
Mechanistic insights into the hydroconversion of cinnamaldehyde using mechanochemically-synthesized Pd/Al-SBA-15 catalysts
Alfonso
Yepez
a,
José M.
Hidalgo
*ab,
Antonio
Pineda
a,
Radek
Černý
b,
Petr
Jíša
b,
Angel
Garcia
a,
Antonio A.
Romero
a and
Rafael
Luque
a
aDepartamento de Química Orgánica, Universidad de Córdoba, Campus de Rabanales, Edificio Marie Curie (C-3), Ctra Nnal IV-A, Km 396, E-14014, Córdoba, Spain. E-mail: q62alsor@uco.es; Fax: +34 957212066; Tel: +34 957211050
bResearch Institute of Inorganic Chemistry (VUAnCh), Department of Efficient Refining Technologies (UniCRE-EFFRET), Chempark Litvínov, 43670 Litvínov, Czech Republic. E-mail: jose.hidalgo@vuanch.cz
First published on 2nd October 2014
Abstract
The hydroconversion of cinnamaldehyde as an α,β-unsaturated compound was studied using a simple and efficient hydrogen-donating protocol catalyzed by mechanochemically synthesized bifunctional Pd/Al-SBA-15 catalysts. Materials were characterized using TGA-TDA, nitrogen physisorption, TEM and EDX analyses. Catalytic results pointed to the presence of competitive pathways that are able to provide a selectivity switch from the expected fully hydrogenated aromatic ring (e.g. cyclohexane) and hydrogenated products (e.g. hydrocinnamaldehyde, cinnamyl alcohol and 3-phenylpropan-1-ol) to the unexpected ethylbenzene and oxalic acid products from hydrodeformylation and hydrocarboxylation reactions of cinnamaldehyde and formic acid, respectively.
Introduction
The selective hydrogenation of α,β-unsaturated aldehydes has attracted significant interest in the production of fine chemicals especially for the fragrances and flavoring industries.1 In particular, the hydroconversion of cinnamaldehyde to various important derivatives including cinnamyl alcohol and hydrocinnamaldehyde has been the subject of mechanistic studies with different types of catalytic systems to investigate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vs. CO hydrogenation selectivities and the potential existence of structure sensitive processes.2–4 In any case, the process also has relevance from the industrial point of view as typically obtained products (e.g. cinnamyl alcohol, hydrocinnamaldehyde) can find applications as additives in the food and pharmaceutical industries.5
C vs. CO hydrogenation selectivities and the potential existence of structure sensitive processes.2–4 In any case, the process also has relevance from the industrial point of view as typically obtained products (e.g. cinnamyl alcohol, hydrocinnamaldehyde) can find applications as additives in the food and pharmaceutical industries.5
Reaction pathways and mechanisms in cinnamaldehyde hydroconversion have been proposed under a rather general consensus that hydrogenations, hydrogenolysis and dehydrations are key processes leading to most compounds illustrated in Scheme 1.2–7 A wide range of conditions and several heterogeneous catalysts have been utilized in the reaction, mostly supported noble metal catalysts (e.g. Pd, Pt, Ru) on porous materials including zeolites, silicates (SBA and MCM-type materials), alumina, etc. at moderate temperatures and high pressures of hydrogen.5,8–12 Interestingly, no literature reports could be found on the use of hydrogen-donating solvents for cinnamaldehyde hydroconversion.
 | ||
| Scheme 1 Reaction scheme of the cinnamaldehyde hydrogenation. Adapted from ref. 3. | ||
Formic acid is a biomass-derived chemical that readily decomposes under certain metal-catalysed and temperature conditions (e.g. microwave-irradiation) into H2 and a mixture of CO + CO2, providing a potentially useful alternative to conventional hydrogen-donors (e.g. isopropanol) in hydrogen-transfer reactions13–15 as well as in nanomaterials synthesis.16,17 Due to its acidic properties, formic acid has additionally been proven to be highly effective in promoting acidolytic cleavage of C–C and C–O bonds in starting materials as recalcitrant as lignin.18
In light of the excelling features of formic acid as a hydrogen-donor and a (co-)catalyst as well as current possibilities to be derived from biomass (concomitantly produced with levulinic acid from cellulose hydrolysis/dehydration19,20), we were prompted to explore a novel cinnamaldehyde hydroconversion strategy using formic acid as both a hydrogen-donating solvent and a co-catalyst under conventional heating and microwave irradiation. Microwave protocols have been extensively reported to accelerate the reaction rates as compared to conventionally heated protocols, often changing selectivities to products, due to the rapid homogeneous heating achieved in the bulk solution as compared to conventional heating.21 Previous work from the group also pointed out a synergetic effect between the use of microwave irradiation and formic acid decomposition catalyzed by supported metal nanoparticles.17,18
Herein, we report the mechanochemical preparation of various Pd-containing Al-SBA-15 with different Pd contents as bifunctional catalysts for the hydroconversion of cinnamaldehyde using formic acid. Results of the activity of supported Pd aluminosilicates were compared to that of a commercial 5% Pd/C.
