
Hydration of alkynes at room temperature catalyzed by gold(I) isocyanide compounds†
Yun
Xu
ab,
Xingbang
Hu
*a,
Jing
Shao
a,
Guoqiang
Yang
a,
Youting
Wu
*a and
Zhibing
Zhang
a
aSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, PR China. E-mail: huxb@nju.edu.cn; ytwu@nju.edu.cn; Fax: +86 2583 336599; Tel: +86 2583 596665
bChina Pharmaceutical University, Nanjing, 211198, PR China
First published on 18th September 2014
Abstract
An effective method using gold(I) isocyanide complexes as catalysts for the transformation of various alkynes to the corresponding ketones is successfully developed. The hydration process proceeds smoothly at room temperature with quite high yield (up to 99%). The catalytic center is the isocyanide-Au(I)+ cation. Further theoretical research reveals a direct hydration mechanism by H2O, and the rate-determining step has an energy barrier of 23.7 kcal mol−1. These results show a good example to reduce unnecessary steps and achieve milder reaction conditions at the same time for the hydration of alkynes.
Introduction
The addition of water to alkynes is an environmentally friendly method to prepare ketones with 100% atomic economy. However, the traditional alkyne hydration was realized using mercury(II) salts, a highly toxic compound, as the catalyst.1 Even though different Brønsted acids were also found to have catalytic activity for the hydration of alkynes,2–4 the processes require, in most cases, stoichiometric or excess amounts of acids and only electron-rich alkynes as the substrates can lead to high conversions.To overcome the disadvantages of mercury(II) salts and Brønsted acids, a variety of organometallic catalysts were explored, such as compounds containing Pt,5 Ag,6,7 Ru,8,9 Au, and even biomimetic catalysts.10 Among them, the unique catalytic ability of gold(I) and gold (III) has attracted much attention, because gold centers show high affinity to activate C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bonds by nucleophilic attack. Though gold compounds were thought to be potential and excellent catalysts for alkyne hydration,11,12 most of the reactions catalyzed by gold compounds require high reaction temperatures, such as 120 °C for the hydration catalyzed by gold N-heterocyclic carbenes (Au-NHC)/AgSbF6,12,13 Au-NHC/HSbF6,11 and Au-NHC@porous organic polymers,14 100–150 °C for bis(phosphane) gold(I),15 100 °C for NaAuCl4,16 80 °C for Au-NHC17 and Au-SH/SO3H-PMO(Et),8 70 °C for (Ph3P)AuCH3 in the presence of H2SO4,18 65 °C for abnormal Au-NHC,19 and reflux temperature.20–24 Obviously, it is still a challenge to develop more effective gold catalysts for alkyne hydration at room temperature.
C triple bonds by nucleophilic attack. Though gold compounds were thought to be potential and excellent catalysts for alkyne hydration,11,12 most of the reactions catalyzed by gold compounds require high reaction temperatures, such as 120 °C for the hydration catalyzed by gold N-heterocyclic carbenes (Au-NHC)/AgSbF6,12,13 Au-NHC/HSbF6,11 and Au-NHC@porous organic polymers,14 100–150 °C for bis(phosphane) gold(I),15 100 °C for NaAuCl4,16 80 °C for Au-NHC17 and Au-SH/SO3H-PMO(Et),8 70 °C for (Ph3P)AuCH3 in the presence of H2SO4,18 65 °C for abnormal Au-NHC,19 and reflux temperature.20–24 Obviously, it is still a challenge to develop more effective gold catalysts for alkyne hydration at room temperature.
In the last decade, an increasing number of studies have focused on the preparation of gold isocyanide complexes (Au-RNC) and the investigation of their structure and properties.25–27 But so far, Au-RNC complexes were mainly used as the starting materials for the synthesis of other novel catalysts.28 Among the complexes obtained via Au-RNC, Au-NHC represents a kind of the most extensively studied compound in organometallic chemistry and catalysis (Scheme 1).29–39 To realize the reported catalytic ability of Au-NHC for the hydration of alkynes, high reaction temperatures are needed to obtain satisfactory conversions.11–14,17,19–23 As far as we know,40 there is almost no report that the Au-RNC complex can be directly used as a catalyst in not only alkyne hydration, but also other reactions.
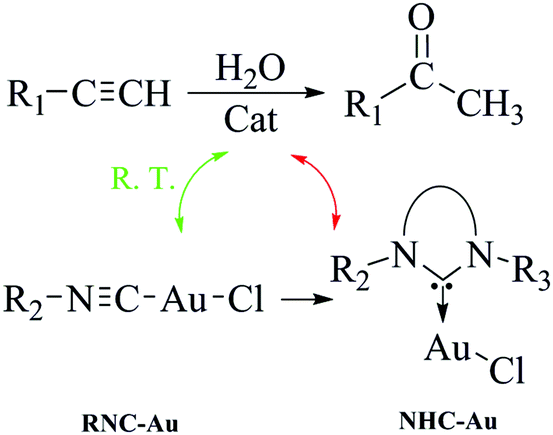 | ||
| Scheme 1 The relationship between Au–RNC and Au–NHC and their catalytic ability for the hydration of alkynes. | ||
Both experimental and theoretical research studies in this paper show, to our surprise, that Au-RNC has excellent activity in catalyzing the hydration of alkynes at room temperature. It is a good example that avoids the use of complicated catalysts and eliminates harsh reaction conditions in the hydration of alkynes (Scheme 1).
