
Selective N-alkylation of indoles with primary alcohols using a Pt/HBEA catalyst†
S. M. A. Hakim
Siddiki
a,
Kenichi
Kon
b and
Ken-ichi
Shimizu
*ab
aElements Strategy Initiative for Catalysts and Batteries, Kyoto University, Katsura, Kyoto 615-8520, Japan. E-mail: kshimizu@cat.hokudai.ac.jp; Fax: +81-11-706-9163
bCatalysis Research Center, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan
First published on 9th September 2014
Abstract
Pt-loaded HBEA (H+-exchanged BEA zeolite) is found to be an effective and reusable heterogeneous catalyst for regioselective N-alkylation of indoles with primary aliphatic and benzylic alcohols under additive-free conditions driven by the borrowing-hydrogen methodology. Structural and mechanistic studies suggest a cooperative mechanism of the Pt0 site on Pt metal clusters and the Brønsted acid site of the zeolite, in which the Pt0 site is responsible for dehydrogenation/hydrogenation steps and the Brønsted acid site is responsible for the regioselective condensation of indoles with aldehydes to the enamine intermediate.
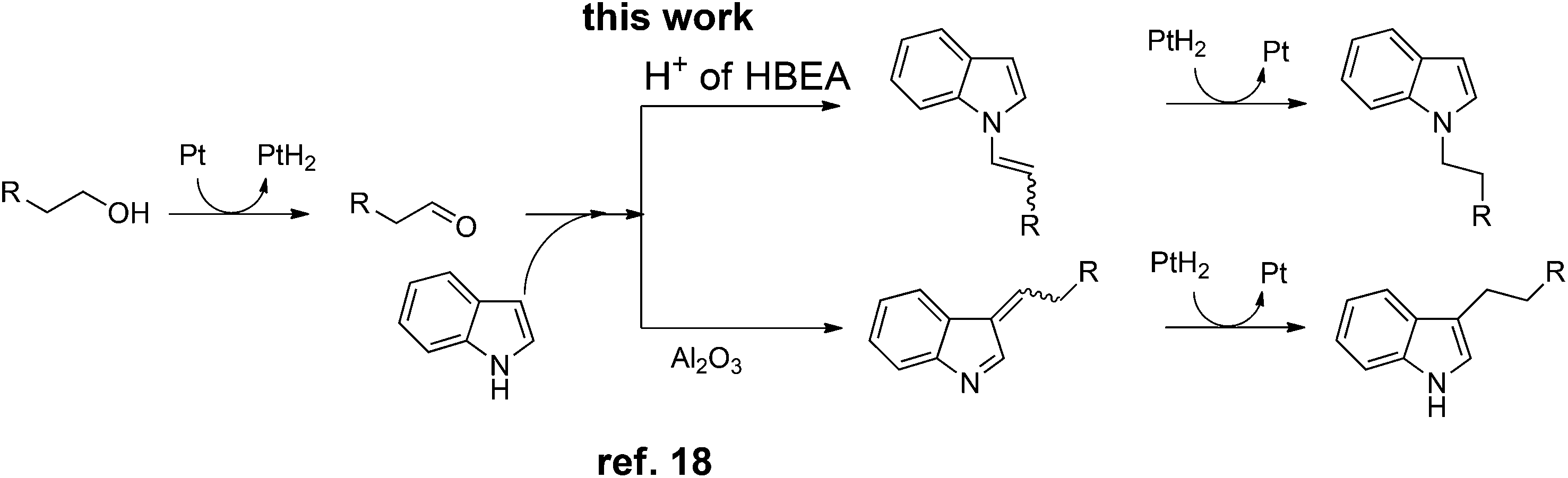
Indoles are relevant structures in heterocyclic natural products, pharmaceutical ingredients, functional materials and agro-chemicals. Hence, attention has been focused on the improved methodologies for synthesis and functionalization of indoles.1–4 Among the indole derivatives, N-substituted indoles are of particular importance for the construction of various biologically active molecules.5–7 Generally, the C3-position of indole has greater nucleophilicity than the N-position, which makes N-selective alkylation of indoles difficult. The classical approach for the N-alkylation of indoles is the base-catalyzed reaction of indole with an alkyl halide,8,9 which suffers from quantitative formation of salt wastes. Use of alcohol as a more benign and abundant alkyl source can overcome this drawback. An earlier example is the N-alkylation of indoles with alcohols by excess amount of RANEY® nickel.10 In contrast to these classical methods, the transition-metal catalyzed hydrogen transfer (borrowing hydrogen) methodology11–15 can provide a more atom-efficient alkylation method using alcohols. As for the C3-alkylation