Experimental
All chemicals and reagents used in this study (at least 99% purity) were purchased from Sigma Aldrich (unless otherwise stated) and were not further purified.Synthesis of supports
Al-SBA-15 materials (Si/Al = 20 ratio) were synthesized according to a previously reported protocol. A solution (300 mL) of hydrochloric acid at pH = 1.5 was prepared, then 8.07 g of the surfactant (poly (ethylene-glycol)-block (P123 Aldrich) was added to this solution. The mixture was stirred until one final transparent solution was obtained. 0.82 g of aluminum isopropoxide 98 wt% (Aldrich) was then added to the mixture followed by 18 mL TEOS (tetraethylortosilicate) added dropwise (time 5–10 min). The final gel was stirred for 24 h and introduced into a furnace at 100 °C, t = 24 h. The products obtained were filtered and calcined in air at 600 °C for 8 h (2 h in a nitrogen atmosphere and 6 h in air).Mechanochemical synthesis of Pd/Al-SBA-15
Pd/Al-SBA-15 materials were synthesized following a previously reported mechanochemical protocol.17,22 Different quantities of a solid Pd precursor [palladium(II) acetate] to reach theoretical Pd contents of 0.5, 1, 2 and 4 wt% Pd in the materials were milled together with the pre-formed Al-SBA-15 in the solid phase in a Retsch PM 100 bioMETA planetary ball mill (optimum conditions: 350 rpm, 10 minutes milling17). Upon milling, the final mechanochemical material obtained was calcined at 450 °C in air (10 °C min−1) for 2 hours. For simplicity, the supported Pd nanoparticle Al-SBA-15 materials have been denoted as PdX-Al where X stands for the Pd content in the materials and Al refers to Al-SBA-15. A commercial 5% Pd/C catalyst (Sigma-Aldrich) has been also employed as a reference catalyst for comparative purposes.Catalyst characterisation
Thermal analysis was performed by simultaneous TG-DTA measurement using a System Setaram Setsys 12 TGA instrument. Samples were heated at a rate of 10 °C min−1, in air (40 mL min−1) at the temperature range 50–1000 °C. The compound α-Al2O3 was used as a reference material.Nitrogen adsorption measurements were carried out at 77 K using an ASAP 2000 volumetric adsorption analyzer from Micromeritics. The samples were outgassed for 2 h at 373 K under vacuum (p < 10−2 Pa) and subsequently analyzed. The linear part of the BET equation (relative pressure between 0.05 and 0.30) was used for the determination of the specific surface area. The pore size distribution was calculated from the adsorption branch of the N2 physisorption isotherms and the Barret–Joyner–Halenda (BJH) formula. The cumulative mesopore volume VBJH was obtained from the PSD curve.
Energy-dispersive X-ray spectroscopy (EDX) measurements were performed using a Scanning Electron Microscope (SEM) JEOL JSM 6300 provided with a microanalysis system Inca Energy 250, with a SiLi detector. Detection interval: from boron to uranium, resolution: 137 eV to 5.9 keV.
Catalytic experiments
Results and discussion
TGA-DTA analyses were carried out to study the desorption–calcination of the parent materials (filtered material during the synthesis, previous to calcination at 600 °C to obtain the SBA-15 solids without adding Pd). The first weight loss at 100 °C (endothermic – TDA signal) of 2–5 wt% was due to the presence of water over the catalyst. The next weight loss (exothermic signals; T = 200–500 °C; 35 wt% mass loss, Fig. 1) can be assigned to the combustion–desorption of carbonaceous species used for the catalyst synthesis (mainly surfactants). From 500 °C only a small amount of weight loss was detected (dehydroxylation of –OH species over the surface of the support). DTA experiments showed an endothermic signal at ca. 100 °C and then two clear exothermic signals (230 and 270 °C) from the desorption–combustion of the surfactant species (Fig. 2). | ||
| Fig. 1 TGA of Al-SBA-15 prior to calcination. | ||
 | ||
| Fig. 2 DTA of parent Al-SBA-15 before calcination. | ||
Textural properties of mechanochemically synthesized supported nanomaterials have been included in Table 1. As expected, a significant decrease in the surface area of all catalysts could be observed upon metal incorporation. Pore sizes were also observed to decrease particularly for samples with higher metal loading (e.g. Pd4-Al = 4% Pd/Al-SBA-15). Interestingly, no significant pore blocking could be observed at low incorporated metal loadings (nanoparticles are distributed within the external surface and the pores of the material as previously demonstrated17,22). Pd4-Al experienced a remarkable reduction in pore size as compared to parent Al-SBA-15 (from 8.9 to 6.8) which may be due to a partial pore blocking upon Pd incorporation within the pores of the material. Pore volumes also followed a similar trend, being significantly reduced upon metal incorporation in Al-SBA-15. Representative TEM images of the metal-containing SBA-15 materials as compared to the parent Al-SBA-15 have been included in Fig. 3. A structurally ordered Al-SBA-15 material can be observed in Fig. 3A, with clearly visible mesopore sizes in good agreement with porosity data (<10 nm, Fig. 3A).
 | ||
| Fig. 3 TEM images of (A) Al-SBA-15 support; (B) Pd1-Al; (C) Pd2-Al; (D) Pd4-Al. | ||
| Catalyst | Si/M molar ratioa (M = Zr, Al) | Metal loadinga (wt%) | S BET (m2 g−1) | D BJH (Å) | V BJH (mL g−1) |
|---|---|---|---|---|---|
| a Measured by EDX. b Pd/C is a microporous material. | |||||
| Al-SBA-15 | — | — | 756 | 8.9 | 0.72 |
| Pd-0.5-Al | 29.5 | 0.4 | 625 | 8.6 | 0.50 |
| Pd-1-Al | 30.2 | 1.2 | 533 | 8.5 | 0.32 |
| Pd-2-Al | 28.2 | 1.9 | 363 | 8.1 | 0.24 |
| Pd-4-Al | 33.5 | 2.9 | 527 | 6.8 | 0.51 |
| 5% Pd/C | — | 5 | 1220 | —b | 1.5 |
Upon Pd incorporation via ball milling, TEM images clearly depict a fairly good dispersion of tiny Pd nanoparticles in the materials, with <10 nm as an average NP size (ca. 7–9 nm). No observable sintering could be found for low loaded materials (e.g. 0.5 to 2% Pd, Fig. 3B and C) although the presence of significantly larger aggregates could be observed at higher loadings (Fig. 3C – bottom of image – and 3D). The structural order in SBA-15 was largely preserved upon milling although the presence of some amorphous silica domains was also evidenced in TEM images (Fig. 3B and C).17
The catalytic activity of mechanochemically synthesized Pd/Al-SBA-15 was subsequently investigated in the hydroconversion of cinnamaldehyde with formic acid under conventional heating using acetonitrile as a solvent under reflux conditions. Formic acid has been previously reported to readily decompose into CO + CO2, hydrogen and water under heating conditions in the presence of noble metals, providing an in situ source of hydrogen for hydrogen-transfer reactions.12
Results of catalytic activity were compared to those of a commercial 5% Pd/C. Most Pd/Al-SBA-15 catalysts exhibited an almost negligible conversion to products (<10%) even after 24 h reaction with the exception of Pd-4-Al for which a 50% conversion was obtained after 24 h (Table 2, Fig. 4). The commercial 5% Pd/C system provided almost quantitative conversion after 24 h, with a remarkably different selectivity to that of Pd/Al-SBA-15 catalysts (Fig. 4 and 5). Taking into account the Pd loadings in the catalysts (2.9 wt% in Pd4-Al and 5 wt% in Pd/C) and the reaction profiles depicted in Fig. 4, it can be clearly visualized that Pd/C possessed almost comparable rates of reaction with respect to Pd4-Al with very slow initial reaction rates, after which Pd/C provided improved reaction rates after prolonged reaction times (>4 h). These results may indicate some diffusional/mass transfer limitations in Pd/C as compared to the lower active Pd/Al-SBA-15 materials (with significantly reduced Pd content) as expected from the inherent microporous nature of Pd/C.
 | ||
| Fig. 4 Reaction profiles for Pd4-Al (dotted line) and Pd/C (solid line) in the hydroconversion of cinnamaldehyde with formic acid under conventional heating. Reaction conditions: 0.5 mL cinamaldehyde, 1.5 mL formic acid, 10 mL acetonitrile, 1 g cat, 83 °C. | ||
 | ||
| Fig. 5 Selectivity comparison (X-axis, mol%) between Pd4-Al containing ca. 2.9 wt% Pd and a commercial 5% Pd/C in the hydroconversion of cinnamaldehyde in formic acid. Reaction conditions: 0.5 mL cinamaldehyde, 1.5 mL formic acid, 10 mL acetonitrile, 1 g cat, 83 °C reaction temperature, 24 h reaction. Products legend: BzProp = hydrocinnamyl alcohol; Oxacid = oxalic acid; MeStyrene = β-methylstyrene; Chxane = cyclohexane; EthylBz = ethylbenzene. | ||
| Catalyst | Time of reaction (h) | Conversion (mol%) |
|---|---|---|
| a Reaction conditions: 0.5 mL cinamaldehyde, 1.5 mL formic acid, 10 mL acetonitrile, 1 g cat, 83 °C reaction temperature. | ||
| Blank | 24 | <1 |
| Pd0.5-Al | 1 | — |
| 4 | <5 | |
| 24 | <5 | |
| Pd1-Al | 1 | — |
| 4 | <5 | |
| 24 | <5 | |
| Pd2-Al | 1 | — |
| 4 | 5 | |
| 24 | 10 | |
| Pd4-Al | 1 | 10 |
| 4 | 17 | |
| 24 | 50 | |
| 5% Pd/C | 1 | 12 |
| 4 | 36 | |
| 24 | 96 | |
Selectivities were significantly different between both systems as illustrated in Fig. 5. The three main competitive and most plausible pathways identified for the products obtained in the hydroconversion of cinnamaldehyde with formic acid have been summarized in Scheme 2. In pathway I, cinammaldehyde undergoes CO hydrogenation to cinnamyl alcohol (Pd sites) followed by dehydration to β-methylstyrene (promoted by acid sites, Scheme 2-I). CC reduction to hydrocinnamaldehyde can further lead to hydrocinnamyl alcohol (Scheme 2-II) which can be followed by subsequent dehydration to β-methylstyrene. Ethylbenzene can also be produced through pathway III via hydrogenation of styrene) produced via cinnamaldehyde hydrodeformylation (Scheme 2-III) or by direct hydrodeformylation of hydrocinnamaldehyde generated in pathway II upon CC bond reduction of cinnamaldehyde.
 | ||
| Scheme 2 Possible competitive pathways (I to III) for the production of ethylbenzene, hydrocinnamaldehyde and β-methylstyrene, main products different from cyclohexane obtained in the hydroconversion of cinnamaldehyde with formic acid. | ||
Results indicated that Pd/C favored typical hydrogenation reactions for the production of major quantities of hydrocinnamaldehyde (pathway II). Since no cinnamyl alcohol was detected as a product for Pd/C and only low selectivities to β-methylstyrene were obtained at short times of reaction (<10 mol%, reaction times under 4–6 h), it is clear that the CC reduction to hydrocinnamaldehyde is the rate determining step in the Pd/C catalyzed hydroconversion of cinnamaldehyde. These results were in good agreement with previous literature reports.4,23 Interestingly, as the reaction progresses, CO reduction leads to the formation of hydrocinnamyl alcohol which undergoes dehydration to β-methylstyrene as the major product (ca. 60 mol% selectivity) observed after 24 h (Fig. 4, mestyrene for Pd/C). Due to the low acidity of Pd/C, dehydration may be promoted by formic acid present in excess in the reaction media.
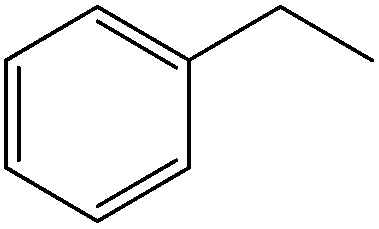
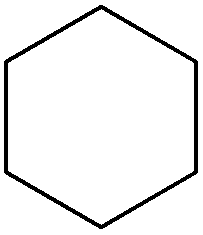
Cyclohexane from hydrogenation of the aromatic ring as well as ethylbenzene and methylstyrene as minor products together with traces of propylbenzene and short amides (e.g. formamide) were also observed in Pd/C catalyzed reactions under conventional heating (Fig. 5). Findings seemed to point out a favored competitive reaction for the production of ethylbenzene to the typical CC and CO reductions steps observed in most reports as proposed in Scheme 2. Ethylbenzene can be most plausibly obtained via hydrodeformylation of hydrocinnamaldehyde in a similar way to previous reports using Ni/Al2O3.23 The catalytic decarbonylation of cinnamaldehyde was also reported many years back using Pd and Pt/C,24 Ni RANEY® catalysts25 and Ru and Rh homogeneous complexes.26,27 Alternatively, hydrodeformylation of cinnamaldehyde could generate styrene and ethylbenzene upon subsequent hydrogenation on the metal sites. This route, however, is not likely to generate ethylbenzene as no traces of styrene could be detected in the reaction under the investigated conditions.
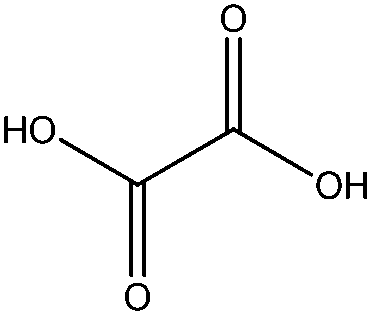
Pd4-Al comparatively exhibited a major hydrogenation selectivity to hydrocinnamaldehyde (CC bond reduction, pathway II) and cyclohexane (aromatic ring hydrogenation). Interestingly, a high selectivity to oxalic acid (15–20 mol%) was observed even after 24 h reaction (Fig. 5). Minor quantities of β-methylstyrene and traces of ethylbenzene were also detected in the catalytic system under conventional heating.
The formation of ethers or acetals from cinnamaldehyde and/or hydrocinnamaldehyde via acid-catalysed reactions (Scheme 1) was not observed in any Pd-containing system even in the presence of Al-SBA-15 as a solid acid support and formic acid under the investigated reaction conditions in conventional heating.
Based on these findings, particularly related to oxalic acid production for Pd/Al-SBA-15 materials, we were prompted to investigate the proposed hydroconversion reaction at shorter reaction times under microwave-irradiation pushing the reaction conditions to maximize product yields.
The main findings for the microwave-assisted hydroconversion of cinnamaldehyde have been summarized in Table 3. Blank runs provided almost negligible activity in the systems (<10%). Interesting catalytic trends were observed in the proposed microwave-assisted system for Pd-containing catalysts (Table 3). Quantitative conversion was achieved for all Pd/Al-SBA-15 materials as compared to a 71% conversion obtained for Pd/C. The activities of Pd-containing systems were remarkably superior to Pd/C as compared to reactions conducted under conventional heating. These findings are in good agreement with previously reported results from the group28 and illustrate the advantages of microwave-activation in mechanochemically synthesized Pd systems as compared to commercial Pd catalysts (e.g. Pd/C). Most importantly, inherent mass transfer and diffusional limitations have already been demonstrated in the Pd/C system under conventional heating present at short reaction times due to its microporous nature (Fig. 4 reaction profiles). Hence, short reaction times under microwave irradiation can justify the reduced activity of Pd/C under the investigated reaction conditions. Previously reported Pd-aluminosilicate interactions as well as the most important preferential external deposition of highly accessible, active and easier to activate nanoparticles in the proposed bifunctional Pd/Al-SBA-15 materials may also partially contribute to the improved activities under microwave irradiation.28
| Catalyst | Conversion (mol%) | Selectivity (mol%) | |||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
||
| a Reaction conditions: 0.1 mL cinnamaldehyde (0.8 mmol), 0.3 mL formic acid (8 mmol), 2 mL acetonitrile, 0.5 g catalyst, microwave irradiation, 200 W (180 °C maximum temperature reached, averaged temperature 150 °C, 250 PSI maximum pressure), 15 min reaction. | |||||||
| Blank | <5 | — | — | — | — | — | — |
| Pd0.5-Al | 97 | <5 | 52 | 26 | 16 | <5 | — |
| Pd1-Al | >99 | <5 | 64 | 11 | 18 | — | — |
| Pd2-Al | 98 | <5 | 66 | 18 | 13 | — | — |
| Pd4-Al | 85 | 13 | 44 | 41 | <5 | — | — |
| 5% Pd/C | 71 | 21 | 48 | 27 | — | <5 | — |



Selectivities in the systems followed the similar trend observed under conventional heating, with expected high activities towards complete aromatic ring hydrogenation as well as hydrogenolysis of the side chain to generate cyclohexane as the final product. The formation of β-methylstyrene, ethylbenzene and oxalic acid accounted for most of the remaining product selectivities. An increase in Pd loading generally favored the formation of β-methylstyrene as a major by-product, with the exception of Pd0.5-Al (Table 3).
The formation of relevant quantities of oxalic acid was repeatedly observed in microwave-assisted reactions for catalysts containing low Pd content (0.5–2 wt%), generally decreasing with Pd content in the materials. Oxalic acid was not detected in blank runs, using similar Pt-based catalysts or in the Pd/C catalyzed hydroconversion of cinnamaldehyde (Table 3). With formic acid decomposing into CO + CO2, hydrogen and water under the investigated microwave-assisted conditions,12 the most plausible mechanism for oxalic acid production is a Pd catalysed hydrocarboxylation. This type of reaction has been previously reported to be catalyzed by various types of Pd catalysts29 including analogous heterogeneous Pd complexes on MCM-41 and MCM-48 materials.30
A plausible explanation for the observed decrease in concentration of oxalic acid at increasing Pd loadings may relate to the presence of large Pd clusters in materials over 2 wt% Pd which favor hydrogenation reactions as compared to hydrocarboxylations promoted in smaller size Pd nanoparticles. The proposed change in selectivity in size-sensitive reactions has been previously reported.4,31 Calculated TON values of the catalysts for microwave-assisted reactions were in the 5–10 range while a maximum catalyst TOF for cinnamaldehyde conversion to products of 42 h−1 (Pd1-Al) and minimum of 15 h−1 (Pd4-Al) were obtained.
Conclusions
Mechanochemically synthesized Pd/Al-SBA-15 materials with different Pd loadings (0.5, 1, 2 and 4 wt%) have been synthesized and characterized using a range of analytical techniques including TG/DTA, N2 physisorption and TEM and subsequently employed as heterogeneous catalysts in the hydroconversion of cinnamaldehyde with formic acid both under conventional heating and microwave irradiation. In contrast to potentially expected products, an interesting selectivity to products including ethylbenzene (via Pd-catalysed hydrodeformylation reactions) and oxalic acid (via Pd-catalysed hydrocarboxylation) were observed. Formic acid was proved to be an effective hydrogen donor in the hydroconversion of cinnamaldehyde under heating (both conventional and microwave). The proposed Pd systems are envisaged to be of potential interest to the extension of the proposed chemistries to related hydro(de)formylation reactions which will be reported in due course.Acknowledgements
Rafael Luque acknowledges the support from the Spanish MICINN via the concession of a Ramon y Cajal contract (ref. RYC 2009-04199) and funding under project CTQ2011-28954-C02-02. A part of this publication was created using the infrastructure supported by the UniCRE project (reg. no. CZ.1.05/2.1.00/03.0071) funded by the EU Structural Funds and the state budget of the Czech Republic.References
- W. Koo-amornpattana and J. M. Winterbottom, Catal. Today, 2001, 66, 277–287 CrossRef CAS.
- A. Y. Hammoudeh, S. M. Sa'ada and S. S. Mahmoud, Jordan J. Chem., 2007, 2, 53–67 CAS.
- J. Hájek, N. Kumara, P. Mäki-Arvela, T. Salmi, D. Yu Murzin, I. Paseka, T. Heikkilä, E. Laine, P. Laukkanen and J. Väyrynen, Appl. Catal., A, 2003, 251, 385–396 CrossRef.
- Y. Zhu and F. Zaera, Catal. Sci. Technol., 2014, 4, 955–962 CAS.
- S. Handjani, E. Marceau, J. Blanchard, J.-M. Krafft, M. Che, P. Mäki-Arvela, N. Kumar, J. Wärnå and D. Yu. Murzin, J. Catal., 2011, 282, 228–236 CrossRef CAS.
- http://lib.hut.fi/Diss/ .
- K.-Y. Jao, K.-W. Liu, Y.-H. Yang and A.-N. Ko, J. Chin. Chem. Soc., 2009, 56, 885–890 CrossRef CAS.
- E. Erasmus, S. Afr. J. Chem., 2013, 66, 216–220 CAS.
- M. Zou, X. Mu, N. Yan and Y. Kou, Chin. J. Catal., 2007, 28, 389–391 CrossRef CAS.
- C. Ge, Y. Li, J. Zhao and R. Zhou, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2010, 49, 281–287 Search PubMed.
- M. Jahjah, B. Caron, S. Menuel, E. Monflier, L. Djakovitch and C. Pinel, ARKIVOC, 2011, 7, 406–415 Search PubMed.
- S. Narayanan, Bull. Catal. Soc. India, 2003, 2, 107–121 Search PubMed.
- D. A. Bulushev, S. Beloshapkin and J. R. H. Ross, Catal. Today, 2010, 154, 7–12 CrossRef CAS.
- D. A. Bulushev and J. R. H. Ross, Catal. Today, 2011, 163, 42–46 CrossRef CAS.
- X. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 CAS.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 CAS.
- A. Pineda, A. M. Balu, J. M. Campelo, A. A. Romero, D. Carmona, F. Balas, J. Santamaria and R. Luque, ChemSusChem, 2011, 4, 1561–1565 CrossRef CAS PubMed.
- (a) A. Toledano, L. Serrano, A. Pineda, A. M. Balu, R. Luque and J. Labidi, ChemSusChem, 2013, 6, 529–536 CrossRef CAS PubMed; (b) A. Toledano, L. Serrano, J. Labidi, A. M. Balu, A. Pineda and R. Luque, ChemCatChem, 2013, 5, 977–985 CrossRef CAS; (c) A. Toledano, L. Serrano, A. Pineda, A. A. Romero, J. Labidi and R. Luque, Appl. Catal., B, 2014, 145, 43–55 CrossRef CAS.
- J. C. Serrano, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- M. Watanabe, T. M. Aida, R. L. Smith and H. Inomata, J. Jpn. Pet. Inst., 2012, 55, 219–228 CrossRef CAS.
- (a) C. Oliver Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef PubMed; (b) C. O. Kappe, B. Pieber and D. Dallinger, Angew. Chem., Int. Ed., 2013, 52, 1088–1094 CrossRef CAS PubMed.
- M. Ojeda, A. M. Balu, V. Barrón, A. Pineda, A. Garcia, A. A. Romero and R. Luque, J. Mater. Chem. A, 2014, 2, 387–393 CAS.
- S.-Y. Chin, F.-J. Lin and A.-N. Ko, Catal. Lett., 2009, 132, 389–394 CrossRef CAS.
- N. E. Hoffmann, A. T. Kanakkanatt and R. F. Schneider, J. Org. Chem., 1962, 27, 2687–2689 CrossRef.
- A. Mailhe, Bull. Soc. Chim. Fr., 1926, 39, 922 CAS.
- A. Mailhe, Bull. Soc. Chim. Fr., 1926, 39, 22 Search PubMed.
- J. G. de Vries, G. Roelfes and R. Green, Tetrahedron Lett., 1998, 39, 8329–8332 CrossRef CAS.
- (a) M. Ojeda, A. Pineda, A. A. Romero, V. Barron and R. Luque, ChemSusChem, 2014, 7, 1876–1880 CrossRef CAS PubMed; (b) M. Ojeda, A. M. Balu, V. Barron, A. Pineda, A. Garcia, A. A. Romero and R. Luque, J. Mater. Chem. A, 2014, 2, 387–393 RSC.
- (a) D. Zargarian and H. Alper, Organometallics, 1993, 12, 712–724 CrossRef CAS; (b) J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254–15255 CrossRef CAS PubMed; (c) M. Al-Masum and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 3809–3810 CrossRef CAS.
- K. Mukhopadhyay, B. R. Sarkar and R. V. Chaudhari, J. Am. Chem. Soc., 2002, 124, 9692–9693 CrossRef CAS PubMed.
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, ACS Catal., 2012, 2, 671–678 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |