
Luminescent dansyl-based ionic liquids from amino acids and methylcarbonate onium salt precursors: synthesis and photobehaviour†
Giulia
Fiorani
*a,
Maurizio
Selva
*a,
Alvise
Perosa
a,
Alvise
Benedetti
ab,
Francesco
Enrichi
c,
Peter
Licence
d and
Timothy L.
Easun
d
aDepartment of Molecular Sciences and Nanosystems, Centre for Sustainable Technologies, University of Ca’ Foscari Venezia, Italy
bINSTN and Centro di Microscopia Elettronica “Giovanni Stevanato”, Calle Larga Santa Marta, Dorsoduro 2137, 30123 Venezia, and Via Torino 155, 30172 Mestre (Venezia), Italy
cVeneto Nanotech, Via delle Industrie 5, 30175 Marghera (Venezia), Italy
dSchool of Chemistry, The University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: selva@unive.it; Tel: +39 (0)041 234 8687
First published on 10th October 2014
Abstract
Five new luminescent ionic liquids (LILs) derived from tryptophan (Trp), phenylalanine (Phe) and the dipeptide Gly-Gly functionalized with a dansyl chromophore moiety, were synthesized by an original protocol involving both green reagents/solvents such as non-toxic dimethyl carbonate (DMC: MeOCO2Me) and 2-propanol, and reaction conditions. In particular, DMC was used for: (i) the synthesis of methyltrioctyl methylcarbonate onium salts [Q1mmn][MeOCO2] (Q = N, m = 1, n = 8; Q = P, n = m = 4, 8) by P- and N-methylation of trioctylphosphine and trioctylamine, respectively, and (ii) acid-catalyzed esterifications of Trp and Phe to produce the corresponding methyl esters (Trp-OMe and Phe-OMe). Both reactions proceeded with >90% isolated yields and a mass index (esterifications) as low as 4.5. 2-propanol was used as the solvent for N-dansylation reactions where Trp-OMe and Gly-Glyethyl ester hydrochloride (Gly-Gly-OEt) were coupled to dansyl chloride (DNS-Cl) as a luminescent precursor. A final anion metathesis step between methylcarbonate onium salts and N-dansyl amino acid derivatives gave desired LILs of general formula [Q1mmn][DNS-X] (X = Trp, Phe, and Gly-Gly) in quantitative yields and with by-products minimization. Upon excitation (λex = 340 nm) in MeCN, all LILs exhibited green luminescence with emission quantum yields in the range of 33–41% and monoexponential emission lifetimes of 12.6 ± 0.5 ns. Moreover, each compound showed a remarkable hypsochromic shift in the peak emission wavelength when dissolved in solvents of decreasing polarity (from water to MeCN, toluene and CH2Cl2, respectively). A photostability test by a 350 nm continuous excitation on thin films of LILs proved that, after 10 min, the GlyGly derivative fully retained its PL intensity, while this (intensity) decreased from 10 to 25% for other LILs.
Introduction
Since 1992, when the first preparation of air and chemically stable second generation ionic liquids (ILs) was established,1 a number of such ionic compounds have become available, triggering a golden age of research for their implementation in innovative reactions and chemical production engineering.2 The key of this success has been mostly due to the virtually indefinite combinations of (organic) cations and anions that may be used to synthesize ILs and to impart to them a range of tunable properties. Accordingly, ILs have been tailored for hundreds of applications, including: (i) solvents for synthetic purposes and separation technologies,3 (ii) task-specific organocatalysts,4 (iii) functional materials for formation/storage/use of nanoparticles;5 (iv) electrolytes for batteries, DSSC and electrolytic devices,6 and more. An emerging field of application also comprises optoelectronics, specifically in the design of light and stimuli responsive materials that, upon excitation by external actions (change of pH, temperature, etc.), are capable of generating luminous and/or electric signals for the control of micro- and nano-devices.7 In this respect, ILs have been mainly used because of their capability to act as solvent/matrix for organic/inorganic components such as metal complexes, nanoparticles and organic chromophores:8 examples include mostly imidazolium and pyridinium salts as solvents/dispersants for various luminescent species and nanoparticles of the class of lanthanide derivatives.9However, ILs also offer an extraordinary potential for the preparation of intrinsically luminescent materials where light emission is allowed by the arrangement of different organic cations and anions.10 This fascinating area is still largely unexplored. Only a few examples of fully organic ILs-based luminescent compounds have been reported. Examples include stimuli responsive conjugated structures of polyamidoamine (PAMAM) dendrimer-like cations exchanged with both bistriflimide11 and oxadiazolecarboxylate anions,12 and polymeric ionic liquids (PILs) based on rhodamine, coumarine and pyranine moieties.13
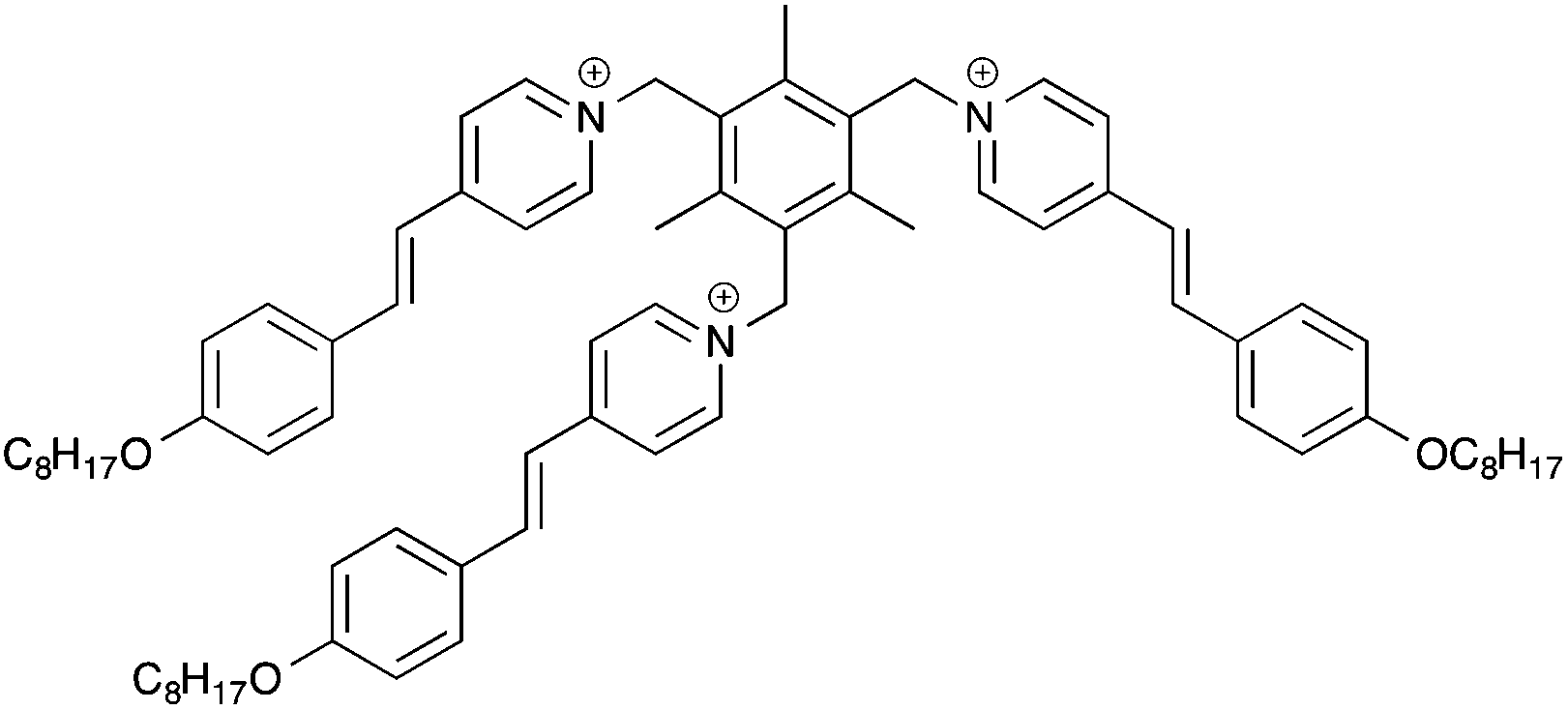
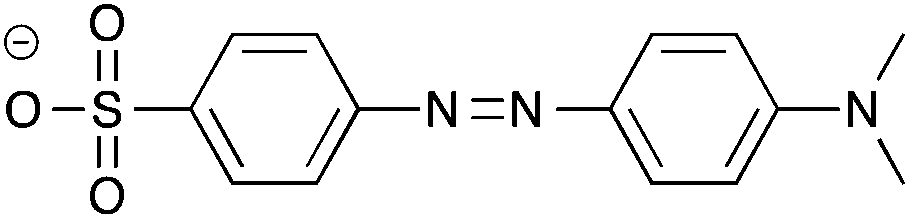
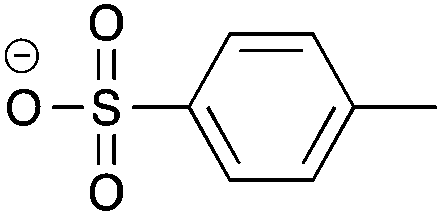
Recently, the concept of “ionic liquidized materials” has been introduced referring to a few recent ionic-type photoluminescent derivatives where small luminescent organic molecules (e.g. dansyl, anthracene) are embedded within an IL structure,14 or ILs and IL crystals are constituted by more complex photoluminescent structures, including tripodal onium salts,15 and conjugated mono and dicationic pyridinium and imidazolium salts16 (Table 1).
