
Oxygen removal from intact biomass to produce liquid fuel range hydrocarbons via fast-hydropyrolysis and vapor-phase catalytic hydrodeoxygenation†
Vinod Kumar
Venkatakrishnan
ab,
W. Nicholas
Delgass
ab,
Fabio H.
Ribeiro
ab and
Rakesh
Agrawal
*ab
aSchool of Chemical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: agrawalr@purdue.edu
bCenter for Direct Catalytic Conversion of Biomass to Biofuels (C3Bio), Bindley Bioscience Center, Discovery Park, Purdue University, West Lafayette, IN 47907, USA
First published on 6th October 2014
Abstract
Proof-of-concept of a novel consecutive two-step biofuel process (H2Bioil), based on fast-hydropyrolysis and downstream vapor-phase catalytic hydrodeoxygenation (HDO), to produce liquid fuel range (C4+) hydrocarbons with undetectable oxygen content, from cellulose and an intact biomass (poplar) is presented. The carbon recovery as C1–C8+ hydrocarbons is ∼73% (C4+ ∼55%) from cellulose and ∼54% (C4+ ∼32%) from poplar. Advantages of independent control of fast-hydropyrolysis and HDO temperatures, along with synergistic process integration aspects are discussed.
The world consumption of liquid fuel was about ∼87 million barrels per day in 2010.1 Presently, most of this liquid fuel is produced from non-renewable fossil-based resources.1 An alternative source of carbon is sustainably available biomass, which has an estimated availability of ∼498 million tons to in excess of a billion tons.2–4 But, the challenge is to develop biomass conversion processes capable of producing the high energy density drop-in hydrocarbons that are required for transportation, with minimal processing steps for large-scale implementation at low cost. One of the viable process options to convert biomass into an oxygenated liquid product is fast-pyrolysis. Fast-pyrolysis involves rapid heating of biomass in an inert environment to temperatures of about 500 °C, with a typical vapor residence time of 1–2 seconds, to produce vapors that are quenched to form a liquid product called bio-oil.5 The bio-oil product is not suitable as a liquid transportation fuel, however, due to its low energy density, high oxygen content, and acidity.6 It needs to be hydrotreated at 100–200 bar pressure,7 to produce a hydrocarbon liquid product with low oxygen content. This bio-oil hydrotreating process is also disadvantageous due to issues of catalyst coking and reactor plugging during revaporization of the condensed bio-oil.8
As an alternative process to produce a hydrocarbon fuel, Agrawal et al.9–12 have proposed the H2Bioil process based on biomass fast-hydropyrolysis (FHP) combined with vapor-phase catalytic hydrodeoxygenation (HDO). In this process, the biomass is rapidly heated in a high-pressure (up to 200 bar) hydrogen environment to produce fast-hydropyrolysis vapors, which are sequentially catalytically hydrodeoxygenated to produce hydrocarbons. The key concept is to upgrade the reactive oxygenate molecules by vapor-phase catalytic hydrodeoxygenation before any undesirable secondary reactions can take place during condensation of the bio-oil mixture. The high pressure of hydrogen was envisioned to be needed for high rates of HDO reaction,12 similar to hydrotreating processes used in a petroleum refinery,13 and would also help in avoiding coking on the catalyst. The H2Bioil process has been modeled and is estimated to have a high carbon efficiency (∼70%) and a high energy efficiency (∼75%), which compares favorably with other traditional biomass conversion processes such as gasification, fermentation etc.12 Economic analysis of this process has revealed that this process can be economically attractive14 and is also favorable for small as well as large scale applications for transportation fuel production due to its ability to produce hydrocarbons in a single step. Clearly, an experimental validation of this process has a potential to have a positive impact on the biofuels sector.
In the literature,15–19 pyrolysis-based processes viz. catalytic pyrolysis and catalytic hydropyrolysis have been reported for producing hydrocarbons from biomass. Catalytic pyrolysis processes using zeolitic catalysts have resulted in carbon recoveries of about ∼25% as hydrocarbons, mainly as light olefins and aromatics, but is limited by coking and concomitant catalyst deactivation.15–17 Recently, catalytic hydropyrolysis studies18,19 have shown the production of hydrocarbons in the gasoline and diesel range, which has partially validated the ideas reported by Agrawal et al.9–12 These studies, however, do not report information about the proprietary catalysts used in the process or the chemical composition of the hydrocarbons. Moreover, catalytic pyrolysis/hydropyrolysis are processes based on ‘in situ’ catalysis due to which the pyrolysis and catalysis processes such as catalytic hydrodeoxygenation are constrained by the same reaction conditions (e.g. same temperature) even though the optimum conditions for each of the process steps might be different. Whereas, in the H2Bioil process, the downstream vapor-phase catalytic hydrodeoxygenation, recently referred to as ‘ex situ’,20,21 allows for independent control of the hydropyrolysis and catalysis at their respective optimum conditions (reaction temperatures, pressures and catalysts) for tailoring the yields and selectivities of the hydrocarbon products.
In this article, we report results from a continuous-flow cyclone-type fast-hydropyrolysis (FHP) reactor system with on-stream vapor-phase catalytic hydrodeoxygenation (HDO).22 We have been able to successfully achieve proof-of-concept of the H2Bioil process using a solid biomass model compound, cellulose, and an intact biomass feedstock, poplar, to produce hydrocarbons by complete HDO. We report experiments at an optimum set of process conditions, tested thus far, with both cellulose and poplar, to demonstrate the capabilities of this process. We also discuss the effects of independent control of fast-hydropyrolysis and HDO temperatures with the cellulose feedstock. In future publications, we plan to report detailed results from the study of the effects of process conditions, such as hydrogen partial pressure and types of biomass on the hydrocarbon yields and distributions from the H2Bioil process.
The continuous-flow experiments were performed with a previously described high-pressure reactor system containing a cyclone-type fast-hydropyrolysis (FHP) reactor with a downstream vapor-phase catalytic HDO reactor (Fig. 1).22 The experimental design for this setup was specially challenging due to the need for a lab-scale high-pressure continuous-flow solids feeder and the necessities of a fast-hydropyrolysis reactor system that could work effectively and safely with high pressures (up to 50 bar) of hydrogen. Furthermore, a catalyst is required that would achieve complete HDO of a mixture of oxygenated molecules with different functional groups to produce a hydrocarbon stream. In this reactor setup, the biomass/cellulose is fed using a high-pressure auger-type screw feeder. The biomass is then entrained with feed gas (hydrogen and nitrogen), at a flow rate of ∼25 std. L min−1, for feeding into the cyclone-type fast-hydropyrolysis reactor. The biomass gets converted to produce fast-hydropyrolysis vapors on contact with the heated inner wall of the cyclone and the char by-product is collected at the bottom of the reactor. The vapor residence time in the fast-hydropyrolysis reactor is ∼3 seconds at the reported process conditions (Table 1). The fast-hydropyrolysis vapors exit from the top of the reactor, move through a transfer section where the temperature of the stream is adjusted and are catalytically upgraded in the downstream fixed-bed HDO reactor. The upgraded products pass through a concentric tube heat exchanger, cooled with a mixture of ethylene glycol and water at 5 °C, to quench the vapors. Two traps, one at room temperature and another cooled with dry ice, are used to collect an aqueous phase of products. The permanent gas products and vapor-phase hydrocarbons pass through the traps and are analyzed with an online gas chromatograph equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID). The permanent gases H2, N2, CO, CO2 and CH4 are separated using 60/80 Supelco Carboxen-1000 packed column (4.6 m × 2.1 mm I.D., catalog no. 12390-U) and analyzed with the TCD. The C1–C8+ hydrocarbons are resolved using an Agilent J&W GS-Gaspro capillary column (60 m × 0.32 mm I.D., catalog no. 113-4362) and analyzed with the FID. Nitrogen was used as an internal standard for the TCD analysis and quantification of methane was used for relating the TCD and FID analysis. The TCD and FID have been calibrated and compound retention times have been identified using calibration gas mixtures.
