
Catalytic conversion of carbohydrate-derived oxygenates over HZSM-5 in a tandem micro-reactor system†
Kaige
Wang‡
ab,
Jing
Zhang‡
c,
Brent
H. Shanks
c and
Robert C.
Brown
*ab
aCenter for Sustainable Environmental Technologies, Iowa State University, Ames, IA 50011, USA
bDepartment of Mechanical Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: rcbrown3@iastate.edu; Fax: +1 515 294 3091; Tel: +1 515 294 7934
cDepartment of Chemical and Biological Engineering, Iowa State University, Ames, IA 50011, USA
First published on 10th October 2014
Abstract
In this study, carbohydrate-derived pyrolysis oxygenates were used as model compounds to investigate the effect of functional group and molecular size on the product formation from their catalytic conversion over HZSM-5. Functional groups in oxygenates were found to strongly affect the oxygen removal pathway, leading to variations in hydrocarbon formation. This study also found that oxygenates of smaller molecular size tended to form more hydrocarbons and less coke. Coking on the external surface of catalysts was greatest for the largest oxygenates. Isotopic labeling experiments demonstrated that the aldehyde group of HMF was cleaved before the furanic ring diffused into the HZSM-5 catalyst. Product distribution from catalytic pyrolysis of glucose was the same as the weighted sum of products obtained by the catalytic pyrolysis of individual oxygenates known to arise from non-catalytic pyrolysis of glucose. This suggests that oxygenates released during pyrolysis of carbohydrate have no significant interaction during their catalytic conversion over HZSM-5.
1. Introduction
Fast pyrolysis has been developed as a promising technology for the production of transportation fuels.1–4 However, the resulting bio-oil has high oxygen content and instability during storage, which impedes its upgrading to transportation fuels. Catalytic pyrolysis, which contacts pyrolysis vapors with deoxygenation catalysts, has emerged as a means to improve the quality of bio-oil.5–10 Among the various catalysts investigated, zeolites are attractive for their ability to generate light olefins and gasoline-range aromatics. Unfortunately, low hydrocarbon yields and excessive coke formation are frequently reported.5,6,11–14 As a result, commercial deployment of catalytic pyrolysis has been hindered.15Catalytic pyrolysis appears to be a process with two stages: thermal decomposition of solid biomass followed by catalytic conversion of the resulting vapors over zeolite. Compared to the relatively well-investigated reaction chemistry of pyrolysis, little is known about the reaction network of catalytic pyrolysis. The theory of indirect hydrocarbon pools in zeolites, originally formulated to explain the methanol to gasoline process via zeolite catalysts, has also been used to explain the complex reaction network inside the zeolite during catalytic pyrolysis.5,6,16 In a study of co-pyrolysis of 12C glucose and 13C glucose over ZSM-5, Carlson et al.16 suggested that all carbon atoms lose their identity in a hydrocarbon pool formed within the zeolite catalyst. They proposed that the oxygenated intermediates diffused into zeolite pores and went through random fragmentation and recombination.
Numerous efforts have been made to improve the production of desirable hydrocarbon products.14,17–21 Changing the properties of zeolite catalyst, such as pore size, crystal sizes, or acidity, have been proposed to enhance the yield of aromatic and olefins.14,17–19 Jae et al.14 investigated the effect of zeolite pore size on the conversion of glucose to aromatics. They found that aromatic yield is a function of pore size in the zeolite catalysts. They concluded that aromatic yields were highest in the medium pore size range of 5.2–5.9 Å. A recent study by Zheng et al.18 suggests that crystal size of HZSM-5 also significantly affects product distribution. Other methods to optimize product distribution for catalytic pyrolysis include changing acidity and increasing mesoporosity.17,19
Several studies20,21 investigated the effect of feedstock properties including the effective hydrogen-to-carbon (H/Ceff) ratio, defined as:
| H/Ceff = (H–2O–3N–2S)/C | (1) |
Competitive reaction pathways have been proposed for fast pyrolysis of hexose-based carbohydrates, which consist of either the release of levoglucosan or the generation of furans and C1 to C3 compounds. A series of oxygenates including acids, aldehydes, furans, and sugars are produced. As shown in previous studies, levoglucosan, furfural, 5-hydroxymethyl furfural (HMF), glycolaldehyde, and acetic acid are the major products.25,26 Although these five compounds all have H/Ceff ratio of zero, they exhibit different structures and functional groups. It is reasonable to hypothesize that their conversion over HZSM-5 varies significantly due to the effect of functional groups, molecular size, and structure. Investigating the conversion of these model compounds may provide a better understanding of the reaction chemistry involved in catalytic pyrolysis of carbohydrates.
Catalytic conversion of oxygenates especially furanic compounds over HZSM-5 have been conducted by several researchers.27–29 Grandmaison et al.30 examined conversion of furfural and furan over H-ZSM5 zeolite in a fixed bed reactor in the temperature range of 350–450 °C. They reported catalytic conversion of furfural undergoes significant decarbonylation, generating furan and formaldehyde but only a limited amount of hydrocarbons. Horne and Williams31 investigated the effect of temperature on conversion of oxygenates over HZSM-5 and found that conversion of furfural required higher temperature than methanol. Carlson et al.6 investigated in situ conversion of furan and furfural using a CDS Pyroprobe, from which 35% carbon yield of aromatic hydrocarbons was achieved for both compounds at 600 °C. Olefins were not reported in their study. Zheng et al.24 reported co-feeding methanol and 2,5-dimethylfuran produced the maximum yields of aromatics and olefins and minimum coke formation compared to other furans.
Relatively few studies have investigated catalytic conversion of HMF, which is a major product from pyrolysis of carbohydrates. To date, only Zhao et al.32 investigated its conversion over zeolite catalyst, which was performed in a fixed bed reactor. HMF produced aromatics with carbon yield as high as 49% using HZSM-5 catalyst.
A few studies investigated catalytic conversion of acetic acid. Fuhse and Bandermann33 reported acetic acid over HZSM-5 at 400 °C only produced acetone and CO2. Carlson et al.6 performed in situ catalytic conversion of acetic acid over HZSM-5 in a CDS Pyroprobe at 600 °C and reported 28% carbon yield of aromatics without reporting olefin formation. Although glycolaldehyde and levoglucosan are major products of the fast pyrolysis of carbohydrate, we are not aware of any studies of their catalytic conversion over zeolites.
The results from previous studies for various oxygenates are sometimes contradictory, probably because experimental conditions varied widely among them. The present study is the first systematic investigation of the catalytic conversion of carbohydrate-derived oxygenates over zeolite catalyst. The effect of functional groups and molecular size on catalytic conversion was investigated. Isotopic labeling was applied to help understand the effect of molecular structure. The interactions among these oxygenates during catalysis process were also explored.
2. Experimental section
2.1. Materials
All the chemicals were purchased from Sigma Aldrich except for isotopically labeled glucose, which was purchased from Cambridge Chemicals. The structure and kinetic diameter of these compounds are summarized in Table 1. The kinetic diameters were estimated from their fluid properties at the critical point, which were used to determine whether molecules could diffuse into zeolite pores.14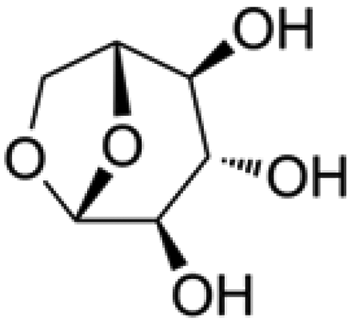
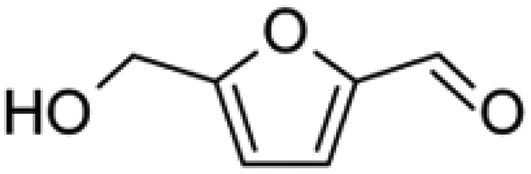
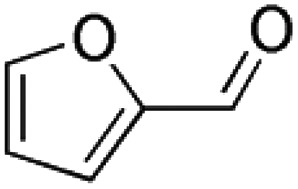

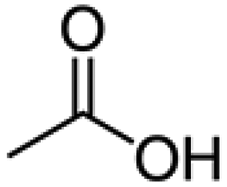
Isotopically labeled HMF was synthesized from D-glucose-1-13C and D-glucose-6-13C (Sigma Aldrich), respectively, in a biphasic solvent system following the procedure described by Wang et al.35 Purity of the synthesized isotopically labeled HMF was further quantified using the Frontier Tandem micro-reactor system described in section 2.2. Details on characterization of the synthesized HMF are found in the ESI.†
2.2. Pyrolysis equipment and analytical instrumentation
A Tandem micro-reactor system (Rx-3050 TR, Frontier Laboratories, Japan) was used for both non-catalytic and catalytic conversion of model compounds. A schematic diagram of the system is shown in Fig. 1. Detailed description about the micro-reactor system can be found in a previous paper.36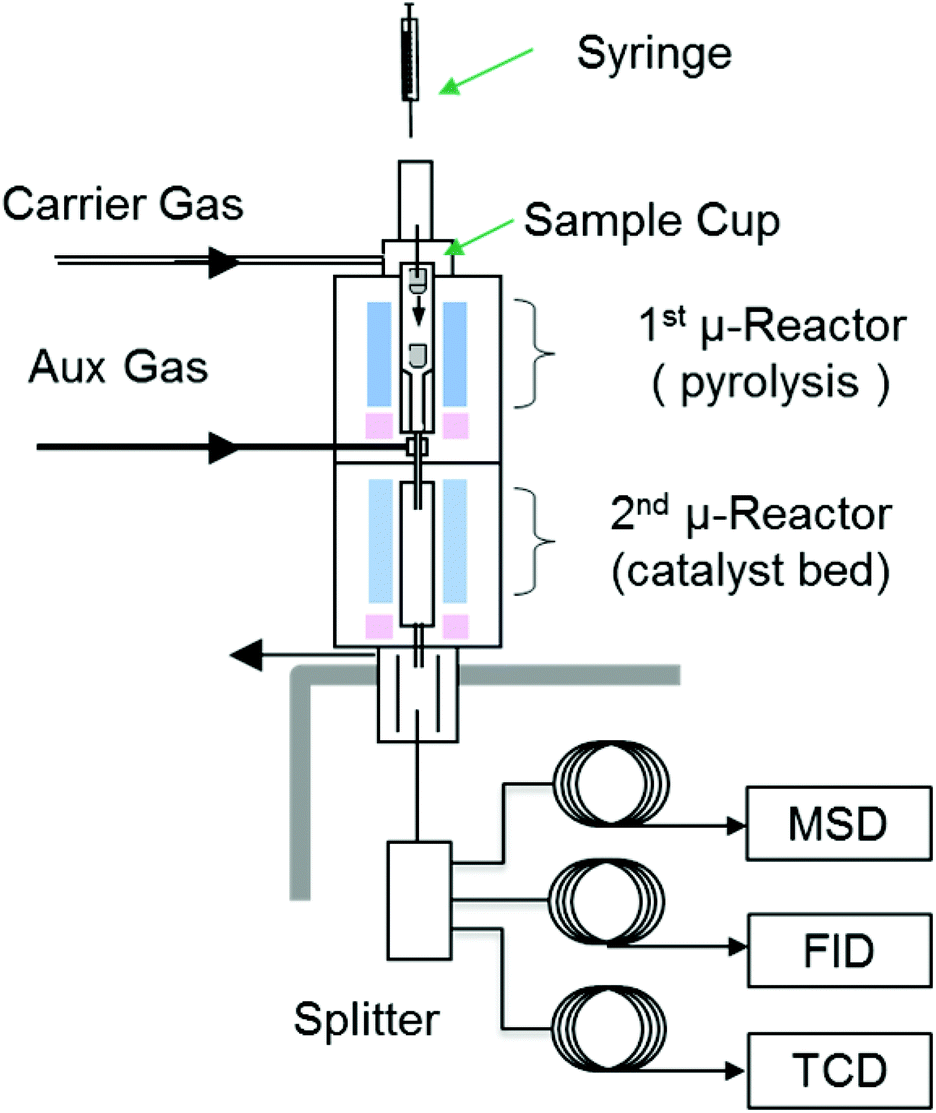 | ||
| Fig. 1 Schematic diagram of micro-pyrolysis system used in this study. | ||
We chose commercially available HZSM-5 catalyst (CBV3024 with SiO2/Al2O3 ratio of 30, Zeolyst, USA) for this study. The as-received catalyst was calcined at 550 °C (5 °C min−1) for 5 hours in a muffle furnace before being pelletized and sieved to 50–70 mesh size. For catalytic conversion experiments, approximately 0.5 mg of sample was rapidly heated in the first reactor, resulting in evaporation of liquid samples or pyrolysis of solid samples. The resulting vapors were transported to the second reactor, which contained the zeolite catalyst. Quartz wool was used to support the catalyst particles and prevent solids from exiting the catalyst bed. The temperature of catalyst bed was held at 600 °C while the first reactor for model compounds evaporation held at 300 °C to assure minimal decomposition before contacting catalyst. The catalyst-to-reactant mass ratio was maintained at 20 to eliminate the influence of catalyst deactivation. No changes in product distribution were observed during triplicate runs for each reactant.
The products formed in the catalyst bed were swept directly to a GC (7890A, Agilent Technologies, USA) installed with a three-way splitter and three detectors. The interface temperature between the catalytic reactor and the GC was set to 350 °C to minimize condensation of products. A mass spectrometer detector (MSD) was used for molecular identification and a flame ionization detector (FID) and a thermal conductivity detector (TCD) were used to quantify the products. Char generated in the pyrolysis reactor and coke deposited on the catalyst bed were separately measured using an elemental analyzer (vario MICRO cube, Elementar, USA). Details of quantification methods can be found in a previous publication.36
All measurements including condensable aromatics, gases and carbonaceous residues, were performed at least in triplicate to check the reproducibility of the data. Final product distributions were reported as molar carbon yields, defined as the molar ratio of carbon in a specific product to the carbon in the feedstock. Selectivity for aromatics in this study was defined as moles of carbon in a specific aromatic hydrocarbon to total moles of carbon in the aromatic products. Selectivity of olefins was similarly defined. The overall carbon balance was performed for each run, which closed at over 95% in most cases. Data was reported as averages with standard deviations.
2.3. Calculation of isotopic content within products
The mass spectra of product from catalytic conversion of isotopically labeled chemicals were used to track the 13C distribution. Relative intensity of molecular ion with or without 13C was deconvoluted after ruling out interference from proton loss.37,38 The contribution of M + 1 peak, which is derived from the presence of natural 13C, was also considered. For a specific product, the relative intensities of deconvoluted molecular ions without 13C, with one 13C, and with multiple 13C were used to determine their distributions in the products. Standard mass spectra for pure chemicals in NIST data base were used.2.4. Catalyst characterization
To test the extent of internal coking from different oxygenates, T-plot micropore volume measurements were performed for fresh and coked catalysts. A gradient of coke deposition was observed in the catalyst bed after experiments, with the entrance region of the bed heavily coked compared to other parts of the bed, as shown in Table S1.† After each test, catalyst was completely removed from the bed and subjected to nitrogen physisorption measurement. The measured micropore volume was the average value for the entire catalyst bed. T-plot micropore volume was analyzed by Micromertitics ASAP 2020. First, fresh and coked catalysts were degassed at 350 °C for 4 h with a ramping rate of 10 °C min−1. Conditions of nitrogen adsorption isotherms at 77 K were used for the measurement. For the HZSM-5, the pores of which are mainly micropores, the difference in T-plot micropore volume between coked and fresh catalyst approximates the volume of internal coke formed during catalysis.5 Elemental analysis was performed with Elementar vario Micro cube to quantify carbon content in coked HZSM-5. Rice flour, purchased from Elemental Microanalysis, was used as a calibration standard.3. Results and discussion
3.1. Catalytic conversion of model compounds
Detailed product distributions for catalytic conversion of furfural, HMF, levoglucosan, acetic acid, and glycolaldehyde are summarized in Table 2. Although the H/Ceff ratio was zero for all of these oxygenates, distinctive product distributions were obtained for each.| Feedstock | HMF | Furfural | Acetic acid | Levoglucosan | Glycolaldehyde |
|---|---|---|---|---|---|
| a C9 aromatics include indanes, indenes, and alkylbenzene. b C10+ aromatics include naphthalenes and higher polyaromatics (≤C15). | |||||
| Overall yield/C (%) | |||||
| CO | 21.9 ± 0.7 | 32.7 ± 0.4 | 8.4 ± 0.3 | 28.6 ± 0.1 | 34.6 ± 0.7 |
| CO2 | 9.7 ± 0.1 | 4.7 ± 0.0 | 26.7 ± 0.6 | 7.5 ± 0.1 | 5.9 ± 0.1 |
| Catalytic coke | 21.1 ± 1.2 | 9.9 ± 1.1 | 6.9 ± 0.9 | 15.8 ± 1.6 | 9.0 ± 0.8 |
| Aromatics | 25.5 ± 0.3 | 35.1 ± 0.8 | 26.8 ± 0.1 | 31.3 ± 0.4 | 33.5 ± 0.3 |
| Olefins | 16.9 ± 1.2 | 16.6 ± 1.2 | 37.5 ± 0.4 | 17.0 ± 0.3 | 22.3 ± 0.3 |
| Total | 95.2 ± 3.5 | 99.0 ± 3.5 | 106.4 ± 2.3 | 100.2 ± 2.5 | 105.3 ± 2.2 |
| Aromatics selectivity (%) | |||||
| Benzene | 24.8 ± 0.1 | 24.2 ± 0.9 | 18.1 ± 0.1 | 27.6 ± 0.0 | 21.9 ± 0.1 |
| Toluene | 29.9 ± 0.5 | 30.2 ± 0.3 | 40.7 ± 0.1 | 36.5 ± 0.2 | 41.8 ± 0.2 |
| Xylene | 11.5 ± 0.4 | 8.7 ± 0.1 | 28.2 ± 0.4 | 11.0 ± 0.0 | 16.2 ± 0.0 |
| C9 aromaticsa | 14.1 ± 0.3 | 13.0 ± 1.4 | 5.9 ± 0.0 | 9.9 ± 0.5 | 9.5 ± 0.3 |
| C10+ aromaticsb | 19.6 ± 0.7 | 23.8 ± 0.3 | 7.1 ± 0.4 | 14.9 ± 0.4 | 10.5 ± 0.4 |
| Olefin selectivity (%) | |||||
| Ethylene | 40.4 ± 0.0 | 47.7 ± 0.4 | 46.9 ± 0.0 | 49.7 ± 0.3 | 63.4 ± 0.2 |
| Propene | 55.7 ± 0.2 | 47.6 ± 1.7 | 43.8 ± 0.2 | 45.8 ± 0.7 | 34.2 ± 0.1 |
| Butene | 4.0 ± 1.2 | 4.7 ± 1.2 | 9.3 ± 0.2 | 4.54 ± 0.4 | 2.40 ± 0.0 |
Carbon yield of aromatic hydrocarbons from furfural was 35.1% compared to only 25.5%for HMF. Both furan-based oxygenates produced similar yield of olefins (∼17%). The only structural difference between furfural and HMF is the inclusion of a hydroxyl group in HMF. Due to the presence of hydroxyl group in HMF, more oxygen in HMF was removed by dehydration,39 which may contribute partly to the relatively lower yield of hydrocarbons from HMF.
It is interesting that acetic acid produced significantly higher yields of hydrocarbons, especially olefins, than other oxygenates. Carbon yield of olefins from acetic acid was 39.0%, compared with 17% for both HMF and furfural. As shown in Table 2, yield of CO2 from acetic acid was 28.9%, which was much higher than for furans, levoglucosan, and aldehyde. In contrast, carbon yield of CO was only 8.7%, which was significantly lower than for other oxygenates. This indicates that decarboxylation primarily contributed to deoxygenation of acetic acid compared to decarbonylation for furanic compounds. The carboxyl group is the major functionality for acetic acid, removal of which released CO2. Decarboxylation is twice as efficient as decarbonylation in removal of oxygen. As shown in Table 2, more oxygen was removed from acetic acid as COx compared to other oxygenates, leading to more hydrogen availability to form hydrocarbons, especially olefins. This indicates that oxygen removal is highly affected by the kind of functional groups associated with oxygenates, which in turn determines the kinds of products formed from catalytic conversion of oxygenates. Aromatic selectivity for benzene, toluene, and xylene from acetic acid were 18.0%, 40.8%, and 28.5%, respectively. The relatively higher BTX selectivity from acetic acid compared to furfural and HMF is also attributed to more abundant hydrogen, which makes formation of hydrogen-deficient polyaromatics less likely.
Glycolaldehyde has the same formula as acetic acid but includes aldehyde and hydroxyl as functional groups. During catalytic conversion of glycolaldehyde, decarbonylation occurred preferentially over decarboxylation by a ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is due to the presence of aldehyde group. Moreover, the hydroxyl group in glycolaldehyde had a strong tendency to remove oxygen in the form of water, as also observed in studies on methanol and glycerol.29,39 The dehydration reaction depletes hydrogen available for hydrocarbon formation. Thus, compared with acetic acid, which contains carboxyl group predominantly leading to CO2 formation, conversion of glycolaldehyde over HZSM-5 resulted in lower yield of hydrocarbons.
:1, which is due to the presence of aldehyde group. Moreover, the hydroxyl group in glycolaldehyde had a strong tendency to remove oxygen in the form of water, as also observed in studies on methanol and glycerol.29,39 The dehydration reaction depletes hydrogen available for hydrocarbon formation. Thus, compared with acetic acid, which contains carboxyl group predominantly leading to CO2 formation, conversion of glycolaldehyde over HZSM-5 resulted in lower yield of hydrocarbons.
Levoglucosan is the most abundant product from pyrolysis of hexose-based carbohydrates.40 Yield of levoglucosan from pyrolysis of cellulose is as high as 58.8 wt%.40 Surprisingly, few researchers have investigated levoglucosan as a model compound in studies of catalytic conversion over HZSM-5. Levoglucosan contains three hydroxyl groups, which would show strong tendency toward dehydration in the presence of strongly acidic HZSM-5. Carbon yields of aromatics and olefins from levoglucosan were 31.3% and 17.0%, respectively. The relatively lower yield of hydrocarbons and higher yield of coke from levoglucosan might be related to the prevailing dehydration reactions, which facilitate coke formation thus depleting the carbon atoms available for hydrocarbon formation.
Calculation of H/Ceff ratios assumes that oxygen is removed from the molecules as water; in fact, dehydration (H2O), decarboxylation (CO2), and decarbonylation (CO) all contribute to deoxygenation. The functional groups of the oxygenates affect the deoxygenation route, which in turn affect the kinds of hydrocarbons produced. Compounds containing aldehyde functionality such as furfural and glycolaldehyde gave higher yield of CO, while carboxyl group gave extremely high yield of CO2. The hydroxyl group showed strong tendency to remove oxygen as water. The method of oxygen rejection during catalysis has a very important impact on the yield and selectivity of hydrocarbon products, especially for the feedstocks with low value of H/Ceff. Ideally, oxygen would be removed through decarboxylation or decarbonylation to preserve hydrogen in the hydrocarbon products.
3.2. Effect of molecular size
As shown in Table 1, acetic acid, glycolaldehyde, and furfural have molecular diameters less than the maximum pore size of HZSM-5. The diameter of levoglucosan is 6.7 Å, which is larger than maximum pore size. For HMF, its kinetic diameter is close to the maximum pore size of HZSM-5 (6.2–6.3 Å).14,41Fig. 2 summarizes the yields of hydrocarbons and coke from these oxygenates. The yield of hydrocarbons and coke was influenced by the molecular diameter of oxygenates during their conversion over HZSM-5. Molecules with relatively smaller diameter shows strong tendency to produce higher yield of hydrocarbons and lower yield of coke. The higher yield of coke was always concurrent with lower yield of hydrocarbons. The higher yield of coke from larger molecules is attributed to geometric hindrance, especially for molecules like levoglucosan and HMF, which have larger diameters than HZSM-5 pores. As shown in Table 1, the kinetic molecular diameter of HMF is 6.2 Å, which is close to the maximum pore size of HZSM-5. Either an aldehyde or hydroxyl group in HMF might have to be cleaved before HMF can enter the pores of HZSM-5 catalyst. This geometric hindrance increases the probability that HMF will polymerize on the external surface of the zeolite, enhancing coke formation. In comparison, furfural has a smaller kinetic diameter of 5.5 Å. Therefore, furfural molecule can more readily diffuse into the pores of zeolite catalyst, resulting less coke formation.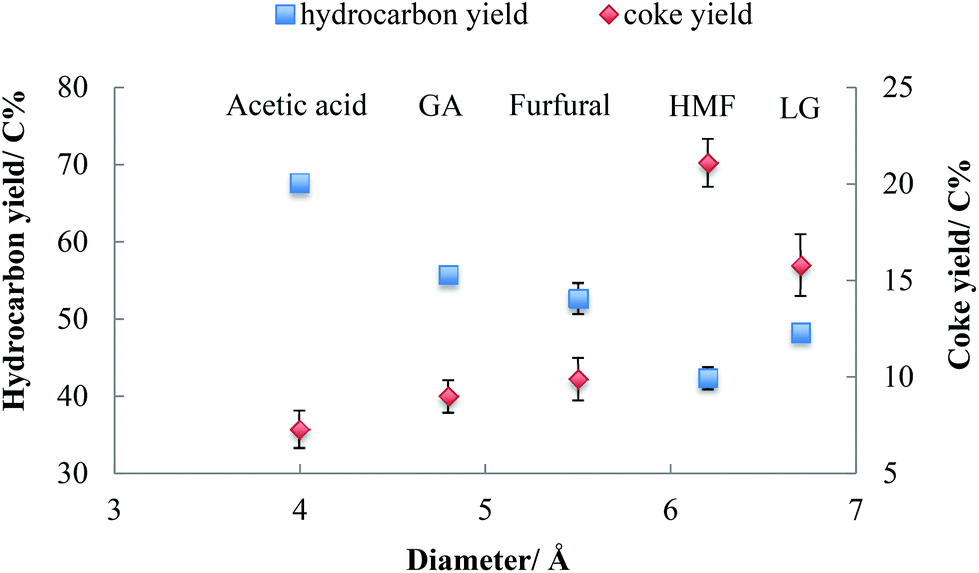 | ||
| Fig. 2 Yield of hydrocarbons (in blue) and coke (in red) from oxygenates as a function of molecular diameter (ex situ catalysis; vaporize temperature = 300 °C; catalyst temperature = 600 °C; reactant loading = 0.5 mg; catalyst CBV 3024 loading = 10 mg; GA = glycolaldehyde; LG = levoglucosan). | ||
According to the literature,42–47 the distribution of coke on HZSM-5 is determined by the molecular size of the reactant and extent of coking on the catalyst. It was reported that for methanol and isobutene, with molecular diameters smaller than the pore size of HZSM-5, initial coking occurred inside the micropores of HZSM-5. Mesitylene, on the other hand, with molecular diameter larger than the pore size of HZSM-5, deposited coke primarily on the external surface of the catalyst. The extent of coking also affects coke distribution. If coking is heavy, even small molecules like methanol produce extensive coking external to the pores.42–47 After three trials in the present study, the catalyst bed contained less than 2 wt% of coke, which is characterized as “initial coking,”42–47 and the distribution of coke would be expected to be determined by the size of reactant molecules relative to HZSM-5 channels.
Geometric hindrance for large molecules might result in extra coke formation outside of zeolite pores through acid promoted dehydration. To test this hypothesis, the extent of internal coking versus external coking was studied by physisorption analysis of HZSM-5 after it was coked by different oxygenates. Catalytic conversion of acetic acid (4.0 Å), furfural (5.5 Å), and HMF (6.2 Å) over identical amounts of catalyst was performed. Since the coke yield for each of these three compounds were different, the amount of compound reacted was varied to achieve similar amounts of coke on the catalyst in each case.
The characterization of fresh and coked HZSM-5 is shown in Table 3. It is clear that more micropore volume was lost when the reactants were smaller molecules. The difference in micropore volume between fresh and coked catalysts roughly represents the volume of internal coke, assuming negligible pore blockage from coking.5 Therefore, data in Table 3 suggests that the extent of internal coking compared to external coking on zeolite decreases for oxygenates in the following order: acetic acid > furfural > HMF. On the other hand, this indicates that higher extent of external coking occurs for larger molecules. Higher effective diffusion coefficient for smaller molecules might alleviate or prevent coke deposition on the outer surface of the zeolites. External coking has been proposed to be more responsible for catalyst deactivation than internal coking.48–50
3.3. Isotopic labeling studies for catalytic fast pyrolysis of HMF
Isotopic labeling has been extensively employed to investigate the mechanism of hydrocarbon pooling.16,49 By using D-glucose-1-13C and D-glucose-6-13C as reactant, the present study proved that both C-1 and C-6 in glucose molecules were randomly distributed in the BTX, as shown in Fig. S2.† Then isotopic labeling was performed to experimentally determine whether C-1 and C-6 in HMF could diffuse into the hydrocarbon pool to randomly appear in aromatic products. Our hypothesis is that HMF molecule is too big to directly enter the zeolite pores. The molecule contains both aldehyde and hydroxyl functional groups. For the HMF molecule, carbon on the aldehyde group is C-1 while the carbon on the hydroxyl group is C-6, as illustrated in Fig. 4. To study the evolution of functional groups during catalytic conversion, 13C was labeled at C-1 or C-6 position on HMF. 1-13C HMF and 6-13C HMF were successfully synthesized from D-glucose-1-13C and D-glucose-6-13C, respectively. The mass spectra of these two isotopically labeled HMF molecules are shown in Fig. S1.† Distribution of 13C in benzene, toluene and p-xylene products for these two forms of HMF is summarized in Fig. 4.As shown in Fig. 3(a), more than 80% benzene, toluene and p-xylene did not contain C-1 from HMF, suggesting few C-1 moieties were involved in reactions within the hydrocarbon pool. Instead, more than 80% of C-1 in HMF ended up producing CO and CO2, as illustrated in Fig. S3(b).† In contrast, Fig. 3(b) shows abundant C-6 in HMF appeared in BTX product. The number of C-6 atoms in BTX product molecules varied from 0 to 3 in a pattern of random distribution, suggesting most C-6 atoms in HMF molecules entered the hydrocarbon pool and underwent random fragmentation and recombination. Accordingly, as suggested in Fig. S3(b),† less than 20% of C-6 ended up forming CO and CO2. Taken together, the isotopic labeling experiments suggest HMF is too large to diffuse into the ZSM-5 catalyst. HMF must first undergo fragmentation to produce molecules small enough to enter zeolite pores. It appears that fragmentation preferentially occurs at the C-1 atom rather than the C-6 atom on the HMF molecule (see Fig. 4), possibly because the aldehyde group is more reactive than the hydroxyl group under the current catalysis conditions. The C-1 atom was primarily released as CO and CO2 outside of the catalyst while C-6 entered the catalyst with the HMF moiety and randomly distributed among the aromatic products.
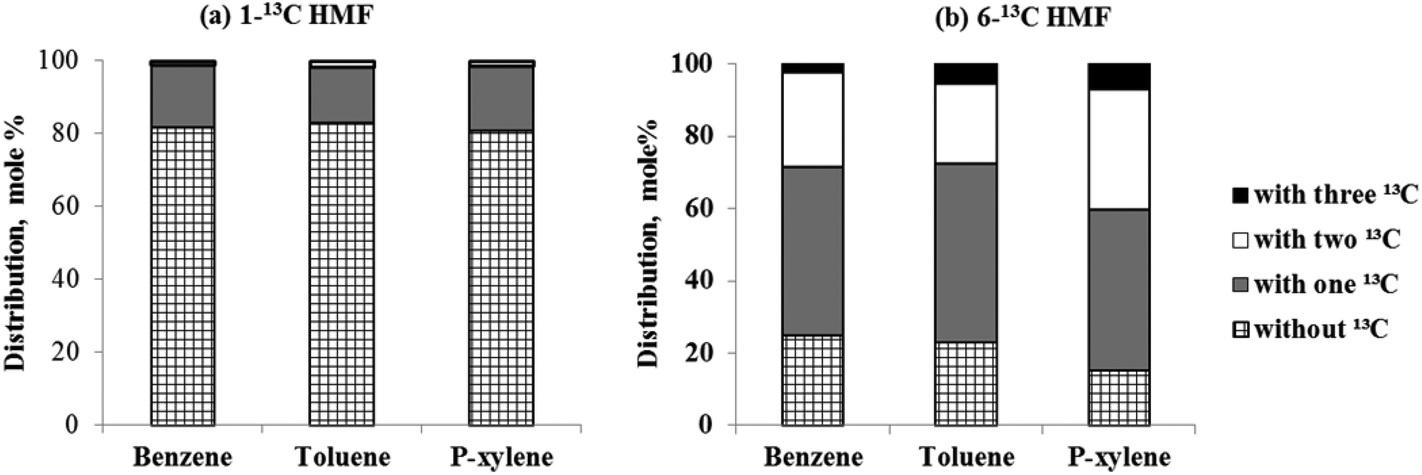 | ||
| Fig. 3 Molar distribution of aromatic products from catalytic reaction of HMF over HZSM-5 according to the number of 13C atoms (a) 13C located at C-1 on HMF and (b) 13C located at C-6 on HMF (ex situ catalysis; vaporization temperature = 300 °C; catalyst temperature = 600 °C; reactant loading = 0.5 mg; CBV 3024 catalyst loading = 10 mg). | ||
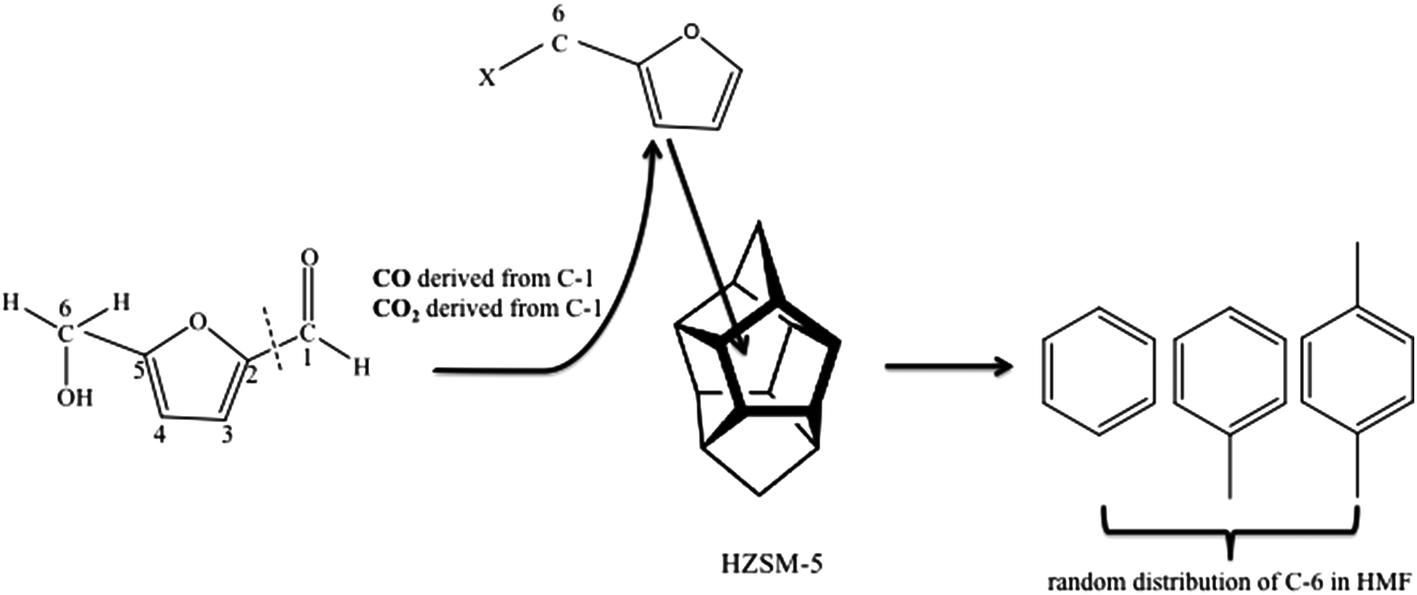 | ||
| Fig. 4 Pyrolysis pathways for HMF over ZSM-5 from isotopic labeling study. | ||
3.4. Interactions between oxygenates during catalytic conversion over HZSM-5
Generally, catalytic pyrolysis is a two-stage process: thermal decomposition of biomass into oxygenates that then undergo catalysis to form aromatics. The interaction among these oxygenates has not been well investigated. To better understand catalytic pyrolysis of glucose, non-catalytic pyrolysis of glucose was performed in the micropyrolyzer at 500 °C. The detailed product yield from glucose pyrolysis is summarized in Table S2.† Acetic acid, glycolaldehyde, furfural, HMF, and LG accounted for over 70% of the products. Other volatile products from glucose pyrolysis include aldehydes, ketones, furans, and sugars. Those minor products were grouped according to functional groups and molecular sizes, as shown in Table S3.† Assuming there are no interactions among those oxygenates during catalytic conversion over HZSM-5, theoretical product yields for catalytic pyrolysis of glucose were calculated using product yield data for glucose pyrolysis shown in Table S2† and product yields from individual oxygenates shown in Table 2. Ex situ catalytic pyrolysis of glucose was also performed in the tandem reactor system. The observed data were compared to the calculated data in Table 4.| Feedstock | Observed | Calculated |
|---|---|---|
| a C9 aromatics include indanes, indenes, and alkylbenzene. b C10+ aromatics include naphthalenes and higher polyaromatics (≤C15). | ||
| Overall yield/C (%) | ||
| CO | 25.5 ± 1.5 | 28.5 ± 0.6 |
| CO2 | 9.7 ± 0.1 | 10.1 ± 0.2 |
| Catalytic coke | 9.4 ± 1.2 | 10.7 ± 0.9 |
| Aromatics | 27.0 ± 0.9 | 25.9 ± 0.3 |
| Olefins | 17.9 ± 0.3 | 15.5 ± 0.3 |
| Aromatic selectivity | ||
| Benzene | 24.5 ± 1.0 | 24.2 ± 0.1 |
| Toluene | 36.5 ± 0.2 | 35.0 ± 0.2 |
| Xylene | 12.5 ± 0.1 | 10.8 ± 0.1 |
| C9 aromaticsa | 11.6 ± 0.5 | 9.2 ± 0.4 |
| C10+ aromaticsb | 14.9 ± 0.2 | 11.9 ± 0.4 |
| Olefin selectivity | ||
| Ethylene | 57.2 ± 0.5 | 48.4 ± 0.1 |
| Propene | 42.8 ± 0.2 | 38.4 ± 0.2 |
| Butene | n.d. | 3.22 ± 0.0 |
It can be seen that the calculated yield is generally consistent with the experimentally observed yield. The observed yields of aromatics and olefins were 27.0% and 17.9%, respectively, while the calculated yields were 25.9% and 15.5%. Observed yield of coke was 9.4%, which is also consistent with the calculated value. Observed yields of CO and CO2 were 25.5% and 9.7% respectively, while the calculated values were 28.5% and 10.1%. Moreover, similar selectivity was also observed within aromatics and olefins between the observed and calculated results, as shown in Table 4. This suggests no significant interaction among oxygenated intermediates during catalytic pyrolysis of glucose. Therefore, product distribution from catalytic pyrolysis of other carbohydrates can be predicted since they produce similar oxygenated intermediates as glucose.
4. Conclusion
This study found that functionality and molecular size of oxygenates play an important role in their catalytic conversion over HZSM-5 catalyst. Functionality highly influences the method of oxygen rejection, which in turn impacts the selectivity and yield of hydrocarbon products, especially for the feedstocks with low H/Ceff ratios. More external coke formation was found for oxygenates of larger molecular size due to relatively lower diffusion rates into pore channels. Isotopic labeling of carbon in HMF suggests that this molecule is too large to diffuse into HZMS-5 pores without first fragmenting outside the catalyst, promoting external coke formation. The study also showed no significant interactions among glucose-derived oxygenates during reaction over HZSM-5 catalyst.Acknowledgements
The authors greatly acknowledge the finical support from Iowa Energy Center (grant no. 13-01) and the National Advanced Biofuels Consortium (grant no. DE-EE0003044).References
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS PubMed.
- R. C. Brown and T. R. Brown, Biorenewable Resources: Engineering New Products from Agriculture, John Wiley & Sons, Ames, IA, 2013 Search PubMed.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- T. R. Carlson, J. Jae, Y.-C. Lin, G. A. Tompsett and G. W. Huber, J. Catal., 2010, 270, 110–124 CrossRef CAS PubMed.
- T. R. Carlson, G. Tompsett, W. Conner and G. Huber, Top. Catal., 2009, 52, 241–252 CrossRef CAS.
- A. A. Lappas, K. G. Kalogiannis, E. F. Iliopoulou, K. S. Triantafyllidis and S. D. Stefanidis, Wiley Interdiscip. Rev.: Energy Environ., 2012, 1, 285–297 CrossRef CAS.
- E. F. Iliopoulou, S. Stefanidis, K. Kalogiannis, A. C. Psarras, A. Delimitis, K. S. Triantafyllidis and A. A. Lappas, Green Chem., 2014, 16, 662–674 RSC.
- T. L. Marker, L. G. Felix, M. B. Linck and M. J. Roberts, Environ. Prog. Sustainable Energy, 2012, 31, 191–199 CrossRef CAS.
- V. K. Venkatakrishnan, J. C. Degenstein, A. D. Smeltz, W. N. Delgass, R. Agrawal and F. H. Ribeiro, Green Chem., 2014, 16, 792–802 RSC.
- Y.-T. Cheng and G. W. Huber, Green Chem., 2012, 14, 3114–3125 RSC.
- Y.-T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 124, 1416–1419 CrossRef.
- K. Wang, K. H. Kim and R. C. Brown, Green Chem., 2014, 16, 727–735 RSC.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS PubMed.
- T. R. Brown and R. C. Brown, Biofuels, Bioprod. Biorefin., 2013, 7, 235–245 CrossRef CAS.
- T. R. Carlson, J. Jae and G. W. Huber, ChemCatChem, 2009, 1, 107–110 CrossRef CAS.
- A. J. Foster, J. Jae, Y.-T. Cheng, G. W. Huber and R. F. Lobo, Appl. Catal., A, 2012, 423, 154–161 CrossRef PubMed.
- A. Zheng, Z. Zhao, S. Chang, Z. Huang, H. Wu, X. Wang, F. He and H. Li, J. Mol. Catal. A: Chem., 2014, 383, 23–30 CrossRef PubMed.
- J. Li, X. Li, G. Zhou, W. Wang, C. Wang, S. Komarneni and Y. Wang, Appl. Catal., A, 2014, 470, 115–122 CrossRef CAS PubMed.
- N. Y. Chen, D. E. Walsh and L. R. Koenig, in Pyrolysis Oils from Biomass, American Chemical Society, 1988, vol. 376, ch. 24, pp. 277–289 Search PubMed.
- H. Zhang, Y.-T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 CAS.
- K. Wang and R. C. Brown, ACS Sustainable Chem. Eng., 2014, 2, 2142–2148 CrossRef CAS.
- H. Zhang, T. R. Carlson, R. Xiao and G. W. Huber, Green Chem., 2012, 14, 98–110 RSC.
- A. Zheng, Z. Zhao, S. Chang, Z. Huang, K. Zhao, H. Wu, X. Wang, F. He and H. Li, Green Chem., 2014 10.1039/c3gc42251h.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, Bioresour. Technol., 2010, 101, 4646–4655 CrossRef CAS PubMed.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 636–643 CrossRef CAS PubMed.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2610–2618 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2619–2626 CrossRef CAS.
- G. Luo and A. G. McDonald, Energy Fuels, 2013, 28, 600–606 CrossRef.
- J.-L. Grandmaison, P. D. Chantal and S. C. Kaliaguine, Fuel, 1990, 69, 1058–1061 CrossRef CAS.
- P. A. Horne and P. T. Williams, Renewable Energy, 1996, 7, 131–144 CrossRef CAS.
- Y. Zhao, T. Pan, Y. Zuo, Q.-X. Guo and Y. Fu, Bioresour. Technol., 2013, 147, 37–42 CrossRef CAS PubMed.
- J. Fuhse and F. Bandermann, Chem. Eng. Technol., 1987, 10, 323–329 CrossRef.
- T. C. Bowen, R. D. Noble and J. L. Falconer, J. Membr. Sci., 2004, 245, 1–33 CrossRef CAS PubMed.
- T. Wang, Y. J. Pagán-Torres, E. J. Combs, J. A. Dumesic and B. H. Shanks, Top. Catal., 2012, 55, 657–662 CrossRef CAS.
- K. Wang, P. A. Johnston and R. C. Brown, Bioresour. Technol., 2014, 173, 124–131 CrossRef CAS PubMed.
- J. B. Paine III, Y. B. Pithawalla, J. D. Naworal and C. E. Thomas Jr., J. Anal. Appl. Pyrolysis, 2007, 80, 297–311 CrossRef PubMed.
- J. B. Paine III, Y. B. Pithawalla and J. D. Naworal, J. Anal. Appl. Pyrolysis, 2008, 82, 10–41 CrossRef PubMed.
- T. Milne, R. Evans and J. Filley, in Research in Thermochemical Biomass Conversion, ed. A. V. Bridgwater and J. L. Kuester, Springer, Netherlands, 1988, ch. 69, pp. 910–926 DOI:10.1007/978-94-009-2737-7_69.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, J. Anal. Appl. Pyrolysis, 2009, 86, 323–330 CrossRef CAS PubMed.
- K. Lourvanij and G. L. Rorrer, J. Chem. Technol. Biotechnol., 1997, 69, 35–44 CrossRef CAS.
- P. Dejaifve, A. Auroux, P. C. Gravelle, J. C. Védrine, Z. Gabelica and E. Derouane, J. Catal., 1981, 70, 123–136 CrossRef CAS.
- D. Bibby, N. Milestone, J. Patterson and L. Aldridge, J. Catal., 1986, 97, 493–502 CrossRef CAS.
- G. McLellan, R. Howe, L. Parker and D. Bibby, J. Catal., 1986, 99, 486–491 CrossRef CAS.
- B. Sexton, A. Hughes and D. Bibby, J. Catal., 1988, 109, 126–131 CrossRef CAS.
- M. Uguina, D. Serrano, R. Van Grieken and S. Venes, Appl. Catal., A, 1993, 99, 97–113 CrossRef CAS.
- T. Behrsing, H. Jaeger and J. Sanders, Appl. Catal., 1989, 54, 289–302 CrossRef CAS.
- N.-K. Bär, F. Bauer, D. M. Ruthven and B. J. Balcom, J. Catal., 2002, 208, 224–228 CrossRef.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef PubMed.
- D. Mores, E. Stavitski, M. H. Kox, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Chem. – Eur. J., 2008, 14, 11320–11327 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01784f |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |