
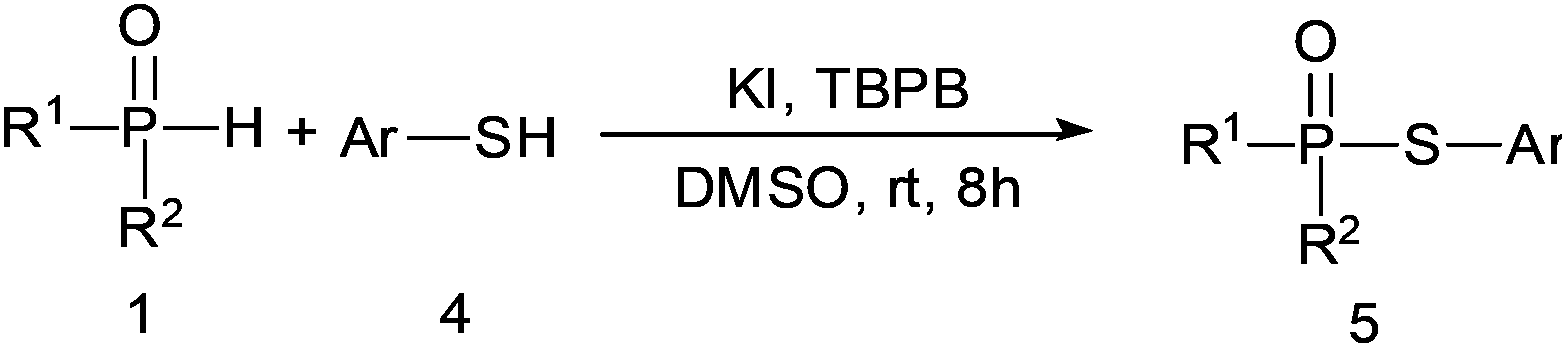
TBPB-promoted metal-free synthesis of thiophosphinate/phosphonothioate by direct P–S bond coupling†
Jichao
Wang
,
Xin
Huang
,
Zhangqin
Ni
,
Sichang
Wang
,
Jun
Wu
and
Yuanjiang
Pan
*
Department of Chemistry, Zhejiang University, Hangzhou, Zhejiang, China. E-mail: cheyjpan@zju.edu.cn
First published on 28th August 2014
Abstract
An efficient method for the direct coupling of thiol/thiophenol with H-phosphine oxides or H-phosphinate esters is reported. Without using any metallic catalyst, the direct sulfur–phosphorus bond coupling reaction was promoted using tert-butyl peroxybenzoate in the presence of KI at room temperature. Consequently, thiophosphinate/phosphonothioate was produced in moderate to excellent yields.
Introduction
Sulfur-containing organophosphorus compounds have received considerable attention for more than 60 years because of their promising bioactivities1 and pest-control application.2 Accordingly, research on the synthesis of these compounds is extensive, especially for the thioesters of dialkyl (or diaryl) phosphites,3 which could be found in organophosphorus pesticides (Fig. 1), such as demeton-S-methyl (1) and iprobenfos (2). Yokomatsu et al. recently reported a CuI-catalyzed synthesis method of thiophosphates through the coupling of diethyl phosphites and benzenethiols.3j Lee et al. developed an NCS promoted thiophosphation reaction between thiols and phosphites.3k The synthesis of thiophosphates based on dialkyl (or diaryl) phosphites is extensively studied, but less attention is paid to the thioester of H-phosphine oxides or H-phosphinate esters,4 which are core structures of certain pesticides, such as inezin (3). These compounds could also be used as phosphoryl transfer reagents in organic syntheses.5 Traditional synthesis work of thiophosphinate often proceeds via the reaction between phosphinic chlorides and thiols [Scheme 1(a)].5a,b,6 Haynes et al. [Scheme 1(b)] reported a synthesis strategy through the coupling of the H-phosphine oxides with thiophenols in the presence of NBS and a base.4c Mioskowski et al. [Scheme 1(c)] developed phosphazene P4-tBu-promoted phosphonothioate formation using alkyl thiocyanates as the thiol source.4d In their work, both H-phosphine oxides and H-phosphinate esters smoothly afford the products in good yields. Yamaguchi et al. [Scheme 1(d)] reported a thiophosphinate synthesis protocol by a rhodium-catalyzed coupling between tetraphenyldiphosphine dioxides and disulfides.4e However, extensive application of this protocol is limited by the use of toxic phosphinic chlorides, strong bases or Lewis acids, complex multiple steps, and expensive transition metals.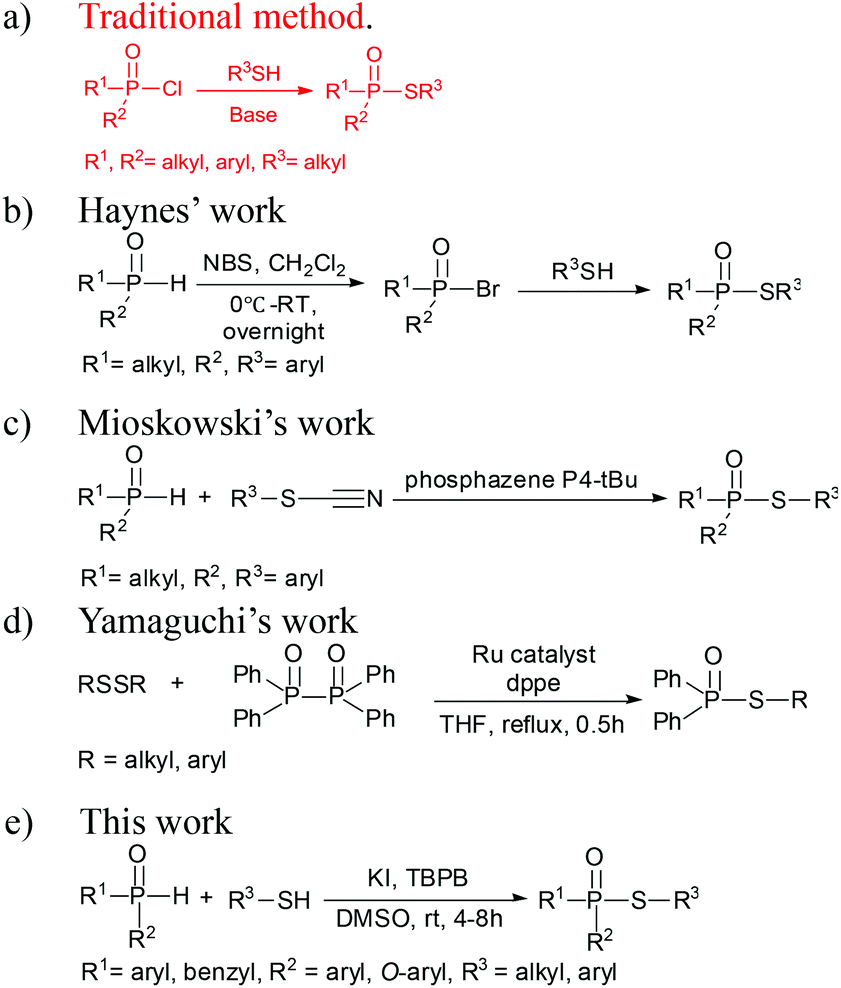 | ||
| Scheme 1 Synthesis of thiophosphates. | ||
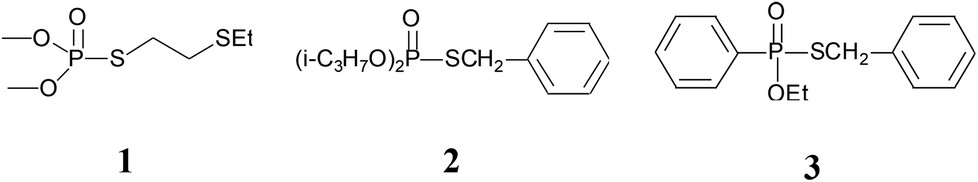 | ||
| Fig. 1 Thiophosphate structures in pesticides. | ||
From the perspective of atom economy, direct coupling between H-phosphine oxides/H-phosphinate esters and thiols is the best option to form thiophosphinates or phosphonothioates. A similar strategy involves C–S bond construction for thioester synthesis.7 Based on our previous work on C–S and S–N bond coupling,8 we present in this work a thiophosphinate/phosphonothioate synthesis method through direct P–S bond coupling using peroxide (TBPB). As a green synthetic method, our work exhibited several advantages, including good atom economy (more than 99%), easy one-step work-up, without any acid or base, avoids the use of any transition metal, relative mild reaction conditions, and most substrates could react at room temperature while with good to excellent yields (up to 99%). Besides, the method has a wide range of functional group compatibilities for H-phosphine oxides/H-phosphinate esters and thiols.
Results and discussion
In our initial work, diphenylphosphine oxide and cyclohexanethiol were used as model reactants to optimize the reaction conditions. First, several oxidants (e.g. peroxides and potassium persulfate) were introduced to the reaction (Table 1, entries 1–5); however, no relative product was detected. Based on Prabhu's work,9 we then added iodine to the reaction and elevated the temperature to 50 °C (Table 1, entries 6–10), and the desired product 3a was thus formed. Screening of the oxidants showed that TBPB had the best reactivity (Table 1, entry 10), although the yield was still unsatisfactory. The poor yield might be attributed to iodine catalyzed disulfide formation.10 We replaced iodine with a less reactive potassium salt (KI) and finally obtained thiophosphinate 3a in nearly quantitative yield at room temperature (Table 1, entry 11). Notably, the decrease in KI loading and oxidant did not affect this result (Table 1, entries 12 and 13). But an increase of the additive amount and temperature results in a reduced yield, which is due to the oxidation of 1a into diphenylphosphinic acid (Table 1, entries 14 and 15). Other iodine sources such as NaI and TBAI were tested, both gave acceptable yields (Table 1, entries 16 and 17). We also studied the effect of solvents, including toluene, CH3CN and water (Table 1, entries 18–20). However, both showed poor yields, and this reaction was ceased when reacted in water.|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Additive | Solvent | Yieldb (%) |
| a Reaction conditions: diphenylphosphine oxide (0.75 mmol), cyclohexanethiol (0.5 mmol), and oxidant (1.0 mmol) were reacted at room temperature for 4 h. b Isolated yield. c Reacted at 50 °C. d 0.5 mmol TBPB was added. (TPHP = tert-Butyl hydroperoxide, DTBP = tert-butyl peroxide, TBPB = tert-butylperoxybenzoate, TBAI = tetrabutylammonium iodide). | ||||
| 1 | H2O2 | None | DMSO | N.R. |
| 2 | TPHP | None | DMSO | N.R. |
| 3 | DTBP | None | DMSO | N.R. |
| 4 | K2S2O8 | None | DMSO | N.R. |
| 5 | TBPB | None | DMSO | N.R. |
| 6c | H2O2 | I2 (0.2 eq.) | DMSO | Trace |
| 7c | TPHP | I2 (0.2 eq.) | DMSO | Trace |
| 8c | K2S2O8 | I2 (0.2 eq.) | DMSO | 13 |
| 9c | DTBP | I2 (0.2 eq.) | DMSO | 10 |
| 10c | TBPB | I2 (0.2 eq.) | DMSO | 21 |
| 11 | TBPB | KI (0.2 eq.) | DMSO | 97 |
| 12 | TBPB | KI (0.1 eq.) | DMSO | 89 |
| 13d | TBPB | KI (0.2 eq.) | DMSO | 85 |
| 14 | TBPB | KI (0.4 eq.) | DMSO | 77 |
| 15c | TBPB | KI (0.2 eq.) | DMSO | 63 |
| 16 | TBPB | NaI (0.2 eq.) | DMSO | 79 |
| 17 | TBPB | TBAI (0.2 eq.) | DMSO | 62 |
| 18 | TBPB | KI (0.2 eq.) | Toluene | 28 |
| 19 | TBPB | KI (0.2 eq.) | CH3CN | 21 |
| 20 | TBPB | KI (0.2 eq.) | H2O | N.R. |
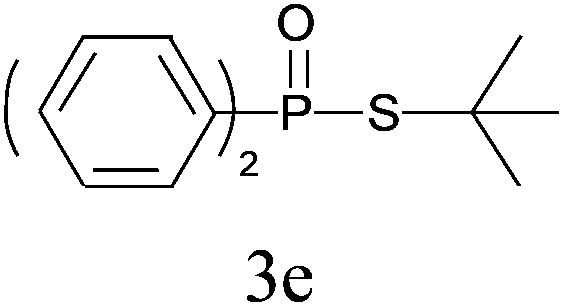
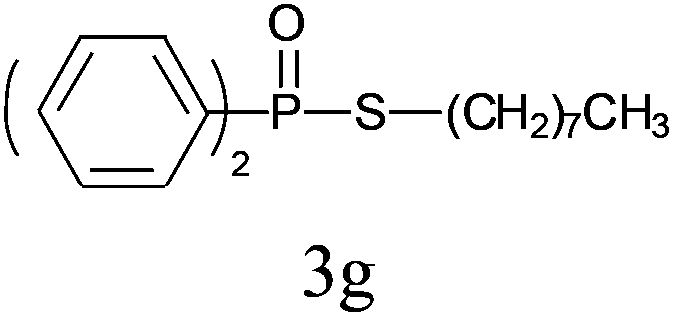
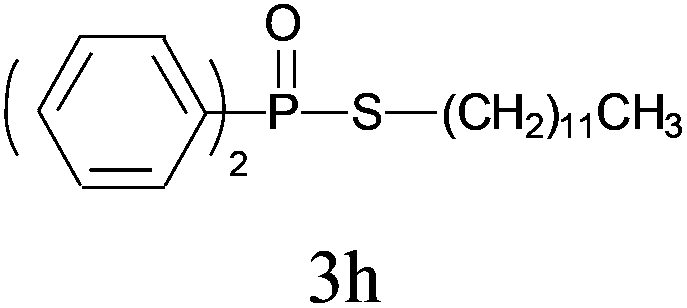
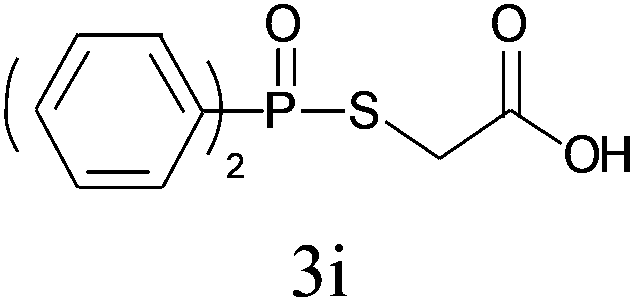
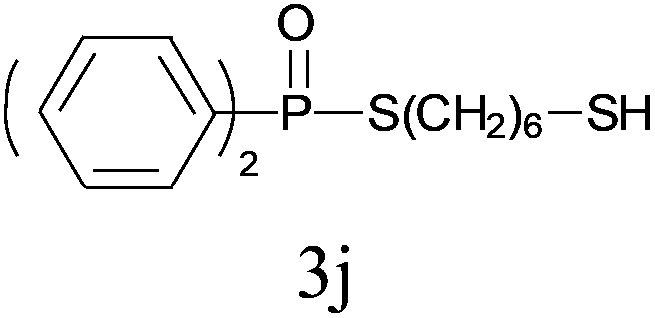
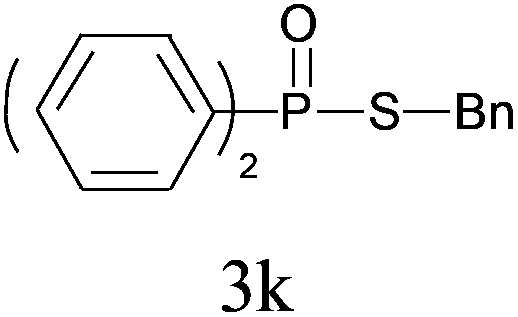
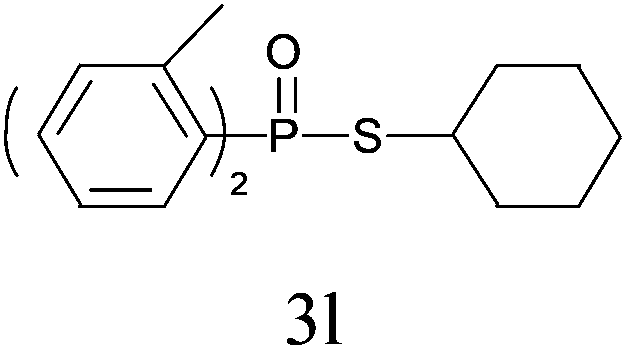
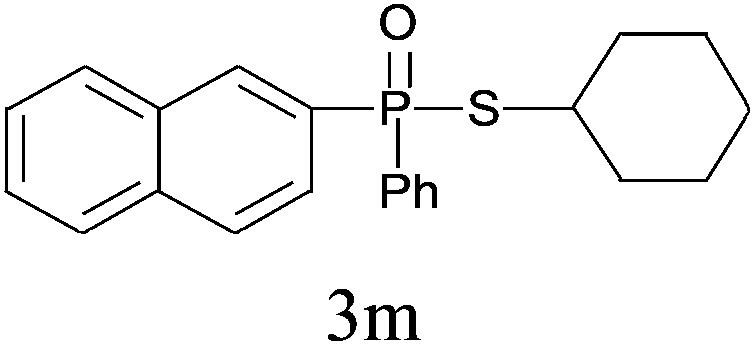
Under the optimized reaction conditions, we then investigated the scope and limitations of the substrate for application to our novel P–S bond coupling method. As summarized in Table 2, a variety of thiols with different chain lengths were readily reacted with H-phosphine oxides or H-phosphinate esters, giving the relative products in moderate to excellent yields. The long-chain thiols such as 1-hexylthiol (Table 2, entry 5), 1-octanethiol (Table 2, entry 6) and 1-dodecanethiol (Table 2, entry 7) were successfully coupled with H-phosphine oxides. Our method exhibited great functionality tolerance, with carboxy (Table 2, entry 8), thiyl (Table 2, entry 9) and benzyl groups (Table 2, entry 10) being well tolerated under our conditions and smoothly forming the products. Other H-phosphine oxides (Table 2, entries 11–13) also efficiently produced P–S coupling products. However, unsymmetric phosphine oxides bearing an alkyl group (Table 2, entry 13) needed more reaction time and still had a decreased yield. According to our method, phosphonothioates (Table 2, entries 14–16) were easily generated in a good yield using simple H-phosphinate esters. Among them, product 3o was named “inezin”, a low toxic pesticide used against rice blast (a serious rice disease).
|
|
||
|---|---|---|
| Entry | 3 | Yieldb (%) |
| a Reaction conditions: H-phosphine oxides/H-phosphinate esters (0.75 mmol), thiols (0.5 mmol), TBPB (1.0 mmol) and KI (0.1 mmol) in 1.5 ml DMSO, reacted at room temperature. b Isolated yield. c Reacted for 18 h. d Reacted in 2 mL EtOAc at 50 °C, 0.6 mmol TBPB was added as an oxidant. | ||
| 1 |

|
97 |
| 2 |

|
99 |
| 3 |
|
97 |
| 4 |
|
88 |
| 5 |

|
90 |
| 6 |
|
98 |
| 7 |
|
96 |
| 8 |
|
66 |
| 9 |
|
82 |
| 10 |
|
95 |
| 11 |
|
81 |
| 12 |
|
76 |
| 13c |
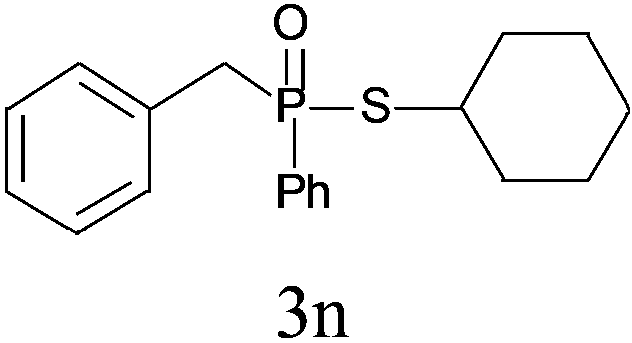
|
65 |
| 14d |
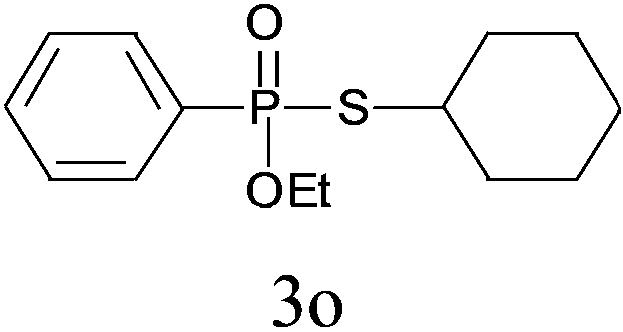
|
73 |
| 15d |
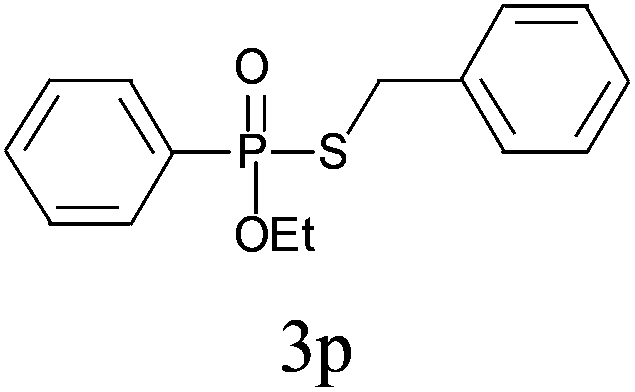
|
81 |
| 16d |
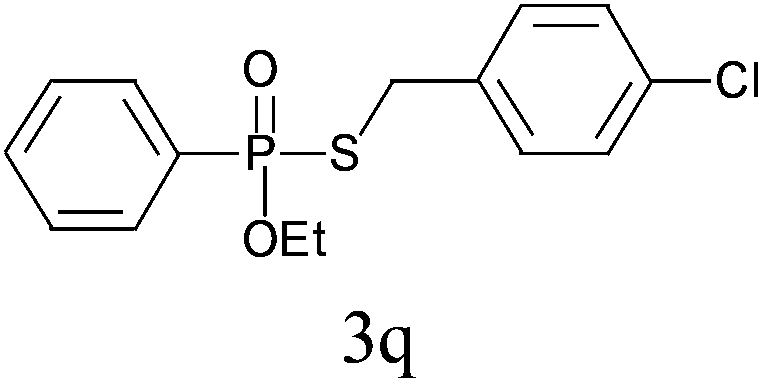
|
84 |

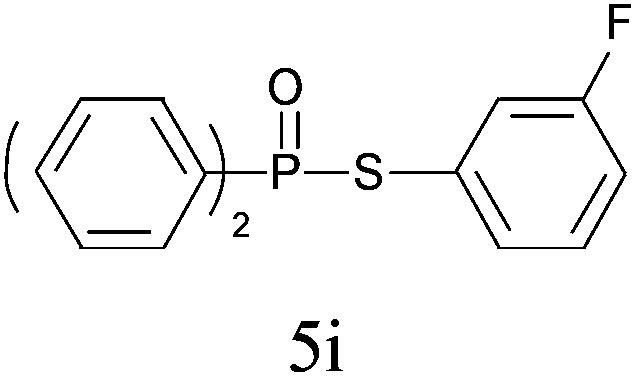
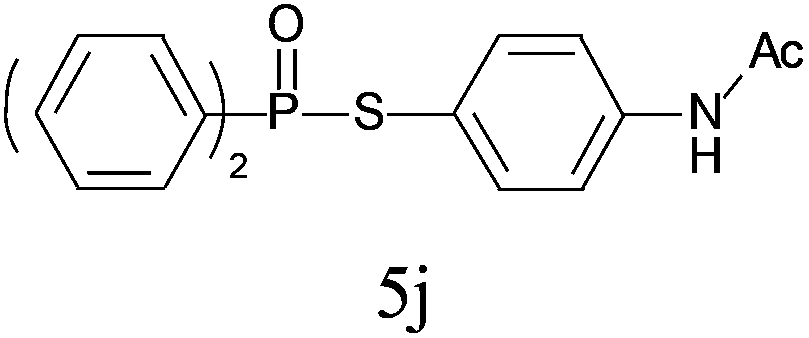
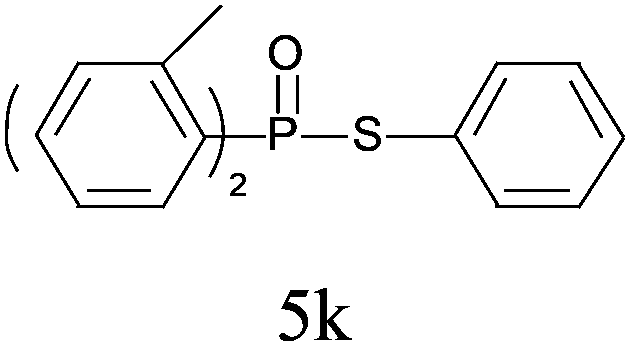
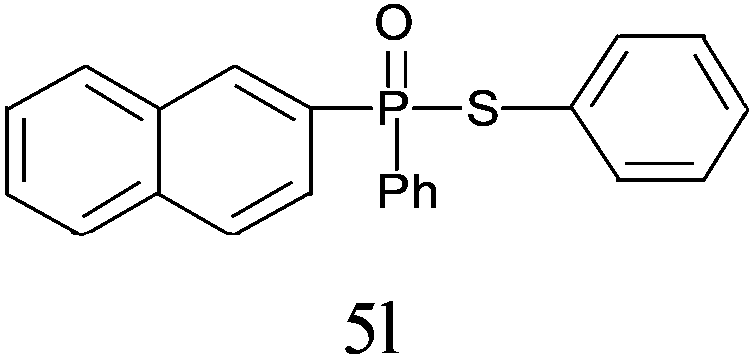
A series of thiophenol substrates was further examined to expand the synthesis utility of our proposed protocol. The coupling reaction between thiophenol and H-phosphine oxide/H-phosphinate ester generally took more time than its thiol counterpart, i.e., at least 8 hours was needed to complete reactant transformation. However, a reduced peroxide amount (1.2 equiv.) was found to be beneficial for this process. A nearly quantitative conversion was obtained for the reaction between substituted thiophenols and diphenylphosphine oxides (Table 3, entries 1–10). Steric hindrance showed little influence on this reaction, and ortho-substituted thiophenols (Table 3, entries 5 and 7) afforded similar yields as their meta- or para- analogs. However, thiophenols bearing strong electron-withdrawing groups (e.g., trifluoromethyl and nitro) yielded no relative products probably because of their higher S–H bond dissociation energies.11 Other functional groups including methoxyl (Table 3, entries 3–5), chloro (Table 3, entries 6 and 7), bromo (Table 3, entry 8), fluoro (Table 3, entry 9) and amide (Table 3, entry 10) were tolerated under our reaction conditions. We also investigated the reactivity of other H-phosphine oxides and H-phosphinate esters (Table 3, entries 11–17), and both were found to provide the target compounds in good to excellent yields.
|
|
||
|---|---|---|
| Entry | 5 | Yieldb (%) |
| a Reaction conditions: H-phosphine oxides/H-phosphinate esters (0.75 mmol), thiophenols (0.5 mmol), TBPB (0.6 mmol) and KI (0.1 mmol) in 1.5 ml DMSO, reacted at room temperature. b Isolated yield. c Reacted for 18 h. | ||
| 1 |
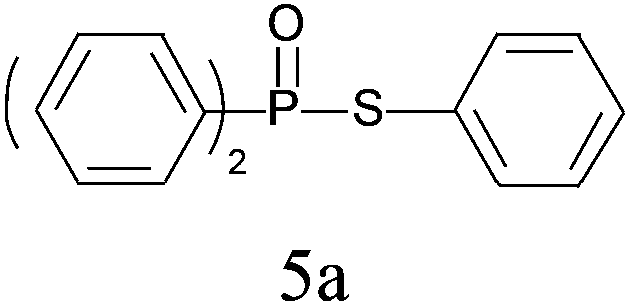
|
96 |
| 2 |
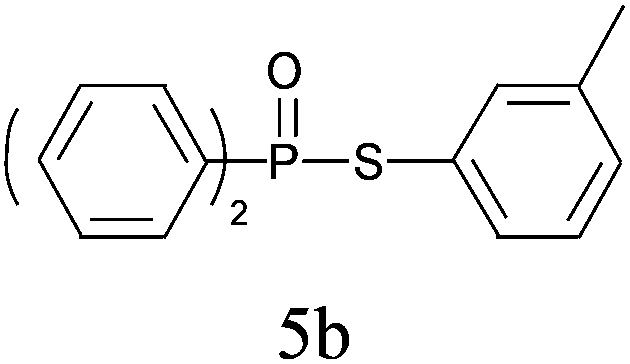
|
99 |
| 3 |
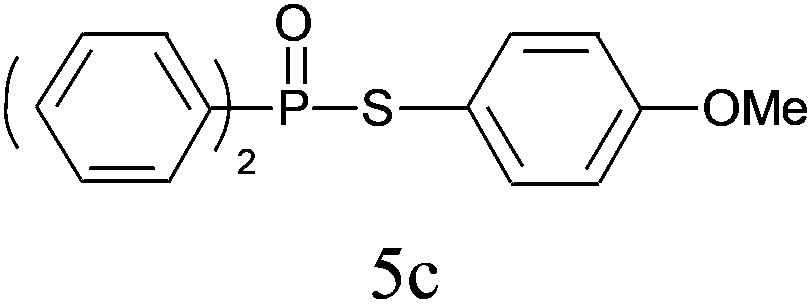
|
98 |
| 4 |
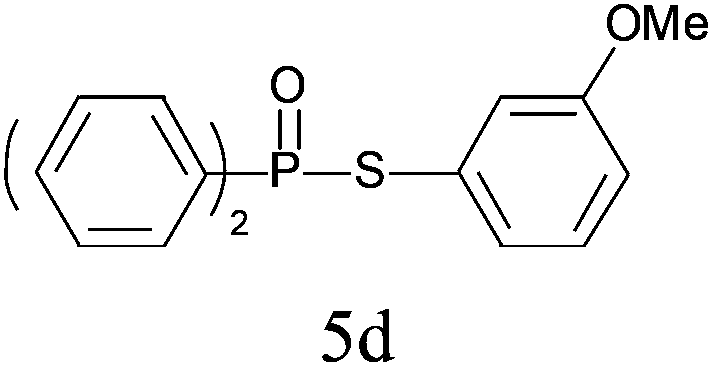
|
99 |
| 5 |
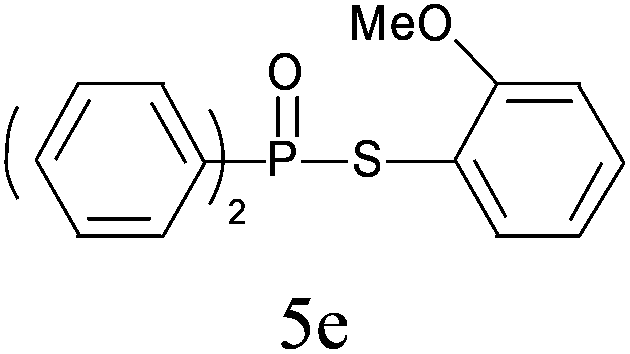
|
99 |
| 6 |
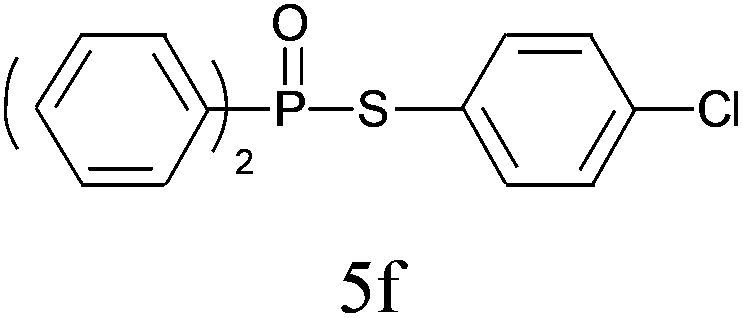
|
99 |
| 7 |
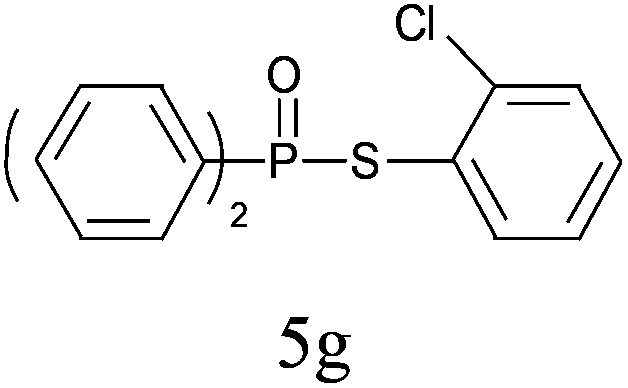
|
95 |
| 8 |
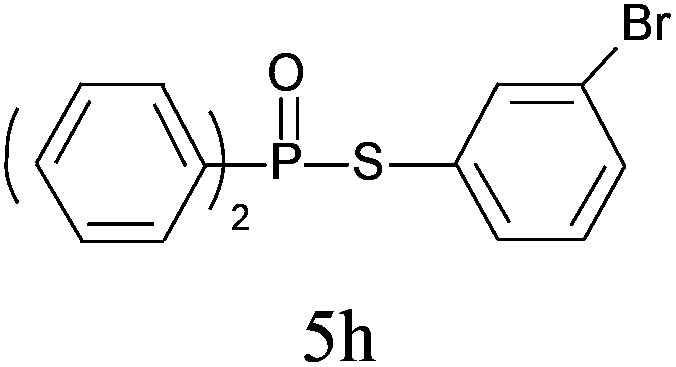
|
96 |
| 9 |
|
97 |
| 10 |
|
51 |
| 11 |
|
87 |
| 12 |
|
97 |
| 13c |
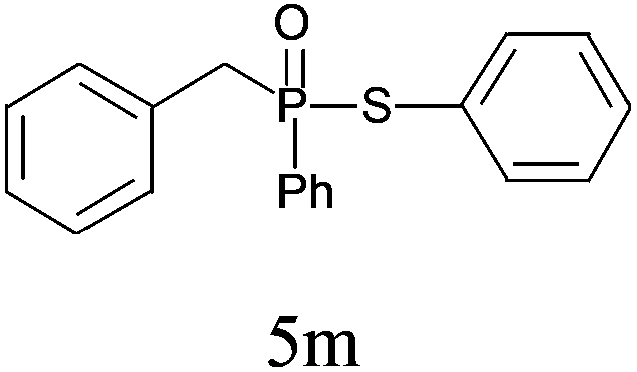
|
71 |
| 14 |

|
99 |
| 15 |
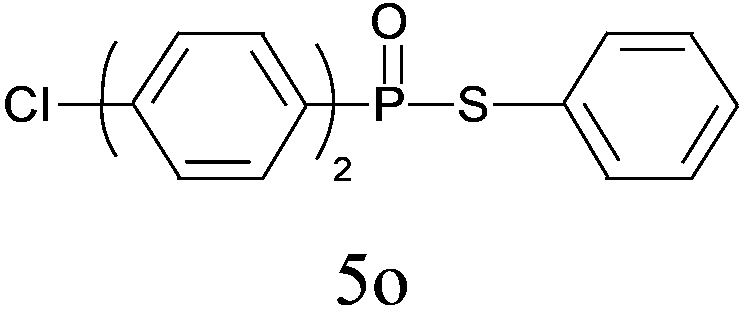
|
90 |
| 16 |
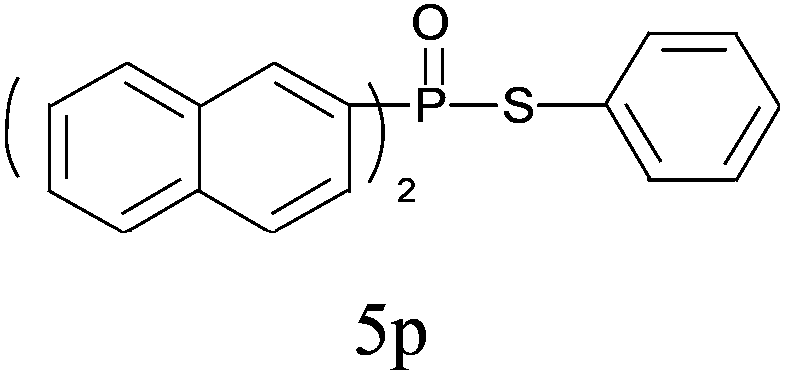
|
99 |
| 17 |
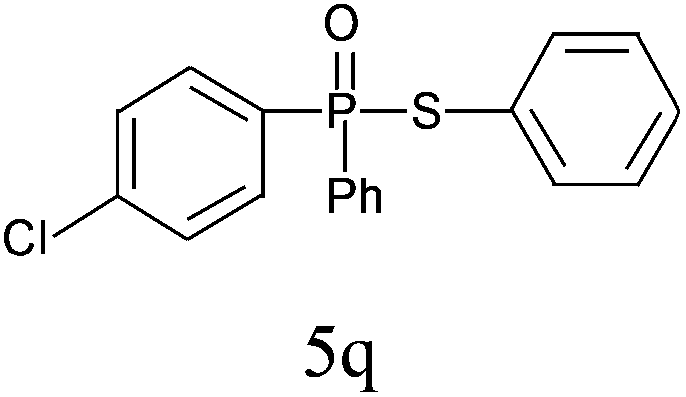
|
95 |
The mechanism of the TBPB promoted P–S coupling reaction was investigated by radical trapping experiments. As shown in Scheme 2, when 2.0 equiv. of TEMPO (a radical scavenger) was added in our reaction system, only a traceable product was detected, which implied that a radical process might be involved. Further experiments suggested that KI may play an important role in catalyzing the oxidation of thiol into the thiyl radical (see ESI†).
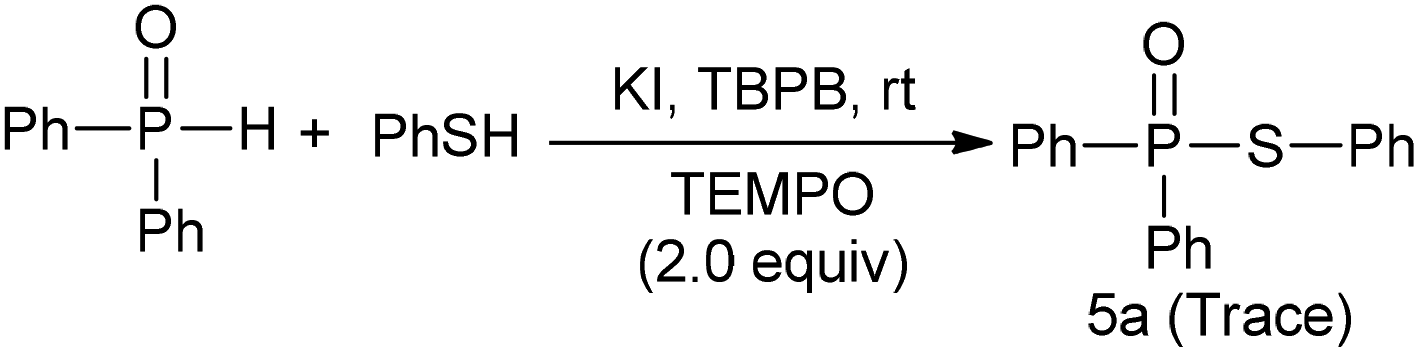 | ||
| Scheme 2 Radical trapping experiments using TEMPO. | ||
Based on the TEMPO trapping experiment results and previous reports,12 a plausible reaction pathway is depicted in Scheme 3. The in situ formed iodine radical (initiated by TBPB) generated thiyl radical A whereas the alkoxyl radical abstracted a hydrogen and produced phosphoryl radical B. Radical B then quickly coupled with the thiyl radical A to yield the thiophosphinate 3a.
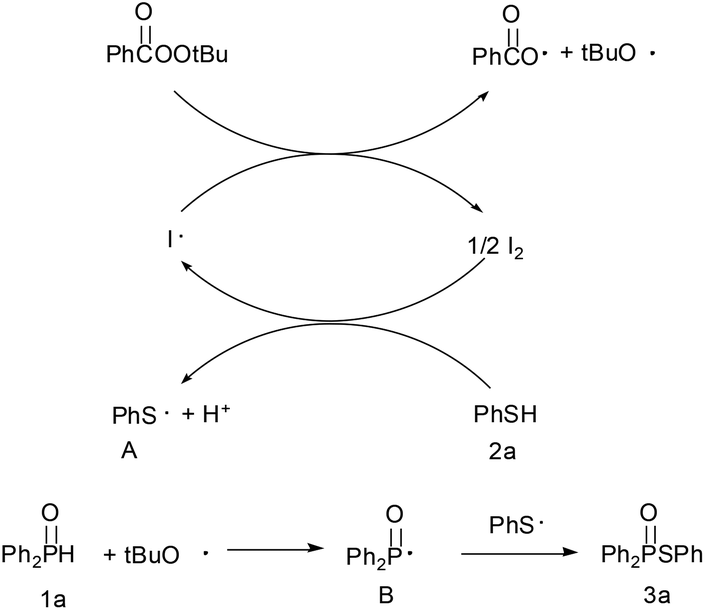 | ||
| Scheme 3 Plausible reaction pathway. | ||
Conclusions
We developed an easy and efficient TBPB-promoted P–S coupling reaction for synthesizing thiophosphinates/phosphonothioates. The reaction was conducted without the presence of metallic catalysts at room temperatures (for H-phosphinate ester 50 °C) and afforded the products in good to excellent yields. The method also exhibited a great functionality tolerance for the substrates and a plausible mechanism was proposed.Experimental
General
All chemicals were obtained from commercial suppliers or prepared according to the former reference (see ESI†). 1H NMR and 13C NMR spectra were recorded on a Bruker ARX-400. Mass spectra were performed on a Bruker Esquire 3000 plus mass spectrometer equipped with ESI interface and ion trap analyzer.General procedure A: for the preparation of products 3a–3q (for thiol substrates)
Thiol (0.5 mmol), H-phosphine oxides/H-phosphinate esters (0.75 mmol) and TBPB (1.0 mmol) were dissolved in 1.5 ml DMSO in a round-bottom flask, then KI (0.1 mmol) was added and reacted at room temperature for 4 h. After reaction completion, the mixture was quenched with 15 ml of Na2S2O4 solution (5%) and extracted with ethyl acetate (3 × 15 ml). The combined organic layer was then dried with anhydrous Na2SO4 and evaporated under vacuum. The crude product was purified by silica gel column chromatography, using PE–EtOAc (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent.
:1) as the eluent.
General procedure B: for the preparation of products 5a–5q (for thiophenol substrates)
Thiophenol (0.5 mmol), H-phosphine oxides/H-phosphinate esters (0.75 mmol) and TBPB (0.6 mmol) were dissolved in 1.5 ml DMSO in a round bottomed flask, then KI (0.1 mmol) was added and reacted at room temperature for 8 h. After reaction completion, the mixture was quenched with 15 ml of Na2S2O4 solution (5%) and extracted with ethyl acetate (3 × 15 ml). The combined organic layer was then dried with anhydrous Na2SO4 and evaporated in a vacuum. The crude product was purified by silica gel column chromatography using PE–EtOAc (3:1) as the eluent.
Acknowledgements
The authors gratefully acknowledge financial support from the National Science Foundation of China (21025207).Notes and references
- (a) J. F. Milligan and O. C. Uhlenbeck, Biochemistry, 1989, 28, 2849–2855 CrossRef CAS; (b) S. H. Vollmer, M. B. Walner, K. V. Tarbell and R. F. Colman, J. Biol. Chem., 1994, 269, 8082–8090 CAS; (c) G. G. Durgam, T. Virag, M. D. Walker, S. Y. Ryoko Tsukahara, K. Liliom, L. A. v. Meeteren, W. H. Moolenaar, X. W. S. Nicole Wilke, G. Tigyi and D. D. Miller, J. Med. Chem., 2005, 48, 4919–4930 CrossRef CAS PubMed; (d) V K. Pandey, A. Dwivedi, O. P. Pandey and S. K. Sengupta, J. Agric. Food Chem., 2008, 56, 10779–10784 CrossRef CAS PubMed; (e) T. Ruman, K. Długopolska, A. Jurkiewicz, D. Rut, T. Frączyk, J. Cieśla, A. Leś, Z. Szewczuk and W. Rode, Bioorg. Chem., 2010, 38, 74–80 CrossRef CAS PubMed; (f) M. W. Loranger, S. A. Beaton, K. L. Lines and D. L. Jakeman, Carbohydr. Res., 2013, 379, 43–50 CrossRef CAS PubMed.
- N. N. Melnilkov, Chemistry of Pesitcides, Springer-Verlag, New York, 1971 Search PubMed.
- For selected reports on thiophosphate synthesis, see: (a) R. G. Harvey, H. I. Jacobson and E. V. Jensen, J. Am. Chem. Soc., 1963, 85, 1623–1626 CrossRef CAS; (b) S. Torii, H. Tanak and N. Sayo, J. Org. Chem., 1979, 44, 2938–2941 CrossRef CAS; (c) B. Kaboudin, Tetrahedron Lett., 2002, 43, 8713–8714 CrossRef CAS; (d) C. M. Timperley, S. A. Saunders, J. Szpalek and M. J. Waters, J. Fluorine Chem., 2003, 119, 161–171 CrossRef CAS; (e) Q. Xu, C. G. Liang and X. Huang, Synth. Commun., 2003, 33, 2777–2785 CrossRef CAS PubMed; (f) Y. X. Gao, G. Tang, Y. Cao and Y. F. Zhao, Synthesis, 2009, 1081–1086 Search PubMed; (g) X. Han and J. Wu, Org. Lett., 2010, 12, 5780–5782 CrossRef CAS PubMed; (h) X. Han, Y. Zhang and J. Wu, J. Am. Chem. Soc., 2010, 132, 4104–4106 CrossRef CAS PubMed; (i) Z. J. Quan, R. G. Ren, Y. X. Da, Z. Zhang and X. C. Wang, Heteroat. Chem., 2011, 22, 653–658 CrossRef CAS; (j) B. Kaboudin, Y. Abedi, J. Kato and T. Yokomatsu, Synthesis, 2013, 45, 2323–2327 CrossRef CAS PubMed; (k) Y. C. Liu and C. F. Lee, Green Chem., 2014, 16, 357–364 RSC.
- (a) W. A. Mosher and R. R. Irino, J. Am. Chem. Soc., 1969, 91, 756–757 CrossRef CAS; (b) C. Yuan, H. Feng and Q. Lin, Synthesis, 1990, 2, 140–141 CrossRef; (c) T. A. Yeung, K. Y. Chan, W. K. Chan, R. K. Haynes, I. D. Williams and L. L. Yeung, Tetrahedron Lett., 2001, 42, 453–456 CrossRef; (d) P. Y. Renard, H. Schwebe, P. Vayron, L. Josien, A. Valleix and C. Mioskowski, Chem. – Eur. J., 2002, 8, 2910–2916 CrossRef CAS; (e) M. Arisawa, T. Ono and M. Yamaguchi, Tetrahedron Lett., 2005, 46, 5669–5671 CrossRef CAS PubMed; (f) Y. Zhou, G. Wang, Y. Saga, R. Shen, M. Goto, Y. Zhao and L. B. Han, J. Org. Chem., 2010, 75, 7924–7927 CrossRef CAS PubMed; (g) M. Arisawa, T. Watanabe and M. Yamaguchi, Tetrahedron Lett., 2011, 52, 2410–2412 CrossRef CAS PubMed.
- (a) P. Carta, N. Puljic, C. Robert, A. L. Dhimane, L. Fensterbank, E. Lacoate and M. Malacria, Org. Lett., 2007, 9, 1061–1063 CrossRef CAS PubMed; (b) P. Carta, N. Puljic, C. Robert, A. L. Dhimane, C. Ollivier, L. Fensterbank, E. Lacote and M. Malacria, Tetrahedron, 2008, 64, 11865–11875 CrossRef CAS PubMed; (c) A. Sato, H. Yorimitsu and K. Oshima, Tetrahedron, 2009, 65, 1553–1558 CrossRef CAS PubMed; (d) N. Hoshi, T. Kashiwabara and M. Tanaka, Tetrahedron Lett., 2012, 53, 2078–2081 CrossRef CAS PubMed.
- (a) L. Hornera and H. W. Flemming, Phosphorus, Sulfur Relat. Elem., 1983, 14, 245–251 CrossRef; (b) R. A. Moss, H. M. Rojas, H. Zhang and B. D. Park, Langmuir, 1999, 15, 2738–2744 CrossRef CAS.
- Selected examples of direct C–S coupling in thioester preparation: (a) S. B. Bandgar, B. P. Bandgar, B. L. Korbad and S. S. Sawant, Tetrahedron Lett., 2007, 48, 1287–1290 CrossRef CAS PubMed; (b) T. Uno, T. Inokuma and Y. Takemoto, Chem. Commun., 2012, 48, 1901–1903 RSC; (c) C. L. Yi, Y. T. Huang and C. F. Lee, Green Chem., 2013, 15, 2476–2484 RSC; (d) Y. Q. Yuan, S. R. Guo and J. N. Xiang, Synlett, 2013, 24, 443–448 CrossRef CAS PubMed; (e) Y. T. Huang, S. Y. Lu, C. L. Yi and C. F. Lee, J. Org. Chem., 2014, 79, 4561–4568 CrossRef CAS PubMed; (f) J. W. Zeng, Y. Liu, P. A. Hsieh, Y. T. Huang, C. L. Yi, S. S. Badsara and C. F. Lee, Green Chem., 2014, 16, 2644–2652 RSC.
- (a) R. S. Xu, J. P. Wan, H. Mao and Y. J. Pan, J. Am. Chem. Soc., 2010, 132, 15531–15533 CrossRef CAS PubMed; (b) Z. Q. Ni, S. C. Wang, H. Mao and Y. J. Pan, Tetrahedron Lett., 2012, 53, 3907–3910 CrossRef CAS PubMed; (c) R. S. Xu, L. Yue and Y. J. Pan, Tetrahedron, 2012, 68, 5046–5052 CrossRef CAS PubMed; (d) X. Huang, J. C. Wang, Z. Q. Ni, S. C. Wang and Y. J. Pan, Chem. Commun., 2014, 50, 4582–4584 RSC.
- J. Dhineshkumar and K. R. Prabhu, Org. Lett., 2013, 15, 6062–6065 CrossRef CAS PubMed.
- (a) C. C. Silveira and S. R. Mendes, Tetrahedron Lett., 2007, 7469–7471 CrossRef CAS PubMed; (b) T. Waag, C. Gelhaus, J. Rath, A. Stich, M. Leippe and T. Schirmeister, Bioorg. Med. Chem. Lett., 2010, 20, 5541–5543 CrossRef CAS PubMed.
- (a) F. G. Bordwell, X. M. Zhang, A. V. Satish and J. P. Cheng, J. Am. Chem. Soc., 1994, 116, 6605–6610 CrossRef CAS; (b) W. M. Nau, J. Org. Chem., 1996, 61, 8312–8314 CrossRef CAS; (c) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- For selected reports on mechanism study, see: (a) Z. J. Liu, J. Zhang, S. L. Chen, E. B. Shi, Y. Xu and X. B. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231–3235 CrossRef CAS PubMed; (b) W. P. Mai, H. H. Wang, Z. C. Li, J. W. Yuan, Y. M. Xiao, L. R. Yang, P. Mao and L. B. Qu, Chem. Commun., 2012, 48, 10117–10119 RSC; (c) W. P. Mai, G. Song, J. W. Yuan, L. R. Yang, G. C. Sun, Y. M. Xiao, P. Mao and L. B. Qu, RSC Adv., 2013, 3, 3869–3872 RSC; (d) Q. C. Xue, J. Xie, P. Xu, K. D. Hu, Y. X. Cheng and C. J. Zhu, ACS Catal., 2013, 3, 1365–1368 CrossRef CAS; (e) Y. H. Lv, Y. Li, T. Xiong, Y. Lu, Q. Liu and Q. Zhang, Chem. Commun., 2014, 50, 2367–2369 RSC; (f) Y. Gao, Q. Song, G. Cheng and X. L. Cui, Org. Biomol. Chem., 2014, 12, 1044–1047 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4gc00944d |
| This journal is © The Royal Society of Chemistry 2015 |