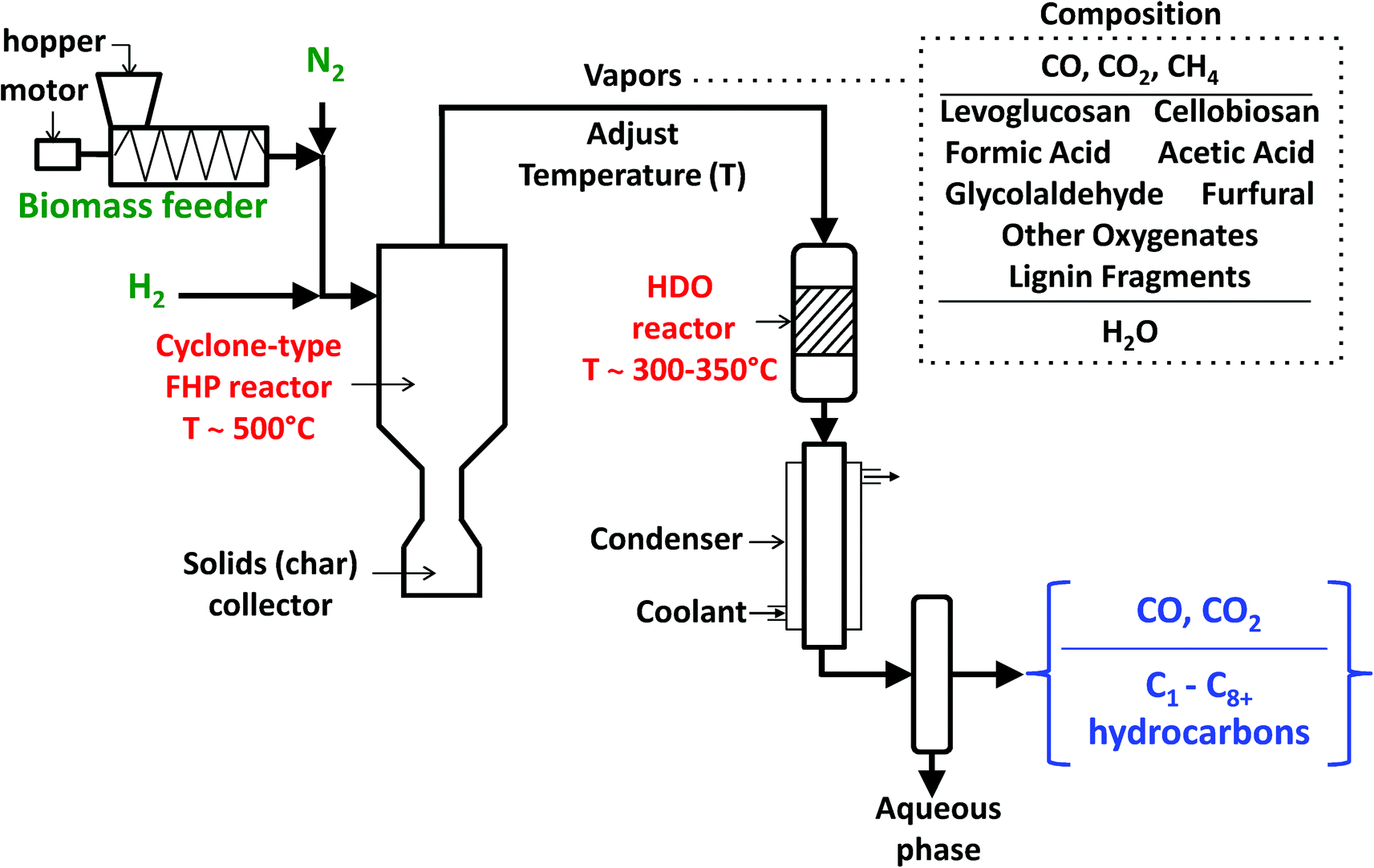 | ||
| Fig. 1 Schematic of the fast-hydropyrolysis (FHP) and catalytic hydrodeoxygenation (HDO) reactor system for experimental proof-of-concept of H2Bioil process. | ||
| Total pressure / bar | 27 |
| Hydrogen partial pressure / bar | 25 |
| Nitrogen partial pressure / bar | 2 |
| Fast-hydropyrolysis temperature / °C | ∼480 |
| HDO catalyst | 5 wt% Pt–2.5 wt% Mo/MWCNT |
| HDO temperature / °C | ∼300 |
| Weight hourly space velocity / h−1 | ∼4 |
The experiments with cellulose and poplar were carried out under similar experimental conditions, as shown in Table 1. The microcrystalline cellulose (50 μm) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The poplar feedstock (termed BESC standard poplar, <53 μm) was a genotype of Populus trichocarpa grown at Oak Ridge National Laboratory.23 The ultimate and proximate analysis of the cellulose and poplar feedstocks, as performed by Hazen Research Inc. (Golden, CO, USA), is shown in Table S.1.† The compositional analysis of poplar feedstock, as provided by NREL, is given in Table S.2.† The cellulose or poplar was fed to the reactor system at the rate of ∼0.1 g min−1 and the total experimental run time was ∼1 hour. After each experiment, the reactor was depressurized, and the aqueous phase and char products along with the remaining feed in the feed hopper were collected and measured for the overall mass balance. The typical experimental error with the reactor system is ∼±5% based on duplicate repeats of experiments. The carbon and hydrogen content of the char and the aqueous phase products (combined from both the traps) and water content of the aqueous phase products was analyzed by Galbraith Laboratories Inc. (Knoxville, TN, USA). The overall carbon balance was ∼95% with both cellulose and poplar. The unaccounted carbon is attributed to product collection losses during and after these high pressure experiments.
Our previous studies22 have shown that, in the absence of a catalyst, high pressures (up to 50 bar) of hydrogen do not have a significant deoxygenation effect on the first stage fast-hydropyrolysis of cellulose. Candidate catalyst testing had shown that the catalyst design is critical for favoring C–O hydrogenolysis over C–C hydrogenolysis for effective HDO.22 Also, non-sulfided catalysts are preferred for this process due to the low content of sulfur in biomass available to retain catalyst activity. Hence, a 5 wt% Pt–2.5 wt% Mo supported on multiwalled carbon nanotubes (MWCNT) catalyst was developed and used for achieving HDO of the cellulose/biomass fast-hydropyrolysis vapors.24 In this catalyst, Pt was chosen for its hydrogenation function, based on the results of candidate catalyst testing,22 and Mo was chosen as an oxophillic promoter, to favor C–O bond scission reactions.25,26 MWCNT were chosen as an inert support, because they are a relatively pure form