
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Renewable polycarbonates and polyesters from 1,4-cyclohexadiene†
Matthias
Winkler
ab,
Charles
Romain
b,
Michael A. R.
Meier
*a and
Charlotte K.
Williams
*b
aLaboratory of Applied Chemistry, Institute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: m.a.r.meier@kit.edu; Web: http://www.meier-michael.com
bDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: c.k.williams@imperial.ac.uk
First published on 12th September 2014
Abstract
Epoxides derived from 1,4-cyclohexadiene (CHD), the latter produced from renewable resources via self-metathesis of plant oil derivatives, are applied as key substrates in ring-opening copolymerizations to produce aliphatic polycarbonates and polyesters. Renewable, unsaturated polycarbonates are prepared by the ring-opening copolymerization of epoxide/CO2; these are catalysed by di-zinc/magnesium complexes previously reported by Williams et al. or by using chromium(III) or cobalt(III) salen complexes. Renewable, unsaturated polyesters, with glass transition temperatures up to 128 °C, were obtained by the ring-opening copolymerization of epoxide/phthalic anhydride. The relative rates of these copolymerizations were monitored using in situ attenuated total reflectance infra-red (ATR-IR) spectroscopy. The polymers were fully characterized using spectroscopy (nuclear magnetic resonance, infra-red), mass spectrometry (matrix assisted laser desorption ionization), and by thermal methods (differential scanning calorimetry and thermogravimetric analysis).
Introduction
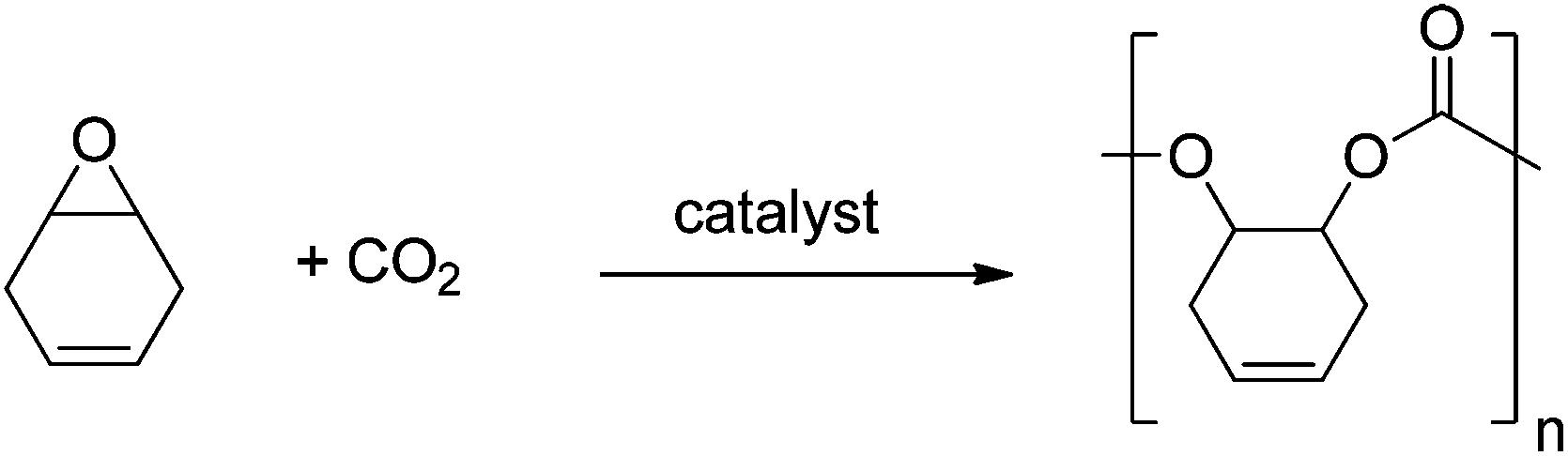
Polycarbonates (PC) are important commodity materials, widely applied in electronics, construction, and as rigid plastics. The most common PC is produced from bisphenol A, which benefits from a high glass transition temperature (149 °C) and good mechanical properties.1 There are, however, concerns associated with the toxicity of reagents, including the possible endocrine disruptor pathways attributed to BPA. Moreover, such classic PCs are produced via polycondensation using phosgene as a co-monomer. This has motivated the quest for alternative materials.Aliphatic polycarbonates, produced by the metal catalysed alternating ring-opening copolymerization (ROCOP) of CO2/epoxides (Fig. 1), are attracting considerable attention as semi-renewable polymers.2 The use of carbon dioxide as a monomer (or generally reagent) is attractive, because it is inexpensive, non-toxic, abundant, renewable and a common waste product of many industrial processes. Although the properties of these materials do not yet match those of PC from bisphenol A, recent research highlights their potential with polymers being produced by this method which show Tg up to 140 °C.3 Another promising application for such polycarbonates is as low molecular weight polyols (hydroxyl terminated polymers), mainly to be used for the production of polyurethanes. Indeed, poly(ethercarbonate) polyols, produced by ROCOP, have been successfully applied to prepare polyurethane foams;4 an in depth life cycle analysis, published this year, found that these materials benefited from reductions in energy use and greenhouse gas emissions of 10–20% compared to conventional polyols.5