of indoles with alcohols, homogeneous Ir catalysts16,17 were first reported by Grigg16 and a heterogeneous Pt/Al2O3 catalyst recently reported by our group18 were shown to be effective. In contrast, N-alkylation of indoles with alcohols has been rarely exploited. Only one successful example is a Ru-based Shvo catalyst with a Brønsted acidic co-catalyst reported by Beller, Williams and co-workers.19 However, this homogeneous method still has some drawbacks such as low turnover number (TON), difficulty in catalyst/product separation, and necessities of an acidic additive (0.025 mol% PTSA) and excess molar amount (2 equiv.) of an alkylating agent. Shi et al. showed N-alkylation of indole with excess amount of benzyl alcohol at 170 °C using an iron oxide immobilized Pd catalyst,20 but the substrate scope was limited to only one example. As a part of our continuing interest in the heterogeneous catalysis for hydrogen-transfer reactions using Pt nanocluster catalysts,18,21–23 we report herein a heterogeneous catalytic system for the selective N-alkylation of indoles with alcohols using Pt nanocluster-loaded HBEA zeolite24 (Pt/HBEA). To our knowledge, this is the first general heterogeneous catalysis of N-alkylation of indole with various aliphatic and benzyl alcohols without any promoter. Additionally, a combination of this work and our previous work18 demonstrates the first example of the support-controlled complete selectivity switch in regioselective alkylation of indole with primary alcohols using heterogeneous Pt cluster catalysts (Scheme 1).
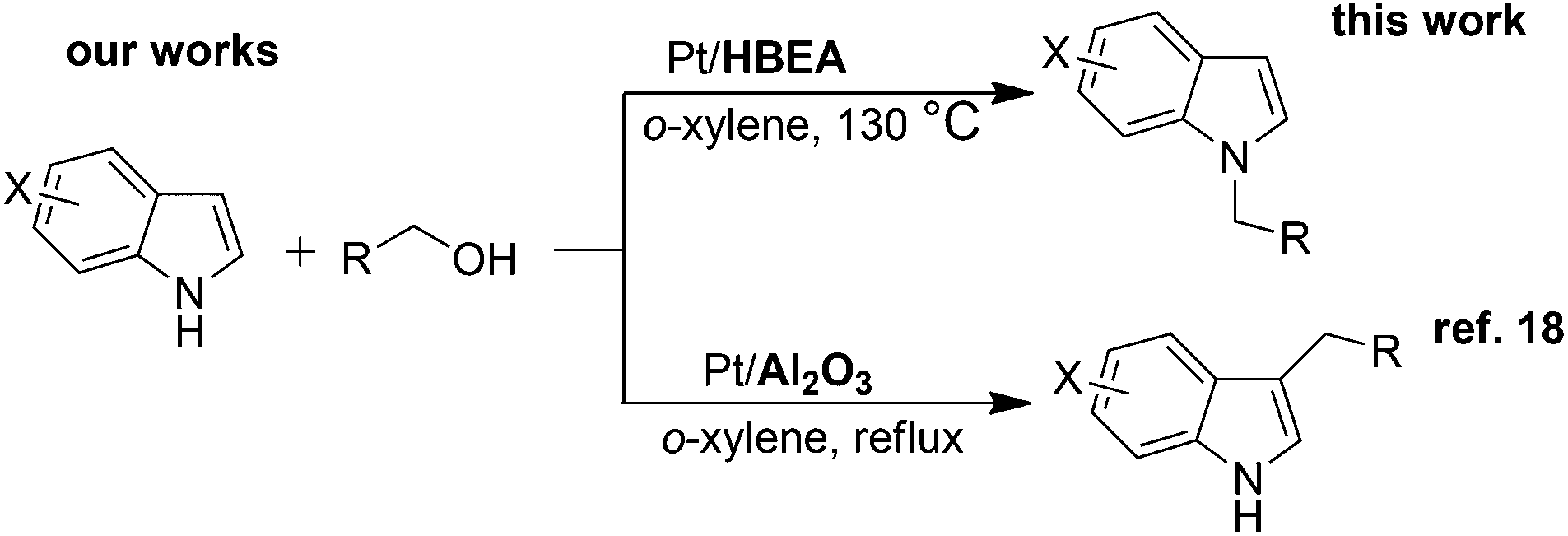 | ||
| Scheme 1 Support-controlled complete selectivity switch in alkylation of indole with primary alcohols using heterogeneous Pt catalysts. | ||
To optimize the catalyst composition, the reaction of equimolar amounts of 1-octanol and indole was tested under N2 at 130 °C for 22 h with various catalysts pre-reduced in H2 at 300 °C (Table 1). Using various transition metals (5 wt% Pt, Ir, Re, Pd, Rh, Ru, Ag, Co, Ni or Cu) supported on HY zeolite (entries 1–10), we carried out a preliminary catalyst screening test. Among the catalysts tested, Pt/HY showed the highest yield of the N-alkylated indole. Then, we studied