of carbon reported to be stable at high temperatures27 and they facilitate catalyst characterization measurements. The MWCNT (10–20 nm outer diameter, >95% purity) support was purchased from Cheap Tubes Inc. (Cambridgeport, VT, USA). The catalyst precursors were purchased from Sigma-Aldrich (St. Louis, MO, USA). The catalyst was prepared by sequential incipient wetness impregnation technique. Platinum was impregnated first on the MWCNT using an aqueous solution of tetraammineplatinum(II) nitrate (Pt(NH3)4(NO3)2) to achieve 5 wt% Pt loading, and then the catalyst was dried in air at 60 °C overnight. Subsequently, molybdenum was impregnated using an aqueous solution of ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24·4H2O)) to achieve Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo atomic ratio of 1:1 (2.5 wt% Mo) loading, and the catalyst was again dried at 150 °C overnight. The dried catalyst was reduced in 5% H2 in balance He with a temperature ramp of 4 °C min−1 to 450 °C and held at 450 °C for 2 hours and cooled in He flow. The catalyst was passivated with 10% air in balance He before removing from the catalyst pretreatment reactor. The catalyst was again reduced in situ in the HDO reactor, at the same catalyst reduction conditions, before the start of all experiments. Catalyst characterization by CO chemisorption is provided in the ESI.†
:Mo atomic ratio of 1:1 (2.5 wt% Mo) loading, and the catalyst was again dried at 150 °C overnight. The dried catalyst was reduced in 5% H2 in balance He with a temperature ramp of 4 °C min−1 to 450 °C and held at 450 °C for 2 hours and cooled in He flow. The catalyst was passivated with 10% air in balance He before removing from the catalyst pretreatment reactor. The catalyst was again reduced in situ in the HDO reactor, at the same catalyst reduction conditions, before the start of all experiments. Catalyst characterization by CO chemisorption is provided in the ESI.†
The product distributions from the experiments with cellulose and poplar are shown in Fig. 2 and Table S.3,† on a percentage carbon in the feed basis. The C1 to C3 hydrocarbons comprise of methane, ethane and propane, respectively. In Fig. 2, the C4–C8+ hydrocarbons are lumped in terms of the carbon number for representation of the hydrocarbon mixture. The C8+ hydrocarbons comprise carbon numbers greater than or equal to 8, with C9 as the highest carbon number observed in the mixture. It is noteworthy that there is no detectable oxygen content in the hydrocarbon product mixture, which consists of saturated hydrocarbons as either straight chain, branched or cyclo-paraffins (alkanes). The detailed hydrocarbon yields, with cellulose and poplar, and estimated hydrogen consumption values are shown in Table S.3.† The elemental compositions of char and aqueous phase products from the experiments are shown in Tables S.4 and S.5,† respectively.
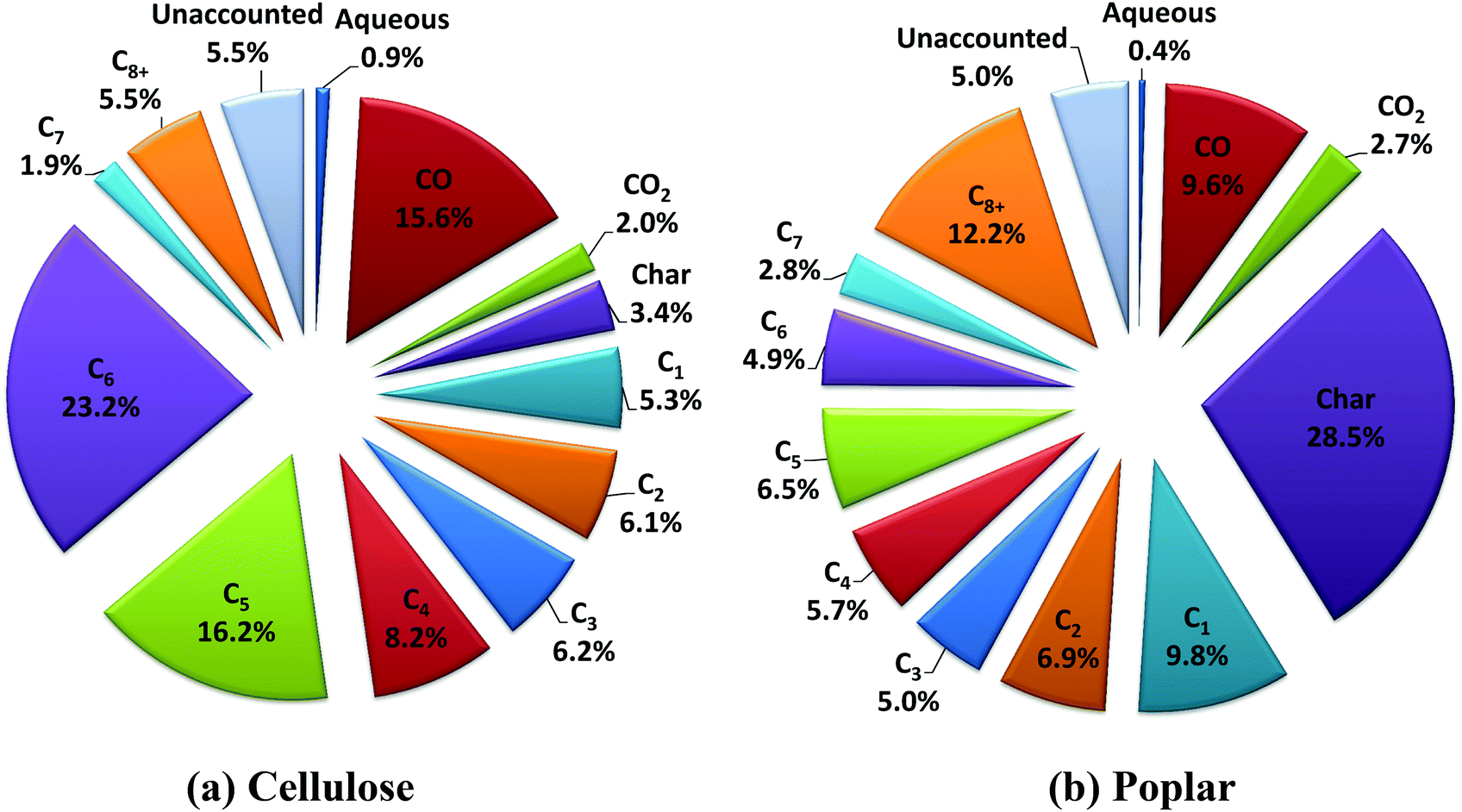 | ||
| Fig. 2 Product distributions (% carbon in the feed basis) from the experiments with cellulose and poplar. | ||
The total C1–C8+ hydrocarbon yield with cellulose (Fig. 2a, Table S.3†) is ∼73% of the carbon fed. The hydrocarbon yield in the liquid fuel range (C4+) is ∼55%. A major fraction of the hydrocarbons falls into the C6 range, mainly due to the complete HDO of levoglucosan (C6H10O5) and its isomers that form a major portion of the vapor mixture from fast-hydropyrolysis of cellulose at these experimental conditions.22 The char yield is low at ∼3% carbon (1.9 wt% of feed) due to the high heat transfer rates that is attributed in part to the relatively small particle size and low flow rate of cellulose. This char yield from cellulose is the lowest reported in the literature, to the best of our knowledge.28,29 The CO yield of ∼16% carbon is mainly due to the C–C bond scission in the second stage HDO using the PtMo/MWCNT catalyst. In contrast, the CO yield is <5% carbon from the first stage fast-hydropyrolysis of cellulose at these experimental conditions.22
The total C1–C8+ hydrocarbon yield with poplar (Fig. 2b, Table S.3†) is ∼54% of the carbon fed. The hydrocarbon yield in the liquid fuel range (C4+) is ∼32%. A major fraction of the hydrocarbons is in the C8+ range, mainly as a result of the HDO of the aromatic oxygenates from the fast-hydropyrolysis of the lignin fraction of biomass.30 The yield of C5 and C6 range hydrocarbons (Fig. 2b, Table S.3†) from poplar show a large difference as compared to cellulose. This is in part due to the lower total C1–C8+ hydrocarbon yield, as compared to cellulose, due to the higher char yield of ∼29% carbon (18.4 wt% of feed) from poplar and due to the presence of hemicellulose and lignin in addition to cellulose in poplar (shown in Table S.2†). The inorganic ash content of the poplar may also contribute to the higher char yield, as