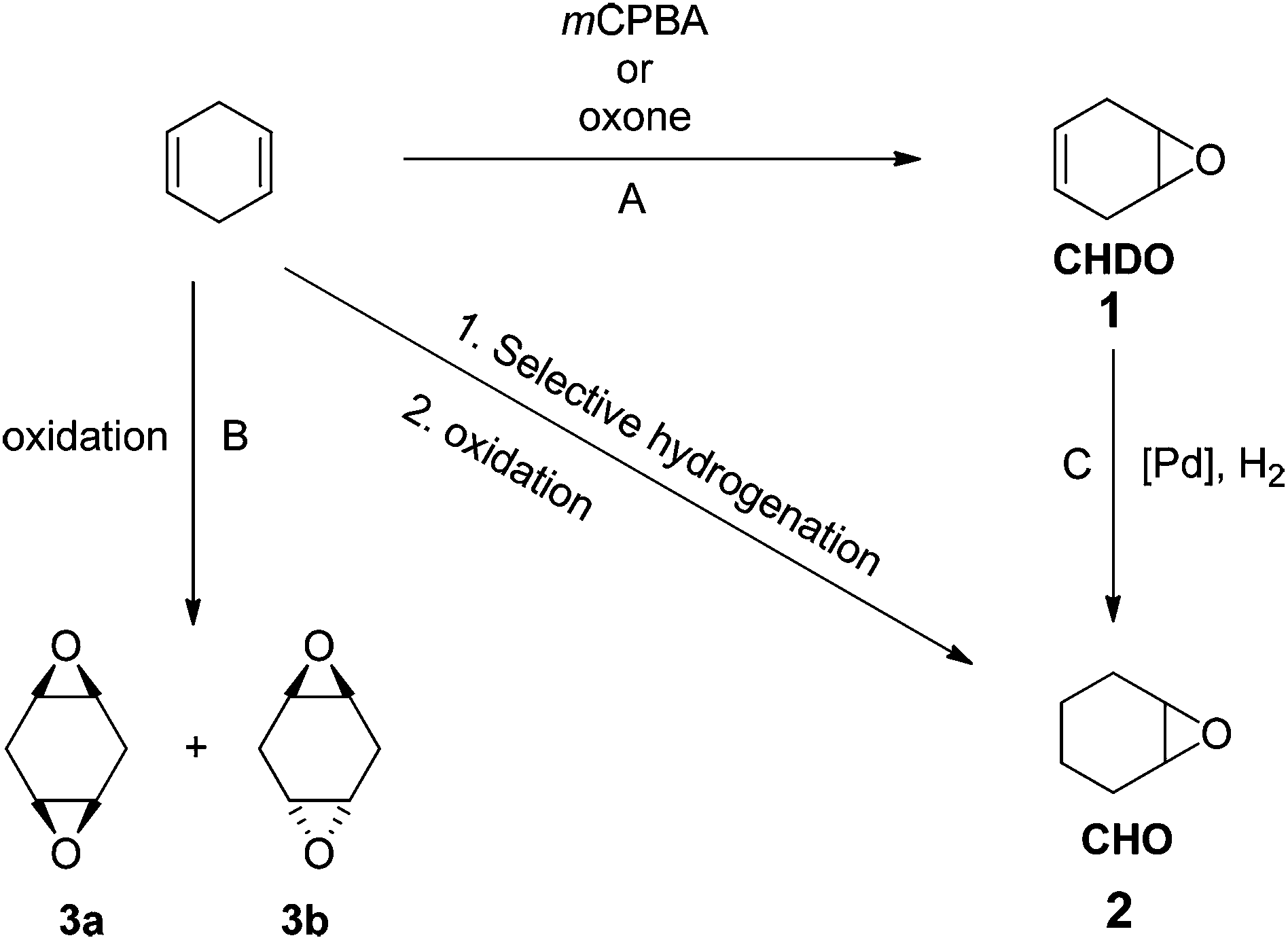 | ||
| Fig. 1 Synthesis of 1,4-cyclohexadiene oxide (CHDO), cyclohexene oxide (CHO), and two bis-epoxides (syn-1,4-cyclohexadiene diepoxide (3a), anti-1,4-cyclohexadiene diepoxide (3b)) from 1,4-cyclohexadiene (CHD). (A) Oxone, DCM/acetone/H2O/NaHCO3, 298 K, 6 h or mCPBA/DCM/H2O/NaHCO3, 298 K, 6 h, (B) same as for A except the slight excess of mCPBA or Oxone used per double bond, (C) Pd/C(en), 1 wt%, H2, 298 K, 24 h. | ||
Considering the sources for the ROCOP monomers, several authors have commented on the ability to use purified carbon dioxide, which might be obtained by carbon capture and sequestration, to prepare the polyols.4–6 Others have reported catalysts that tolerate high levels of water, a common contaminant in captured carbon dioxide.7 In terms of epoxides, the majority of studies apply cyclohexene oxide (CHO) or propylene oxide (PO),2b,d,8 both of which are usually derived by oxidation of petrochemicals. There remains just a single report of the application of a renewable epoxide, limonene oxide, to produce a fully renewable polycarbonate.9 It is worth also mentioning that even since 1970, many groups have investigated protected glycidyl ethers, which could, in principle, be derived from glycerol.10
Epoxides are also applied in ROCOP with anhydrides, utilizing similar catalysts as for epoxide/CO2 ROCOP.11 This allows the production of polyesters and, in particular, is an attractive controlled polymerization route to semi-aromatic polyesters with improved thermal properties (i.e. higher glass transition temperatures).11f Various authors have reported the ROCOP using anhydrides from renewable resources, including maleic or succinic anhydride.11b,d,f,g,12 Furthermore, phthalic anhydride, which is particularly attractive as it yields thermally resistant aromatic moieties in the polymer backbone, can be produced from carbohydrate derived biomass.13 In terms of epoxides, so far the only renewable options reported for this ROCOP are limonene oxide and pinene oxide.11g,12a
Thus, there is an impetus to consider routes to prepare epoxides from renewable resources as this will allow the production of 100% renewable polyesters/carbonates and is furthermore expected to expand the property profiles for this class of material. In this context, plant oils are an interesting feedstock, as they are produced on sufficient scale and at relatively low cost. Already today, they are used as biofuels and as a source of renewable monomers.14 A range of transformations of such triglycerides and fatty acids, including polymerizations, have already been reported.15 Of these methods, olefin metathesis is particularly interesting, as it can be used to produce long-chain carboxylic acids/esters.16 These are interesting monomers to produce materials with properties approaching some of those of polyethylene.17Inter alia, during oleochemical olefin metathesis reactions, a common by-product is 1,4-cyclohexadiene (CHD), if polyunsaturated fatty acids (i.e. linoleic and/or linolenic acid) are present in the fatty acid mixture.18 For instance, the self-metathesis of polyunsaturated fatty acid methyl esters was used to prepare long-chain diesters, which are important AA-type monomers.17b,19 In these metathesis processes, CHD is obtained as a waste by-product, as recently reported by Meier et al.20 Cole-Hamilton and co-workers reported the metathesis of cardanol and ethylene, also yielding CHD as a by-product,21 offering an alternative route to renewable CHD. In this context, the valorisation of CHD would be particularly beneficial to the biorefinery concept, where all products of a feedstock should be utilized. Recently, Meier et al. described the synthesis of substituted caprolactones and their polymers, prepared via the modification of cyclohex-2-en-1-one, which was obtained from CHD.22 Moreover, 1,3-cyclohexadiene, obtained via isomerization of CHD, can directly be used in polymerization reactions involving anionic, cationic, and free-radical mechanisms.23 The poly(cyclohexadiene)s are of interest, since these polymers display good properties and can be transformed into conducting polymers and proton conductors.1,24 Mathers and co-workers presented a very attractive strategy using 1,4-CHD, obtained from un-purified plant oils, to prepare poly(cyclohexadiene)s via a one- or two-step isomerization polymerization cascade.25 Here, the strategy to valorise CHD is to investigate its transformation into epoxides and the subsequent ROCOP reactions using these bio-derived epoxides.
Results and discussion
Epoxides from 1,4-cyclohexadiene
Epoxides are interesting and important monomers to prepare polycarbonates or polyesters by ROCOP methods. Given the widespread application of cyclohexene oxide (CHO) in ROCOP polymerizations, CHD should be a promising substrate to produce both new and known materials that are fully renewable. We observed that CHD can be converted into various epoxides, including 4,5-epoxycyclohex-1-ene (cyclohexadiene oxide 1 = CHDO), cyclohexene oxide (CHO, 2) and two diastereomeric bis-epoxides (3a, 3b) using the reagents and conditions shown in Fig. 1.The oxidation of CHD was accomplished using meta-chloroperbenzoic acid (mCPBA),26 in dichloromethane at 298 K, to yield monomer 1 in 82% yield. Alternatively, a more sustainable oxidation of CHD was successfully performed using Oxone® leading to the production of 1 in 65% yield. Interestingly, cyclohexene oxide (CHO) 2 was also prepared from CHD by a selective bi-phasic hydrogenation leading to cyclohexene, and its subsequent oxidation.27 For the hydrogenation reaction using 1, the use of Pd/C under 1 atm of H2 was unsuccessful, but the use of Pd/C(en),28 in THF under 1 atm of H2 for 24 h, enabled formation of CHO, together with the formation of side-products such as, among others, the corresponding vicinal diols (as determined by 1H NMR). Thus, this route allows access to CHO from renewable resources as an alternative to the more usual routes to petrochemicals (from benzene).
Finally, in order to explore the range of epoxides accessible from CHD, a complete oxidation of 1 to the bis-epoxides 3a and 3b was investigated. Thus, the oxidation of 1 was performed using a slight excess of mCPBA (1.05 equivalents per double bond) at 298 K in dichloromethane to yield the corresponding bis-epoxides (3a (20%), 3b (65%)). Alternatively, Oxone can also be used as the oxidant leading to variable proportions of the bis-epoxides (depending on the quantity of Oxone® applied, for more details see ESI†).
ROCOP using bio-derived epoxides
The bio-derived epoxides were applied in ROCOP reactions, using either CO2 or phthalic anhydride as a co-monomer. These polymerizations all require catalysts; four different homogeneous catalysts with a good track record in these fields were compared (the structures of which are illustrated in Fig. 2 and 3).7a,11c,12b,29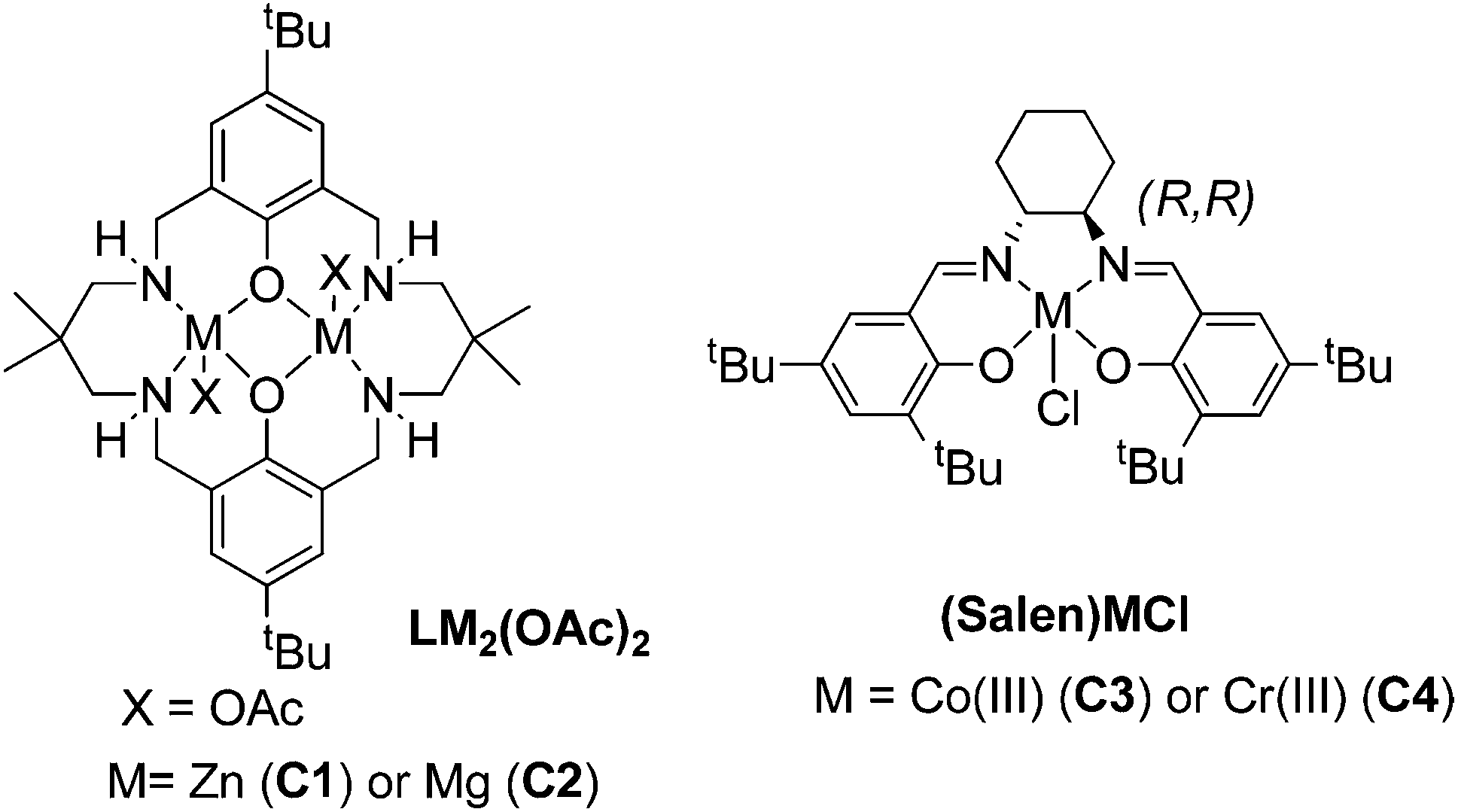 | ||
| Fig. 2 Structures of the four homogeneous catalysts investigated for ROCOP (for catalyst synthesis methods, see the ESI†).7a,11c,12b,29 | ||
 | ||
| Fig. 3 ROCOP of monomer 1 utilizing catalysts C1–C3. Catalyst C3 was applied in combination with a co-catalyst (PPN-Cl). | ||
ROCOP using monomer 1, catalysed by the di-zinc complex C1, did not result in polymer formation, even under the conditions previously successfully used for the ROCOP of CHO (80 °C, atmospheric CO2 pressure). Increasing the temperature to 100 °C led to some formation of the cyclic carbonate, but no polymer. These results prompted us to investigate the magnesium analogue C2, which had previously been found to be both more active and more selective than C1.7a Thus, ROCOP using 1, catalysed by C2 (0.2 mol%) under atmospheric CO2 pressure, afforded well-defined, perfectly alternating polycarbonates (i.e. no formation of ether linkages or cyclic carbonate was observed (see Fig. S3†), albeit with considerably lower activity than the analogous copolymerizations using 2.7a The 1H NMR spectrum of the crude product (Fig. S2†) was used to determine the monomer conversion; specifically this was achieved by comparison of the relative, normalized integrals for the cyclohexyl resonances (3.24 ppm 1, 4.96 ppm polymer) and for the olefin signals (5.43 ppm 1, 5.57 ppm polymer). In both cases, the same conversions were obtained, suggesting that the double bond was unreactive and that there was no formation of cross-linked polymers. In addition, monitoring the reaction by in situ ATR-IR spectroscopy using 1 showed linear formation of the polymer with time, in accordance with results previously obtained with C1 for CHO/CO2 copolymerization (see Fig. S4†).7b Gel permeation chromatography (GPC) analysis of the polymers exhibited molecular weight distributions with a small shoulder to higher molecular weights (see Fig. S5†), similar to previous obtained ROCOP results using these types of catalysts.7b The structure of the polymers was confirmed by MALDI-ToF mass spectrometry. The MALDI-ToF spectrum (see ESI, Fig. S6†) exhibited two series of chains, corresponding to chains end-capped with α-acetate-ω-hydroxyl and α,ω-dihydroxyl moieties.
In order to further investigate the reactivity of 1 in ROCOP, a well-known CHO/CO2 catalyst, (1R,2R)SalcyCo(III)-Cl complex C3, was applied. Thus, the ROCOP using 1, catalysed by C3 (0.2 mol%) and with PPN-Cl (0.2 mol%) as co-catalyst, under 20 bar CO2 pressure, also afforded polycarbonate without formation of any cyclic carbonate or ether linkages, albeit once again with a lower turn-over frequency than the analogous polymerizations using 2.12b In addition, the 1H NMR spectra of both the crude and purified polymer showed that the double bond remains unreacted after polymerization, allowing access to unsaturated CO2-derived polycarbonates. The polymers prepared using the cobalt catalyst showed slightly higher molecular weights (12.9 kg mol−1), bimodal molecular weight distributions and narrow dispersities (1.18) (see Fig. S7†). Furthermore, ROCOP occurred in a controlled manner, as evidenced by a linear correlation between the monomer conversion and the polymer molecular weight (see ESI, Fig. S8†).
Thus, applying a range of different copolymerization catalysts (C2, C3) enabled the successful copolymerization of 1/CO2, although the rates were lower than the analogous polymerizations run using cyclohexene oxide 2/CO2.7a,12b,29 This likely relates to different metal binding energies of the two epoxides.
Terpolymerizations of 1/CHO/CO2 were also investigated (Fig. 4); the incorporation of 1 was attractive as a means to introduce unsaturation into the polymer backbone, allowing for further post-polymerization modifications.30 Different catalysts and ratios of monomers 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were investigated (see Table 1). The relative monomer conversions were determined by 1H NMR spectroscopy by integrating the signals of the cyclohexyl moiety in the polymer (4.96 ppm for incorporated 1, 4.66 ppm for incorporated 2) and monomer (3.24 ppm 1, 3.11 ppm 2), respectively. Once again, the terpolymerizations using di-magnesium C2 are significantly slower than those with the cobalt salen complex C3. GPC analysis revealed polycarbonates with moderate molecular weights of (4–11.5 kg mol−1) and narrow dispersities (<1.15).
:2 were investigated (see Table 1). The relative monomer conversions were determined by 1H NMR spectroscopy by integrating the signals of the cyclohexyl moiety in the polymer (4.96 ppm for incorporated 1, 4.66 ppm for incorporated 2) and monomer (3.24 ppm 1, 3.11 ppm 2), respectively. Once again, the terpolymerizations using di-magnesium C2 are significantly slower than those with the cobalt salen complex C3. GPC analysis revealed polycarbonates with moderate molecular weights of (4–11.5 kg mol−1) and narrow dispersities (<1.15).
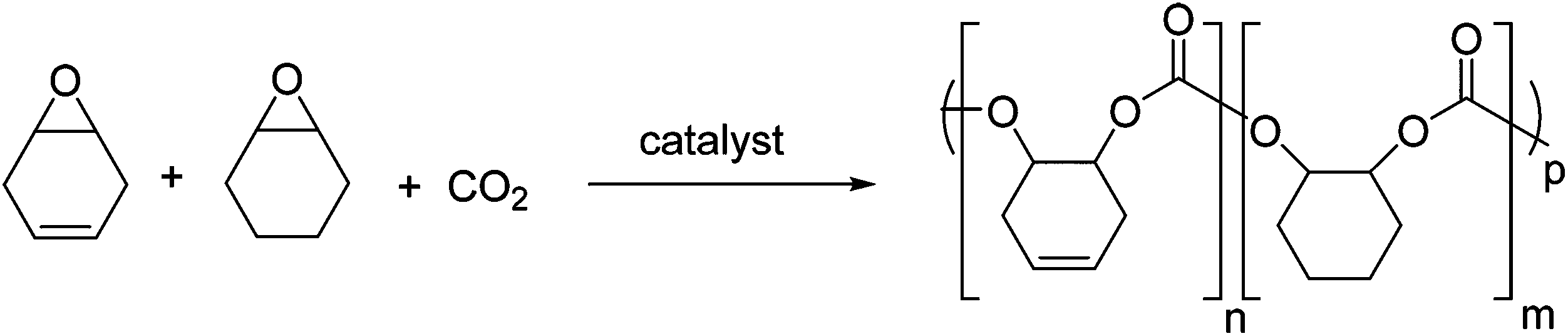 | ||
| Fig. 4 The terpolymerizations of 1, CHO and CO2 so as to produce partially unsaturated polycarbonates. | ||
| Polymer | Catalyst | Co-Cat. | CO2 [bar] | [M]:[Cat.] |
T [°C] | Conv.d [%] |
TOF [h−1] |
M
ne [kg mol−1] |
Đ |
|---|---|---|---|---|---|---|---|---|---|
|
a The only product of this reaction was cyclic carbonate.
b Mixtures of monomers 1 and 2 were applied such that the ratio of 1:2 was 4:1 and the conversion is given for the respective monomer.
c Mixtures of monomers 1 and 2 were applied such that the ratio of 1:2 was 1:1 and the conversion is given for the respective monomer.
d Conversion was determined by 1H-NMR spectrum of the crude product by comparison of the relative normalized integrals for the cyclohexyl resonances (3.24 ppm 1, 4.96 ppm polymer) and for the olefin signals (5.43 ppm 1 and 5.57 ppm polymer).
e Molecular weights and polymer dispersities were determined by GPC calibrated with polystyrene standards.
|
|||||||||
| P1 | C1 | — | 1 | 500:1 |
80 | 0 | — | — | — |
| P2 | C1 | — | 1 | 500:1 |
100 | 12 | 4 | — | — |
| P3 | C2 | — | 1 | 500:1 |
80 | 25 | 5 | 2.9 | 1.17 |
| P4 | C2 | — | 1 | 250:1 |
80 | 20 | 3 | 2.5 | 1.08 |
| P5 | C2 | — | 1 | 500:1 |
100 | 21 | 6 | 2.8 | 1.15 |
| P6 | C3 | — | 20 | 200:1 |
20 | 15 | 1 | 5.3 | 1.43 |
| P7 | C3 | PPN-Cl | 20 | 500:1 |
28 | 78 | 65 | 12.9 | 1.18 |
| P8 | C2 | — | 1 | 500:1 |
80 | (11/56)b | 21 | 4.0 | 1.15 |
| P9 | C3 | PPN-Cl | 20 | 500:1 |
28 | (22/72)c | 118 | 11.5 | 1.12 |
Finally, the bis-epoxides 3a and 3b were selected to prepare branched or cross-linked polycarbonates, however, in all cases completely insoluble material was produced which limited further analysis but was indicative of the formation of cross-linked products.
Unsaturated polyesters by ROCOP
Monomer 1 was also used for the synthesis of unsaturated polyesters via ROCOP with phthalic anhydride (Fig. 5). An attractive feature of the products would be the facility to introduce unsaturation into the polymer backbone. It is notable, that many researchers have reported problems in copolymerizing maleic anhydride (a similarly unsaturated co-monomer) and thus novel routes to unsaturated polyesters are of interest.11c,12b | ||
| Fig. 5 ROCOP using epoxide 1 and phthalic anhydride (PA). | ||
Catalyst C1 did not afford any polymerization, whereas C2 enabled the slow formation of perfectly alternating polyesters. Similarly, catalysts C3 and C4 were effective and yielded unsaturated polyester. In each case, the monomer conversion was estimated using 1H NMR spectroscopy. In particular, the relative integrals were compared for the aromatic signals (for PA at 7.81–7.67 ppm and for polymer at 7.66–7.36 ppm, see Fig. S11,†Table 2). Utilizing the salen based catalysts yielded polyesters of moderate molecular weights (7.5 kg mol−1) and narrow dispersities (1.20) with TOF values up to 246 h−1. The chromium salen catalyst (C4) showed the best performance at a reaction temperature of 110 °C in presence of PPN-Cl as co-catalyst. Polymerizations in bulk were hampered by viscosity increases; however, using monomer solutions (2.5 M in toluene) led to conversions of 91% within 3 h. ROCOP occurred in a controlled manner, as evidenced by a linear correlation between the monomer conversion and the polymer molecular weight (see Fig. S13†). Interestingly, for both C3 and C4, the use of PPN-Cl as the co-catalyst was required to increase the activity. It is notable that compared to maleic anhydride/epoxide copolymerization, where the co-catalyst was purported to react with the olefin moiety,12b in this case the double bonds remained unreacted in the polymer.
| Polymer | Catalyst | [1]:[PA]:[Cat.] |
T [°C] | Conv.c [%] |
TOF |
M
nd [kg mol−1] |
Đ |
|---|---|---|---|---|---|---|---|
| a Reaction was performed without co-catalyst. b Reaction was conducted in toluene (2.5 M). c Conversion was determined from the 1H-NMR spectrum of the crude product by comparison of the relative, normalized integrals for the cyclohexyl resonances (4.96 ppm polymer, 4.64 ppm polymer, 3.24 ppm 1, 3.11 ppm 2). d Molecular weights and polymer dispersities were determined by GPC calibrated with polystyrene standards. | |||||||
| P10 | C2 | 250:250:1 |
120 | 10 | 6.25 | 1.6 | 1.18 |
| P11 | C3 | 250:250:1 |
110 | 48 | 60 | 4.3 | 1.23 |
| P12 | C4 | 250:250:1 |
110 | 74 | 246 | 4.5 | 1.34 |
| P13 | C4 | 250:250:1 |
110 | 91 | 75.8 | 7.5 | 1.17 |
Thermal properties of polyesters and polycarbonates
The thermal properties of the unsaturated polycarbonates were studied by TGA and DSC analysis (Table 3). The polycarbonates prepared from 1 showed glass-transition temperatures of approximately 115 °C (depending on the molecular weight), a value close to that reported for PCHC (polycyclohexene carbonate, the polymer produced by copolymerization of 2/CO2).1,31 The thermal degradation temperature (10% weight loss) of P7 was 255 °C, whereas that of P9 was higher at 285 °C. Both values are somewhat lower than that reported for poly(cyclohexene carbonate) (Td ∼ 300 °C32), suggesting that the olefin group slightly lowers the thermal stability. Terpolymers, especially P8 (Tg = 106 °C), showed slightly lower glass transition temperatures than PCHC or P3–P7 (Tg = 115 °C), which is mainly due to the low molecular weight of P8. The thermal properties of the polyesters derived from 2/phthalic anhydride showed better thermal properties, with glass transition temperature of 128 °C and a thermal decomposition temperature (10% weight loss) of 325 °C. It is noteworthy that polyesters derived from CHO/phthalic anhydride typically show a Tg below 100 °C.11e,29
Conclusions
The preparation of a series of epoxides from 1,4-cyclohexadiene, which is itself a by-product of the self-metathesis of triglycerides and other fatty acid derivatives, are presented. The diene was epoxidized, using various oxidants, to yield cyclohexadiene oxide or a bis-epoxide product. The diene could also be partially hydrogenated to cyclohexene and then oxidized to yield the well-known monomer cyclohexene oxide. The development of a bio-derived route to cyclohexene oxide is of interest, as the monomer is widely applied in various ring-opening copolymerization reactions, including epoxide/CO2 to produce polycarbonates or epoxide/anhydride to produce polyesters. Thus, the method presented here provides an alternative route to prepare cyclohexene oxide.Furthermore, the preparation of the bio-derived, unsaturated epoxide is also of high interest as it enables the preparation of partially unsaturated polycarbonates and polyesters. For the co-polymerization with CO2 and cyclohexadiene oxide, the readily available cobalt salen complex provided the best results and yielded a polycarbonate with closely related thermal properties to the well-known poly(cyclohexene carbonate). Terpolymerizations of CO2, cyclohexene oxide, and cyclohexadiene oxide were also successful yielding polycarbonates with controllable quantities of unsaturation. Copolymerization of cyclohexadiene oxide and phthalic anhydride leads to renewable, unsaturated polyesters having high glass-transition and decomposition temperatures. These unsaturated polycarbonates and polyesters offer the possibility for further modification of the double bond, which would be expected to alter the polymer properties. Future research will focus on further development of the synthesis of epoxides from biomass as well as the post-polymerization functionalization and application of such unsaturated polyesters and polycarbonates.
Acknowledgements
MW acknowledges the DAAD for financial support. CKW and CR acknowledge the EPSRC for financial support (EP/L017393/1, EP/K014070/1).References
- C. Koning, J. Wildeson, R. Parton, B. Plum, P. Steeman and D. J. Darensbourg, Polymer, 2001, 42, 3995–4004 CrossRef CAS.
- (a) K. Nozaki, Pure Appl. Chem., 2004, 76, 541–546 CrossRef CAS; (b) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed; (c) D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC; (d) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (e) X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed.
- (a) D. J. Darensbourg and S. J. Wilson, Macromolecules, 2013, 46, 5929–5934 CrossRef CAS; (b) Y. Liu, W.-M. Ren, J. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2013, 52, 11594–11598 CrossRef CAS PubMed; (c) Y. Liu, M. Wang, W.-M. Ren, K.-K. He, Y.-C. Xu, J. Liu and X.-B. Lu, Macromolecules, 2014, 47, 1269–1276 CrossRef CAS; (d) D. J. Darensbourg and S. J. Wilson, J. Am. Chem. Soc., 2011, 133, 18610–18613 CrossRef CAS PubMed.
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- D. J. Darensbourg, W.-C. Chung, K. Wang and H.-C. Zhou, ACS Catal., 2014, 4, 1511–1515 CrossRef CAS.
- (a) M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed; (b) F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed; (c) A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS; (d) M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196–1201 RSC; (e) C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed; (f) P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 4164–4167 RSC; (g) G.-P. Wu, D. J. Darensbourg and X.-B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS PubMed; (h) D. J. Darensbourg and G.-P. Wu, Angew. Chem., Int. Ed., 2013, 52, 10602–10606 CrossRef CAS PubMed.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- (a) H. Koinuma, S. Inoue and T. Tsuruta, Makromol. Chem., 1970, 136, 65–71 CrossRef CAS; (b) S. Inoue, J. Macromol. Sci., 1979, 13, 651–664 CrossRef; (c) W. Kuran and A. Niestochowski, Polym. Bull., 1980, 2, 411–416 CrossRef CAS; (d) J. Łukaszczyk, K. Jaszcz, W. Kuran and T. Listos, Macromol. Rapid Commun., 2000, 21, 754–757 CrossRef; (e) J. Łukaszczyk, K. Jaszcz, W. Kuran and T. Listoś, Macromol. Biosci., 2001, 1, 282–289 CrossRef; (f) J. Geschwind and H. Frey, Macromolecules, 2013, 46, 3280–3287 CrossRef CAS; (g) J. Geschwind and H. Frey, Macromol. Rapid Commun., 2013, 34, 150–155 CrossRef CAS PubMed; (h) J. Geschwind, F. Wurm and H. Frey, Macromol. Chem. Phys., 2013, 214, 892–901 CrossRef CAS; (i) J. Hilf and H. Frey, Macromol. Rapid Commun., 2013, 34, 1395–1400 CrossRef CAS PubMed; (j) H. Zhang and M. W. Grinstaff, J. Am. Chem. Soc., 2013, 135, 6806–6809 CrossRef CAS PubMed; (k) J. Hilf, A. Phillips and H. Frey, Polym. Chem., 2014, 5, 814–818 RSC; (l) J. Hilf, P. Schulze, J. Seiwert and H. Frey, Macromol. Rapid Commun., 2014, 35, 198–203 CrossRef CAS PubMed; (m) H. Zhang and M. W. Grinstaff, J. Appl. Polym. Sci., 2014, 131 DOI:10.1002/app.39893; (n) D. J. Darensbourg and A. D. Yeung, Green Chem., 2014, 16, 247–252 RSC.
- (a) T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef CAS; (b) R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed; (c) A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed; (d) S. Huijser, E. HosseiniNejad, R. Sablong, C. d. Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS; (e) D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS; (f) E. H. Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC; (g) E. H. Nejad, A. Paoniasari, C. G. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- (a) C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2, 586 CrossRef PubMed; (b) E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef; (c) J. Liu, Y.-Y. Bao, Y. Liu, W.-M. Ren and X.-B. Lu, Polym. Chem., 2013, 4, 1439–1444 RSC.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167–175 RSC.
- (a) M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC; (b) J. O. Metzger, Eur. J. Lipid Sci. Technol., 2009, 111, 865–876 CrossRef CAS; (c) S. Lestari, P. Mäki-Arvela, J. Beltramini, G. Q. M. Lu and D. Y. Murzin, ChemSusChem, 2009, 2, 1109–1119 CrossRef CAS PubMed.
- (a) D. Quinzler and S. Mecking, Angew. Chem., Int. Ed., 2010, 49, 4306–4308 CrossRef CAS PubMed; (b) U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- S. Chikkali and S. Mecking, Angew. Chem., Int. Ed., 2012, 51, 5802–5808 CrossRef CAS PubMed.
- (a) D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2010, 49, 8564–8566 CrossRef CAS PubMed; (b) J. Trzaskowski, D. Quinzler, C. Bährle and S. Mecking, Macromol. Rapid Commun., 2011, 32, 1352–1356 CrossRef CAS PubMed; (c) I. van der Meulen, E. Gubbels, S. Huijser, R. l. Sablong, C. E. Koning, A. Heise and R. Duchateau, Macromolecules, 2011, 44, 4301–4305 CrossRef CAS; (d) F. Stempfle, D. Quinzler, I. Heckler and S. Mecking, Macromolecules, 2011, 44, 4159–4166 CrossRef CAS; (e) N. Kolb, M. Winkler, C. Syldatk and M. A. R. Meier, Eur. Polym. J., 2014, 51, 159–166 CrossRef CAS PubMed.
- R. T. Mathers, U.S. Provisional Application no. 61/654,589, WO2013180837 A1, 2013.
- P. Ortmann and S. Mecking, Macromolecules, 2013, 46, 7213–7218 CrossRef CAS.
- H. Mutlu, Hofsa, R. E. Montenegro and M. A. R. Meier, RSC Adv., 2013, 3, 4927–4934 RSC.
- J. A. Mmongoyo, Q. A. Mgani, S. J. M. Mdachi, P. J. Pogorzelec and D. J. Cole-Hamilton, Eur. J. Lipid Sci. Technol., 2012, 114, 1183–1192 CrossRef CAS.
- M. Winkler, Y. S. Raupp, L. A. M. Köhl, H. E. Wagner and M. A. R. Meier, Macromolecules, 2014, 47, 2842–2846 CrossRef CAS.
- (a) D. T. Williamson, J. F. Elman, P. H. Madison, A. J. Pasquale and T. E. Long, Macromolecules, 2001, 34, 2108–2114 CrossRef CAS; (b) X. Li, J. Baldamus, M. Nishiura, O. Tardif and Z. Hou, Angew. Chem., Int. Ed., 2006, 45, 8184–8188 CrossRef CAS PubMed.
- (a) D. G. H. Ballard, A. Courtis, I. M. Shirley and S. C. Taylor, Macromolecules, 1988, 21, 294–304 CrossRef CAS; (b) I. Natori, K. Imaizumi, H. Yamagishi and M. Kazunori, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 1657–1668 CrossRef CAS; (c) T. Huang, H. Zhou, K. Hong, J. M. Simonson and J. W. Mays, Macromol. Chem. Phys., 2008, 209, 308–314 CrossRef CAS.
- R. T. Mathers, M. J. Shreve, E. Meyler, K. Damodaran, D. F. Iwig and D. J. Kelley, Macromol. Rapid Commun., 2011, 32, 1338–1342 CrossRef CAS PubMed.
- (a) Q. Tan and M. Hayashi, Org. Lett., 2009, 11, 3314–3317 CrossRef CAS PubMed; (b) Y. N. Belokon, D. Chusov, A. S. Peregudov, L. V. Yashkina, G. I. Timofeeva, V. I. Maleev, M. North and H. B. Kagan, Adv. Synth. Catal., 2009, 351, 3157–3167 CrossRef CAS.
- (a) J. Dupont, P. A. Z. Suarez, A. P. Umpierre and R. F. de Souza, J. Braz. Chem. Soc., 2000, 11, 293–297 CrossRef CAS PubMed; (b) J.-Q. Yu and E. J. Corey, Org. Lett., 2002, 4, 2727–2730 CrossRef CAS PubMed; (c) E. V. Starokon, K. A. Dubkov, D. E. Babushkin, V. N. Parmon and G. I. Panov, Adv. Synth. Catal., 2004, 346, 268–274 CrossRef CAS; (d) X.-M. Lv, L.-J. Kong, Q. Lin, X.-F. Liu, Y.-M. Zhou and Y. Jia, Synth. Commun., 2011, 41, 3215–3222 CrossRef CAS.
- H. Sajiki, K. Hattori and K. Hirota, Chem. – Eur. J., 2000, 6, 2200–2204 CrossRef CAS.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014 10.1039/c1034py00748d.
- A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11350–11351 CrossRef CAS PubMed.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 1999, 33, 303–308 CrossRef.
- (a) L. Jianxin, Z. Min, Z. Zhiqiang, L. Baohua and C. Liban, e-Polymers, 2009, 9, 1420–1432 CrossRef; (b) S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01353k |
| This journal is © The Royal Society of Chemistry 2015 |