| Cation | Anion | |
|---|---|---|
|
|
|
|
[PF6]− | |
| [BF4]− | ||
| [Tf2N]− | ||
| HFPSI | ||
| PFOS | ||
|
|
|
|
||
The fabrication of such compounds is not only opening a frontier for materials where luminescence can be tuned through the flexible design of organic salts, but also allows fundamental studies to elucidate mechanisms of solvent-dependent photoluminescence and of IL solvation.
These considerations along with our long-standing interest in both the preparation/applications of ionic liquids4b,17 and nanostructured luminescent ensembles,18 prompted us to investigate the synthesis of novel fully organic ionic compounds with a built-in luminescent core.
In this paper, we wish to report on a library of five new such products obtained by a strategy which was partly developed starting from a methodology recently implemented by us.17a Accordingly, three sequential processes were set up: n-trioctylphosphine, n-tributyl phosphine and dimethyloctylamine were first subjected to P- and N-methylation by reactions with dimethyl carbonate (DMC). Three methylcarbonate ionic liquids, [P1888][MeOCO2], [P1444][MeOCO2], and [N1118][MeOCO2] were so obtained (Scheme 1, left). In the second step, the luminescent anionic precursors were synthesized. Significant effort was aimed at identifying green solutions for the preparation of salts of aromatic amino acids and acidic peptides that were functionalized with a dansyl (5-(dimethylamino)naphthalene-1-sulfonyl, DNS) chromophore moiety on the N-terminus. Both safer reagents and solvents including dimethylcarbonate and i-propanol were successfully applied for esterification and dansylation protocols. Three luminescent anionic precursors were prepared from tryptophan (Trp), phenylalanine (Phe), and glycylglycine (GlyGly): they bore not only the luminescent dansyl core, but also possessed the suitable acidity to combine with the weakly basic methylcarbonate ionic liquids (Scheme 1, middle). In the last (third) step, acid–base anion exchange reactions took place quantitatively to produce the desired luminescent ionic liquids (LILs) along with the simultaneous decomposition of the methylcarbonate anion to CO2 and methanol (Scheme 1, right).
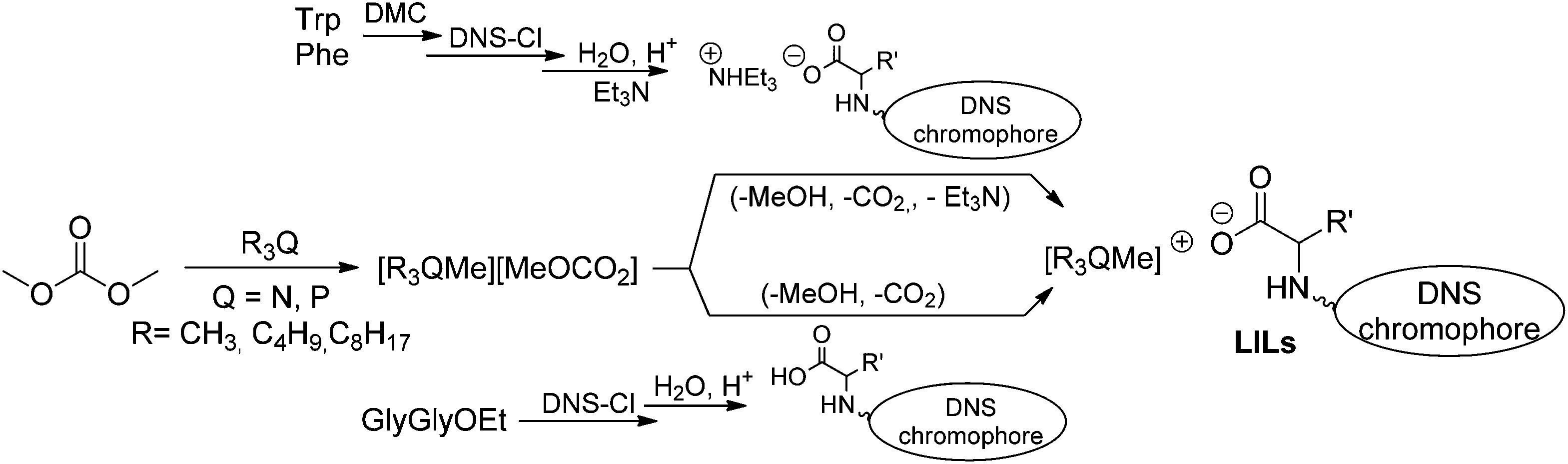 | ||
| Scheme 1 Synthetic strategy for the preparation of DNS-bearing LILs. | ||
The overall synthesis was also aimed at improving the efficiency and sustainability of the transformations used to build up the dansyl-bearing amino acids and peptides: both reaction and purification steps were carried out under very mild conditions, favoring the use of non-toxic reagents and low carbon footprint solvents, including dimethyl carbonate and light alcohols.19 Finally, luminescent properties of onium (N-dansylated)-aminocarboxylate derivatives were investigated upon light excitation between 280 and 350 nm. All compounds proved to be robust luminescent materials with light emission between 400 and 600 nm. The most satisfactory performance was observed with the glycylglycine product (3d) that displayed a sharp luminescence, stable light emission over time and minimal concurrent photobleaching processes.
