
Dioxomolybdenum(VI) complex immobilized on ascorbic acid coated TiO2 nanoparticles catalyzed heterogeneous oxidation of olefins and sulfides
Maasoumeh
Jafarpour
*,
Abdolreza
Rezaeifard
*,
Mahboube
Ghahramaninezhad
and
Fahimeh
Feizpour
Catalysis Research Laboratory, Department of Chemistry, Faculty of Science, University of Birjand, Birjand, 97179-414 Iran. E-mail: mjafarpour@birjand.ac.ir; rrezaeifard@birjand.ac.ir; rrezaeifard@gmail.com; Fax: +98 561 2502515; Tel: +98 561 2502516
First published on 8th September 2014
Abstract
Addition of MoO2 (acac)2 to TiO2 coated with ascorbic acid (AA) under ultrasonic agitation resulted in a nanohybrid (TiO2/AA/MoO2) with size ranging between 20–25 nm. The structural and morphological characterization of the as-prepared nanohybrid was carried out by different techniques such as XRD, FT-IR, TGA and transmission electron microscopy (TEM). The catalytic performance of the TiO2/AA/MoO2 nanocomplex in the heterogeneous oxidation of olefins and sulfides using H2O2 in ethanol as a safe solvent was exploited. Our results clearly demonstrated the efficiency, selectivity and oxidative stability of the heterogeneous nanocatalyst providing its effective reusability and removing by-products. The catalytic activity of the TiO2/AA/MoO2 nanocomplex was strikingly different from other nanometer sized metal oxides such as m-ZrO2, MoO3, Fe3O4 and TiO2, as well as their nanocomposites such as TiO2/AA, ZrO2/AA/MoO2, MoO3/AA/MoO2 and Fe3O4/AA/MoO2.
Introduction
In recent years, heterogenization of homogeneous catalysts has attracted much attention because they combine the best properties of both homogeneous and heterogeneous catalysts.1 Among the various approaches for immobilizing soluble catalysts, covalent attachment has been the most frequently used strategy, as the resulting heterogeneous catalysts have good stability during the course of catalytic reactions. The stable covalent bonds experience no leaching from the support and thus provide an efficient approach to prepare site isolated catalysts.Epoxides, as precious precursors in organic synthesis, can be easily obtained from alkenes by the use of strong organic oxidants (m-CPBA, NaClO),2 or smoother oxidants (THBP, H2O2),3 with the assistance of metal-based catalysts (Fe, Mn, Re, Mo, V, and W).3,4 Among these metals, molybdenum attracts most attention.
The enthusiasm shown in the coordination chemistry of molybdenum followed the discovery of molybdenum in a number of redox enzymes, such as aldehyde oxidase, sulphite oxidase, xanthine oxidase, nitrate reductase and nitrogenase.5 Dioxomolybdenum(VI)-complexes are important catalysts as well as catalyst-precursors for oxygen-transfer-reactions in chemical and biological systems.6 Considerable efforts devoted to the preparation and investigation of the catalytic properties of dioxomolybdenum complexes under both homogeneous and heterogeneous conditions are a testimony that they are valuable catalysts.7 Most of the investigated processes are performed in chlorinated solvents with catalyst loadings as high as 1% Mo. Solvent recovery requires extra efforts and costs that could be eliminated by the application of greener processes.
With the aim of developing cleaner processes and in continuation of our ongoing research on the catalytic activity of Mo catalysts,8 in this work, we incorporated simple and easily available MoO2 (acac)2 into ascorbic acid-coated TiO2 nanoparticles under ultrasonic agitation to obtain a novel heterogeneous molybdenum nanocatalyst for the oxidation of olefins and sulfides using H2O2 in ethanol as a safe solvent. The catalyst showed high thermal and oxidation stability and desired activity and selectivity in the oxidation of olefins and sulfides using hydrogen peroxide as an industrially and environmentally important oxidant (Scheme 1). The additional advantage of this catalytic system is facile and efficient reusability of the solid catalyst at the end of the reaction.
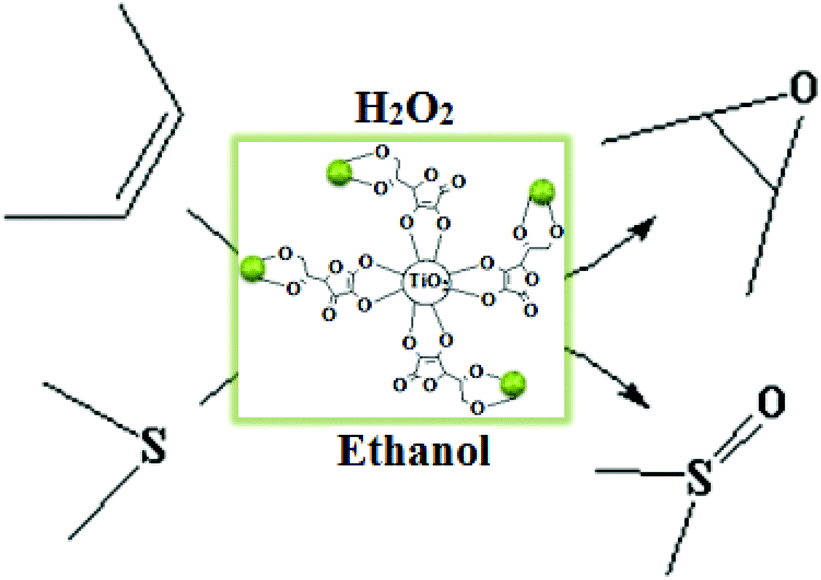 | ||
