
Metal-free hydroacyloxylation and hydration reactions of ynamides: synthesis of α-acyloxyenamides and N-acylsulfonamides†
Shijie
Xu
a,
Jianquan
Liu
b,
Donghua
Hu
*a and
Xihe
Bi
*bc
aSchool of Pharmaceutical Sciences, Changchun University of Chinese Medicine, Changchun 130117, China. E-mail: hudonghua8888@gmail.com
bDepartment of Chemistry, Northeast Normal University, Changchun 130024, China. E-mail: bixh507@nrenu.edu.cn
cState Key Laboratory and Institute of Elemento-Organic Chemistry, Tianjin 300071, China
First published on 16th September 2014
Abstract
Two types of highly efficient and operationally simple reactions of ynamides under metal-free conditions are developed, including hydroacyloxylation and hydration, which afforded highly functionalized α-acyloxyenamides and pharmaceutically important N-acylsulfonamides in good to excellent yields.
Ynamides are a class of electron-deficient ynamines with the initial report dating far back to the pioneering work of Viehe in 1972.1 Substitution of a nitrogen atom for electronegative elements, such as acyl and sulfonyl groups, has realized an exceptionally fine balance between the stability and the reactivity of ynamines, thus providing a workable solution to the long-term problem of the sensitivity of these compounds toward hydrolysis.2 In the last decade, exploration on the synthetic utility of ynamides captivated the attention of the scientific community and hence resulted in a great number of transformations.3 Among these, the regioselective addition of a nucleophilic moiety to the reactive keteniminium intermediates, generated from ynamides due to the electron-donating ability of the nitrogen, constitutes an attractive strategy to prepare the functionalized enamides.4 However, this strategy remains largely underexploited, especially under metal-free conditions.

 | ||
| Fig. 1 Hydroacyloxylation and hydration reactions of ynamides. | ||
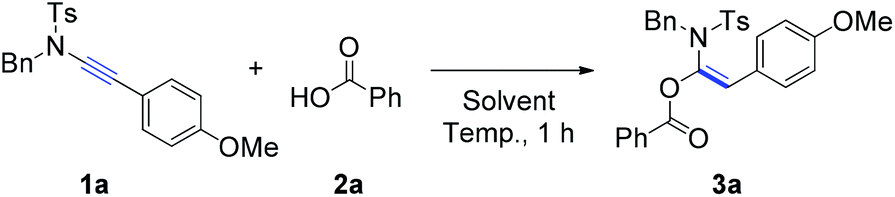
Initially, the reaction of ynamide (1a) with benzoic acid (2a) was selected as a model for optimization of the reaction conditions (Table 1). The effect of the reaction temperature was first examined using toluene as the solvent for a reaction period of 1 hour. Delightfully, the desired product, α-acyloxyenamide 3a, was obtained in 25% yield at room temperature (ca. 20 °C) in toluene (Table 1, entry 1). Encouraged by this promising result, the reaction temperature was increased gradually, and a nearly quantitative yield (98%) was obtained at 100 °C (Table 1, entries 2 and 3). However, as the temperature was increased to 120 °C, the ynamide yield decreased to 85% (Table 1, entry 4). Notably, the hydroacyloxylation of benzoic acid 2a with ynamide 1a was regio- and stereoselective. The structure of product 3a was established by comparing its 1H/13C NMR spectra with those of similar compounds reported by Lam and co-workers.6 Next, we evaluated the solvent effect at a fixed temperature of 100 °C. Surprisingly, no reaction took place when solvents such as DMF, DMSO, and 1,4-dioxane were used (Table 1, entries 5–7). Therefore, the reaction conditions listed in entry 3 were found to be optimal and selected for further investigations.
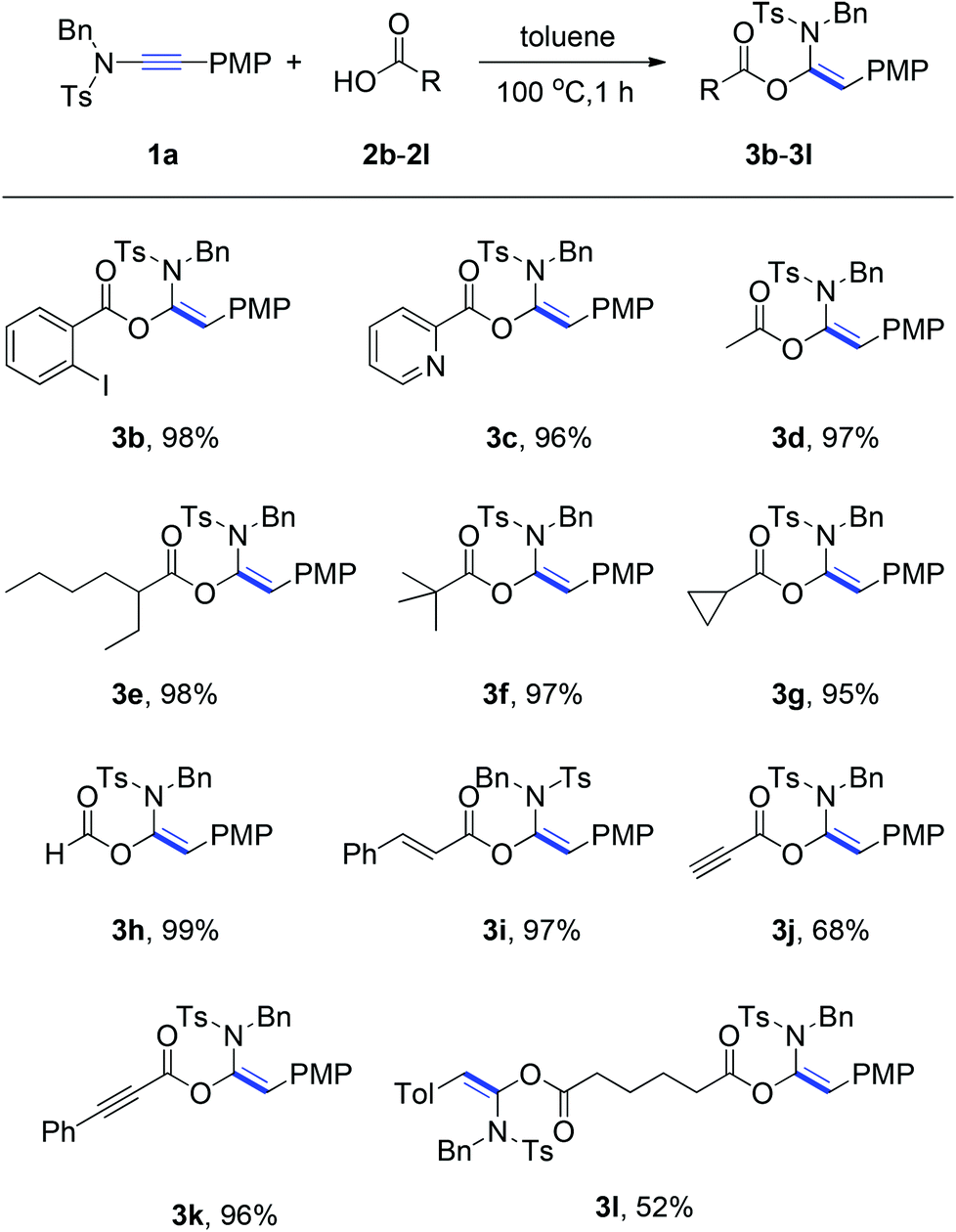
With the optimized reaction conditions in hand (100 °C in toluene), the substrate scope of the metal-free hydroacyloxylation reaction was next investigated using diverse carboxylic acids with different substitution patterns. The experimental results from these studies are summarized in Scheme 1. In general, all reactions proceeded smoothly affording the corresponding α-acyloxyenamides (3b–3l) in good to excellent yields (52–99%) with high level regio- and stereo-control. The stereochemistry of α-acyloxyenamides was further unambiguously confirmed by X-ray diffraction (XRD) analysis of product 3b (CCDC 1010978) (Fig. 2). A possible rationalization of this high stereoselectivity might be attributed to the steric repulsion in the transition state and the stability of the E-configuration product. The scope of carboxylic acids’ adaptability to the hydroacyloxylation reaction with ynamides was quite broad. For example, the aromatic carboxylic acids such as 2-iodobenzoic acid (2b) and picolinic acid (2c) efficiently reacted with ynamide 1a to afford the desired products 3b and 3c in 98 and 96% yields, respectively. Similarly, a broad scope of aliphatic carboxylic acids, including diverse alkyl, alkenyl, and alkynyl carboxylic acids, could be applied to this hydroacyloxylation reaction with ynamide 1a, allowing the synthesis of functionalized α-acyloxyenamides (3d–3l). It is noteworthy that the reaction of ynamide 1a with formic acid (2h), the simplest carboxylic acid, afforded the corresponding product 3h in nearly quantitative yield (99%). In addition, the presence of functional groups, such as cyclopropyl (3g), alkenyl (3i), and alkynyl (3j and 3k), was well tolerated, thus providing a group of synthetically valuable intermediates. Furthermore, bis-α-acyloxyenamide 3l could be synthesized in a moderate yield (52%) through a one-step reaction with 0.5 equivalent amount of adipic acid.
