
Selective catalytic oxidation of sugar alcohols to lactic acid†
Michael G.
Manas
,
Jesús
Campos
 ,
Liam S.
Sharninghausen
,
Elisa
Lin
and
Robert H.
Crabtree
*
,
Liam S.
Sharninghausen
,
Elisa
Lin
and
Robert H.
Crabtree
*
Department of Chemistry, Yale University, P.O. Box 208107, New Haven, CT 06520-8107, USA. E-mail: robert.crabtree@yale.edu
First published on 11th November 2014
Abstract
Sorbitol and xylitol obtained from biomass are considered promising potential sources of both carbon building blocks and energy. We report the efficient and selective conversion of sorbitol, xylitol and other polyols into lactic acid as the major product through homogeneous iridium-NHC catalyzed dehydrogenative processes. The proposed reaction mechanism involves base-driven hydrolysis of simple sugars which accounts for the catalyst selectivity observed. In addition, catalyst deactivation pathways are explored and rational catalyst optimization is attempted through fine tuning of the complex.
1. Introduction
Global fossil fuel production is environmentally unsound in the long term and alternative, renewable sources for both carbon containing chemical feedstocks and for energy production need to be identified.1 A highly promising candidate in the search for alternatives is plant biomass.2 This has a number of major strengths: It is renewable, sustainable and, if used as an energy source through combustion, carbon neutral.3 Through photosynthesis, plants provide an efficient means of converting the simple C1-building block, carbon dioxide, into organic molecules such as glucose, fructose (C6) and xylose (C5), as well as polymers and oligomers, such as lignin, cellulose and hemicellulose. Mixtures of these biomass materials can be derived from agricultural residues or young growth lumber4 and processed through hydrogenolysis to the monomeric materials.5 There is a growing field of research involving processing these simple sugars into value-added organic molecules for use as alternative fuels or as simple carbon building blocks. One common upgrading strategy is a tandem hydrolysis/hydrogenation reaction to yield sugar alcohols (polyols) such as sorbitol and xylitol from cellulose.6,7Sorbitol (C6), as well as its lower analogues xylitol (C5), erythritol (C4) and glycerol (C3) (Fig. 1), have been identified by the US Department of Energy to be among the most promising renewable carbon sources for the future.8 A number of catalytic transformations of these polyols are known to give value-added products such as alkanes,9 alcohols10 and functionalized short-chain hydrocarbons, some of which can act as polymer building blocks.
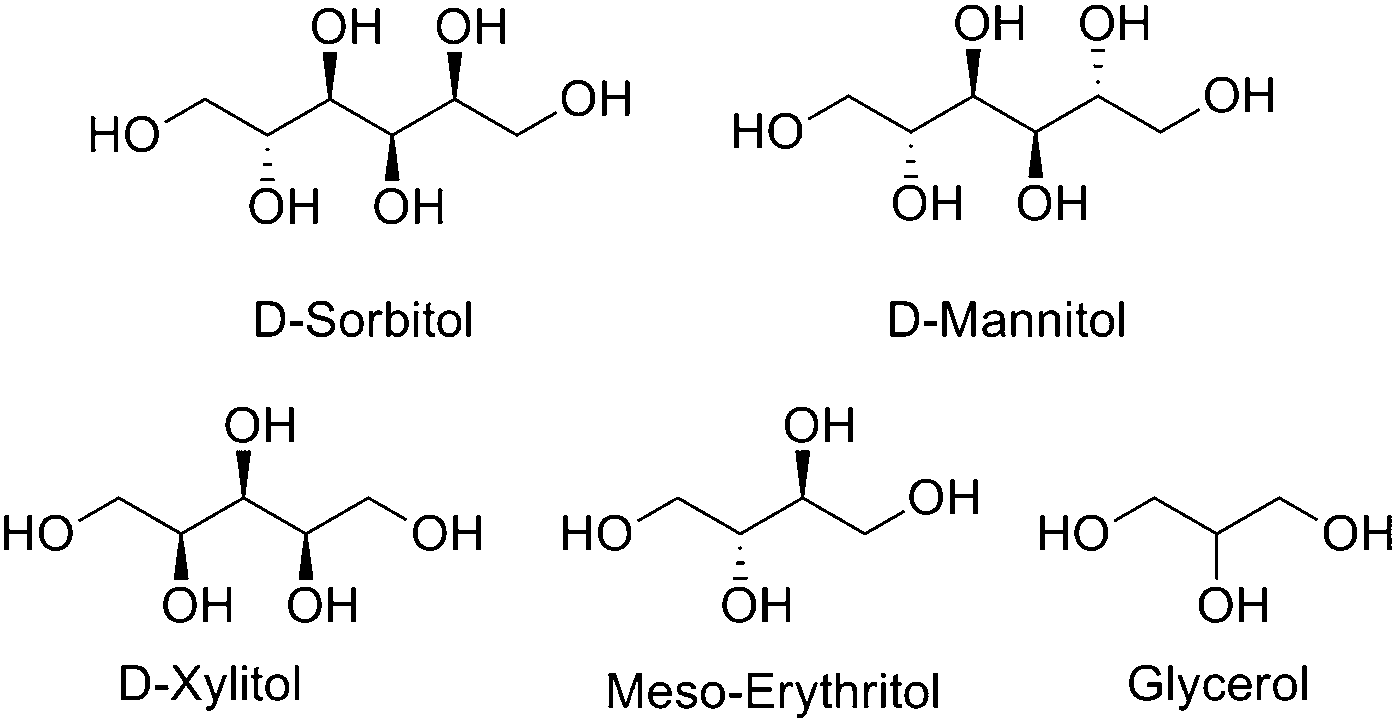 | ||
| Fig. 1 Common polyol substrates that have been identified as promising future feedstock carbon sources by the US DOE. | ||
One biomass-derived product of increasing interest is lactic acid.11 The catalytic conversion of polyols to lactic acid (LA) or lactate would be of general interest due to the variety of applications LA has as a platform chemical.11 Lactic acid can be used as a feedstock for the production of the biodegradable polyester, poly(lactic acid) (PLA) as well as a number of green solvents and commodity chemicals. In addition, LA itself has value in the pharmaceutical, food and detergent industries. Even though large quantities of LA are currently produced (260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year, much of it through biological processes) alternative methods of production are sought after to meet projected increasing demand.11,12 Non-biological means of producing LA are also attractive as they might be efficient sources of racemic or D-LA while biological sources produce almost exclusively L-LA. Additionally, any product of value that could be extracted from a waste material or low value biomass refuse would be advantageous.
000 tons per year, much of it through biological processes) alternative methods of production are sought after to meet projected increasing demand.11,12 Non-biological means of producing LA are also attractive as they might be efficient sources of racemic or D-LA while biological sources produce almost exclusively L-LA. Additionally, any product of value that could be extracted from a waste material or low value biomass refuse would be advantageous.
2. Results and discussion
2.1 Catalytic condition screening
Recently, our group has reported a family of homogeneous Ir-(NHC)2-based catalysts for the efficient conversion of glycerol to lactic acid and an equivalent of hydrogen through dehydrogenation under alkaline conditions (Fig. 2).13 This process was highly selective and was even capable of converting industrial biofuel waste glycerol into lactic acid in high yields. Since a number of heterogeneous systems give similar transformations of larger sugars,10,11,14,15 and in a few cases of the more difficult polyols,16,17 we have now explored the reactivity of our optimized glycerol oxidation precatalyst with such substrates.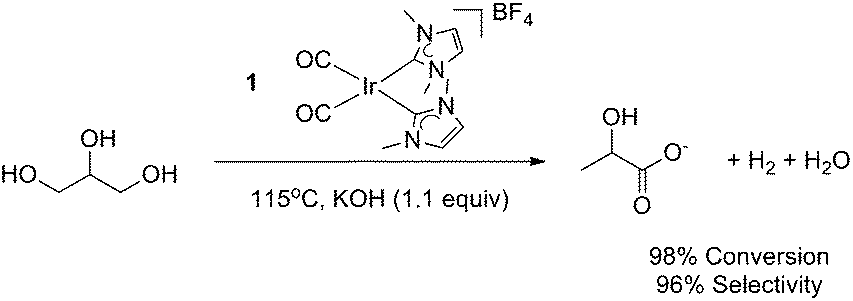 | ||
| Fig. 2 Selective conversion of glycerol to LA catalyzed by 1. | ||
Catalytic dehydrogenation of sorbitol was initially explored under conditions similar to those previously used for glycerol upgrading. An aqueous solution of sorbitol and potassium hydroxide in a 1:1.1 mol ratio with 0.1 mol% precatalyst (1) loading gave conversion to LA at reflux over 5 h.
Although the yield in aqueous conditions was poor (∼8%, Table 1, entry 1), the reaction was relatively selective for LA with the only minor products of note being acetate, formate and ethylene glycol (<5% each). By analogy to our results with glycerol, the LA yield increased dramatically in neat sorbitol (entry 6, m.p. 95 °C, b.p. 296 °C), heated to 160 °C, giving rise to 28% yield of LA corresponding to 280 turnover numbers (TON). The ability of this system to operate both in water and solvent-free is attractive, broadening its potential applicability. Despite the increased size of the polyol, a substrate:KOH ratio of 1:1.1 remained ideal. In the absence of KOH there was no activity. We believe that in order for 1 to form the active catalytic species, the base is required and, as expected, no dehydrogenation is observed in the presence of base alone. Thus, both base and iridium precatalyst are necessary for the reaction to proceed. As in the prior glycerol reaction, the addition of elemental mercury did not hinder the reaction while the addition of triphenylphosphine (1:1 PPh3:Ir) did quench lactate formation, consistent with the active catalytic species in solution being homogeneous. We did not observe any first step dehydrogenation products such as glucose or fructose in the product mixtures.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | 1 | KOH (equiv.) | 5 h LA yield (%) | 24 h LA yield (%) |
| a Yield for LA calculated by 1H NMR spectroscopy using an internal standard (10 mol% sodium trimethylsilyl propionate-d4). Yield based on 1 LA equiv. per sorbitol. Reactions carried out under N2 atmosphere. b Reaction run at 115 °C. c Reaction run at 160 °C. | |||||
| 1 | H2Ob | 0.1 mol% | 1.1 | 8 | 10 |
| 2 | H2Ob | — | 1.1 | 0 | 0 |
| 3 | Neatc | 0.1 mol% | — | 0 | 0 |
| 4 | Neatc | 0.1 mol% | 0.25 | 6 | — |
| 5 | Neatc | 0.1 mol% | 0.5 | 12 | — |
| 6 | Neatc | 0.1 mol% | 1.1 | 28 | 40 |
| 7 | Neatc | 0.1 mol% | 2.2 | 21 | — |
| 8 | Neatc | 0.1 mol% | 3.3 | 16 | — |
| 9 | Neatc | — | 1.1 | 0 | 0 |
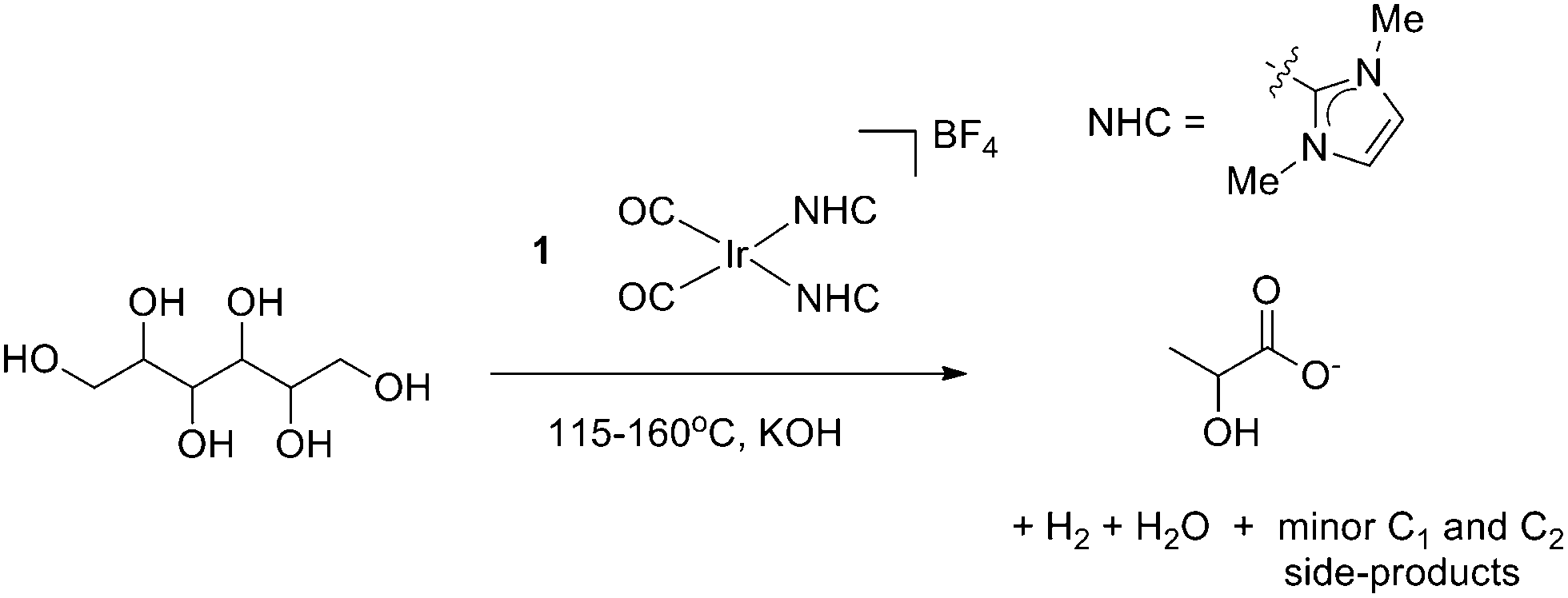
2.2 Substrate screening