Results and discussion
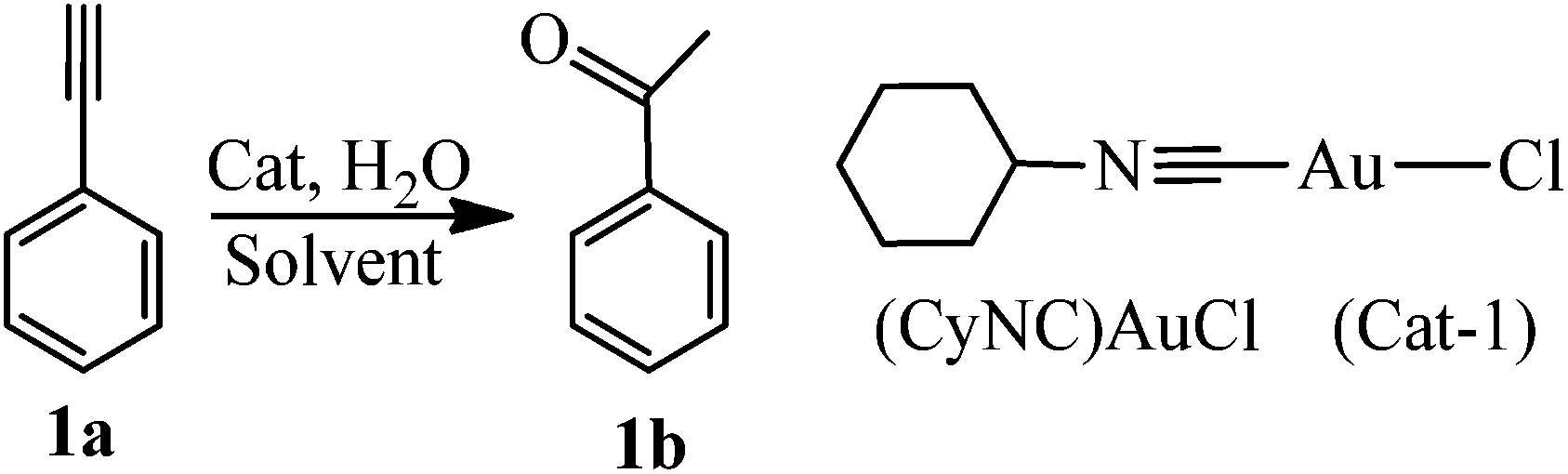
Hydration of phenylacetylene catalyzed by gold isocyanide
Two simple gold isocyanide complexes (RNC)AuCl (R = Cy, 2,6-Me2C6H3) were synthesized according to the literature procedures (see ESI† for detailed synthetic procedures and characterization).31,34,38 The hydration of phenylacetylene (1a) catalyzed by (CyNC)AuCl (Cat-1) was selected as a probe to investigate the activity of the catalyst (Table 1). No acetophenone (1b) was observed when Cat-1 was used as the catalyst alone (entry 1). As we know, the active center of gold(I) complexes in many reactions should be the gold(I) cation.41 To release the gold cation, some weakly coordinating anions (such as SbF6−, BF4−, B(C6H5)4−, and B(C6F5)4−) were added to the reaction system (entries 2–5). Though AgSbF6 itself can be used as the catalyst for the hydration of alkynes,7 almost no ketone was produced in our research when using Cat-1/AgSbF6 as the catalyst because the reaction temperature is far lower than that in ref. 7. Among these anions (SbF6−, BF4−, B(C6H5)4−, and B(C6F5)4−), only B(C6F5)4− which has the weakest coordination ability gives satisfactory results (entry 5). To exclude the possibility of KB(C6F5)4 acting as a catalyst, a reaction with only KB(C6F5)4 was also performed and no product was observed (entry 6). All the results mentioned above indicated that the active center in this reaction system should be the (CyNC)Au+ cation (Cat-1+).|
|
|||
|---|---|---|---|
| Entry | Catalystb (mol) | Solvent | Conv.c (%) |
| a Reaction conditions: phenylacetylene (0.5 mmol), solvent (0.5 ml), H2O (0.5 ml), room temperature, 24 h. b The catalyst amount is based on the molar percent to 1a. c Conversion of 1a determined by GC. d Methanol (0.5 ml) and H2O (1.0 ml). e Methanol (1.0 ml) and H2O (0.5 ml). f Methanol (2.0 ml) and H2O (0.5 ml). | |||
| 1 | 5% Cat-1 | Methanol | NR |
| 2 | 5% Cat-1 + 5% AgSbF6 | Methanol | 0.6 |
| 3 | 5% Cat-1 + 5% KBF4 | Methanol | 3.7 |
| 4 | 5% Cat-1 + 5% KB(C6H5)4 | Methanol | 1.5 |
| 5 | 5% Cat-1 + 5% KB(C6F5)4 | Methanol | 99.9 |
| 6 | 5% KB(C6F5)4 | Methanol | NR |
| 7d | 5% Cat-1 + 5% KB(C6F5)4 | Methanol | 99.9 |
| 8e | 5% Cat-1 + 5% KB(C6F5)4 | Methanol | 32.2 |
| 9f | 5% Cat-1 + 5% KB(C6F5)4 | Methanol | 5.0 |
| 10 | 1% Cat-1 + 1% KB(C6F5)4 | Methanol | 20.2 |
| 11f | 1% Cat-1 + 1% KB(C6F5)4 | Methanol | <1.0 |
| 12 | 5% Cat-1 + 5% KB(C6F5)4 | Ethanol | 27.2 |
| 13 | 5% Cat-1 + 5% KB(C6F5)4 | Hexane | 25.7 |
| 14 | 5% Cat-1 + 5% KB(C6F5)4 | Dioxane | 10.1 |
| 15 | 5% Cat-1 + 5% KB(C6F5)4 | Acetone | 3.4 |
| 16 | 5% Cat-1 + 5% KB(C6F5)4 | THF | 0.8 |
| 17 | 5% Cat-1 + 5% KB(C6F5)4 | None | 57.0 |
Using an environmentally preferable solvent is quite important for a green process. The solvents screening experiments on methanol, ethanol, hexane, dioxane, acetone and THF show that the reaction performed in methanol gives the best result (entries 5, 12–16 in Table 1). Using methanol as a solvent is quite green according to the framework for the environmental assessment of solvents,42 Sanofi's solvent selection guide,43 and the EHS assessment for alternative solvents.44 The amount of solvent has an obvious influence on the conversion of phenylacetylene. Comparing with the standard conditions, increasing the amount of methanol results in low conversion of the reactants (entries 5, 7–9).
It has been found that some alkynes can be converted into the vinyl ether intermediate by the addition of methanol to the CC bond, and vinyl ether can then be hydrolyzed into ketone.11,12 The catalysis reported here should not proceed in such a pathway, because no vinyl ethers were detected by GC-MS during the reactions of all the substrates investigated here (Tables 1 and 2). Furthermore, when only H2O was used as both the solvent and the reactant, the reaction conversion still can reach 57.0% in 24 h (entry 17 in Table 1).
| Entry | Reactant | Product | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: alkyne (1.0 mmol), Cat-1 (0.05 mmol), KB(C6F5)4 (0.05 mmol), H2O (1.0 ml), methanol (1.0 ml), room temperature, 24 h. b Yield determined by GC with biphenyl as the internal standard. Isolated yields are shown in parenthesis. c 10% mol catalyst, 48 h. d Reaction time: 48 h [24 h]. | |||
| 1 |
|
|
>99 |
| 2 |
|
|
>99 (96) |
| 3 |
|
|
>99 |
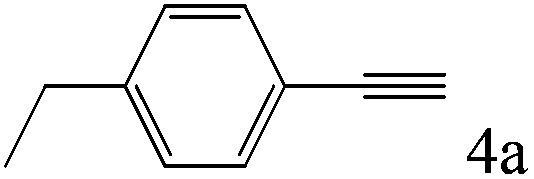
| 4 |
|
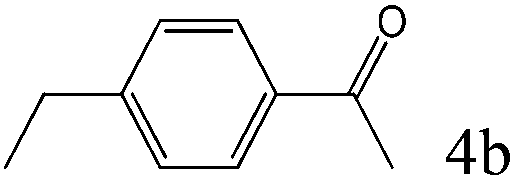
|
>99 (95) |
| 5 |
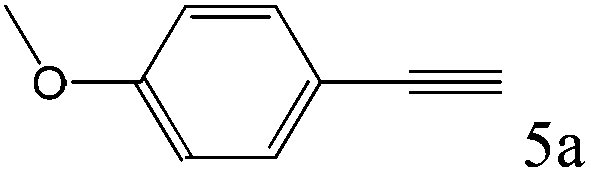
|
|
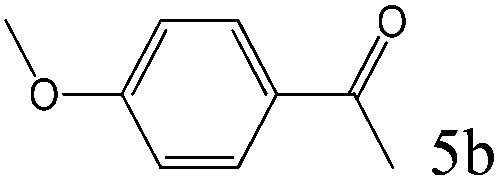
>99 (97) |
| 6c |
|
|
63 |
| 7c |
|
|
8 [5]d |
| 8 |
|
|
>99 (99) |
| 9 |
|
|
>99 (95) |
| 10 |
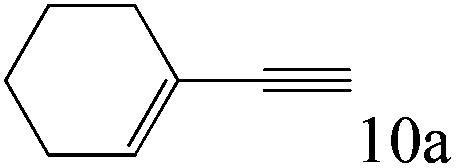
|
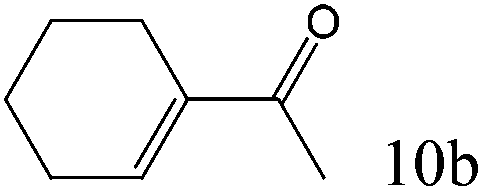
|
>99 (98) |
| 11 |

|
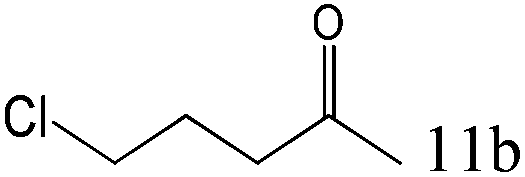
|
>99 |
| 12 |
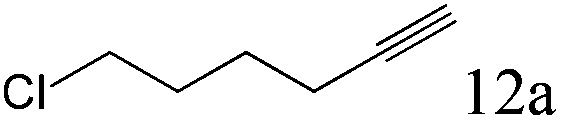
|
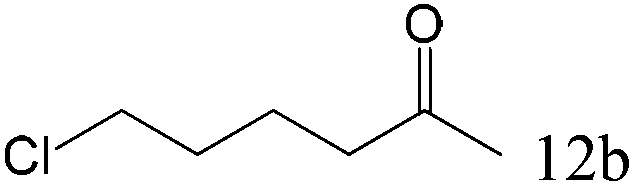
|
>99 (99) |
| 13 |
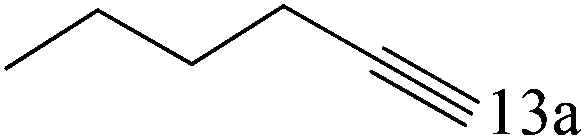
|
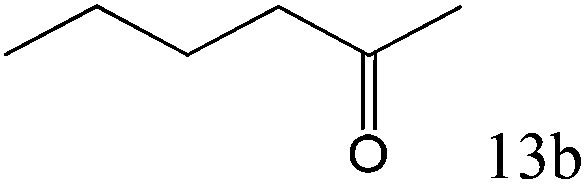
|
>99 (98) |
| 14 |
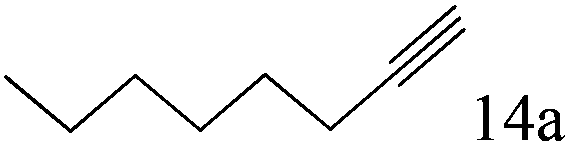
|
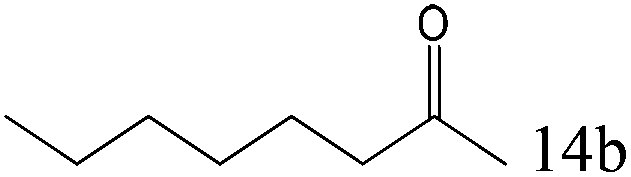
|
97 |
| 15 |
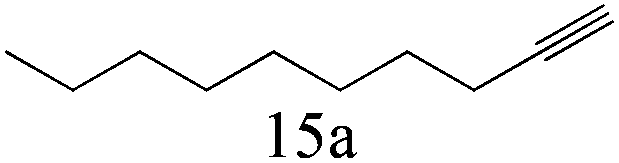
|
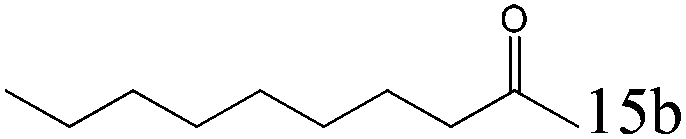
|
>99 (99) |
| 16c |
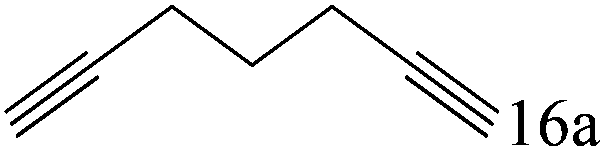
|
|
97 (95) |
| 17c |
|
|
98 (97) |
Substrate scope of the gold isocyanide catalysts
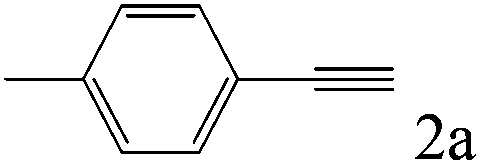
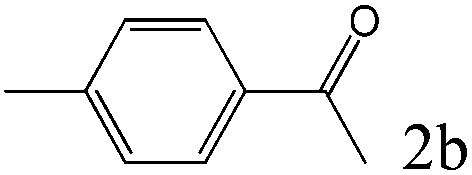
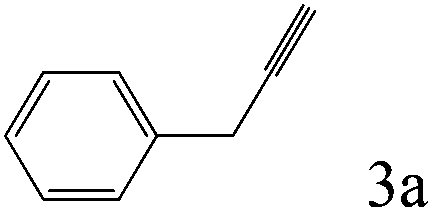
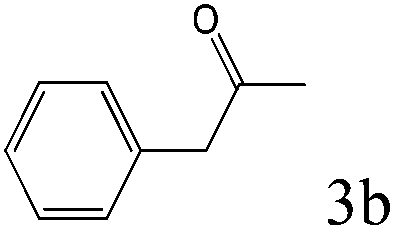
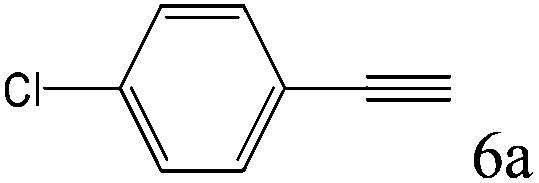
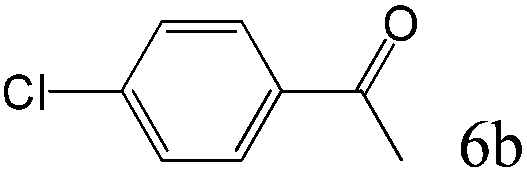
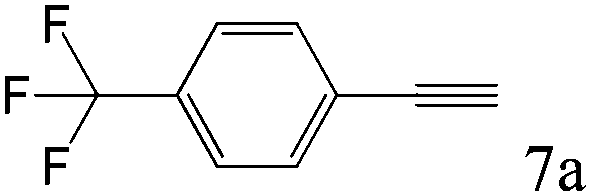
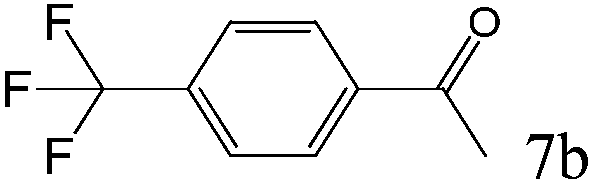
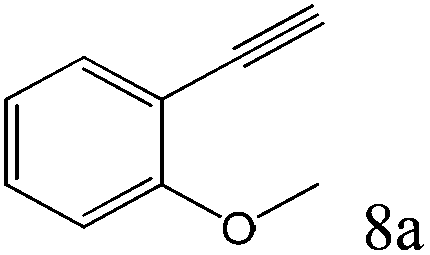
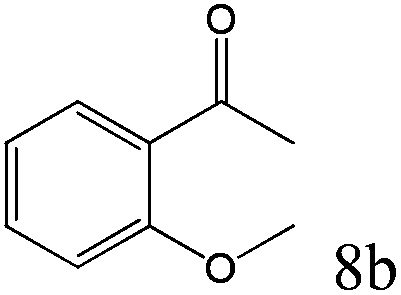
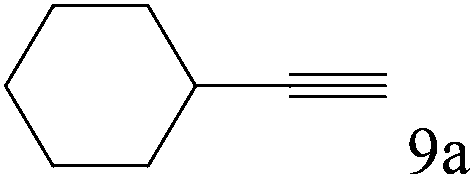
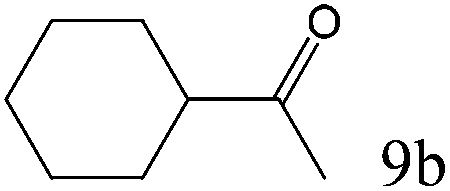
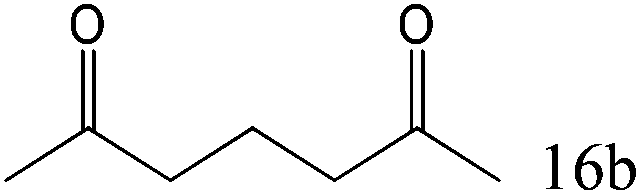
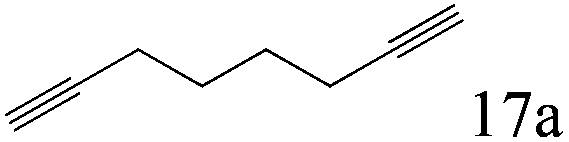
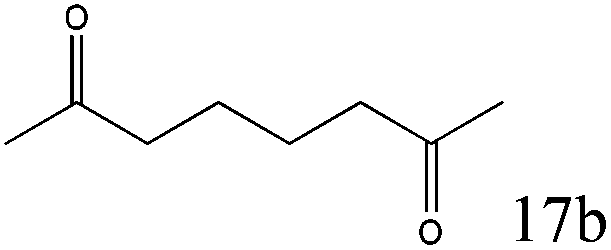
To explore the substrate scope of this catalytic system, the hydration of different alkynes was investigated under the standard conditions (Table 2). Cat-1 shows excellent catalytic reactivity at room temperature for a wide range of substrates, including electron-rich and electron-deficient alkynes, aromatic and non-aromatic alkynes. In total seventeen alkynes were investigated, and the isolated or GC yields of fifteen alkynes are larger than 95%. For relatively electron-deficient alkynes, such as 6a, only 63% yield was obtained (entry 6). Further increase in the electron withdrawing ability of the substituent by CF3 results in a quite slow reaction rate. The GC yield of 7b is only 5% in 24 hours and 8% in 48 hours (entry 7). For electron-donating alkynes, 5a and 8a, satisfactory isolated yields (>97%) were obtained, especially when the methoxyl is located at the ortho-position (entries 5 and 8). No matter the CC triple bond binds with the aromatic ring or alkane, the reactions perform quite well. Besides alkynes, the hydration of diynes, such as 16a and 17a, can also be well catalyzed by gold isocyanide with high yields at room temperature (entries 16 and 17).
In addition to Cat-1, another gold isocyanide (2,6-Me2C6H3-NC)AuCl (Cat-2) was also investigated and its catalytic ability was compared with that of Cat-1 (Fig. 1). Though the activity of Cat-2 is a little weaker than that of Cat-1, the reaction catalyzed by Cat-2 can still proceed quite well at room temperature (Table S1 in ESI†).
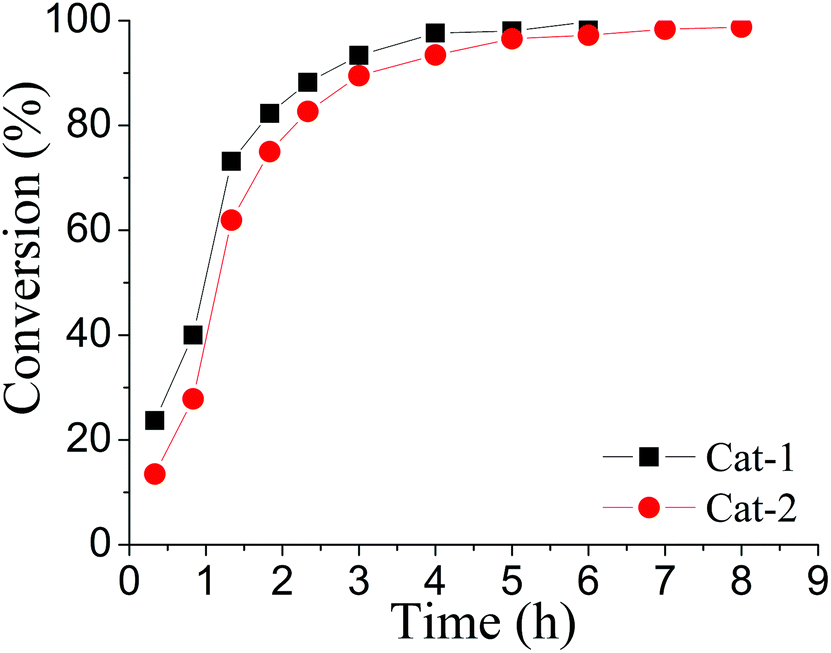 | ||
| Fig. 1 Kinetic plot of conversion of 5a with respect to time under standard conditions. | ||
Another route to synthesize these ketones is the catalytic aerobic oxidation of alcohols.45–53 It should be noticed that the only by-product is water for an ideal oxidation process, and some of these oxidations can be performed at room temperature.45–49 However, NaNO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48,49 or excess amounts of bases45–47,50,51 are usually indispensable in these aerobic oxidations. Though some of the alcohols used in the oxidation routes are cheaper than the alkynes in the hydration routes, the following characteristics of the route reported here make itself a more green choice for the synthesis of ketones: 100% atomic economy, mild reaction conditions, an environmentally preferable solvent, no need of acids or bases.
48,49 or excess amounts of bases45–47,50,51 are usually indispensable in these aerobic oxidations. Though some of the alcohols used in the oxidation routes are cheaper than the alkynes in the hydration routes, the following characteristics of the route reported here make itself a more green choice for the synthesis of ketones: 100% atomic economy, mild reaction conditions, an environmentally preferable solvent, no need of acids or bases.
Hydration mechanism of the reaction catalyzed by gold isocyanide
It has been found in the literature that alkynes with methanol as a solvent can be hydrated to the expected ketones via vinyl ether intermediates.11,12 However, for the hydration catalyzed by the gold isocyanide complexes (Cat-1 and Cat-2), no signal of vinyl ether intermediates was detected by GC-MS analysis. Besides, the reaction still can perform well without the addition of methanol (entries 13 and 14, Table 1). These two evidences mentioned above imply a direct hydration of alkynes by H2O.7 To further understand the hydration mechanism catalyzed by the gold isocyanide, a DFT based theoretical investigation was performed (see ESI† for computational details) (Fig. 2). According to the known knowledge of other Au(I) catalysts41 and results listed in Table 1, the active center in the reaction should be the (Cy–NC)Au+ (Cat-1+) cation.
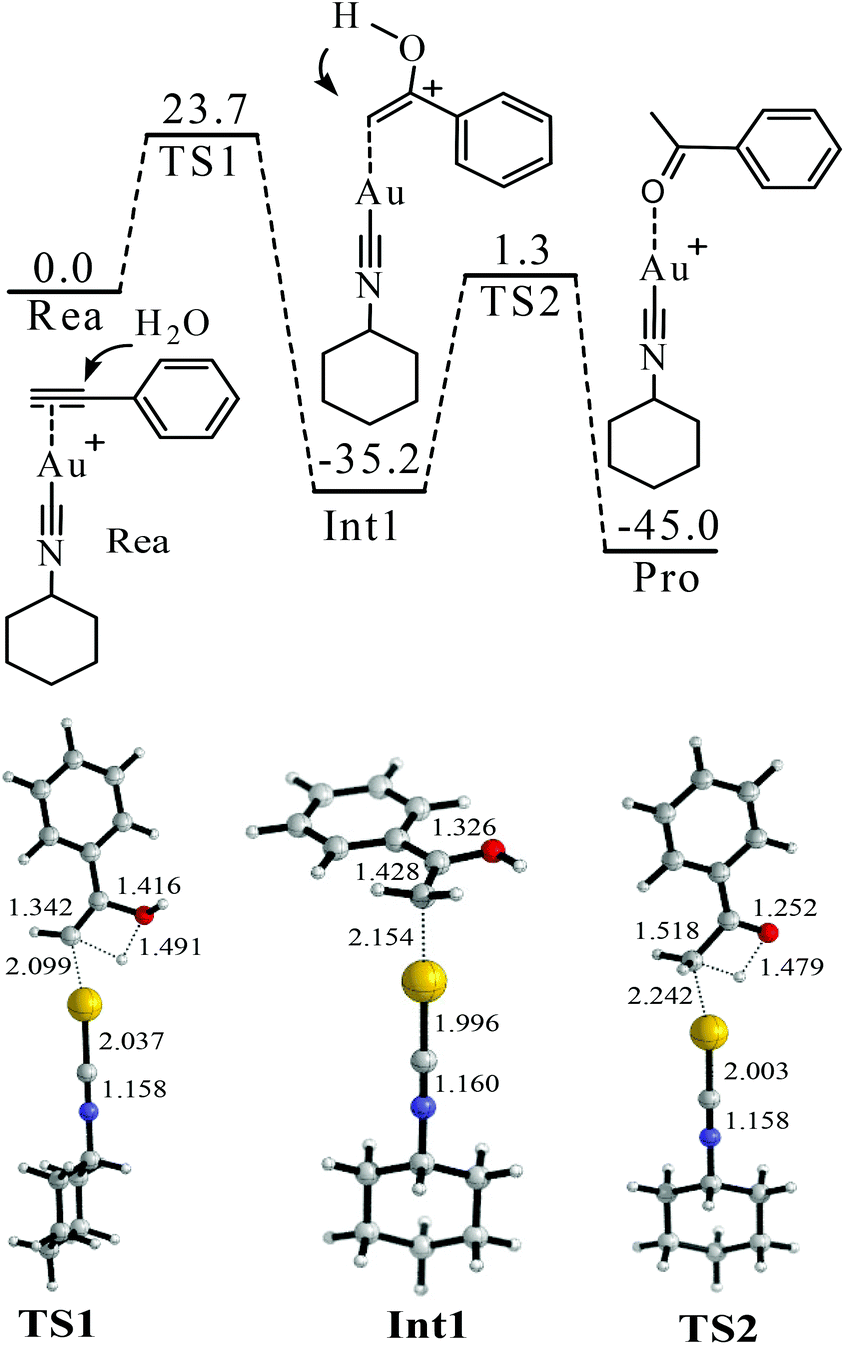 | ||
| Fig. 2 Proposed mechanism of the hydration of phenylacetylene. The values in the reaction pathway are in kcal mol−1. The other values are bond lengths in Å. | ||
The binding between Cat-1+ and 1a is found to be highly exothermic (−47.3 kcal mol−1). With the assistance of Cat-1+, H2O can attack 1avia a transition state TS1 (Fig. 2). The barrier of TS1 is 23.7 kcal mol−1. The CC triple bond of 1a becomes almost a double bond in TS1 (length increasing from 1.212 to 1.342 Å). An enol form of the intermediate (Int1 or Int1′) can be produced via TS1 (Fig. 2 and 3). After that, enol–keto isomerisation via a proton transfer process (TS2 or TS2′) takes place to generate the final product with a total energy change of −45.0 kcal mol−1. However, the barrier of TS2 or TS2′ is too high (36.5 or 37.6 kcal mol−1) to support the room temperature hydration of 1a. Considering that the microenvironment of the reaction is full of water, a water assisting enol–keto isomerisation process should be more feasible.54–58 In this process, the proton of C–O–H transfers to an explicit H2O, and meanwhile, one proton of this explicit H2O transfers to the terminal carbon of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. In this case, the transition from the Int1′ hydrate (Int1′-W) to the final product only needs to overcome a quite low barrier (22.5 kcal mol−1 of TS2′-W). Of course, if there are two or more explicit water molecules to join the process and assist the proton transfer, the barrier should be further reduced to smaller than 22.5 kcal mol−1.54–58 Thus, the rate-determining step of this hydration process should be H2O attacking the CC triple bond with a barrier of 23.7 kcal mol−1 (Fig. 2).
C bond. In this case, the transition from the Int1′ hydrate (Int1′-W) to the final product only needs to overcome a quite low barrier (22.5 kcal mol−1 of TS2′-W). Of course, if there are two or more explicit water molecules to join the process and assist the proton transfer, the barrier should be further reduced to smaller than 22.5 kcal mol−1.54–58 Thus, the rate-determining step of this hydration process should be H2O attacking the CC triple bond with a barrier of 23.7 kcal mol−1 (Fig. 2).
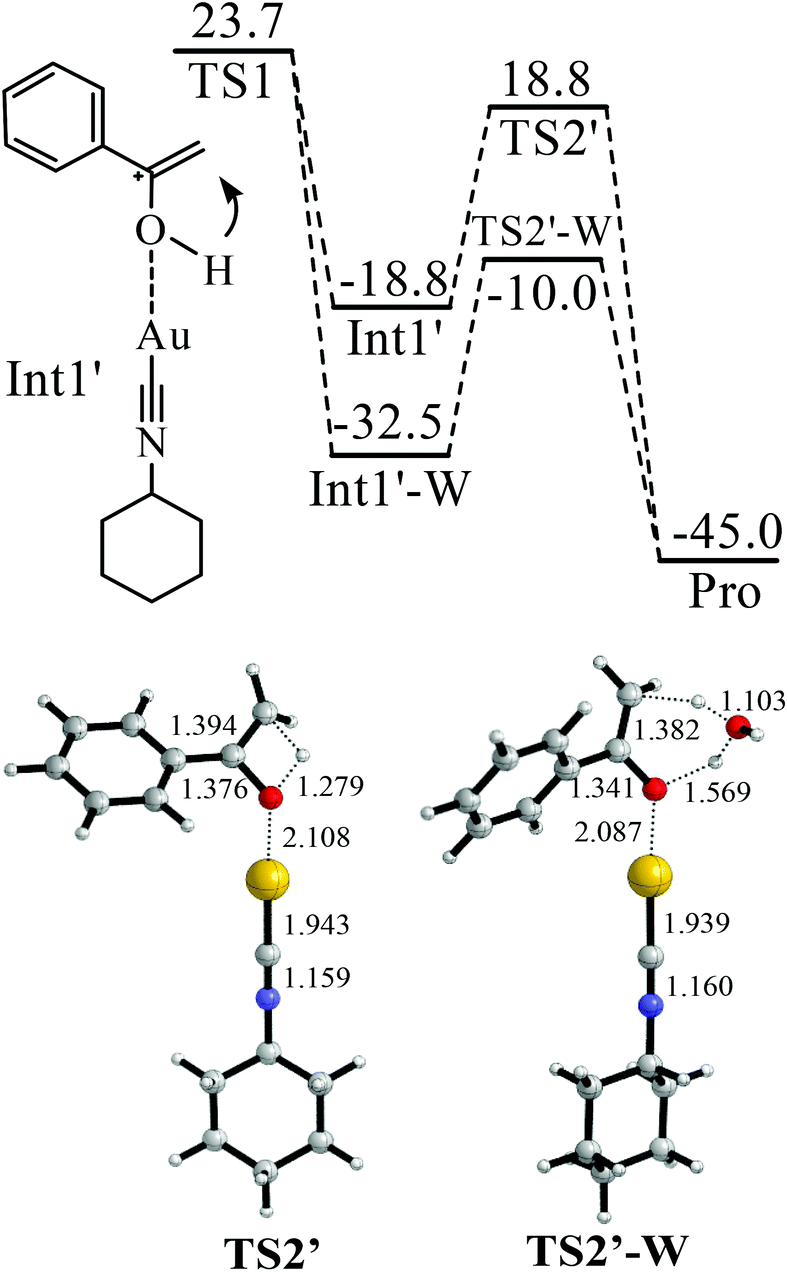 | ||
| Fig. 3 Water-assisted enol–keto isomerisation in the hydration of phenylacetylene. The values in the reaction pathway are in kcal mol−1. The other values are bond lengths in Å. | ||
Another interesting pathway for the reaction of the terminal alkyne using the gold catalyst as gold(I) may react with the terminal alkyne to produce a gold vinylidene (LAuCCR2) intermediate and this intermediate can further react with other functional groups, such as the cycloisomerization of alkyne.59,60 The possibility of similar gold vinylidene (Rea″) reacting with H2O was further investigated (Fig. 4). It was found that the addition of H2O to Rea″ is not easy because of a quite high barrier of TS1″ (51.4 kcal mol−1). This energy barrier is 27.7 kcal mol−1 higher than that of TS1. Furthermore, the energy barrier for the proton transfer from C–O–H to the terminal carbon is 45.9 kcal mol−1 (TS2″), which is 9.4 kcal mol−1 higher than that of TS2. Hence, for the hydration of alkynes catalyzed by gold(I) isocyanide compounds, the reaction mechanism is more likely to be the pathway presented in Fig. 2 and 3. The barrier height of the rate-determining step for the pathway in Fig. 2 and 3 is only 23.7 kcal mol−1, and this barrier height is reasonable for the reaction to proceed readily at room temperature.61
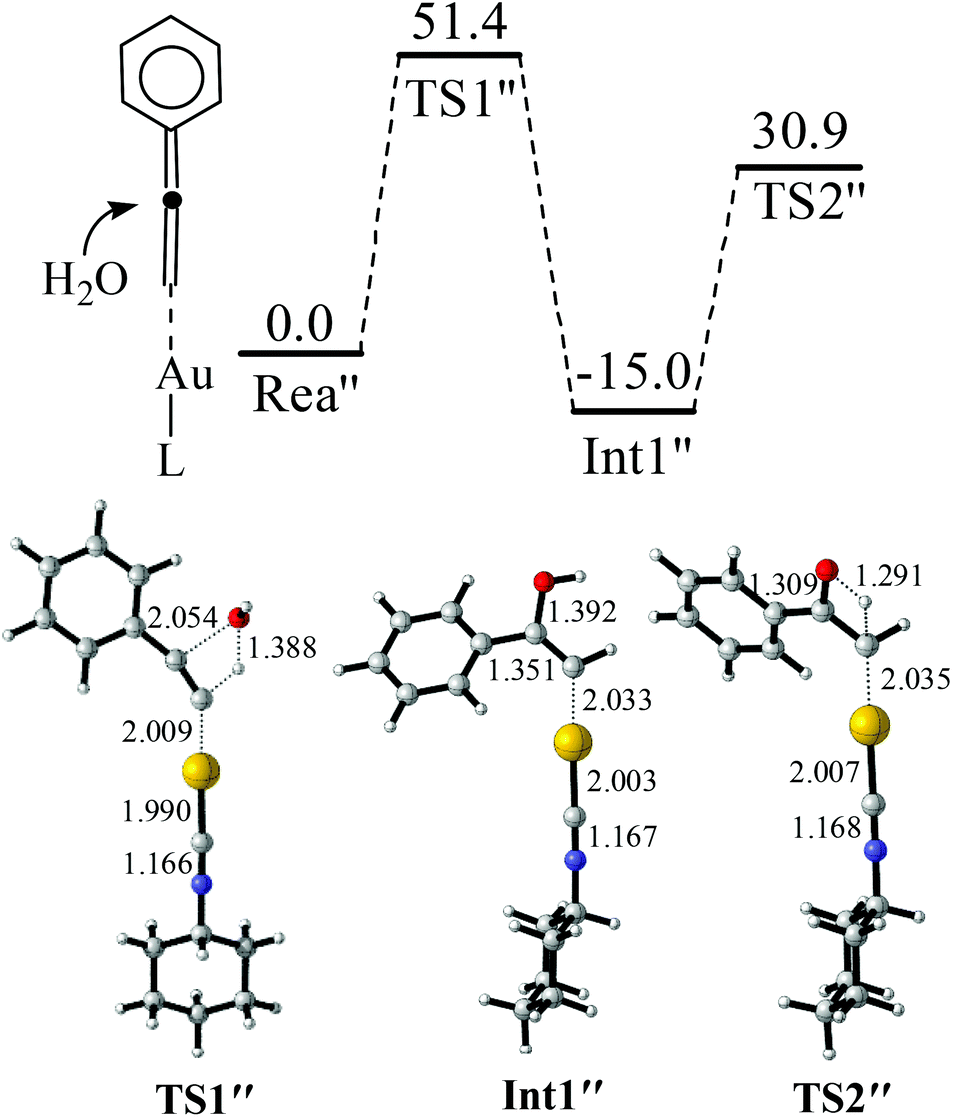 | ||
| Fig. 4 Reaction between gold vinylidenes and H2O. The values in the reaction pathway are in kcal mol−1. The other values are bond lengths in Å. | ||
Conclusions
In summary, the hydration of different alkynes was successfully performed at room temperature in the presence of gold isocyanide complexes. The substrates investigated here include electron-rich and electron-deficient alkynes, aromatic and non-aromatic alkynes. The isocyanide Au(I)+ cation is extrapolated to be the active center that shows quite high catalytic ability for most of the substrates. Theoretical research further reveals the hydration mechanism which includes two steps: H2O attacking the CC triple bond and explicit H2O assisting the proton transfer to the CC double bond.
Au-RNC, as a precursor for the synthesis of Au-NHC,29–39 shows comparable catalytic ability to Au-NHC in the hydration of alkynes. This is a rare example that Au-RNC is used as the catalyst directly to obtain satisfactory hydration results. Considering the simple structure and high reactivity of gold isocyanide, it deserves to receive more attention in other reactions. Because gold isocyanide is a homogeneous catalyst, developing a method to recycle it will bring a more green process. Supporting the gold isocyanide on zeolite62 or ionic liquids63–65 will be an effective method to reuse this compound.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21176110 and 21376115) and Jiangsu Province Natural Science Foundation (BK20141311).Notes and references
- M. Kutscheroff, Chem. Ber., 1881, 14, 1540–1542 CrossRef.
- K. Rao, P. Prasad and N. Lingaiah, Green Chem., 2012, 14, 1507–1514 RSC.
- Z. Nairoukh, D. Avnir and J. Blum, ChemSusChem, 2013, 6, 430–432 CrossRef CAS PubMed.
- W. L. Wong, K. P. Ho, L. Y. Lee, K. M. Lam, Z. Y. Zhou, T. H. Chan and K. Y. Wong, ACS Catal., 2011, 1, 116–119 CrossRef CAS.
- F. Trentin, A. M. Chapman, A. Scarso, P. Sgarbossa, R. A. Michelin, G. Strukul and D. F. Wass, Adv. Synth. Catal., 2012, 354, 1095–1104 CrossRef CAS.
- H. T. He, C. R. Qi, X. H. Hu, Y. Q. Guan and H. F. Jiang, Green Chem., 2014, 16, 3729–3733 RSC.
- B. Mathieu, A. Mann and A. Wagner, Chem. Commun., 2012, 48, 434–436 RSC.
- F. X. Zhu, W. Wang and H. X. Li, J. Am. Chem. Soc., 2011, 133, 11632–11640 CrossRef CAS PubMed.
- F. Chevallier and B. Breit, Angew. Chem., Int. Ed., 2006, 45, 1599–1602 CrossRef CAS PubMed.
- T. Tachinami, T. Nishimura, R. Ushimaru, R. Noyori and H. Naka, J. Am. Chem. Soc., 2013, 135, 50–53 CrossRef CAS PubMed.
- P. Nun, R. S. Ramon, S. Gaillard and S. P. Nolan, J. Organomet. Chem., 2011, 696, 7–11 CrossRef CAS PubMed.
- N. Marion, R. S. Ramon and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448–449 CrossRef CAS PubMed.
- N. Marion, R. Gealageas and S. P. Nolan, Org. Lett., 2008, 10, 1037–1037 CrossRef CAS.
- W. L. Wang, A. M. Zheng, P. Q. Zhao, C. Xia and F. W. Li, ACS Catal., 2014, 4, 321–327 CrossRef CAS.
- G. A. Carriedo, S. Lopez, S. Suarez-Suarez, D. Presa-Soto and A. Presa-Soto, Eur. J. Inorg. Chem., 2011, 9, 1442–1447 CrossRef.
- N. A. Romero, B. M. Klepser and C. E. Anderson, Org. Lett., 2012, 14, 874–877 CrossRef CAS PubMed.
- G. A. Fernandez, A. S. Picco, M. R. Ceolin, A. B. Chopa and G. F. Silbestri, Organometallics, 2013, 32, 6315–6323 CrossRef CAS.
- E. Mizushima, K. Sato, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2002, 41, 4563–4565 CrossRef CAS.
- X. Y. Xu, S. H. Kim, X. Zhang, A. K. Das, H. Hirao and S. H. Hong, Organometallics, 2013, 32, 164–171 CrossRef CAS.
- C. E. Czegeni, G. Papp, A. Katho and F. Joo, J. Mol. Catal. A: Chem., 2011, 340, 1–8 CrossRef CAS PubMed.
- P. de Fremont, R. Singh, E. D. Stevens, J. L. Petersen and S. P. Nolan, Organometallics, 2007, 26, 1376–1385 CrossRef CAS.
- A. Almassy, C. E. Nagy, A. C. Benyei and F. Joo, Organometallics, 2010, 29, 2484–2490 CrossRef CAS.
- R. Casado, M. Contel, M. Laguna, P. Romero and S. Sanz, J. Am. Chem. Soc., 2003, 125, 11925–11935 CrossRef CAS PubMed.
- S. Sanz, L. A. Jones, F. Mohr and M. Laguna, Organometallics, 2007, 26, 952–957 CrossRef CAS.
- S. Coco, C. Cordovilla, P. Espinet, J. Martin-Alvarez and P. Munoz, Inorg. Chem., 2006, 45, 10180–10187 CrossRef CAS PubMed.
- M. Benouazzane, S. Coco, P. Espinet, J. M. Martin-Alvarez and J. Barbera, J. Mater. Chem., 2002, 12, 691–696 RSC.
- R. L. White-Morris, M. Stender, D. S. Tinti, A. L. Balch, D. Rios and S. Attar, Inorg. Chem., 2003, 42, 3237–3244 CrossRef CAS PubMed.
- G. F. Warnock and N. J. Cooper, Organometallics, 1989, 8, 1826–1827 CrossRef CAS.
- W. F. Gabrielli, S. D. Nogai, J. M. McKenzie, S. Cronje and H. G. Raubenheimer, New J. Chem., 2009, 33, 2208–2218 RSC.
- R. Manzano, F. Rominger and A. Hashmi, Organometallics, 2013, 32, 2199–2203 CrossRef CAS.
- A. Hashmi, T. Hengst, C. Lothschutz and F. Rominger, Adv. Synth. Catal., 2010, 352, 1315–1337 CrossRef CAS.
- E. Gonzalez-Fernandez, J. Rust and M. Alcarazo, Angew. Chem., Int. Ed., 2013, 52, 11392–11395 CrossRef CAS PubMed.
- C. Bartolome, Z. Ramiro, D. Garcia-Cuadrado, P. Perez-Galan, M. Raducan, C. Bour, A. M. Echavarren and P. Espinet, Organometallics, 2010, 29, 951–956 CrossRef CAS.
- A. Hashmi, Y. Yu and F. Rominger, Organometallics, 2012, 31, 895–904 CrossRef CAS.
- L. Canovese, F. Visentin, C. Levi and C. Santo, Inorg. Chim. Acta., 2012, 391, 141–149 CrossRef CAS PubMed.
- A. Hashmi, D. Riedel, M. Rudolph, F. Rominger and T. Oeser, Chem. – Eur. J., 2012, 18, 3827–3830 CrossRef CAS PubMed.
- C. Dominguez, B. Donnio, S. Coco and P. Espinet, Dalton Trans., 2013, 42, 15774–15784 RSC.
- A. Hashmi, C. Lothschutz, C. Bohling, T. Hengst, C. Hubbert and F. Rominger, Adv. Synth. Catal., 2010, 352, 3001–3012 CrossRef CAS.
- R. Dopp, C. Lothschutz, T. Wurm, M. Pernpointner, S. Keller, F. Rominger and A. Hashmi, Organometallics, 2011, 30, 5894–5903 CrossRef.
- V. G. Nenajdenko, Isocyanide Chemistry, Wiley-VCH Verlag GmbH &Co. KGaA, 2012 Search PubMed.
- D. Zhang, X. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913–924 CrossRef CAS PubMed.
- C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927–934 RSC.
- D. Prat, O. Pardigon, H. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- F. M. Kerton and R. Marriott, Alternative solvents for green chemistry, Green Chemistry Series, RSC Publishing, Cambridge, 2nd edn, 2013, p. 7 Search PubMed.
- K. Kaizuka, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2010, 132, 15096–15098 CrossRef CAS PubMed.
- B. Karimi and F. K. Esfahani, Adv. Synth. Catal., 2012, 354, 1319–1326 CrossRef CAS.
- J. Zheng, S. Lin, X. Zhu, B. Jiang, Z. Yang and Z. Pan, Chem. Commun., 2012, 48, 6235–6237 RSC.
- M. B. Lauber and S. S. Stahl, ACS Catal., 2013, 3, 2612–2616 CrossRef CAS.
- L. Wang, J. Li, X. Zhao, Y. Lv, H. Zhang and S. Gao, Tetrahedron, 2013, 69, 6041–6045 CrossRef CAS PubMed.
- P. Das, N. Aggarwal and N. R. Guha, Tetrahedron Lett., 2013, 54, 2924–2928 CrossRef CAS PubMed.
- B. Karimi, A. Zamani, S. Abedi and J. H. Clark, Green Chem., 2009, 11, 109–119 RSC.
- T. Hara, M. Ishikawa, J. Sawada, N. Ichikuni and S. Shimazu, Green Chem., 2009, 11, 2034–2040 RSC.
- K. Nagashima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2010, 12, 2142–2144 RSC.
- A. Michalkova, D. Kosenkov, L. Gorb and J. Leszczynski, J. Phys. Chem. B, 2008, 112, 8624–8633 CrossRef CAS PubMed.
- M. K. Shukla and J. Leszczynski, J. Phys. Chem. A, 2005, 109, 7775–7780 CrossRef CAS PubMed.
- Z. Smedarchina, W. Siebrand, A. Fernandez-Ramos, L. Gorb and J. Leszczynski, J. Chem. Phys., 2000, 112, 566–573 CrossRef CAS PubMed.
- X. B. Hu, H. R. Li, W. C. Liang and S. J. Han, J. Phys. Chem. B, 2004, 108, 12999–13007 CrossRef CAS.
- X. B. Hu, H. R. Li, W. C. Liang and S. J. Han, J. Phys. Chem. B, 2005, 109, 5935–5944 CrossRef CAS PubMed.
- L. W. Ye, Y. Z. Wang, D. H. Aue and L. M. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef CAS PubMed.
- L. M. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS PubMed.
- D. C. Young, Computational chemistry: A practical guide for applying techniques to real-world problems, John Wiley & Sons, Inc., 2001, p. 147 Search PubMed.
- A. Molnar, Chem. Rev., 2011, 111, 2251–2320 CrossRef CAS PubMed.
- W. Miao and T. H. Chang, Acc. Chem. Res., 2006, 39, 897–908 CrossRef CAS PubMed.
- G. Q. Yang, X. B. Hu, Y. T. Wu, C. Y. Liu and Z. B. Zhang, Catal. Commun., 2012, 26, 132–135 CrossRef CAS PubMed.
- X. B. Hu, J. Y. Mao, Y. Sun, H. Chen and H. R. Li, Catal. Commun., 2009, 10, 1908–1912 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization results (1H-NMR, 13C-NMR, elemental analysis, and MS); hydration of various alkynes catalyzed by Cat-2; computational methods and Cartesians coordinates of the optimized structures. See DOI: 10.1039/c4gc01322k |
| This journal is © The Royal Society of Chemistry 2015 |