
Understanding the destructuration of starch in water–ionic liquid mixtures†
L. S.
Sciarini
*ac,
A.
Rolland-Sabaté
bc,
S.
Guilois
bc,
P.
Decaen
ab,
E.
Leroy
ab and
P.
Le Bail
bc
aLUNAM Université, CNRS, GEPEA, UMR 6144, CRTT, 37, Boulevard de l'Université, 44606 St Nazaire Cedex, France. E-mail: losciarini@agro.unc.edu.ar
bUR1268 Biopolymères Interactions Assemblages, INRA, F-44300 Nantes, France
cStructure Fédérative IBSM, INRA Nantes-Angers, Rue de la Géraudière, BP 71627, 44316 Nantes cedex 3, France
First published on 9th September 2014
Abstract
The destructuration of native maize starch in mixtures of water and ionic liquids (ILs) containing acetate anions was studied in dynamic heating conditions, combining calorimetry, rheology, microscopy and chromatographic techniques. A phase diagram of starch in water–IL solutions was established. The phase transitions undergone by starch include the typical endothermic gelatinization phenomenon for IL–water ratios lower than 0.5, while for mixtures with a higher ionic liquid content, a complex exothermic phenomenon combining mild degradation and solubilization takes place. This results in an optimum destructuration temperature as low as 40–50 °C for an IL–water ratio close to 0.7. In addition, specific macromolecular chain breaking reactions appear to take place, depending on the nature of the cations present, resulting in different macromolecular structures of the recovered starch. These results suggest the possibility of using solvent media design for a controlled modification of starch macromolecular characteristics.
1. Introduction
As renewable resources become essential for industry, the creative design and application of innovative technology for the optimization of such resources is a research topic raising huge interest in the past decade.1 Among all polysaccharides investigated as potential alternatives to conventional oil based plastics, starch has attracted a large amount of attention.2 Starch is one of the most abundant biopolymers in nature, and considering its low cost, renewability and biodegradability, it can be considered as a raw material for the fabrication of biologically degradable materials.Starch is composed of two different glucose polymers: amylose, a predominantly linear macromolecule formed from α(1→4) linkages with a molar mass of ∼105–106 g mol−1, and amylopectin, a massive multiply branched polymer containing both α(1→4) and α(1→6) linkages with a molar mass of ∼107–109 g mol−1. Starch is synthesized in the form of densely packed granules, containing both amorphous and crystalline regions.3 Given its granular structure, starch shows low solubility in any conventional solvent despite being highly hydrophilic. However, when suspended and heated in excess water, starch undergoes an order–disorder transition called gelatinization. During this phenomenon, starch granules swell and amylose progressively leaches out of the granules, and the semi-crystalline structure is disrupted. Although some starch molecules are readily solubilized in water, some granule remnants may still be present even after gelatinization has occurred. Thus, starch insolubility represents a problem when trying to obtain homogeneous amorphous materials.
In recent years, the performance of ionic liquids (ILs) as solvents for biopolymers has generated lots of interest. ILs are room-temperature molten salts; since they present high thermal stability and are not volatile, it has been reported that they offer an alternative to common organic solvents. For this reason and because they are easily recyclable, and even some of them are biodegradable,4 they have been classified as ‘green solvents.’ For these reasons, they have attracted enormous attention over the past decade, becoming a very important area of research.5 Over the last few years, the use of ILs to dissolve and process starch has been reported.6–10 While the first reports focusing on ionic liquids containing chlorine anions showed strong depolymerisation of starch, thus limiting potential applications,9 more recent works on acetate based ionic liquids are more promising,8 despite the fact that no clear evaluation of starch degradation in such systems has been communicated by the authors. In the presence of chlorine anions, the macromolecular degradation of starch has been related to the acidic hydrolysis of glycosidic bonds.6,9,11–13 According to Mateyawa et al.8 the presence of acetate based ionic liquids is likely useful for avoiding such phenomena. The same authors also reported an exothermic transition when starch was heated in pure and concentrated 1-ethyl-3-methylimidazolium acetate (EMIMAc)–water solutions and it was proposed that this exothermic transition was due to starch dissolution, without gelatinization. Stevenson et al.9 reported no enthalpic transition when analysing recovered starch, previously treated with 1-butyl-3-methylimidazolium chloride, suggesting that after being heated in ILs, the starch is destructured and no further gelatinization can be observed when re-heating in water.
As recalled by Brennecke et al.14 one of the major advantages of ILs is that they offer the possibility of being tailored by modifying the chemical structure of the cation and anion moieties. Consequently, in the present work, we focus on the influence of the cation by comparing the thermal destructuration of starch mixed with water and two acetate based ionic liquids: EMIMAc and cholinium acetate. The latter presents the advantage of very low toxicity, choline being an essential nutrient and thus biocompatible.
2. Experimental
2.1. Materials
Regular corn starch (Maritena 100) was purchased from Tate & Lyle (Paris, France) with an initial moisture content of 12%. EMIMAc was produced by BASF and supplied by Sigma-Aldrich. Before use, both materials were dried with P2O5 under vacuum at room temperature for one week. After this time, the starch moisture was lower than 3%. Cholinium acetate (CholAc) was synthesized by a metathesis reaction.15 Equivalent amounts (0.06 mol each) of cholinium chloride and potassium acetate (both purchased from Aldrich) were dissolved in absolute ethanol, and mixed and stirred for 1 hour at room temperature. A white precipitate of potassium was formed and removed by filtration. The ethanol was evaporated on a rotary evaporator. The CholAc thus produced was freeze-dried prior to use. After purification and freeze-drying, the melting point of CholAc was 83 °C.For analysis, the starch was suspended in aqueous solutions of varying IL concentrations (from 0% w/w to 100% w/w IL). Since different concentrations of starch were also studied, a phase diagram was prepared.
ILs are known to be highly hygroscopic, thus the sample preparation was carried out in a glove box under dry gas purge.