Results and discussion
The synthesis of luminescent ionic liquids was comprised of three phases [(a)–(c)], which included the preparation of methylcarbonate-onium salts and dansyl-bearing amino acids, and a final anion metathesis reaction.Methylcarbonate onium salts
Tributyl methyl phosphonium methylcarbonate [P1444][H3COCO2], methyl trioctylphosphonium methylcarbonate [P1888][H3COCO2], and trimethyloctyl ammonium methylcarbonate [N1118][H3COCO2] were used as precursors of luminescent compounds. The three methylcarbonate salts were obtained through a procedure recently described by us and reported elsewhere.17a In a typical reaction, a mixture of trialkylphosphine or trialkyl amine (R3P: R = n-C8H17, n-C4H9; R2NR′: R = n-C8H17, R′ = Me; 50 mmol), dimethyl carbonate (DMC, 30 mL) as a methylating agent, and MeOH (30 mL) as a co-solvent, was set to react in a stainless steel autoclave at 150 °C. The use of non-toxic DMC not only allowed efficient P- and N-alkylation processes, but also improved the safety of the overall method since harmful halogenated derivatives (the most common alkylating reagents employed in the synthesis of -onium salts) were avoided. Remarkably: (i) the target products were isolated by simple distillation of residual DMC and MeOH, without any further purification required; (ii) the synthesis of [N1118][H3COCO2] has not previously been reported by this or other protocols. All compounds were obtained in substantially quantitative yields. 1H and 13C NMR analyses confirmed that they were highly pure ionic liquids, not hygroscopic, and stable on the shelf for months.Dansyl-bearing amino acids and peptides
A straightforward convergent strategy was devised for the preparation of dansyl-bearing amino acids and dipeptides. In particular, aromatic amino acids such as tryptophan (Trp: 1) and phenylalanine (Phe: 2), and the dipeptide glycylglycine ethyl ester (Gly-GlyOEt: 3a) were used as starting reactants. These compounds are robust, commercially available and cheap, and most importantly, they possess a double functionality that is suited to our purpose: the amine group can be used to anchor the luminescent dansyl core (for example, via a strong amide bond), while the (carboxylic) acid function is necessary to allow the above described metathetic acid–base exchange (Scheme 1). The synthesis of luminescent anionic moieties was also aimed at improving the overall sustainability of the involved reactions. Catalytic transformations were therefore developed to operate under mild conditions, increase the safety, and prevent the formation of harmful contaminated streams. Moreover, green solvents were used throughout reaction and purification steps. The protocol for luminescent dansyl-based anions ILs followed the retrosynthetic approach illustrated in Scheme 2.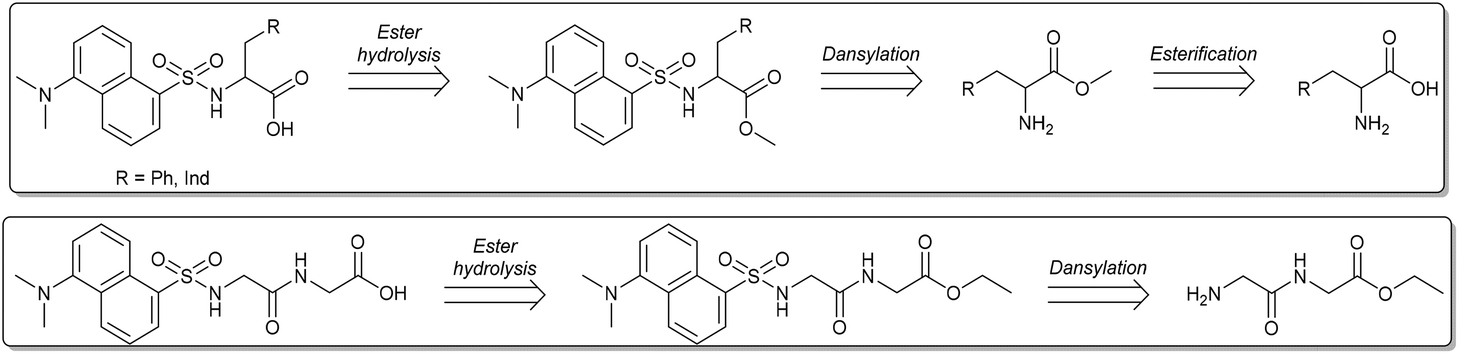 | ||
| Scheme 2 Retrosynthetic analysis of the ILs anionic moieties: L-Phe and L-Trp amino acids (top), dipeptide Gly-Gly-OEt (bottom). | ||
As described above, the introduction of the luminescent core in each anion was conceived through the reaction of dansyl chloride (DNS-Cl) and an amine group. Such transformation (N-dansylation) could take place in a basic aqueous environment (pH > 9 was compulsory to prevent the concurrent dansylation of the –CO2H groups of the reactants).20 Under these conditions however, a hydrolytic conversion of DNS-Cl to the corresponding dansyl acid took place: not only an excess reagent was required, but also expensive work-up/extraction steps for the dansyl acid disposal were necessary.21
To circumvent these drawbacks, the first step of our strategy considered the protection of the –CO2H group of the chosen amino acids (L-Trp and L-Phe) via their conversion into methyl esters (Scheme 2, top). The literature reports that typical esterification protocols of amino acids include using an excess of thionyl chloride as an activator of the carboxylic function, and CH3OH as a solvent and a reagent:22 furthermore, a toxic reagent (SOCl2) and over stoichiometric quantities of HCl and SO2 as by-products are involved. Dimethyl carbonate was (once again) of help for our implementation of a greener procedure to obtain amino acid methyl esters.23,24 Experiments were carried out through an adjustment of a method recently reported by us:25 a mixture of Trp or Phe (10 mmol; 1.65 and 2.04 g, respectively) and dimethylcarbonate (DMC, 5 mL, 52 mmol; DMC served both as a safe reagent and solvent) was set to react at T = 90 °C, in the presence of H2SO4 as a catalyst (12 mmol, 750 μL). This sequence successfully gave the corresponding methyl esters hydrogensulfate salts [RCH2CH(NH3·HSO4)CO2Me; R = 3-indolyl, Ph] in almost quantitative yields. Treatment of such salts with aqueous sodium hydroxide afforded the methyl esters as free bases [RCH2CH(NH2)CO2Me; R = 3-indolyl: 1a; R = Ph: 2a] in yields of 90 and 93%, respectively (Compounds were fully characterized by NMR. Further details are in the Experimental section). This setup greatly simplified the reaction work-up since the produced sulphate wastes are substantially innocuous effluents26 and the excess of DMC could be recycled. It should also be noted that, for the model esterification of Trp, the mass index of SOCl2 and DMC-based methods is 17.9 and 4.5, respectively:27 again, from this simple comparison, the novel DMC-based esterification method stands out as a cheap and sustainable alternative to classic methodologies.
The esterification reaction was not necessary for the Gly-Gly derivative which was commercially available as an ethyl ester (Scheme 4, bottom).
