
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient and selective hydrogen peroxide-mediated oxidation of sulfides in batch and segmented and continuous flow using a peroxometalate-based polymer immobilised ionic liquid phase catalyst†
S.
Doherty
*a,
J. G.
Knight
a,
M. A.
Carroll
a,
J. R.
Ellison
a,
S. J.
Hobson
a,
S.
Stevens
a,
C.
Hardacre
*b and
P.
Goodrich
b
aNUCAT, School of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: simon.doherty@ncl.ac.uk; Fax: +44 (0) 191 208 6929; Tel: +44 (0) 191 208 6537
bThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast BT9 5AG, UK
First published on 8th December 2014
Abstract
The peroxometalate-based polymer immobilized ionic liquid phase catalyst [PO4{WO(O2)2}4]@PIILP has been prepared by anion exchange of ring opening metathesis-derived pyrrolidinium-decorated norbornene/cyclooctene copolymer and shown to be a remarkably efficient system for the selective oxidation of sulfides under mild conditions. A cartridge packed with a mixture of [PO4{WO(O2)2}4]@PIILP and silica operated as a segmented or continuous flow process and gave good conversions and high selectivity for either sulfoxide (92% in methanol at 96% conversion for a residence time of 4 min) or sulfone (96% in acetonitrile at 96% conversion for a residence time of 15 min). The immobilized catalyst remained active for 8 h under continuous flow operation with a stable activity/selectivity profile that allowed 6.5 g of reactant to be processed (TON = 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 428) while a single catalyst cartridge could be used for the consecutive oxidation of multiple substrates giving activity-selectivity profiles that matched those obtained with fresh catalyst.
428) while a single catalyst cartridge could be used for the consecutive oxidation of multiple substrates giving activity-selectivity profiles that matched those obtained with fresh catalyst.
Introduction
The selective oxidation of sulfides is a challenging and technologically important process as sulfoxides and sulfones are versatile intermediates used in the synthesis of fine chemicals, bioactive compounds, agrochemicals,1 as chiral auxiliaries2 and most recently as ligands for transition metal asymmetric catalysis.3 Moreover, sulfide oxidation is also the basis for the catalytic oxidative desulfurisation of crude oil in which sulfur compounds are removed by selective extraction of their sulfones into a polar solvent under much milder conditions than typically required for classical industrial catalytic hydrodesulfurization.4 Sulfoxidations have traditionally employed either strong oxidants, (e.g. nitric acid or KMnO4) which suffer numerous drawbacks such as low yields, extreme reaction conditions and poor E-factors,5 or reagents such as m-chloroperbenzoic acid,6 UHP,7 NaClO,8 NaIO4,9 oxone10 and dimethyldioxirane11 which are expensive, must be used in excess, generate stoichiometric amounts of by-product and involve protocols that require long reaction times, high temperatures and complicated handling procedures. Since hydrogen peroxide is economical, more environmentally benign and relatively atom efficient it is considered the oxidant of choice and as such numerous organocatalysts,12 enzymes13 as well as transition metal catalysts based on iron,14 manganese,15 vanadium,16 titanium,17 ruthenium,18 molybdenum,19 tungsten,20 and zinc21 have been developed for this process. In this regard, an efficient catalyst must be highly selective for either sulfone or sulfoxide, inexpensive, easy to prepare and handle, operate under mild conditions across a wide range of substrates and functionality, have long term stability and be easy to recover and recycle. While several of these criteria have been successfully realized there is still a demand to identify and develop alternative systems to address remaining drawbacks such as long reaction times, protracted and tedious isolation and catalyst recovery protocols and leaching of the active components under liquid–liquid biphasic conditions while also being suitable for use in a continuous flow process.22To this end, oxidation catalysts have been immobilized on the surface of porous supports or polymers to improve separation and recovery as well as recycling but such systems often suffer from slow reaction rates as well as leaching.23 Ionic liquids (ILs) are a fascinating class of solvent that have been widely embraced by the catalysis community as a method for immobilization of catalysts under homogeneous, liquid–liquid biphasic and liquid–solid (SILP) biphasic conditions.24 Recent relevant examples include an efficient protocol for the oxidation of sulfides to sulfones catalyzed by V2O5 in [C12mim][HSO4],25 highly selective sulfoxidation of sulfides to sulfoxides catalysed by the bifunctional IL combination [SO3HC3py][TiF6]/[C4py][BF4],26 a selective catalyst-free oxidation of sulfides to sulfoxides that is faster in ionic liquid than conventional solvents,27 and, most recently, heterogeneous selective oxidation of sulfides catalysed by ionic liquid–based polyoxometalates28 or tungstate-based poly(ionic)liquid entrapped magnetic nanoparticles.29 While good conversions and high selectivities have been obtained in IL media, surprisingly few recycle studies have been reported.
Taking SILP catalysis as a lead, we have been exploring the concept of Polymer Immobilized Ionic Liquid Phase (PIILP) catalysis30 whereby an ionic liquid is immobilized in the form of a cation-decorated co-polymer. This is an intriguing concept with a great deal of design potential as it aims to combine the favourable properties of ILs with the advantages of a solid support. Such a catalytic process will minimize the amount of ionic liquid in much the same manner as a SILP system, eliminate undesired leaching of ionic liquid, and facilitate catalyst recovery and recycling while also being ideally suited to liquid and gas phase continuous flow processing. Thus, we reasoned that the use of a well-behaved living polymerization should enable surface properties, the ionic microenvironment and porosity of the polymer to be modified in a rational and systematic manner and thereby catalyst–surface interactions, substrate accessibility and efficiency to be optimized and new activity–selectivity relationships to be established. Our initial foray in this area demonstrated that peroxophosphotungstate [PO4{WO(O2)2}4]3− immobilized on a ROMP-derived cation-decorated polymer ([PO4{WO(O2)2}4]@PIILP) is an efficient catalyst for H2O2-mediated epoxidations.30a Herein, we report that the same system catalyses the oxidation of sulfides under mild conditions, both in batch and in a continuous flow process with short residence times and that high selectivity for sulfoxide and sulfone can be obtained in segmented and continuous flow using methanol and acetonitrile, respectively, as the mobile phase. Moreover, continuous flow operation can be maintained over 8 h with only a minor reduction in performance and the same resin can also be used consecutively with different substrates to give yields and selectivities that match those obtained when a fresh batch of catalyst is used for each substrate.
Results and discussion
Catalyst synthesis, batch and recycle studies
Peroxometalate-based PIILP [PO4{WO(O2)2}4]@PIILP (2) was prepared by stoichiometric exchange of the bromide anions in pyrrolidinium-based ROMP-derived polymer 1 with [PO4{WO(O2)2}4]3− (Scheme 1)30a and the resulting amorphous white solid that precipitated was characterized by a combination of solid state P-31 NMR spectroscopy, ICP-OES (tungsten content), nitrogen sorption analysis, TEM and TGA/DSC. A series of catalytic reactions was first conducted under batch conditions to evaluate the efficiency of 2 as a catalyst for sulfide oxidation and to undertake preliminary optimization studies and recycle experiments.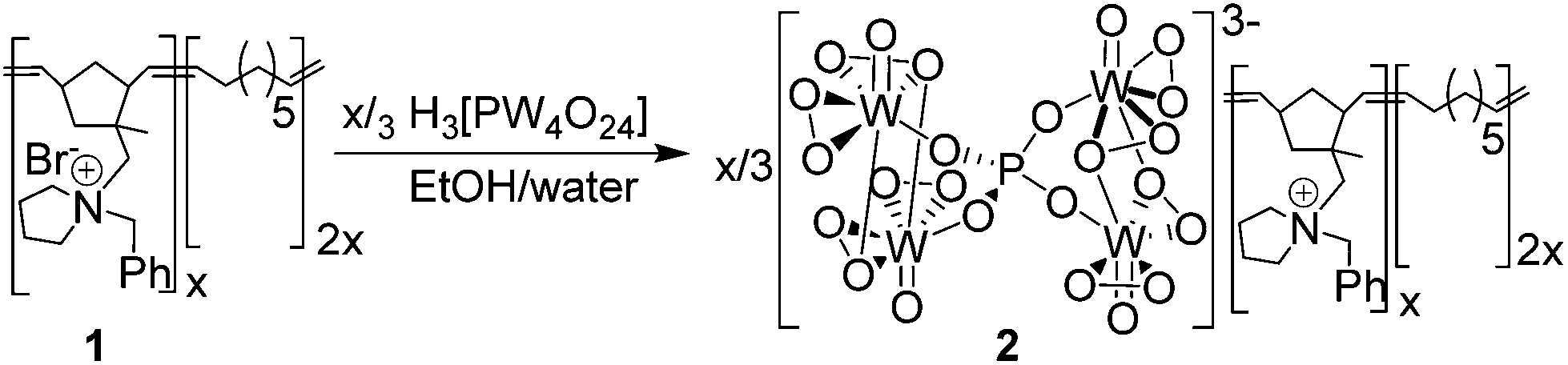 | ||
| Scheme 1 | ||
Our initial catalyst evaluation, comparison and optimization focused on thioanisole as the benchmark substrate as this oxidation has recently been catalysed by peroxometalate-based systems immobilized on the surface of ionic liquid modified silica,23b,c ionic liquid-based polyoxometalates salts,28 a temperature responsive phase transfer catalyst based on a Keggin-type lacunary anions20b as well as tungstate ions embedded in mesochannels of SBA-1523a and poly(ionic) liquid entrapped magnetic nanoparticles.29 A series of batch reactions were first conducted to explore the effect of the H2O2:substrate mole ratio on conversion and selectivity in acetonitrile and methanol, details of which are presented in Table 1.
|
|
||||||
|---|---|---|---|---|---|---|
| Solvent | H2O2 equiv. | Conv.b | % Sulfoxideb | % Sulfoneb | Sulfoxide selectivityb,c | |
| a Reaction conditions: 0.5 mol% 2, 1 mmol substrate, 1.0–3.0 mmol 35% H2O2, 3 mL solvent, 20 °C, 15 minutes. b Determined by 1H NMR spectroscopy. c Sulfoxide selectivity = [%sulfoxide/%sulfoxide + %sulfone] × 100%. d Reactions conducted under the same conditions using 0.5 mol% [nBu4]3[PO4{WO(O2)2}4] as catalyst. e Reaction conducted without catalyst using 0.5 mol% 1. f EG = ethylene glycol. g PC = propylene carbonate. | ||||||
| 1 | MeCN | 1.0 | 54 | 52 | 2 | 96 |
| 2 | MeCN | 2.0 | 68 | 65 | 3 | 96 |
| 3 | MeCN | 2.5 | 88 | 74 | 14 | 84 |
| 4 | MeCN | 3.0 | 100 | 80 | 20 | 80 |
| 5 | MeCN | 5.0 | 100 | 53 | 47 | 53 |
| 6 | MeCNd | 2.5 | 100 | 5 | 95 | 5 |
| 7 | MeCNe | 2.5 | 2.2 | — | — | — |
| 8 | MeOH | 1.0 | 70 | 70 | 0 | 100 |
| 9 | MeOH | 2.0 | 90 | 88 | 2 | 98 |
| 10 | MeOH | 2.5 | 95 | 91 | 4 | 96 |
| 11 | MeOH | 3.0 | 100 | 95 | 5 | 95 |
| 12 | MeOH | 5.0 | 100 | 85 | 15 | 85 |
| 13 | MeOHd | 2.5 | 100 | 93 | 7 | 93 |
| 14 | MeOHe | 2.5 | 0 | — | — | — |
| 15 | EtOH | 2.5 | 85 | 71 | 14 | 84 |
| 16 | i-PrOH | 2.5 | 32 | 52 | 0 | 100 |
| 17 | EGf | 2.5 | 21 | 21 | 0 | 100 |
| 18 | PCg | 2.5 | 72 | 56 | 16 | 78 |
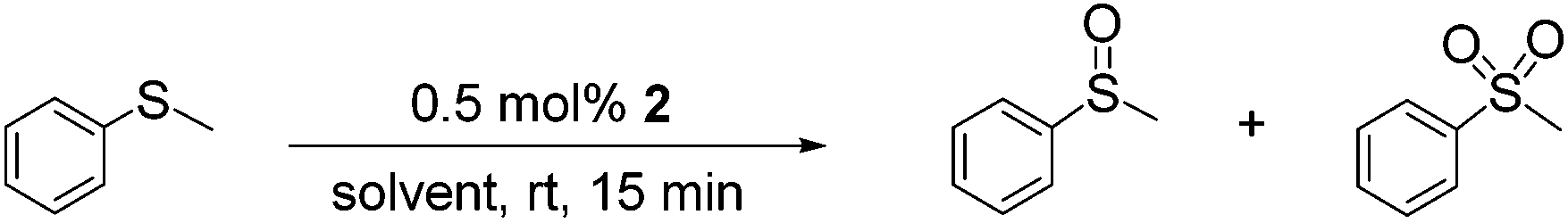
High sulfoxide selectivity (96%) and moderate to good conversions were obtained in acetonitrile using a 0.5 mol% loading of 2 to catalyse the oxidation of thioanisole at room temperature for 15 min with H2O2:S mole ratios of either one or two while selectivity dropped to 80% when the peroxide to substrate ratio reached three, albeit at complete conversion (entries 1–4). In contrast, a parallel series of reactions conducted in methanol showed that sulfoxide selectivity remained high even at a H2O2:S mole ratio of three (entries 8–11). In this regard, the solvent has previously been reported to have a dramatic effect on the selectivity of sulfide oxidation with protic solvents such as methanol and ethanol favouring formation of sulfoxide which in some cases was obtained as the exclusive product at short reaction times; this has been attributed to the high hydrogen bonding capacity of these solvents.23d,31 for example, high sulfoxide and sulfone selectivity has been obtained with relatively low loadings of Merrifield resin supported peroxomolybdenun(VI) compounds23g and polymer-immobilised peroxotungstates23d under mild conditions while peroxotungstates immobilised on multilayer ionic liquid brushes-modified silica23c or ionic liquid modified silica23a and Keggin heteropolycompounds19f required elevated temperatures, higher catalyst loading and/or markedly longer reaction times to reach comparable conversions. The data in Table 1 also shows that high sulfoxide selectivity is retained at high conversion for reactions conducted in methanol (entries 10–11) whereas selectivity appears to drop with increasing conversion in acetonitrile (entries 3–4) and that sulfoxide selectivity decreases much more dramatically in acetonitrile compared with methanol as the hydrogen peroxide ratio increases (entries 5 and 12). In stark contrast, under the same reaction conditions the [nBu4]3[PO4{WO(O2)2}4]-catalysed sulfoxidation of thioanisole with 2.5 equivalents of H2O2 in acetonitrile resulted in quantitative conversion to afford sulfone as the major product in 95% selectivity (entry 6), whereas the corresponding reaction in methanol gave sulfoxide as the major product in 93% selectivity (entry 13), again with complete consumption of sulfide. Even though heterogenisation of [PO4{WO(O2)2}4]3− in the form of [PO4{WO(O2)2}4]@PIILP results in an inversion of selectivity to favour sulfoxide, high yields of sulfone have been obtained with 2 using excess H2O2 at a higher reaction temperature (vide infra). The efficiency of 2 enabled the catalyst loading to be reduced to 0.025 mol% and under otherwise identical conditions the oxidation of thioanisole using 2.5 equivalents of H2O2 in acetonitrile reached 74% conversion with a sulfoxide selectivity of 96% after 2 h at 25 °C, which corresponds to a total TON of 2960 mole sulfoxide per mole cat−1 and a TOF of 1480 mole sulfoxide per mole cat−1 h−1. This TOF compares favourably with those of 1470 h−1 and 1235 h−1 reported for a Merrifield resin supported dioxomonoperoxomolybdenun(VI)23g and a polymer-immobilised peroxotungstate,23d respectively. Control reactions for the oxidation of thioanisole conducted in methanol and acetonitrile in the absence of the tungstate catalyst but with 0.5 mol% polymer immobilized ionic liquid 1 and 2.5 equivalents of H2O2 gave 0% and 2.2% conversion, respectively, which confirmed the active role of the catalyst (Entries 7 and 14). A survey of the influence of solvent on conversion and selectivity revealed that reactions conducted in ethanol and propylene carbonate gave high conversions and good sulfoxide selectivity (entries 15 and 18), albeit slightly lower than that obtained in methanol. Both solvents will be promising candidates to develop greener processes and, in this regard, studies are currently underway to improve selectivity. Although reactions conducted in isopropanol and ethylene glycol gave sulfoxide as the sole product a marked improvement in conversion will be required if these solvents are to be considered as viable replacements for methanol.
Encouraged by the efficacy of 2 for the selective oxidation of thioanisole, catalyst testing was extended to a range of sulfides, full details of which are summarized in Table 2. Gratifyingly, high conversions were obtained across the range of substrates examined and in each case selectivity for sulfoxide was higher in methanol than in acetonitrile ranging from 89–98% in the former compared with 58–98% in the latter. Oxidation of allylphenyl sulfide and homoallylphenyl sulfide occurred with complete chemoselectivity for sulfoxide and sulfone with no evidence for epoxidation of the double bond, presumably due to the mild conditions and short reaction times.19b,20c,23a–d The moderate conversion obtained for the [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidation of DBT at room temperature in acetonitrile is entirely consistent with the proposed electrophilic pathway, the lower nucleophilicity of this substrate and previous reports that the rate of oxidation increases with increasing nucleophilicity of the sulfide.20b,23d,31c,32 As expected, the same oxidation occurred more rapidly at higher temperatures such that a conversion of 95% was obtained after 15 min at 65 °C, albeit at the expense of sulfoxide selectivity as the sulfone was obtained as the major product in 81% selectivity. Unfortunately, it was not possible to obtain reliable data for the sulfoxidation of dibenzothiophene in methanol, possibly due to its low solubility in this solvent.
|
|
||||||
|---|---|---|---|---|---|---|
| Substrate | Solvent | % Conversionb | % Sulfoxideb | % Sulfoneb | Sulfoxide selectivityb,c | TONd |
| a Reaction conditions: 0.5 mol% 2, 1 mmol substrate, 2.5 mmol 35% H2O2, 3 mL MeCN, room temperature, 15 minutes. b Determined by 1H NMR spectroscopy. c sulfoxide selectivity = [%sulfoxide/%sulfoxide + %sulfone] × 100%. d TOF = moles sulfide consumed per mole catalyst per hour. e Determined by 13C NMR spectroscopy. f Reaction conducted at 65 °C. g Reactions conducted in deuteriated solvent for 2 minutes and monitored by NMR spectroscopy. | ||||||
|
MeCN | 88 | 74 | 14 | 84 | 704 |
| MeOH | 95 | 91 | 4 | 96 | 760 | |
|
MeCN | 92 | 74 | 18 | 80 | 736 |
| MeOH | 92 | 86 | 6 | 93 | 736 | |
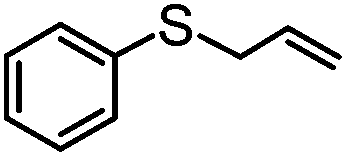
|
MeCN | 98e | 64e | 34e | 65 | 784 |
| MeOH | 99 | 97 | 2 | 98 | 792 | |
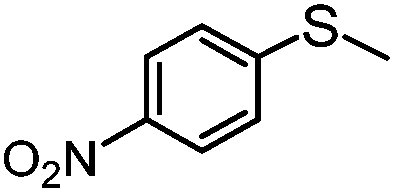
|
MeCN | 89 | 78 | 11 | 88 | 712 |
| MeOH | 56 | 53 | 3 | 95 | 448 | |
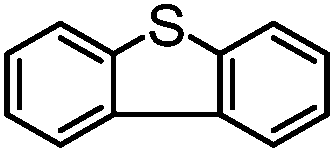
|
MeCN | 24 | 21 | 3 | 88 | 192 |
| MeCNf | 95 | 18 | 77 | 19 | 760 | |
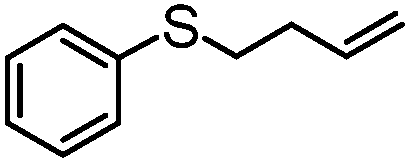
|
MeCN | 98 | 58 | 40 | 58 | 792 |
| MeOH | 62 | 56 | 6 | 90 | 456 | |
|
MeCN | 99 | 75 | 24 | 76 | 792 |
| MeOH | 92 | 90 | 2 | 98 | 736 | |
|
MeCNg | 59 | 58 | 1 | 98 | 472 |
| MeOHg | 99 | 77 | 22 | 78 | 792 | |
As sulfones are a useful class of compound, the conversion-selectivity profile was also monitored as a function of time and temperature in methanol and acetonitrile using a catalyst loading of 0.5 mol% and a peroxide to substrate ratio of 5 with the aim of identifying optimum conditions for the formation of methyl phenyl sulfone. Not surprisingly, selectivity for sulfone increased with increasing temperature and reached a maximum at 45 °C for a reaction time of 15 min, irrespective of the solvent, however, the data in Fig. 1a also highlights the disparate solvent dependent selectivity as reactions conducted in acetonitrile consistently gave a higher selectivity for sulfone, under comparable conditions. This solvent dependent selectivity is also evident in Fig. 1b which shows the selectivity as a function of time at 45 °C; in acetonitrile sulfone is obtained as the exclusive product after only 30 min whereas in methanol a reaction time of 60 min is required to obtain sulfone with 100% selectivity. High selectivity for methyl phenyl sulfone has previously been reported for the oxidation of thioanisole in methanol catalysed by [C4mim]3[PMo12O40] and, moreover, conditions were similar to those identified above.28 Under optimum conditions 0.025 mol% of 2 catalysed the oxidation of thioanisole in acetonitrile at 45 °C to give a total turnover number of 3960 mole sulfone per mole cat−1, albeit after 6 h.