the effect of support materials on 5 wt% Pt loaded catalysts (entries 1, 11–20). As reported in our previous study on C-3 alkylation of indole,18 the use of an acid–base bifunctional support, Al2O3, resulted in selective formation of the C-3 adduct with 56% yield (entry 14). In contrast, Pt catalysts on acidic supports such as HY zeolite (entry 1), TiO2 (entry 15), silica-alumina (entry 16), and HBEA (entry 20) selectively catalyzed the N-alkylation of indole, and the yield of N-adduct was 19–87%. Pt/HBEA (entry 20) showed the highest yield of N-alkylated product (87%) with no formation of the C3-adduct. Pt catalysts on HMFI zeolite with narrow pore size (<0.56 nm), basic (MgO, CeO2), and neutral supports (SiO2, C) were nearly inactive, and the yield of C3- and N-adducts were below 1%. HBEA itself (entry 22) is inactive, indicating that the Brønsted acid of HBEA itself does not catalyze the N-alkylation of indole. To verify the necessity of the Brønsted acid site of Pt/HBEA we studied the poisoning effect of a basic additive (pyridine) which is known to neutralize the Brønsted acid sites of HBEA. The presence of 0.05 mmol of pyridine in the reaction mixture resulted in complete deactivation of Pt/HBEA (entry 21). From these results, we can conclude that the Brønsted acid site of Pt/HBEA is an important co-catalyst.
|
|
||||
|---|---|---|---|---|
| Entry | Catalysts | Conv. (%) | 3 Yield (%) | 4 Yield (%) |
| a Conversion of 1 and yields were determined by GC. b In the presence of pyridine (0.05 mmol). c 39 mg of HBEA. | ||||
| 1 | Pt/HY | 51 | 40 | 0 |
| 2 | Ir/HY | 8 | 0 | 0 |
| 3 | Re/HY | 6 | 0 | 0 |
| 4 | Pd/HY | 8 | 0 | 0 |
| 5 | Rh/HY | 4 | 0 | 0 |
| 6 | Ru/HY | 8 | 0 | 0 |
| 7 | Ag/HY | 8 | 0 | 0 |
| 8 | Co/HY | 9 | 0 | 0 |
| 9 | Ni/HY | 6 | 0 | 0 |
| 10 | Cu/HY | 4 | 0 | 0 |
| 11 | Pt/MgO | 10 | 0 | 0 |
| 12 | Pt/CeO2 | 15 | 0 | 0 |
| 13 | Pt/ZrO2 | 13 | 5 | 0 |
| 14 | Pt/Al2O3 | 66 | 3 | 56 |
| 15 | Pt/TiO2 | 68 | 54 | 2 |
| 16 | Pt/SiAl | 30 | 19 | 1 |
| 17 | Pt/SiO2 | 8 | 0 | 1 |
| 18 | Pt/C | 7 | 0 | 0 |
| 19 | Pt/HMFI | 5 | 0 | 0 |
| 20 | Pt/HBEA | 100 | 87 | 0 |
| 21b | Pt/HBEA | 5 | 0 | 0 |
| 22c | HBEA | 6 | 0 | 0 |
| 23 | PtOx/HBEA | 12 | 0 | 0 |
| 24 | Pt/HBEA-air | 65 | 46 | 0 |

To study the structure of Pt species, we carried out spectroscopic characterizations of Pt/HBEA. The X-ray absorption near edge structure (XANES) of Pt/HBEA was almost identical to that of the Pt foil (Fig. S1 in the ESI†), indicating the metallic state of the Pt species in Pt/HBEA. The analysis of the Pt L3-edge extended X-ray absorption fine structure (EXAFS) showed the Pt–Pt coordination number of 10.2 at a distance of 2.74 Å (Fig. S1 and Table S1†). This result also shows the metallic state of the Pt species in Pt/HBEA. Fig. S2† shows the size distribution of the Pt particle on the outer surface of Pt/HBEA observed by TEM. The mean diameter from TEM analysis (3.3 ± 0.7 nm) was larger than that from the CO adsorption experiment (1.8 nm). Considering that small Pt clusters inside zeolite cannot be observed by TEM, the results suggest that small Pt clusters are present inside the micropores of HBEA zeolite. From these results, it can be concluded that dominant Pt species in Pt/HBEA is the metallic Pt nanocluster with an average size of 1.8 nm.
Next we discuss the structure of the active Pt species. Platinum oxides-loaded HBEA (PtOx/HBEA, entry 23) showed no activity, indicating that oxides of Pt are inactive. Pt/HBEA-air (entry 24), prepared by exposing the reduced Pt/HBEA to the ambient conditions for 0.5 h, showed lower yield (46%) than the as-reduced Pt/HBEA (87%). Fig. S3† compares IR spectra of CO adsorbed on Pt/HBEA and Pt/HBEA-air. The spectrum for Pt/HBEA shows a band at 2080 cm−1 due to the linear coordination of CO on a Pt atom25 and a weak band at 1835 cm−1 due to bridged CO on two Pt atoms. The spectrum for Pt/HBEA-air showed a band at 2080 cm−1 with lower intensity than Pt/HBEA. This indicates that the surface metallic Pt0 sites of Pt/HBEA are partially oxidized by air even at room temperature. Combined with the catalytic result, it is shown that the surface metallic Pt0 sites are the active species. Taking into account that the Brønsted acid site of HBEA is also required for the catalytic system, we can conclude that the co-presence of Pt0 sites and Brønsted acid sites of HBEA is indispensable to the catalytic system.