reported in earlier studies.31 Also, within the experimental time period used in these studies, due to the high partial pressure (25 bar) of hydrogen used in the experiments, no coking, measurable by weight gain of the used catalyst, is observed with either cellulose or poplar. We do note that for long-term catalyst stability testing, time-on-stream studies would need to be done with the intact biomass feed. Also, in case of catalyst coking during long term testing, other more refractory catalyst supports that can withstand high temperature calcination to regain activity would be needed for the PtMo catalysts, but should not be difficult to develop.
To study the effect of HDO temperature, an experiment where cellulose fast-hydropyrolysis was conducted at the process conditions shown in Table 1 but the HDO reactor operated at a higher temperature of 350 °C was performed. The product distributions under these conditions are compared with those for earlier 300 °C HDO in Fig. S.1.† The comparison shows that increasing the HDO temperature leads to higher C–C bond scission, resulting in a higher proportion of CO and lower proportion of C4+ range hydrocarbons in the product. Our previous studies22 on cellulose fast-hydropyrolysis have shown that the optimal temperature for pyrolysis is in the neighborhood of 480 °C to minimize yields of CO, CO2, CH4 and light oxygenates. Hence, the independent control of the fast-hydropyrolysis and catalytic HDO temperatures in our vapor-phase upgrading approach enables operation of each stage at its optimal temperature. This independent control of the fast-hydropyrolysis and catalytic HDO, using separate process conditions and catalysts, could also help in tailoring the hydrocarbon distribution to higher carbon number hydrocarbons in the diesel range and to produce olefins and aromatics from biomass, which would lower hydrogen consumption and be useful for high-octane fuel or chemicals applications.