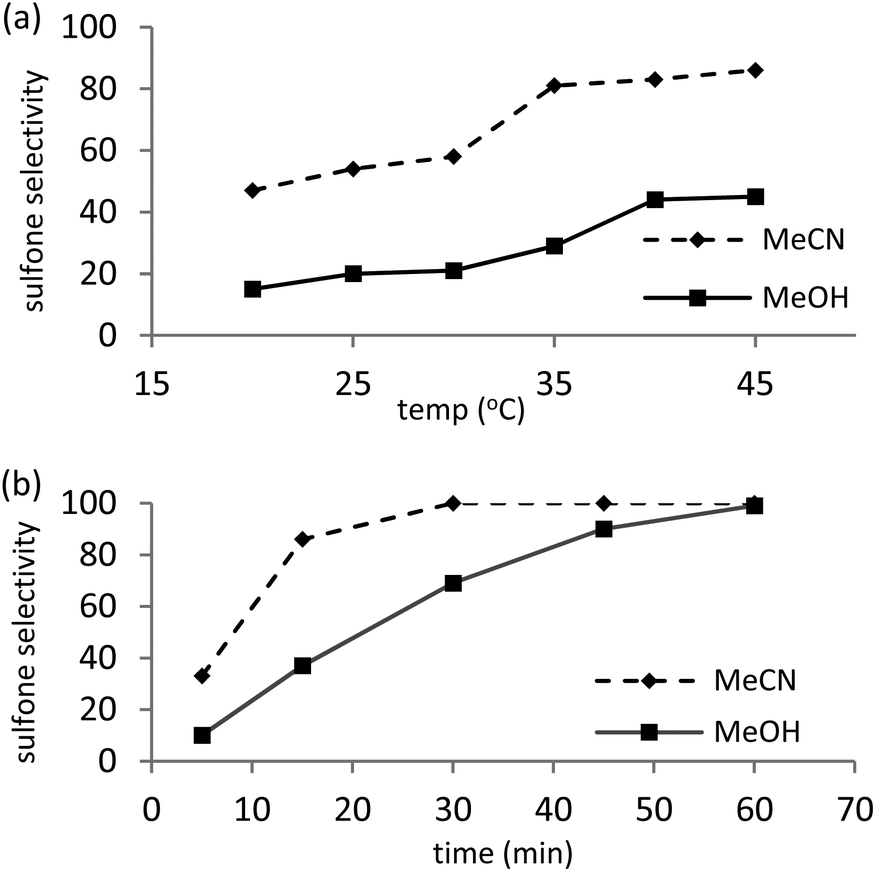 | ||
| Fig. 1 Influence of reaction conditions on the selectivity for methyl phenyl sulfone for the oxidation of thioanisole with H2O2 using 0.5 mol% [PO4{WO(O2)2}4]@PIILP. Influence of (a) temperature at a reaction time of 15 min (b) reaction time at 45 °C. Reaction conditions: 0.5 mol% catalyst, 1 mmol substrate, 5 mmol 35% H2O2, 3 mL solvent. All reactions reached 100% conversion under these conditions. | ||
The favourable formation of sulfone when the sulfoxidation was conducted in acetonitrile is consistent with previous reports23d and having identified optimum conditions for the formation of sulfone catalyst testing was extended to include a selection of aryl alkyl sulfides, details of which are summarized in Table 3. Under these conditions, a 0.5 mol% loading of 2 catalysed the sulfoxidation to give complete conversion after 15 min with high selectivity for the corresponding sulfone (77–100%) with the exception of dibenzothiophene which only reached 68% conversion and 51% selectivity for its sulfone; however, for all other substrates tested complete conversion to sulfone was achieved when the reaction time was extended to 60 min. Even though the TOF of 280 h−1 obtained for the sulfoxidation of dibenzothiophene under these conditions is lower than that for the other substrates examined, the data in Table 2 shows that high TOFs and high sulfone selectivity can be obtained at 65 °C; the TOF of 760 h−1 is a marked improvement on that obtained with the temperature responsive phase transfer system [(C18H37)2(CH3)2N]7[PW11O39] (248 h−1 at 60 °C),20b polymer immobilised peroxotungstates (9 h−1 at 78 °C),23d V2O5/[C12mim][HSO4] (ca. 4 h−1 at 45 °C)25 and Merrifield resin supported peroxomolybdenum(VI) compounds (7.2 h−1 at 78 °C).23g Even at 45 °C the sulfoxidation of allyl and homoallylphenyl sulfone occurred with complete chemoselectivity to the sulfoxide and sulfone with no evidence for epoxidation of the double bond.
|
|
|||||
|---|---|---|---|---|---|
| Substrate | Conv.b | % Sulfoxideb | % Sulfoneb | Sulfone selectivityb,c | TOFd |
| a Reaction conditions: 0.5 mol% 2, 1 mmol substrate, 5 mmol 35% H2O2, 3 mL MeCN, 45 °C, 15 minutes. b Determined by 1H NMR spectroscopy. c Sulfone selectivity = [%sulfone/%sulfone + %sulfoxide] × 100%. d TOF = mole sulfone generated per mole catalyst per hour. e Determined by 13C NMR spectroscopy. | |||||
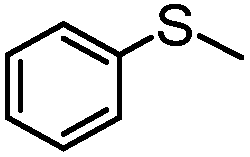
|
100 | 14 | 86 | 86 | 688 |
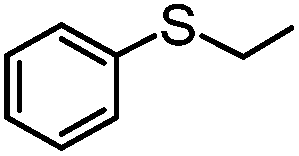
|
100 | 0 | 100 | 100 | 800 |
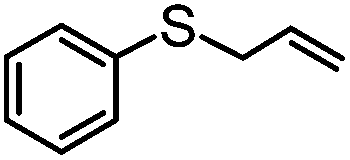
|
99e | 1e | 98e | 99e | 792 |
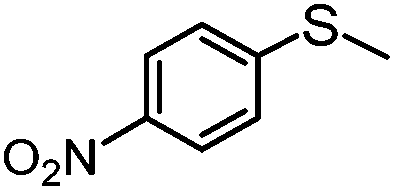
|
100 | 33 | 77 | 77 | 616 |
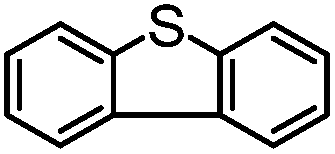
|
68 | 33 | 35 | 51 | 280 |
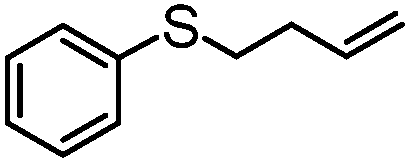
|
100 | 3 | 97 | 97 | 776 |
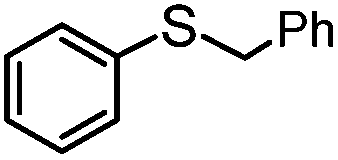
|
100 | 4 | 96 | 96 | 768 |
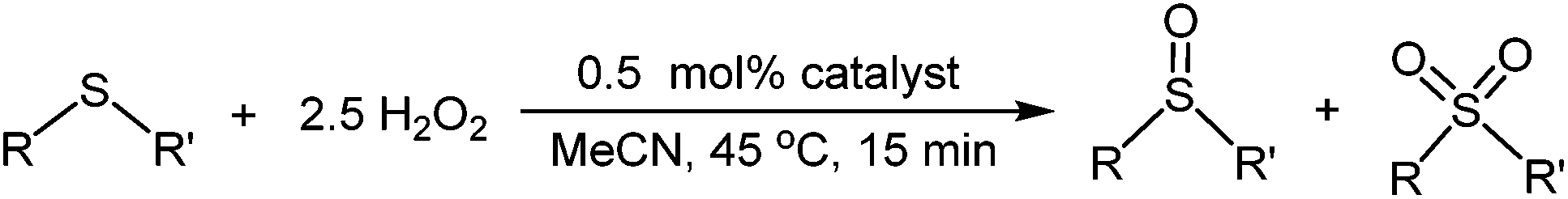
Reasoning that the polymer immobilized ionic liquid should efficiently retain the peroxometalate, catalyst recycle experiments on the sulfoxidation of thioanisole were undertaken to assess the robustness and longevity of the catalyst and the potential for incorporation into a continuous flow process. Methanol was identified as the solvent of choice for the initial recycle experiments on the basis that conditions for the selective oxidation to sulfoxide and sulfone had already been identified (vide supra). The catalyst was recovered in an operationally straightforward filtration, washed with methanol, dried and reused directly without being replenished or reconditioned. A comparative study of the conversion and sulfoxide selectivity for the [PO4{WO(O2)2}4]@PIILP-catalyzed sulfoxidation of thioanisole in methanol as a function of time across two runs is presented in Fig. 2a. The data clearly shows that selectivity reduces gradually from 99% to 90% with increasing conversion and that the conversion and selectivity-time profiles for the two runs map closely to each other. For comparison, a parallel recycle experiment in acetonitrile revealed a much more dramatic drop in selectivity, from 98% to 54%, as a function of conversion/time than the corresponding reaction in methanol (Fig. 2b). Thus, methanol was chosen for a more extensive recycle study which showed that [PO4{WO(O2)2}4]@PIILP recycled efficiently across six runs with only a minor reduction in conversion and no significant change in selectivity (Table 4).