2.2. Methodology
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n, w, the dispersity (w/n), and the radius of gyration,
n, w, the dispersity (w/n), and the radius of gyration, ![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) G (nm), were established using ASTRA® software from WTC (version 6.1 for PC), as previously described by Rolland-Sabaté et al.16,18 A value of 0.146 mL g−1 was used as the refractive index increment (dn/dc) for glucans and the normalization of photodiodes was achieved using a low molar mass pullulan standard (P20).
G (nm), were established using ASTRA® software from WTC (version 6.1 for PC), as previously described by Rolland-Sabaté et al.16,18 A value of 0.146 mL g−1 was used as the refractive index increment (dn/dc) for glucans and the normalization of photodiodes was achieved using a low molar mass pullulan standard (P20).
3. Results and discussion
3.1. Differential scanning calorimetry (μDSC)
The thermal behaviour of the starch mixed with aqueous IL solutions of different concentrations was monitored by μDSC (Fig. 1). Increasing concentrations of the ILs were used from the bottom to the top of the figure. When the IL concentration was low, the starch underwent a typical gelatinization, represented by an endothermic transition (a second endothermic transition can be observed at around 100 °C, and is ascribed to the melting of amylose–lipid complexes). The fact that the gelatinization shifts to higher temperatures with increasing IL concentration is consistent with the previously described effect due to the presence of different salts in aqueous solution.19,20 The effect of the salts on starch gelatinization has been found to follow the Hofmeister series, with kosmotropes (structure making, salting-out) delaying gelatinization and chaotropes (structure breaking, salting-in) accelerating it. Acetate is a well-known kosmotrope, so it was expected to produce a shift of gelatinization toward higher temperatures. Nevertheless, a further increase in IL concentration (up to 50% for EMIMAc and 60% for CholAc) led to a decrease in the gelatinization temperature. This trend will be discussed below (see Macromolecular characteristics).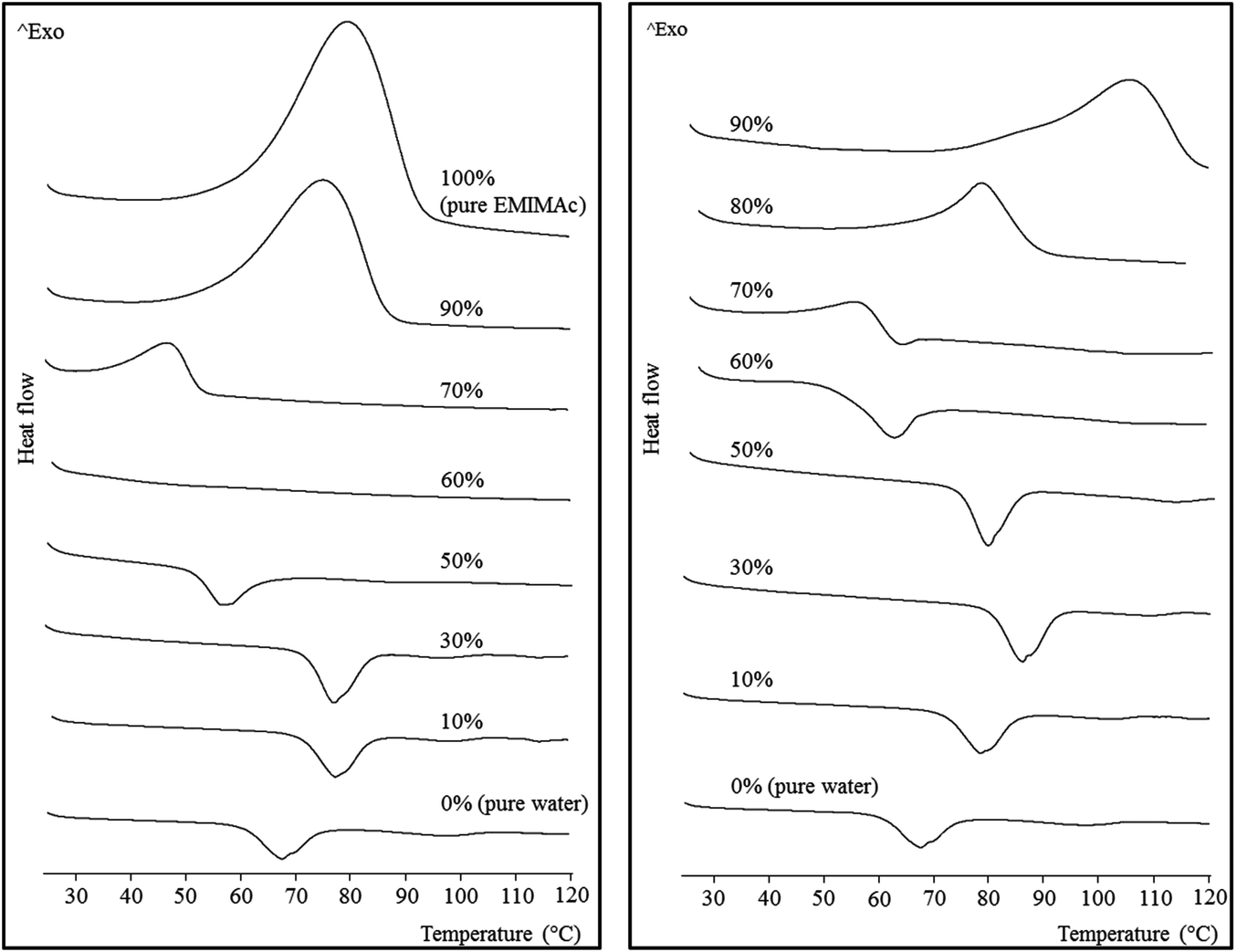 | ||
| Fig. 1 Micro differential scanning calorimetry thermograms for regular corn starch heated in EMIMAc–water (left) and CholAc–water (right) solutions. | ||
If we now consider high IL concentrations (from the top to the bottom, Fig. 1) an exothermic transition is present. The highest concentration of CholAc studied was 95%, but these results were not included in the figure since the exothermic peak was not complete at 120 °C, which is the upper limit for the μDSC; the peak onset temperature is presented in Table 1. This exotherm starts at lower temperatures when water is added and the heat released (ΔH) is also decreased. There is a critical concentration (depending on the IL used) at which both transitions (exo- and endothermic) take place: 70% CholAc and 60% EMIMAc. In the former case, both transitions can be observed, but in the latter both phenomena seem to happen at very close temperatures thus probably cancelling one another.
| Solvent | Transition | ΔH (J g−1) | T o (°C) | T p (°C) | T c (°C) |
|---|---|---|---|---|---|
| a ΔH: transition enthalpy; To: onset temperature; Tp: peak temperature; Tc: conclusion temperature. Values followed by different lowercase letters in the same column are significantly different (p < 0.05). Values followed by different uppercase letters in the same column are significantly different (p < 0.05). | |||||
| 0% IL (pure water) | Endo | 11.9 ± 0.2a | 60.3 ± 0.1b | 67.2 ± 0.0b | 73.3 ± 1.4d |
| 10% CholAc | Endo | 13.7 ± 0.0b | 72.5 ± 0.5c | 78.4 ± 0.1c | 84.6 ± 0.4ef |
| 20% CholAc | Endo | 14.3 ± 0.9bc | 77.5 ± 0.2de | 82.7 ± 0.1d | 86.7 ± 3.4fg |
| 30% CholAc | Endo | 16.2 ± 0.0d | 80.3 ± 0.5e | 85.7 ± 0.6d | 89.8 ± 0.1g |
| 50% CholAc | Endo | 16.2 ± 0.5d | 73.9 ± 0.4cd | 78.7 ± 0.2c | 83.1 ± 0.1e |
| 60% CholAc | Endo | 14.4 ± 0.9bc | 58.7 ± 4.7b | 65.1 ± 3.4b | 69.6 ± 2.6c |
| 70% CholAc | Exo + endo | nd | 46.7 ± 0.2B | nd | 66.9 ± 0.8b |
| 80% CholAc | Exo | 39.5 ± 1.4B | 68.2 ± 2.4E | 78.4 ± 1.4CD | 88.4 ± 1.9CD |
| 90% CholAc | Exo | 67.2 ± 12.7C | 85.9 ± 1.1F | 103.7 ± 2.7E | 114.3 ± 3.4E |
| 95% CholAc | Exo | nd | 97.8 ± 2.5G | nd | nd |
| 10% EMIMAc | Endo | 14.0 ± 0.3b | 70.7 ± 1.0c | 76.6 ± 0.9c | 82.9 ± 0.8d |
| 20% EMIMAc | Endo | 14.7 ± 0.4bc | 73.1 ± 0.4cd | 78.4 ± 0.2c | 84.8 ± 0.2ef |
| 30% EMIMAc | Endo | 15.5 ± 0.3cd | 72.9 ± 0.4c | 77.7 ± 0.5c | 83.7 ± 0.3ef |
| 50% EMIMAc | Endo | 12.5 ± 0.6a | 51.5 ± 0.3a | 56.4 ± 0.3a | 62.9 ± 0.9a |
| 60% EMIMAc | None | nd | nd | nd | nd |
| 70% EMIMAc | Exo | 17.3 ± 4.5A | 36.2 ± 0.8A | 46.7 ± 0.9A | 52.2 ± 0.6A |
| 80% EMIMAc | Exo | 63.3 ± 2.3C | 48.9 ± 1.7B | 65.0 ± 1.9B | 72.5 ± 0.8B |
| 90% EMIMAc | Exo | 110.8 ± 5.3D | 56.2 ± 0.5C | 74.8 ± 0.5C | 85.7 ± 0.8C |
| 100% EMIMAc | Exo | 180.7 ± 20.8E | 62.3 ± 0.6D | 80.1 ± 0.3D | 92.4 ± 0.3D |
The same behaviour was also observed by Mateyawa et al.8 working with EMIMAc and by Koganti et al.21 using N-methyl morpholine N-oxide (NMMO). These authors attributed the exothermic transition to starch dissolution in these solvents. Enthalpy values for the exotherm of normal corn starch in NMMO were 17.5 J g−1 (no enthalpy change was observed when increasing NMMO concentration from 70 to 78%), whereas Mateyawa et al.8 did not provide any ΔH value. In the present study, ΔH of the exothermic transition ranged between 17.3 J g−1 (70% EMIMAc) and 180.7 J g−1 (100% EMIMAc) (Table 1). Moreover, when heated at low rates (0.1 °C min−1), two peaks were clearly observed by μDSC (data presented in the ESI, Fig. S1†); this finding indicates that more than a single phenomenon would be responsible for the exothermic transition. The same trends were observed at different starch–solvent ratios (data not shown).
3.2. Macromolecular characterization of treated starches
In order to understand the phenomena underlying the exothermic transition, the macromolecular properties of starch treated with ILs were studied using the HPSEC-MALLS system. To this end, the starch was treated with different IL–water solutions, recovered and pre-treated with DMSO. The DMSO pre-treatment recoveries were between 95% and 100% for all samples. DMSO pre-treatment is known to remove the polysaccharide oligomers with degree of polymerization (DP) lower than 12. Thus, this high recovery percentage shows that the heating of samples in different IL solutions does not induce the apparition of sugars with DPs smaller than 12. The solubilisation recovery rates and the elution recoveries of the starches were higher than 90%. The high sample recovery values obtained here indicate that the fractionation response was quantitative for all the samples. Overall, this solubilisation procedure was thus considered to enable the structural characterization of these samples.Fig. 2a presents chromatograms for starch heated in pure water and in EMIMAc solutions. When considering starch heated in pure water, two peaks were observed for the differential refractive index signal (corresponding to the concentration of the chains). The first and bigger one (peak I, 5.8 mL) corresponds to amylopectin population, while the second, and smaller, to amylose (peak II, 6.6 mL). When analysing the starch–EMIMAc 100% chromatogram, two peaks are also observed; nevertheless, some important features can be highlighted: (1) the first peak started to elute at higher volumes, indicating a lower size for these molecules as the elution volume is inversely proportional to the molecular size, (2) the second peak is bigger than the first one, and (3) no evident shift in peak II is observed. In addition the molar mass is clearly lower for each fraction of starch–EMIMAc 100% compared to starch–pure water solutions. Overall, these features indicate that amylopectin is depolymerized when heated in EMIMAc, which explains the shift of the amylopectin peak (which accounts for a smaller size), while there is co-elution of the depolymerisation products and amylose, thus explaining the increased area of the second peak. Finally, no evidence of amylose depolymerisation is found (no shift of the peak II). For the samples treated with EMIMAc 70%, amylopectin also eluted at higher volumes, although the overall profile and molar mass distribution are more similar to that of pure water. For EMIMAc 50%, no shift of the amylopectin peak was observed, but the area of peak II is bigger than for starch treated with water, indicating the presence of amylopectin depolymerisation products. Nevertheless, since mild depolymerisation occurs under these conditions (Table 2) the detector response to amylopectin is still high.
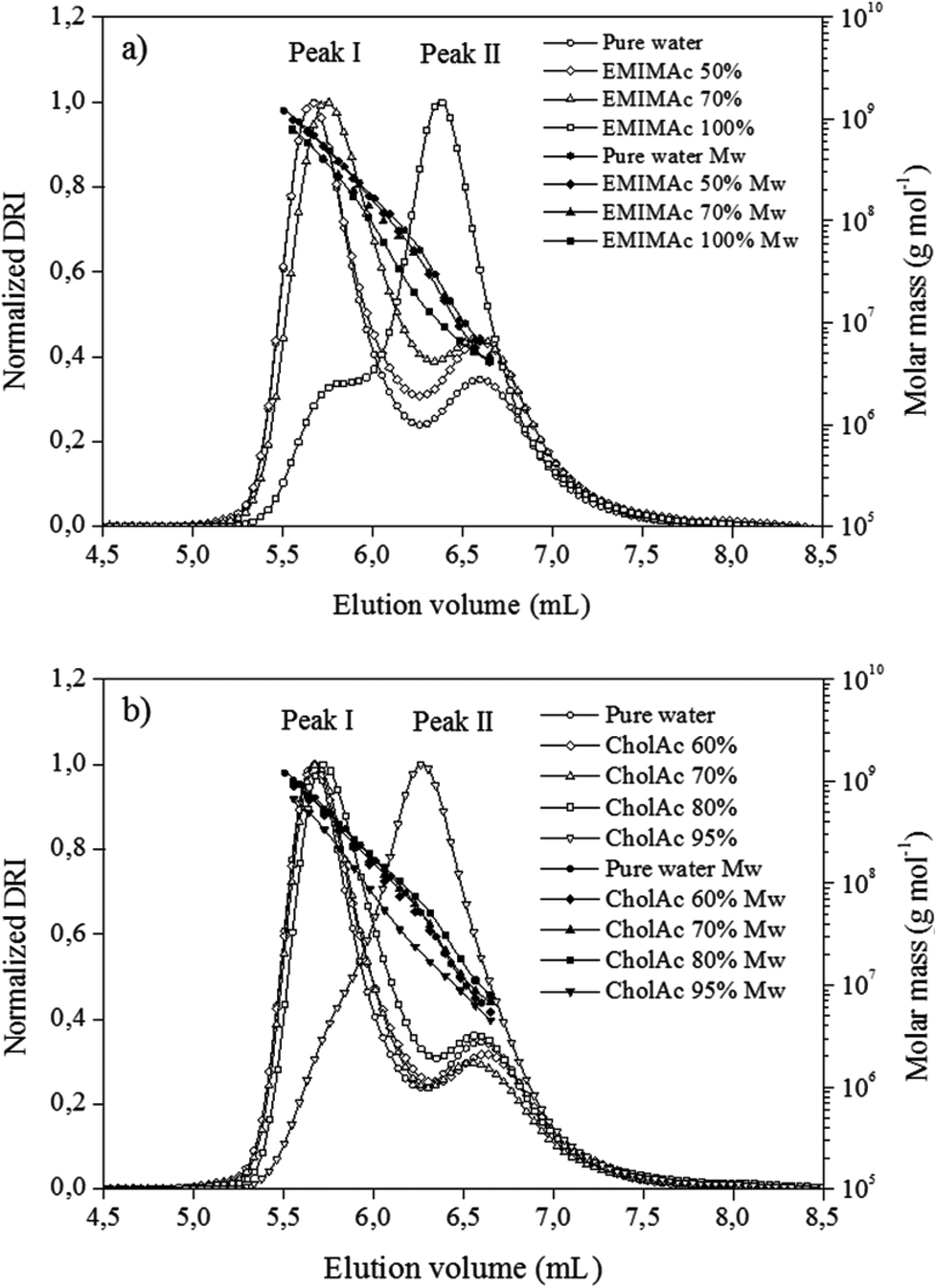 | ||
| Fig. 2 Chromatograms of regular corn starch treated with EMIMAc (a) and CholAc (b) solutions. Differential refractive index (DRI) and molar mass (Mw) versus elution volume. | ||
w), dispersity (w/n) and z-average radius of gyration (G) determined by HPSEC-MALLS for corn starch samples treated in ILs solutions
| Solvent | Whole population | ||
|---|---|---|---|
|
w (107) (g mol−1) |
w/n |
G (nm) |
|
| Pure water | 44.36 ± 0.63 | 7.56 ± 0.49 | 302.8 ± 1.8 |
| EMIMAc 50% | 31.43 ± 2.93 | 10.59 ± 0.31 | 275.5 ± 10.7 |
| EMIMAc 70% | 23.08 ± 0.15 | 6.65 ± 1.37 | 247.6 ± 1.4 |
| EMIMAc 100% | 8.78 ± 0.02 | 7.10 ± 0.04 | 225.8 ± 1.1 |
| CholAc 60% | 33.13 ± 0.18 | 11.09 ± 0.37 | 279.2 ± 0.6 |
| CholAc 70% | 33.57 ± 2.69 | 8.44 ± 0.89 | 268.6 ± 9.5 |
| CholAc 80% | 32.3 ± 0.07 | 5.05 ± 0.06 | 282.4 ± 2.3 |
| CholAc 95% | 8.11 ± 0.00 | 4.48 ± 0.05 | 208.5 ± 0.0 |
For the starch heated in 95% CholAc, only one peak could be clearly detected, while the amylopectin fraction is represented by a shoulder (Fig. 2b) and the molar mass is smaller for each elution volume. This accounts for the depolymerisation of amylopectin by CholAc as well. When water was added to CholAc, a shift of amylopectin elution toward higher volumes is still present, but again the overall behaviour is more similar to that of pure water.
Table 2 shows w and G values obtained by integrating the signals for the whole population of molecules present in the sample. A progressive and linear reduction in w is observed when the EMIMAc concentration is increased, with a reduction of 29%, 48% and 80% for EMIMAc 50%, EMIMAc 70% and EMIMAc 100%, respectively, compared to starch heated in pure water. Interestingly, when treated with CholAc, the reduction in w is non-linear, and reductions are 25%, 24%, 27% and 81% for CholAc 60%, CholAc 70%, CholAc 80% and CholAc 95%, respectively. The same trend is observed for G. This indicates that both ILs have a different response in the presence of water, with rather small quantities of water (20%) significantly reducing the depolymerisation caused by CholAc.
The dispersity (w/n) decrease for the samples treated with ILs (from 7.56 to 7.10 and 4.48 for EMIMAc 100% and CholAc 95%, respectively) is linked to the reduction of the overall peak broadness, and explained by the reduction of the amylopectin molar mass.
Although the two acetate based ionic liquids tested do not completely avoid starch depolymerisation, the reductions of the molar masses found in this study are very different to those obtained after treating starches with halide based imidazolium IL, where reductions of 1–3 orders of magnitude can be observed.6,9,12
From Table 2 it can be observed that a slight depolymerisation takes place when treating starch with EMIMAc 50% and CholAc 60%, even though no exothermic transition was observed by μDSC. This finding may explain why the gelatinization shifts to lower temperatures and ΔH decreases when the starch is heated in μDSC with these IL solutions, since this mild depolymerisation may facilitate starch swelling, shifting gelatinization toward lower temperatures.
Moreover, the significantly lower molar masses observed for the amylopectin populations (peak I, Fig. 2) in EMIMAc 100% and particularly in CholAc 95% treated samples compared to starch–pure water solutions account for a less dense structure (as these fractions exhibit the same size because elution volume is proportional to size in HPSEC). One can deduce that the original amylopectin population is linearized after heating in the ILs, and further in CholAc.
To summarize, it is clear that the starch is depolymerized when heated in IL and that the depolymerization pattern varies according to the cation nature, not only to anion characteristics. Though at present it is not possible to propose a mechanistic explanation for this differential behavior, these results suggest the possibility of tailoring the ionic liquid for a controlled modification of the macromolecular characteristics of starch through mild depolymerisation during destructuration.
The possible interaction between the starch and the IL during heating that could lead to the formation of new molecular species was monitored by FTIR and NMR. No significant changes were found between starch heated in water or the ILs. These results are presented in the ESI (Fig. S2 and S3†).
It is also possible that the mechanism involves not only starch and the IL, but also water molecules. For future studies, a possibly fruitful approach for trying to understand the interactions between these three components and their influence on the destructuration mechanism would be the use of molecular simulation. A recent paper showed the particular interest in this tool for understanding the interactions in the case of cellulose dissolution in IL–water and IL–DMSO mixtures.22 It would also be interesting to study the destructuration of starch in IL–DMSO mixtures, since these simulations show that the co-solvent nature plays an important role in cellulose dissolution by IL.22
3.3. Microstructure of starch suspensions
Fig. 3 presents some representative images of corn starch heated in the IL solutions under light and polarized-light microscopy (Fig. 3a), and differential interference contrast and polarized-light microscopy (Fig. 3b and c). Fig. 3a presents images of the samples before the enthalpic transition (endothermic or exothermic depending on the water content) for the starch suspensions heated in EMIMAc solutions where a well-defined granular structure can be observed (similar images were obtained for CholAc, data not shown). Images of the starch suspensions after the enthalpic transition when heated in EMIMAc and CholAc solutions are presented in Fig. 3(b and c). From these images, it can be observed that, when heated in pure water, starch granules are gelatinized: swelled and deformed with no remaining crystallinity. However, when EMIMAc is added (30 and 50%), gelatinization is not complete, since some granules are still birefringent as a result of their crystallinity. When the amount of EMIMAc reached 70%, however, no polarization was evident after heating, nor the presence of granular remnants; the same was observed for EMIMAc 100%. The absence of granular remnants may indicate that starch is not only depolymerized in concentrated EMIMAc solutions, but is also solubilized. Both phenomena, leading to an overall starch destructuration, may account for the exothermic transitions observed by μDSC. Fig. 3c shows the same behaviour when CholAc was used, but 80% of CholAc was necessary to achieve complete destructuration, since at a lower concentration (70%) granular remnants were present. | ||
| Fig. 3 Light, polarized-light and differential interference contrast images for starch treated in ILs. (a) Light and polarized-light images of starch heated in EMIMAc solutions (To); (b) differential interference contrast and polarized-light images of starch heated in EMIMAc solutions (Tc), and (c) differential interference contrast and polarized-light images of starch heated in CholAc solutions (Tc). Bar scale: 20 μm. | ||
Interestingly, starch suspended in EMIMAc 70% and CholAc 80% is completely destructured at 56 °C and 92 °C, respectively, and under these conditions, only mild depolymerisation occurs (Table 2).
When EMIMAc 100% and CholAc 95% were used, a few gas bubbles were observed under the microscope. These gas bubbles may indicate the formation of volatile products, but could not be identified.
These results are supported by images obtained with an Environmental Scanning Electron Microscope (ESEM) for starch heated in pure water, pure EMIMAc and CholAc 95%. In these images, the destructuration/solubilisation process is evidenced (Fig. S4†).
3.4. Rapid Visco Analyser (RVA)
RVA is an empirical study commonly performed on starch slurries to follow the viscosity behaviour as the sample is heated. While heated, the starch granules start to retain solvent and swell, which results in a concomitant increase in viscosity, i.e. viscosity onset temperature. The viscosity of the suspension increases to the point where the number of swollen intact starch granules is maximal. The peak viscosity (PV) is indicative of the solvent-binding capacity. When the temperature increases and the granule absorbs as much solvent as to achieve its rupture point, the viscosity decreases to a minimum. This decrease in viscosity is called breakdown (BD). When the starch suspension cools, the amylose retrogrades (re-crystallizes), resulting in an increase in viscosity named setback (SB), until a gel is formed at the end of the test. In this study, the viscosity onset temperature correlated with the μDSC onset temperature, although the former was higher than the latter (Tables 1 and 3).| Solvent | Peak viscosity (cP) | Trough (cP) | Breakdown (cP) | Final viscosity (cP) | Setback (cP) | Pasting temperature (°C) |
|---|---|---|---|---|---|---|
| a Values followed by different lowercase letters in the same column are significantly different (p < 0.05). | ||||||
| 0% IL (pure water) | 1444 ± 119a | 1049 ± 42ab | 374 ± 9b | 2673 ± 271b | 1623 ± 229a | 72.5 ± 0.5c |
| 30% CholAc | 2700 ± 41b | 2264 ± 18d | nd | 1740 ± 27a | nd | 90.9 ± 0.8f |
| 50% CholAc | 5473 ± 399d | 4754 ± 339e | nd | 4332 ± 721c | nd | 84.9 ± 0.1e |
| 70% CholAc | nd | nd | nd | nd | nd | nd |
| 95% CholAc | nd | nd | nd | nd | nd | nd |
| 30% EMIMAc | 3568 ± 76c | 1387 ± 38bc | 2181 ± 38c | 3994 ± 76c | 2607 ± 38b | 80.6 ± 0.5d |
| 50% EMIMAc | 8249 ± 20e | 1548 ± 20c | 6701 ± 0d | 4289 ± 62c | 2742 ± 42b | 62.6 ± 0.4b |
| 70% EMIMAc | 3071 ± 95b | 815 ± 16a | 2256 ± 79c | 3868 ± 36.8c | 3053 ± 21c | 55.7 ± 0.9a |
| 100% EMIMAc | 2849 ± 391b | 2681 ± 412d | 168 ± 22a | 9939 ± 308.6d | 7255 ± 105d | 78.8 ± 2.1d |
It has been established that the viscosity onset temperature is higher than the gelatinization onset temperature,23 since different techniques detect starch transitions in different ways, giving slight differences in the determined parameters.
Fig. 4 presents the viscosity profiles of starch in EMIMAc (Fig. 4a) and CholAc (Fig. 4b) solutions. The ILs alone were also analysed: their viscosity was near zero and no change in viscosity was observed during the heating and cooling processes. From Fig. 1 it can be seen that the starch heated in concentrated EMIMAc solutions (100% and 70%) undergoes an exothermic transition, related to starch depolymerisation/solubilisation; when the amount of EMIMAc is 50% or lower, an endothermic transition – ascribed to gelatinization – takes place. Fig. 4a shows that when EMIMAc 100% is used, the viscosity increases as depolymerisation/solubilisation take place. During the cooling stage, the viscosity increases significantly, probably as a consequence of the interaction between the products of depolymerisation, which are smaller and less branched than amylopectin, favouring their association, and also as a consequence of amylose retrogradation. As water is added (EMIMAc 70%), the viscosity onset temperature and peak viscosity are lower (in agreement with μDSC results, Table 1). The decrease in the viscosity onset temperature is related to the lower viscosity of the solvent when compared to pure EMIMAc, and its diffusion into the granule would be faster facilitating depolymerisation. However, starch depolymerisation is lower in EMIMAc 70%, explaining the lower viscosity value during heating when compared to pure EMIMAc (Table 3). On increasing the water content to 50% (EMIMAc 50%), solvent diffusion into the granules increases and the amount of water is enough to gelatinize the starch. In addition, a slight depolymerisation is also observed with EMIMAc 50% (Table 2). Both phenomena may explain the higher viscosity shown by this sample (Table 3). When EMIMAc 30% is used, the pasting behaviour is closer to that of starch in pure water, although the overall viscosity is higher. These results are in good agreement with Mateyawa et al.8
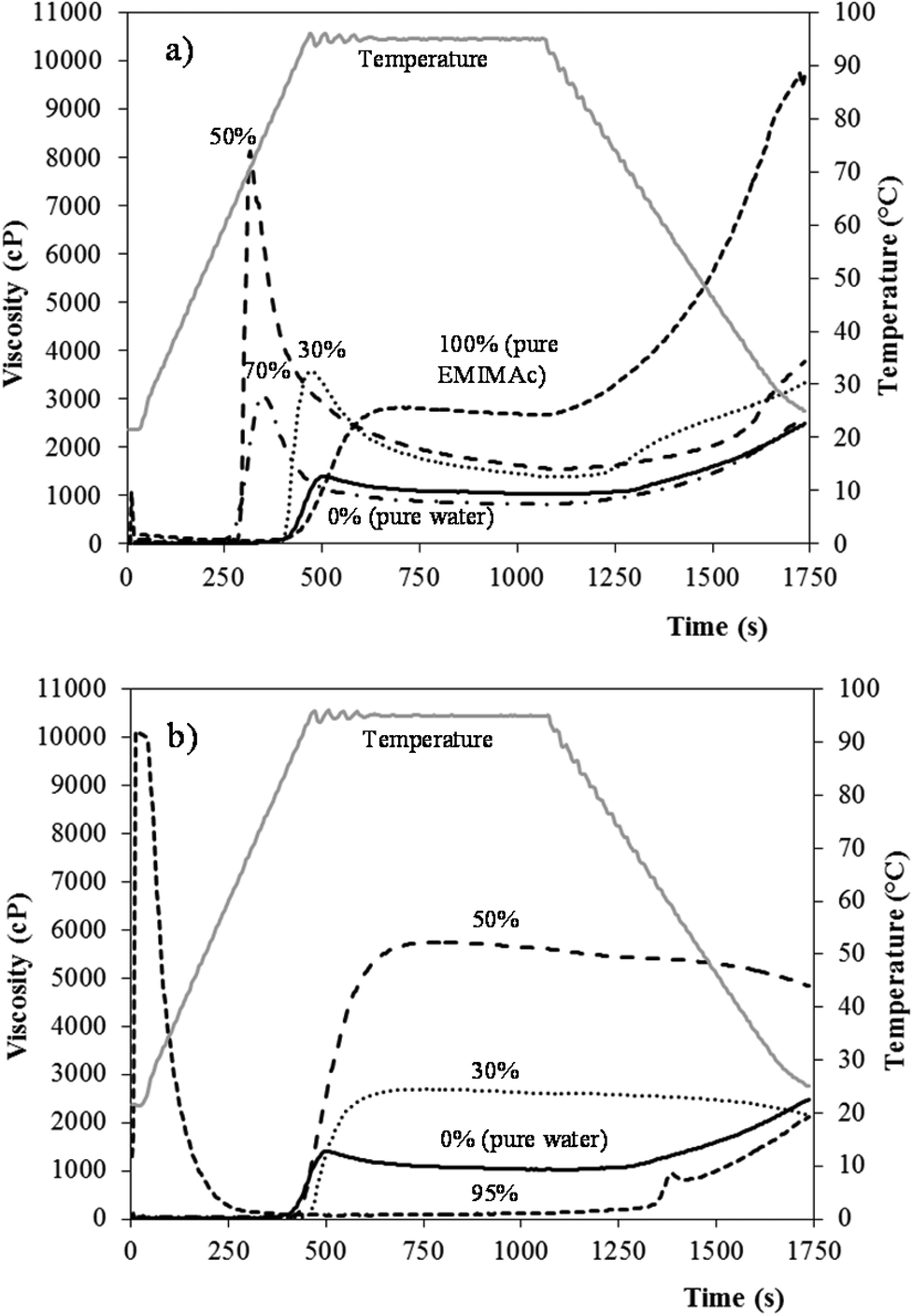 | ||
| Fig. 4 RVA curves for starch heated in EMIMAc (a) and CholAc (b) solutions. | ||
Fig. 4b shows that the starch in CholAc solutions has a different behaviour than in EMIMAc solutions. When heated in the concentrated CholAc solution (CholAc 95%), two peaks are present: at the beginning of the test, CholAc is in the solid state – explaining the high viscosity of the sample at this point – but as the temperature increases, it melts; a second increase in the viscosity is observed during the cooling period. The exothermic peak (observed by μDSC) for this sample starts at around 97 °C (Table 1); this may explain the absence of a viscosity peak during heating. However, some depolymerisation may have occurred during the heating at 95 °C, forming smaller and more linear molecules from amylopectin, explaining the slight increase in viscosity during cooling. CholAc 70% could not be analysed since the viscosity exceeded the RVA limit (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cP).
000 cP).
For CholAc 50% and 30%, an increase in the viscosity was observed between 85 and 90 °C (Table 3), and no viscosity breakdown was found. The viscosity increase started late during heating, while the maximum temperature reached by the RVA is 95 °C. This temperature may not be sufficient to completely disrupt the starch granular structure, although granules may swell and some amylose may leach out, resulting in a viscosity increase.
3.5. Phase diagram
In summary, Fig. 5 shows the corn starch phase diagrams when treated in different IL solutions. When the IL concentration is high, a complete loss of granular structure is observed (results are supported by microscopy images) and this destructuration is accompanied by starch depolymerisation (HPSEC-MALLS results); the degree of depolymerisation depends on the water amount. When the water content is sufficiently high, gelatinization occurs, instead of destructuration/solubilisation. Under these conditions, granular remnants are still observed after heating. When EMIMAc 60% is used, a partial gelatinization followed by partial destructuration/solubilisation takes place, and the same is true for CholAc 70%.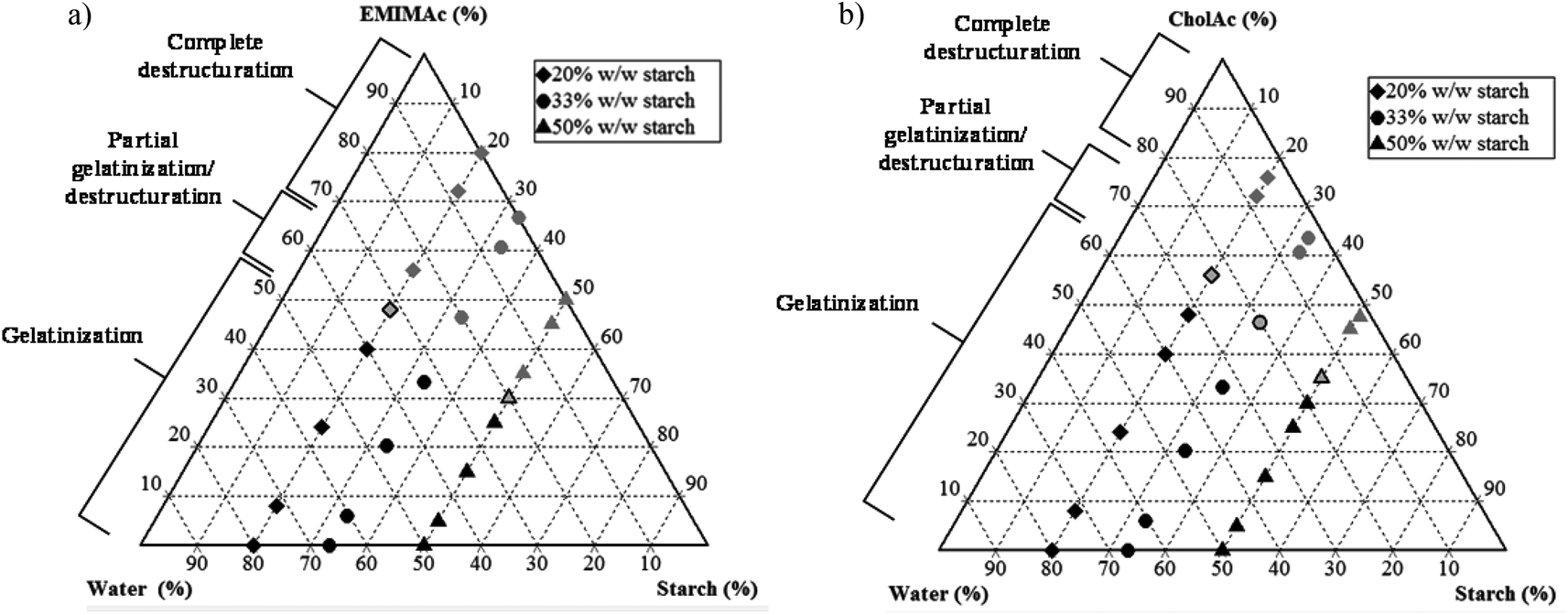 | ||
| Fig. 5 Phase diagrams for starch treated in solution with different IL concentrations. (a) EMIMAc treated starch, (b) CholAc treated starch. | ||
4. Conclusions
The results presented in this study show that two different phenomena take place when starch is heated in EMIMAc and CholAc solutions: when the concentration of both ILs is low enough, gelatinization is the dominant phenomenon, whereas when the concentration is higher, depolymerisation and dissolution of the starch take place. The effect of both ILs on gelatinization corresponds to that of stabilizing salts.EMIMAc and CholAc have been shown to be appropriate solvents for starch destructuration when mixed with the correct amount of water (30% water for EMIMAc and 20% water for CholAc). At these concentrations, destructuration (depolymerisation and dissolution) starts at temperatures as low as 36 °C and 68 °C, respectively and, after heating at 120 °C, the average molar mass of starch is reduced by 27% and 48% when heated in CholAc 80% and EMIMAc 70%, respectively. This suggests that specific starch chain breaking reactions may occur depending on the cation present in the IL, which could open the possibility of solvent media design for a controlled modification of the macromolecular characteristics of the starch.
Acknowledgements
Authors gratefully thank the Région des Pays de la Loire for financial support. We would also like to thank Bérénice Houinsou-Houssou for her kind technical support, and Brigitte Bouchet, Bénédicte Bakan and Guillaume Roelens for their support in microscopy, FTIR and ESEM analysis, respectively.Notes and references
- S. H. Ghaffar and M. Fan, Int. J. Adhes. Adhes., 2014, 48, 92 CrossRef CAS PubMed.
- R. L. Shogren, G. F. Fanta and W. M. Doane, Starch/Staerke, 1993, 45, 276 CrossRef CAS.
- A. Buléon, P. Colonna, V. Planchot and S. Ball, Int. J. Biol. Macromol., 1998, 23, 85 CrossRef.
- N. Gathergood, P. J. Scammells and M. T. Garcia, Green Chem., 2006, 8, 156 RSC.
- T. G. A. Youngs, C. Hardacre and J. D. Holbrey, J. Phys. Chem. B, 2007, 111, 13765 CrossRef CAS PubMed.
- K. Lappalainen, J. Kärkkäinen and M. Lajunen, Carbohydr. Polym., 2013, 93, 89 CrossRef CAS PubMed.
- W. Liu and T. Budtova, Carbohydr. Polym., 2013, 93, 199 CrossRef CAS PubMed.
- S. Mateyawa, D. Fengwei Xie, R. W. Truss, P. J. Halley, T. M. Nicholson, J. L. Shamshina, R. D. Rogers, M. W. Boehm and T. McNally, Carbohydr. Polym., 2013, 94, 520 CrossRef CAS PubMed.
- D. G. Stevenson, A. Biswas, J.-L. Jane and G. E. Inglett, Carbohydr. Polym., 2007, 67, 21 CrossRef CAS PubMed.
- A. Biswas, R. L. Shogren, D. G. Stevenson, J. L. Willett and P. K. Bhowmik, Carbohydr. Polym., 2006, 66, 546 CrossRef CAS PubMed.
- K. Lappalainen, J. Kärkkäinen, J. Panula-Perälä and M. Lajunen, Starch/Staerke, 2012, 64, 263 CrossRef CAS.
- J. Kärkkäinen, K. Lappalainen, P. Joensuu and M. Lajunen, Carbohydr. Polym., 2011, 84, 509 CrossRef PubMed.
- V. Myllymäki and R. Aksela, WO Pat. No2005/066374A1, 2005 Search PubMed.
- J. F. Brennecke, R. D. Rogers and K. R. Seddon, in ACS symposium series, American Chemical Society, Washington DC, 2007, vol. 975 Search PubMed.
- P. Nockemann, B. Trijs, K. Driesen, C. Janssen, K. Van Hecke, L. Van Meervelt, S. Kossmann, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2007, 111, 5254 CrossRef CAS PubMed.
- A. Rolland-Sabaté, N. G. Amani, D. Dufour, S. Guilois and P. Colonna, J. Sci. Food Agric., 2003, 83, 927 CrossRef.
- V. Planchot, P. Colonna and L. Saulnier, in Guide Pratique d'Analyses dans les Industries des Céréales, ed. B. Godon and W. Loisel, Lavoisier, Paris, France, 1997, p. 341 Search PubMed.
- A. Rolland-Sabaté, P. Colonna, M. G. Mendez-Montealvo and V. Planchot, Biomacromolecules, 2008, 9, 1719 CrossRef PubMed.
- J.-L. Jane, Starch/Staerke, 1993, 45, 161 CrossRef CAS.
- F. B. Ahmad and P. A. Williams, J. Agric. Food Chem., 1999, 47, 3359 CrossRef CAS PubMed.
- N. Koganti, J. R. Mitchell, R. N. Ibbett and T. J. Foster, Biomacromolecules, 2011, 12, 2888 CrossRef CAS PubMed.
- J.-M. Andanson, E. Bordes, J. Devémy, F. Leroux, A. A. H. Pádua and M. F. Costa Gomes, Green Chem., 2014, 16, 2528 RSC.
- C. G. Biliaderis, in Starch: Chemistry and Technology, ed. J. N. BeMiller and R. L. Whistler, Academic Press, New York, USA, 2009, p. 293 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: μDSC, FTIR, 13C CP/MAS NMR results and ESEM images are provided. See DOI: 10.1039/c4gc01248h |
| This journal is © The Royal Society of Chemistry 2015 |