
Structure–performance relations of molybdenum- and tungsten carbide catalysts for deoxygenation†
Daniel R.
Stellwagen
*a and
Johannes H.
Bitter
ab
aDepartment of Inorganic Chemistry and Catalysis, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: d.r.stellwagen@uu.nl
bBiobased Chemistry and Technology, Wageningen University, Bornse Weilanden 9, 6708WG, Wageningen, The Netherlands. E-mail: harry.bitter@wur.nl; Tel: +31(0)317480303
First published on 23rd October 2014
Abstract
This work demonstrates for the first time that carbide particle size is a critical factor for the activity and stability of carbon supported tungsten- and molybdenum carbide catalysts in (hydro-)deoxygenation reactions. The stability of the catalyst was shown to increase for larger particles due to the improved resistance of the metal carbide phase against full oxidation to crystalline metal oxides under reaction conditions. In addition to the improved catalyst stability, supported molybdenum carbides were found to more than double their weight-based catalytic activity upon increasing carbide particle size from 2 to 10 nanometers. The strongly improved (de-)hydrogenation activity of these larger carbide particles also facilitated a new deoxygenation pathway for fatty acids, in which an initial hydrogenation to fatty-aldehyde is combined with a decarbonylation step. This is the first time in which this deoxygenation pathway is observed over supported tungsten- or molybdenum carbide catalysts.
Introduction
The possibility of petroleum scarcity has been an incentive in the recent decades to explore alternative sources for oil-derived products. A resource that can function as a partial petroleum replacement is biomass. Triglyceride and fatty acid feeds are most easily utilized, since their structural similarity to diesel-range hydrocarbons allows them to be used in the current infrastructure with only minimal adaptation. In particular jatropha- or microalgae oil are considered promising in this regard, since they are non-edible and thus not in competition with existing food consumption.1 These triglyceride feeds can be upgraded to transportation fuels via transesterification, yielding fatty-acid methyl ester (FAME) type biodiesel. Alternatively, the biomass derived oils can be fully deoxygenated to produce alkanes or alkenes. This (hydro-)deoxygenation (HDO) process has received a lot of attention in recent years.1–4 Most studies have focused on the use of existing hydro-desulfurization (HDS) catalysts, or the use of palladium based catalysts.3,4 However, the high cost of noble metal catalyst and the issues related to the use of sulfur in the HDS catalysts have prompted a search for new types of deoxygenation catalysts. Promising results have recently been obtained with supported tungsten- and molybdenum carbides. These materials were shown be active for the deoxygenation of triglycerides and fatty acids to linear alkanes, while product selectivity could be further tuned to alkenes and branched alkanes if desired.5–9The latter significantly improve the quality of the diesel fuel, while the alkenes might find direct application in cosmetics, or be used as bulk chemical precursors. Though these transition metal carbides appear to be versatile deoxygenation catalysts, there are still unsolved issues regarding the activity and stability of such catalysts. Supported molybdenum carbide catalysts have been reported to be able to produce alkanes from fatty acids in high yields, but the deoxygenation reaction required either high reaction temperatures and long reaction times,6 or use of a supercritical solvent as reaction medium.7,8 Tungsten carbides have been reported to be highly selective for the production of alkenes from fatty acid.5,9 However, catalyst activity was reported to be slightly lower than that of supported molybdenum carbides, while the tungsten carbides additionally suffered from deactivation due to oxidation of the active carbide phase.6
In order to design carbon nanofiber (CNF) supported carbide catalysts with an improved performance in deoxygenation reactions, supported tungsten- and molybdenum carbides of different crystallite sizes were examined in this study for the hydro-deoxygenation of stearic acid to hydrocarbons. To vary carbide particle size while maintaining equal metal loadings, the distribution of the metal precursor over the carbon nanofiber support after impregnation was altered by changing the surface properties of the CNF support. Surface oxygen groups were introduced to the CNF by treatment with nitric acid (‘CNF-ox’), while a non-oxidizing treatment with hydrochloric acid instead resulted in a largely oxygen-free surface (‘CNF-hcl’).10 The surface oxygen groups of the CNF-ox material are known to interact with the poly-molybdate or -tungstate complexes that are present in the aqueous impregnation solutions. Therefore, impregnation of CNF-ox results in a higher metal dispersion than impregnation of the CNF-hcl support.11,12 Conversion of the tungsten- and molybdenum ammonium salts to their respective carbides was achieved using a carbothermal reduction process, in which the material is heated under N2 flow after impregnation and drying, resulting in reduction and carburization of the metal by the carbon support.13,14 The preferential stearic acid deoxygenation pathway was investigated for each of the catalysts by comparing the differences in product yield at different reaction times. In addition, the conversion of key intermediates such as octadecanol and octadecanal was also examined over these carbide catalysts. The obtained insights allowed us to design a process which optimized alkene yield in the deoxygenation of stearic acid.
Results and discussion
Catalyst preparation and characterization
The molybdenum- and tungsten carbide catalysts examined in this work were prepared by impregnation and subsequent heat treatment using two different types of carbon nanofiber supports. Details on the catalyst preparation method used in this work can be found in the ESI.† The samples that were prepared using a polar CNF-ox support are denoted X2C/CNF, while the samples that were prepared using an a-polar CNF-hcl support are denoted b-X2C/CNF, with X = Mo,W in both cases.Fig. 1 shows the XRD patterns of the supported carbide catalysts used in this work. Diffractions corresponding to the Mo2C and W2C carbide phases (shown as Δ in Fig. 1) are observed at 40, 44, 46, 62 and 73 degrees 2θ for the b-Mo2C/CNF and b-W2C/CNF catalysts.
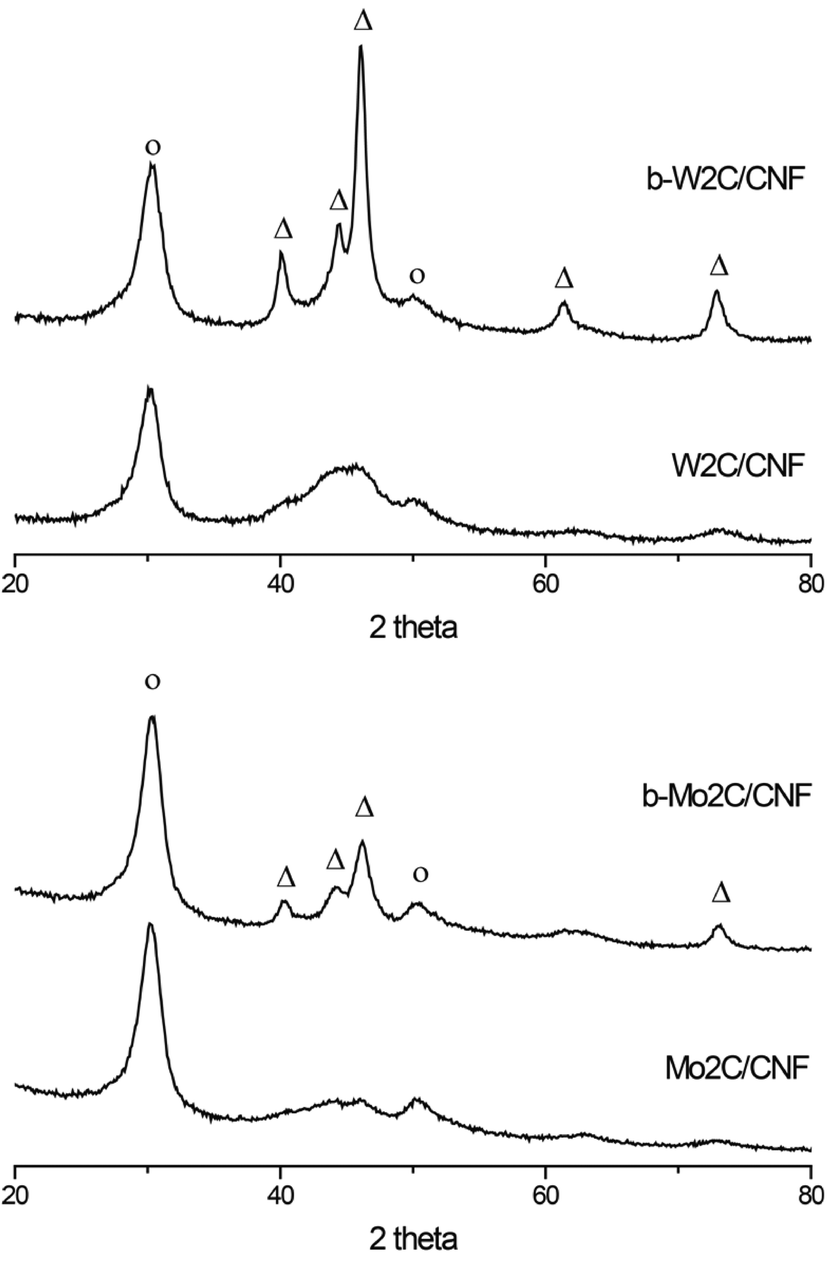 | ||
| Fig. 1 XRD diffraction patterns of the CNF supported tungsten (top) and molybdenum (bottom) carbides used in this work. Symbols: Δ, diffractions for X2C phase (X = Mo,W); o, diffraction for the CNF support. | ||
For these samples a carbide crystallite size of 10–12 nm was estimated from the line broadening of the three carbide reflections between 35–50 degrees 2θ. The bottom two diffraction patterns in Fig. 1, corresponding to Mo2C/CNF and W2C/CNF, are less well defined and show much broader peaks in the carbide region. Particle size of these catalysts was calculated to be below 3 nm. The extent of metal carburization achieved during the heat treatment step was also verified with TGA (Fig. S1†).