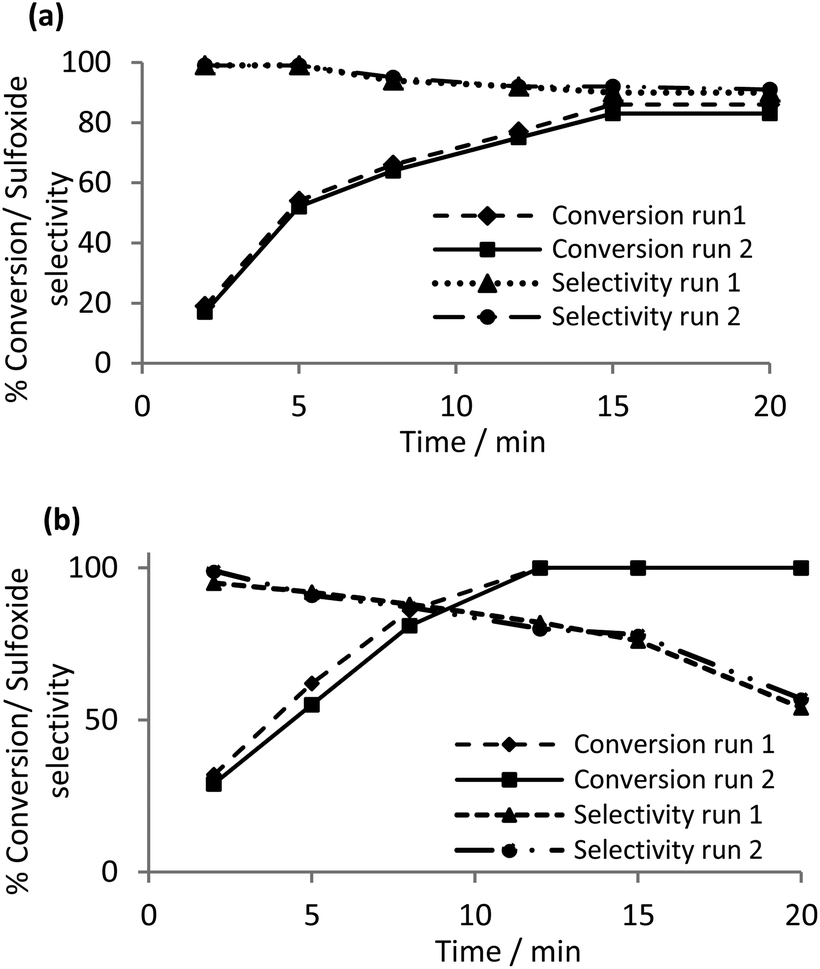 | ||
| Fig. 2 (a) Conversion and sulfoxide selectivity profiles as a function of time for a recycle [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidation of thioanisole in (a) methanol at room temperature (b) acetonitrile at room temperature, using 0.5 mol% 2 and a substrate:H2O2 ratio 1:2.5. | ||
|
|
||||
|---|---|---|---|---|
| Run | [PO4{WO(O2)2}4]@PIIP | [NBu4]3[PO4{WO(O2)2}4]/SiO2 | ||
| Conversionb | Sulfoxide selectivityb,c | Conversionb | Sulfoxide selectivityb,c | |
| a Reaction conditions: 0.5 mol% 2, 1 mmol substrate, 2.5 mmol 35% H2O2, 3 mL MeOH, 30 °C, 20 minutes. b Determined by 1H NMR spectroscopy. c Sulfoxide selectivity = [%sulfoxide/%sulfone + %sulfoxide] × 100%. | ||||
| 1 | 88 | 92 | 99 | 97 |
| 2 | 87 | 92 | 44 | 97 |
| 3 | 88 | 92 | 14 | 96 |
| 4 | 86 | 91 | ||
| 5 | 86 | 90 | ||
| 6 | 84 | 88 | ||
Analysis of the solvent after filtration and recovery of the catalyst from the first two runs of the recycle experiment revealed that the tungsten content was too low to be detected by ICP-OES (i.e. < 1 ppm), confirming that the peroxotungstate was efficiently retained by the polymer immobilized ionic liquid and potentially suitable for application in a continuous flow process (vide infra). Moreover, analysis of catalyst recovered after the 6th run gave a tungsten content of 25.84% which is close to that of unused catalyst, again a strong indication that leaching was negligible. In a direct comparative study, [nBu4N]3[PO4{WO(O2)2}4], immobilised on Geduran® Si60 (43–60 μm) by wet impregnation (6.5 wt%), was shown to be an efficient catalyst for the sulfoxidation of thioanisole in methanol and a 0.5 mol% loading gave 99% conversion and 97% selectivity for phenyl methyl sulfoxide after 15 min at room temperature. However, the system recycled very poorly as evidenced by a dramatic drop in conversion to 44% after the first run and a subsequent drop to only 14% for the third run, although the selectivity remained constant at 97% across the three runs (Table 4). ICP-OES analysis of the combined aqueous and organic phases collected after work-up and analysis of the first run revealed that 23% of the tungsten had been removed or extracted, a strong indication that the drop in conversion was due to efficient leaching of the active tungstate from the SILP based catalyst, which also highlights the advantage of the PIILP methodology. The IR spectrum of 2 contains characteristic bands at 1079 cm−1ν(P–O), 957 cm−1ν(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 585 cm−1νasym(W–O2) and 531 cm−1νsym(W–O2) which are entirely consistent with those for related supported peroxometalates reported in the literature.33 Catalyst recovered after the 6th run was found to contain IR bands similar to that for 2 with no evidence for oxidation of the benzylic group to the corresponding benzoyl pyrrolidinium cation strongly suggesting that the catalyst remains intact; a comparison of these IR spectra are provided in the ESI (Fig. S19†). A benchmark oxidation of thioanisole in the presence of ethylbenzene also confirmed that benzylic oxidation did not occur under these mild conditions and short reaction times.
O), 585 cm−1νasym(W–O2) and 531 cm−1νsym(W–O2) which are entirely consistent with those for related supported peroxometalates reported in the literature.33 Catalyst recovered after the 6th run was found to contain IR bands similar to that for 2 with no evidence for oxidation of the benzylic group to the corresponding benzoyl pyrrolidinium cation strongly suggesting that the catalyst remains intact; a comparison of these IR spectra are provided in the ESI (Fig. S19†). A benchmark oxidation of thioanisole in the presence of ethylbenzene also confirmed that benzylic oxidation did not occur under these mild conditions and short reaction times.
Segmented and continuous flow processing
The short reaction times and mild conditions required for [PO4{WO(O2)2}4]@PIILP catalysed oxidations and the recyclability of the system prompted us to explore the potential applications of this system in a continuous flow protocol and to establish how residence time, substrate/peroxide mole ratio and temperature influence selectivity. The flow set-up for sulfoxidation of thioanisole is shown in Fig. 3 and is based on a Uniqsis FlowSyn reactor. Preliminary evaluation and optimization studies were conducted using segmented flow in which either methanol or acetonitrile solutions of sulfide (0.2 M) and 30% hydrogen peroxide (0.6 M) are simultaneously pumped through a cartridge reactor packed with 2.0 g of silica (Geduran® Si60 43–60 μm) mixed with 0.1 g of [PO4{WO(O2)2}4]@PIILP with precise control of the flow rates; the residence times were calculated based on these flow rates and methanol or acetonitrile was used as the carrier solvent. The exiting product stream was collected in triplicate as 2 mL aliquots, subjected to an aqueous work-up and analysed by either 1H or 13C NMR spectroscopy to determine conversion and selectivity.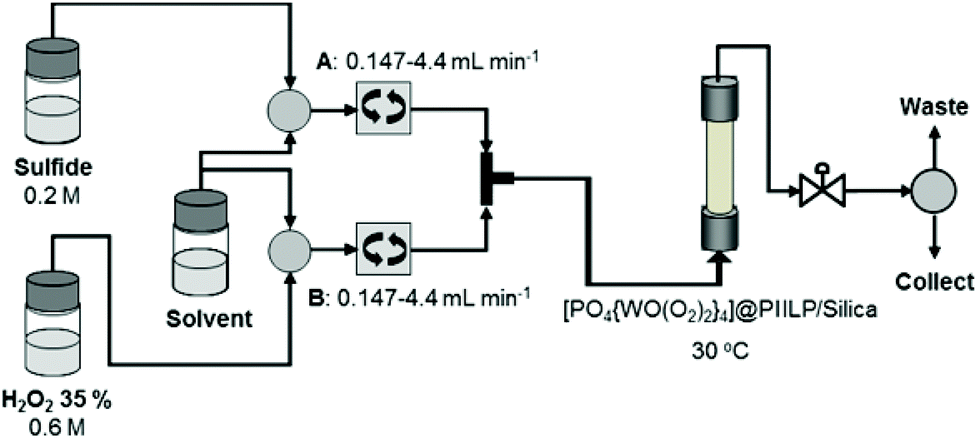 | ||
| Fig. 3 Schematic representation of the set-up for segmented and continuous flow sulfoxidations catalysed by [PO4{WO(O2)2}4]@PIILP (2). | ||
Using the optimized batch conditions as a lead, we first explored the effect of residence time, temperature and the amount of hydrogen peroxide on selectivity and conversion for the sulfoxidation of thioanisole in acetonitrile and methanol. Preliminary studies examined the effect of temperature on the conversion and selectivity profile at a fixed flow rate of 1.1 mL min−1, corresponding to a residence time (Rt) of 4 min, and in acetonitrile conversions increased smoothly from 44% at 20 °C to 100% above 45 °C while sulfoxide selectivity decreased across this temperature range from 88% to 21% (Fig. 4a). For comparison, the corresponding conversion-selectivity profile obtained using methanol as the mobile phase revealed that conversions increased steadily from 24% at 20 °C to 88% at 50 °C while sulfoxide selectivity remained high and decreased only slightly from 100% to 90% within this temperature range (Fig. 4b); a similar solvent dependent selectivity profile was obtained in batch wherein the sulfoxide selectivity obtained at high conversion in acetonitrile was lower than that in methanol under the same conditions. Taking both studies into account, a reaction temperature of 30 °C gave the optimum balance/compromise between conversion and sulfoxide selectivity and as such this temperature was chosen to examine a range of substrates (vide infra). A corresponding study to explore the influence of residence time on the conversion-selectivity profile at 30 °C with acetonitrile as the mobile phase revealed that conversions increased gradually with increasing residence time from 21% for a residence time of 0.5 min to 90% when this was increased to 4 min and that high selectivity for sulfoxide was maintained across this range of retention times; however, selectivity decreased quite dramatically at longer residence times such that sulfone was obtained as the major product in 96% selectivity after 15 min (Fig. 4c). For comparison, the conversion–selectivity profile in methanol was qualitatively similar although interestingly the optimum balance of conversion and sulfoxide selectivity of 96% and 92%, respectively, obtained at a residence time of 5 min was a significant improvement on that in acetonitrile while the selectivity for sulfone only reached 63% for a residence time of 15 min; this is markedly lower than the 96% obtained under the same conditions in acetonitrile (Fig. 4d). Thus, the data in Fig. 4 clearly shows that high selectivity for either sulfoxide or sulfone can be obtained under continuous flow processing through a judicious choice of solvent, residence time and temperature.
