
Pd/C-catalyzed reactions of HMF: decarbonylation, hydrogenation, and hydrogenolysis†
Joyee
Mitra
,
Xiaoyuan
Zhou
and
Thomas
Rauchfuss
*
School of Chemical Sciences, University of Illinois at Urbana-Champaign, Illinois 61801, USA. E-mail: rauchfuz@illinois.edu
First published on 16th September 2014
Abstract
The diverse reactivity of 5-hydroxymethylfural (HMF) in Pd/C-catalyzed reactions is described with emphasis on the role of additives that affect selectivity. Three broad reactions are examined: decarbonylation, hydrogenation, and hydrogenolysis. Especially striking are the multiple roles of formic acid in hydrogenolysis/hydrogenation and in suppressing decarbonylation, as illustrated by the conversion of HMF to DMF. Hydrogenation of the furan ring is suppressed by CO2 and carboxylic acids. These results emphasize the utility of Pd/C as a convenient catalyst for upgradation of cellulosic biomass.
Introduction
Methods for the conversion of lignocellulosic biomass to commodity chemicals have escalated due to the interest in carbon-neutral fuels, as well as energy security.1–5 Cellulose and hemicelluloses are abundant precursors to liquid fuels and chemicals, and the principal pathway for this upconversion involves deoxygenation and hydrogenolysis, usually sequentially. Major routes from sugars to liquid fuels involve furanic intermediates. For glucose-based feedstocks, 5-hydroxymethylfurfural (HMF) is of central importance.6–9Recent years have witnessed major advances in the production of HMF. The imminent availability of cheap HMF places greater emphasis on its process chemistry, i.e., conversion to liquid fuels and chemicals.1–5 HMF can be oxidized to dicarboxylic acids that are promising precursors to polymers.10–12 It can also be deoxygenated to 2,5-dimethylfuran (DMF) and 2,5-dimethyltetrahydrofuran (DMTHF) with further hydrogenation, which may be suitable fuel additives owing to their high energy density, volatility, and solubility.13–15 Hydrogenation of HMF produces bis-(hydroxymethyl)tetrahydrofuran (BHMTHF), which is a promising monomer.8 Heterogeneous catalysts have been used for decarbonylation, decarboxylation, hydrogenation, and hydrogenolysis.16
One of the key challenges for upgrading HMF and furfural is product selectivity. For example, hydrogenation of HMF results in a mixture of side chain- and ring-hydrogenated products along with ring-opening products.17–19 Although a large variety of catalysts have been investigated for the conversion of HMF,16 we have found 5 weight% palladium on carbon (henceforth abbreviated Pd/C) to be especially versatile. Indeed, palladium-based catalysts have been widely examined to reduce oxygen content of HMF and carbohydrates under reducing conditions, but in most cases, a mixture of products is obtained.20–24 This report describes new insights that allow the use of Pd/C for more selective transformations of HMF: decarbonylation, hydrogenolysis, and hydrogenation (Scheme 1). Furthermore, owing to the mild operating conditions permitted by Pd/C, formation of humins could be minimized if carbohydrates are used as precursors to HMF.
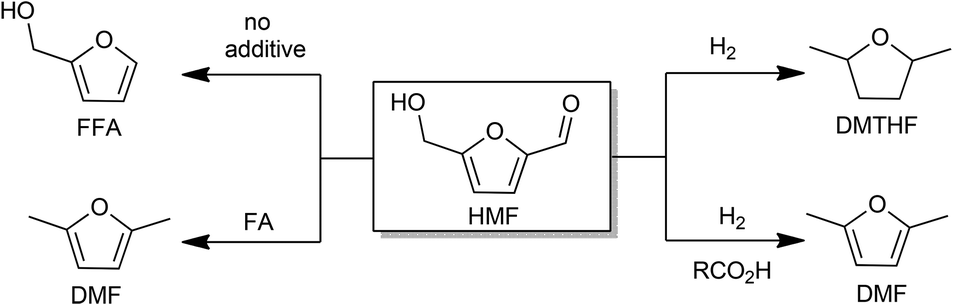 | ||
| Scheme 1 Reactions of HMF catalyzed by Pd/C with and without additives. HMF: 5-hydroxymethylfurfural; FFA: furfuryl alcohol; DMF: dimethylfuran; FMF: 5-(formyloxymethyl)furfural; DMTHF: dimethyltetrahydrofuran. | ||
Hydrogenolysis of the side chains of HMF is competitive with ring hydrogenation.13,18,20,25–28 Decoupling those processes is highly relevant to the use of furans as fuels. Supercritical CO2–H2O mixtures favor formation of DMF from HMF, although the product distribution is highly sensitive on the reaction parameters and involves the use of high pressure H2 or CO2.29,30
In parallel with hexose-derived HMF, furfural (FF) is the principal dehydration product of pentoses. FF is a precursor to furfuryl alcohol (FFA), which is used commercially.31 We were initially interested in developing an approach to produce FFA from HMF. The decarbonylation of HMF was realized decades ago with Pd-based catalysts under harsh reaction conditions.32 Recently, the Leitner group reported the decarbonylation of HMF with iridium phosphine catalysts under high pressure CO2 (50 bar).33 Maiti and Fu carried out the same reaction using Pd(OAc)2 and Pd/SBA-15 respectively, in the presence of molecular sieves.34,35 These decarbonylation reactions, however, require high temperatures, prolonged reaction times, and the presence of CO scavengers.
Results
Decarbonylation of HMF
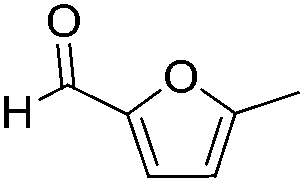
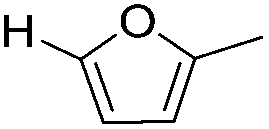
Decarbonylation experiments were carried out in dioxane solution, a relatively high boiling point that is useful in processing of lignocellulose.36,37 The effect of temperature on the decarbonylation of HMF is dramatic (Table 1). When the reaction was conducted at 100 °C, no conversion of HMF was observed. In contrast, the yield of FFA reached 90% when the temperature was elevated to 120 °C. In contrast to experiments conducted in open flasks, conversion to FFA is inefficient (32%) in a sealed reactor, an effect attributed to catalyst poisoning by in situ generated CO (Table 1, entry 6). Solvent is also necessary for this reaction to proceed with high efficiency (Table 1, entry 7). The generality of this method was probed with diverse HMF derivatives (Table 2).38 5-Methylfurfural (5-MF), which can be derived from fructose,39 gave 2-methylfuran in nearly quantitative yield. The formate ester of HMF (FMF) was decarbonylated with excellent selectivity to afford the activated ester of FFA. This transformation is noteworthy because the ester of FFA cannot be readily synthesized from FFA. Instead FA induces oligomerization of FFA owing to the high reactivity of the 5-CH center.40 Further illustrating the protection afforded by the formyl group, 5-(mesitylmethyl)furfural, derived from the alkylation of mesitylene with HMF,38 also cleanly decarbonylated. In contrast, 5-chloromethylfurfural was unreactive (Table 2, entry 5), probably due to catalyst deactivation by this reactive alkyl chloride.41|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | t (h) | Conversion (%) | Selectivity (%) |
| a Reaction conditions: HMF (1.0 mmol), substrate/Pd = 20 molar ratio, the mixture was refluxed in air. b This reaction was conducted in a closed reactor. | |||||
| 1 | Dioxane (5 mL) | 120 | 2 | 12 | >95 |
| 2 | Dioxane (5 mL) | 120 | 8 | 56 | >95 |
| 3 | Dioxane (5 mL) | 120 | 15 | 90 | >95 |
| 4 | Dioxane (5 mL) | 120 | 20 | >95 | >95 |
| 5 | Dioxane (5 mL) | 100 | 15 | Trace | >95 |
| 6b | Dioxane (5 mL) | 120 | 15 | 32 | >95 |
| 7 | No solvent | 120 | 45 | Trace | >95 |
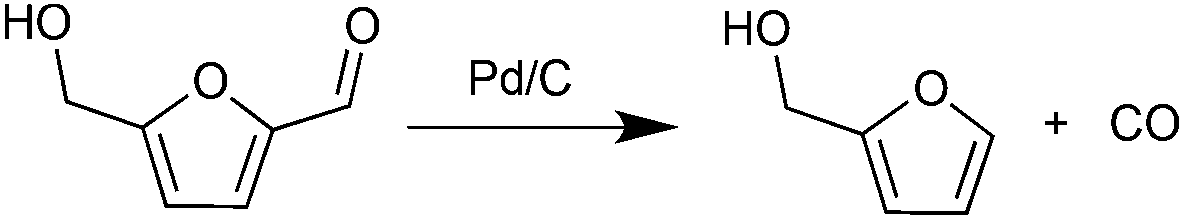
| Entry | Substrate | Product | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|
| a Reaction conditions: substrate (1.0 mmol), substrate/Pd = 20 molar ratio, dioxane (5 mL), 15 h. | ||||
| 1 |
|
|
95 | >95 |
| 2 |
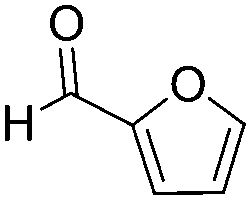
|
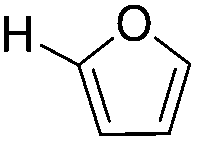
|
83 | >95 |
| 3 |
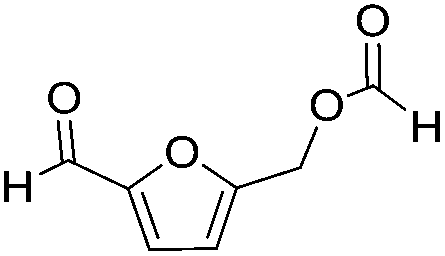
|

|
>95 | >95 |
| 4 |
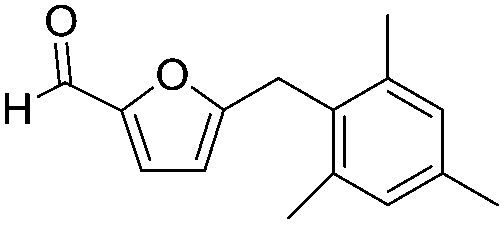
|
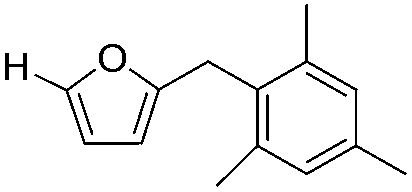
|
>95 | >95 |
| 5 |
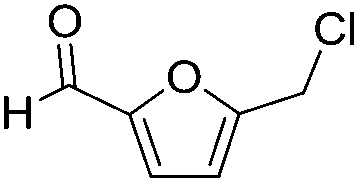
|
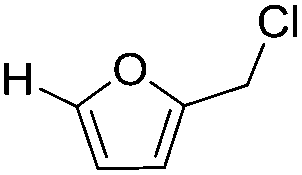
|
0 | N/A |
Hydrogenolysis of HMF with Pd/C and formic acid
In the presence of formic acid (FA), a commonly used reagent in lignocellulose processing,42 the Pd/C-catalyzed decarbonylation of HMF is suppressed. Instead, 2,5-dimethylfuran (DMF) is produced as the exclusive product (Scheme 2).43 For such reactions a closed reactor (glass autoclave) is required. Such conversion entails both hydrogenation of the formyl group and hydrogenolysis of the hydroxymethyl groups. Notably, the furan ring is not hydrogenated, which is commonly observed when H2 is used as the hydrogen source.13,18,20,25–29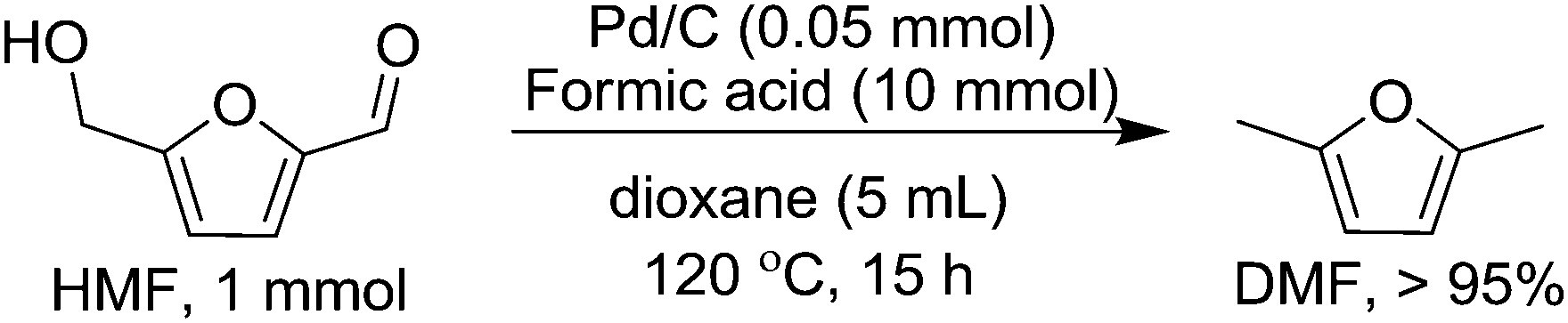 | ||
| Scheme 2 Conversion of HMF to DMF with Pd/C and FA. | ||
In terms of mechanism, the FA-promoted conversion of HMF to DMF was envisioned to proceed via either of two pathways, which differ in the sequence of hydrogenation of the formyl group and hydrogenolysis of the C–OH bond (Scheme 3). These two pathways would produce distinctive intermediates: bis(hydroxymethyl)furan (BHMF) for Path A and 5-MF for Path B. By 1H NMR spectroscopy of the reaction mixtures, BHMF was indeed observed at a low (∼5%) concentration during the reaction while no 5-MF was detected (Fig. 1(a)). This result suggests that hydrogenation of the formyl group precedes hydrogenolysis, implicating Path A. Control experiments starting with BHMF yielded DMF quantitatively under these reaction conditions.
 | ||
| Scheme 3 Pathways for the conversion of HMF and FMF to DMF. | ||
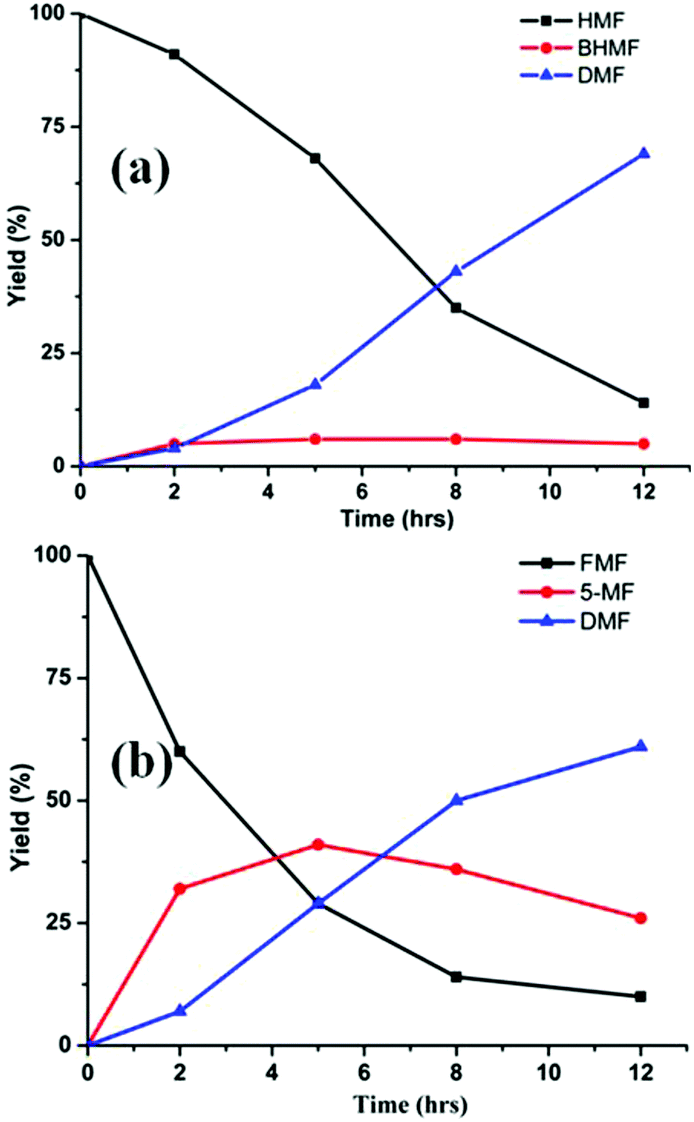 | ||
| Fig. 1 Time course of hydrogenolysis of HMF (a) and FMF (b) to DMF. Reaction conditions (see Scheme 2): 0.5 mmol substrate, substrate/Pd = 10 molar ratio, FA (10 equiv.), dioxane (5 mL), 120 °C. | ||
We also investigated the role of formate esters in the conversion of HMF to DMF. 2-(Formoxymethyl)furfural (FMF) and 2,5-bis(formoxymethyl)furan (BFMF) are efficiently generated by dissolving of HMF and BHMF, respectively, in FA at room temperature.43 These esters are poised to undergo Pd/C-catalyzed conversion to DMF. Our experiments showed that in dioxane solution, neither FMF nor BFMF are reactive toward Pd/C, i.e., they do not undergo decarboxylation in contrast to literature claims.43,44In the presence of FA and Pd/C, however, both FMF and BFMF readily convert to DMF (conditions in Scheme 2).
Detailed studies revealed that the pathways for conversion of FMF and HMF under our standard conditions differ in the sequence of C–O bond hydrogenolysis vs. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond hydrogenation (Fig. 1(b)). In the case of FMF, 5-MF was observed as the major intermediate, indicating that hydrogenolysis is faster for the RCH2–OCHO bond than the hydrogenation of the formyl group. Under comparable conditions, HMF converts to BHMF, indicating that the hydrogenation of the formyl group is faster than hydrogenolysis of the RCH2–OH bond.
O bond hydrogenation (Fig. 1(b)). In the case of FMF, 5-MF was observed as the major intermediate, indicating that hydrogenolysis is faster for the RCH2–OCHO bond than the hydrogenation of the formyl group. Under comparable conditions, HMF converts to BHMF, indicating that the hydrogenation of the formyl group is faster than hydrogenolysis of the RCH2–OH bond.
The reactivity of the formate leaving group was further confirmed by comparing hydrogenolysis rates for BHMF vs. BFMF (Fig. 2). Under our conditions, DMF was generated ca. 3× faster from BFMF than from BHMF. The advantage of formate esters was further confirmed by the low reactivity of (acetoxymethyl)furfural (AcMF) toward hydrogenolysis. Under standard conditions (0.5 mmol substrate, substrate/Pd = 10, 10 equiv. FA, 5 mL dioxane, 120 °C), AcMF converted to DMF at half the rate observed for FMF. The hydrogenolysis of formate esters using FA as a hydrogen source in principle is catalytic in FA since it regenerates FA (eqn (1)).43
| OC4H2(CH2OCHO)2 → OC4H2(CH3)2 + 2CO2 | (1) |
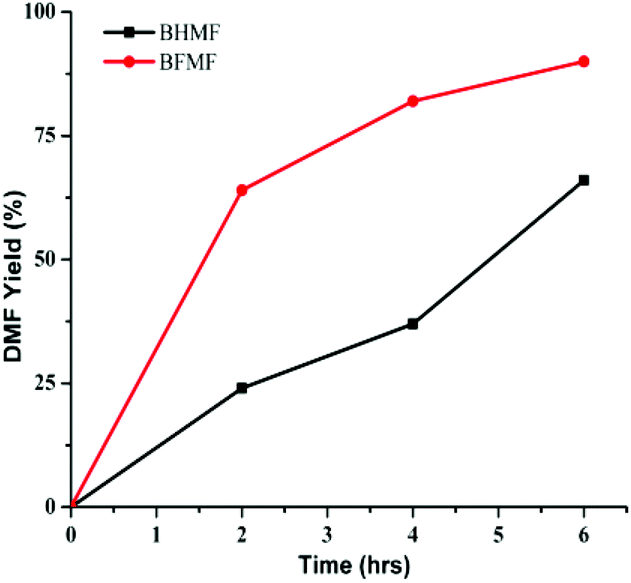 | ||
| Fig. 2 Time course of conversion of BHMF and BFMF to DMF. Reaction conditions: substrate (0.5 mmol), substrate/Pd = 10 molar ratio, FA (10 equiv.), dioxane (5 mL), 120 °C. | ||
The effect of catalyst loading varied with the furanic substrate (ESI†). BFMF was found to be an ideal substrate: with only 1% catalyst loading, it converted in 78% yield to DMF after 15 h.
Tuning the selectivity of the hydrogenolysis of HMF
In the preceding section, FA was shown to facilitate the conversion of HMF to DMF by (i) serving as a hydrogen source and (ii) inhibiting decarbonylation. Fruitful experiments were conducted to probe the influence of FA on the hydrogenation (using H2) of furanic substrates catalysed by Pd/C. Results are summarized in Table 3. Under 30 psi of H2, Pd/C catalyzes the conversion of HMF to a mixture of ring hydrogenated species, (bis-hydroxymethyl)tetrahydrofuran (BHMTHF), dimethyldihydrofuran (DMDHF), and dimethyltetrahydrofuran (DMTHF) (Table 3, entry 1). DMF was not observed. These results are in accordance with literature reports that document the tendency of Pd/C to catalyze hydrogenation of the furan ring.20,22,29,45–51 The selectivity of this reaction was, however, favorably affected by the addition of various acids. As shown in Table 3, addition of FA results in excellent yield of DMF, with only 12% of ring-hydrogenation products. Acetic acid (pKa 4.74, vs. 3.77 for FA) shows similar favorable effect, although not as significantly (Table 3, entry 3). Monitoring of the time course of the reaction under H2 reveals the significant effect of acid (Fig. 3). In the absence of acid, BHMF appears as the initial intermediate, followed by the formation of DMF. Under these conditions, DMF undergoes further hydrogenation to DMDHF and finally to DMTHF (Scheme 4). In contrast, with the addition of acid, DMF and BHMF accumulate, suggesting that ring-hydrogenation is inhibited by the acid.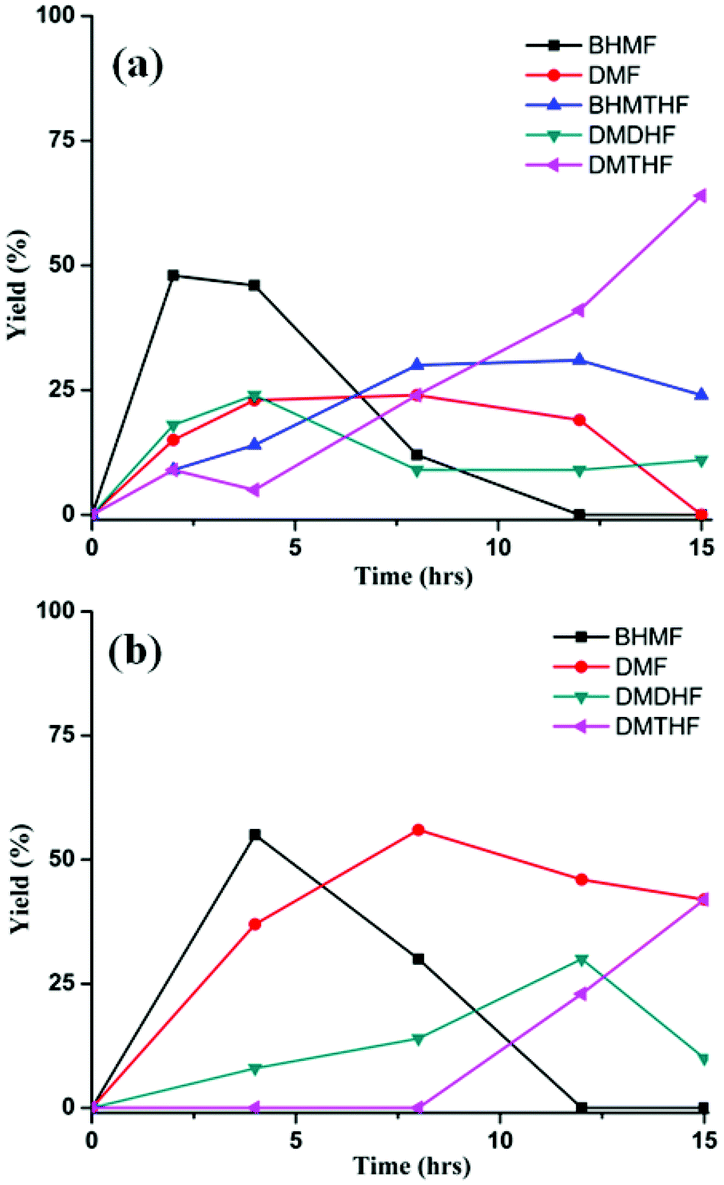 | ||
| Fig. 3 Time course of the Pd/C catalyzed reaction of HMF under H2 (30 psi). (a): w/o acid. (b): CH3CO2H (10 equiv.). Reaction conditions: substrate (0.5 mmol), substrate/Pd = 10 molar ratio, dioxane (5 mL), 120 °C. | ||
 | ||
| Scheme 4 Hydrogenation/Hydrogenolysis of HMF under H2 (30 psi) with Pd/C. | ||
| Entry | Additive | Yield (%) | Conversion (%) | |||
|---|---|---|---|---|---|---|
| DMF | BHMTHF | DMDHF | DMTHF | |||
| a Reaction conditions: HMF (0.5 mmol), substrate/Pd = 10 molar ratio, H2 (30 psi), dioxane (5 mL), 120 °C, 15 h. b 0.5 mL H2O and 4.5 mL dioxane, total solvent volume = 5 mL. | ||||||
| 1 | None | 0 | 24 | 11 | 64 | >95 |
| 2 | HCO2H (5 mmol) | 85 | 0 | 8 | 4 | >95 |
| 3 | CH3CO2H (5 mmol) | 42 | 0 | 10 | 42 | >95 |
| 4 | CO2 (30 psi)/H2Ob | 37 | 0 | 50 | 12 | >95 |
| 5 | CO2 (30 psi) | 0 | 57 | 0 | 42 | >95 |
| 6 | [(CH3OC(O)]2O (5 mmol)/H2Ob | 52 | 0 | 40 | 7 | >95 |
| 7 | [(CH3OC(O)]2O (5 mmol) | 0 | Trace | 0 | >95 | >95 |
More interestingly, the combination of CO2 and H2O strongly affects the product distribution. Selectivity to DMF increased from 0 to 38% in presence of CO2 (30 psi). A small amount of H2O is essential as a co-solvent, implicating a role for carbonic acid (Table 3, entries 4 and 5). Dimethyldicarbonate ((CH3OC(O))2O), a source of CO2, showed similar beneficial effect on the selective production of DMF from HMF, but only in the presence of water required for its hydrolysis (Table 3, entries 6 and 7). The influence of CO2–H2O on the reactivity of HMF has also been observed by Chatterjee.29
Product distribution is also highly dependent on reactant concentration and catalyst loading. Increasing the concentration of HMF by 4-fold led to a decrease in ring hydrogenation products and improved the selectivity of DMF under both CH3CO2H and CO2/H2O conditions (Table 4). In particular, DMF was produced in 85% yield with trace amount of ring hydrogenation products when CO2/H2O was used. Hydrogenation of HMF was equally facile with a decreased catalyst loading (Table 4, entries 5 and 6). Selectivity of hydrogenation/hydrogenolysis over ring hydrogenation was strongly dependent on the catalyst loading. In fact, it decreased the tendency for ring hydrogenation prior to hydrogenolysis. BHMTHF was not observed with 5 mol% catalyst loading even in absence of additives, though DMDHF and DMTHF were the dominant products. Gratifyingly, a decreased catalyst loading in presence of additives resulted in absence of any ring hydrogenation product in the reaction mixture after 15 h (Fig. 5).
| Entry | Catalyst | Additive | Yieldc (%) | Convc (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Loading (mol%) | BHMF | DMF | BHMTHF | DMDHF | DMTHF | |||
| a Reaction conditions: HMF (2 mmol), H2 (30 psi), dioxane (5 mL), the mixture was heated in a pressure reactor at 120 °C for 15 h. b 5% unconverted HMF. c Yields were determined by 1H NMR with nitromethane as the internal standard. | ||||||||
| 1 | 10 | None | 0 | 26 | 7 | 23 | 43 | >95 |
| 2b | 10 | CH3CO2H (20 mmol) | 9 | 85 | 0 | 0 | 0 | ∼95 |
| 3 | 10 | CO2 (30 psi)/H2O (0.5 mL) | 0 | 62 | 0 | 32 | 5 | >95 |
| 4 | 5 | None | 0 | 39 | 0 | 25 | 35 | >95 |
| 5b | 5 | CH3CO2H (20 mmol) | 8 | 75 | <5% | Trace | Trace | ∼95 |
| 6 | 5 | CO2 (30 psi)/H2O (0.5 mL) | 43 | 56 | 0 | 0 | 0 | >95 |
Conclusions
This work illustrates the diverse reactivity of HMF in the presence of Pd/C and how this reactivity can be fine-tuned with simple bio-derived additives. Three broad reactions are demonstrated: decarbonylation to furfuryl derivatives, conversion to DMF, and hydrogenation/hydrogenolysis to tetrahydrofurans. Because of its easy decarbonylation, HMF is useful for the synthesis of FFA derivative that cannot be conveniently generated from FFA owing to its sensitivity towards acids and electrophiles.Striking in this work is the influence of formic acid, which serves multiple roles: a mild source of hydrogen, a precursor to formate esters that are activated toward hydrogenolysis, and a catalyst moderator that suppresses decarbonylation and ring hydrogenation. Our results revealed the following relative rates for 2-furanyl substituents: hydrogenolysis of C–OC(O)H > hydrogenation of CO > hydrogenolysis of C–OH bond. Collectively these results highlight beneficial effects of renewable additives on transformations of cellulosic derivatives.
Experimental section
Reactions were carried out in air unless otherwise noted. All reagents were commercially supplied. Yields and selectivities were assayed by 1H NMR spectroscopy using mesitylene (Tables 1 and 2) and nitromethane (Tables 3 and 4, and Fig. 1–5) as the internal standards. See ESI† for details. Pd/C indicates 5 wt% Pd on carbon (Aldrich).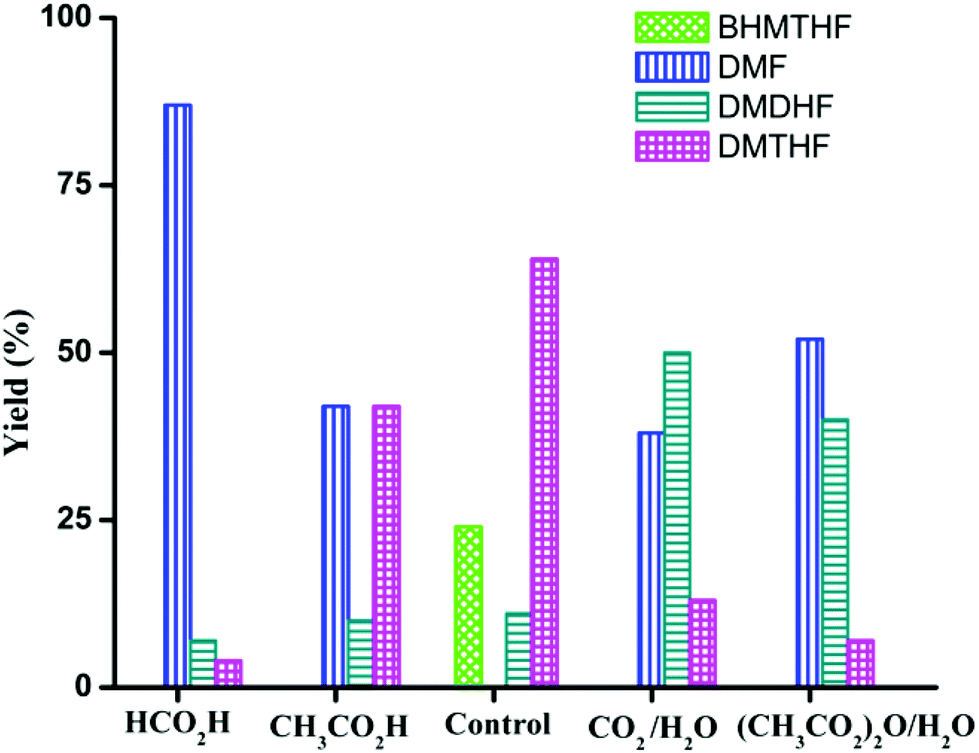 | ||
| Fig. 4 Effect of additives on the conversion of HMF under H2 (30 psi) with Pd/C catalyst. Control: no additive. Reaction conditions: substrate (0.5 mmol), substrate/Pd = 10 molar ratio, dioxane (5 mL), 120 °C. | ||
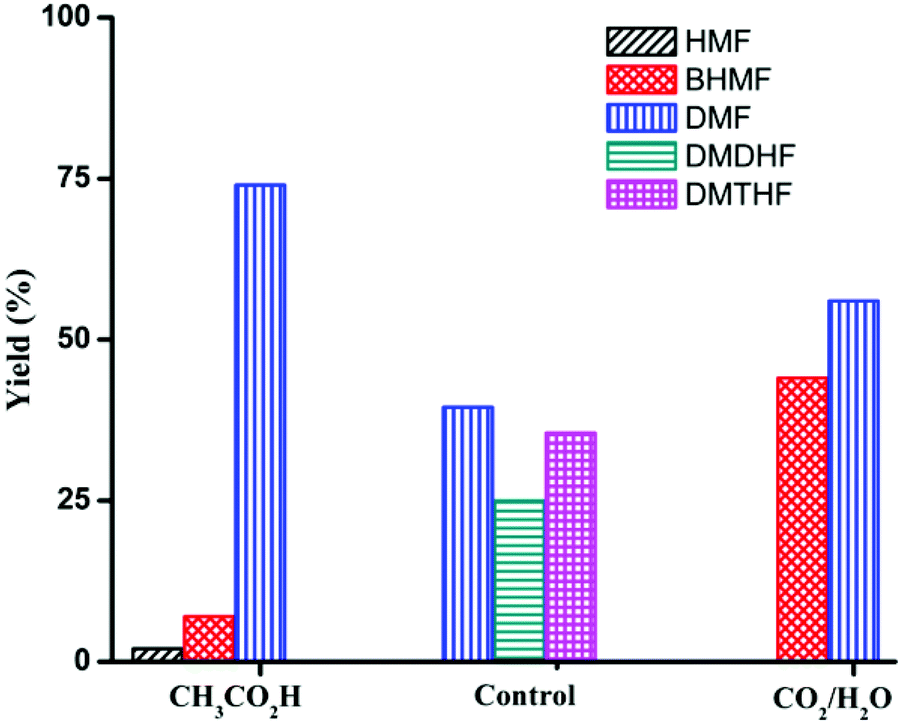 | ||
| Fig. 5 Product distribution for the reaction of HMF with Pd/C (substrate/Pd = 20 molar ratio). Control: no additive Reaction conditions: substrate (2 mmol), dioxane (5 mL), 120 °C, 15 h. | ||
Representative procedure for decarbonylation
A suspension of 0.125 g (1.0 mmol) of HMF and 0.1 g of 5 wt% Pd/C in 5 mL of dioxane was heated at reflux (oil bath temperature: 120 °C) in air for 15 h with stirring.Representative procedure for DMF synthesis
A pressure reactor was charged with HMF (0.5 mmol, 63 mg), 5 wt% Pd/C (100 mg, 10 mol%), dioxane (5 mL), and formic acid (190 μL). The mixture was stirred at 120 °C (oil bath temperature) for 15 h and cooled to room temperature. A known amount of MeNO2 was added to the reaction mixture as internal standard and the product yield was assayed by 1H NMR spectroscopy.Representative procedure for hydrogenation/hydrogenolysis of HMF
In a typical experiment, a Fisher–Porter reactor (150 mL) was charged with HMF (0.50 mmol, 63 mg), 5 wt% Pd/C (100 mg, 10 mol%), and dioxane (5 mL). The reactor was refilled with H2 (30 psi) and the closed system was stirred at 120 °C (oil bath temperature). After 15 h, the mixture was cooled to room temperature. A known amount of MeNO2 was added as internal standard and an aliquot was taken for analysis. Conversion and selectivity of the product mixture were determined by 1H NMR.Acknowledgements
This research was conducted under contract DEFG02-90ER14146 with the U.S. Department of Energy by its Division of Chemical Sciences, Office of Basic Energy Sciences. Support for JM and XY was provided in part by the Energy Biosciences Institute, funded by British Petroleum.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Corma, O. de la Torre and M. Renz, ChemSusChem, 2011, 4, 1574–1577 CrossRef CAS PubMed.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- A. V. Subrahmanyam, S. Thayumanavan and G. W. Huber, ChemSusChem, 2010, 3, 1158–1161 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- (a) R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed; (b) B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24 RSC.
- S. P. Teong, G. S. Yi and Y. G. Zhang, Green Chem., 2014, 16, 2015–2026 RSC.
- F. M. Jin and H. Enomoto, Energy Environ. Sci., 2011, 4, 382–397 CAS.
- S. Dutta, S. De and B. Saha, ChemPlusChem, 2012, 77, 259–272 CrossRef CAS PubMed.
- G. Yi, S. P. Teong, X. Li and Y. Zhang, ChemSusChem, 2014, 7, 2131–2137 CrossRef CAS PubMed.
- W. Yang and A. Sen, ChemSusChem, 2010, 3, 597–603 CrossRef CAS PubMed.
- J. Lewkowski, ARKIVOC, 2001, 2, 17–54 Search PubMed.
- M. R. Grochowski, W. Yang and A. Sen, Chem. – Eur. J., 2012, 18, 12363–12371 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- J. D. Garber, R. E. Jones and U. Torleif, US3083236A, 1963.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. Dumesic, Green Chem., 2012, 14, 1413 RSC.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253–1262 RSC.
- Y. Kwon, E. de Jong, S. Raoufmoghaddam and M. T. M. Koper, ChemSusChem, 2013, 6, 1659–1667 CrossRef CAS PubMed.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154–156 CrossRef CAS PubMed.
- S. Nishimura, N. Ikeda and K. Ebitani, Catal. Today, 2014, 232, 89–98 CrossRef CAS PubMed.
- J. Tuteja, H. Choudhary, S. Nishimura and K. Ebitani, ChemSusChem, 2014, 7, 96–100 CrossRef CAS PubMed.
- T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457 RSC.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS PubMed.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- Y. Yang, Z. Du, J. Ma, F. Lu, J. Zhang and J. Xu, ChemSusChem, 2014, 7, 1352–1356 CrossRef CAS PubMed.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 1543 RSC.
- F. Liu, M. Audemar, K. De Oliveira Vigier, J.-M. Clacens, F. De Campo and F. Jérôme, ChemSusChem, 2014, 7, 2089–2093 CrossRef CAS PubMed.
- H. E. Hoydonckx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000 Search PubMed.
- L. D. Lillwitz, US4089871A, 1978 (The Quaker Oats Company).
- F. M. A. Geilen, T. vom Stein, B. Engendahl, S. Winterle, M. A. Liauw, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2011, 50, 6831–6834 CrossRef CAS PubMed.
- Y. B. Huang, Z. Yang, M. Y. Chen, J. J. Dai, Q. X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1348–1351 CrossRef CAS PubMed.
- A. Modak, A. Deb, T. Patra, S. Rana, S. Maity and D. Maiti, Chem. Commun., 2012, 48, 4253–4255 RSC.
- F. Hu, S. Jung and A. Ragauskas, ACS Sustainable Chem. Eng., 2012, 1, 62–65 Search PubMed.
- X. Zhou, J. Mitra and T. B. Rauchfuss, ChemSusChem, 2014, 7, 1623–1626 CrossRef CAS PubMed.
- X. Zhou and T. B. Rauchfuss, ChemSusChem, 2013, 6, 383–388 CrossRef CAS PubMed.
- W. Yang and A. Sen, ChemSusChem, 2011, 4, 349–352 CrossRef CAS PubMed.
- W. R. Edwards and L. H. Reeves, J. Am. Chem. Soc., 1942, 64, 1583–1584 CrossRef CAS.
- CMF is however known to oxidatively add to Pd(0): Y. Shi, P. Brenner, S. Bertsch, K. Radacki and R. D. Dewhurst, Organometallics, 2012, 31, 5599–5605 CrossRef CAS.
- J. J. Villaverde, J. Li, M. Ek, P. Ligero and A. de Vega, J. Agric. Food Chem., 2009, 57, 6262–6270 CrossRef CAS PubMed.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2010, 49, 6616–6618 CrossRef CAS PubMed.
- J. S. Matthews, D. C. Ketter and R. F. Hall, J. Org. Chem., 1970, 35, 1694–1695 CrossRef CAS.
- K. Yoshii and A. Yamada, WO2013133208A1, 2013 (Ube Industries, Ltd).
- H. Cai, C. Li, A. Wang and T. Zhang, Catal. Today, 2014, 234, 59–65 CrossRef CAS PubMed.
- V. Schiavo, G. Descotes and J. Mentech, Bull. Soc. Chim. Fr., 1991, 704–711 CAS.
- M. A. Lilga, R. T. Hallen, T. A. Werpy, J. F. White, J. E. Holladay, J. G. Frye and A. H. Zacher, US20070287845A1, 2007 (Battelle Memorial Institute).
- P. D. Bloom and A. J. Sanborn, WO2010062689A2, 2010 (Archer Daniels Midland Company).
- W. Dedsuksophon, K. Faungnawakij, V. Champreda and N. Laosiripojana, Bioresour. Technol., 2011, 102, 2040–2046 CrossRef CAS PubMed.
- Y. Wang, Q. Xia, G. Lu, X. Liu, J. Ren, X. Gong, Y. Wang, Y. Guo, Y. Guo and Z. Zhang, CN102850157A, 2013 (East China University of Science and Technology).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01520g |
| This journal is © The Royal Society of Chemistry 2015 |