For the reaction of indole with 1-octanol, Pt/HBEA was removed from the reaction mixture at 3 h (3 yield = 20%). Then, further heating of the filtrate at 130 °C for 21 h did not increase the yield. ICP-AES analysis of the filtrate confirmed that the content of Pt in the solution was below the detection limit (10 ppb). Fig. 1 shows the result of catalyst reuse. After the reaction, the catalyst was separated by centrifugation, followed by washing with acetone, and drying at 90 °C for 3 h, and H2-reduction at 300 °C for 0.5 h. The catalyst was reused at least three times without a marked loss of its activity. These results indicate that Pt/HBEA acts as a reusable heterogeneous catalyst.
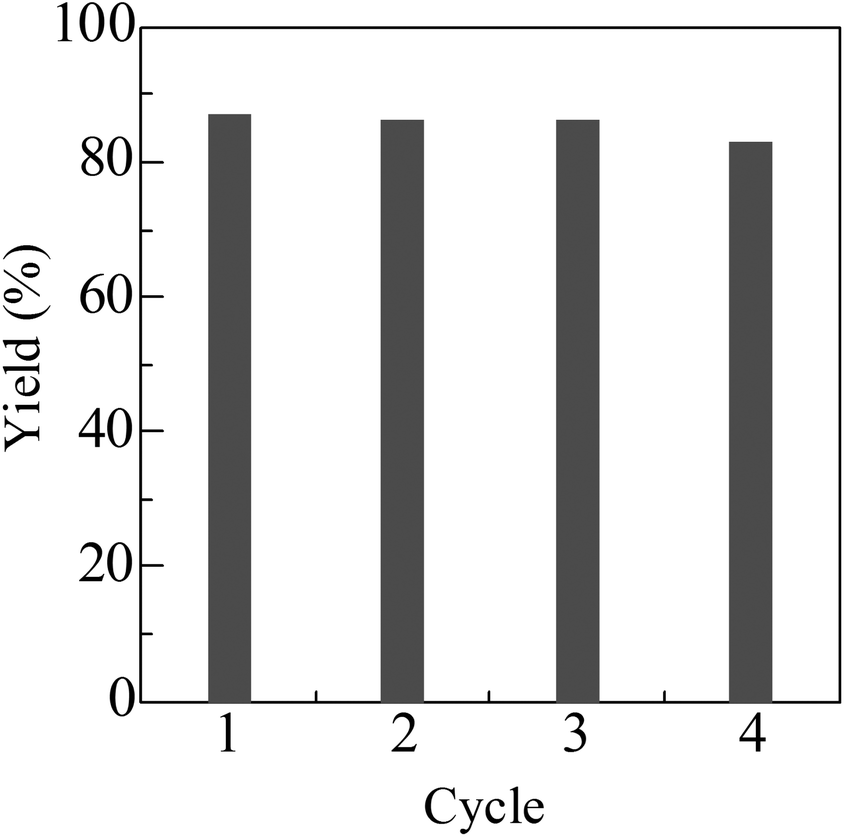 | ||
| Fig. 1 Catalyst reuse for N-alkylation of indole with 1-octanol using Pt/HBEA. Conditions are shown in Table 2. | ||
Table 2 shows the substrate scope for the reaction of equimolar amounts of indoles and alcohols by 1 mol% of Pt/HBEA. Various aliphatic alcohols including linear and branched aliphatic alcohols (entries 1–10) were completely converted to yield the N-alkylated indoles with good isolated yields (73–87%). Benzylalcohol (entry 11) was converted to the N-alkylated indole in 77% yield.26 Benzyl alcohols with electron-withdrawing and electron-donating substituents (entries 13–15) were also converted to N-alkylated indoles in 55–66% yields.27 Note that the previous method19 was only applicable to the reaction of an activated (methoxide substituted) indole with benzyl alcohols. N-alkylation of 3-methyl indole and 5-methyl indole with 1-octanol (entries 16 and 17) resulted in excellent yields (85% and 93%). N-Alkylation of indole with a secondary alcohol (2-octanol) resulted in 5% yield. For all the reactions shown in Table 2, C3-alkylated indoles were not observed in GCMS analyses. This indicates that the present method is completely regioselective. Using a small amount of the catalyst (0.1 mol%), N-alkylation of indole with 1-octanol (entry 2), 1-hexanol (entry 4) and benzylalcohol (entry 12) gave 80, 85 and 70% yields, corresponding to TON of 800, 850, and 700 respectively. TON for the reaction of indole with 1-hexanol is 4 times higher than that of a Ru complex (TON of 213 per Ru atom),19 and TON for reaction of indole with benzyl alcohol is 3 times higher than that of Pd/Fe2O3 (TON of 213).20
| Entry | Alcohols | Products | Isolated yield (%) |
|---|---|---|---|
| a Conditions: 0.01 mmol Pt (1 mol%), 1 mmol indole, 1 mmol alcohol, 1 g o-xylene, 130 °C, 22 h. b GC yield. c 0.001 mmol Pt (0.1 mol%), 90 h. d 1.5 g o-xylene. | |||
| 1 |