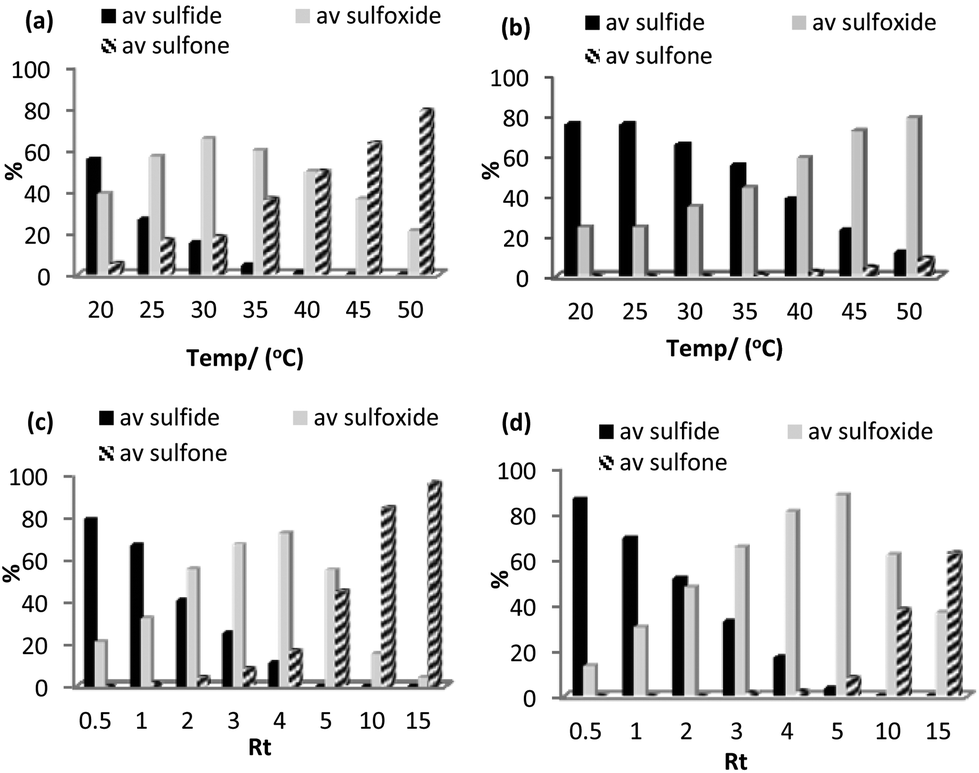 | ||
| Fig. 4 Conversion-selectivity profile as a function of temperature for the continuous flow [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidation of thioanisole in (a) acetonitrile (b) methanol for a residence time of 4 min. Conversion-selectivity profile as a function of residence time (Rt) for the [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidation of thioanisole in (c) acetonitrile (d) methanol at 30 °C. Reaction conditions: 0.1 g catalyst/2.0 g silica, 3 equiv. 35% H2O2, solvent, residence time 0.5–15 min or temperature 20–50 °C. | ||
A survey of the influence of the amount of H2O2 on conversion and selectivity as a function of residence time for the oxidation of thioanisole revealed a disparate solvent dependent behaviour. In the case of acetonitrile, a strong dependence on the rate was observed up to 12 equivalents of H2O2. For both 12 and 20 equivalents similar profiles with respect to Rt were observed. In comparison a much weaker dependence on [H2O2] was observed in methanol and approximately zero order behaviour was found. Details of the composition-time profile and analysis are provided in the ESI (Fig. S49–S62†). Approximate rate constants for the formation of methyl phenyl sulfoxide (ka) and methyl phenyl sulfone (kb) in acetonitrile and methanol were extracted by fitting the concentration-time profile for the consumption of sulfide and the formation of product using pseudo steady state analysis. As 2.5 equivalents of H2O2 are consumed during the reaction the rate constants derived were only calculated for the reaction with 20 equivalents of H2O2. Table 5 compares the effects of the solvent on the two rates assuming a first order dependence on the substrate and sulfoxide. Interestingly, the solvent has a much less significant effect on the second oxidation compared with the first. The strong dependence on peroxide concentration in acetonitrile may reflect a reduced solubility of the substrate compared with methanol which blocks the surface sites on the catalyst and/or hydrogen bonding between H2O2 and the substrate impeding access to the catalyst. In contrast, the increased H-bonding capacity of methanol could result in a greater degree of solvation of the substrate which would reduce the coverage of substrate at the catalyst and thereby allow peroxide to bind effectively. This may also be the reason why methanol has a reduced overall rate compared with acetonitrile. The possible effect of the additional water content on the conversion-selectivity profiles arising from the increasing mole ratio of aqueous hydrogen peroxide was also investigated by doping experiments based on three equivalents of hydrogen peroxide with the corresponding amount of water that would be present with six equivalents of peroxide; the associated conversion-selectivity profiles as a function of residence time are provided in the ESI (Fig. S63–S64†). Interestingly, for reactions conducted in acetonitrile the first oxidation is more rapid in the water-doped system while the second oxidation appears to be slightly slower. In contrast, the profile for the water-doped methanol system maps more closely, across the range of retention times, to the data for the undoped system with three mole equivalents of hydrogen peroxide. The slight increase in rate for the water doped-acetonitrile system compared with methanol may be due to interaction of the water with the catalyst-support surface facilitating access of the substrate to the catalyst, possibly through a network of hydrogen bonds. High selectivity for sulfoxide was retained when the water content was increased further by generating the segmented flow with equal volumes of aqueous hydrogen peroxide and methanolic thioanisole. Under these conditions the combination of 93% sulfoxide selectivity and 92% conversion at a flow rate of 0.88 mL min−1 (Rt = 5 min) was a marked improvement on that obtained by varying the hydrogen peroxide concentration; the associated conversion-selectivity profile as a function of residence time is presented in the ESI (Fig. S25†). A comparison with the corresponding batch reaction in 1:1 methanol/water revealed a further potential benefit of continuous flow as 0.5 mol% catalyst gave a conversion of only 22% in batch after 15 min, albeit with high sulfoxide selectivity (91%); such a poor conversion may well be due to agglomeration of the catalyst.
| MeCN | MeOH | ||||
|---|---|---|---|---|---|
| Entry | H2O2 | k a | k b | k a | k b |
| a Data obtained using 0.2 M solution of thioanisole and a 4.0 M solution of H2O2 with flow rates between 0.293 mL min−1 and 8.8 mL min−1 at 30 °C. | |||||
| 1 | 20 | 2.68 | 0.17 | 0.55 | 0.11 |
In addition to the obvious advantages associated with continuous flow operation such as simple product catalyst separation and ease of scale-up due to the linear increase in TON with time, our preliminary studies revealed that high selectivity for sulfoxide or sulfone can also be obtained by changing the solvent and residence time. For example, a sulfoxide selectivity of 98% at 83% conversion was obtained for the sulfoxidation of thioanisole at a residence time of 4 min under segmented flow in methanol, which is a marked improvement on the corresponding selectivity of 89% at 75% conversion in acetonitrile under otherwise identical conditions. For comparison, the corresponding sulfoxidation conducted in methanol under batch conditions gave 96% conversion but with slightly lower selectivity of 95%. In contrast, complete conversion and a sulfone selectivity of 96% was obtained in acetonitrile by extending the residence time to 15 min; this is significantly higher than the 62% selectivity obtained either under segmented flow in methanol or under batch conditions in both solvents (Table 1). With the optimized conditions in hand the substrate range was extended to establish the scope and efficiency of this system. As for thioanisole, the selectivity for sulfoxide obtained in methanol at any given residence time was higher than that in acetonitrile for each substrate studied and in the majority of cases the optimum compromise between conversion and selectivity appeared to peak at a flow rate of 1.1 mL min−1 (Rt = 4 min) with the exception of 4-nitrothioanisole which required a slightly longer residence time. Under these conditions sulfoxide selectivities as high as 90% were obtained with reasonable to good conversions (61–93%). While sulfone selectivity increased with increasing residence time and reached 75–89% at complete conversion, the optimum selectivity was solvent dependent. Full details of the conversion-selectivity profile as a function of residence time for the [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidation of each sulfide in methanol and acetonitrile are reproduced in the ESI (Fig. S47–S48 and S66–S72†) and a summary of the optimum selectivity and conditions is given in Table 6.
| Substrate | Rt | Solvent | % Conv.b | Sulfoxide selectivityb,c | Sulfone selectivityb,d |
|---|---|---|---|---|---|
| a Reaction conditions: 0.1 g 2/2.0 g silica, 3 equivalents 35% H2O2, solvent, residence time 0.5–15 min, 30 °C. b Determined by 1H NMR spectroscopy. c Sulfoxide selectivity = [%sulfoxide/%sulfone + %sulfoxide] × 100%. d Sulfone selectivity = [%sulfone/%sulfone + %sulfoxide] × 100%. | |||||
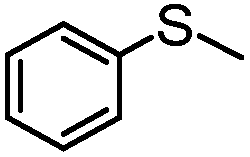
|
4 | MeOH | 83 | 98 | — |
| 15 | MeCN | 100 | — | 96 | |
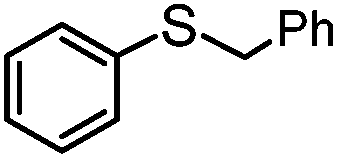
|
4 | MeOH | 93 | 90 | — |
| 15 | MeCN | 100 | — | 79 | |
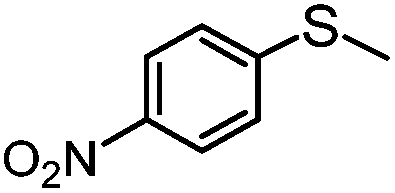
|
4 | MeOH | 61 | 90 | — |
| 15 | MeCN | 100 | — | 75 | |
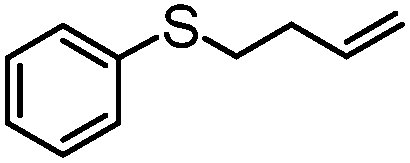
|
4 | MeOH | 80 | 89 | — |
| 15 | MeCN | 100 | — | 89 | |
|
— | MeOH | — | — | — |
| 15 | MeCN | 76 | — | 76 | |