Total weight loss corrected for weight loss of the bare support in the same temperature region confirms the target loadings of 7.8 wt% molybdenum and 14.6 wt% tungsten when assuming the final phase is Mo2C or W2C respectively, indicating that full carburization was achieved in 3 hours at 900 °C in all cases. It should be noted that the TGA profiles of the bare support materials confirmed that the surface oxygen groups on the CNF-ox support were completely removed upon heat treatment at 900 °C, and thus play no role in the performance of the final CNF supported carbide catalysts.
Fig. 2 shows HAADF-TEM and bright field TEM images of the samples. It can be seen that the W2C/CNF and Mo2C/CNF catalysts consist of highly dispersed, small carbide particles. Average particle size was approximately 2 nm for both catalysts. Measurements at larger magnification (Fig. 2, middle) showed that the catalysts contained a significant number of smaller carbide particles, with a size of around 1 nm. The bright field TEM images of the b-Mo2C/CNF and b-W2C/CNF catalysts, which were prepared using an a-polar CNF-hcl support, are shown at the bottom part of Fig. 2. Average particle size was determined to be 11 nm for b-Mo2C/CNF and 14 nm for the b-W2C/CNF catalyst, which is in good agreement with the XRD estimates. The characteristics of the different types of CNF supported carbide catalysts used in this work are summarized in Table 1.
 | ||
| Fig. 2 HAADF-TEM images of W2C/CNF (top; middle right) and Mo2C-CNF (top; middle left) at different magnifications. Bright field TEM images are shown for b-W2C (bottom right) and b-Mo2C/CNF (bottom left). | ||
| Catalyst | Supporta | Particle size (XRD)b | Particle size (TEM) | Surface area (m2 g−1) |
|---|---|---|---|---|
| a CNF-ox = HNO3-pretreated carbon nanofibers. CNF-hcl = HCl-pretreated carbon nanofibers. b Estimated using Scherrer equation. | ||||
| Mo2C/CNF | CNF-ox | <3 nm | 2 (±1) nm | 120 |
| b-Mo2C/CNF | CNF-hcl | 10 nm | 11 (±2) nm | 107 |
| W2C/CNF | CNF-ox | <3 nm | 1.5 (±0.5) nm | 118 |
| b-W2C/CNF | CNF-hcl | 12 nm | 14 (±4) nm | 110 |
The tungsten- and molybdenum carbide catalysts were further examined using XPS to investigate the extent of surface oxidation of the carbide catalysts. Previous work6 on CNF supported tungsten- and molybdenum carbides showed that the surface of small (<4 nm) particles was severely oxidized upon short exposure to air at ambient conditions. XPS spectra of these sub-4 nm sized W2C and Mo2C particles were shown to have a large (>75%) surface contributions of W6+ (‘WOx’) and Mo6+ (‘MoOx’) species, with the tungsten carbide sample showing more extensive oxidation. In the current work, the supported tungsten- and molybdenum carbides with a larger particle size (10–12 nm) were examined using the same XPS setup. The XPS spectra of these b-W2C/CNF and b-Mo2C/CNF catalysts are shown in Fig. 3 and Table 2 below. The W-4f region shows signals at 32.2 eV (W-4f7/2) and 34.4 eV (W-4f5/2), corresponding to tungsten carbide, which are in good agreement with literature data.16 Signals observed at 35.9 eV (W-4f7/2) and 38.0 eV (W-4f5/2) are related to tungsten oxide (W6+).
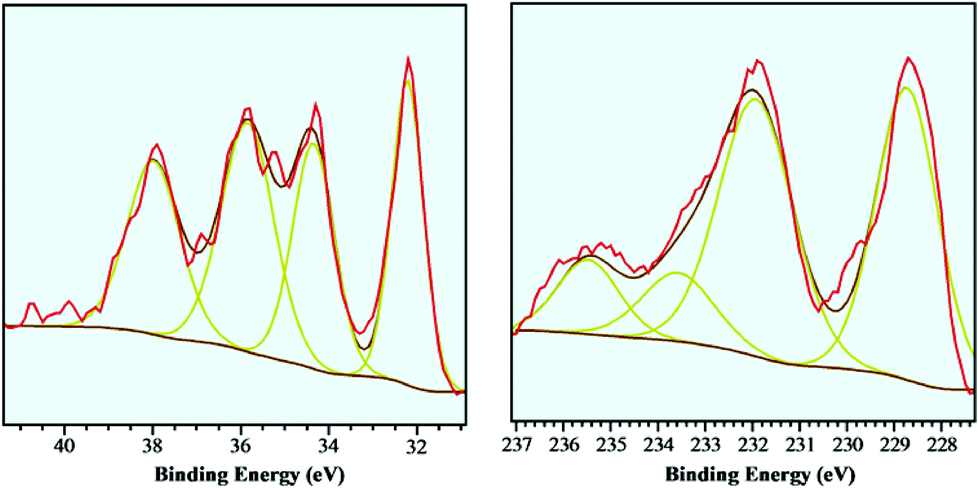 | ||
| Fig. 3 XPS spectra of b-W2C/CNF, W-4f edge (left) and b-Mo2C/CNF, Mo-3d edge (right). | ||
| Mo-C (3d-3/2) | Mo-C (3d-5/2) | Mo4+ (3d-5/2) | Mo6+ (3d-5/2) | |
|---|---|---|---|---|
| Position (eV) | 228.7 | 231.9 | 233.5 | 235.5 |
| fwhma (eV) | 1.5 | 1.8 | 1.8 | 1.5 |
| Concentration (at%) | 38.7 | 41 | 10.6 | 9.7 |
From the data of the W-4f edge shown in Fig. 3 and Table 2, the tungsten carbide/oxide surface ratio was estimated to be 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49, indicating the surface oxidation of the large tungsten carbide particles was significant, though less than that of their small counterparts.6 The exposure to ambient air that caused the initial oxidation of the carbide surfaces occurred during the passivation treatment in a flow of ambient air after carburization of the samples, though brief contact with ambient air could not be excluded in the sample preparation prior to the XPS measurements.
:49, indicating the surface oxidation of the large tungsten carbide particles was significant, though less than that of their small counterparts.6 The exposure to ambient air that caused the initial oxidation of the carbide surfaces occurred during the passivation treatment in a flow of ambient air after carburization of the samples, though brief contact with ambient air could not be excluded in the sample preparation prior to the XPS measurements.
The Mo-3d region shows two distinctive contributions at 228.7 eV (Mo-3d5/2) and at 231.9 eV (Mo-3d3/2) which originate from molybdenum carbide. A shoulder at higher energies (233.5 eV) is related to molybdenum(IV)-oxides or oxo-carbides, while the peak at 235.5 eV could be assigned to Mo(VI) oxide species.17
The molybdenum carbide/oxide surface ratio was estimated to be 80:20, indicating that the surface oxidation of the molybdenum carbide sample b-Mo2C/CNF was less pronounced than that of the tungsten carbide sample b-W2C/CNF discussed above.
The difference in metal-carbide/oxide surface ratio between the b-W2C/CNF and b-Mo2C/CNF catalysts is consistent with the previously reported results for small carbide particles supported on carbon nanofibers,6 indicating that tungsten carbides are in general more susceptible to oxidation in ambient air than molybdenum carbides. Both the tungsten- and molybdenum carbides showed a decrease in oxidation of the carbide surface in ambient conditions upon an increase of their particle size. The oxide species (‘WOx’ and ‘MoOx’) that were observed in the XPS measurements on the tungsten- and molybdenum carbide surfaces have previously been shown to have acidic properties.18,21,22 The carbides used in this work are thus expected to behave as bi-functional catalysts, possessing both (de-)hydrogenation activity due to the surface carbide species and acidic properties due to the surface oxide domains.