The product distributions from both cellulose and poplar show the opportunities for synergistic process integration for improving the utilization of the feed carbon into liquid fuel range (C4+) hydrocarbons. For achieving the overall objective of “no carbon left behind”, one of the process options for synergistic utilization of CO, CO2, C1–C3 hydrocarbons and char could be a combination of heat assisted gasification and reforming (Fig. 3a).9,10 This synergistic process integration could be tailored for hydrogen supply to the fast-hydropyrolysis versus the conversion of the syngas stream to liquid fuels through a Fischer–Tropsch process, versus conversion to methanol or dimethyl ether. Combustion of the non-CO2 components (Fig. 3b) could also be used as an option for providing the process heat based on the acceptable CO2 emissions from the process. The H2Bioil process is envisioned to be deployed as mobile biofuel processing unit close to the biomass sources, which would aid the process economics by reducing biomass transportation costs.2 For these small-scale mobile applications, a combination of char combustion and reforming of CO, C1–C3 hydrocarbons along with hydrogen separation (using selective membranes) and recycle would be a promising synergistic integration.
 | ||
| Fig. 3 Synergistic process integrations of fast-hydropyrolysis and HDO along with gasification, combustion and reforming. | ||
Conclusions
Overall, the experiments for proof-of-concept of the H2Bioil process have shown the potential positive impact of this process on the biofuels sector. Using a 5 wt% Pt–2.5 wt% Mo/MWCNT catalyst, HDO of cellulose and intact biomass (poplar) fast-hydropyrolysis vapors is achieved. The carbon recoveries as C1–C8+ hydrocarbons is ∼73% with cellulose and ∼54% with poplar, with liquid fuel range (C4+) hydrocarbon recoveries as ∼55% and ∼32%, respectively. Independent control of the fast-hydropyrolysis and HDO temperatures is necessary to increase the overall C4+ hydrocarbon yields by minimizing C–C bond scission. Synergistic process integration with gasification, reforming and combustion of CO, C1–C3 hydrocarbons and char would aid in improving overall carbon and energy efficiency of an integrated biorefinery process.Acknowledgements
This research was supported as part of the Center for Direct Catalytic Conversion of Biomass to Biofuels (C3Bio), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES), under Award #DE-SC0000997 (graduate staff salary for Vinod Kumar Venkatakrishnan) and by the U.S. Department of Energy (DOE) under Award #DE-FG36-08GO18087 (catalyst development). Authors are sincerely grateful to Dr. Bryon Donohoe from National Renewable Energy Laboratory (NREL) for providing the BESC (BioEnergy Science Center) standard poplar feedstock and compositional analysis. Catalyst development work by Sara Yohe, Harshavardhan Choudhari, Dhairya Mehta and Paul Dietrich is gratefully acknowledged. Discussions with Emre Gencer and John Degenstein are also gratefully acknowledged.References
- U.S. Energy Information Administration, International Energy Outlook 2013, 2013 Search PubMed.
- R. Agrawal and N. R. Singh, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 343–364 CrossRef CAS PubMed.
- America's Energy Future Panel on Alternative Liquid Transportation Fuels, National Academy of Sciences, National Academy of Engineering and National Research Council, Liquid Transportation Fuels from Coal and Biomass: Technological Status, Costs, and Environmental Impacts, The National Academies Press, Washington, D.C., 2009 Search PubMed.
- U.S. Department of Energy, U.S. Billion-Ton Update: Biomass Supply for a Bioenergy and Bioproducts Industry, Oak Ridge National Laboratory, Oak Ridge, TN, 2011 Search PubMed.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- A. H. Zacher, M. V. Olarte, D. M. Santosa, D. C. Elliott and S. B. Jones, Green Chem., 2014, 16, 491–515 RSC.