Encouraged by the recyclability of 2 in batch and the conversion-selectivity profile obtained under segmented flow conditions, comparative scale-up continuous flow studies were conducted in methanol and acetonitrile using a mixture of [NBu4]3[PO4{WO(O2)2}4] and silica as the benchmark to assess the robustness, longevity and relative merits of the PIILP-based system. The efficiency of [PO4{WO(O2)2}4]@PIILP for the catalytic oxidation of thioanisole under continuous flow conditions (1.1 mL min−1, 30 °C, Rt = 4 min) in acetonitrile was monitored over 8 h and the resulting time-performance profile (Fig. 5a) revealed a slight decrease in activity with time, as evidenced by the decrease in conversion from 89% to 79%, with a parallel decrease in sulfone formation from 29% to 16% while the concentration of sulfoxide remained constant. Reasoning that these changes may be due to either deactivation or leaching of the catalyst and/or decomposition of peroxide the experiment was repeated and the reservoir replenished with fresh hydrogen peroxide after 4 h. The resulting system showed a marked improvement in the activity/selectivity profile as the concentration of sulfide, sulfoxide and sulfone remained constant and stable (Fig. 5b), indicating that the change in performance with time is unlikely to be due to leaching and more likely associated with the efficacy of catalyst activation and/or decomposition of hydrogen peroxide. In this regard, ICP-OES analysis of aliquots collected every hour revealed that 4.3% of the tungsten leached from [PO4{WO(O2)2}4]@PIILP over an 8 h period. Interestingly, the corresponding scale-up study in methanol showed a much more dramatic variation in the activity/selectivity profile which was manifested by a marked reduction in conversion from 85% to just 55% after 8 h accompanied by a concomitant reduction in formation of sulfoxide from 77% to 53% and a minor reduction in sulfone (Fig. 5c). Although stability, as measured by the absolute change in conversion and selectivity, improved noticeably after replenishing the peroxide a gradual decrease in conversion was paralleled by a decrease in the amount of sulfoxide (Fig. 5d). The contrasting behaviour of this system may be due, at least in part, to catalyst leaching as ICP-OES analysis of samples collected every hour indicated that 21% of the tungsten leached over an 8 h run. A comparative set of life-time studies was also conducted using a cartridge packed with [nBu4N]3[PO4{WO(O2)2}4]/SiO2 in order to assess the effectiveness and influence of the PIILP-based approach. Under the same flow conditions a cartridge packed with [nBu4N]3[PO4{WO(O2)2}4]/SiO2 was more active than its polymer immobilized ionic liquid-based counterpart for the oxidation of an acetonitrile solution of thioanisole, as judged by the quantitative conversion of sulfide during the first 3 hours of operation. However, in addition to a minor but steady increase in the amount of unreacted sulfide after 3 h there was a smooth and significant change in the sulfoxide selectivity from 60% to 90% over the same time (Fig. 5e); the tungsten leaching of 7% over the 8 h run is higher than that determined for the [PO4{WO(O2)2}4]@PIILP/SiO2 system under the same conditions. The corresponding comparison with [nBu4N]3[PO4{WO(O2)2}4]/SiO2 and methanol as the mobile phase resulted in a rapid and dramatic drop in activity such that the system was essentially inactive after the first hour (Fig. 5f); in this case ICP analysis of the methanol collected over the first 60 min contained 14.9 ppm tungsten, which corresponds to 21% leaching after only 1 h thereby accounting for the dramatic loss in activity. A semi-quantitative scale-up flow experiment conducted in acetonitrile at 30 °C with a flow rate of 1.1 mL min−1 (Rt = 4 min) processed 6.5 g of sulfide over the course of an 8 hour period (0.81 g h−1) with 88% conversion and a sulfoxide selectivity of 78% while a similar scale-up in methanol only gave 52% conversion but with excellent sulfoxide selectivity (98%). For comparison, a polystyrene supported benzenesulfonic acid Amberlite IR 120 H has recently been reported to catalyse the oxidation of sulfides under continuous flow with high sulfoxide selectivity and excellent long term stability at 22 °C, however, the system required a catalyst to substrate ratio of 0.3.34
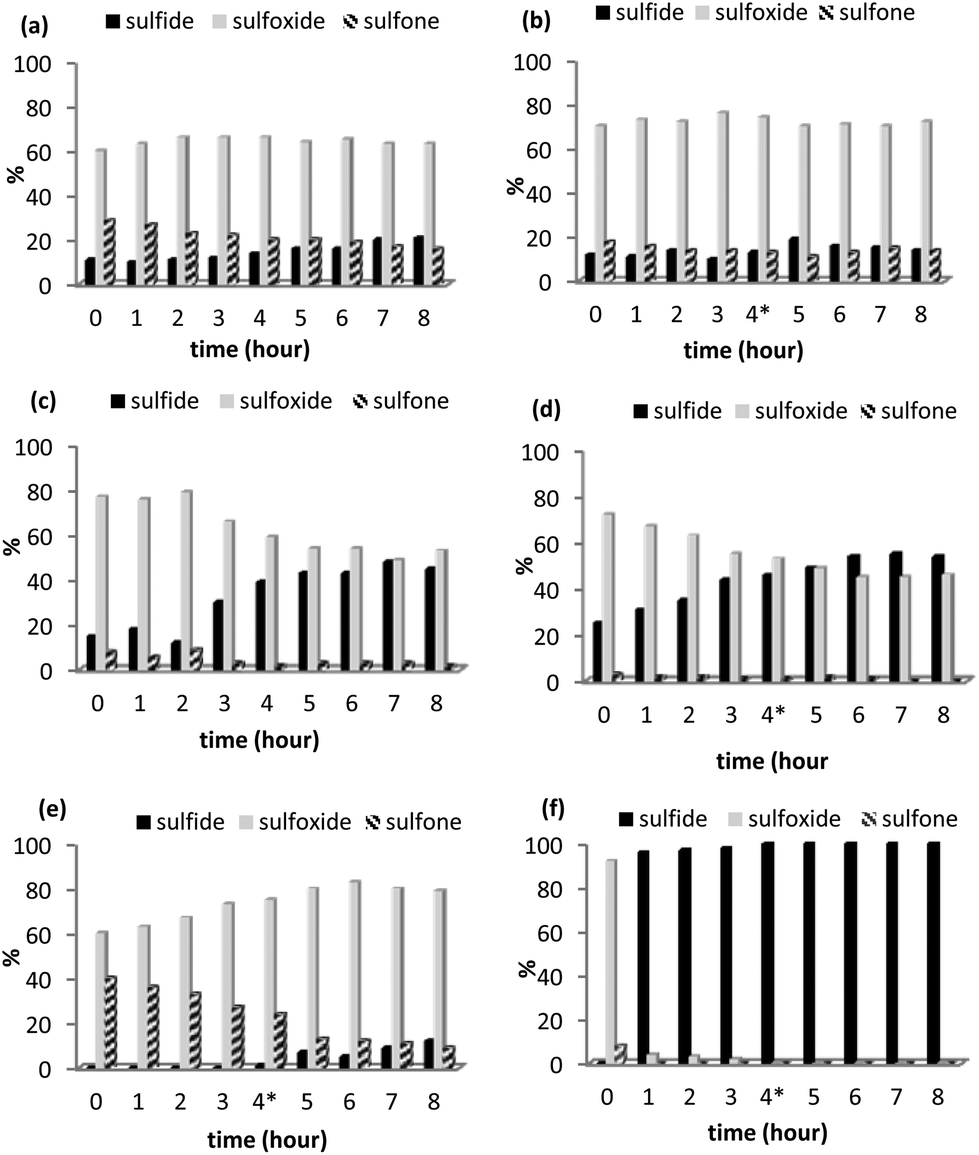 | ||
| Fig. 5 Conversion-selectivity profile as a function of time for an 8 h continuous flow [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidation of thioanisole at 30 °C with a residence time of 4 min in (a) acetonitrile, (b) acetonitrile with the H2O2 reservoir replenished after 4 h (*), (c) methanol, (d) methanol with the H2O2 reservoir replenished after 4 h (*). Conversion-selectivity profile as a function of time for an 8 h continuous flow sulfoxidation of thioanisole using [nBu4N]3[PO4{WO(O2)2}4] as catalyst at 30 °C with a residence time of 4 min in (e) acetonitrile and (f) methanol. | ||
The promising long term stability of [PO4{WO(O2)2}4]@PIILP for the continuous flow oxidation of thioanisole in acetonitrile provided the driver to examine whether the same resin-packed column could be used consecutively for the oxidation of several different substrates. In a proof of principle exercise, a column packed with [PO4{WO(O2)2}4]@PIILP and silica was operated under continuous flow (1.1 mL min−1, 30 °C, Rt = 4 min, MeCN) for the sequential oxidation of thioanisole, 4-nitrothioanisole and dibenzothiophene. Gratifyingly the conversion-selectivity profile for residence times of 5 and 15 min for the substrates run sequentially closely matched that obtained for the oxidation of each substrate conducted with fresh catalyst under the same conditions (Fig. 6a–b), provided the cartridge was purged with methanol between different substrates.
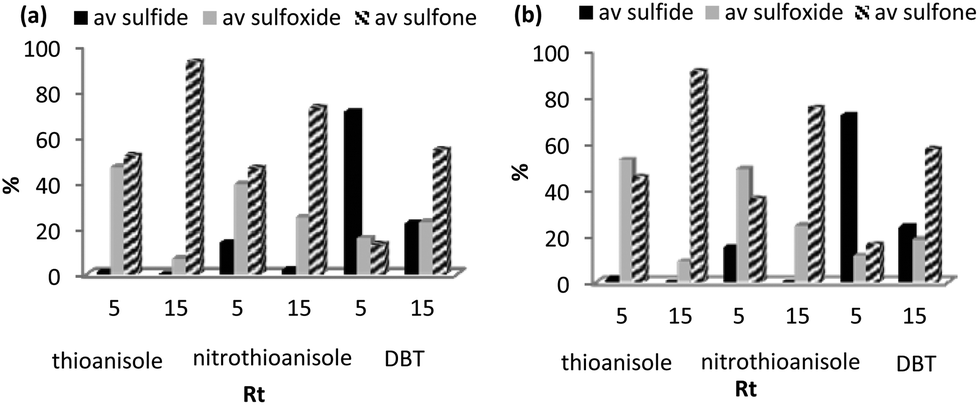 | ||
| Fig. 6 (a) Conversion-selectivity profile at residence times of 5 and 15 min for the continuous flow [PO4{WO(O2)2}4]@PIILP-catalysed consecutive sulfoxidation of three different substrates (i) thioanisole, (ii) 4-nitrothioanisole and (iii) dibenzothiophene with the same catalyst cartridge. (b) Conversion-selectivity profile at residence times of 5 and 15 min for the continuous flow [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidation using a fresh catalyst cartridge for each substrate. Reaction conditions: 0.1 g catalyst/2.0 g silica, 3 equiv. 35% H2O2, MeCN, 30 °C, residence times 5 and 15 min. | ||
Conclusions
The peroxophosphotungstate-based pyrrolidinium-decorated polymer immobilized ionic liquid [PO4{WO(O2)2}4]@PIILP catalyses the oxidation of sulfides with remarkable efficacy under mild conditions and in short reaction times using hydrogen peroxide as oxidant. High selectivity for sulfoxide can be obtained at room temperature in methanol while the sulfone can be obtained in acetonitrile at higher temperature with an excess hydrogen peroxide. The catalyst could be recovered in a straightforward filtration procedure with no evidence for leaching and recycle studies gave good conversions and a stable selectivity profile over several runs. A segmented flow process based on a catalyst cartridge packed with a mixture of [PO4{WO(O2)2}4]@PIILP and silica gave good conversions and high selectivity for sulfoxide in methanol at short residence times while sulfone was obtained with high selectivity in acetonitrile at longer residence times. The catalyst is remarkably robust and ideally suited for scale-up and could be operated under continuous flow conditions for 8 h with a stable activity-selectivity profile, allowing 6.5 g of material to be processed (TON = 46428). A single cartridge was also used for the consecutive oxidation of three different substrates and resulting conversion and selectivity profiles closely matched those obtained with fresh catalyst. Future studies are underway to develop robust adaptable multifunctional support materials based on polymer immobilized ionic liquids and to this end we are currently (i) introducing functionality onto the polymer chain and modifying hydrophilicity to explore what factors influence catalyst performance in order to identify an optimum catalyst-support combination to improve the selectivity profile, stability and recyclability, (ii) exploring whether phase separated polymers can be used for catalyst isolation, (iii) extending the concept of PIILP catalysis to a wider range of transformations as well as the stabilization of nanoparticles and (iv) developing peroxometalate@PIILP systems for the oxidative desulfurization of crude oil in batch and under continuous flow as well as the capture and removal/scavenging of toxic/odorous sulfur compounds.