| Scheme 1 Catalytic activity of the TiO2/AA/MoO2 nanocomplex in oxidation of olefins and sulfides. | ||
Results and discussion
Synthesis and characterization of the catalyst
In the first step of this investigation, TiO2 nanoparticles of approximately 18–20 nm diameter were prepared by the PC sol–gel method (Scheme 2).8f The particles were subsequently coated with an ascorbic acid layer followed by complexation with MoO2 (acac)2 to yield the corresponding supported cis-dioxomolybdenum nanocomplex (Scheme 3). Inductively coupled plasma atomic emission (ICP) analysis revealed that the content of the supported Mo was 2.78 wt%. Therefore, each gram of the heterogeneous catalyst contains 29 μmol Mo or MoO2 group.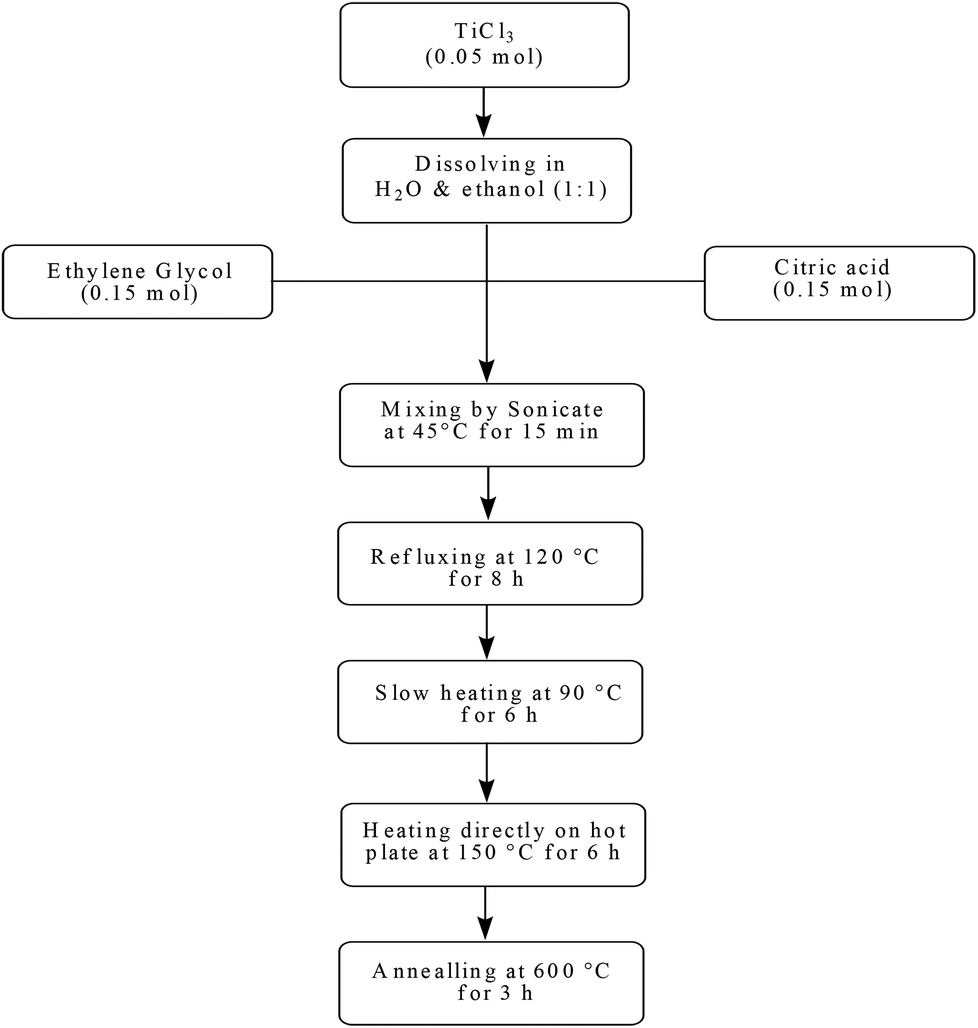 | ||
| Scheme 2 The flowchart of the preparation of nanoTiO2 by the sol–gel process. | ||
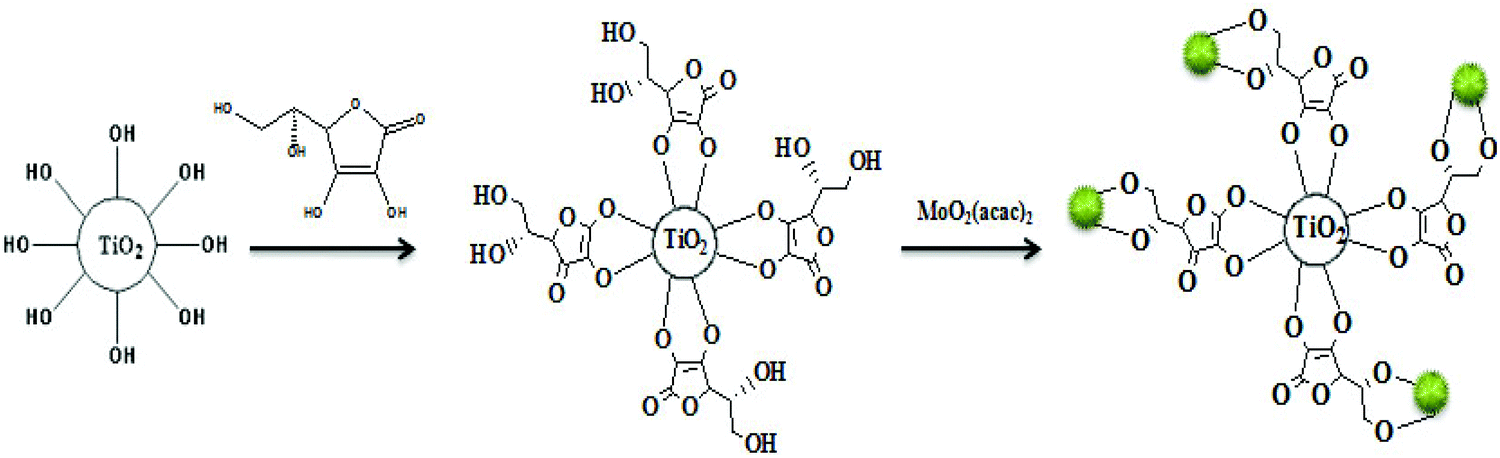 | ||
| Scheme 3 Schematic representation of the procedure for the fabrication of the TiO2/AA/MoO2 nanocomplex. | ||
The spectral and analysis data confirmed successful synthesis of the title nanocatalyst. The XRD pattern of TiO2 nanoparticles exhibited two different phases of TiO2 such as anatase (tetragonal, a = b = 3.782 Å, c = 9.502 Å) and rutile (tetragonal, a = b = 4.584 Å, c = 2.953 Å)9 (Fig. 1). The strong diffraction peaks observed at 2θ = 25, 38, 48, 54, 55, 63, 69, 70, 75 were assigned to the (101), (004), (200), (105), (211), (204), (116), (220), (215) reflection planes of tetragonal crystals of anatase TiO2, respectively (JCPDS no. 21-1272).10 The other diffraction peaks observed at 2θ = 27, 35, 41, 56 were assigned to the (110), (101), (111), (220) reflection planes of tetragonal crystals of rutile TiO2 respectively (JCPDS no. 21-1276).11 The size of particles was estimated to be 20 nm according to the Debye Sherrer formula (D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ). It is well known that TiO2 containing both the rutile and anatase phases exhibit higher photocatalytic activity in visible light than either pure phase alone.12
cosθ). It is well known that TiO2 containing both the rutile and anatase phases exhibit higher photocatalytic activity in visible light than either pure phase alone.12
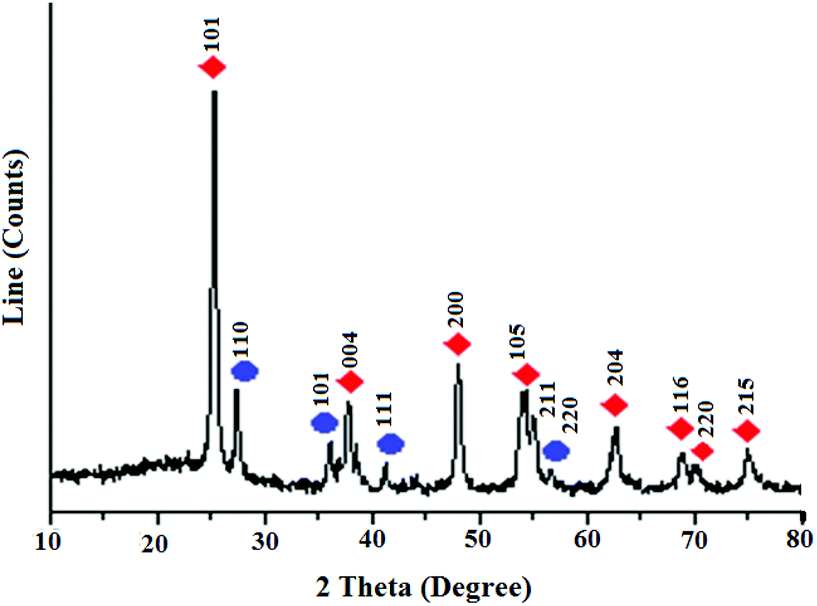 | ||
Fig. 1 XRD pattern of nanoTiO2:  (anatase), (anatase),  (rutile). (rutile). | ||
The FT-IR spectrum of TiO2 nanoparticles (Fig. 2a) reveals the presence of major bands at 450–775 cm−1 which are attributed to the stretching vibrations of the Ti–O group. It can be observed that there are broad peaks at 3400 and 1638 cm−1, which correspond to the surface adsorbed water and hydroxyl groups.13Fig. 2b confirms the successful fabrication of TiO2/AA composite particles. ortho-substituted hydroxyl groups of the furan ring binding to AA act as bidentate ligands to attach to surface Ti atoms forming chelate-type coordination in the desired complexes.14 The appearance of peaks in the range of 927 and 1010 cm−1 rationalized to C–O stretching vibration of Ti–O–C.15 The strong peak at 1320 cm−1 corresponding to the (O–H) enediol for the free acid disappeared in the spectra of the complexes showing the coordination of the OC3 and OC2 atoms to the Ti atoms.16
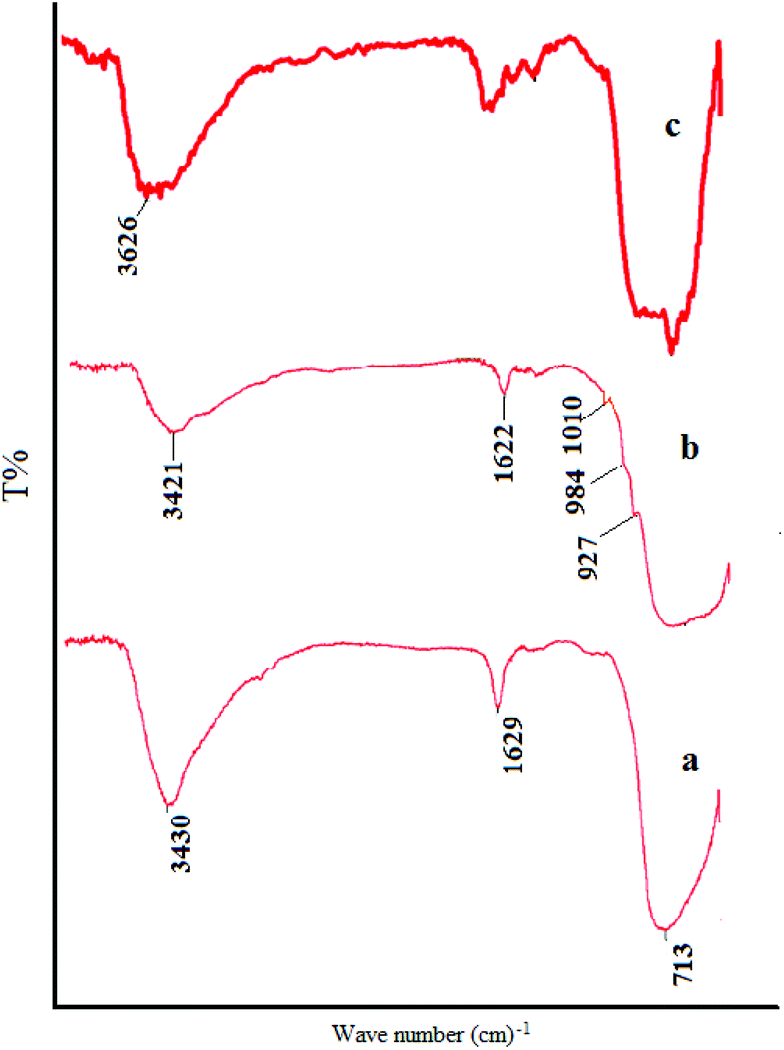 | ||
| Fig. 2 FT-IR spectra of (a) nanoTiO2, (b) the TiO2/AA nanocomposite and (c) the TiO2/AA/MoO2 nanocomplex. | ||
Apparently, AA coating on the surface of TiO2 particles contributes to the decrease of the number of –OH on the TiO2 surface. The vibration absorption at low frequencies, such as that observed at 720 cm−1, shows the existence of the nanoTiO2 core in TiO2/AA nanocomposites.
Comparison of the FT-IR spectra of the TiO2/AA/MoO2 nanocomplex (Fig. 2c) with those of TiO2 and the TiO2/AA composite supports the formation of the respective composite since significant spectral changes are observed. It revealed the presence of major bands at 500–750 cm−1 which are attributed to the stretching vibrations of the Ti–O group and the symmetric and asymmetric stretching vibrations of the cis-MoO2 at 800–950 (Fig. 2c).
Transmission electron microscopy (TEM) observations clearly revealed spherical nanoparticles of TiO2 and the TiO2/AA/MoO2 composite with size ranging between 18–20 nm and 20–25 nm, respectively (Fig. 3).
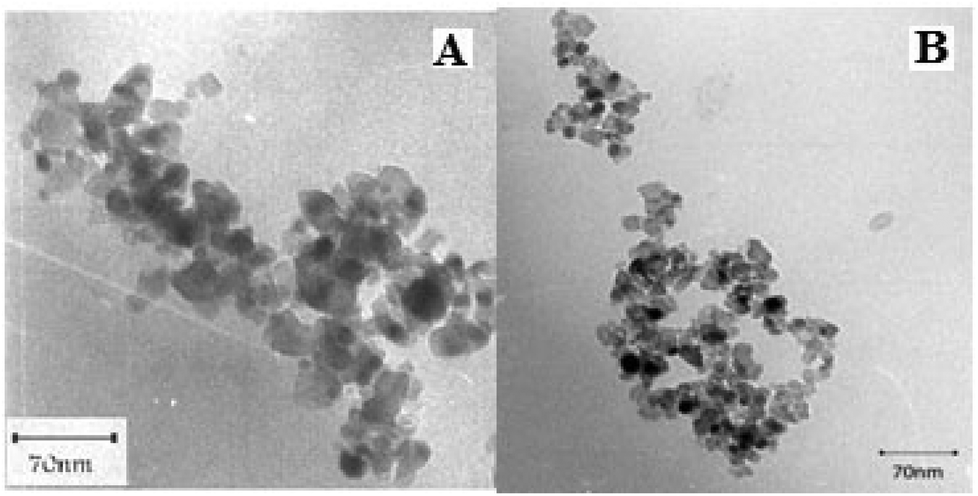 | ||
| Fig. 3 TEM of (A) nanoTiO2 and (B) the TiO2/AA/MoO2 nanocomplex. | ||
Thermal behavior of the synthesized amorphous powder dried at 150 °C was analyzed through TGA (thermogravimetric analysis) from ambient temperature to 800 °C. The results showed that 600 °C was an optimum calcination temperature for the synthesis of TiO2 nanopowders (Fig. 4).
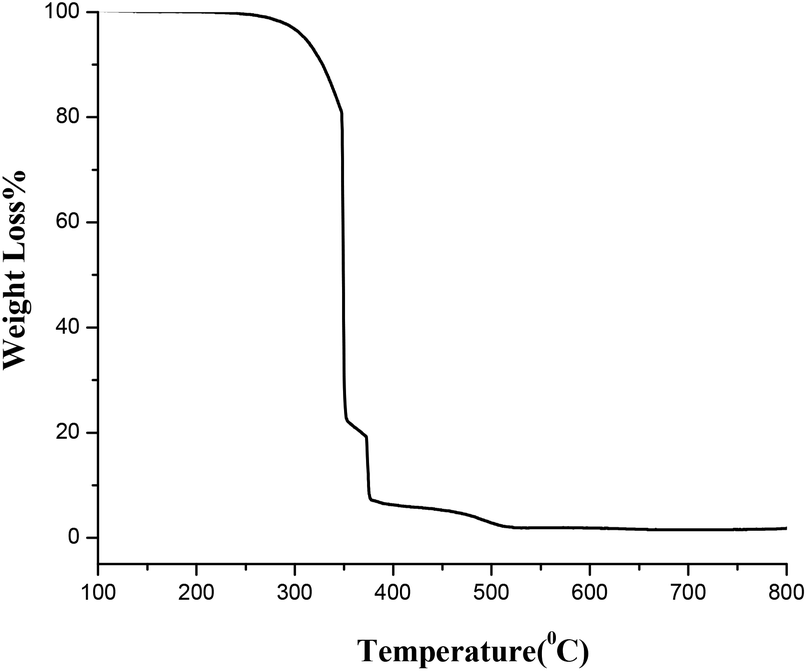 | ||
| Fig. 4 The TGA of TiO2 powder. | ||
The TGA curve of the TiO2/AA/MoO2 nanocomplex (Fig. 5) demonstrates its degradation at 817 °C, which indicates the high thermostability of the catalyst. The organic parts decomposed completely at 877 °C.
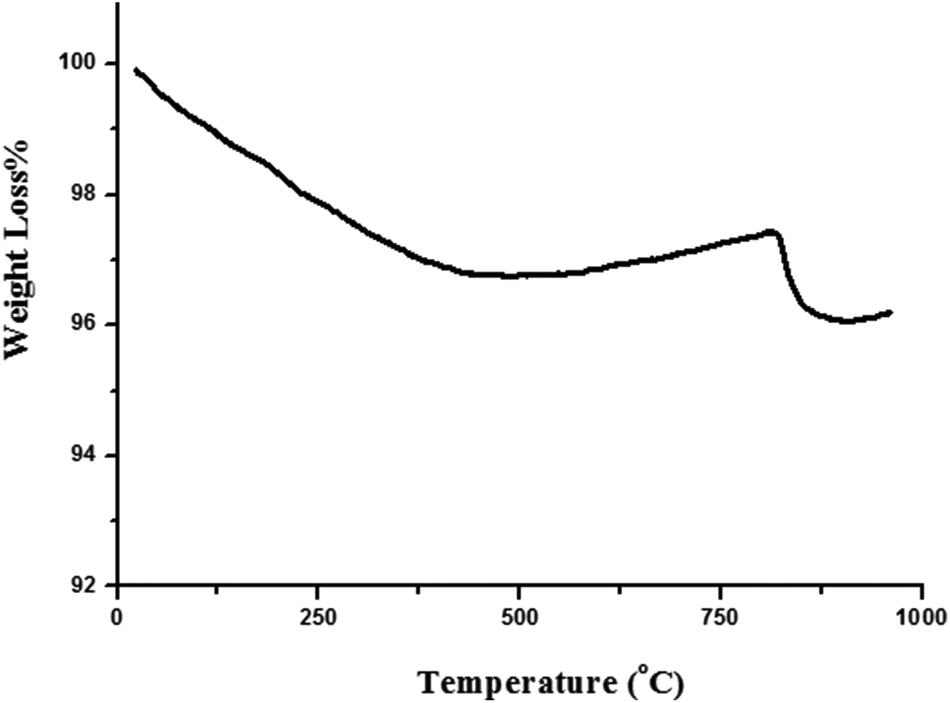 | ||
| Fig. 5 The TGA of the TiO2/AA/MoO2 nanocomplex. | ||
Catalytic activity of the TiO2/AA/MoO2 nanocomplex
A systematic examination of the solvent nature was performed in various solvents such as chloroform, dichloroethane (DCE), acetonitrile, methanol, ethanol and water using 0.03 mol% of the TiO2/AA/MoO2 nanocomposite catalyst (Fig. 6) at different temperatures (Fig. 7). The best yield and conversion rate were obtained in ethanol at 70 °C.
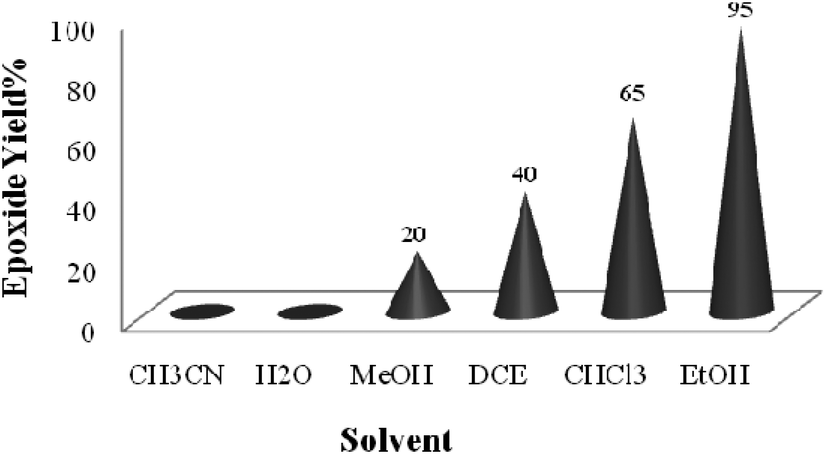 | ||
| Fig. 6 The screening of the solvent nature (1 mL) on oxidation of cyclooctene (1 mmol) using H2O2 (2 mmol) catalyzed by the TiO2/AA/MoO2 nanocomplex (0.03 mol%) at 70 °C except for solvents having lower b.p. which were studied at reflux temperature. | ||
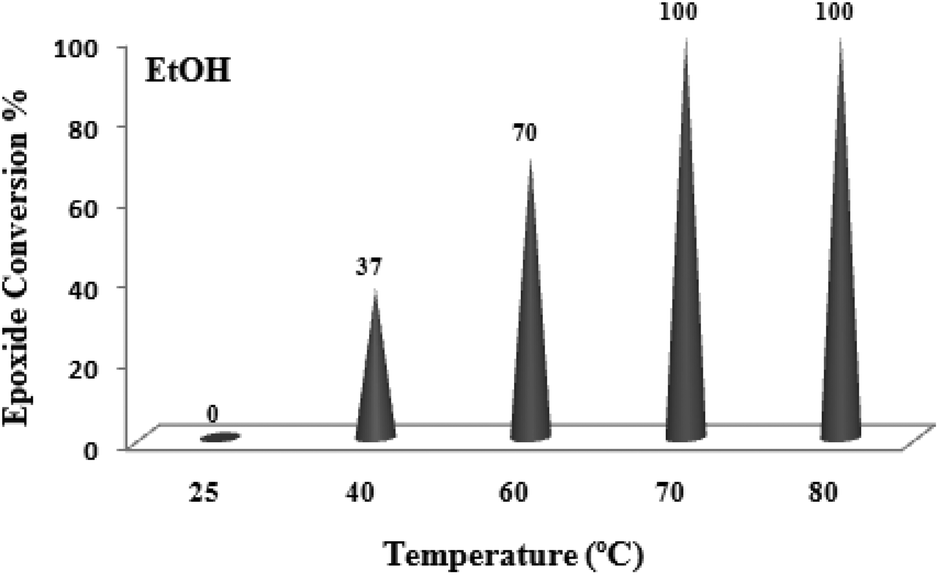 | ||
| Fig. 7 The screening of temperature on oxidation of cyclooctene (1 mmol) using H2O2 (2 mmol)–EtOH (1 mL) catalyzed by the TiO2/AA/MoO2 nanocomplex (0.03 mol%) after 6 h. | ||
The reaction was further optimized with respect to the catalyst (Fig. 8) and oxidant amounts (Fig. 9). It was observed that full conversion of cyclooctene required 0.03 mol% of the nanocatalyst and two equivalents of H2O2 within 6 h, and an increase in any of these ratios did not affect noticeably the reaction rate.
 | ||
| Fig. 8 The screening of the catalyst amount on the epoxidation of cyclooctene (1 mmol) using H2O2 (2 mmol)–EtOH (1 mL) catalyzed by the TiO2/AA/MoO2 nanocomplex at 70 °C. | ||
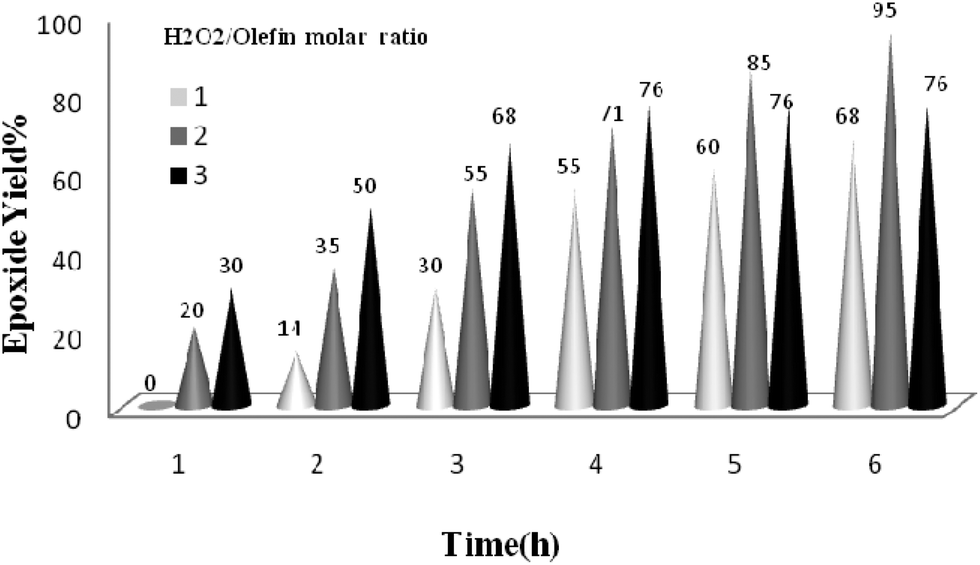 | ||
| Fig. 9 The screening of H2O2 amount on the epoxidation of cyclooctene (1 mmol) in EtOH (1 mL) catalyzed by the TiO2/AA/MoO2 nanocomplex (0.03 mol%) at 70 °C after 6 h. | ||
To evaluate the oxidizing potential of other common oxidants, cyclooctene was subjected to the oxidation protocol using TBHP, NaIO4 and Oxone® under the catalytic influence of TiO2/AA/MoO2 nanocomposites in ethanol at 70 °C (Fig. 10). Only small amounts of the oxidation products were observed.
 | ||
| Fig. 10 The screening of different oxidants (2 mmol) on oxidation of cyclooctene (1 mmol) in EtOH (1 mL) catalyzed by the TiO2/AA/MoO2 nanocomposite (0.03 mol%) at 70 °C after 6 h. | ||
Under the optimized conditions (10000:20000:3 molar ratio for olefin–H2O2–catalyst in ethanol at 70 °C), cyclooctene converted completely within 6 h and 95% of the corresponding epoxide was secured as the sole product.
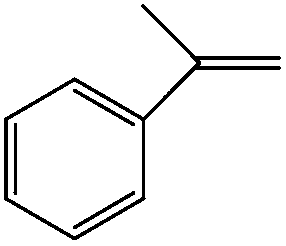
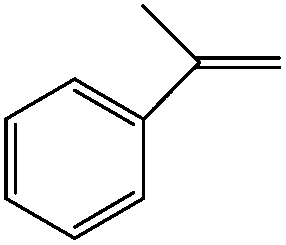
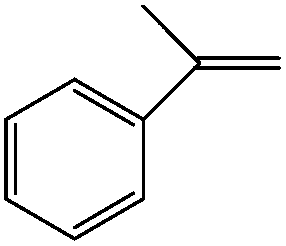
Moreover, prolonged reaction resulted for cyclooctene oxidation when the TiO2/AA/MoO2 nanocomplex was replaced by other nanooxometals such as MoO3,8gm-ZrO2, Fe3O4 and TiO2, as well as their nanocomposites such as TiO2/AA, ZrO2/AA/MoO2, MoO3/AA/MoO2 and Fe3O4/AA/MoO2 under the same conditions. Photocatalytic properties of TiO2 at the core of the title nanocomplex may be an acceptable reason for the great improvement of catalytic activity (Fig. 11). To support this claim, oxidation of α-methyl styrene and diphenyl sulfide under UV light, room light lamps as well as in the dark was investigated (Table 1). Accelerated reactions under light radiation, particularly UV light, demonstrated the photocatalytic activity of the TiO2 core on the oxidation efficiency of the TiO2/AA/MoO2 nanocomplex.
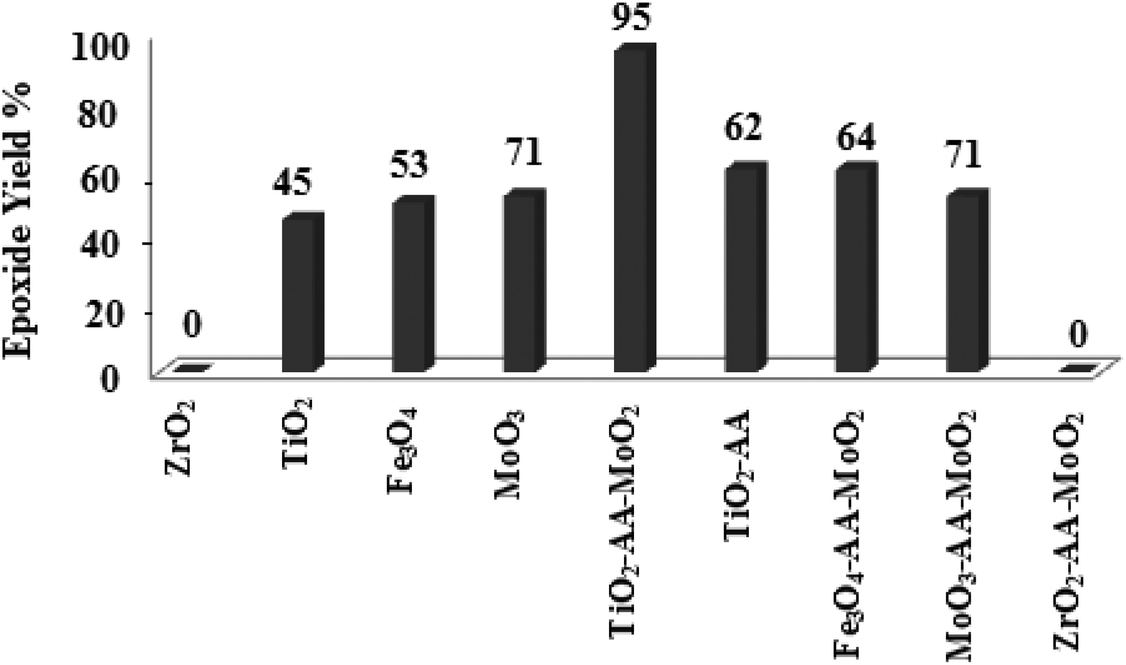 | ||
| Fig. 11 The comparison of the catalytic activity of the TiO2/AA/MoO2 nanocomplex with other nanooxometals and nanocomposites in the epoxidation of cyclooctene (1 mmol) in EtOH (1 mL) at 70 °C after 6 h. | ||
| Substrate | Condition | Product (selectivity %) | Conversion % (time) |
|---|---|---|---|
|
a The molar ratio of the substrate–oxidant–catalyst was 10000:20000:3. The reactions were run under air at 70 °C.
|
|||
|
Room light lamps | a-Methyl styrene oxide (75) | 100 (10 h) |
|
UV light | a-Methyl styrene oxide (70) | 100 (8 h) |
|
Darkness | a-Methyl styrene oxide (60) | 100 (12 h) |

|
Room light lamps | Diphenyl sulfoxide (90) | 80 (30 min) |
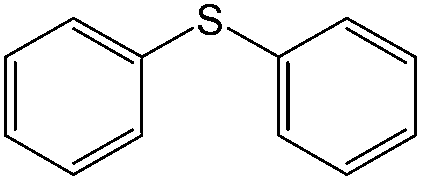
|
UV light | Diphenyl sulfoxide (90) | 80 (20 min) |
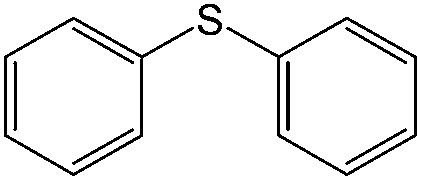
|
Darkness | Diphenyl sulfoxide (90) | 50 (45 min) |
Considering the results presented in Fig. 11 and Table 1 the catalytic activity of the TiO2/AA/MoO2 nanocomplex can be related to the Lewis acid catalyst activity of Mo(VI) centers7c,17 combined with photocatalytic activity of the TiO2 core which also acts as a support. It is noteworthy that one of the most widely used methods to provide the selectivity is encapsulation of TiO2 particles into substances.18
In order to establish the general applicability of the method, various olefins were subjected to the oxidation protocol under the catalytic influence of the TiO2/AA/MoO2 nanocomplex (Table 2).
| Entry | Alkene | Conversion % (isolated yield %) | Productb | Selectivityb % | Time (h) |
|---|---|---|---|---|---|
|
a The molar ratio of the substrate–H2O2–catalyst was 10000:20000:3. The reactions were run under air at 70 °C.
b The products were identified by 1H NMR or by comparison with authentic samples retention times of GC analysis.19
|
|||||
| 1 |

|
90 (82) |

|
100 | 6 |
| 2 |
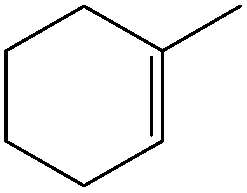
|
95 (85) |

|
100 | 6 |
| 3 |
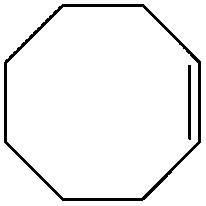
|
100 (95) |
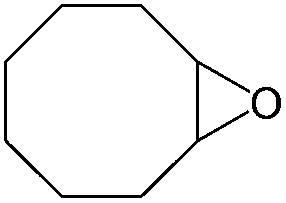
|
100 | 6 |
| 4 |
|
90 (83) |
|
100 | 12 |
| 5 |

|
15 |
|
100 | 24 |
| 6 |
|
100 (93) |

|
75 | 16 |
|
25 | ||||
| 7 |
|
100 (94) |
|
75 | 10 |
|
25 | ||||
| 8 |
|
80 (72) |
|
85 | 20 |
|
15 | ||||
| 9 |
|
100 |

|
100 | 12 |
Several useful features of this catalytic method can be seen in Table 2. Different olefins were generally good substrates for this catalyst. It led to high conversions of cyclooctene, norbornene and cyclohexenes with the formation of the corresponding epoxides as sole products (entries 1–4).
It is worth mentioning that cyclic olefin conjugated with a phenyl ring produced exclusively the pertinent epoxide, albeit with low yield (entry 5).
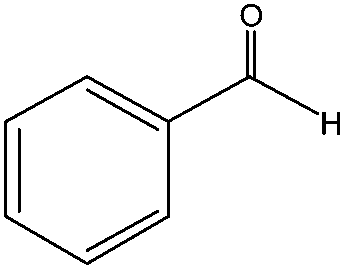
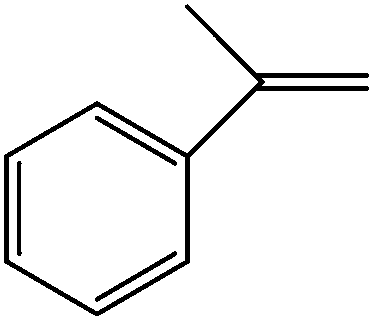
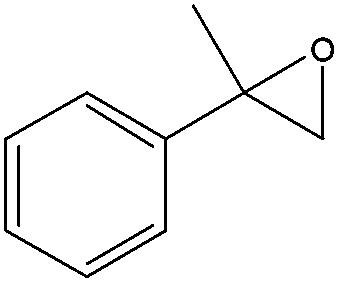
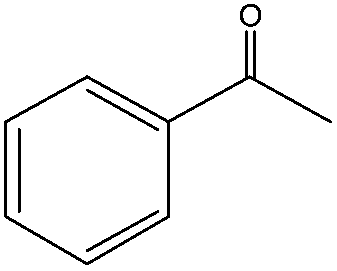
When the terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond was conjugated with an aromatic ring, the major product was the corresponding epoxide and the related carbonyl compound was achieved as a byproduct resulting from a ring-opening reaction of styrene oxide derivatives (entries 6–8).
C double bond was conjugated with an aromatic ring, the major product was the corresponding epoxide and the related carbonyl compound was achieved as a byproduct resulting from a ring-opening reaction of styrene oxide derivatives (entries 6–8).
The chemoselectivity of the procedure was notable. The catalyst was able to oxidize 2-cyclohexene-1-ol as an allylic alcohol to the related unsaturated carbonyl compound in high yield and selectivity (entry 9).
Oxidation of sulfides to sulfoxides
To extend the generality and scope of the present methodology, oxidation of sulfides was investigated. Oxidation of thioanisole using 2.0 equivalents of H2O2 in the presence of 0.03 mol% of the TiO2/AA/MoO2 nanocomposite in ethanol produced the related sulfoxide as the sole product in excellent yield within 30 min. Accordingly, different sulfides were subjected to the reaction system in the presence of the title nanocatalyst (Table 3). The results given in Table 3 illustrate the high efficiency of this protocol for the oxidation of structurally different sulfides. All substrates could be smoothly converted into sulfoxides with excellent conversion rates, and excellent selectivities were obtained under mild conditions.| Entry | Substrate | Conversion % (isolated yield %)b | Sulfoxide selectivity % |
|---|---|---|---|
|
a The reactions were run at 70 °C and the molar ratio of the sulfide–H2O2–catalyst was 10000:20000:3 in 1 mL ethanol within 30 min.
b The products were identified by comparison with authentic samples.20
c The remainder is the related sulfone.
|
|||
| 1 |
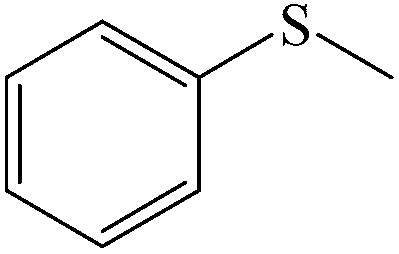
|
100 (96) | 100 |
| 2 |
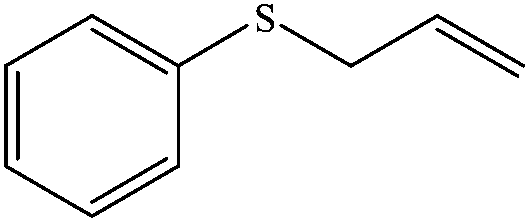
|
100 (95) | 90c |
| 3 |
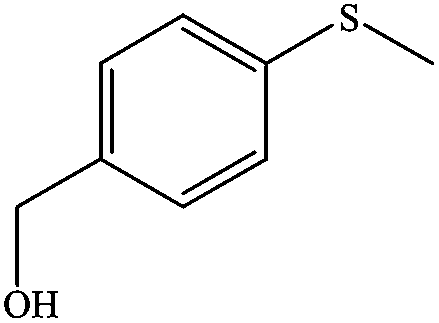
|
100 (96) | 100 |
| 4 |
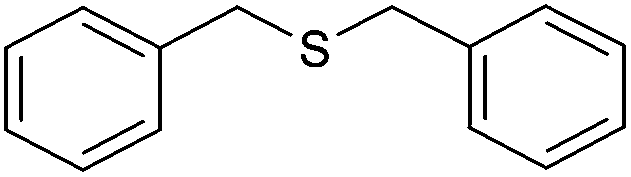
|
80 (72) | 95c |
| 5 |
|
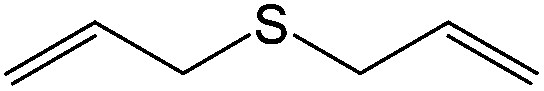
80 (73) | 90c |
| 6 |
|
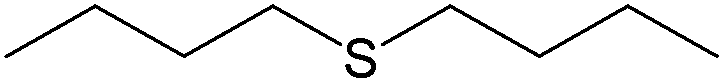
100 (96) | 100 |
| 7 |
|
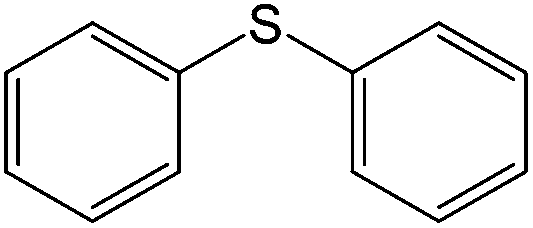
90 (80) | 95c |
| 8 |
|
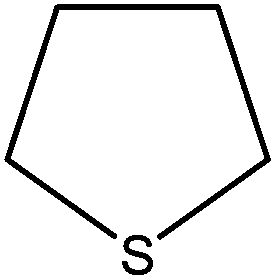
100 (96) | 100 |
The chemoselectivity of the method is noteworthy, as exemplified by the sulfide containing hydroxyl group (Table 3, entry 3) or CC double bond (Table 3, entries 2 and 6). While the sulfide oxidized completely, the alcohol and olefin moieties remained intact. In addition, dibenzyl sulfide (Table 3, entry 4) was selectively oxidized to its corresponding sulfoxide without the formation of any benzylic oxidation byproducts.
Recovery of the catalyst
The good/excellent yields of epoxides and sulfoxides obtained using these new catalytic methods demonstrated the high stability and catalytic activity of the prepared TiO2/AA/MoO2 nanocomplex. This was further supported by the evaluation of the recovery potential of the catalyst in the oxidation of both olefins and sulfides under different conditions mentioned in Tables 2 and 3.Recovery of the TiO2/AA/MoO2 nanocomplex catalyst was easy and efficient. The catalyst was recovered by centrifuging and decantation of the reaction mixture. It was then washed with ethanol as safe solvents, dried under vacuum, and used directly for the next round of reaction without further purification. The ease of recovery, combined with the intrinsic stability of the TiO2/AA/MoO2 nanocomplex, allows the catalyst to be recovered efficiently over at least five times in the oxidation of olefins and sulfides under different conditions used in this study (Fig. 12).
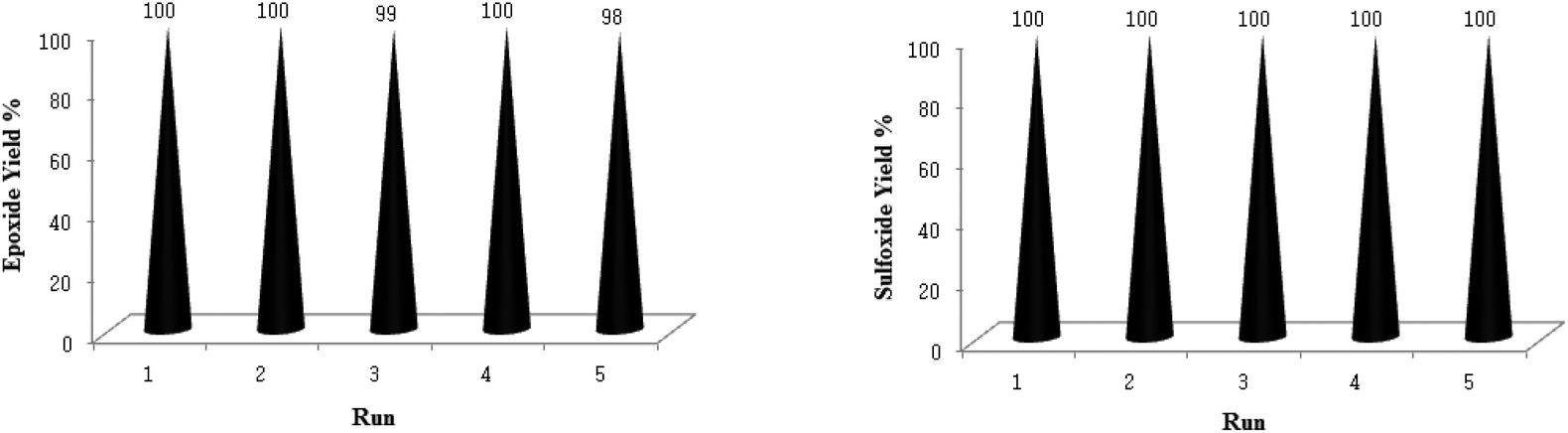 | ||
| Fig. 12 Recycling of the catalytic system for oxidation of cyclooctene (left) and thioanisole (right) using H2O2 by the TiO2/AA/MoO2 nanocomplex at 70 °C according to procedures mentioned in the Experimental section. | ||
The comparison of FT-IR and TEM images of the used TiO2/AA/MoO2 nanocomplex (Fig. 13) with the fresh one showed that the structure, size and morphology of the catalyst remained almost intact after five times recovering.
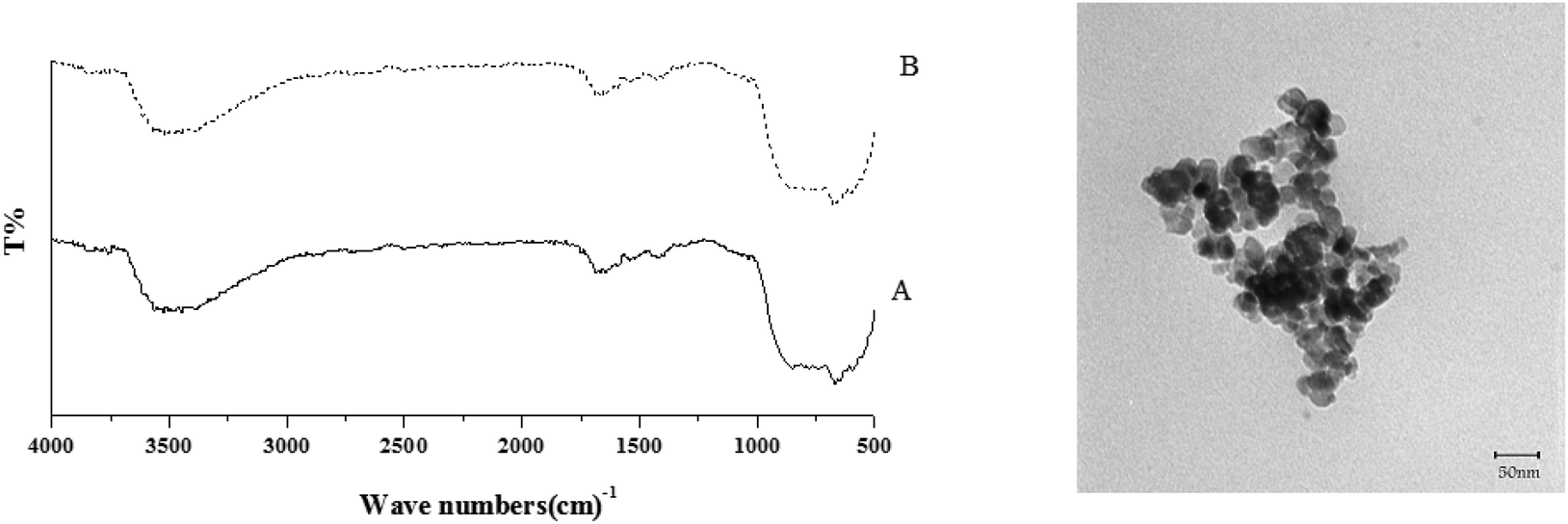 | ||
| Fig. 13 (Left) FT-IR spectra of the fresh TiO2/AA/MoO2 nanocomplex (A) and after 5 times reuse (B) in the oxidation of cyclooctene; (right) TEM image of the TiO2/AA/MoO2 nanocomplex after 5 times reuse in the oxidation of cyclooctene. | ||
Table 4 shows the merit of this operationally simple catalytic protocol for epoxidation of cyclooctene as a model substrate in comparison with the other catalysts in terms of oxygen source, conversion rate, catalyst loading and especially the conditions used in the reactions.
| Entry | Catalyst | Catalyst amount | Conditions | Time (h) | Conversion (%) | Ref. |
|---|---|---|---|---|---|---|
| a This work. b Unpublished results. The experiments were performed under the same conditions in our laboratory. c Manganese complexes of 1,4,7-trimethyl-1,4,7-triazacyclononane, in the presence of trichloroacetic acid. d Diol selectivity. e The Keggin-type di-vanadium-substituted silicotungstate [γ-1,2-H2SiV2W10O40]4− with the {VO-(μ-H)2-VO} core. f Host (nanocavity of zeolite-X)–guest (manganese(III)tetrakis[4-N-methylpyridinium]porphyrin). | ||||||
| 1 | TiO2/AA/MoO2 nanocomplex | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 100 | |
| 2 | m-ZrO2 | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 0 | |
| 3 | TiO2 | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 45 | |
| 4 | Fe3O4 | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 53 | |
| 5 | MoO3 | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 71 | |
| 6 | TiO2/AA | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 62 | |
| 7 | Fe3O4/AA/MoO2 | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 64 | |
| 8 | MoO3/AA/MoO2 | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 71 | |
| 9 | ZrO2/AA/MoO2 | 0.03 mol% | Ethanol–H2O2 (70 °C) | 6 | 0 | |
| 10 | Dititanium-containing 19-tungstodiarsenate(III) | 0.002 M | CH3CN–H2O2 (50 °C) | 5 | 70 | 21 |
| 11 | Mn(II)/pyridine-2-carboxylic Acid | 0.01 mol% | CH3CN–H2O2 (0–25 °C) | 0.5–2 | 95 | 22 |
| 12 | [Ga(phen)2Cl2]Cl | 5 mM | CH3CN–PAA (0 °C) | 1 | 77 | 23 |
| 13 | Mn–TMTACNc [MnIV,IV2(μ-O)(μ-RCO2)2(TMTACN)2]2+ | 0.1 mol% | CH3CN–H2O2 (0 °C) | 1 | 71 | 24 |
| 14 | [p-C5H5N(CH2)15CH3]3[PW4O32] | 30 mg | EtOAc–H2O2 (65 °C) | 1 | 98 | 25 |
| 15 | Ti–Fe3O4@MCM-41 | 0.1 g | Toluene–TBHP (80 °C) | 6 | 91 | 26 |
| 16 | Ti–Fe3O4@MCM-41 | 0.1 g | CH3CN–H2O2 (80 °C) | 6 | 24 | 27 |
| 17 | [Fe(BMIPnPr)2](OTf)2 | 0.1 mol% | CH3CN–H2O2 (r.t.) | 24 | 25 | 27 |
| 18 | Manganese complexes of (tmtacn) | 0.3 mM | CH3CN–H2O2 (0 °C) | 5 | 54d | 28 |
| 19 | Bis(μ-hydroxo) bridged di-vanadiume | 1.67 mM | CH3CN–tBuOH–H2O2 (20 °C) | 24 | 93 | 29 |
| 20 | Polyoxotungstate Na9[SbW9O33] [MTCA]+Cl− | 0.09 mmol | S.F–H2O2 (60 °C) | 6 | 97 | 30 |
| 21 | Mn (TMPyP)-NaXf | 0.007 mmol | CH3CN–H2O–NaIO4 (r.t.) | 10 | 94 | 31 |
| 22 | Mn-beta-1 Mn2+-exchanged zeolites | 100 mg | DMF–NaHCO3–H2O2 (r.t) | 4 | 13.5 | 32 |
| 23 | NH TiO2-SiO2 | 50 mg | CH3CN–H2O2 (60 °C) | 6 | 67 | 33 |
| 24 | [MnL(OTf)2] | 0.5 mol% | CH3CN–PAA(0 °C) | Over 3 min | 89 | 34 |
Therefore, title methodologies are cost effective and industrially important because of reusing the catalyst and using H2O2 as an environmental oxidant, especially in ethanol as a safe reaction media. These advantages for high yielding oxidation methods offered ready scalability. For example, the use of a semi-scale-up procedure (10 mmol) for epoxidation of cyclooctene and oxidation of thioanisole in the presence of the TiO2/AA/MoO2 nanocomplex led to the isolation of the related epoxide and sulfoxide in 95 and 96% yield, respectively.
Conclusions
In conclusion, ascorbic acid coated TiO2 nanoparticles provided a favored solid support for heterogenization of dioxomolybdenum(VI) centers under ultrasonic agitation producing a novel nanohybrid (TiO2/AA/MoO2) with size ranging between 20–25 nm. The TiO2/AA/MoO2 nanocomplex demonstrated the desired activity and selectivity in the epoxidation of olefins and oxidation of sulfides to sulfoxides using H2O2 as an environmentally benign oxidant in ethanol as a green solvent. These favorable characteristics for a heterogeneous catalytic system, along with the easy and safe workup procedure and excellent reusability of the catalyst, are environmentally benign and cost-effective. Thus, our method possesses high generality which makes it suitable for industrial goals.Experimental
General remarks
All chemicals were purchased from Merck and Fluka Chemical Companies. Powder X-ray diffraction (XRD) was performed on a Bruker D8-advance X-ray diffractometer with Cu Kα (λ = 1.54178 Å) radiation. The FT-IR spectra were recorded on a NICOLET system. Raman spectra were recorded on a Bruker senterra (2009) model with the spectral range 200–3500 cm−1 and a laser wavenumber of 785 nm. Thermogravimetric analysis (TGA) of nanopowders was performed in air using Shimadzu 50. TEM images were obtained by TEM instrumentation (Philips CM 10). Progress of the reactions was monitored by TLC using silica-gel SIL G/UV 254 plates and by GC-FID on a Shimadzu GC-16A instrument using a 25 m CBP1-S25 (0.32 mm ID, 0.5 μm coating) capillary column. NMR spectra were recorded on Bruker Avance DPX 250 and 400 MHz instruments.Fabrication of TiO2 nanoparticles
To a solution of TiCl3 (0.05 mol) in a mixture of double-distilled water and absolute ethanol (1:1) was added citric acid (0.15 mol) and ethylene glycol (0.15 mol). The resulting mixture was dissolved at 45 °C under ultrasound for 15 min to give a clear violet solution. The solution was refluxed at 120 °C for 8 h which turned into a metal-citrate homogeneous complex with a little color change from clear violet to black-violet. After cooling down, in order to bring about the required chemical reactions for the development of polymerization and evaporation of the solvent, the sol was further slowly heated at 90 °C for 6 h in an open bath until a beige wet gel was obtained. During continuous heating at this temperature, the polymerization between citric acid, ethylene glycol and complexes was developed and ultimately the sol became more viscous as a wet gel. In the final step of the sol–gel process, the wet gel was fully dried by direct heating on the hot plate at 150 °C for 6 h. The resulting product was a black powder. Then it was calcined in a furnace at 600 °C for 3 h at a rate of 5 °C min−1. Finally TiO2 nanoparticles with white color were obtained (Scheme 2).
Preparation of TiO2/AA nanocomposite particles
To 1.0 g TiO2 particles was gradually added 10.0 mL of 0.01 M ascorbic acid in water over a period of 20 min at room temperature under ultrasonic agitation. Then, the reaction mixture was stirred at room temperature for 8 h. Afterwards, the product was centrifuged and washed with distilled water. Finally, TiO2/AA nanocomposites were obtained after drying for 4 h at 100 °C.Preparation of the TiO2/AA/MoO2 nanocomplex
To 0.2 g of the TiO2/AA nanocomposite was gradually added 1.0 mmol MoO2(acac)2 dissolved in ethanol over a period of 20 min at room temperature under ultrasonic agitation. Then, the as-obtained mixture was refluxed for 12 h. Afterwards, the product was centrifuged and washed with ethanol. Finally, the TiO2/AA/MoO2 nanocomplex was obtained after drying for 12 h at 80 °C (Scheme 3).General procedure for catalytic oxidation of olefins
To a mixture of olefin (1.0 mmol) and the TiO2/AA/MoO2 nanocomplex (0.03 mol%, 0.01 g) in ethanol (1.0 mL) was added 30% H2O2 (2.0 mmol), and the reaction mixture was stirred in air at 70 °C for the required time. The reaction progress was monitored by GC, and the yields of the products were determined by GC and NMR analyses. After the completion of the reaction, the TiO2/AA/MoO2 nanocomplex (solid phase) was separated by centrifugation followed by decantation (3 × 5 mL ethanol). Then, the desired product (liquid phase) was extracted by plate chromatography and eluted with n-hexane–EtOAc (10/1). Assignment of the products was done by IR, 1H NMR and MS spectral data in comparison with authentic samples.General procedure for the oxidation of sulfides
To a mixture of the sulfide (1.0 mmol) and the TiO2/AA/MoO2 nanocomplex (0.03 mol%, 0.01 g) in ethanol (1.0 mL) was added H2O2 (2.0 mmol), and the reaction mixture was stirred in air at 70 °C for 30 min. The reaction progress was monitored by TLC, and the yields of the products were determined by GC analysis. After the completion of the reaction, the TiO2/AA/MoO2 nanocomplex (solid phase) was separated by centrifugation followed by decantation (3 × 5 mL ethanol). Then the desired product (liquid phase) was extracted by plate chromatography and eluted with n-hexane–EtOAc (10/3). Assignment of the products was done by IR, 1H NMR and MS spectral data in comparison with authentic samples.Reusability of the catalyst
To a mixture of the cyclooctene (10.0 mmol) and the TiO2/AA/MoO2 nanocomplex (0.3 mol%, 0.1 g) in ethanol (10.0 mL) was added H2O2 (20.0 mmol), and the reaction mixture was stirred in air at 70 °C for 4 h. After completion of the reaction, the TiO2/AA/MoO2 nanocomplex was separated by centrifugation followed by decantation (3 × 5 mL ethanol). The isolated solid phase (TiO2/AA/MoO2 nanocomplex) was dried under reduced pressure and reused for the next runs. Catalyst recovery was also investigated in the oxidation of sulfides with H2O2 in ethanol according to the above mentioned procedure.Acknowledgements
Support for this work by the Research Council of University of Birjand is highly appreciated.References
- (a) J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS; (b) D. Trong, D. Desplantier-Giscard, C. Danumah and S. Kaliaguine, Appl. Catal., A, 2001, 222, 299–357 CrossRef; (c) D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed; (d) M. H. Valkenberg and W. F. Holderich, Catal. Rev., 2002, 44, 321 CrossRef CAS PubMed; (e) A. Choplin and F. Quignard, Coord. Chem. Rev., 1998, 178–180, 1679–1702 CrossRef CAS; (f) M. Masteri-Farahani, F. Farzaneh and M. Ghandi, J. Mol. Catal. A: Chem., 2003, 192, 103–111 CrossRef CAS.
- A. Corma, I. Dominguez, A. Domenech, V. Fornes, C. J. Gomez-Garcia, T. Rodenas and M. J. Sabater, J. Catal., 2009, 265, 238–244 CrossRef CAS PubMed.
- (a) P. M. Reis, C. A. Gamelas, J. A. Brito, N. Saffon, M. Gomez and B. Royo, Eur. J. Inorg. Chem., 2011, 5, 666–673 CrossRef; (b) C. Dinoi, M. Ciclosi, E. Manoury, L. Maron, L. Perrin and R. Poli, Chem. – Eur. J., 2010, 16, 9572–9584 CrossRef CAS PubMed; (c) C. Cordelle, D. Agustin, J. C. Daran and R. Poli, Inorg. Chim. Acta, 2010, 364, 144–149 CrossRef CAS PubMed.
- (a) R. Sanz and M. R. Pedrosa, Curr. Org. Chem., 2009, 6, 239–263 CAS; (b) F. E. Kühn, A. M. Santos and M. Abrantes, Chem. Rev., 2006, 106, 2455–2475 CrossRef PubMed.
- R. C. Bray, Enzymes, 1975, 12, 299 CAS.
- (a) R. Hille, Eur. J. Inorg. Chem., 2006, 1913–1926 CrossRef CAS; (b) M. J. Romao, Dalton Trans., 2009, 4053–4068 RSC; (c) M. J. Romao, M. Archer, I. Moura, J. J. G. Moura, J. LeGall, R. Engh, M. Schneider, P. Hof and R. Huber, Science, 1995, 270, 1170–1176 CAS; (d) R. Hille, Arch. Biochem. Biophys., 2005, 433, 107–116 CrossRef CAS PubMed.
- (a) M. Masteri-Farahani, F. Farzaneh and M. Ghandi, J. Mol. Catal. A: Chem., 2006, 248, 53–60 CrossRef CAS PubMed; (b) M. Masteri-Farahani, F. Farzaneh and M. Ghandi, Catal. Commun., 2007, 8, 6–10 CrossRef CAS PubMed; (c) M. Amini, M. M. Haghdoost and M. Bagherzadeh, Coord. Chem. Rev., 2013, 257, 1093–1121 CrossRef CAS PubMed; (d) M. Masteri-Farahani, J. Mol. Catal. A: Chem., 2010, 316, 45–51 CrossRef CAS PubMed; (e) C. D. Nunes, M. Pillinger, A. A. Valente, A. D. Lopes and I. S. Goncalves, Inorg. Chem. Commun., 2003, 6, 1228–1233 CrossRef CAS; (f) M. Jia and W. R. Thiel, Chem. Commun., 2002, 2392–2393 RSC.
- (a) A. Rezaeifard, I. Sheikhshoaie, N. Monadi and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 2010, 799–806 CrossRef CAS; (b) I. Sheikhshoaie, A. Rezaeifard, N. Monadi and S. Kaafi, Polyhedron, 2009, 28, 733–738 CrossRef CAS PubMed; (c) A. Rezaeifard, I. Sheikhshoaie, N. Monadi and M. Alipour, Polyhedron, 2010, 29, 2703–2709 CrossRef CAS PubMed; (d) A. Rezaeifard, M. Jafarpour, H. Raissi, M. Alipour and H. Stoeckli-Evans, Z. Anorg. Allg. Chem., 2012, 638, 1023–1030 CrossRef CAS; (e) A. Rezaeifard, R. Haddad, M. Jafarpour and M. Hakimi, J. Am. Chem. Soc., 2013, 135, 10036–10039 CrossRef CAS PubMed; (f) M. Jafarpour, A. Rezaeifard, M. Ghahramaninezhad and T. Tabibi, New J. Chem., 2013, 37, 2087–2095 RSC; (g) M. Jafarpour, M. Ghahramaninezhad and A. Rezaeifard, RSC Adv., 2014, 4, 1601–1608 RSC; (h) M. Jafarpour, M. Ghahramaninezhad and A. Rezaeifard, New J. Chem., 2014, 38, 2917–2926 RSC.
- (a) G. V. Samsonov, The Oxide Handbook, IFI/Plenum Press, New York, 1982 Search PubMed; (b) U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- H. Peng, X. Wang, G. Li, H. Pang and X. Chen, Mater. Lett., 2010, 64, 1898–1901 CrossRef CAS PubMed.
- M. Ye, Z. Chen, W. Wang, L. Zhen and J. Shen, Mater. Lett., 2008, 62, 3404–3406 CrossRef CAS PubMed.
- G. S. Mital and T. Manoj, Chin. Sci. Bull., 2011, 56, 1639–1657 CrossRef.
- (a) S. C. Pillai, P. P. Periyat, R. George, D. E. McCormack, M. K. Seery, H. Hayden, J. Colreavy, D. Corr and S. J. Hinder, J. Phys. Chem. C, 2007, 111, 1605–1611 CrossRef CAS; (b) J. Yang, J. Zhang, L. W. Zhu, S. Y. Chen, Y. M. Zhang, Y. Tang, Y. L. Zhu and Y. W. Li, J. Hazard. Mater. B, 2006, 137, 958–952 Search PubMed.
- T. Rajh, J. M. Nedeljkovic and M. C. Thurnauer, J. Phys. Chem. B, 1999, 103, 3515 CrossRef CAS.
- Y. Ou, J. D. Lin, H. M. Zou and D. W. Liao, J. Mol. Catal. A: Chem., 2005, 241, 59–64 CrossRef CAS PubMed.
- D. A. Köse and B. Zümreoglu-Karan, New J. Chem., 2009, 33, 1874–1881 RSC.
- (a) A. O. Chong and K. B. Sharpless, J. Org. Chem., 1977, 42, 1587 CrossRef CAS; (b) S. Patai, in The Chemistry of Peroxides, ed. S. Patai, Wiley, Chichester, 1983 Search PubMed; (c) R. Sheldon and J. Kochi, Metal-catalyzed Oxidations of Organic Compounds, Academic Press, 1981 Search PubMed; (d) J. E. Backvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, Germany, 2nd edn, 2010 Search PubMed; (e) H. Srour, P. Le Maux, S. Chevance and G. Simonneaux, Coord. Chem. Rev., 2013, 257, 3030–3050 CrossRef CAS PubMed.
- (a) S. Ikeda, Y. Kowata, K. Ikeue, M. Matsumura and B. Ohtani, Appl. Catal., A, 2004, 265, 69 CrossRef CAS PubMed; (b) K. Inumaru, T. Kasahara, M. Yasui and S. Yamanaka, Chem. Commun., 2005, 2131 RSC; (c) J. Seok Lee, K. Hwan You and Ch. Beum Park, Chem. Phys. Chem., 2005, 6, 714–718 CrossRef PubMed; (d) K. Raghava Reddy, V. G. Gomes and M. Hassan, Mater. Res. Express, 2014, 1, 015012 CrossRef; (e) U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- (a) A. Rezaeifard, M. Jafarpour, A. Naeimi and S. Kaafi, Catal. Commun., 2011, 12, 761–765 CrossRef CAS PubMed; (b) A. Rezaeifard, M. Jafarpour, A. Naeimi and K. Mohammadi, J. Mol. Catal. A: Chem., 2012, 357, 141–147 CrossRef CAS PubMed; (c) Y. Ding, W. Zhao, H. Hua and B. Ma, Green Chem., 2008, 10, 910–913 RSC; (d) E. G. Ankudey, H. F. Olivo and T. L. Peeples, Green Chem., 2006, 8, 923–926 RSC; (e) F. Bruyneel, C. Letondor, B. Bastürk, A. Gualandi, A. Pordea, H. Stoeckli-Evans and R. Neier, Adv. Synth. Catal., 2012, 354, 428–440 CrossRef CAS.
- (a) F. Rajabi, S. Naserian, A. Primo and R. Luque, Adv. Synth. Catal., 2011, 353, 2060–2066 CrossRef CAS; (b) L. Xu, J. Cheng and M. L. Trudel, J. Org. Chem., 2003, 68, 5388–5391 CrossRef CAS PubMed; (c) J. M. Samanen and E. Brandeis, J. Org. Chem., 1988, 53, 561–569 CrossRef CAS; (d) M. H. Ali and G. J. Bohnert, Synth. Commun., 1998, 28, 2983–2998 CrossRef CAS.
- B. G. Donoeva, T. A. Trubitsina, N. S. Antonova, J. J. Carbó, J. M. Poblet, Gh. Al-Kadamany, U. Kortz and O. A. Kholdeeva, Eur. J. Inorg. Chem., 2010, 5312–5317 CrossRef CAS.
- J. Dong, P. Saisaha, T. G. Meinds, P. L. Alsters, E. G. Ijpeij, R. P. van Summeren, B. Mao, M. Fananas-Mastral, J. W. de Boer, R. Hage, B. L. Feringa and W. R. Browne, ACS Catal., 2012, 2, 1087–1096 CrossRef CAS.
- W. Jiang, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2012, 51, 2725–2727 CrossRef CAS PubMed.
- P. Saisaha, J. W. de Boerb and W. R. Browne, Chem. Soc. Rev., 2013, 42, 2059–2074 RSC.
- Y. Ding, W. Zhao, H. Hua and B. Ma, Green Chem., 2008, 10, 910–913 RSC.
- A. Jha, Ch. R. Patil, A. C. Garade and Ch. V. Rode, Ind. Eng. Chem. Res., 2013, 52, 9803–9811 CrossRef CAS.
- M. A. H. Moelands, S. Nijsse, E. Folkertsma, B. de Bruin, M. Lutz, A. L. Spek and R. J. M. Klein Gebbink, Inorg. Chem., 2013, 52, 7394–7410 CrossRef CAS PubMed.
- N. J. Schoenfeldt, Zh. Ni, A. W. Korinda, R. J. Meyer and J. M. Notestein, J. Am. Chem. Soc., 2011, 133, 18684–18695 CrossRef CAS PubMed.
- N. Mizunoa, Y. Nakagawa and K. Yamaguchi, J. Mol. Catal. A: Chem., 2006, 251, 286–290 CrossRef PubMed.
- R. H. Ingle and N. K. Kala Raj, J. Mol. Catal. A: Chem., 2008, 294, 8–13 CrossRef CAS PubMed.
- M. Moghadam, Sh. Tangestaninejad, V. Mirkhani, I. Mohammadpoor-Baltork and M. Moosavifar, J. Mol. Catal. A: Chem., 2009, 302, 68–75 CrossRef CAS PubMed.
- B. Qi, X.-H. Lu, D. Zhou, Q.-H. Xia, Z.-R. Tang, S.-Y. Fang, T. Pang and Y.-L. Dong, J. Mol. Catal. A: Chem., 2010, 322, 73–79 CrossRef CAS PubMed.
- A. M. Cojocariua, P. H. Mutin, E. Dumitriu, F. Fajula, A. Viouxc and V. Hulea, Appl. Catal., B, 2010, 97, 407–413 CrossRef PubMed.
- G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722–1760 RSC.
| This journal is © The Royal Society of Chemistry 2015 |