- D. C. Elliott, T. R. Hart, G. G. Neuenschwander, L. J. Rotness and A. H. Zacher, Environ. Prog. Sustain. Energy, 2009, 28, 441–449 CrossRef CAS.
- R. Agrawal, M. Agrawal and N. R. Singh, US Pat, 8217211, 2012 Search PubMed.
- R. Agrawal and N. R. Singh, US Pat, 8217210, 2012 Search PubMed.
- R. Agrawal and N. R. Singh, AIChE J., 2009, 55, 1898–1905 CrossRef CAS.
- N. R. Singh, W. N. Delgass, F. H. Ribeiro and R. Agrawal, Environ. Sci. Technol., 2010, 44, 5298–5305 CrossRef CAS PubMed.
- J. A. Melero, J. Iglesias and A. Garcia, Energy Environ. Sci., 2012, 5, 7393–7420 CAS.
- N. R. Singh, D. S. Mallapragada, R. Agrawal and W. E. Tyner, Biomass Convers. Biorefin., 2012, 2, 141–148 CrossRef PubMed.
- A. A. Lappas, K. G. Kalogiannis, E. F. Iliopoulou, K. S. Triantafyllidis and S. D. Stefanidis, Wiley Interdiscip. Rev.: Energy Environ., 2012, 1, 285–297 CrossRef CAS.
- H. Zhang, Y.-T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 CAS.
- H. Zhang, T. R. Carlson, R. Xiao and G. W. Huber, Green Chem., 2012, 14, 98–110 RSC.
- D. C. Dayton, J. Carpenter, J. Farmer, B. Turk and R. Gupta, Energy Fuels, 2013, 27, 3778–3785 CrossRef CAS.
- T. L. Marker, L. G. Felix, M. B. Linck and M. J. Roberts, Environ. Prog. Sustain. Energy, 2012, 31, 191–199 CrossRef CAS.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- R. M. Baldwin and C. J. Feik, Energy Fuels, 2013, 27, 3224–3238 CrossRef CAS.
- V. K. Venkatakrishnan, J. C. Degenstein, A. D. Smeltz, W. N. Delgass, R. Agrawal and F. H. Ribeiro, Green Chem., 2014, 16, 792–802 RSC.
- M. H. Studer, S. Brethauer, J. D. DeMartini, H. L. McKenzie and C. E. Wyman, Biotechnol. Biofuels, 2011, 4, 19 CrossRef CAS PubMed.
- W. N. Delgass, R. Agrawal, F. H. Ribeiro, S. Yohe, M. Abu-Omar, T. Parsell and P. J. Dietrich, US Provisional Patent Application, 61/896110, 2013 Search PubMed.
- P. J. Dietrich, R. J. Lobo-Lapidus, T. Wu, A. Sumer, M. C. Akatay, B. R. Fingland, N. Guo, J. A. Dumesic, C. L. Marshall, E. Stach, J. Jellinek, W. N. Delgass, F. H. Ribeiro and J. T. Miller, Top. Catal., 2012, 55, 53–69 CrossRef CAS.
- P. J. Dietrich, F. G. Sollberger, M. C. Akatay, E. A. Stach, W. N. Delgass, J. T. Miller and F. H. Ribeiro, Appl. Catal., B, 2014, 156–157, 236–248 CrossRef CAS PubMed.
- X. Wang, N. Li, J. A. Webb, L. D. Pfefferle and G. L. Haller, Appl. Catal., B, 2010, 101, 21–30 CrossRef CAS PubMed.
- M. S. Mettler, S. H. Mushrif, A. D. Paulsen, A. D. Javadekar, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 5414–5424 CAS.
- P. R. Patwardhan, D. L. Dalluge, B. H. Shanks and R. C. Brown, Bioresour. Technol., 2011, 102, 5265–5269 CrossRef CAS PubMed.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2013, 7, 103–129 Search PubMed.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, Bioresour. Technol., 2010, 101, 4646–4655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01746c |
| This journal is © The Royal Society of Chemistry 2015 |