Experimental
Synthesis of polymer immobilised peroxophosphotungstate 2
A hydrogen peroxide solution (35% w/w, 9.7 mL, 100 mmol) was added to phosphotungstic acid (1.73 g, 0.6 mmol) dissolved in a minimum volume of water and stirred at room temperature for 30 min. After this time, pyridine (0.145 mL, 1.8 mmol) was added followed by a solution of 1 (0.877 g, 1.8 mmol) in the minimum volume of ethanol, which resulted in the immediate precipitation of an amorphous white solid. The mixture was cooled to 0 °C, filtered through a sintered glass frit and the precipitate washed with water (2 × 10 mL) and diethyl ether (3 × 75 mL) and dried under vacuum to afford 6 in 84% yield. FT-IR (KBr plates, cm−1):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1087, 1058, 1028 (P–O), 956 (WO), 837 (O–O), 585, 535 W(O2)asym,sym: Anal. Calc. for C108H168N3O24PW4: N, 1.58; W, 27.66. Found: N, 1.46; W, 26.31. Loading of tungsten 1.4 mmol g−1.
= 1087, 1058, 1028 (P–O), 956 (WO), 837 (O–O), 585, 535 W(O2)asym,sym: Anal. Calc. for C108H168N3O24PW4: N, 1.58; W, 27.66. Found: N, 1.46; W, 26.31. Loading of tungsten 1.4 mmol g−1.
Synthesis of [nBu4N]3[PO4{WO(O2)2}4]/SiO2
A flame-dried Schlenk flask was charged with [nBu4N]3[PO4{WO(O2)2}4] (0.094 g, 0.05 mmol) and dichloromethane (6 mL) and the mixture stirred at room temperature for 15 min after which time 1.0 g of Geduran® Si60 (43–60 μm) was added and stirring continued for a further 2 h. The dichloromethane was removed under vacuum to afford a free flowing powder. FT-IR (KBr plates): = 1087, 1058, 1028 (P–O), 956 (WO), 837 (O–O), 585, 535 W(O2)asym,sym. The tungsten loading was confirmed to be 0.19 mmol of W g−1 of silica by ICP-OES analysis.
General procedure for catalytic sulfoxidation in batch
An oven-dried Schlenk flask was allowed to cool to room temperature and charged sequentially with sulfide (1.0 mmol), catalyst (0.013 g, 0.005 mmol) and solvent (3 mL), the reaction was then activated by the addition of 35% H2O2 (0.24 mL, 2.5 mmol) and allowed to stir at room temperature for 15 min. The reaction mixture was diluted with dichloromethane (25 mL), washed with water (ca. 50 mL) and the organic extract dried over MgSO4 and the solvent removed under reduced pressure. The resulting residue was analysed by either 1H or 13C{1H} NMR spectroscopy to quantify the composition of starting material and products; for each substrate tested an internal standard of 1,3-dinitrobenzene was initially employed to ensure mass balance.General procedure for catalytic sulfoxidation recycle studies
An oven-dried Schlenk flask was allowed to cool to room temperature and charged sequentially with sulfide (3.0 mmol), polymer immobilised catalyst 2 (0.039 g, 0.015 mmol) or [nBu4N]3[PO4{WO(O2)2}4]/SiO2 (100 mg, 6.5 wt% W) and solvent (9 mL). The reaction was initiated by addition of 35% H2O2 (0.72 mL, 7.5 mmol) and the resulting mixture allowed to stir at room temperature for 20 min. After this time the solution was centrifuged (10 min, 12000 rpm), decanted using a pipette and the remaining catalyst washed with the reaction solvent and dried prior to reuse under the same conditions. The remaining solution was subject to the same work-up and analysis as described above. ICP analysis of a portion of the organic and aqueous phases between recycles was conducted using a using a Perkin Elmer Optima 4300DV ICP-OES (Inductively Coupled Plasma Optical Emission Spectrometer).
General procedure for catalytic sulfoxidation kinetic studies
An oven-dried Schlenk flask was allowed to cool to room temperature and charged sequentially with sulfide (3.0 mmol), polymer immobilised catalyst 2 (0.039 g, 0.005 mmol) and solvent (9 mL). The reaction was initiated by addition of 35% H2O2 (0.72 mL, 7.5 mmol) and the resulting mixture stirred at room temperature for the 20 min during which time 0.5 mL aliquots were removed for work-up (as above) and analysis by 1H NMR spectroscopy. After the final aliquot had been removed the remaining reaction mixture was decanted, the catalyst washed with diethyl ether, dried and replenished to account for losses arising from the sampling. The recycle kinetic experiment was conducted by charging the Schlenk with fresh sulphide (3.0 mL), solvent (9 mL) and 35% H2O2 (0.72 mL, 7.5 mmol).General procedure for segmented and continuous flow catalytic sulfoxidation
Two reservoirs were charged with sulfide (5.0 mmol) dissolved in the appropriate solvent (25 mL, 0.2 M) and hydrogen peroxide (1.29 mL, 35%) in the same (25 mL, 0.6 M). A Uniqsis FlowSyn reactor was used to pump 1 mL of each reagent at total flow rates that varied between 0.293 mL min−1 and 8.8 mL min−1 through a T-piece mixer to combine the two streams; in the case of segmented flow an additional reservoir of carrier solvent was also employed. The reaction stream was then flowed through a OMNIFIT® glass column reactor cartridge (10 mm id × 100 mm) packed with 0.1 g of [PO4{WO(O2)2}4]@PIILP and 2.0 g of SiO2 (Geduran® Si 60) and mounted in a FlowSyn column heater. The exiting stream was passed through a back pressure regulator (BPR) and 2 mL fractions were collected into separate vials followed by a 2 mL post-collect. Each sample was diluted with dichloromethane (10 mL), washed with water (ca. 15 mL), the organic extract dried over MgSO4, the solvent removed under reduced pressure and the resulting residue analysed by either 1H or 13C NMR spectroscopy to quantify the composition of starting material and products.Acknowledgements
We gratefully acknowledge Newcastle University for financial support (J. R. E.). Solid-state 31P NMR spectra were obtained at the EPSRC UK National Solid-State NMR Service at Durham and high resolution mass spectra were obtained at the EPSRC National Mass Spectrometry Service Centre in Swansea.Notes and references
- (a) M. C. Carreno, Chem. Rev., 1995, 95, 1717–1760 CrossRef CAS; (b) I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651–3706 CrossRef CAS PubMed; (c) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed; (d) P. Metzner and A. Thuillier, Sulfur Reagents in Organic Synthesis, Academic Press, London, 1994 Search PubMed; (e) Synthesis of Sulfones, Sulfoxides and Cyclic Sulfides, ed. S. Patai and Z. Rappoport, John Wiley, Chichester, U.K., 1994 Search PubMed; (f) R. Bentley, Chem. Rev., 2005, 105, 609–624 RSC.
- L. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef PubMed.
- (a) L. Du, P. Cao, J. Xing, Y. Lou, L. Jiang, L. Li and J. Liao, Angew. Chem., Int. Ed., 2013, 52, 4207–42011 CrossRef CAS PubMed; (b) W.-Y. Qi, T.-S. Zhu and M.-H. Xu, Org. Lett., 2011, 13, 3410–3413 CrossRef CAS PubMed; (c) S.-S. Jin, H. Wang and M.-H. Xu, Chem. Commun., 2011, 47, 7230–7232 RSC; (d) J. Chen, J.-M. Chen, F. Lang, X.-Y. Zhang, L.-F. Cun, J. Zhu, J.-G. Deng and J. Liao, J. Am. Chem. Soc., 2010, 132, 4552–4553 CrossRef CAS PubMed; (e) Q.-A. Chen, X. Dong, M.-W. Chen, D.-S. Wang, Y.-G. Zhou and Y.-X. Li, Org. Lett., 2010, 12, 1928–1931 CrossRef CAS PubMed; (f) F. Lang, D. Li, J.-M. Chen, J. Chen, L.-C. Li, L.-F. Cun, J. Zhu, J.-G. Deng and J. Liao, Adv. Synth. Catal., 2010, 352, 843–846 CrossRef CAS.
- (a) H. Li, L. He, J. Lu, W. Zhu, X. Jiang, Y. Wang and Y. Tan, Energy Fuels, 2009, 23, 1354–1357 CrossRef CAS; (b) W. Zhu, H. Li, X. Jiang, Y. Yan, J. Lu, L. He and J. Xia, Green Chem., 2008, 10, 641–646 RSC; (c) L. He, H. Li, W. Zhu, J. Guo, X. Jiang, J. Lu and Y. Yan, Ind. Eng. Chem. Res., 2008, 47, 6890–6895 CrossRef CAS; (d) H. Li, X. Jiang, W. Zhu, J. Lu, H. Shu and Y. Yan, Ind. Eng. Chem. Res., 2009, 48, 9034–9039 CrossRef CAS; (e) W. Huang, W. Zhu, H. Li, H. Shi, G. Zhu, H. Li and G. Chen, Ind. Eng. Chem. Res., 2010, 49, 8998–9003 CrossRef CAS; (f) Y. Ding, W. Zhu, H. Li, W. Jeng, M. Zhang, Y. Duan and Y. Chang, Green Chem., 2011, 13, 1210–1216 RSC; (g) W. Zhu, Y. Ding, H. Li, J. Qin, Y. Yanhong, J. Xiong, Y. Xu and H. Liu, RSC Adv., 2013, 3, 3893–3898 RSC; (h) E. Lissner, W. F. de Souza, B. Ferrera and J. Dupont, ChemSusChem, 2009, 2, 962–964 CrossRef CAS PubMed.
- (a) T. Durst, J. Am. Chem. Soc., 1969, 91, 1034–1035 CrossRef CAS; (b) D. Edwards and J. B. Stenlake, J. Chem. Soc., 1954, 3272–3278 RSC; (c) G. W. Gokel, H. M. Gerdes and D. M. Dishong, J. Org. Chem., 1980, 45, 3634–3639 CrossRef CAS.
- (a) N. K. Jana and J. G. Verkade, Org. Lett., 2003, 5, 3787–3790 CrossRef CAS PubMed; (b) R. J. Griffin, A. Henderson, N. J. Curtin, A. Echalier, J. A. Endicott, I. R. Hardcastle, D. R. Newell, N. E. M. Noble, L. Z. Wang and B. T. Golding, J. Am. Chem. Soc., 2006, 128, 6012–6013 CrossRef CAS PubMed.
- R. S. Varma and K. P. Naicker, Org. Lett., 1999, 1, 189–192 CrossRef CAS.
- N. Fukuda and T. Ikemoto, J. Org. Chem., 2010, 75, 4629–4631 CrossRef CAS PubMed.
- R. S. Varma, R. K. Saini and H. M. Meshram, Tetrahedron Lett., 1997, 38, 6525––66528 CrossRef CAS.
- B. Yu, A. H. Liu, L. N. He, B. Li, Z. F. Diao and Y. N. Li, Green Chem., 2012, 34, 957–962 RSC.
- P. Hanson, R. A. A. J. Hendrickx and J. R. L. Smith, New J. Chem., 2010, 34, 65–84 RSC.
- A. A. Lindén, M. Johansson, N. Hermanns and J.-E. Bäckvall, J. Org. Chem., 2006, 71, 3869–3853 CrossRef PubMed.
- F. Van de Velde, I. W. C. E. Arends and R. A. Sheldon, J. Inorg. Biochem., 2000, 80, 81–89 CrossRef CAS PubMed.
- (a) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2003, 42, 5487–5489 CrossRef CAS PubMed; (b) J. Legros and C. Bolm, Angew. Chem., Int. Ed., 2004, 43, 4225–4228 CrossRef CAS PubMed; (c) M. Bagherzadeh and M. Amini, Inorg. Chem. Commun., 2009, 21–25 CrossRef CAS; (d) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–4941 CrossRef CAS PubMed; (e) B. Li, A.-H. Liu, L.-N. He, Z.-Z. Yang, J. Gao and K.-H. Chen, Green Chem., 2012, 14, 130–135 RSC.
- (a) M. Bagherzadeh, R. Latifi, L. Tahsini and M. Amini, Catal. Commun., 2008, 10, 196–200 CrossRef CAS; (b) M. Bagherzadeh, L. Tahsini and R. Latifi, Catal. Commun., 2008, 9, 1600–1609 CrossRef CAS; (c) F. Xie, Z. Fu, H. S. Zhong, Z. P. Ye, X. P. Zhou, F. L. Liu, C. Y. Rong, L. Q. Mao and D. L. Yin, J. Mol. Catal. A: Chem., 2009, 307, 93–97 CrossRef CAS.
- (a) A. V. Anisimov, E. V. Fedorova, A. Z. Lesnugin, V. M. Senyavi, L. A. Aslanov, V. B. Rybakov and A. V. Tarakanova, Catal. Today, 2003, 78, 319–325 CrossRef CAS; (b) V. Conte, F. Fabbianesi, B. Floris, P. Galloni, D. Sordi, I. W. C. E. Arends, M. Bonchio, D. Rehder and D. Bogdal, Pure Appl. Chem., 2009, 81, 1265–1277 CrossRef CAS; (c) S. Barroso, P. Adao, F. Madeira, M. T. Duarte, J. C. Pessoa and A. M. Martins, Inorg. Chem., 2010, 49, 7452–7463 CrossRef CAS PubMed.
- (a) A. M. Cojocariu, P. H. Mutin, E. Dumitriu, F. Fajula, A. Vioux and V. Hulea, Chem. Commun., 2008, 5357–5359 RSC; (b) M. Mba, L. J. Prins, C. Zonta, M. Cametti, A. Valkonen, K. Rissanen and G. Licini, Dalton Trans., 2010, 39, 7384–7392 RSC; (c) W. Al-Maksoud, S. Daniele and A. B. Sorokin, Green Chem., 2008, 10, 447–451 RSC; (d) S. K. Karmee, L. Greiner, A. Kraynov, T. E. Müller, B. Niemeijera and W. Leitner, Chem. Commun., 2010, 6705–6707 RSC.
- T. Soundiressane, S. Selvakumar, S. Menage, O. Hamelin, M. Fontecave and A. P. Singh, J. Mol. Catal. A, 207, 270, 132–143 CrossRef.
- (a) C. Yang, Q. P. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. G. Deng, Green Chem., 2009, 11, 1401–1405 RSC; (b) R. D. Chakravarthy, V. Ramkumar and D. K. Chand, Green Chem., 2014, 16, 2190–2196 RSC; (c) K. Jeyakumar and D. K. Chand, Tetrahedron Lett., 2006, 47, 4573–4576 CrossRef CAS; (d) T. Cavattoni, T. D. Giacco, O. Lanzalunga, M. Mazzonna and P. Mencarelli, J. Org. Chem., 2013, 78, 4886–4894 CrossRef CAS PubMed; (e) P. W. Davies and S. J.-C. Albrecht, Angew. Chem., Int. Ed., 2009, 48, 8372–8375 CrossRef CAS PubMed; (f) G. P. Romanelli, P. I. Villabrille, C. V. Cáreres, P. G. Vázquez and P. Tundo, Catal. Commun., 2011, 12, 726–730 CrossRef CAS.
- (a) M. Ciclosi, C. Dinoi, L. Gonsalvi, M. Peruzzini, E. Manoury and R. Poli, Organometallics, 2008, 27, 2281–2286 CrossRef CAS; (b) X. Xue, W. Zhao, B. Ma and Y. Ding, Catal. Commun., 2012, 29, 73–76 CrossRef CAS; (c) K. Kamata, T. Hiranoa and N. Mizuno, Chem. Commun., 2009, 3958–3960 RSC.
- X.-F. Wu, Tetrahedron Lett., 2012, 53, 4328–4331 CrossRef CAS.
- (a) L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680–3704 RSC; (b) T. Noël and S. L. Buchwald, Chem. Soc. Rev., 2011, 40, 5010–5029 RSC; (c) C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC; (d) V. Hessel, D. Kralisch, N. Kockmann, T. Noel and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed; (e) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC; (f) C. J. Smith, N. Nikbin, S. V. Ley, H. Lange and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1938–1947 RSC.
- (a) B. Karimi and M. Khorasani, ACS Catall., 2013, 3, 1657–1664 CrossRef CAS; (b) X.-Y. Shi and J.-F. Wei, J. Mol. Catal. A, 2008, 280, 142–147 CrossRef CAS; (c) X. Shi, X. Han, W. Ma, J. Wei, J. Li, Q. Zhang and Z. Chen, J. Mol. Catal. A, 2008, 341, 57–62 CrossRef; (d) S. P. Das, J. J. Boruah, N. Sharma and N. S. Islam, J. Mol. Catal. A: Chem., 2012, 356, 36–45 CrossRef CAS; (e) B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS PubMed; (f) F. Rajabi, S. Naserian, A. Primo and R. Luque, Adv. Synth. Catal., 2011, 353, 2060–2066 CrossRef CAS; (g) J. J. Boruah, S. P. Das, S. R. Ankireddy, S. R. Gogoi and N. S. Islam, Green Chem., 2013, 15, 2944–2959 RSC.
- S. Doherty, in Homogenous Catalysis in Ionic Liquids in Catalysis in Ionic Liquids: From Catalyst Synthesis to Application, ed. C. Hardacre and V. Parvulescu, RSC Catalysis Series, 2014, pp 44–308 Search PubMed.
- Y.-L. Hu, X.-B. Liu and D. Fang, Catal. Sci. Technol., 2014, 4, 38–42 CAS.
- S. Wang, L. Wang, M. Đaković, Z. Popović, H. Wu and Y. Liu, ACS Catal., 2012, 2, 230–237 CrossRef CAS.
- (a) B. Zhang, M.-D. Zhou, M. Cokaja, J. Mink, S.-L. Zang and F. E. Kuhn, RSC Adv., 2012, 2, 8416–8420 RSC; (b) F. Liu, Z. Fu, Y. Liu, C. Lu, Y. Wu, F. Xie, Z. Ye, X. Zhou and D. Yin, Ind. Eng. Chem. Res., 2010, 49, 2533–2536 CrossRef CAS.
- P. Zhao, M. Zhang, Y. Wu and J. Wang, Ind. Eng. Chem. Res., 2012, 51, 6641–6647 CrossRef CAS.
- A. Pourjavadi, S. H. Hosseini, F. M. Moghaddam, B. Koushki and C. Bennett, Green Chem., 2013, 15, 2913–2919 RSC.
- (a) S. Doherty, J. G. Knight, J. R. Ellison, D. Weekes, R. W. Harrington, C. Hardacre and H. Manyar, Green Chem., 2012, 14, 925–929 RSC; (b) S. Doherty, J. G. Knight, J. R. Ellison, P. Goodrich, L. Hall, C. Hardacre, M. J. Muldoon, S. Park, A. Ribeiro, C. A. N. de Castro, M. J. Lourenço and P. Davey, Green Chem., 2014, 16, 1470–1479 RSC.
- (a) F. Gregori, I. Nobili, F. Bigi, R. Maggi, G. Predieri and G. Sartori, J. Mol. Catal. A, 2008, 286, 124–127 CrossRef CAS; (b) E. Baciocchi, M. F. Gerini and A. Lapi, J. Org. Chem., 2004, 69, 3586–3589 CrossRef CAS PubMed; (c) B. M. Choudary, B. Bharathi, C. V. Reddy and M. L. Kantam, J. Chem. Soc., Perkin Trans., 2002, 1, 2069–2074 RSC.
- (a) V. Hulea, A.-L. Maciuca, F. Fajula and E. Dumitriu, Appl. Catal., A, 2006, 313, 200–207 CrossRef CAS; (b) A. L. Maciuca, C. E. Ciocan, E. Dumitriu, F. Fajula and V. Hulea, Catal. Today, 2008, 138, 33–37 CrossRef CAS; (c) M. Bonchio, V. Conti, M. A. De Conciliis, F. Di Furia, F. P. Ballistreri, G. A. Tomaselli and R. M. Toscano, J. Org. Chem., 1995, 60, 4475–4480 CrossRef CAS.
- (a) N. J. Cambell, A. C. Dengal, C. J. Edwards and W. P. Griffith, J. Chem. Soc., Dalton Trans., 1989, 1203–1207 RSC; (b) A. J. Bailey, W. P. Griffith and B. C. Parkin, J. Chem. Soc., Dalton Trans., 1995, 1833–1837 RSC; (c) A. C. Dengel, W. P. Griffith and B. C. Parkin, J. Chem. Soc., Dalton Trans., 1993, 2683–2688 RSC; (d) E. Radkov and R. H. Beer, Polyhedron, 1995, 14, 2139–2143 CrossRef CAS; (e) C. Rocchiccioli-Deltcheff, M. Fournier, R. Franck and R. Thouvenot, Inorg. Chem., 1983, 22, 207–216 CrossRef CAS.
- R. Maggi, S. Chitsaz, S. Loebbecke, C. G. Piscopo, G. Sartori and M. Schwarzer, Green Chem., 2011, 13, 1121–1123 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of polymer 1, polymer immobilized peroxometalate 2, TGA and DSC curves for 1, TEM images and FTIR traces of 2, characterisation data, NMR spectra and mass spectroscopic data for sulfoxides and sulfones, details of catalysis, recycle experiments and graphs showing conversion-selectivity profiles as a function of residence time for [PO4{WO(O2)2}4]@PIILP-catalysed sulfoxidations. See DOI: 10.1039/c4gc01770f |
| This journal is © The Royal Society of Chemistry 2015 |