|

|
82 (87)b |
| 2c | 80b | ||
| 3 |

|
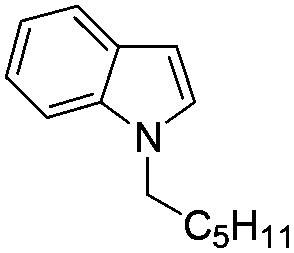
|
87 |
| 4c | 85b | ||
| 5 |

|

|
85 |
| 6 |
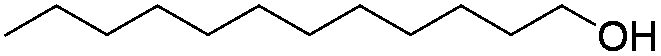
|
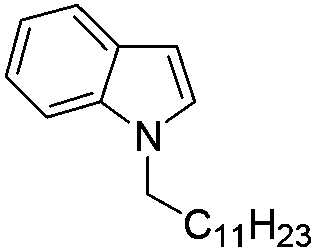
|
87 |
| 7d |
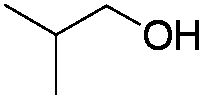
|
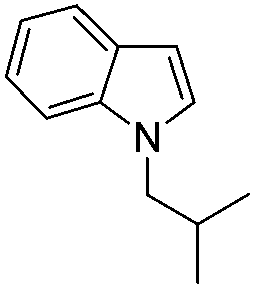
|
77 |
| 8d |

|

|
82 |
| 9 |
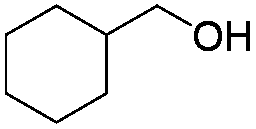
|

|
73 |
| 10 |

|

|
81 |
| 11 |
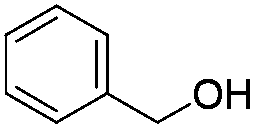
|
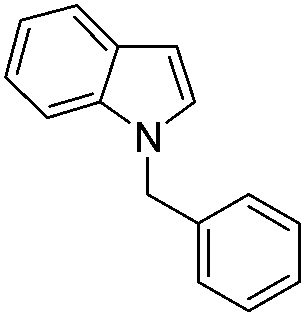
|
77 |
| 12c | 70b | ||
| 13 |
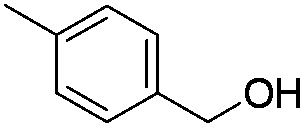
|
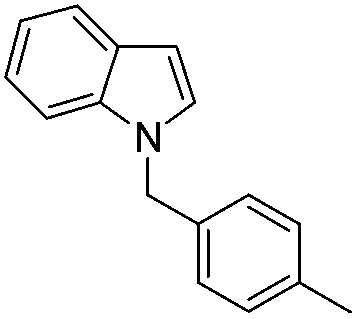
|
60 |
| 14 |

|

|
55 |
| 15 |
|
|
66 |
| 16 |

|
|
85 |
| 17 |
|

|
93 |
Similar to the proposed mechanism for the C3-alkylation of indole with alcohols using Pt/Al2O3 (Scheme 2, lower part),18 the present reaction may proceed through the hydrogen-borrowing pathway, which starts with dehydrogenation of the primary alcohol. In the present system with Pt/HBEA, nucleophilic attack of the N atom of indoles to aldehydes can result in the formation of an enamine intermediate (iminium ion intermediate for benzylic alcohol),19 which is re-hydrogenated using Pt–H species (Scheme 2). The presumed pathway is supported by the following results. The reaction of n-octanal and indole (eqn (1)) with Pt/HBEA under N2 resulted in the formation of the enamine 3a in 61% yield because of the absence of PtH species. In the presence of a hydrogen donor (2-propanol), the same reaction (eqn (2)) resulted in 60% yield of the N-alkylated indole, which can be formed by transfer hydrogenation28,29 of the enamine intermediate 3a using 2-propanol.
 | (1) |
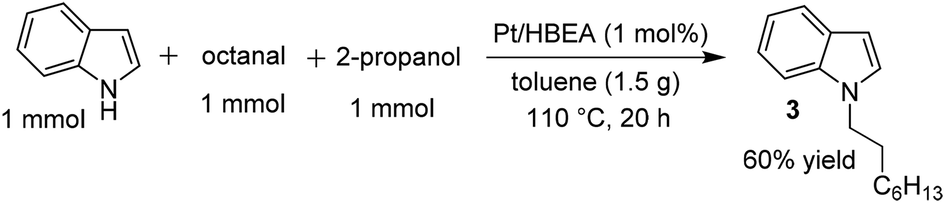 | (2) |
 | ||
| Scheme 2 Presumable pathways of N-alkylation (by Pt/HBEA) and C3-alkylation (by Pt/Al2O3)18 of indole with primary alcohols. | ||
To discuss the origin of the support-dependent switch of the regioselectivity between Pt/HBEA (to N-adduct) and Pt/Al2O3 (to C3-adduct), we conducted the test reaction of n-octanal with indole with Pt-unloaded support materials (HBEA or Al2O3). The result showed that the regioselectivity completely depended on the support; HBEA gave only the enamine 3a (eqn (3)), whereas Al2O3 gave only the C3-adduct 3b (eqn (4)). Interaction between the Brønsted acid site of HBEA and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of indole may reduce the nucleophilicity of C3 carbon, and instead nucleophilic attack of N atom of indole to aldehyde results in the formation of the enamine intermediate.
C bond of indole may reduce the nucleophilicity of C3 carbon, and instead nucleophilic attack of N atom of indole to aldehyde results in the formation of the enamine intermediate.
 | (3) |
To get further insight into the mechanism of the present catalytic reaction, the kinetic isotopic effect (KIE) was investigated by the reaction of indole and α-deutero benzylalcohol (benzyl-α,α-d2 alcohol), and a moderate kH/kD value of 2.3 was obtained (Fig. S4†). The relative rates of N-alkylation from indole and para-substituted benzylalcohols (X = OMe, Me, H, F, Cl, and CF3) were also examined. A reasonable linearity between the log(kX/kH) values and the Hammett parameters (σ) was obtained (Fig. S5†), suggesting that the transition state in the rate-determining step of the alkylation reaction involves a positive charge at the α-carbon atom adjacent to the phenyl ring, which is stabilized by electron-donating substituents. The results of KIE and the Hammett plot indicate that the dissociation of the α-C–H bond of benzylalcohol is a relatively slow step.
In summary, we have developed the first additive-free, heterogeneous and reusable catalytic method for regioselective N-alkylation of indoles with various primary alcohols using a Pt/HBEA catalyst driven by the borrowing-hydrogen mechanism. Compared with the previous homogeneous catalytic system, our method has the following advantages: (1) easy catalyst/product separation, (2) catalyst reusability, (3) higher TON, (4) additive-free conditions, and (5) no need of excess amount of alcohol. Structural and mechanistic studies suggest that this system is driven by cooperation of Pt0 sites on the surface of the Pt cluster and the Brønsted acid site in the zeolite in which Pt0 sites are responsible for dehydrogenation/hydrogenation steps and the Brønsted acid site is responsible for the regioselective condensation of indoles with aldehydes to the enamine intermediate.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Nano Informatics” (25106010) from JSPS and a MEXT program “Elements Strategy Initiative to Form Core Research Center”.Notes and references
- R. J. Sundberg, Indoles, Academic Press, San Diego, 1996 Search PubMed.
- K. Krger, A. Tillack and M. Beller, Adv. Synth. Catal., 2008, 350, 2153 CrossRef.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed.
- S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed.
- M. Negwer and H.-G. Scharnow, Organic-Chemical Drugs and Their Synonyms, Wiley-VCH, Weinheim, 9th edn, 2007, vol. 3, p. 1915 Search PubMed.
- M. Negwer and H.-G. Scharnow, Organic-Chemical Drugs and Their Synonyms, Wiley-VCH, Weinheim, 9th edn, 2007, vol. 4, p. 2631 Search PubMed.
- M. Negwer and H.-G. Scharnow, Organic-Chemical Drugs and Their Synonyms, Wiley-VCH, Weinheim, 9th edn, 2007, vol. 4, p. 2632 Search PubMed.
- (a) G. Vavilina, A. Zicmanis, S. Drozdova, P. Mekss and M. Klavins, Chem. Heterocycl. Compd., 2008, 44, 530 CrossRef CAS; (b) A. V. Karchava, F. S. Melkonyan and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2012, 48, 391 CrossRef CAS PubMed.
- Y. R. Jorapur, J. M. Jeongb and D. Y. Chi, Tetrahedron Lett., 2006, 47, 2435 CrossRef CAS PubMed.
- F. De Angelis, M. Grasso and R. Nicoletti, Synthesis, 1977, 335 CrossRef CAS.
- T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753 RSC.
- G. Guillena, D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358 CrossRef CAS PubMed.
- R. Yamaguchi, K. Fujita and M. Zhu, Heterocycles, 2010, 81, 1093 CrossRef CAS PubMed.
- Y. Obora and Y. Ishii, Synlett, 2011, 30 CrossRef CAS PubMed.
- F. Alonso, F. Foubelo, J. C. González-Gómez, R. Martínez, D. J. Ramón, P. Riente and M. Yus, Mol. Diversity, 2010, 14, 411 CrossRef CAS PubMed.
- S. Whitney, R. Grigg, A. Derrick and A. Keep, Org. Lett., 2007, 9, 3299 CrossRef CAS PubMed.
- A. E. Putra, K. Takigawa, H. Tanaka, Y. Ito, Y. Oe and T. Ohta, Eur. J. Org. Chem., 2013, 6344 CrossRef CAS.
- S. M. A. H. Siddiki, K. Kon and K. Shimizu, Chem. – Eur. J., 2013, 19, 14416 CrossRef CAS PubMed.
- S. Bahn, S. Imm, K. Mevius, L. Neubert, A. Tillack, J. M. J. Williams and M. Beller, Chem. – Eur. J., 2010, 16, 3590 CrossRef PubMed.
- Y. Zhang, X. Qi, X. Cui, F. Shi and Y. Deng, Tetrahedron Lett., 2011, 52, 1334 CrossRef CAS PubMed.
- K. Kon, S. M. A. H. Siddiki and K. Shimizu, J. Catal., 2013, 304, 63 CrossRef CAS PubMed.
- C. Chaudhari, S. M. A. H. Siddiki and K. Shimizu, Tetrahedron Lett., 2013, 54, 6490 CrossRef CAS PubMed.
- S. M. A. H. Siddiki, K. Kon, A. S. Touchy and K. Shimizu, Catal. Sci. Technol., 2014, 4, 1716 Search PubMed.
- Catalysis Society of Japan supplies the HBEA sample (JRC-Z-HB25), which is originally supplied as B-25 from Clariant Catalysts.
- K. Shimizu, K. Ohshima, Y. Tai, M. Tamura and A. Satsuma, Catal. Sci. Technol., 2012, 2, 730 CAS.
- Michael addition adduct (3,3′-bisindolyl phenylmethane) was observed as a byproduct (8% yield).
- GCMS analysis showed that relatively low yields for benzyl alcohols were due to the formation of Michael addition adducts.
- F. Alonso, P. Riente, F. Rodriguez-Reinoso, J. Ruiz-Martinez, A. Sepulveda-Escribano and M. Yus, J. Catal., 2008, 260, 113 CrossRef CAS PubMed.
- F. Alonso, P. Riente, F. Rodriguez-Reinoso, J. Ruiz- Martinez, A. Sepffllveda-Escribano and M. Yus, ChemCatChem, 2009, 1, 75 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01419g |
| This journal is © The Royal Society of Chemistry 2015 |