Indeed, NH3-TPD measurements done previously on similar molybdenum carbides have qualitatively shown that exposure to oxygen under ambient conditions generates (weak) acid sites, related to the formation of MoO3 on the catalyst surface.25
Catalytic performance of tungsten- and molybdenum carbides in the deoxygenation of stearic acid
Tungsten- and molybdenum carbides supported on carbon nanofibers were investigated in the deoxygenation of stearic acid. Reactions were performed in a batch autoclave at an initial H2 pressure of 30 bar and a temperature of 350 °C.Representative plots showing product yields over time for the Mo2C/CNF and W2C/CNF catalysts, both consisting of 2 nm particles, are presented in Fig. 4. Under these conditions, stearic acid was previously reported to be converted to hydrocarbon products via the hydrodeoxygenation (HDO) route over both tungsten- and molybdenum carbide catalysts, yielding octadecane as the final product.5–9Fig. 4 shows octadecane yield are identical at 85% for both Mo2C/CNF and W2C/CNF catalysts after 300 minutes of reaction, which suggests that the deoxygenation proceeds via a similar pathway on both catalysts. However, Fig. 4 also shows that the liquid phase product distribution at intermediate reaction times (i.e. 30–180 minutes) was clearly different for the two catalysts, which demonstrates that the deoxygenation pathway is in fact different for both catalysts, even though the final product is the octadecane in both cases. In the molybdenum carbide catalyzed conversion of stearic acid (Fig. 4, left) a large fraction of the C18-oxygenate intermediates octadecanol and octadecanal was observed in the reaction mixture at short reaction times (60% yield after 60 minutes). These intermediates were obtained by hydrogenation of the stearic acid substrate.3,5 The build-up of C18-oxygenates in the reaction mixture indicates that stearic acid hydrogenation was fast compared to the subsequent conversion of these reaction intermediates. Conversion of the octadecanol intermediate can occur via a dehydration step to yield the C18-alkene octadecene, which is in turn hydrogenated to the C18-alkane octadecane.5,19,20 C17-alkanes were also observed in small quantities (<5%) in the reaction mixture. These C17 hydrocarbons were previously reported to be produced via a direct decarboxylation or decarbonylation (DCO) of the stearic acid substrate.5,6
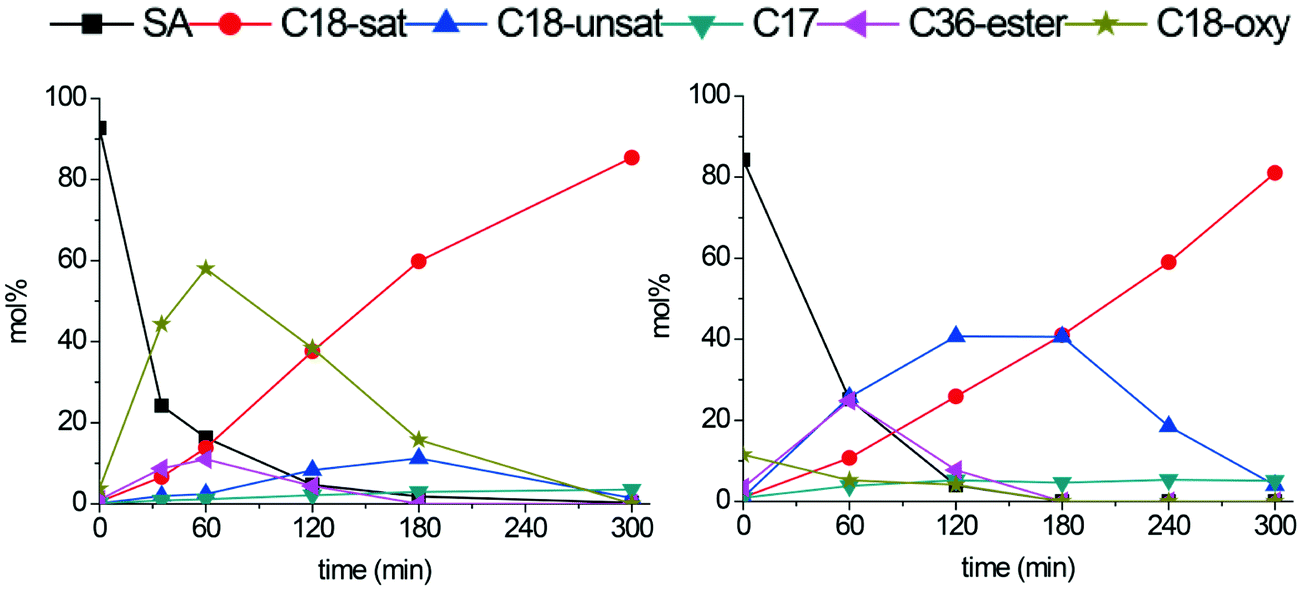 | ||
| Fig. 4 Stearic acid deoxygenation over Mo2C/CNF (left) and W2C/CNF (right). Reaction temperature: 350 °C. H2 pressure: 30 bar. | ||
The tungsten carbide catalyst showed only trace amounts of C18-oxygenate intermediates in the first 60 minutes of reaction (Fig. 4, right). Instead this catalyst selectively produced C18-alkenes and stearyl-stearate intermediates (both 25% after 60 minutes). Stearyl-stearate was produced by esterification of stearic acid with the octadecanol that is present in the reaction mixture. The acid catalyzed esterification can take place on small tungsten- or molybdenum oxide domains (‘WOx’ or ‘MoOx’), which are known to have acidic properties.18,21,22 At longer reaction times the stearyl-stearate ester was converted to a C18-alkene by hydrogenation/dehydration. After 300 minutes of reaction the octadecene intermediate was fully hydrogenated to the final product octadecane. Similar to the molybdenum carbide catalyzed deoxygenation of stearic acid, small amounts (5%) of DCO products were observed in the reaction mixture after 300 minutes.
The difference in intermediate product selectivity between the two carbides can be explained by the more oxophilic nature of tungsten carbide (W2C) surfaces compared to molybdenum carbide (Mo2C) surfaces. Since both carbide catalysts were shortly exposed to air flow after synthesis to passivate the surface, interaction between the carbide surface and the oxygen in the air can be assumed. Tungsten carbides were previously shown to readily form bi-functional oxo-carbide type species upon exposure to oxygen, while oxygen modification of a molybdenum carbide surface proceeds more slowly.6,18,22 The XPS measurements on the CNF supported molybdenum- and tungsten carbides show that the respective ‘MoOx’ and ‘WOx’ metal oxide domains are present in both supported carbides, but the extent of surface oxidation was found to be higher on the tungsten carbides.6
The stability of the W2C/CNF and Mo2C/CNF catalysts was investigated by performing recycle tests using spent catalysts from the initial run shown in Fig. 4. Changes in the structure of the spent catalysts were monitored using XRD. Diffraction patterns of the fresh and spent W2C/CNF catalysts are shown in Fig. 5. A change in tungsten phase can be clearly observed going from the fresh to the spent W2C/CNF catalyst.
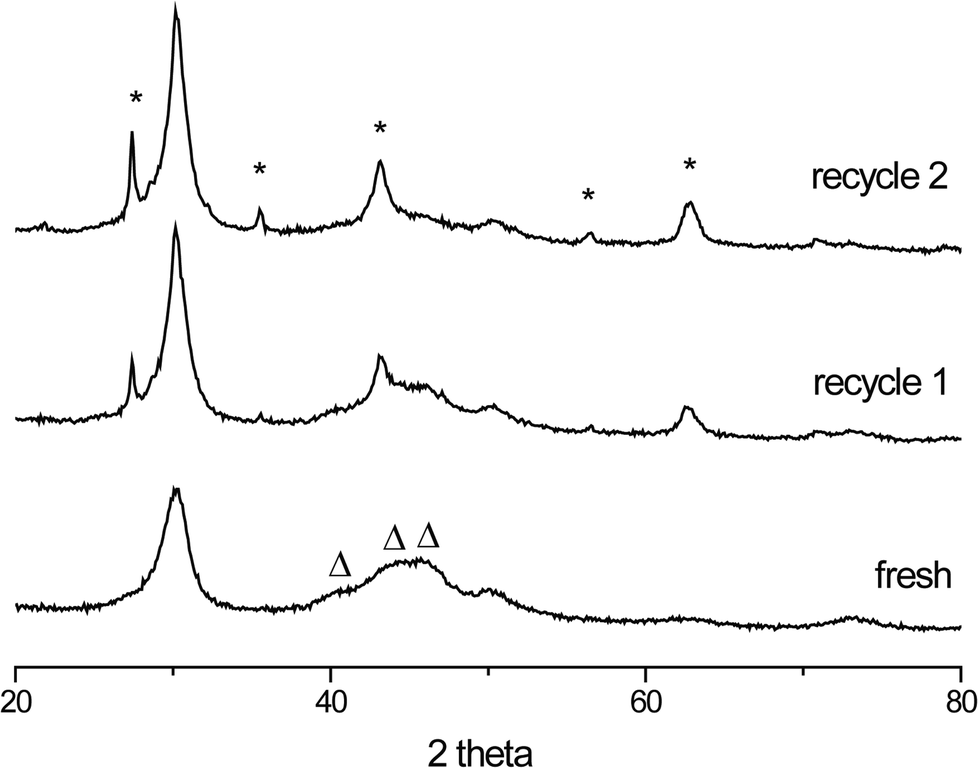 | ||
| Fig. 5 Fresh and spent W2C/CNF catalysts. New peaks in the spent catalysts (*) can be identified as WO2 and WO2.72 crystallites. The original W2C phase (Δ) disappears upon recycle. | ||
The spent catalysts (recycle 1 and 2) showed sharp peaks corresponding to crystalline WO2 (43, 56, and 63 degrees 2θ) and WO2.72 (27 and 36 degrees 2θ) phases. The formation of these partially reduced oxides comes at the expense of the tungsten carbide phase, of which the diffraction peaks decreased in intensity.
Another WO2 diffraction line at 30 degrees 2θ coincides with a different diffraction originating from the graphitic CNF support. Due to formation of the WO2 crystal phase, this CNF reflection thus appeared to become sharper and increase in intensity upon recycle of the catalyst. The change in catalyst structure can be correlated to a decrease in catalyst activity. The W2C/CNF catalyst lost approximately half of its activity upon re-use. The 2nd run showed that only 70% of the stearic acid was converted after 300 minutes of reaction, with the corresponding C18-hydrocarbon yield being 58%. The decrease in the ability of the catalyst to convert stearic acid is consistent with the disappearance of the tungsten carbide phase observed in XRD, while conversion of the C18-oxy intermediates proceeded faster due to the appearance of acidic tungsten oxide phases such as WO2.72. A similar result was obtained for the Mo2C/CNF catalyst (not shown), in which a decrease in intensity of the molybdenum carbide diffractions in XRD corresponded to a loss in catalyst activity of 30% upon recycle. These results are consistent with previous work that showed that the small, 1–2 nanometer sized particles present in these W2C/CNF and Mo2C/CNF catalysts are not stable under these reaction conditions due to oxidation of the carbide particles, resulting in a decrease in catalyst activity.6 Both the W2C/CNF and the Mo2C/CNF catalysts used in this work similarly showed a decrease in activity upon recycle, mainly due to formation of crystalline reduced oxide phases (i.e. WO2 and MoO2) that were previously shown to be inactive in deoxygenation reactions.5 In order to improve catalyst stability, the carbide phase is required to improve its resistance against oxidation to crystalline oxides. To this end, both molybdenum- and tungsten carbide catalysts with larger particle size were synthesized as described above (denoted b-Mo2C/CNF and b-W2C/CNF). The carbide particle size was increased from approximately 2 nm to 10–12 nm for the b-Mo2C/CNF and b-W2C/CNF catalysts. The larger carbide particles were tested for the deoxygenation of stearic acid.
Recycle tests were performed to investigate the stability of the new carbide catalysts. XRD diffractograms of the fresh and spent (recycle 1 and 2) b-Mo2C/CNF and b-W2C/CNF catalysts are shown in Fig. 6. No change in the diffraction patterns of the fresh and spent b-Mo2C/CNF catalysts (Fig. 6, top) was observed. Similarly, formation of a WO2 phase was almost fully suppressed upon reuse of the b-W2C/CNF catalyst (Fig. 6, bottom). This is a large difference compared to the results shown in Fig. 5, where the small W2C/CNF particles were almost completely oxidized to crystalline WO2 after the first recycle test. As indicated by the XRD patterns in Fig. 6, the larger carbide particles were more stable during reaction than their smaller counterparts. No loss in catalyst activity was observed in upon reuse of the supported large particle tungsten- and molybdenum carbide catalysts.
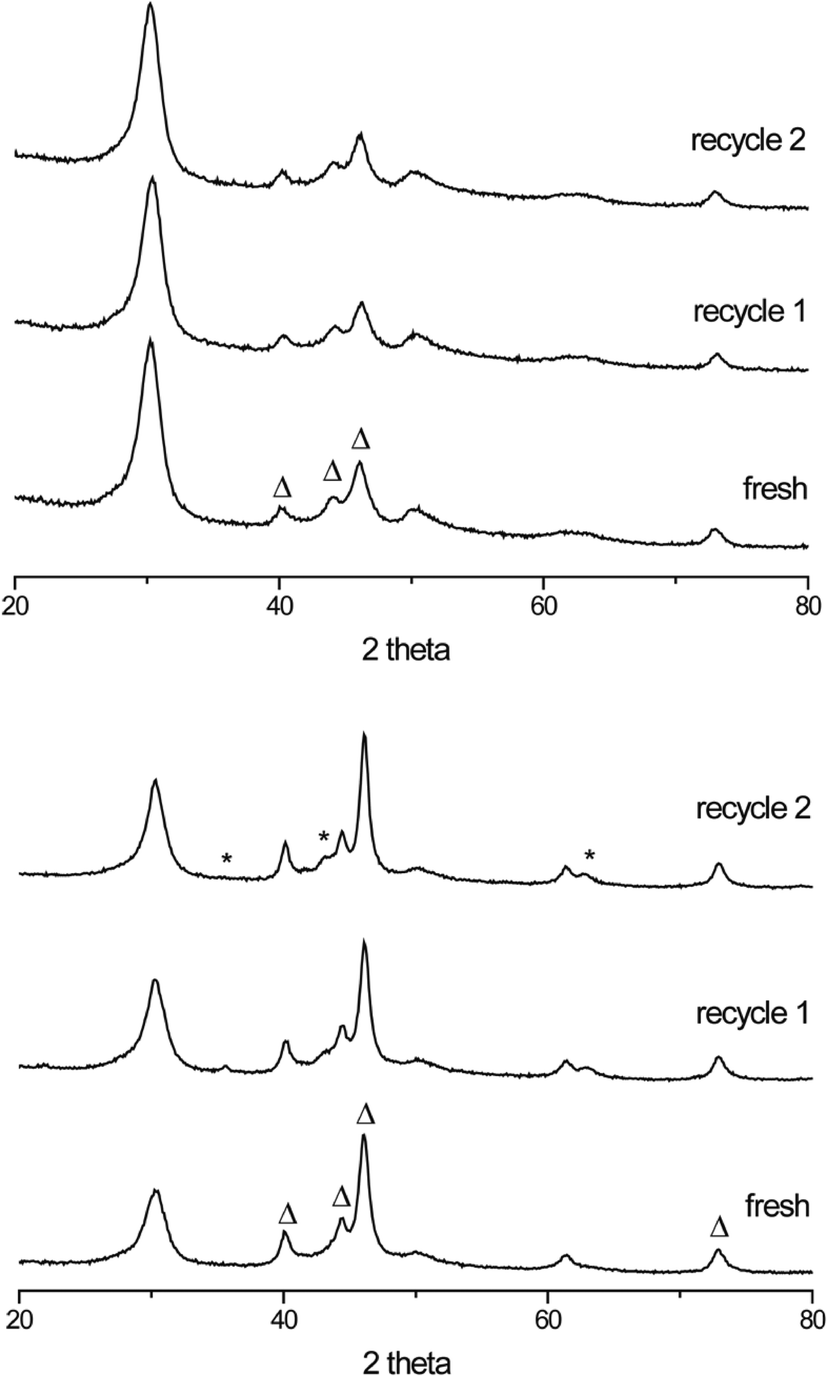 | ||
| Fig. 6 XRD of b-Mo2C/CNF (top) and b-W2C/CNF (bottom). The carbide structure (Δ) is observed in all diffractograms. Peaks related to trace WO2 (*) can also be observed. | ||
To further verify the oxidative stability of the larger carbide particles, we have performed a reference oxidation experiment in which the b-Mo2C/CNF catalyst was exposed to ambient air for an additional 6 h after the initial carburization and passivation procedure described in the Experimental section. This reference test shows that the product distribution over time, shown in Table S1,† is nearly identical to the fresh b-Mo2C/CNF catalyst. This indicates that the large molybdenum carbide particles are more resistant to oxidation than their smaller counterparts, which were previously shown to degrade upon exposure to ambient air.6
Conversion and product yields over time for the fresh b-Mo2C/CNF and b-W2C/CNF catalysts are shown in Fig. 7. Remarkably, the larger particles not only showed an increase in stability, but also significant changes in activity and product selectivity. Full deoxygenation to hydrocarbons was now achieved in 90 minutes with the b-Mo2C/CNF catalyst. This is a significant increase compared to the original Mo2C/CNF catalyst, over which full deoxygenation could only be achieved after 180 minutes (Fig. 4). Over the b-W2C/CNF catalyst, full deoxygenation of stearic acid to hydrocarbons was completed after 180 minutes, which is similar to what was found for the original W2C/CNF catalyst shown in Fig. 4. A comparison of Fig. 4 and 7 shows that the original W2C/CNF catalyst gives high selectivity to stearyl-stearate and octadecene at intermediate conversion levels, while the new b-W2C/CNF catalyst instead gives a build-up of C18-oxygenates octadecanol and octadecanal. This build-up of C18-oxygenates at the cost of octadecene and stearyl-stearate intermediates indicates a decrease in the amount of acidic ‘WOx’ domains and an increased hydrogenation capability for the b-W2C/CNF catalyst compared to the original W2C/CNF catalyst. This is consistent with the XPS and XRD results, which indicate that the larger tungsten carbides particles are less susceptible to (surface) oxidation. The data in Fig. 7 shows that a large amount of the C17-alkane heptadecane was formed over both b-W2C/CNF and b-Mo2C/CNF catalysts. While the presence of C17 hydrocarbons implies that direct decarboxylation and/or decarbonylation (DCO) of stearic acid occurred, the production of high amounts of C18-oxygenates prior to the genesis of the C17-alkane seems to indicate that the heptadecane was in fact not formed by direct DCO of stearic acid. Instead, octadecanol and/or octadecanal appear to be converted to both C18- and C17 hydrocarbons in parallel reaction pathways. To investigate the formation of these C17 hydrocarbons in more detail and to explore the effect of reaction temperature on the deoxygenation pathway, the stearic acid deoxygenation was also performed at lower reaction temperature (300 °C).
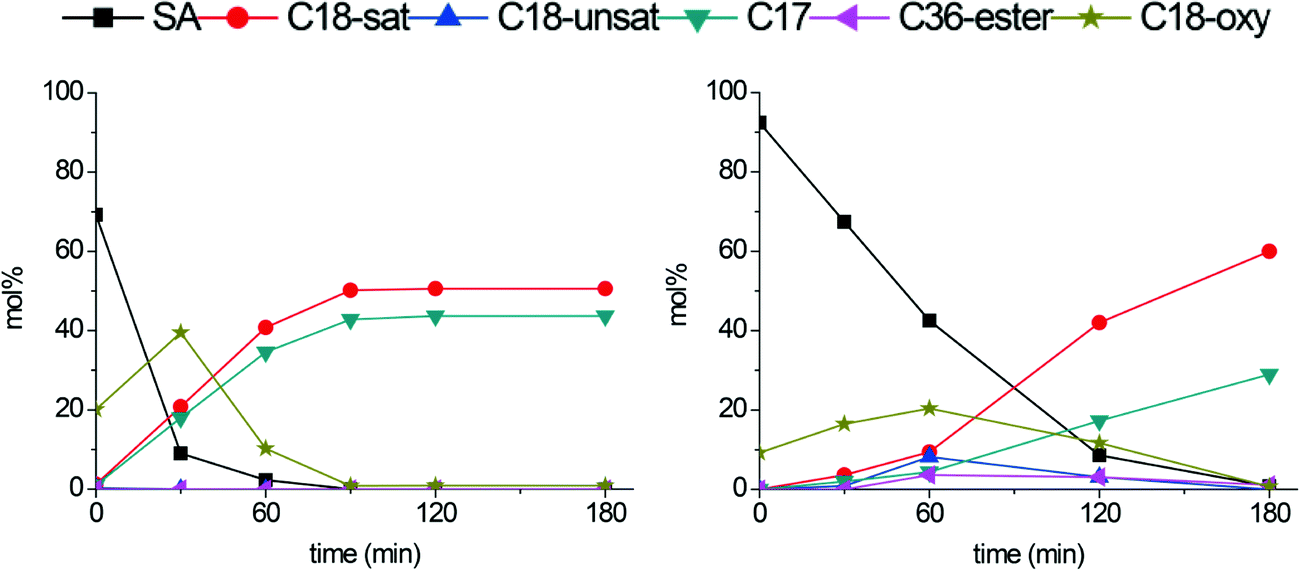 | ||
| Fig. 7 Stearic acid deoxygenation over b-Mo2C/CNF (left) and b-W2C/CNF (right). Reaction temperature: 350 °C. H2 pressure: 30 bar. | ||
Fig. 8 shows the conversion plots for stearic acid deoxygenation at 300 °C catalyzed by Mo2C/CNF (left) and b-Mo2C/CNF (right) catalysts. The Mo2C/CNF catalyst (left) achieved full conversion of stearic acid in 300 minutes. The main products were C18-oxygenates (octadecanol and octadecanal) and the C36-ester (stearyl-stearate), while only C18 hydrocarbon products were observed.
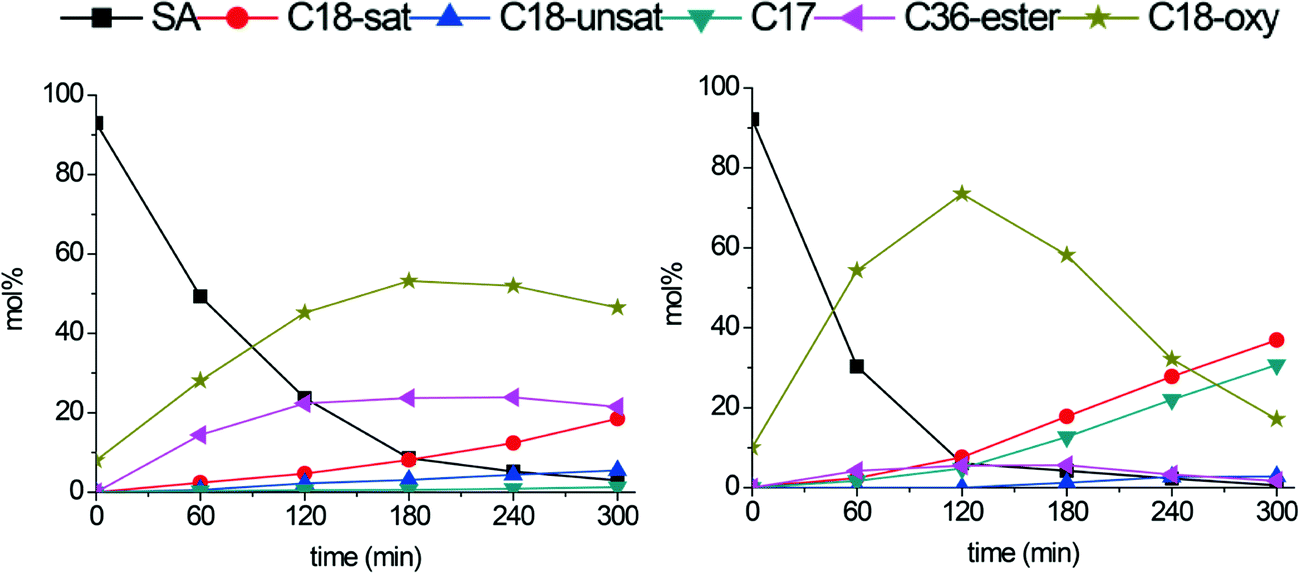 | ||
| Fig. 8 Stearic acid deoxygenation over Mo2C/CNF (left) and b-Mo2C/CNF (right). Reaction temperature: 300 °C. H2 pressure: 30 bar. | ||
The b-Mo2C/CNF catalyst (right) was more active and achieved full conversion of stearic acid after 120 minutes of reaction, producing C18-oxygenates octadecanal (22%) and octadecanol (51%) in high yields. These C18-oxy intermediates were subsequently converted to both C17- and C18 alkanes in parallel reaction pathways. The conversion of C18-oxygenates to C18 hydrocarbons occurred via dehydration of the C18-alcohol, as already mentioned above. The conversion of C18-oxygenates to C17-hydrocarbons can proceed via decarbonylation of the octadecanal intermediate, possibly in combination with a dehydrogenation step, to yield either heptadecane or heptadecene.19,20 An overview of the different deoxygenation pathways of stearic acid over these supported carbide catalysts is shown in Scheme 1.
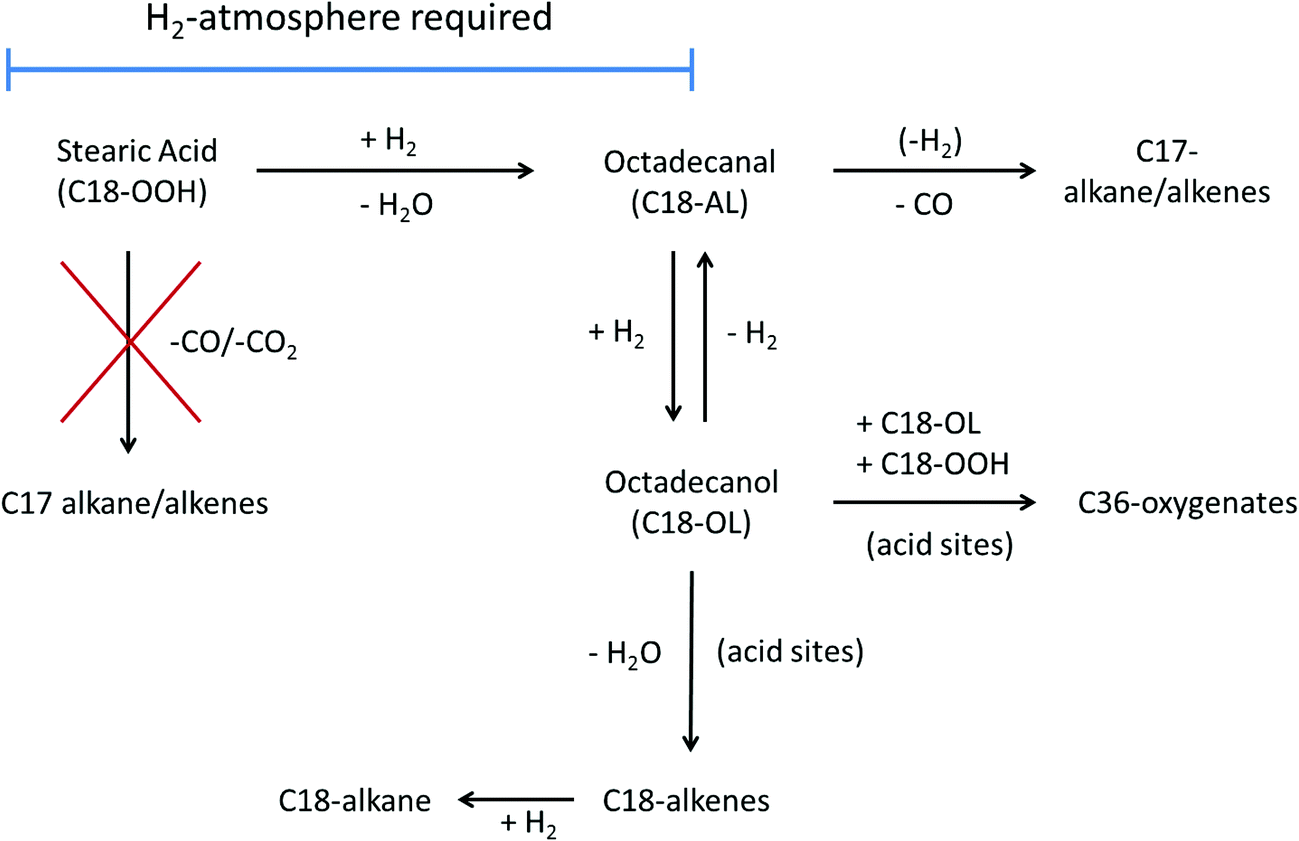 | ||
| Scheme 1 Reaction pathways for stearic acid (hydro)deoxygenation over supported tungsten- and molybdenum carbides. | ||
The conversion pathways of C18-oxygenate intermediates over the different carbide catalysts were further investigated by studying the conversion of C18-alcohol octadecanol over both small and large tungsten- and molybdenum carbides particles in a hydrogen-free atmosphere. The absence of external hydrogen prevented excess hydrogenation of formed alkenes, while a possible dehydrogenation pathway to form octadecanal was facilitated. It is important to note that stearic acid could not be converted by any of the carbide catalyst under these H2-free conditions, indicating direct decarboxylation and/or decarbonylation of the acid functional group was not possible on the carbide surfaces used in this work. The performance of the various carbide catalysts in the conversion of octadecanol under N2 atmosphere is reported in Table 3.
| Catalyst | Conversiona (%) | C18-AL (%) | C18-sat (%) | C18-unsat (%) | C17-sat (%) | C17-unsat (%) | C36-Ob (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 2.0 g octadecanol, 36 g dodecane, 1.0 g tetradecane (IS), 0.25 g catalyst. T = 300 °C, 30 bar N2 pressure, 360 minutes. b Higher (C36-) oxygenates. c Catalyst: WO3/CNF.5 Conversion after 120 minutes. d Substrate: octadecanal. 0.125 g catalyst. Conversion after 120 minutes. e Substrate: stearic acid. No conversion observed after 360 minutes of reaction. | |||||||
| Mo2C/CNF | 66 | 24 | 12 | 11.5 | 4 | 1 | 12 |
| b-Mo2C/CNF | 65 | 18 | 7.8 | 7.1 | 17.6 | 8.5 | 3.5 |
| W2C/CNF | >99 | 6.5 | 2.6 | 81 | — | <1 | 5.5 |
| b-W2C/CNF | 96 | 22.5 | 3.8 | 60.4 | — | 3.9 | 5.3 |
| WO3/CNFc | >99 | 12 | <1 | 81 | — | — | 4 |
| b-Mo2C/CNFd | 32 | — | — | — | — | 25 | 4 |
| b-Mo2C/CNFe | <1 | <1 | — | — | — | — | <1 |
The results listed in Table 3 show that the tungsten carbide catalysts predominantly enabled dehydrogenation of the octadecanol substrate, yielding octadecene as the main product. Dehydrogenation of octadecanol to the C18-aldehyde octadecanal was also observed, thus producing in situ hydrogen that allowed for the partial hydrogenation of the octadecene product. The formed C18-aldehyde was decarbonylated over the b-W2C/CNF catalyst, yielding heptadecene products. Compared to the tungsten carbides, the molybdenum carbide catalysts had a lower activity for the dehydration of octadecanol under these conditions. Instead, (de-)hydrogenation reactions dominated, leading to an increase in aldehyde- and saturated hydrocarbon products. For the b-Mo2C/CNF catalyst, decarbonylation of the aldehyde intermediate was a major reaction pathway, giving high yields of C17-hydrocarbons. This was further verified by testing the b-Mo2C/CNF catalyst for the deoxygenation of octadecanal in N2 atmosphere. Table 3, entry 6, shows that octadecanal was selectively decarbonylated to heptadecenes on b-Mo2C/CNF. Formation of higher C36-oxygenates via condensation of C18-oxygenates was observed to be a minor reaction pathway over all of the tested carbide catalysts when starting from the C18-alcohol.
As mentioned earlier, the reason for the different behavior of the molybdenum- and tungsten carbide catalysts is most likely the more oxophilic nature of the latter. Thus, the presence of acidic WOx-domains on the tungsten carbide surface facilitated dehydration of the C18-alcohol, while hindering (de-)hydrogenation reactions. For this reason, the product distribution for the deoxygenation of octadecanol over the W2C/CNF catalyst is comparable to that over the pure tungsten oxide catalyst WO3/CNF.
A similar explanation can be given for the difference in product selectivity between the Mo2C/CNF and b-Mo2C/CNF catalyst (and to a lesser extent between the W2C/CNF and b-W2C/CNF catalysts). The smaller carbide particles are more susceptible to oxidation upon exposure to air, thereby increasing the amount of weakly acidic oxide domains and thus increasing the selectivity towards dehydration products and higher oxygenates.
The intermediate tests reported in Table 3 indicate there is a possibility to efficiently convert stearic acid to alkenes via a two-step process. The initial hydrogenation step, converting stearic acid to octadecanol, is performed in a H2 atmosphere, while further conversion of C18-oxygenates to alkenes can be efficiently performed in a hydrogen free environment, thus minimizing H2 usage in the deoxygenation process. To test this hypothesis, stearic acid deoxygenation was performed over a b-Mo2C/CNF catalyst at 300 °C. After 120 minutes, the reaction was quenched by rapidly cooling the reactor. The temperature of the reaction mixture was then kept at 60 °C, during which the reactor was purged of hydrogen gas by three subsequent pressurization–depressurization cycles with nitrogen. After this, the reaction mixture was re-pressurized with nitrogen and re-heated to 300 °C. The reaction was then continued for an additional 180 minutes. The product distribution over time is plotted in Fig. 9. After the first 120 minutes of reaction (with H2 pressure) near full conversion of stearic acid was obtained. The main products were octadecanol (56%) and octadecanal (11%, not shown). Other products were stearyl-stearate (9.5%, not shown), octadecane (9.5%) and heptadecane (9.4%). The product distribution in the reaction mixture essentially remained unchanged upon switching from H2 to N2 atmosphere. The only notable change was the partial dehydrogenation of octadecanol to octadecanal. However, in the next 180 minutes the product distribution changed significantly. The C18-oxygenates were converted to C17-hydrocarbons, similar to the results in the intermediate tests shown above (Table 3). After 180 minutes of reaction in N2 atmosphere, the main products are heptadecane and heptadecene, while the dehydration product octadecene was formed in small quantities (<5%). Octadecane was no longer produced in significant amounts, in contrast to the reaction in H2 atmosphere. For comparison, product distributions after 300 minutes for both the reactions in H2 and N2 atmosphere are shown in Table 4.
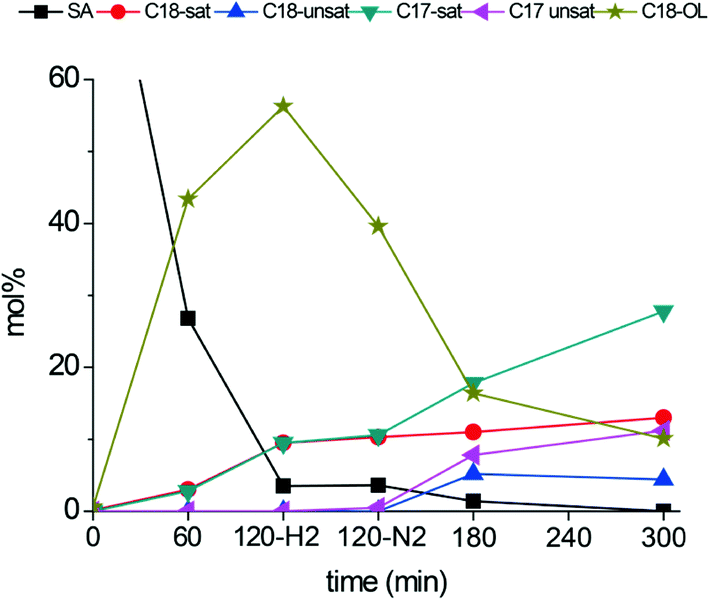 | ||
| Fig. 9 Reaction temperature: 300 °C. Catalyst: b-Mo2C/CNF. H2-pressure in 0–120 minutes: 30 bar. N2-pressure in 120–300 minutes: 30 bar. | ||
| Catalysts | Gas atmosphere | SAa (%) | C18-sat (%) | C18-unsat (%) | C17-sat (%) | C17-unsat (%) | C18-OL (%) | C18-AL (%) | C36-oxyb (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Stearic Acid. b Stearyl-stearate. c Reaction conditions: 2.0 g stearic acid, 36 g dodecane, 1.0 g tetradecane (IS), 0.25 g catalyst. Reaction temperature: 300 °C. 30 bar gas pressure. d Reaction initially in H2, temporarily quenched after 120 minutes. Reaction continued after purging and re-pressurizing with N2 gas. e Reaction initially in H2, quenched after 90 minutes. Catalysts changed to WO3/CNF and reaction continued under N2 atmosphere. | |||||||||
| b-Mo2C/CNF (120 min)c | H2 | 3.5 | 9.5 | — | 9.4 | — | 56.3 | 11.2 | 9.5 |
| b-Mo2C/CNF (300 min)c | H2 | — | 40.1 | 3 | 33.3 | 1 | 9.6 | 9 | 1.8 |
| b-Mo2C/CNF (300 min)d | N2 | — | 12.9 | 4.4 | 27.8 | 11.2 | 10.1 | 17.9 | 14.3 |
| b-Mo2C/CNF (90 min)e | H2 | 14.7 | 4.4 | — | 1.5 | — | 51.7 | 16.1 | 11.7 |
| WO3/CNF (180 min)e | N2 | 9.6 | 5.1 | 48 | 1.9 | — | — | 4 | 31 |
When the stearic acid deoxygenation is separated in two individual reaction steps, product selectivity can be further tuned to maximize yield to octadecene by using a different catalyst for the conversion of the C18-oxygenates under H2 free conditions. In a separate experiment, stearic acid deoxygenation over b-Mo2C/CNF at 300 °C was quenched after 90 minutes of reaction in H2 atmosphere. The reaction was rapidly cooled and the molybdenum carbide catalyst was separated by hot-filtration.
A new CNF supported tungsten trioxide catalyst (WO3/CNF), prepared as described in previous work,5 was added to the reaction mixture, which was then re-pressurized with N2 and heated to 300 °C. Upon reaching the desired temperature the reaction was allowed to continue for an additional 90 minutes. As shown in Table 4, this process allows for a 48% yield to octadecene from stearic acid, with stearyl-stearate (31%) being the main side-product. Octadecene and stearyl-stearate can be easily separated, and both are valuable products with applications in cosmetics or as bulk chemical precursors. It should be noted that the 2-step process proposed above is not yet optimized for maximum octadecene production. One way to obtain higher octadecene yields in the final reaction mixture would be to increase yield of the stearic acid conversion to octadecanol in the first reaction step. The b-Mo2C/CNF catalyst reported in this work shows catalytic activity and selectivity in the hydrogenation of stearic acid to the fatty alcohol that outperforms the commercially used non-noble metal copper-chromite catalysts.23 In industrial processes fatty acid hydrogenations are typically performed at high (>100 bar) hydrogen pressures in order to maximize fatty alcohol yields. Using such high H2 pressures for the b-Mo2C/CNF catalyzed reaction would most likely lead to complete hydrogenation of the C18-aldehyde that currently still present in the reaction mixture. Unfortunately this could not be verified experimentally in this work due to the limitations of our reactor setup. Another possible way to improve overall octadecene yield could be the use of a different type of solid acid catalyst in the second reaction step, the dehydration of octadecanol in N2 atmosphere. The WO3/CNF catalyst used here was chosen since the material was previously shown to be active under these reaction conditions, while cracking of the octadecene product was not observed for this catalyst.5 However, both the reaction time and reaction temperature might be significantly reduced when using a catalyst with a higher amount of weak acid sites, such as phosphorus modified ZSM-5.24
Kinetics of stearic acid deoxygenation over supported tungsten- and molybdenum carbide catalysts
To investigate the effect of reaction temperature of the catalytic performance of b-W2C/CNF, the material was tested for stearic acid deoxygenation at 300, 325 and 350 °C.The product distributions in the b-W2C/CNF catalyzed deoxygenation of stearic acid are compared at approximately 50% stearic acid conversion at different reaction temperatures in Table 5. No stearic acid conversion was observed at reaction temperatures below 300 °C. The rate of stearic acid hydrogenation on the b-W2C/CNF catalyst increased upon increase of the reaction temperature. The produced C18-oxy intermediates were converted to either stearyl-stearate via esterification at 300 °C, or to hydrocarbons via dehydration and decarbonylation routes at 350 °C.
| Catalyst | Temperature (°C) | Time (min) | Conversiona (%) | C18-oxyb (%) | C36-oxyc (%) | HC-yieldd (%) | Initial ratee (mmol g−1 h−1) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 2.0 g stearic acid, 0.25 g catalyst, 36 g dodecane. 30 bar H2. b Sum of octadecanol and octadecanal yield. c Stearyl-stearate. d Total hydrocarbon yield (C17 + C18). e Initial stearic acid conversion rate in mmol g−1 h−1, determined using conversion after 60 minutes of reaction. | |||||||
| b-W2C/CNF | 300 | 300 | 44 | 12 | 27 | 3 | 1.9 |
| b-W2C/CNF | 325 | 120 | 49 | 9 | 28 | 12 | 6.8 |
| b-W2C/CNF | 350 | 60 | 53 | 22 | 4 | 24 | 14.6 |
Both esterification and hydrocarbon formation occurred simultaneously at 325 °C. By comparing the initial rates of stearic acid hydrogenation at different reaction temperatures, an estimate of the activation energy of this reaction step can be given. The activation energy for stearic acid hydrogenation on these tungsten carbide catalysts was found to be 121 kJ mol−1. The corresponding Arrhenius plot is shown in Fig. 10.
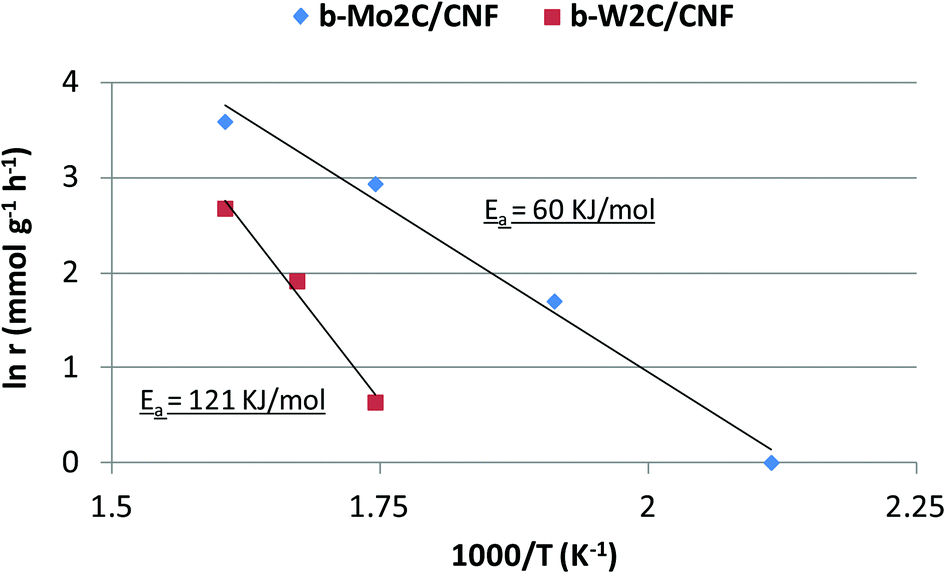 | ||
| Fig. 10 Arrhenius plot for stearic acid conversion over b-Mo2C/CNF and b-W2C/CNF catalysts. The activation energy for the hydrogenation of stearic acid was found to be 60 kJ mol−1 over the b-Mo2C/CNF catalyst and 121 kJ mol−1 over the b-W2C/CNF catalyst. | ||
The performance of b-Mo2C/CNF catalyst in the deoxygenation of stearic acid was also examined at different reaction temperatures. Results are shown in Table 6. It can be seen that the molybdenum carbide catalyst was very active in the hydrogenation of stearic acid. Full conversion of stearic acid could be achieved in 360 minutes at a reaction temperature of 250 °C. The b-Mo2C/CNF catalyst still showed activity for the hydrogenation of stearic acid at a reaction temperature as low as 200 °C, although the rate of stearic acid conversion was very low in this case (1.0 mmol g−1 h−1). The Arrhenius plots for b-Mo2C/CNF and b-W2C/CNF are shown in Fig. 10.
| Catalyst | Temperature (°C) | Time (min) | Conversiona (%) | C18-oxyb (%) | C36-oxyc (%) | HC-yieldd (%) | Initial ratee (mmol g−1 h−1) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 2.0 g stearic acid, 36 g dodecane, 1.0 g tetradecane (IS), 0.25 g catalyst, 30 bar H2. b Sum of octadecanol and octadecanal yield. c Stearyl-stearate. d Total hydrocarbon yield (C17 + C18). e Initial stearic acid conversion rate in mmol g−1 h−1, determined using SA conversion after 60 minutes of reaction. f Estimated by using SA conversion in the first 30 minutes of reaction at 350 °C. | |||||||
| b-Mo2C/CNF | 200 | 480 | 18 | 11.2 | 7 | <1 | 1.0 |
| b-Mo2C/CNF | 250 | 360 | 96 | 67.7 | 24.5 | 1.8 | 5.5 |
| b-Mo2C/CNF | 300 | 120 | 94 | 73.5 | 5.5 | 12.4 | 19 |
| b-Mo2C/CNF | 350 | 30 | 90 | 43.9 | — | 43 | 36f |
The apparent activation energy for the hydrogenation of stearic acid over the supported large molybdenum carbide particles of the b-Mo2C/CNF catalyst was calculated to be 60 kJ mol−1, half of the 121 kJ mol−1 found for their tungsten carbide analogs. This difference in activation energy between both catalysts is most likely caused by the difference in surface oxygen content of both catalysts, shown in the XPS results discussed earlier in this work.
The data in Table 6 shows that conversion of C18-oxygenates to hydrocarbons did not occur over b-Mo2C/CNF catalysts at temperatures below 250 °C. Instead octadecanol and stearic acid were partially converted to stearyl-stearate, similar to the low temperature reactions catalyzed by b-W2C/CNF (Table 5, T = 300 °C). At a reaction temperature of 350 °C, hydrocarbon production was rapid over the b-Mo2C/CNF catalyst, as indicated by the high hydrocarbon yield of 43% that was obtained after 30 minutes of reaction. Stearyl-stearate was only observed in trace amounts under these reaction conditions. An intermediate reaction temperature of 300 °C showed buildup of both stearyl-stearate and hydrocarbon products. The C36-ester was converted to C17- and C18-hydrocarbon products at longer reaction times, indicating the ester intermediate reverts back to stearic acid and octadecanol over time under these conditions.
Experimental
Carbon nanofibers (CNF) were grown in-house from a mixture of hydrogen and carbon monoxide at 3 bar total pressure over a reduced 5 wt% Ni/SiO2 catalyst, using a previously established protocol.10 After the CNF synthesis, the carbon fibers were treated three times with a boiling 1 M KOH solution to dissolve the silica remaining from the growth catalyst. After the alkali treatment, the CNF were treated with hot concentrated acid to remove the remaining nickel. Surface oxygen group were introduced on the CNF by treatment with concentrated nitric acid, yielding CNF-ox. When the CNF were treated with concentrated hydrochloric acid instead, the carbon surface remained fully hydrophobic (i.e. negligible amount of surface oxygen functionalities), yielding CNF-hcl as the final material.Supported carbide catalysts were prepared by incipient wetness impregnation of a carbon nanofiber (CNF) support (90–212 μm) with aqueous ammonium molybdate or ammonium metatungstate solution, followed by drying using a rotary evaporator to ensure deposition of the metal on the outside of the CNF bodies. The beneficial effect of this drying procedure, previously reported by Qin et al.7 was confirmed in this work. Details can be found in ESI.† Molar loadings of the metal were kept constant (0.8 mmol g−1) for all catalysts used in this work to allow a comparison between the different carbide catalysts. After impregnation and drying, the material (0.5 gram) was carburized for 360 minutes at 900 °C under nitrogen gas flow (50 ml min−1). The reduction and carburization steps are performed by carbon originating from the support material, i.e. using a carbothermal reduction method. Details on this carbothermal reduction process can be found in the ESI.† After the heat treatment, the catalyst material was cooled down to room temperature and exposed to ambient air for 10 minutes. The synthesized supported carbide catalyst was either used directly in a deoxygenation reaction, or stored under inert atmosphere to prevent additional oxidation of the carbide surface.
Supported carbide catalysts synthesized from a hydrophobic CNF-hcl support are named “b-X2C/CNF”, while catalysts that were prepared using an oxygen containing, hydrophilic CNF-ox support are named “X2C-CNF”, with X = Mo, or W, in both cases.
Tungsten tri-oxide supported CNF (‘WO3/CNF’) was synthesized by adaptation of the procedure described in previous work.5 A CNF-ox support was impregnated with an aqueous ammonium metatungstate solution and dried using a rotary evaporator to obtain a final tungsten loading of 15 wt% (0.8 mmol g−1). The sample (0.5 gram) was then treated at 600 °C for 180 minutes under a N2 gas flow (50 ml min−1).
Deoxygenation reactions of the model compound stearic acid were carried out in a 100 mL EZE Autoclave engineer batch reactor system. Dodecane was used as a solvent and tetradecane was used as an internal standard in all reactions. In a typical procedure, 2.0 g stearic acid, 1.0 g tetradecane, 36 g dodecane and 0.25 g catalyst were added to the reactor. During transfer of the catalyst to the reactor setup, contact of the catalyst with ambient air was minimized as much as possible, but could not be avoided completely. Upon addition of the reactants and catalyst, the system was stirred at 1000 rpm and flushed with N2 (100 ml min−1) for 10 minutes, after which it was flushed with H2 for another 10 minutes. The reactor was then pressurized with 30 bar of hydrogen, while heating to the desired reaction temperature at 5 °C min−1. Final pressure in the reactor varied between 40–50 bar, depending on the reaction temperature. Product distribution was monitored overtime by taking liquid samples (approximately 1 ml) from the reactor at regular time intervals. Samples were diluted with a 2:1 CHCl3–MeOH mixture and methylated by addition of a tri-methylsulfonium hydroxide (TMSH) solution prior to GC analysis.15 The molar balance of the liquid phase products is between 95–100% in all liquid samples. The molar balance is defined as the sum of the number of moles of liquid products (excluding the solvent dodecane and the internal standard tetradecane) in the reaction mixture divided by the initial number of moles of stearic acid substrate. Reaction rates are expressed as mmol [substrate] converted per gram catalyst per hour of reaction time (i.e. mmol g−1 h−1).
XRD measurements were performed on a Bruker-AXS D2 phaser powder X-ray diffractometer using Co-Kα1,2 with λ = 1.79026 Å. Measurements were carried out at ambient conditions. Scans were performed between 20–80 degrees 2 theta, using a step size of 0.09 degrees 2θ and a scan speed of 2 seconds.
N2-physisorption measured on a Micromeretics Tristar 7000 at 77 K. The samples were dried for 16 hours at 473 K in N2 flow. The total pore volume was defined as the single-point pore volume at P/P0 = 0.97.
TGA (Q50, TA instruments) was measured by heating approximately 15 mg of sample to 900 °C in N2 flow (50 ml min−1) with a ramp of 5 °C min−1. The sample was held at 900 °C while continuing the measurement for another 3 hours. The measured samples were prepared by impregnation of the CNF support with an ammonium metal salt solution, followed by drying using a rotary evaporator. See above for details.
TEM images were acquired using a FEI TECNAI20F transmission electron microscope operated at 200 keV. The samples were ground in a mortar and deposited on a copper grid as dry powder prior to the measurements.
XPS measurements were performed using a JPS-9200 photoelectron spectrometer. Spectra were collected using Al Kα X-ray radiation at 10 kV and 20 mA with analyzer pass energy of 50 eV for wide scans and 10 eV for narrow scans. Samples were pressed into an indium foil in ambient conditions prior to being transferred to the vacuum chamber. Exposure of the samples to ambient air was minimized as much as possible during preparation of the XPS samples though it could not be completely avoided for practical reasons. Exposure time to ambient air was less than 10 minutes for all samples studied in this work. Analysis of the data was performed using the CasaXPS software. The C1s at 284.7 eV of graphitic carbon was used as a reference.
Conclusions
The deoxygenation of stearic acid and related intermediates over various carbon nanofiber supported molybdenum- and tungsten carbides has been studied in detail.It has been shown that carbide particle size is an important factor in the activity and stability of the catalyst in (hydro-)deoxygenation reactions. When carbide crystallite size increased, catalyst stability was clearly improved for both the molybdenum- and tungsten carbides supported on CNF. The increase in catalyst stability for larger particles was shown to be due to the improved resistance of the metal carbide phase against full oxidation to crystalline metal oxides. In addition to the improved catalyst stability, supported molybdenum carbides were found to more than double their weight-based catalytic activity upon increasing carbide particle size from 2 to 10 nanometers. The surface-specific activity (TOF) is estimated to be a factor of 10 higher for the larger metal carbide particles. The activation energy for the initial hydrogenation of stearic acid to octadecanol was found to be a 60 kJ mol−1 for the large molybdenum carbide particles supported on CNF and 121 kJ mol−1 for the respective CNF supported tungsten carbide catalyst. This large difference in hydrogenation capability between these two group 6 metal carbide catalysts most likely originates from the difference in surface oxidation between the two carbides. XPS measurements showed that the surface of the tungsten carbide catalyst contained a higher fraction of metal-oxide species than that of the molybdenum carbide.
Carbide particle size was also found to influence the reaction pathway of the conversion of fatty alcohol intermediates that are produced by hydrogenation of the stearic acid substrate. The reaction mechanism was shown to be related to the (de-)hydrogenation ability of the catalyst, which increases when the amount of metal-oxide domains on the catalyst surface decreases. Besides the typical C18-hydrocarbon HDO products that have been previously reported in carbide catalyzed deoxygenation, the larger molybdenum- and tungsten carbide particles were shown to produce C17-hydrocarbons from C18-oxygenate intermediates by a dehydrogenation–decarbonylation pathway. Direct decarbonylation of stearic acid was not observed on any of the carbide catalysts. The improved understanding of the interplay between the catalyst structure and the related deoxygenation pathways allowed for the design of an alternative process for fatty acid deoxygenation, in which high-value unsaturated hydrocarbon products can be obtained with high selectivity. The initial hydrogenation of fatty acid to fatty alcohol can be efficiently performed by the new carbide catalysts developed in this work, while further conversion of the C18-oxy intermediates can be performed in H2-free conditions to selectively obtain high yields of high-value alkenes and/or fatty acid esters.
List of abbreviations
| CNF-hcl | Carbon nanofibers treated with concentrated HCl |
| CNF-ox | Carbon nanofibers treated with concentrated HNO3 |
| Cn-sat | Saturated hydrocarbon with chain length n |
| Cn-unsat | Unsaturated hydrocarbon with chain length n |
| C18-AL | Octadecanal |
| C18-OL | Octadecanol |
| C18-oxy | Octadecanal and/or Octadecanol |
| C36-ester | Stearyl-stearate |
| DCO | Decarboxylation/Decarbonylation |
| HDO | Hydrodeoxygenation |
| SA | Stearic acid |
Acknowledgements
This research has been performed in the framework of the Catchbio program. The authors gratefully acknowledge the support of the Smart Mix program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. The authors would like to thank Krijn de Jong, Stefan Hollak, Rob Gosselink, Charlotte Paesen and Tomas van Haasterecht for the fruitful discussions about this work. The authors further thank Marjan Versluijs-Helder for the TGA and SEM measurements, Cor van der Spek and Jingxiu Xie for the TEM measurements and Barend van Lagen (Wageningen University) for the XPS measurements.Notes and references
- C. Zhao, T. Brück and J. A. Lercher, Green Chem., 2013, 15, 1720 RSC.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147 CrossRef CAS.
- B. Donnis, R. G. Egeberg, P. Blom and K. G. Knudsen, Top. Catal., 2009, 52, 229 CrossRef CAS.
- M. Snåre, I. Kubličková, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708 CrossRef.
- R. W. Gosselink, D. R. Stellwagen and J. H. Bitter, Angew. Chem., Int. Ed., 2013, 52, 5089 CrossRef CAS PubMed.
- S. A. W. Hollak, R. W. Gosselink, D. S. van Es and J. H. Bitter, ACS Catal., 2013, 3, 2837 CrossRef CAS.
- Y. Qin, P. Chen, J. Duan, J. Han, H. Lou, X. Zheng and H. Hong, RSC. Adv., 2013, 3, 17485 RSC.
- J. Han, J. Duan, P. Chen, H. Lou, X. Zheng and H. Hong, Green Chem., 2011, 13, 2561 RSC.
- H. Ren, Y. Chen, Y. Huang, W. Deng, D. Vlachos and J. G. Chen, Green Chem., 2014, 16, 761 RSC.
- M. L. Toebes, M. K. Van der Lee, L. M. Tang, M. H. Huis in ‘t Veld, J. H. Bitter, J. van Dillen and K. P. de Jong, J. Phys. Chem. B, 2004, 108, 11611 CrossRef CAS.
- X. Li, D. Ma, L. Chen and X. Bao, Catal. Lett., 2007, 116, 63 CrossRef CAS PubMed.
- G. de la Puente, A. Centeno, A. Gil and P. Grange, J. Colloid Interface Sci., 1998, 202, 155 CrossRef CAS.
- W. Gruner, S. Stolle and K. Wetzig, Int. J. Refract. Mat. Hard Mater., 2000, 18, 137 CrossRef CAS.
- D. S. Venables and M. E. Brown, Thermochim. Acta, 1996, 282/283, 251 CrossRef CAS.
- W. Butte, J. Chromatogr., 1983, 261, 142 CrossRef CAS.
- G. Leclercq, M. Kamal, J. M. Giraudon, P. Devassine, L. Feigenbaum, L. Leclercq, A. Frennet, J. M. Bastin, A. Löfberg and M. Dufour, J. Catal., 1996, 158, 142 CrossRef CAS.
- M. J. Ledoux and C. Pham-Huu, Catal. Today, 1992, 15, 263 CrossRef CAS.
- E. Iglesia, F. H. Ribeiro, M. Boudart and J. E. Baumgartner, Catal. Today, 1992, 15, 307 CrossRef CAS.
- W. Song, C. Zhao and J. A. Lercher, Chem. – Eur. J., 2013, 19, 9833 CrossRef CAS PubMed.
- B. Peng, C. Zhao, S. Kasakov, S. Foraita and J. A. Lercher, Chem. – Eur. J., 2013, 19, 4732 CrossRef CAS PubMed.
- F. H. Ribeiro, R. A. Dalla Betta, M. Boudart, J. Baumgartner and E. Iglesia, J. Catal., 1991, 130, 86 CrossRef CAS.
- E. A. Blekkan, C. Pham-Huu, J. M. Ledoux and J. Guille, Ind. Eng. Chem. Res., 1994, 33, 1657 CrossRef CAS.
- R. D. Rieke, D. S. Thakur, B. D. Roberts and G. T. White, J. Am. Oil Chem. Soc., 1997, 74, 333 CrossRef CAS.
- K. Ramesh, C. Jie, Y.-F. Han and A. Borgna, Ind. Eng. Chem. Res., 2010, 49, 4080 CrossRef CAS.
- Y. Qin, L. He, J. Duan, P. Chen, H. Lou, X. Zheng and H. Hong, ChemCatChem, 2014, 6, 2698–2705 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the preparation of carbon nanofiber supported carbides via the carbothermal reduction pathway. See DOI: 10.1039/c4gc01831a |
| This journal is © The Royal Society of Chemistry 2015 |