 | ||
| Scheme 1 Scope of carboxylic acids in the hydroacyloxylation of ynamides. PMP = 4-methoxyphenyl. | ||
 | ||
| Fig. 2 Single crystal structure of 3b. | ||
Following an illustration of the compatibility of ynamide 1a with a broad variety of carboxylic acids, investigation was furthered on the scope of the ynamides and the experimental results are summarized in Scheme 2. The R3 group of ynamides was first systemically varied. We were pleased to find that aryl- (e.g., phenyl, 4-chlorophenyl), fused aryl- (e.g., naphthyl), and heteroaryl- (e.g., 2-thienyl, 3-pyridyl) substituted ynamides were all suitable substrates for the hydroacyloxylation, affording the corresponding α-acyloxyenamides (3m–3q) in 63–96% yields. The aliphatic R3 group containing functionalities such as ether and hydroxyl groups has no influence on the reaction outcome. In each case, the functionalized α-acyloxyenamides 3r and 3s were obtained in 99% and 87% yields. Notably, when the ynamide 1i that had a terminal alkyne unit (R3 = H) was subjected to the reaction with benzoic acid under the standard conditions, the desired product 3t was obtained in 96% yield. Furthermore, the electron-withdrawing group on the nitrogen center of ynamides was varied. For example, oxazolidinone-substituted ynamides 1j and 1k were found to be effective substrates, providing products 3u and 3v in 98% and 97% yields.
 | ||
| Scheme 2 Scope of ynamides in the hydroacyloxylation of ynamides. PMP = 4-methoxyphenyl. | ||
Due to the biological and pharmaceutical importance of N-acylsulfonamides8 as well as the lack of a systemic study on the hydration of ynamides,3 we next focused on the search for the conditions of the hydrolysis of ynamides. After several trials, we ultimately realized that two equivalent amounts of acetic acid in H2O and acetone as the mixture solvent can offer a clean and complete conversion from ynamides to N-acylsulfonamides. Some selected results are shown in Scheme 3. This hydration reaction of ynamides all smoothly proceeded and afforded the corresponding N-acylsulfonamides (4a–4f) in 70–96% yields. In addition, the variation of N-substituents has no influence on the reaction outcome, for example imides 4g and 4h were obtained in excellent yields under the hydrolysis conditions. Consequently, an efficient and operationally simple access to pharmaceutically important N-acylsulfonamides was developed. Notably, for most reactions listed in Schemes 1–3, pure products can be obtained simply by filtering the reaction mixture, without the need for tedious chromatographic purification steps.
 | ||
| Scheme 3 Hydration reaction of ynamides to N-acylsulfonamides. PMP = 4-methoxyphenyl. | ||
In summary, we have developed two types of highly efficient metal-free reactions of ynamides, hydroacyloxylation and hydration reactions, which afforded a variety of highly functionalized α-acyloxyenamides and pharmaceutically important N-acylsulfonamides in good to excellent yields.
Financial support by the NNSFC (21172029, 21202016, 21372038), the Ministry of Education of the People's Republic of China (NCET-13-0714), and the Jilin Provincial Research Foundation for Basic Research (20140519008JH).
Notes and references
- Z. Janousek, J. Collard and H. G. Viehe, Angew. Chem., Int. Ed. Engl., 1972, 11, 917 CrossRef CAS.
- For recent reviews on ynamine chemistry, see: (a) C. A. Zificsak, J. A. Mulder, C. Rameshkumar, L.-L. Wei and R. P. Hsung, Tetrahedron, 2001, 57, 7575 CrossRef CAS; (b) J. A. Mulder, K. C. M. Kurtz and R. P. Hsung, Synlett, 2003, 1379 CAS; (c) A. R. Katritzky, R. Jiang and S. K. Singh, Heterocycles, 2004, 63, 1455 CrossRef CAS PubMed.
- For reviews, see: (a) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064 CrossRef CAS PubMed; (b) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840 CrossRef CAS PubMed; (c) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Fadel, F. Legrand, G. Evano and N. Rabasso, Adv. Synth. Catal., 2011, 353, 263 CrossRef CAS; (b) N. Saito, K. Saito, M. Shiro and Y. Sato, Org. Lett., 2011, 13, 2718 CrossRef CAS PubMed; (c) J. A. Mulder, K. C. M. Kurtz, R. P. Hsung, H. Coverdale, M. O. Frederick, L. Shen and C. A. Zificsak, Org. Lett., 2003, 5, 1547 CrossRef CAS PubMed; (d) Y. Zhang, Tetrahedron Lett., 2005, 46, 6483 CrossRef CAS PubMed; (e) Y. Zhang, Tetrahedron, 2006, 62, 3917 CrossRef CAS PubMed.
- E. Genin, P. Y. Toullec, S. Antoniotti, C. Brancour, J.-P. Genêt and V. Michelet, J. Am. Chem. Soc., 2006, 128, 3112 CrossRef CAS PubMed.
- D. L. Smith, W. R. F. Goundry and H. W. Lam, Chem. Commun., 2012, 48, 1505 RSC.
- For related examples on hydrothiolation of ynamides, see: (a) H. Yasui, H. Yorimitsu and K. Oshima, Chem. Lett., 2008, 37, 40 CrossRef CAS; (b) S. Kanemura, A. Kondoh, H. Yasui, H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2008, 81, 506 CrossRef CAS.
- (a) T. Hasegawa and H. Yamamoto, Bull. Chem. Soc. Jpn., 2000, 73, 423 CrossRef CAS; (b) M. G. Banwell, C. F. Crasto, C. J. Easton, A. K. Forrest, T. Karoli, D. R. March, L. Mensah, M. R. Nairn, P. J. O'Hanlon, M. D. Oldham and W. Yue, Bioorg. Med. Chem. Lett., 2000, 10, 2263 CrossRef CAS; (c) L. L. Chang, W. T. Ashton, K. L. Flanagan, T.-B. Chen, S. S. O'Malley, G. J. Zingaro, P. K. S. Siegl, S. D. Kivlighn, V. J. Lotti, R. S. L. Chang and W. J. Greenlee, J. Med. Chem., 1994, 37, 4464 CrossRef CAS.
- F. Wang, H. Liu, H. Fu, Y. Jiang and Y. Zhao, Adv. Synth. Catal., 2009, 351, 246 CrossRef CAS.
- For a recent review, see: G. Evano, K. Jouvin and A. Coste, Synthesis, 2013, 17 CAS.
- (a) N. Ghosh, S. Nayak and A. K. Sahoo, Chem. – Eur. J., 2013, 19, 9428 CrossRef CAS PubMed; (b) G. Compain, K. Jouvin, A. Martin-Mingot, G. Evano, J. Marrot and S. Thibaudeau, Chem. Commun., 2012, 48, 5196 RSC.
- For examples, see: (a) Z. Fang, J. Liu, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2014, 53, 7209 CrossRef CAS PubMed; (b) Z. Liu, J. Liu, L. Zhang, P. Liao, J. Song and X. Bi, Angew. Chem., Int. Ed., 2014, 53, 5305 CrossRef CAS PubMed; (c) J. Liu, Z. Liu, N. Wu, P. Liao and X. Bi, Chem. – Eur. J., 2014, 20, 2154 CrossRef CAS PubMed; (d) J. Liu, Z. Fang, Q. Zhang, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2013, 52, 6953 CrossRef CAS PubMed.
- For related synthesis of N,O-ketene aminals, see: (a) J. Sun and S. A. Kozmin, Angew. Chem., Int. Ed., 2006, 45, 4991 CrossRef CAS PubMed; (b) D. Fujino, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2014, 136, 6255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, and spectral copies of all compounds. CCDC 1010978. For ESI and crystallographic data in CIF or other electronic format. see DOI: 10.1039/c4gc01328j |
| This journal is © The Royal Society of Chemistry 2015 |