A wider range of polyols including mannitol (C6), xylitol (C5) and erythritol (C4) were then tested for similar reactivity (Table 2). Under similar conditions, all substrates gave rather clean conversion to LA, with the exception of erythritol, which led to more complex mixtures; in all cases, a number of smaller breakdown products were also seen including appreciable amounts of ethylene glycol (EGO) (∼1:4, EGO:LA). In contrast to our findings for glycerol conversion, the reaction was still incomplete after 24 h despite a relatively higher loading of precatalyst 1. This is especially noteworthy in the case of C6 substrates as they could potentially split into two C3 LA molecules giving yields greater than one LA per polyol. Selectivity for these reactions was difficult to determine by 1H-NMR integration of remaining starting material due to overlapping chemical shifts with very minor side products. It was instead determined by 1H-NMR integration of all hydrocarbon products, accounting for the number of carbon atoms per side product as a close approximation of the selectivity of the reaction for lactic acid. Increasing the catalyst loading (entries 7 and 8) gave no noticeable increase in yield, and in some cases an unexpected detrimental effect was seen. Interestingly, decreasing the catalyst loading below 0.1 mol% seemed to have a negligible effect in most cases suggesting the existence of a concentration dependent deactivation pathway leading to deactivation at higher loadings. We have observed the same effect in our previous studies on the related precatalyst [(η5-C5Me5)Ir(IMe)2Cl]BF4 for transfer hydrogenation from iso-propanol to ketones.18 Letting the reactions run for longer did not lead to an increase over the 24 h yield; indeed, the reaction had essentially stopped after ∼20 h, when gas formation ceased. We ascribe this to catalyst deactivation.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | 1 | 5 h LA yield (%) select. [X] | 24 h LA yield (%) select. [X] |
| a Yield for LA calculated by 1H NMR spectroscopy using internal standard (10 mol% NaTMSP-d4). Yield based on 1 LA equiv. per 1 equiv. substrate. Reactions carried out under N2 atmosphere. Selectivity calculated as [mol LA/(mol LA + formate + ethylene glycol + acetate]. Mass balance as approximated by LA + formate + ethylene glycol + acetate + discernible starting material >95% calculated by 1H NMR. | ||||
| 1 | Sorbitol | 1.0 mol% | 26 [84] | 28 [84] |
| 2 | Sorbitol | 0.1 mol% | 28 [83] | 40 [82] |
| 3 | Sorbitol | 0.01 mol% | 32 [88] | 34 [84] |
| 4 | Sorbitol | — | 0 | 0 |
| 5 | Mannitol | 1.0 mol% | 32 [85] | 32 [83] |
| 6 | Mannitol | 0.1 mol% | 31 [84] | 43 [80] |
| 7 | Mannitol | 0.01 mol% | 28 [90] | 29 [89] |
| 8 | Mannitol | — | 0 | 0 |
| 9 | Xylitol | 1.0 mol% | 39 [86] | 40 [84] |
| 10 | Xylitol | 0.1 mol% | 44 [86] | 49 [88] |
| 11 | Xylitol | 0.01 mol% | 36 [92] | 42 [90] |
| 12 | Xylitol | — | 0 | 0 |
| 13 | Erythritol | 1.0 mol% | 6 [62] | 6 [60] |
| 14 | Erythritol | 0.1 mol% | 12 [74] | 12 [74] |
| 15 | Erythritol | 0.01 mol% | 13 [76] | 13 [75] |
| 16 | Erythritol | — | 0 | 0 |
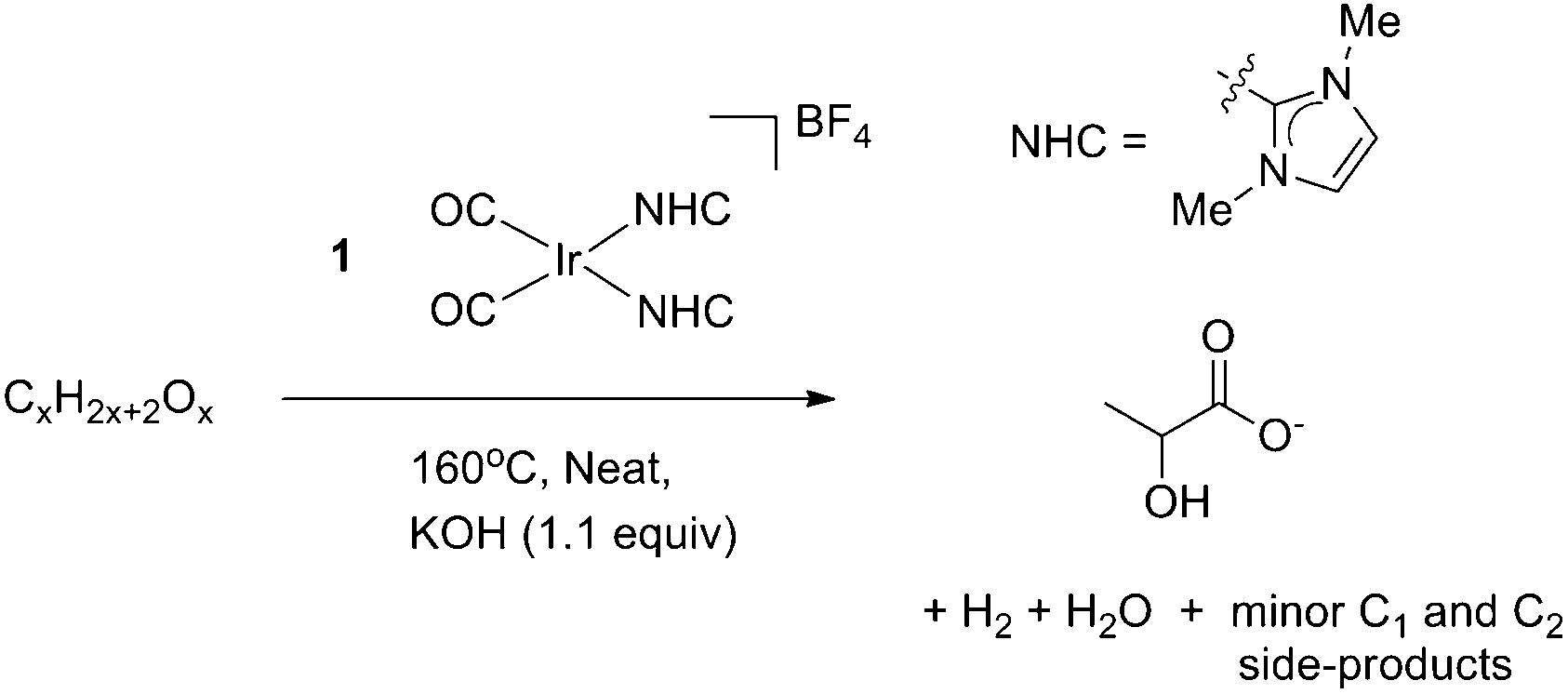
The detrimental effect of increasing catalyst loading and the negligible influence of its decrease in this study suggest that multi-iridium species are likely being formed and constitute undesired deactivation pathways that are largely concentration-dependent. This is supported by our recent characterization of a catalyst deactivation product, an [Ir6(IMe)8(CO)2H14]2+ cluster that is formed during glycerol dehydrogenation catalysis.19 There is precedent for deactivated iridium cluster formation for complexes of this general class. The [Ir(cod)(PCy3)(pyridine)]PF6 catalyst forms trinuclear iridium polyhydrides on deactivation20 and related tri- and tetranuclear clusters were seen for related cases.21 Similarly, under catalytic conditions for sorbitol we confirmed the formation of the same type of dinuclear hydrides already reported in our glycerol work.13 As expected, increasing the catalyst to substrate ratio for 1 leads to greater deactivation.
2.3 Catalyst optimization
In light of this proposed cluster-based catalyst deactivation, we thought that replacing the N-methyl by N-ethyl and N-n-butyl substituents on the NHC would prevent or at least reduce deactivation as a result of the increased steric bulk of the new ligands. Thus compounds 2 and 3 were synthesized in ∼50% yield by the reaction of [Ir(cod)(μ-Cl)]2 with Ag[IEt]2BF4 (IEt = 1,3-diethylimidazol-2-ylidene) or Ag[IBu]2BF4 (IBu = 1,3-dibutylimidazol-2-ylidene), respectively (Fig. 3; see ESI† for details). Subsequent treatment of dichloromethane solutions of these cyclooctadiene precursors with carbon monoxide resulted in the formation of the desired bis-carbonyl compounds 4 and 5 in ∼70% yields. The four species were characterized by 1H and 13C{1H} NMR spectroscopy, as well as by HRMS, and the molecular structure of 5 was confirmed by X-Ray diffraction (Fig. S1†).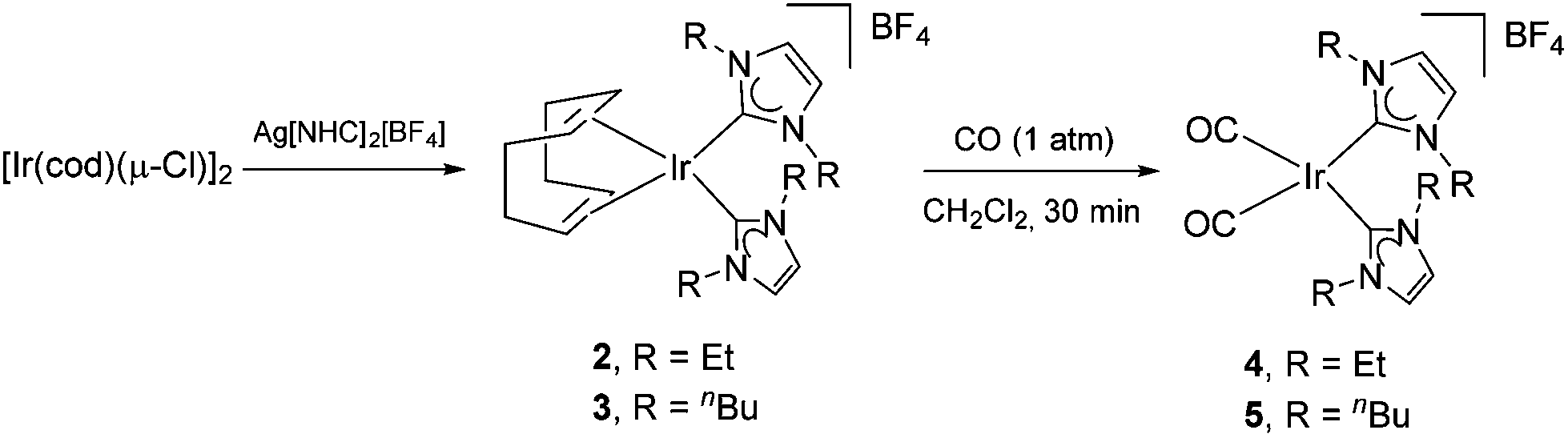 | ||
| Fig. 3 Synthesis of modified iridium pre-catalysts. | ||
Unfortunately, the activity was found to be generally lower for these new iridium analogues (see Table 3). We ascribe this to Hofmann elimination of the N-alkyl groups under basic conditions leading to the cleavage of the wingtip alkyl group from the imidazolium ligand.22 Since a β-H elimination is impossible in the IMe ligand, this potential decomposition pathway would not be viable, explaining the higher activity exhibited by precatalyst 1. Attempts were made to employ bulkier NHC ligands, but juxtaposing two such NHCs in a cis-arrangement using a similar procedure was not viable, perhaps due to steric clash. Further studies on these deactivation pathways have been carried out and the isolation and characterization of a number of multi-iridium clusters resulting from reactions using 1 have been reported by our group.13,19
|
|
||||
|---|---|---|---|---|
| Entry | R | Ir (mol%) | KOH | 5 h LA yield (%) |
| a Yield for LA calculated by 1H NMR spectroscopy using internal standard (10 mol% NaTMSP-d4). Yield based on 1 LA equiv. per sorbitol. Reactions carried out under N2 atmosphere. | ||||
| 1 | Me | 0.1 mol% | 1.1 equiv. | 28 |
| 2 | Et | 0.1 mol% | 1.1 equiv. | 13 |
| 3 | n-Bu | 0.1 mol% | 1.1 equiv. | 5 |
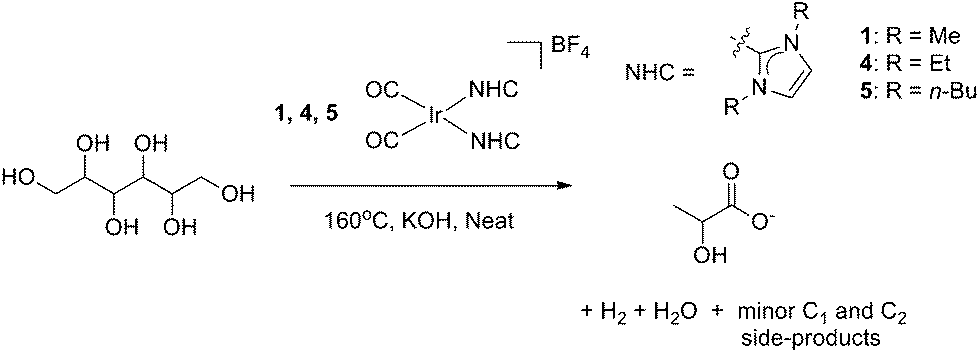
2.4 Mechanistic discussion
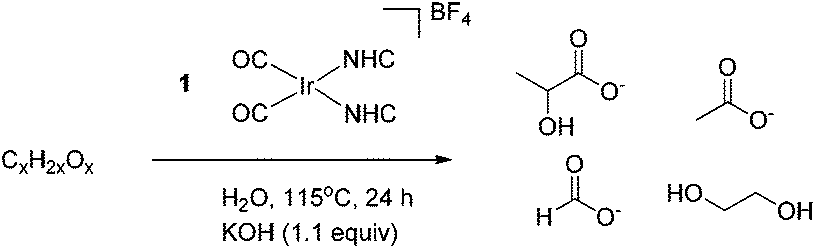
A mechanism by which precatalyst 1 converts glycerol to LA was proposed in our earlier work.13 In the presence of glycerol and base, precatalyst 1 is thought to form an active species that dehydrogenates glycerol to give either dihydroxyacetone or glyceraldehyde which interconvert under the basic reaction conditions.23,24 Glyceraldehyde can then undergo an intramolecular, base catalyzed internal Cannizzaro reaction, in effect undergoing a dehydration followed by a rehydration to form LA. By analogy, we believe that a similar mechanism is active for the longer polyols. Sorbitol, for example, can dehydrogenate at one of three positions, yielding glucose (an aldose), fructose (a 2-ketose) or galtose (a 3-ketose) as initial intermediates. These products are known to undergo isomerization under basic conditions,25 as in the glycerol case. The long chain polyols are then thought to give a retro-aldol-like cleavage under basic conditions to form the shorter chain acids. This base catalyzed cleavage explains why we do not observe these sugar intermediates in any of the product mixtures we reported above.In order to test whether these proposed dehydrogenation products are valid intermediates in the decomposition pathway from sorbitol to LA, glucose, fructose and galtose were screened for reactivity under conditions similar to those for the polyols. Sugars were found to be less amenable to the neat conditions used for the polyols and were more prone to burning, caramelization and other side processes. In light of this, experiments with sugars were instead carried out under the less concentrated aqueous conditions originally used in our screening for polyol dehydrogenation. After 24 h with 1.1 equivalents of KOH, both with and without precatalyst 1, LA was observed by 1H NMR spectroscopy in the reaction mixtures for all sugars. However, the selectivity for LA was greatly decreased and acetate, rather than lactate, became the major product in these reactions. In addition, more formate was seen as well as an appreciable amount of ethylene glycol. In reactions run without 1, trace amounts of methanol were also observed in the 1H NMR spectra. This loss of selectivity conforms with our previous glycerol studies where glyceraldehyde and dihydroxyacetone also gave lower selectivity.13 The production of acetate with loss of selectivity occurred both with and without iridium, indicating that after the initial dehydrogenation, iridium does not appear to play any major role. The isomerization and degradation of all C4, C5 and C6 products appears to be completely driven by base catalyzed organic reactions, independent of the iridium catalyst. Thus, the catalyst is only important for the initial dehydrogenation of the sugar alcohols, and plays no substantial role thereafter. All C6 sugars gave nearly identical yields of all four major products as evidenced by identical multiplet patterns in the relevant region of the 1H NMR spectrum. This agrees with base catalyzed isomerization of each of these sugars giving a common tautomeric mixture. Interestingly, the loss of selectivity could be partially combated by slow addition of a solution of the sugar substrate to a stirring solution of base. Under these conditions, the majority product observed was lactate and the relative amounts of formate and acetate decreased. When the reaction was run with an overall lower amount of sugar, keeping all other variables constant, it was also found that the selectivity shifted heavily in favor of lactate over the other products and that overall conversion was vastly increased, likely due to the higher amount of base versus substrate. The experiments with lower sugar concentration with respect to the base as well as the slow addition experiments could be simulating conditions more similar to the kinetic conditions from the dehydrogenation of sorbitol by 1 and therefore it is logical that they exhibit similar selectivity. The dehydrogenation products from mannitol and xylitol, mannose and xylose, gave similar results to sorbitol: selectivity for LA was lost and larger amounts of acetate and formate were observed (Table 4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | 1 | LA (%) | Formate (%) | Acetate (%) | EGO (%) |
|
a Yield for LA calculated by 1H NMR spectroscopy using internal standard. Yield based on 1 LA:1 substrate. Methanol was observed as a very minor product (<1%) in all entries without 1.
b Sugar solution added via syringe pump over 6 h to stirring solution of KOH–H2O.
c 20% of standard substrate loading.
d 10% of standard substrate loading.
|
||||||
| 1 | Glucose | 1.0 mol% | 5 | 2 | 21 | 4 |
| 2 | Glucose | — | 6 | 12 | 23 | 8 |
| 3 | Fructose | 1.0 mol% | 5 | 2 | 21 | 4 |
| 4 | Fructose | — | 7 | 14 | 23 | 8 |
| 5 | Galtose | 1.0 mol% | 6 | 2 | 21 | 4 |
| 6 | Galtose | — | 5 | 14 | 24 | 6 |
| 7 | Mannose | 1.0 mol% | 5 | 2 | 21 | 4 |
| 8 | Mannose | — | 6 | 11 | 28 | 8 |
| 9 | Xylose | 1.0 mol% | 4 | 2 | 20 | 8 |
| 10 | Xylose | — | 5 | 10 | 23 | 10 |
| 11 | Glucoseb | — | 17 | 6 | 7 | 7 |
| 12 | Fructoseb | — | 13 | 3 | 5 | 5 |
| 13 | Glucosec | — | 53 | 4 | 12 | 10 |
| 14 | Glucosed | 70 | 9 | 4 | 3 | |
In light of these observed product mixtures, a mechanistic scheme has been proposed as illustrated in Fig. 4 and 5 starting with sorbitol. Beginning with an initial iridium catalyzed dehydrogenation at any of the three potential positions, glucose, fructose and galtose can all be produced as a tautomeric mixture under the basic conditions.25 Each of these sugars can undergo a retro-aldol condensation, cleaving C–C bonds, forming two smaller carbohydrate molecules. Fructose can undergo such a cleavage at only one point, forming two C3 molecules, dihydroxyacetone and glyceraldehyde (Fig. 4). These have previously been proposed as direct precursors to lactate via an intramolecular Cannizzaro reaction.26 Glucose can undergo dehydration in one of two ways yielding two potential enol molecules. The 2-hydroxy enol can convert to a 1,2-di-ketone which can then cleave to two C3 molecules which both can then convert to lactate. These pathways are well documented.15,27
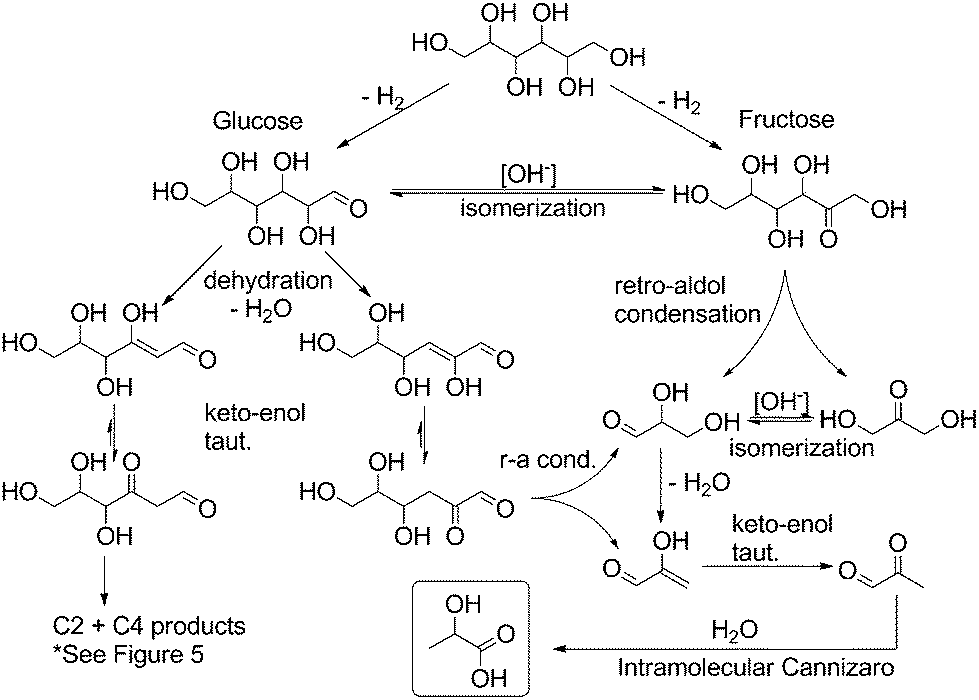 | ||
| Fig. 4 Proposed mechanistic pathway from sorbitol to lactate through glucose and fructose (r-a = retro-aldol; taut. = tautomerization). | ||
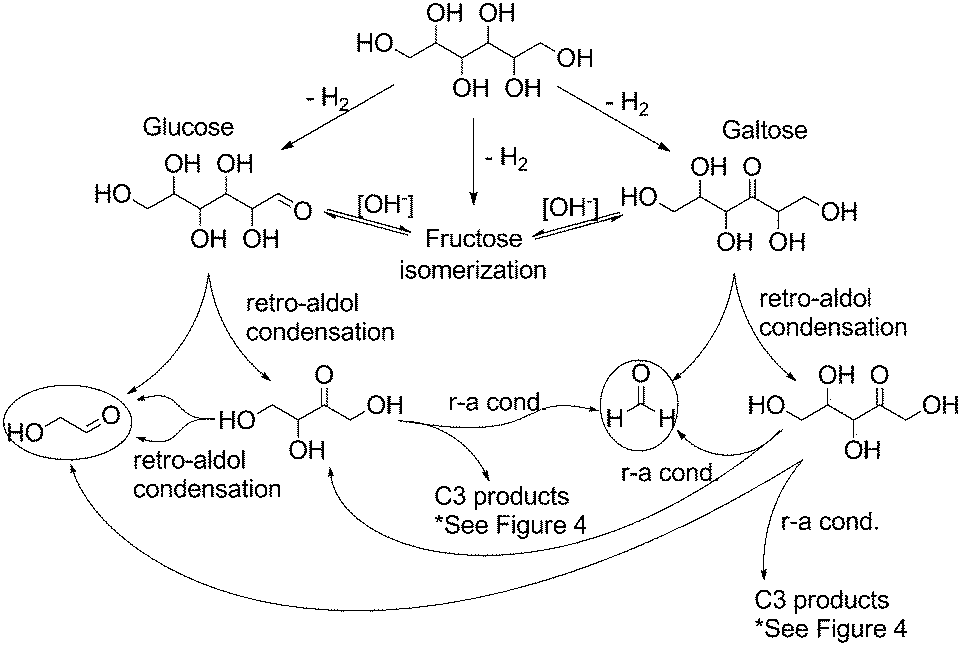 | ||
| Fig. 5 Proposed mechanistic pathway from sorbitol to C1 and C2 products through glucose and galtose (r-a = retro-aldol). | ||
Glucose and galtose can both undergo retro-aldol condensations (Fig. 5), converting into the C5, C4, C2 and C1 products: xylose and erythrose (and their isomers) as well as glycoaldehyde and formaldehyde. Xylose and erythrose isomers can undergo further degradation forming additional equivalents of glycoaldehyde and formaldehyde in addition to glyceraldehyde.
None of these species are observed in the final mixture of any of our reactions as they are readily converted into the products we do observe under our reaction conditions (see Fig. 6). Formaldehyde readily undergoes a disproportionation, via an intermolecular Cannizzaro reaction, forming an equivalent each of formate and methanol. The lack of methanol and decrease in observed formate when reactions were run in the presence of 1 might be explained in terms of iridium catalyzed dehydrogenation of formic acid and of methanol. These reactions are precedented for a range of catalysts28 and are the object of current investigations in our research group. Glycolaldehyde can form acetate through hydration/dehydration and tautomerization steps, but can also react with an equivalent of formaldehyde and hydroxide in an intermolecular Cannizzaro reaction to form ethylene glycol. Through these steps, the presence of not only lactate but also acetate, formate, methanol and ethylene glycol in the reactions starting with sugars can be rationalized.
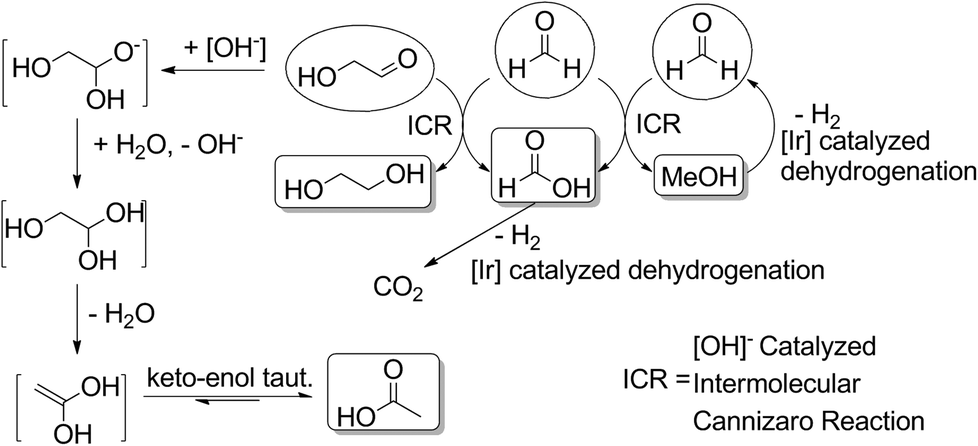 | ||
| Fig. 6 Proposed mechanistic pathway from C1 and C2 products to methanol, formate, ethylene glycol and acetate. | ||
Considering all these findings, the selectivity for lactate in the dehydrogenation of polyols is likely a kinetic effect resulting from selective dehydrogenation at one of the three positions to form either glucose, fructose or galtose derivatives. If the reaction proceeds through glucose or fructose, it would be expected that the overall mass balance would favor the formation of more than one lactate per starting sorbitol molecule. If the reaction proceeds through galtose and the retro-aldol cleavages happen through a sequence of terminal cleavages, we would expect to see only a single lactate per substrate molecule. So far all attempts at determining an overall mass balance has proven very difficult. Identifying the remaining sorbitol in 1H or 13C NMR spectra has been shown to be inaccurate due to overlap with other product peaks. Integration of the characteristic starting material peaks gave selectivity figures in close agreement with cumulative integration values outlined in Table 2. Separation of remaining sorbitol from the crude mixture through chromatography has given inconclusive results due to streaking of the mixture, attributed to the presence of many –OH groups. Protection of the sorbitol prior to chromatography has been attempted but recovered yields of the product have been too inconsistent to make any conclusions. Alternatively, the consumption of sorbitol could also be tracked via gas burette by measuring the volume of H2 gas produced. In this way, we were able to conclude that ∼1.2 equivalents of hydrogen were produced per lactate formed. This would seem to indicate that one equivalent of lactate was formed per equivalent of dehydrogenated product with the additional 0.2 equivalents coming from the potential dehydrogenation of the formate side product. The only other piece of evidence from which we can suggest a probable mechanism is by comparison of the yields attained for sorbitol with those for xylitol. Xylitol can only yield one equivalent of lactate but under identical conditions we find that both xylitol and sorbitol yield comparable amounts of lactate. This in conjunction with the volumetric H2 measurements seems to suggest that a pathway through galtose, sequentially cleaving the terminal carbon by retro aldol-condensation would be the most likely mechanism for the degradation of sorbitol.
3. Conclusions
The catalytic dehydrogenation of sorbitol, mannitol, xylitol and erythritol was carried out using a series of Ir-NHC complexes under basic conditions. The resulting dehydrogenated products rearranged and/or cleaved to form lactic acid as well as an equivalent of hydrogen. The homogeneous nature of the catalyst was suggested from studies with elemental mercury and triphenylphosphine additives. Attempts to optimize the reaction conditions by increasing the catalyst loading were unsuccessful because of deactivation, likely through the formation of inactive cluster species. In order to prevent the formation of such clusters, 4 and 5 were synthesized and screened but it was found that these catalysts were less active than 1 likely due to instability under the basic reaction conditions. Plausible mechanistic pathways have been proposed to describe the conversion from these polyols to lactate as well as a number of potential side-products. These proposals are supported by preliminary experiments in which likely intermediate dehydrogenated species were exposed to the basic conditions present in the standard reaction conditions.4. Experimental
4.1 General methods
The syntheses of the metal complexes were conducted under nitrogen using dry degassed solvents unless otherwise indicated. Solvents were dried and degassed prior to use. (Et2Im)BF4,29 Ag(IBu)2BF430 and [Ir(cod)Cl]231 and 113 were synthesized using literature procedures. All other reagents were commercially available from Airgas, Sigma-Aldrich, Alfa-Aesar and Strem Chemicals and used as received unless otherwise indicated. NMR spectra were recorded at room temperature on a 400 or 500 MHz Bruker or Varian spectrometer. Chemical shifts are reported in ppm with respect to residual internal protio solvent for 1H and 13C{1H} NMR spectra. NMR coupling constants (J) are given in Hz.
4.2 General procedure for catalytic conversion of sorbitol, mannitol, xylitol and erythritol to lactic acid under optimized neat conditions
To a pear shaped flask equipped with a stir bar was added 1 (5 mg, 9.43 μmol), sugar alcohol (94.3 mmol/9.43 mmol/0.943 mmol) and KOH (5.81 g, 103.8 mmol/581 mg, 10.38 mmol/58.1 mg, 1.038 mmol). The flask was evacuated and refilled with dry nitrogen. The flask was then heat in an oil bath at 160 °C for 5/24 h under positive pressure of nitrogen. The flask was removed from heat and before the molten substrate could solidify, deionized water was added to dissolve the products. NaTMSP-d4 (sodium 3-(trimethylsilyl)-2,2′,3,3′-tetradeuteropropionate) was added to this solution as a 1H NMR standard and then the water is removed in vacuo. The resulting brown solid was taken back up in D2O and yield was determined by 1H NMR without additional workup.4.3 General procedure for base catalyzed hydrolysis of sugars
To a Schlenk flask equipped with a stir bar was added sugar (1.11 mmol), KOH (68.4 mg, 1.22 mmol) and 4 (5.86 mg, 0.0111 mmol) when appropriate and the vessel was evacuated and refilled with dry N2. Degassed, deionized water (4 ml) was added to the flask via syringe. The solution was heated to 115 °C and left to stir for 24 h. The solution was cooled and NaTMSP-d4 was added as an internal standard. The water was removed in vacuo and the sample was redissolved in D2O. Yield of products was determined by 1H NMR spectroscopy and the identity of each product was confirmed by spiking the samples with authentic, commercially available samples. For reactions using slow addition of the sugar, a solution of sugar (1.11 mmol in 2 ml deionized water) was added via syringe pump over 6 h to a stirring solution of KOH (68.4 mg, 1.22 mmol) in deionized water (4 ml).:1 mixture of CH2Cl2–Et2O, giving compound 2 as an orange oil (84 mg, 57%). 1H NMR (400 MHz, CD2Cl2, 25 °C): ∂ = 7.05 (s, 4H, CHAr), 4.52 (m, 4H, NCH2(α), 4.11 (m, 4H, NCH2(α), 3.82 (br. s, 4H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 2.25 (m, 4H, CH2), 1.93 (m, 4H, CH2), 1.42 (t, 12H, 3JHH = 7.2 Hz, CH3(β)). 13C{1H} NMR (125 MHz, CD2Cl2, 25 °C): ∂ = 176.2 (IrC), 121.0 (CHAr), 76.3 (CH2), 45.9 (NCH2(α)), 31.8 (CH2), 16.2 (CH3(β)). HRMS (FT-ICR): calcd for C22H36IrN4 ([M]+): 549.2569. Found: m/z = 549.2573. Using the same procedure compound 3 was isolated as an orange powder in 48% yield. 1H NMR (500 MHz, CDCl3, 25 °C): ∂ = 7.05 (s, 4H, CHAr), 4.51 (m, 4H, NCH2(α), 3.89 (m, 4H, NCH2(α), 3.77 (br. s, 4H, CCH), 2.19 (m, 4H, CH2), 1.94–1.83 (m, 8H, 2CH2(COD), 2CH2(β)), 1.66 (m, 4H, CH2(β)), 1.43 (m, 8H, CH2(γ)), 1.01 (t, 12H, 3JHH = 7.5 Hz, CH3(δ)). 13C{1H} NMR (125 MHz, CDCl3, 25 °C): ∂ = 175.5 (IrC), 120.7 (CHAr), 75.5 (CH2(COD)), 50.6 (NCH2(α)), 32.8 (CH2(β)), 31.2 (CH2(COD)), 29.5 (CH2(COD)), 20.3 (CH2(γ), 13.5 (CH3(δ)). HRMS (FT-ICR): calcd for C30H52IrN4 ([M]+): 661.3821. Found: m/z = 661.3732.
C), 166.1 (C
CH), 2.25 (m, 4H, CH2), 1.93 (m, 4H, CH2), 1.42 (t, 12H, 3JHH = 7.2 Hz, CH3(β)). 13C{1H} NMR (125 MHz, CD2Cl2, 25 °C): ∂ = 176.2 (IrC), 121.0 (CHAr), 76.3 (CH2), 45.9 (NCH2(α)), 31.8 (CH2), 16.2 (CH3(β)). HRMS (FT-ICR): calcd for C22H36IrN4 ([M]+): 549.2569. Found: m/z = 549.2573. Using the same procedure compound 3 was isolated as an orange powder in 48% yield. 1H NMR (500 MHz, CDCl3, 25 °C): ∂ = 7.05 (s, 4H, CHAr), 4.51 (m, 4H, NCH2(α), 3.89 (m, 4H, NCH2(α), 3.77 (br. s, 4H, CCH), 2.19 (m, 4H, CH2), 1.94–1.83 (m, 8H, 2CH2(COD), 2CH2(β)), 1.66 (m, 4H, CH2(β)), 1.43 (m, 8H, CH2(γ)), 1.01 (t, 12H, 3JHH = 7.5 Hz, CH3(δ)). 13C{1H} NMR (125 MHz, CDCl3, 25 °C): ∂ = 175.5 (IrC), 120.7 (CHAr), 75.5 (CH2(COD)), 50.6 (NCH2(α)), 32.8 (CH2(β)), 31.2 (CH2(COD)), 29.5 (CH2(COD)), 20.3 (CH2(γ), 13.5 (CH3(δ)). HRMS (FT-ICR): calcd for C30H52IrN4 ([M]+): 661.3821. Found: m/z = 661.3732.
C), 166.1 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O), 122.6 (CHAr), 46.9 (NCH2(α)), 16.0 (CH3(β)). HRMS (FT-ICR): calcd for C16H24IrN4O2 ([M]+): 497.1529. Found: m/z = 497.1522. Using the same procedure compound 5 was isolated as an orange powder in 78% yield. 1H NMR (400 MHz, CDCl3, 25 °C): ∂ = 7.26 (s, 4H, CHAr), 4.11 (br. s, 4H, NCH2(α)), 3.93 (br. s, 4H, NCH2(α)), 1.77 (quint, 8H, 3JHH = 7.6 Hz, CH2(β)), 1.37 (h, 8H, 3JHH = 7.4 Hz, CH2(γ)), 1.37 (t, 12H, 3JHH = 7.4 Hz, CH2(δ)). 13C{1H} NMR (150 MHz, CDCl3, 25 °C): ∂ = 179.8 (IrC), 165.4 (CO), 122.6 (CHAr), 51.1 (NCH2(α)), 32.7 (CH2(β)), 19.8 (CH2(γ)), 13.4 (CH3 (δ)). HRMS (FT-ICR): calcd for C24H40IrN4O2 ([M]+): 609.2781. Found: m/z = 609.2718.
O), 122.6 (CHAr), 46.9 (NCH2(α)), 16.0 (CH3(β)). HRMS (FT-ICR): calcd for C16H24IrN4O2 ([M]+): 497.1529. Found: m/z = 497.1522. Using the same procedure compound 5 was isolated as an orange powder in 78% yield. 1H NMR (400 MHz, CDCl3, 25 °C): ∂ = 7.26 (s, 4H, CHAr), 4.11 (br. s, 4H, NCH2(α)), 3.93 (br. s, 4H, NCH2(α)), 1.77 (quint, 8H, 3JHH = 7.6 Hz, CH2(β)), 1.37 (h, 8H, 3JHH = 7.4 Hz, CH2(γ)), 1.37 (t, 12H, 3JHH = 7.4 Hz, CH2(δ)). 13C{1H} NMR (150 MHz, CDCl3, 25 °C): ∂ = 179.8 (IrC), 165.4 (CO), 122.6 (CHAr), 51.1 (NCH2(α)), 32.7 (CH2(β)), 19.8 (CH2(γ)), 13.4 (CH3 (δ)). HRMS (FT-ICR): calcd for C24H40IrN4O2 ([M]+): 609.2781. Found: m/z = 609.2718.
Acknowledgements
This material is based in part upon work supported by the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center funded by the U.S. Department of Energy (DoE), Office of Science, Office of Basic Energy Sciences, under award no. DE-SC0001298 (R.H.C. and J.C., synthesis and crystallography), by the U.S. Department of Energy (DoE), Office of Science, Office of Basic Energy Sciences catalysis award (M.G.M., L.S., J.C., DE-FG02-84ER13297, catalysis).References
- J. Fargione, J. Hill, D. Tilman, S. Polasky and P. Hawthorne, Science, 2008, 319, 1235–1238 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- (a) E. Thomas, Science, 1999, 5431, 1209–1209 Search PubMed; (b) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed; (c) Z. Zhang, B. Du, L. J. Zhang, Y. X. Da, Z. J. Quan, L. J. Yang and X. C. Wang, RSC Adv., 2013, 3, 9201–9205 RSC.
- Y. Q. Pu, D. C. Zhang, P. M. Singh and A. J. Ragauskas, Biofuel, Bioprod. Biorefin., 2008, 1, 58–73 CrossRef.
- W. Zhu, H. Yang, J. Chen, C. Chen, L. Guo, H. Gan, X. Zhao and Z. Hou, Green Chem., 2014, 16, 1534–1542 RSC.
- L. N. Ding, A. Q. Wang, M. Y. Zheng and T. Zhang, ChemSusChem, 2010, 7, 818–821 CrossRef PubMed.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 31, 5161–5163 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- L. Vilcocq, R. Koerin, A. Cabiac, C. Especiel, S. Lacombe and D. Duprez, Appl. Catal., B, 2014, 27, 499–508 CrossRef PubMed.
- N. Li and G. W. Huber, J. Catal., 2010, 1, 48–59 CrossRef PubMed.
- M. Dusselier, P. van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS.
- F. A. Castillo Martinez, E. M. Balciunas, J. M. Salgado, J. M. Domínguez González, A. Converti and R. P. DeSouza Oliveira, Trends Food Sci. Technol., 2013, 30, 70–83 CrossRef PubMed.
- L. S. Sharninghausen, J. Campos, M. G. Manas and R. H. Crabtree, Nat. Commun., 2014, 5 Search PubMed , 5084.
- W. Deng, Q. Zhang and Y. Wang, Catal. Today, 2014, 234, 31–41 CrossRef CAS PubMed.
- X. Wang, Y. Song, C. Huang, F. Liang and B. Chen, Green Chem., 2014, 16, 4234–4240 RSC.
- L. Vilcoq, A. Cabiac, C. Especel, E. Guillon and D. Duprez, Oil Gas Sci. Technol., 2013, 68, 841–860 CrossRef PubMed.
- (a) H. Zhou, F. Jin, B. Wu, J. Cao, X. Duan and A. Kishita, J. Phys.: Conf. Ser., 2010, 215, 012125 CrossRef; (b) C. A. Ramirez-Lopez, J. R. Ochoa-Gomez, S. Gil-Rio, O. Gomez-Jimenez-Aberasturi and J. Torrecilla-Soria, J. Chem. Technol. Biotechnol., 2011, 86, 867–874 CrossRef CAS.
- (a) U. Hintermair, J. Campos, T. P. Brewster, L. M. Pratt, N. D. Schley, C. D. Incarvito and R. H. Crabtree, ACS Catal., 2014, 4, 99–108 CrossRef CAS; (b) J. Campos, U. Hintermair, T. P. Brewster, M. K. Takase and R. H. Crabtree, ACS Catal., 2014, 4, 973–985 CAS.
- J. Campos, L. S. Sharninghausen, R. H. Crabtree and D. Balcells, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201407997.
- D. F. Chodosh, R. H. Crabtree, H. Felkin, S. Morehouse and G. E. Morris, Inorg. Chem., 1982, 21, 1307–1311 CrossRef CAS.
- Y. Xu, M. A. Celik, A. L. Thompson, H. Cai, M. Yurtsever, B. Odell, J. C. Green, D. Michael, P. Mingos and J. M. Brown, Angew. Chem., Int. Ed., 2009, 48, 582–585 CrossRef CAS PubMed.
- (a) A. W. Salman, R. A. Haque, S. Budagumpi and H. Zetty Zulikha, Polyhedron, 2013, 49, 200–206 CrossRef CAS PubMed; (b) S. Budagumpi, R. A. Haque, A. Rosenani, A. W. Salman and M. Z. Ghdhayeb, Inorg. Chim. Acta, 2012, 392, 61–72 CrossRef CAS PubMed.
- J. C. Sowden and E. K. Pohlen, J. Am. Chem. Soc., 1958, 80, 242–244 CrossRef CAS.
- C. Appayee and R. J. Breslow, Am. Chem. Soc., 2014, 136, 3720–3723 CrossRef CAS PubMed.
- M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- (a) C. Montassier, J. C. Menezo, L. C. Hoang, C. Renaud and J. Barbier, J. Mol. Catal., 1991, 70, 99–110 CrossRef CAS; (b) E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS PubMed; (c) E. P. Maris, W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 251, 281–294 CrossRef CAS PubMed.
- (a) Y. B. Yang and R. Montgomery, Carbohydr. Res., 1996, 280, 47–57 CrossRef; (b) F. Clippel, M. Dusselier, R. V. Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef PubMed.
- (a) M. Nielsen, E. Ablerico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed; (b) T. Yumura, T. Amenomori, Y. Kagawa and K. Yoshizawa, J. Phys. Chem. A, 2002, 106, 621–630 CrossRef CAS.
- M. D. Bala and M. I. Ikhile, J. Mol. Catal. A: Chem., 2014, 385, 98–105 CrossRef CAS PubMed.
- U. Hintermair, U. Englert and W. Leitner, Organometallics, 2011, 30, 3726–3731 CrossRef CAS.
- R. H. Crabtree, J. M. Quirk, H. Felkin and T. Fillebeen-khan, Synth. React. Inorg. Met.-Org. Chem., 1982, 12, 407–413 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1021458. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4gc01694g |
| This journal is © The Royal Society of Chemistry 2015 |