, 476, 205–208 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc02052a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence