DOI:
10.1039/C4GC00997E
(Paper)
Green Chem., 2015,
17, 435-441
Oxidation of tetrahydrofuran to butyrolactone catalyzed by iron-containing clay
Received
10th June 2014
, Accepted 10th September 2014
First published on 12th September 2014
Abstract
Thermally treated iron-containing clay was used as a greener oxidation catalyst for the conversion of tetrahydrofuran (THF) to butyrolactone (BTL). Mild liquid phase reactions were tested at 50–66 °C using hydrogen peroxide (H2O2) as an oxidizing agent. XRD, TGA, ESR, DR-UV, and FTIR revealed the dislodged iron oxide species formed by treating at ≥500 °C. Formation of active oxidizing species on the surface occurs on contact the dislodged Fe(III) oxide with H2O2. Such active species can promote the oxidation of THF, giving high yield and selectivity of BTL, whereas the iron-containing clay treated at lower temperatures (<500 °C) perform Fenton-like oxidation with lower THF conversions and non-selective products. 2-Hydroxytetrahydrofuran (THF-2-ol) was primarily produced and further oxidized to BTL with a small amount of 4-hydroxybutyric acid as a minor product. Minimal H2O2/THF ratio of 1.0 is sufficient for the production of BTL. Deactivation can be observed presumably due to deposition of the products despite slight leaching of the active iron species.
1. Introduction
Because of factors such as finite petroleum reserves, unequal geographic distributions, and an environmentally motivated desire to decrease CO2 emissions, renewable feedstocks, such as lingo-cellulosic biomass, have become promising candidates for the production of renewable fuels and chemicals. An attractive biomass-derived chemical feedstock is butyrolactone (BTL), and its demand is steadily growing due to its major use as an important intermediate for biodegradable polymer and direct use as a dielectric material or solvent. BTL is typically produced using two processes: (i) the Davy McKee process based on the hydrogenation of diethyl or dimethyl maleate and (ii) the Reppe process based on acetylene and formaldehyde.1 Alternatively, the oxidation of tetrahydrofuran (THF) can be applied for the production of BTL. THF can be simply obtained by decarbonylation and hydrogenation of furfural derivatives,2 found mainly in biomass pyrolysis oil.
Selective oxidation3–10 is one of the key approaches for the production of BTL from THF. The earlier reports describe the synthesis of BTL by the organometallic-catalyzed oxidation of THF, which was performed homogeneously with BTL selectivity of 22%–100%, depending on the oxidant types.11–14 Although molecular oxygen (O2) is a cheap oxidant that does not generate waste, its oxidation potential is limited due to a long reaction time (>24 hours). Although H2O2 is more expensive than the molecular oxygen, it can serve as the oxidant of choice for fine chemicals because of its operational simplicity.11,12 Salavati-Niasari et al.9 reports that bis(macrocyclic) copper(II) complexes containing aromatic linkers can catalyze the oxidation of THF to afford BTL under mild conditions. However, the homogeneous process has some disadvantages such as the use of an expensive catalyst. As a comparison, heterogeneous catalytic processes serve as an environmentally benign alternative for easy catalyst separation, regeneration, and relatively low processing costs. Encapsulated complexes ([Cu(R2[16]aneN6)]2+/Zeolite Y)14 are examples of early attempts for heterogeneously catalytic systems for this approach. However, it mainly yields 2-hydroxytetrahydrofuran (THF-2-ol) and insignificant amounts of BTL, whereas, with their homogeneous counterparts, selectivity to BTL is higher.14 Titanosilicates (TS-1) can be used as heterogeneous catalysts for the oxidation of THF to BTL in moderate to good yields in the presence of dilute aqueous H2O2 as an oxidant.10 However, such catalysts in use today are based on expensive components, and methods of preparing catalysts continue to be both complicated and laborious. Iron is the element of choice that performs a number of chemically challenging oxidative processes. Fenton's reagent,15,16 a mixture of Fe(II) ions and H2O2, is widely applied to oxidize organic substrates. In the last decade, several systems based on heterogeneous Fenton-like catalysts (Fe-supported solids), such as iron oxides, zeolites, pillared clays, and alumina, have been investigated for use in environmental remediation processes.15–18 Some preliminary studies using iron-containing clay-based catalysts have shown the promising possibilities of this kind of catalysts because they are natural, abundant, inexpensive, and environmentally friendly.
In this research, greener oxidation using earth-abundant iron-containing clay as a catalyst and H2O2 as an oxidizing agent is investigated for BTL production under mild conditions. The heterogeneous oxidizing sites can be generated by simple thermal treatment at high temperatures. The ESR, DR-UV and FTIR study permits us to detect the active oxidizing iron species. Effects of thermal treatment and role of the oxidizing iron species on BTL production from THF are discussed. The reaction pathway for selective oxidation is also generalized.
2. Experimental
2.1. Catalyst preparation and characterization
The iron-containing clay was obtained from the SCG Chemicals. The sample was brown in color and used as received. Such parent clay possesses a relatively high wt% of Fe (∼5%), which makes it ideal as an oxidation catalyst. The sample was treated at 100–700 °C for 5 hours and designated as Clay-Temperature, according to their treatment temperature. XRD and TGA provided information on the catalyst structure and phase transformation upon thermal treatment. BET surface areas of all samples were also determined. The ESR spectra was taken in the X-band at 20 °C and registered at 1 mW microwave power in the field range of 10–810 mT (one scan with a sweep time of 4 min). The diffuse reflectance UV-VIS spectra of the clay samples in the form of self-supporting pellets were recorded with BaSO4-coated integration spheres. FTIR spectra were acquired in the transmission mode at room temperature over the wavenumber range of 4000–650 cm−1.
2.2. Catalytic testing
In a typical procedure for oxidation of THF, a mixture of iron-containing clay as a catalyst (0.05–0.20 g) and THF (0.05 mol) was stirred for 30 min in a 100 mL round-bottomed, three-necked flask equipped with a condenser. Then, 0.05 mol of the H2O2 (30% in H2O) was added via the dropping funnel. The mixture was then heated under reaction temperatures (50 °C and 66 °C) for 1–8 hours. At the end of reaction, the mixture was quenched in an ice bath, and the solid catalyst was separated from the solution by cold filtration. The remaining H2O2 residual was analyzed using the iodometric titration method.19 The cold mixture was promptly analyzed by GC-FID to avoid further oxidation, and the products are identified using GC-MS. The products containing the H2O2 residual was disposed based on previously reported methods.20
3. Results and discussion
3.1. Catalytic characterization
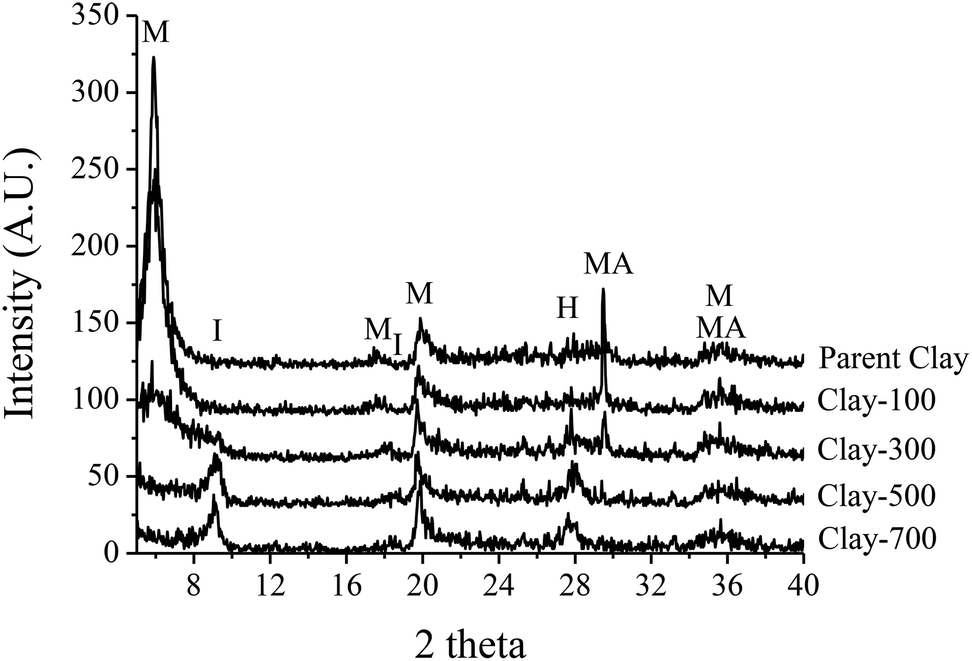
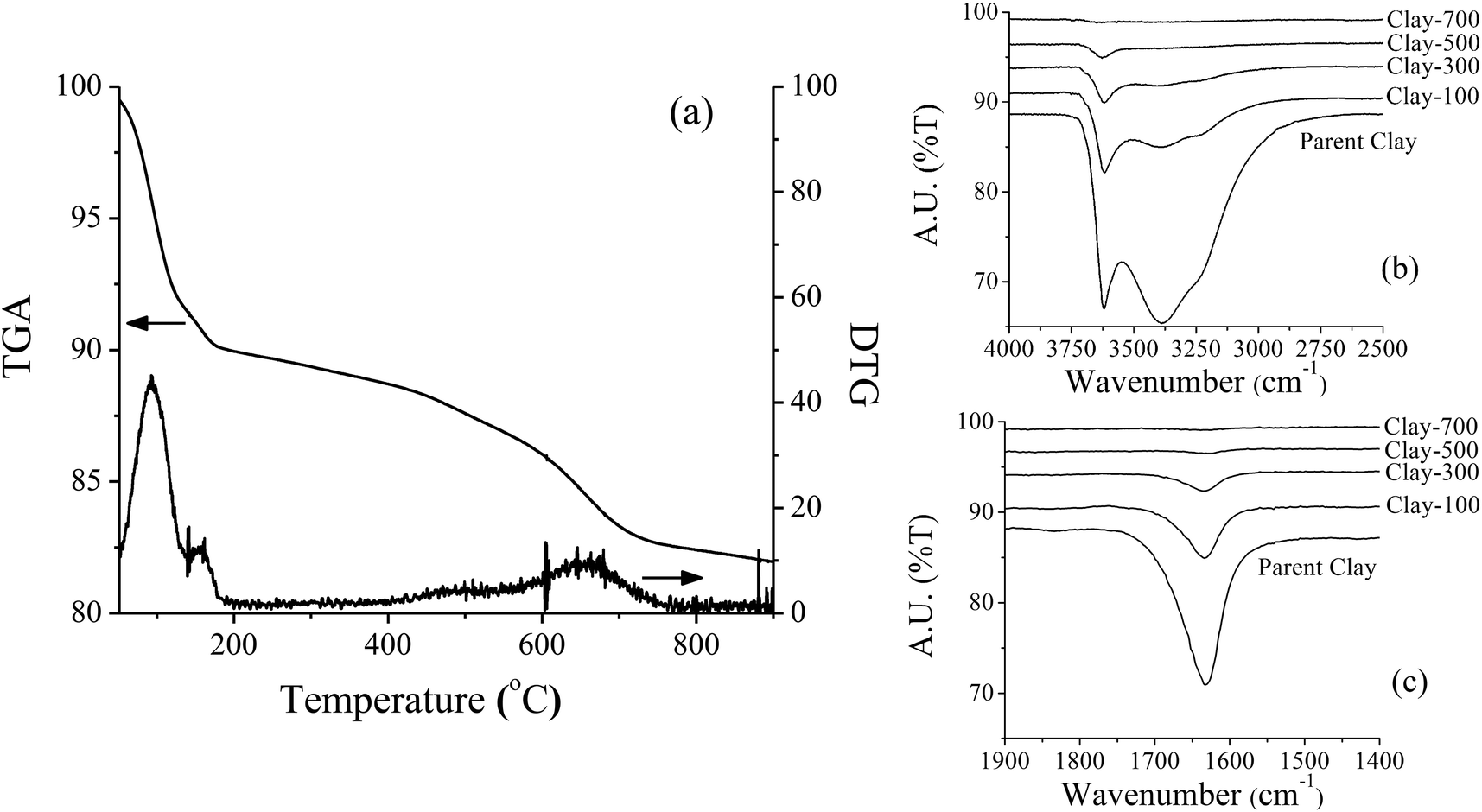
The parent iron-containing clay shows a strong diffraction peak corresponding to montmorillonite and magnetite (Fe3O4 or FeO·Fe2O3) crystallite (Fig. 1). Because it is a mineral, it is expected that these two phases are separately aggregated and exist as a physical mixture. Under thermal treatment ≥500 °C, such an iron-containing clay catalyst was completely transformed into illite and hematite (Fe2O3). The TGA and DTG profiles of the catalyst sample showed major weight-loss at 60–180 °C, corresponding to desorption of the physisorbed water and the minor weight loss at 450–700 °C, denoting the dehydroxylation of iron hydroxide and/or the clay layer (Fig. 2a). Consistent with TGA, a noticeable decrease in the terminal hydroxyl group (Si–OH, Al–OH) is observed by FTIR at ∼3630 cm−1, together with a lower intensity of bands at ∼3500–3250 cm−1 (Fig. 2b) and ∼1630 cm−1 (Fig. 2c), corresponding to the adsorbed water. A significant change in BET surface area is in line with the observed phase change by thermal treatment, whereas Fe content remains similar for all samples (Table 1).
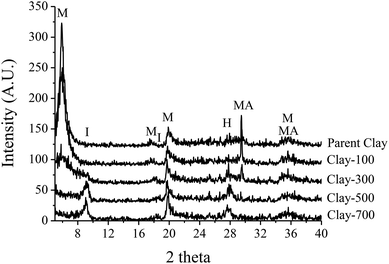 |
| | Fig. 1 XRD patterns of treated-clay (M = Montmorillonite, I = Illite, H = Hematite, MA = Magnetite). | |
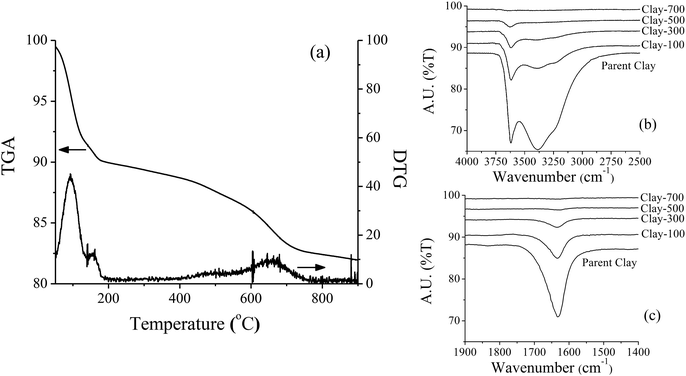 |
| | Fig. 2 TGA/DTG thermogram (a) and FTIR spectra of parent and treated-clay (b) and (c). | |
Table 1 BET surface area and Fe content
| Type of catalyst |
BET surface area (m2 g−1) |
Fe contenta (wt%) |
|
Elemental analysis of Fe done by AAS.
|
| Clay-100 |
85 |
5.4 |
| Clay-300 |
84 |
5.3 |
| Clay-500 |
72 |
5.1 |
| Clay-700 |
43 |
5.1 |
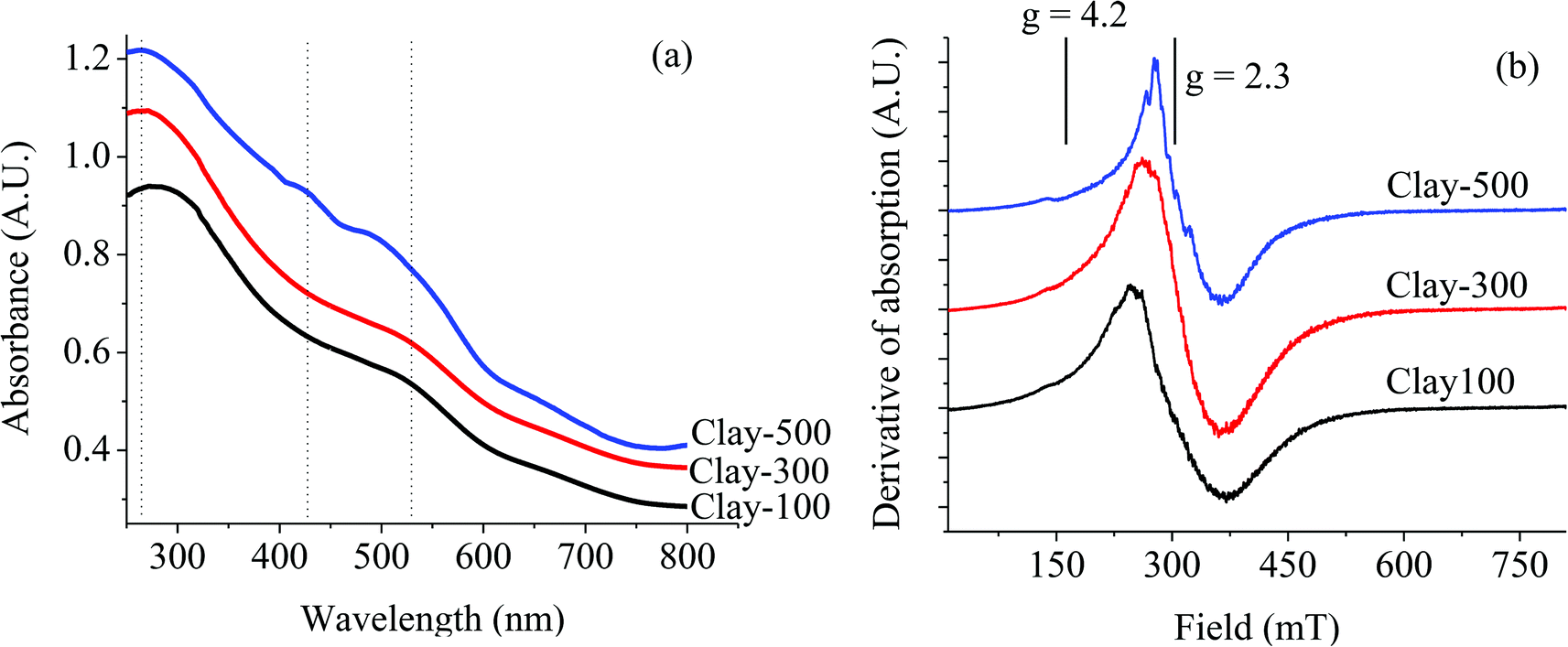
As phase transformation takes place upon calcination at ≥500 °C, DR-UV reveals different coordination of the Fe species, as shown in Fig. 3a. It can be seen that Clay-100 and Clay-300 exhibit only two absorption edges at about λ ∼263 nm and 525 nm, corresponding to octahedral Fe(III) substituted for Al(III)21 and larger particles of Fe2O3 aggregates,22 respectively. This suggests that a certain amount of Fe is also present in the clay structure, and not only as separated magnetite aggregates. However, the additional signal of the 425 nm band ascribed to the oligomeric octahedrally coordinated Fe(III) species22 can be observed on the Clay-500. This Fe species is presumably formed by the dislodgement of the substituted Fe(III) in the clay structure upon dehydroxylation, and then segregated within the clay layer. The presence of this oligomeric Fe species is also demonstrated as a series of ESR signals superimposed on the major resonance of Fe oxide clusters at g = 2.3,23,24 as shown in Fig. 3b. Experimentally, the intensity of the ESR line at g = 2.3 increased significantly when the parent clay was thermally treated due to the transformation of Fe3O4 to Fe2O3. However, such a series of superimposed signals are only pronounced when the sample was treated at 500 °C, consistent with an additionally observed UV signal at 425 nm, as previously discussed.
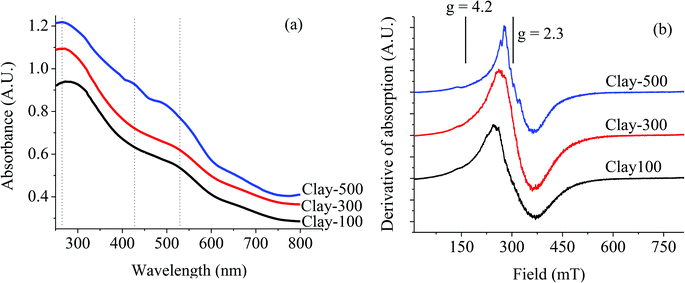 |
| | Fig. 3 DR-UV (a) and ESR (b) of thermally treated iron-containing clay. | |
It is noteworthy that the weak ESR signal at g = 4.2, which can be assigned to the presence of isolated Fe(III)23,24 in the tetrahedral or octahedral coordination, is observed for all samples. For the clay minerals, this corresponds to the Fe located in the interior of the clay sheets (iron substituting for aluminum in the octahedral layers).
3.2. Oxidation of tetrahydrofuran
Table 2 shows the activity of various thermally treated iron-containing clay catalysts in the oxidation of THF to BTL. The Clay-500 exhibits relatively higher activity, as compared to those from the Clay-100, Clay-300, and Clay-700. It is clear that the oxidation activity is generally increased with the treatment temperature. However, the lower activity observed in the reaction using Clay-700 as the catalyst is presumably due to the significant loss of catalyst surface area (Table 1) by further dehydroxylation at 700 °C. This is consistent with the TGA/DTG and FTIR results (Fig. 2) that show the dehydroxylation of the clay layer.
Table 2 Type of catalyst and reaction temperaturea
| Type of catalyst |
Clay-100 |
Clay-300 |
Clay-500 |
Clay-700 |
Clay-500 |
| Reaction temperature (°C) |
66 °C |
66 °C |
66 °C |
66 °C |
50 °C |
|
Reaction condition: reaction temperature = 50, and 66 °C, reaction time = 4 h, catalyst type = Clay-100, Clay-300, Clay-500, and Clay-700, catalyst content = 0.1 g, H2O2 = 0.05 mol, THF = 0.05 mol.
|
|
Conversion (%)
|
14.81 |
19.22 |
26.91 |
11.54 |
8.45 |
|
Yield (%)
|
| Butyrolactone |
6.41 |
9.78 |
16.65 |
6.70 |
5.08 |
| 2-Hydroxytetrahydrofuran |
3.69 |
4.52 |
6.85 |
2.60 |
2.53 |
| 4-Hydroxybutyric acid |
1.61 |
2.03 |
1.22 |
1.02 |
0.35 |
| Others |
3.10 |
2.89 |
2.19 |
1.22 |
0.49 |
| |
|
Selectivity (%)
|
| Butyrolactone |
43.28 |
50.88 |
61.87 |
58.06 |
60.12 |
| 2-Hydroxytetrahydrofuran |
24.92 |
23.52 |
25.46 |
22.53 |
29.94 |
| 4-Hydroxybutyric acid |
10.87 |
10.56 |
4.53 |
8.84 |
4.14 |
| Others |
20.93 |
15.04 |
8.14 |
10.57 |
5.80 |
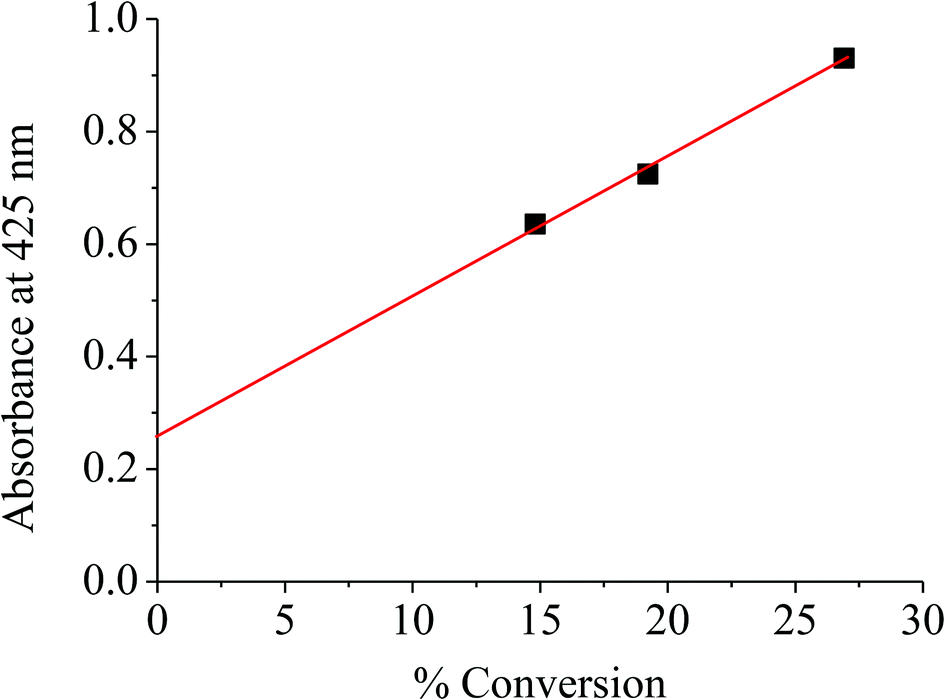
With respect to the same range of catalyst surface area (72–85 m2 g−1), the increase in activity with the treatment temperature can account for the degree of dehydroxylation and hence the formation of oligomeric octahedrally coordinated Fe(III) species formed by dislodgement of the substituted Fe(III) in the clay structure, as demonstrated by TGA, FTIR, DR-UV, and ESR. This is speculated from a linear relationship between the catalyst activity (conversion) and the UV absorbance at 425 nm for such Fe(III) species, as shown in Fig. 4. It is expected that the dislodged Fe(III) oxide species would possess higher degrees of coordinative unsaturation, as compared to those in the bulk oxide. Hence, H2O2 can readily react with such Fe(III) species forming high valence iron-oxo, iron-peroxo or iron-superoxo species (Surface-Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, Surface-Fe(O–O) or Surface-FeOO−˙),25–28 which act as oxidizing species. As higher extents of dehydroxylation was observed when treated at 500 °C, higher fractions of dislodged Fe(III) oxide species, and hence higher activity could be expected.
O, Surface-Fe(O–O) or Surface-FeOO−˙),25–28 which act as oxidizing species. As higher extents of dehydroxylation was observed when treated at 500 °C, higher fractions of dislodged Fe(III) oxide species, and hence higher activity could be expected.
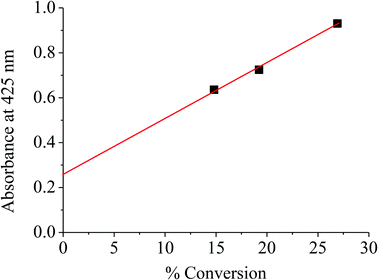 |
| | Fig. 4 % Conversion versus absorbance of catalyst at 425 nm. | |
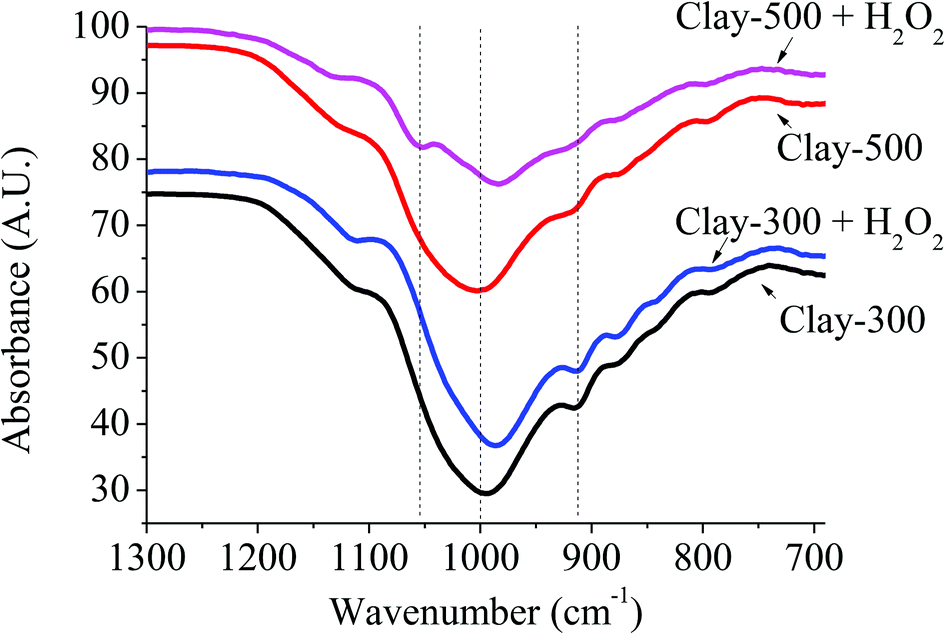
The presence of active oxidizing species generated by the reaction of H2O2 and the dislodged Fe(III) oxide can be further supported by FTIR spectroscopy. From Fig. 5, the band at ∼1000 cm−1 was assigned to Fe–O–H overlapped with Si–O–Al in montmorillonite,29 whereas the signal at 916 cm−1 can be referred to Al2–OH.30 Interestingly, a new signal at 1050 cm−1 became pronounced when H2O2 is introduced into Clay-500. This relatively high-frequency signal is generally attributed to O–O stretching of end-on superoxo or peroxo complexes.31–34 Hence, it is proposed in this work that the new signal at 1050 cm−1 is derived from the formation of high-valence iron-superoxo or peroxo species (Fe–O–O−˙ or Fe(O–O)) on the surface of dislodged Fe oxide via surface oxidation by H2O2. This species may well be an active oxidizing species or may also be a precursor of a hydroxyl and hydroperoxyl Fe species (Fe(OH)(OOH))28,35 that promote the oxidation of THF. It is important to emphasize that this additional signal at 1050 cm−1 is particularly obtained only for the clay treated at high temperatures (Clay-500 and Clay-700) without a certain degree of dehydroxylation; hence, the presence of dislodged Fe oxide species, the bulk Fe oxide and substituted Fe in the clay structure may be too stable to interact with H2O2 to form such Fe superoxo or peroxo species. The formation of the active site can be proposed as follows:
| | | Fe(III) oxide in the framework → Surface-Fe(III) (Dislodged species) | (1) |
| | | Surface-Fe(III) + H2O2 → Surface-Fe(III)–O2−˙ | (2) |
| | | Surface-Fe(III)–O2−˙ + H2O → Surface-Fe(IV)(OH)(OOH) | (3) |
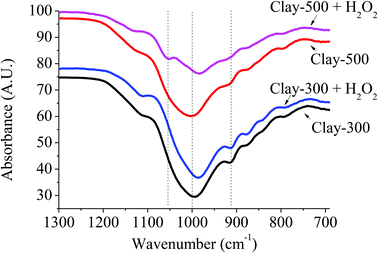 |
| | Fig. 5 FTIR of thermally treated iron-containing clay with and without H2O2. | |
It could be expected that the Fenton-type oxidation36,37 would be also responsible for the observed activity. This may be true in the case of iron-containing clay treated at low temperatures (Clay-100 or Clay-300) because a larger fraction of Fe(II) oxide species is present in these samples. However, a relatively lower conversion of THF was obtained from the reaction over that of Clay-100 or Clay-300. This is because the Fenton process involves the reaction of Fe(II) with H2O2, giving rise to hydroxyl radicals (˙OH) (eqn (4)). This catalytic cycle can be completed by the reduction of Fe(III) to Fe(II) (eqn (5)):37
| | | Surface-Fe(II) + H2O2 → Surface-Fe(III) + ˙OH + OH− | (4) |
| | | Surface-Fe(III) + H2O2 → Surface-Fe(II) + ˙OOH + H+ | (5) |
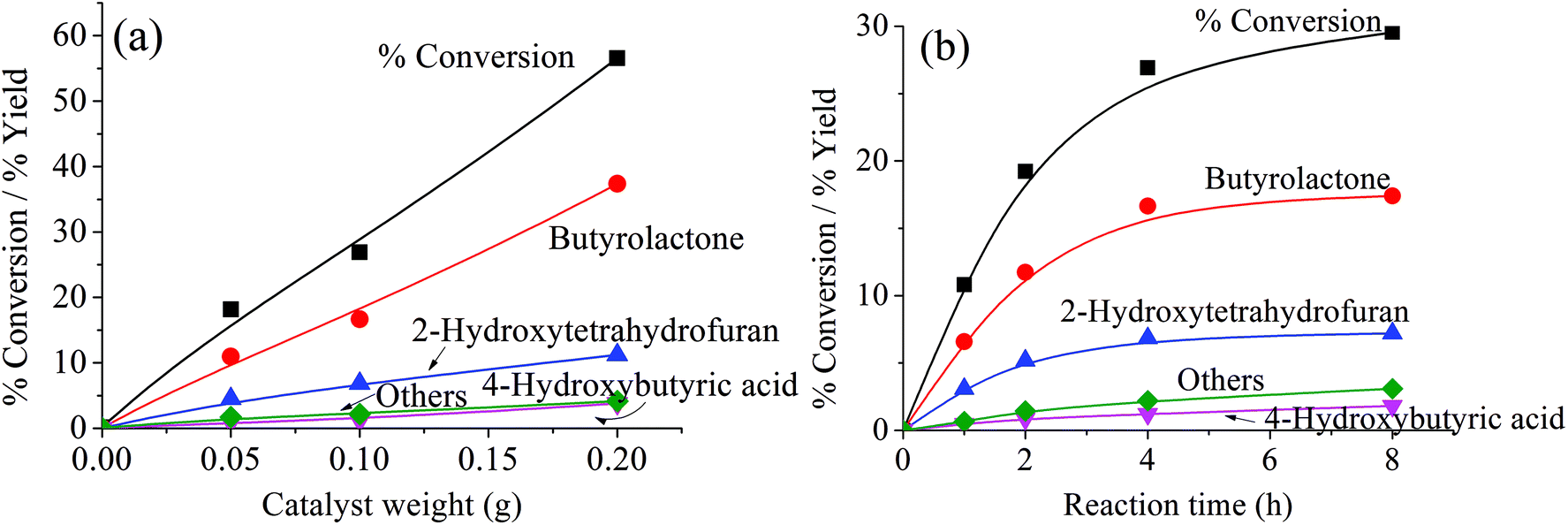
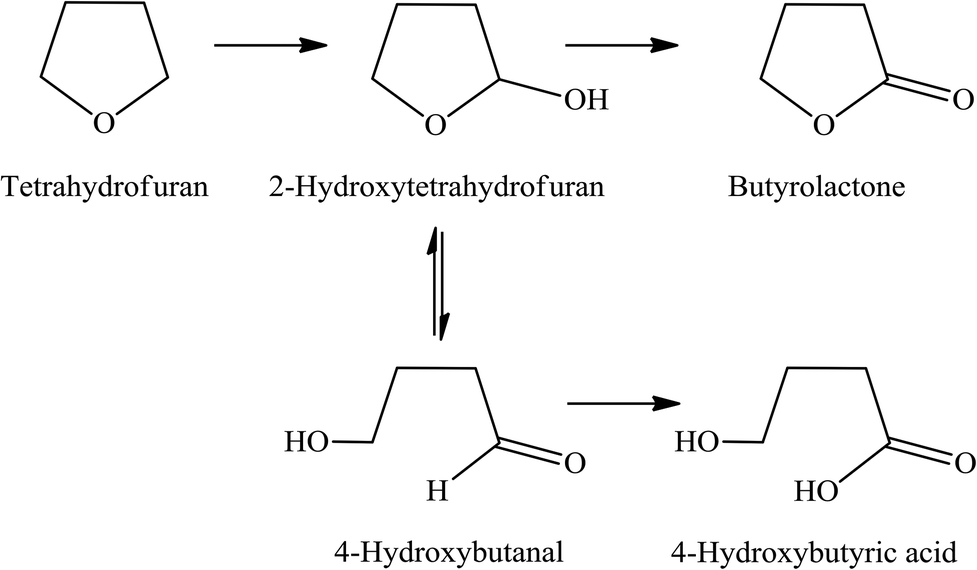
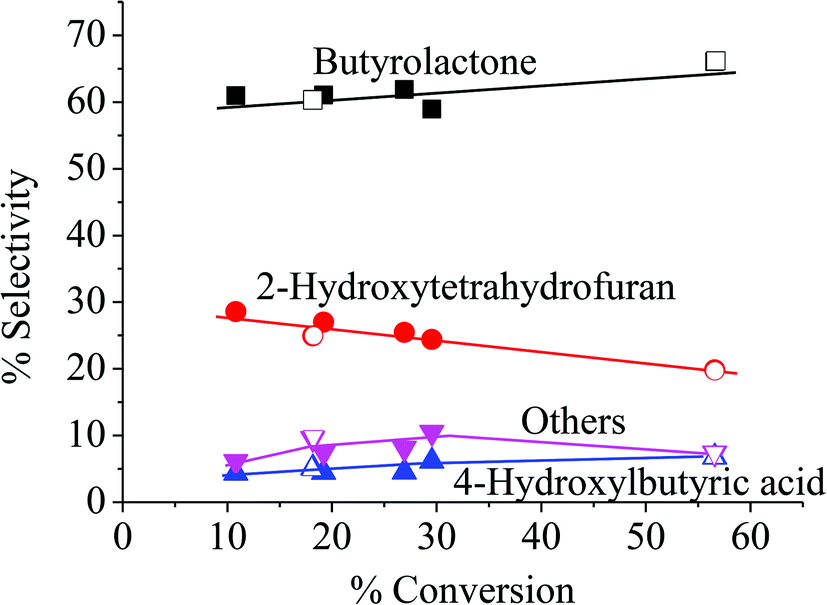
Because the reduction of Fe(III) to Fe(II) by H2O2 is extremely slow,38–40 the Fe(III) would be accumulated in the system and would result in low activity.41 Therefore, the observed higher activity for the catalyst treated at higher temperatures (Clay-500) suggests that the active oxidizing center would be derived from the high-valence iron-oxo species that is formed by interaction of H2O2 with the dislodged Fe(III) oxides rather than the Fenton-type species. For all catalysts, butyrolactone (BTL) is the main product from THF oxidation. Major co-products include 2-hydroxytetrahydrofuran (THF-2-ol) and 4-hydroxybutyric acid (4-HBA). Furanone and polyhydroxy derivatives of the aforementioned products are also observed. It is clear from Fig. 6 that the conversion is increased together with yields of all products when both catalyst content and reaction time is increased, suggesting that the reaction is catalytically controlled. As BTL and THF-2-ol appears to form in parallel, these products may well be derived from the decomposition of the parent hydroperoxide intermediate (in a manner similar to ketone-alcohol from the decomposition of alkyl hydroperoxide). However, it is observed that THF-2-ol yield started to level off, whereas yields of BTL and 4-HBA steadily increased with conversion. This also suggests that THF-2-ol may well be primarily produced from the oxidation of THF and could become a precursor of BTL and 4-HBA (Scheme 1). From the mechanistic point of view, the H-abstraction at the carbon adjacent to the oxygen in THF can be most likely promoted, and hydroxylation at this position readily leads to formation of THF-2-ol. This product is prone to be further oxidized to BTL. It is possible that the BTL is formed either by (i) the direct oxidation of THF-2-ol, whereas the ring is kept intact or by (ii) ring opening of THF-2-ol to 4-hydroxybutanal that is subsequently oxidized to 4-HBA. This ω-hydroxy acid can also undergo self-esterification to form BTL. Although 4-HBA is actually found as evidence for the ring opening-oxidation (ii), it is unlikely that the self-esterification of 4-HBA to form BTL is the case in the reaction with large amount of water present. Moreover, selectivity of 4-HBA is relatively small and is increased together with BTL selectivity, whereas THF-2-ol selectivity is reduced with increased conversion (Fig. 7). This suggests that BTL and 4-HBA may be produced in parallel, presumably from the oxidation of THF-2-ol. It is also noteworthy that BTL is relatively stable under the reaction condition, and no hydrolysis of BTL to 4-HBA was observed when BTL was used as feed in a separate test (not shown). The overall reaction pathway can be proposed as shown in Scheme 1.
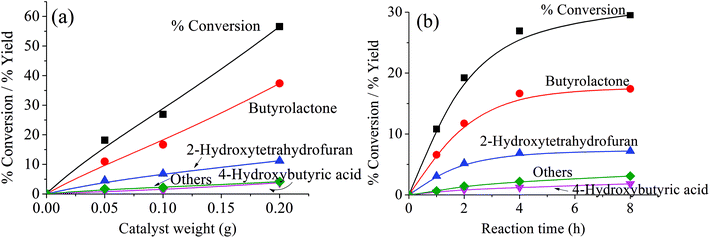 |
| | Fig. 6 Product distribution as a function of catalyst weight and reaction time. Reaction condition: reaction temperature = 66 °C, reaction time = 4 h for (a) and 1–8 h for (b), catalyst = Clay-500, catalyst content = 0.05–0.20 g for (a) and 0.10 g for (b), H2O2 = 0.05 mol, THF = 0.05 mol. | |
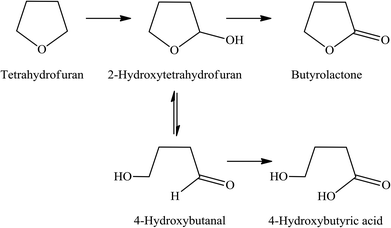 |
| | Scheme 1 Proposed reaction mechanism of THF oxidation over Clay-500 catalyst. | |
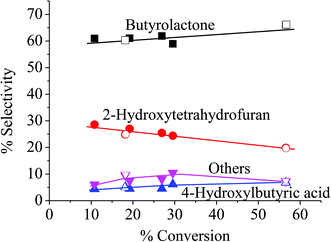 |
| | Fig. 7 Product distribution. Reaction condition: reaction temperature = 66 °C, reaction time = 1–8 h, catalyst type = Clay-500, catalyst content = 0.05–0.20 g, H2O2 = 0.05 mol, THF = 0.05 mol. | |
As also seen in Fig. 6, small amounts of other products, such as 2-furanone and 3,4-dihydroxytetrahydrofuran, were obtained with increased catalyst content and time. However, no significant change in the selectivity can be observed at lower temperatures (Table 2). These co-products may be the result of further oxidation of C–H bond at different positions on the major products. However, the selectivity of these products is particularly high in the reaction using Clay-100 and Clay-300 as catalysts despite the lower conversions that were obtained (Table 2). This is presumably because there is a much smaller amount of dislodged Fe(III) oxide present in these samples, and hence a smaller number of active Fe superoxo or peroxo species as previously discussed. Nevertheless, the oxidation of THF can be slightly facilitated via an ˙OH radical mechanism that is promoted by the decomposition of H2O2 over Fe(II) oxide surface. Although they are less active as compared to the Clay-500, the ˙OH radical initiates a radical chain reaction by randomly abstracting hydrogen atoms from THF. This leads to the generation of carbon radicals that undergo oxidation to various oxygenated products.
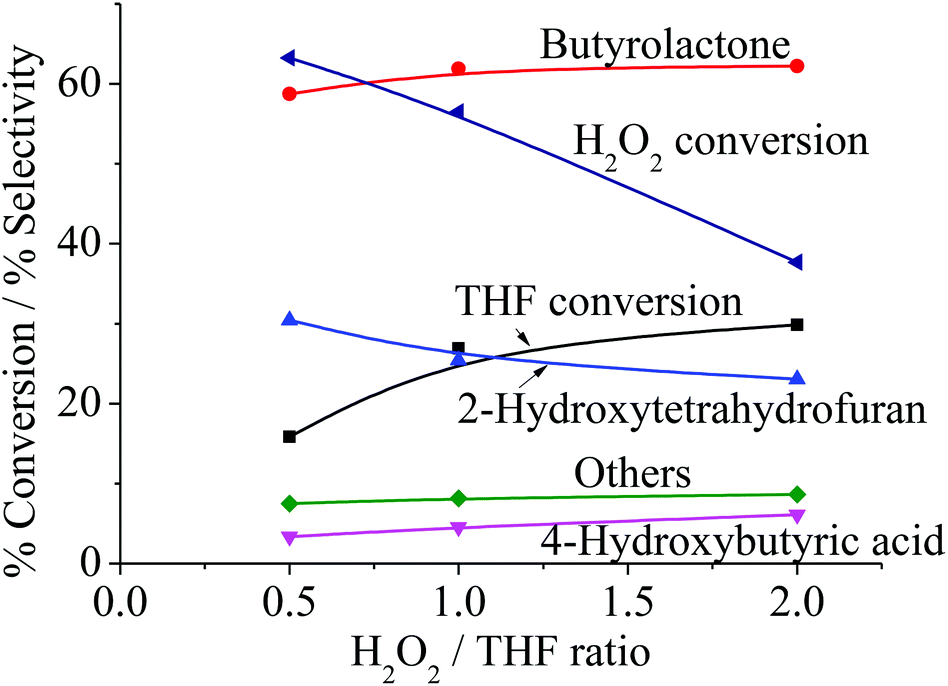
From Fig. 6b, it can be observed that the activity gradually declines over the course of the reaction. This can be attributed either to (i) the limit of the oxidant, H2O2, or (ii) the deactivation of the catalysts. Accordingly, the effect of H2O2/THF ratio was investigated, and it is clear that the decline in activity observed in Fig. 6b is not due to the limit of H2O2. As seen from Fig. 8, no significant increase in activity is obtained when the H2O2/THF ratio was raised to 2, which suggested that H2O2/THF ratio of 1 is sufficient for the oxidation of THF to BTL and shall not restrain the activity as observed in Fig. 6b. Although the excess H2O2 does not strongly affect the conversion, the selectivity of 4-HBA and other products are slightly increased with H2O2/THF ratio, whereas THF-2-ol selectivity is decreased. This indicates that further oxidation of THF-2-ol can be promoted when H2O2 is in excess.
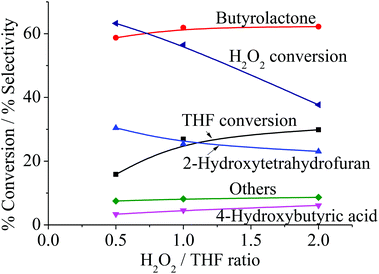 |
| | Fig. 8 Product distribution as a function of H2O2/THF ratio. Reaction condition: reaction temperature = 66 °C, reaction time = 4 h, catalyst type = Clay-500, catalyst content = 0.1 g, H2O2 = 0.025–0.10 mol, THF = 0.05 mol. | |
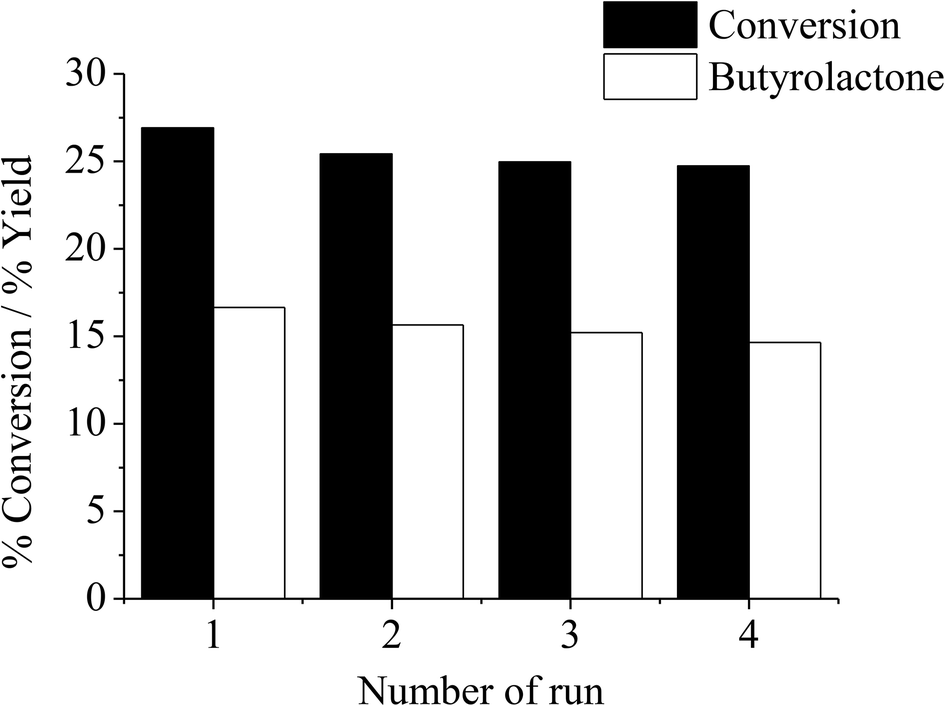
According to the study on the effects of H2O2, it appears that the observed decline in activity after 4 hours would result from the catalyst deactivation. This may be due to the adsorption of products on the catalyst surface or leaching of the active iron species. However, it is clear from Fig. 9 that severe catalyst leaching is not the case because as the catalyst recovered from THF oxidation experiment was tested under similar conditions for an additional three cycles, the catalytic activity is reproducible in consecutive experiments without a remarkable drop in efficiency. As iron leaching is not evidenced, the observed deactivation is presumably due to the adsorption of products, and the washing with THF or an appropriate solvent can easily regenerate the catalyst.
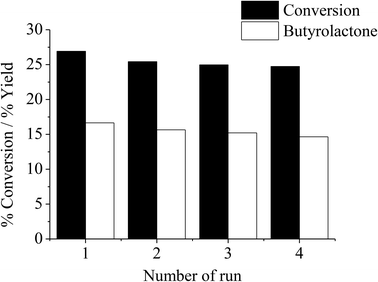 |
| | Fig. 9 Reusability of thermally treated iron-containing clay catalyst. Reaction condition: reaction temperature = 66 °C, reaction time = 4 h, catalyst type = Clay-500, catalyst content = 0.1 g, H2O2 = 0.05 mol, THF = 0.05 mol. | |
4. Conclusions
Butyrolactone can be successfully produced via a greener oxidation of THF using earth-abundant iron-containing clay as catalysts. The active oxidizing species can be obtained by thermal treatment at temperatures ≥500 °C. The dehydroxylation of the clay structure leads to partial dislodgement of the iron oxide species that can react with hydrogen peroxide. Spectroscopic evidences suggest the formation of Fe superoxo or peroxo upon contacting hydrogen peroxide with the dislodged Fe oxide species, which is also well correlated with the observed activity. It appears that 2-hydroxytetrahydrofuran (THF-2-ol) is primarily a product from THF oxidation. This product is further obtained by oxidation of butyrolactone (BTL), 4-hydroxybutyric acid (4-HBA) and other oxidized products. Up to 70% selectivity of BTL is obtained over the Clay-500 catalyst. H2O2/THF ratio of 1.0 is sufficient for BTL production, and no significant leaching of the active species can be observed. The catalyst can be simply regenerated by washing with the feed, THF, and re-used for at least 4 cycles without a significant drop in activity.
Acknowledgements
This work was financially supported by the Thailand Research Fund (grant no. TRG-5780177).
References
- U. R. Pillai, E. S. Demessie and D. Young, Appl. Catal., B, 2003, 43, 131–138 CrossRef CAS.
- S. Sitthisa, T. Pham, T. Prasomsri, T. Sooknoi, R. G. Mallinson and D. E. Resasco, J. Catal., 2011, 280, 17–27 CrossRef CAS PubMed.
- C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34(8), 633–639 CrossRef CAS PubMed.
- R. A. Periana, O. Mironov, D. Taube, G. Bhalla and C. J. Jones, Science, 2003, 301, 814–818 CrossRef CAS PubMed.
- S. Pradhan, J. K. Bartley, D. Bethell, C. A. Frederick, C. Marco, G. Stanislaw, H. Matthew, J. R. Leyshon, L. Rhys and H. G. John, Nat. Chem., 2012, 4, 134–139 CrossRef CAS PubMed.
- P. Stavropoulos, R. Celenligil-Cetin and A. E. Tapper, Acc. Chem. Res., 2001, 34, 745–752 CrossRef CAS PubMed.
- D. H. R. Barton, Tetrahedron, 1998, 54, 5805–5817 CrossRef CAS.
- D. H. R. Barton and D. Doller, Acc. Chem. Res., 1992, 25, 504–512 CrossRef CAS.
- M. Salavati-Niasari and M. Bazarganipour, Inorg. Chem. Commun., 2006, 9, 332–336 CrossRef CAS PubMed.
- M. Sasidharan and A. Bhaumik, J. Mol. Catal. A: Chem., 2011, 338, 105–110 CAS.
- M. Sommovigo and H. Alper, J. Mol. Catal., 1994, 88, 151–158 CrossRef CAS.
- P. Li and H. Alper, J. Mol. Catal., 1992, 72, 143–152 CrossRef CAS.
- L. Metsger and S. Bittner, Tetrahedron, 2000, 56, 1905–1910 CrossRef CAS.
- M. Salavati-Niasari, Inorg. Chem. Commun., 2006, 9, 628–633 CrossRef CAS PubMed.
- H. J. H. Fenton, Chem. News, 1876, 33, 190–200 Search PubMed.
- H. J. H. Fenton, J. Chem. Soc. Proc., 1894, 10, 157–158 Search PubMed.
- J. H. Ramirez, F. J. Maldonado-Hodar and A. F. Perez-Cadenas, Appl. Catal., B, 2007, 75, 312–323 CrossRef CAS PubMed.
- G. Calleja, J. A. Melero, F. Martinez and R. Molina, Water Res., 2005, 39, 1741–1750 CrossRef CAS PubMed.
- C. Kormann, D. W. Bahnemann and M. R. Hoffmann, Environ. Sci. Technol., 1988, 22, 798–806 CrossRef CAS PubMed.
- W. Liu, S. A. Andrews, M. I. Stefan and J. R. Bolton, Water Res., 2003, 37, 3697–3703 CrossRef CAS.
- M. N. Timofeeva, S. T. Khankhasaeva, Y. A. Chesalov, S. V. Tsybulya, V. N. Panchenko and E. T. Dashinamzhilova, Appl. Catal., B, 2009, 88, 127–134 CrossRef CAS PubMed.
- S. Caudo, G. Centi, C. Genovese and S. Perathoner, Appl. Catal., B, 2007, 70, 437–446 CrossRef CAS PubMed.
- A. Kucherov and M. Shelef, J. Catal., 2000, 195, 106–112 CrossRef CAS.
- E. Gue'lou, J. Barrault, J. Founier and J. M. Tatibouët, Appl. Catal., B, 2003, 44, 1–8 CrossRef.
- W. C. Bray and M. H. Gorin, J. Am. Chem. Soc., 1932, 54, 2124–2125 CrossRef CAS.
- E. Kim, M. E. Helton, I. M. Wasser, K. D. Karlin, S. Lu, H. Huang, P. Moenne-Loccoz, C. D. Incarvito, A. L. Rheingold, M. Honecker, S. Kaderli and A. D. Zuberbuhler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(7), 3623–3628 CrossRef CAS PubMed.
- M. Newcomb, P. F. Hollenberg and M. J. Coon, Arch. Biochem. Biophys., 2003, 409, 72–79 CrossRef CAS.
- H. Mimoun, I. Seree de Rochm and L. Sayas, Bull. Soc. Chim. Fr., 1969, 5, 1481–1492 Search PubMed.
- P. Yuan, F. Annabi-Bergaya, Q. Tao, M. Fan, Z. Liu, J. Zhu, H. He and T. Chen, J. Colloid Interface Sci., 2008, 324, 142–149 CrossRef CAS PubMed.
- J. Madejova, Vib. Spectrosc., 2003, 31, 1–10 CrossRef CAS.
- Y. M. Lee, S. Hong, Y. Morimoto, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2010, 132, 10668–10670 CrossRef CAS PubMed.
- J. Annaraj, Y. Suh, M. S. Seo, S. O. Kim and W. Nam, Chem. Commun., 2005, 4529–4531 RSC.
- J. Cho, S. Jeon, S. A. Wilson, L. V. Liu, E. A. Kang, J. J. Braymer, M. H. Lim, B. Hedman, K. O. Hodgson, J. S. Valentine, E. I. Solomon and W. Nam, Nature, 2011, 478, 502–505 CrossRef CAS PubMed.
- J. U. Rohde, J. H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que Jr., Science, 2003, 299, 1037–1039 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codola’, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- R. Matta, K. Hanna and S. Chiron, Sci. Total Environ., 2007, 385(1–3), 242–251 CrossRef CAS PubMed.
- W. P. Kwan and B. M. Voelker, Environ. Sci. Technol., 2003, 37, 1150–1158 CrossRef CAS.
- W. G. Barb, J. H. Baxendale, P. George and K. R. Hargrave, Nature, 1949, 163, 692–694 CrossRef CAS.
- W. G. Barb, J. H. Baxendale, P. George and K. R. Hargrave, Trans. Faraday Soc., 1951, 47, 462–500 RSC.
- W. G. Barb, J. H. Baxendale, P. George and K. R. Hargrave, Trans. Faraday Soc., 1951, 47, 591–616 RSC.
- C. Jiang, Z. Gao, H. Qu, J. Li, X. Wang, P. Li and H. Liu, J. Hazard. Mater., 2013, 250–251, 76–81 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.