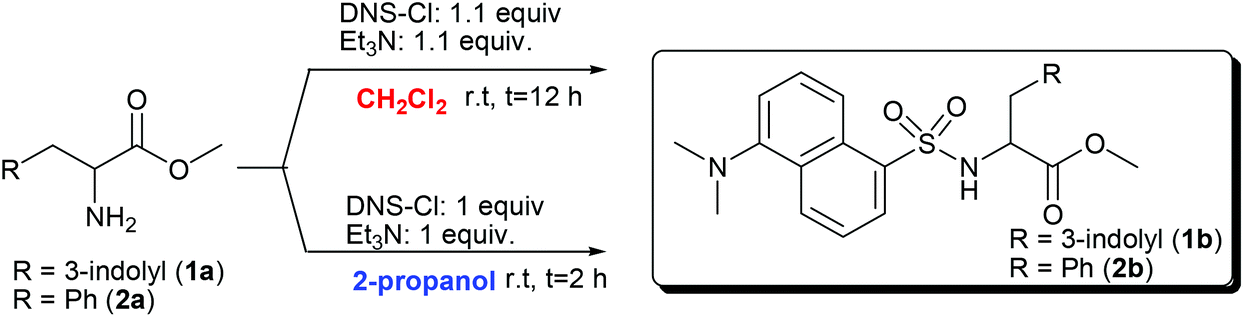
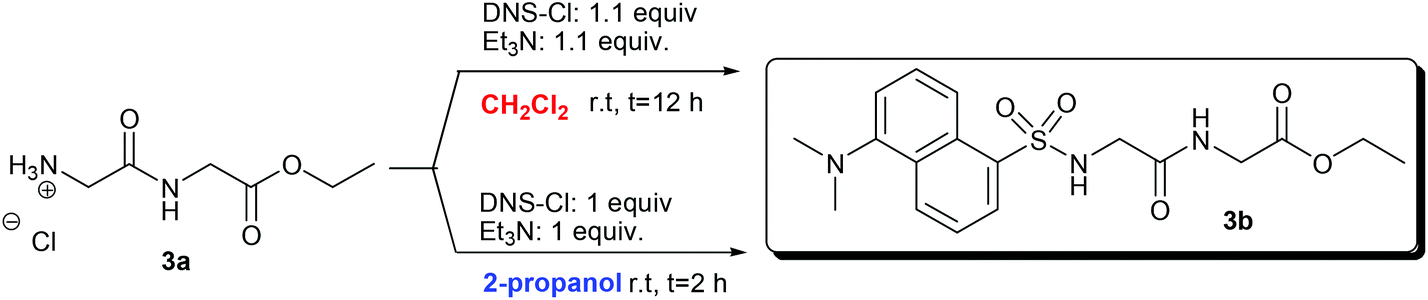
In the second step, the N-dansylation of amino acid esters 1a, 2a, and 3a was investigated. Conventional procedures for this reaction claim the use of DNS-Cl in the presence of toxic chlorinated solvents, of which the most commonly used is dichloromethane (DCM).28 A screening of greener media including alcohols, ethers, and carbonates with low carbon footprints allowed us to identify 2-propanol as a promising candidate to replace DCM.29Table 2 compares experimental conditions and yields of the N-dansylation of esters of Trp, Phe, and GlyGly carried out with both DCM and 2-propanol.
| CH2Cl228 | 2-Propanol | |
|---|---|---|
| Yield (%) | ||
|
1b: 99 | 1b: 99 |
| 2b: 79 | 2b: n.d. | |
|
3b: 93 | 3b: 96 |
Experiments were performed at rt, using a 0.12 M (in CH2Cl2, 25 mL) or a 0.25 M (in 2-propanol, 2 mL) solution of amino acid esters 1a, 2a, and 3a. The N-dansylation of 1a and 3a proceeded smoothly in the two chosen solvents and isolated yields of the corresponding products 1b and 3b were excellent in both cases (93–99%). However, compared to DCM, the use of 2-propanol was advantageous since it allowed shortening remarkably the reaction time (from 12 to 2 hours) and to reduce the amounts of DNS-Cl and Et3N (necessary for HCl neutralization) to the exact molar equivalents required.
Reactions of Table 2 proceeded through an acyl nucleophilic substitution like-mechanism. Since a protic solvent was expected to provide a not negligible nucleophilic solvation, the observed rate enhancement was probably due to a stabilization of the leaving group induced by 2-propanol.30
Due to the poor solubility of compound 2a in 2-propanol, the corresponding reaction was run only in DCM: product 2b was obtained in a 79% yield. The structures of 1b–3b were confirmed by 1H and 13C NMR analyses.
Esters 1b–3b were then subjected to hydrolysis. Although this third step was apparently simple, the amphoteric nature and solubility features of products made their separation tricky and a careful pH control was necessary. A screening of conditions proved that: (i) the hydrolysis reaction was most conveniently carried out using alkali hydroxides as catalysts; (ii) esters 1b–2b required a different treatment with respect to 3b. The optimized procedure was the following.
Compounds 1b–3b were initially dissolved in THF (0.1 M; 10 mL). At 0 °C, aq. LiOH (0.34 M; 40 mL) was added. Reactions were complete in 2–12 h. Then, the pH of the mixtures was adjusted to 5 by adding aliquots of aq. HCl: this allowed the separation of N-dansylated amino acid 3c in a 61% yield (Scheme 3, bottom). The corresponding derivatives 1c and 2c however, were deliquescent compounds that could not be isolated in pure form. These products (approximately 1.0 and 0.4 g of 1c and 2c, respectively) were taken up in a water–acetone mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v; 70 and 20 mL) and treated with Et3N (1.1 mol). The expected triethylammonium salts 1′c and 2′c were obtained in 83 and 90% yields, respectively (Scheme 3, top right). 1′c and 2′c were not only easy-to-handle solids, but they displayed acid–base properties suited for the metathesis reaction outlined in Scheme 1 (Further synthetic details as well as the NMR characterization analyses are in the Experimental section).
:1 v/v; 70 and 20 mL) and treated with Et3N (1.1 mol). The expected triethylammonium salts 1′c and 2′c were obtained in 83 and 90% yields, respectively (Scheme 3, top right). 1′c and 2′c were not only easy-to-handle solids, but they displayed acid–base properties suited for the metathesis reaction outlined in Scheme 1 (Further synthetic details as well as the NMR characterization analyses are in the Experimental section).
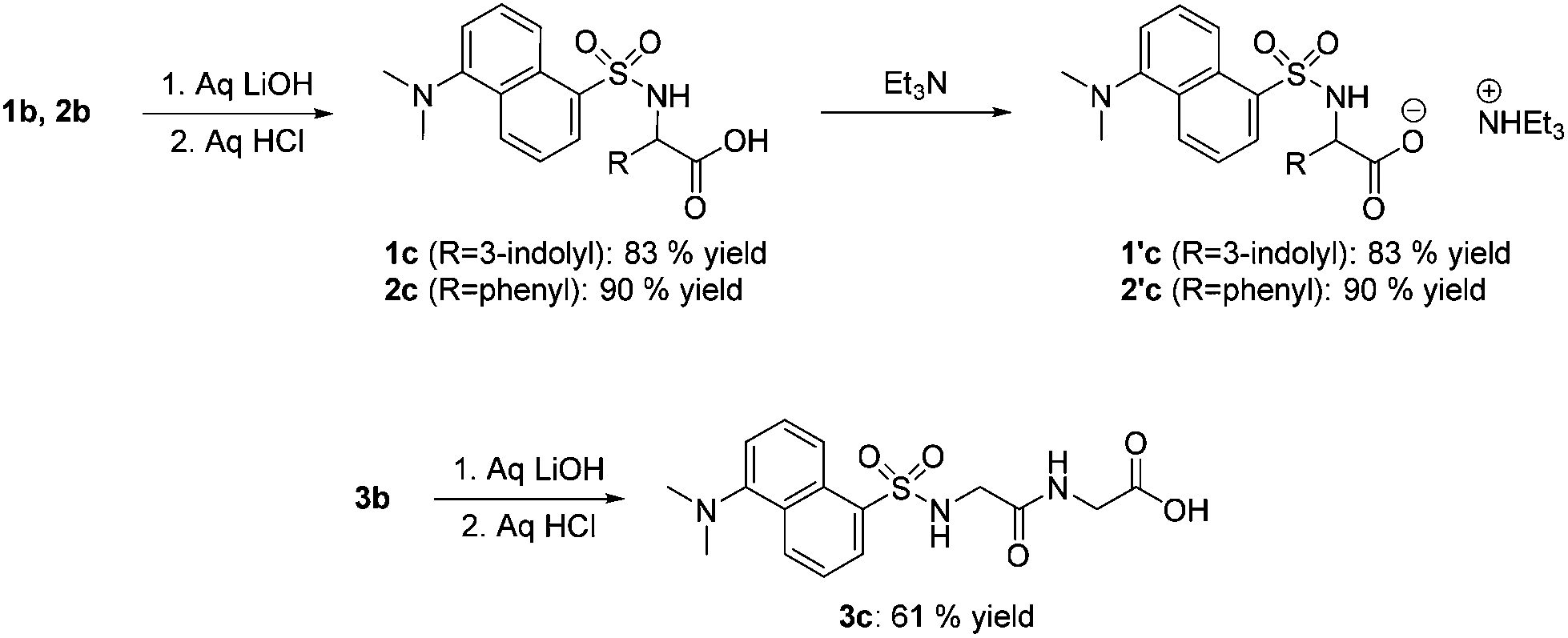 | ||
| Scheme 3 Luminescent acid precursors from Trp (1), Phe (2) (top), and GlyGly (3) (bottom). | ||
Although direct hydrolysis was not possible, conditions of Scheme 3 were still acceptable in the green context of the overall procedure.
The acid–base metathesis reaction
Basic methylcarbonate onium salts ([P1444][H3COCO2], [P1888][H3COCO2], and [N1118][H3COCO2]) underwent a simple and very efficient anion-exchange reaction with compounds 1′c, 2′c and 3c. In a model example, an equimolar mixture of a methyl trioctylphosphonium methylcarbonate ([P1888][H3COCO2], 445 mg, 0.90 mmol) and triethylammonium 2-(N-dansyl)amino-3-indolyl propanoate (1′c, 484 mg, 0.90 mmol) was set to react at 50 °C in the presence of MeOH (0.5 mL) to ensure a complete solubilisation of both reactants. The incipient formation of bubbles (CO2) proved the progress of the anion metathesis reaction that was complete within 1 hour. MeOH and the co-product Et3N were then removed under vacuum, and the desired product (1d) was isolated in a quantitative yield and a purity >99% (by NMR) (Scheme 4, top). The same procedure was successfully used for both [P1444][MeOCO2] and [N1118][MeOCO2] salts: three other ILs bearing a luminescent dansyl core were prepared in quantitative yields (1e–f and 2d: Scheme 4, middle). Finally, the direct reaction of acid 3c with [P1888][H3COCO2] gave the expected Gly-Gly derivative 3d (Scheme 4, bottom). All products were new compounds whose structures were proven by NMR and ESI analyses. Further characterization of LILs was carried out by thermal analyses (DSC and TGA, respectively. See ESI†). Results are summarized in Table 3 which details the glass transition and decomposition temperatures, and the physical state of products 1d–e, 2d and 3d.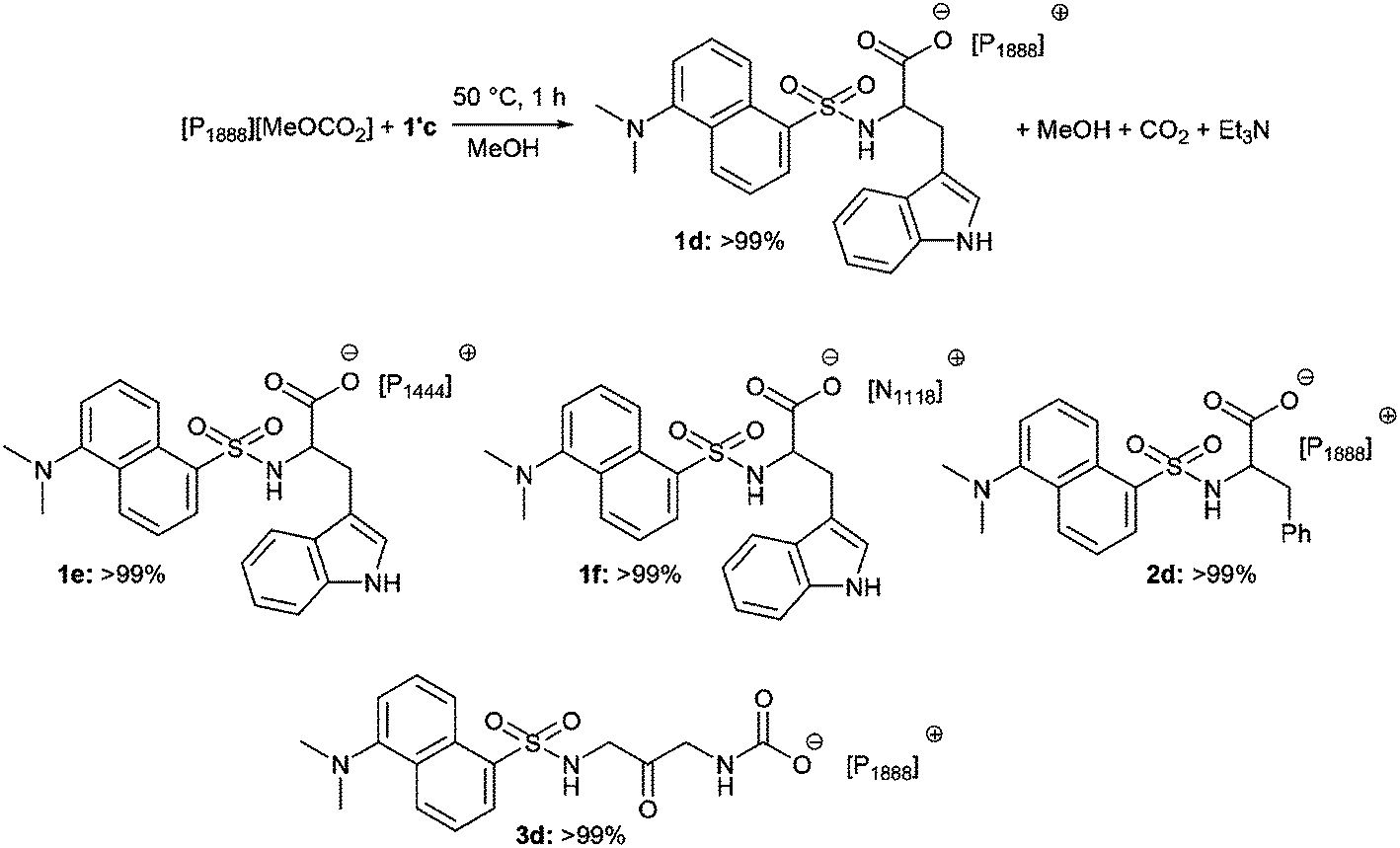 | ||
| Scheme 4 Dansylated ILs based on onium salts. | ||
| Compound | Physical state (rt) | T g (°C) | Decomposition temperature (Td, °C) |
|---|---|---|---|
| 1d | Viscous liquid | −33 | 250 |
| 1e | Viscous liquid | −38 | 250 |
| 1f | Sticky, very viscous liquid | −29 | 180 |
| 2d | Very viscous liquid | −24 | 240 |
| 3d | Liquid | −23 | 220 |
Compared to solid triethyl ammonium derivatives 1′c–2′c (Scheme 3), compounds 1d–f, 2d and 3d are viscous room temperature ionic-liquids. They all display good solubility in conventional organic solvents. Of note, such properties (due to the presence of bulky phosphonium and ammonium cations) are quite desirable in view of the investigation of salts 1d–f, 2d and 3d as luminescent species.
The Td values (180–250 °C) were comparable to those of amino acid-based ILs reported previously.31
Overall, the synthesis of the (luminescent) ILs of interest was achieved through a modular and sustainable strategy. Genuine green benefits could be recognized mostly in two aspects: (i) the use of non-toxic compounds such as dimethyl carbonate and 2-propanol, which were reagents and solvents in the synthesis of methyl carbonate-onium salts, the esterification of Trp and Phe, and the N-dansylation protocols above described; (ii) the setup of an anion metathesis reaction (Scheme 4) that coupled a high efficiency to an exceptionally easy and cheap reaction work-up. Accordingly, drawbacks of typical procedures due to the use and formation of halogenated (particularly chlorides) precursors, solvents, and by-products were minimized, if not prevented at all.
Luminescent behavior of onium salts exchanged with (N-dansyl)aminoacid anions
All compounds 1d–f, 2d and 3d displayed luminescence when irradiated by a UV light source at 365 nm (Fig. 1). The initial investigations of the photophysical behavior of such products were carried out using 10−4–10−6 M solutions (3 mL) in CH3CN as a solvent. Under such conditions, a detailed study was undertaken to measure key properties of each LIL, including absorption maximum (λabs), molar extinction coefficient (ε), maximum emission wavelength (λem), and the radiative process quantum efficiency (Φf). Results are summarized in Table 4. | ||
| Fig. 1 Luminescence of the synthesised ILs under UV irradiation (λ = 365 nm) (∼10−3 M in CH3CN). | ||
The overall luminescence properties (λex and λem) observed for all the synthesized LILs could be ascribed to the presence of a dansyl unit within the structure (vide infra, the relative absorption maxima at λ ∼ 280 nm did not contribute substantially to the resulting emission spectra).
The UV/vis absorption spectra of 1d, 1e, 1f, 2d and 3d in acetonitrile are shown in Fig. 2. The primary feature of all the compounds is a broad absorption band centred at approximately 335 nm with extinction coefficients ranging from ∼ 5600–8400 M−1 cm−1, typical of dansyl chromophore derivatives.32 On excitation into this band (λex = 340 nm) all compounds exhibit green luminescence characterized by broad emission spectra (Fig. 1 and 3), with significant Stokes shifts in the region of ∼10500 cm−1 in each case. The peak maxima of the emission spectra are all centred around ca. 520 nm, with the exception of compound 3d which exhibits the largest Stokes shift (λem = 534 nm). Quantum yield calculations show these compounds emit with efficiencies in the range 31–44%, and the lifetime of emission is monoexponential and strikingly similar in all cases (12.6 ± 0.5 ns).
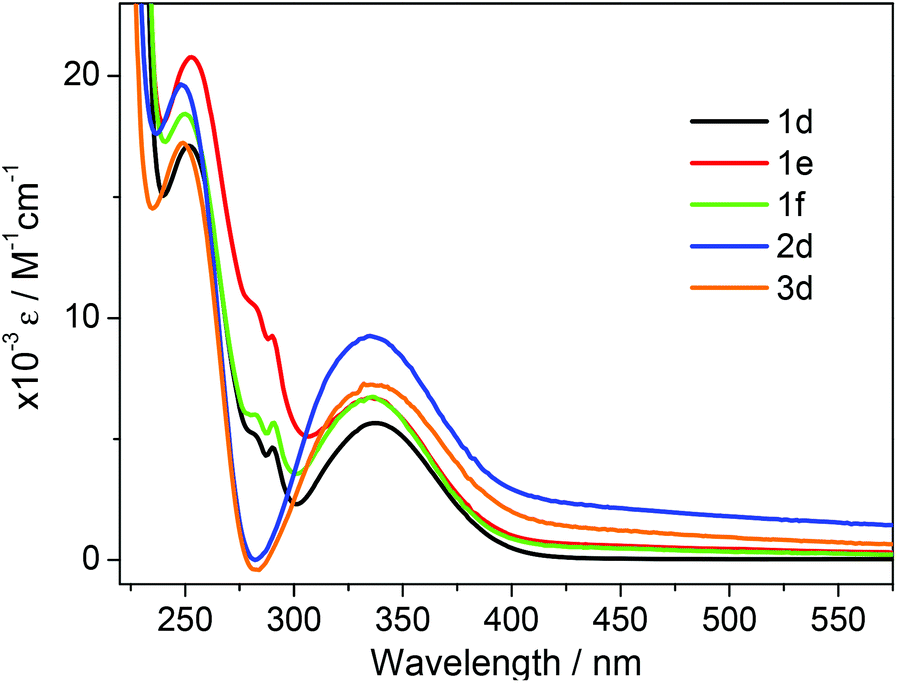 | ||
| Fig. 2 UV/vis absorption spectra of compounds 1d–f, 2d and 3d recorded in acetonitrile at concentrations ∼1 × 10−5 M. | ||
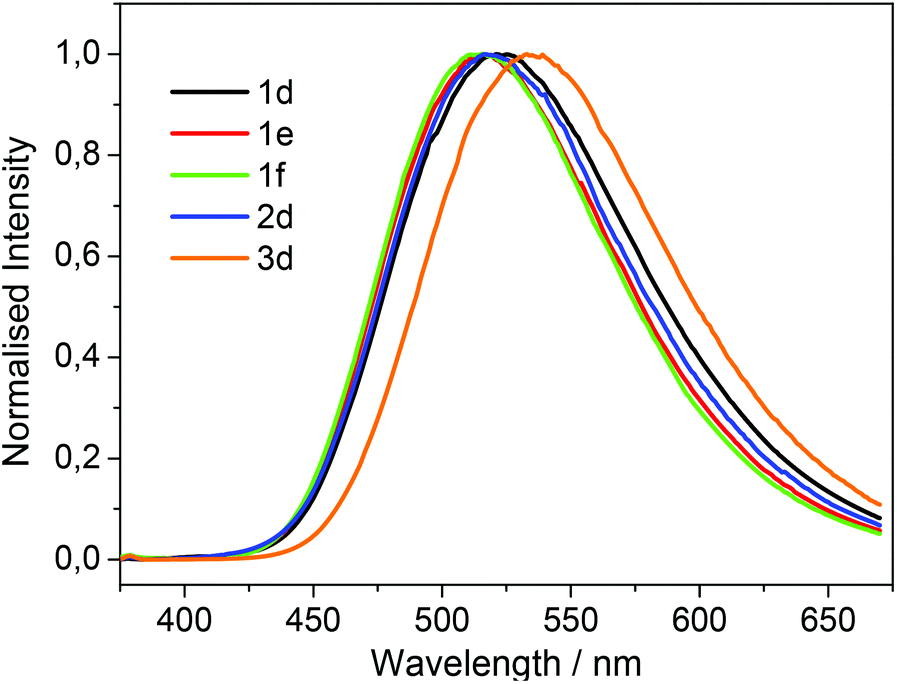 | ||
| Fig. 3 Normalized emission spectra of 1d–f, 2d and 3d recorded in acetonitrile with excitation at 340 nm. | ||
The fluorescent behavior of methyltrioctylphosphonium based ILs in solution was further examined by expanding the range of solvents. Four solvents with different polarities, including water, acetonitrile, dichloromethane, and toluene were considered. The absorption and emission maxima of compounds 1d–f, 2d, and 3d are reported in Table 5.
| Ionic liquid | Solvent | ET(30)b, (kcal mol−1) | DNc,14b | ε | λ abs (nm) | λ em (nm) | Stokes shifte (cm−1/103) |
|---|---|---|---|---|---|---|---|
a 5 × 10−6 M solutions (2 mL) of 1e–3e in any given solvent were used.
b ET(30) was the electronic transition energy of the betaine dye 4-(2,4,6-triphenylpyridinium)-2,6-diphenylphenoxide in a specific solvent.
c DN was the donor number measured at 25 °C.
d
ε was the dielectric constant of the solvent, measured at 25 °C.
e
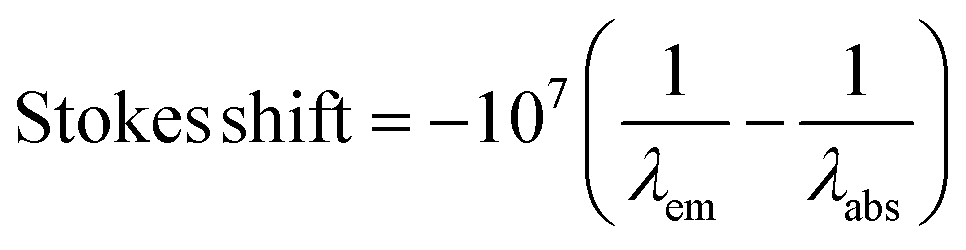 .
f Estimated position as a result of very poor sample solubility in H2O.
g Position uncertain due to overlap with solvent absorption. .
f Estimated position as a result of very poor sample solubility in H2O.
g Position uncertain due to overlap with solvent absorption.
|
|||||||
| 1d | H2O | 63.1 | 33 | 78.4 | 330f | 562 | 12.5 |
| CH3CN | 45.6 | 14.1 | 37.5 | 338 | 523 | 10.5 | |
| CH2Cl2 | 39.1 | 1 | 8.93 | 339 | 511 | 9.93 | |
| Toluene | 33.9 | 0.01 | 2.38 | 336g | 490 | 9.35 | |
| 1e | H2O | 63.1 | 33 | 78.4 | 327 | 571 | 13.1 |
| CH3CN | 45.6 | 14.1 | 37.5 | 335 | 516 | 10.5 | |
| CH2Cl2 | 39.1 | 1 | 8.93 | 336 | 504 | 9.92 | |
| Toluene | 33.9 | 0.01 | 2.38 | 335g | 490 | 9.44 | |
| 1f | H2O | 63.1 | 33 | 78.4 | 328 | 572 | 13.0 |
| CH3CN | 45.6 | 14.1 | 37.5 | 334 | 513 | 10.5 | |
| CH2Cl2 | 39.1 | 1 | 8.93 | 337 | 503 | 9.79 | |
| Toluene | 33.9 | 0.01 | 2.38 | 336g | 491 | 9.40 | |
| 2d | H2O | 63.1 | 33 | 78.4 | 327 | 554 | 12.5 |
| CH3CN | 45.6 | 14.1 | 37.5 | 334 | 517 | 10.6 | |
| CH2Cl2 | 39.1 | 1 | 8.93 | 339 | 504 | 9.66 | |
| Toluene | 33.9 | 0.01 | 2.38 | 333g | 486 | 9.45 | |
| 3d | H2O | 63.1 | 33 | 78.4 | 328 | 578 | 13.2 |
| CH3CN | 45.6 | 14.1 | 37.5 | 333 | 534 | 11.3 | |
| CH2Cl2 | 39.1 | 1 | 8.93 | 338 | 515 | 10.2 | |
| Toluene | 33.9 | 0.01 | 2.38 | 333g | 494 | 9.79 | |
There was no significant shift in absorption peak maxima for any of the samples on changing solvent but a marked hypsochromic shift in the peak emission wavelength was observed as the solvent polarity was reduced, consistent with decreasing stabilization of the emissive excited state. Of note, the decrease in Stokes shift of each compound was almost linear with decreasing solvent dielectric constant, again congruent with a decrease in solvent-stabilisation of the emissive excited state on changing from polar aqueous solvent to non-polar toluene. Fig. 4 exemplifies the model case of compound 1e.
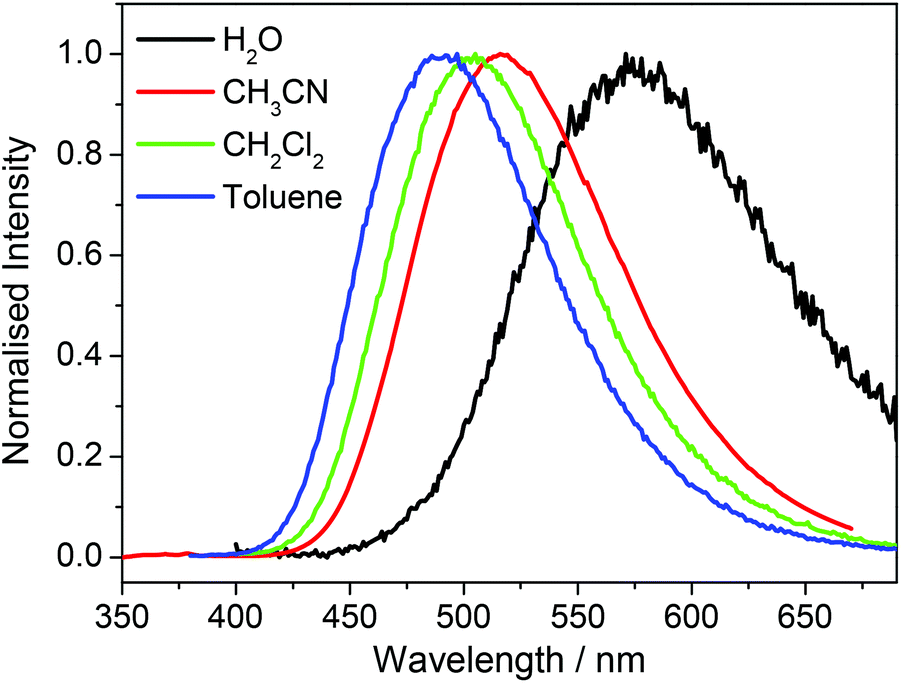 | ||
| Fig. 4 Normalized emission spectra of 1e recorded in H2O, CH3CN, CH2Cl2 and toluene with excitation at 350 nm. | ||
Both hypsochromic and Stokes shift trends were compatible with the behavior of dansyl (5-(dimethylamino)naphthalene-1-sulfonyl) chromophores in solution or in motionally restricted macromolecular environments.32 In fact, energy absorption of 1-aminonaphthalenes is typically described by a high energy absorption band associated to a non-charged transfer (non-CT) state, and, therefore, mainly unaffected by solvent polarity, and a lower energy absorption band associated to a twisted intramolecular charged transfer state (TICT). TICT is observed only with fluorophores where the donor and acceptor parts are connected by a single bond as in the dansyl specie. When excited, the donor may rotate around the bond to transfer an electron to the acceptor. As a result, the fluorophore undergoes a change of its molecular structure from a planar to a perpendicular conformation and two photoinduced excited modes can be envisaged corresponding to a simple charge transfer (CT) and a TICT excited state.
Since the newly synthesized dansyl based products of Scheme 3 were all liquids at room temperature, an investigation of optical properties was also carried out on thin films of compounds 1d–f, 2d and 3d as such. In Fig. 5 the PL and PLE normalized spectra of the concentrated LILs samples are reported. Both the excitation and emission curves for compounds 1e and 1f showed a blue shift with respect to samples 1d–3d. It was also worth noting that LILs 1e and 2d displayed a second contribution around 460 nm which was in agreement with the existence of two distinct emission bands.33 This contribution remains hidden for other samples in relation to the higher intensity of the main band.
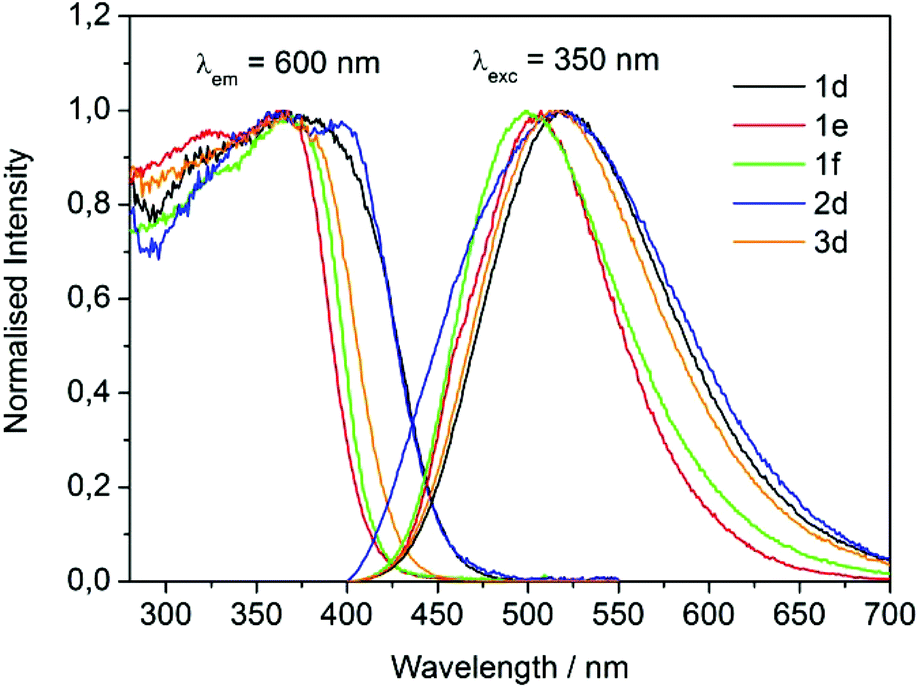 | ||
| Fig. 5 Normalized PL and PLE spectra of 1d–f, 2d and 3d non-diluted samples. | ||
Time-resolved luminescence of LILs was also investigated under 373 nm pulsed excitation (Table 6). For a good fitting of PL decay curves, measured at their emission maxima in neat liquid samples, a double-exponential decay was necessary (eqn (1)): this was compatible with the presence of two bands observed in the emission spectra of LILs as such (Fig. 5). The average photoluminescence lifetimes (τav in Table 6) were derived from the best fitting parameters by eqn (2) following a standard procedure.18
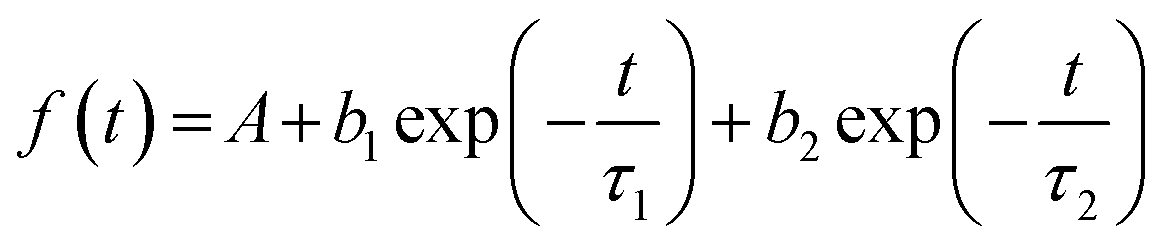 | (1) |
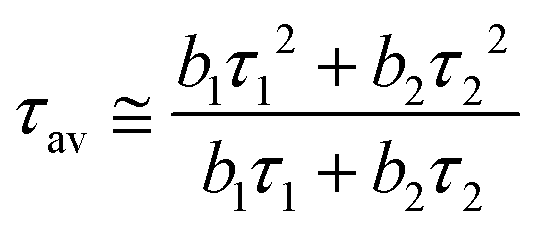 | (2) |
| LIL | λ em (nm) | τ av (ns) |
|---|---|---|
| 1d | 520 | 15.9 |
| 1e | 505 | 13.2 |
| 1f | 500 | 14.2 |
| 2d | 517 | 12.4 |
| 3d | 515 | 14.8 |
The fluorescence bi-exponential decay (with lifetimes comparable for both the excited states) of neat liquid LILs 1d–f, 2d and 3d was rather different from the behavior in acetonitrile solution (Table 4), and it has seldom been reported for dansyl chromophores (especially when included in rigid scaffolds).32 The above mentioned TICT (twisted intramolecular charged transfer state) phenomenon may offer an explanation for the results obtained in neat, viscous IL solutions.
A more general hypothesis for the scenario outlined by Fig. 5 and Table 6 should consider the different structure of the associated counter-cations: LILs 1e and 1f bear smaller aliphatic cations ([P1444]+ and [N1118]+, respectively) within their structure, while LILs 1d–2d are characterized by bulkier [P1888]+ counter-cations. A more sterically hindered cationic centre, as in the case of [P1888]+ based LILs, results in a poorer interaction with the carboxylic acid based anionic moieties, which may be susceptible to give dimeric or oligomeric anion–anion interactions, particularly in protic environment.34 Therefore, for the [P1888]+ containing LIL 1d–2d, the emission maxima observed in neat solution are similar, in energy and hence wavelength, to a moderately polar environment (cf.Fig. 4, CH3CN curve). Conversely, when the anion–cation interaction is sterically more favored, as in the case of 1e and 1f, the observed emission patterns is similar in energy to that observed in low polar solutions of CH2Cl2 (cf. Table 5). The observed trend suggests the possibility of formation of polar and nonpolar domains within the LIL bulk,35 or of dimeric and oligomeric anion aggregates in a more protic environment, like the one investigated in non-diluted solution of LILs.
Notwithstanding the presence of [P1888] cation, the emission maximum of neat liquid 3d closely resembles that observed in CH2Cl2 (520 and 515 nm, respectively: Tables 5 and 6). This incongruity with respect to other LILs is plausibly due to the (larger) size and the dipeptide nature of the anion of salt 3d, the behavior of which may be dominated by intra- and inter-H bonding networks.36
A photostability test was finally carried out by monitoring the PL intensity at the emission maximum of each neat compound (thin film) under 350 nm continuous excitation. Fig. 6 reports the results. After 10 minutes, the loss of PL intensity was ∼25% for compounds 1d and 1f, while it (intensity loss) decreased to less than 10% for 1e and 2d. A remarkably better performance was shown by the GlyGly derivative (3d), the intensity of which was fully retained.
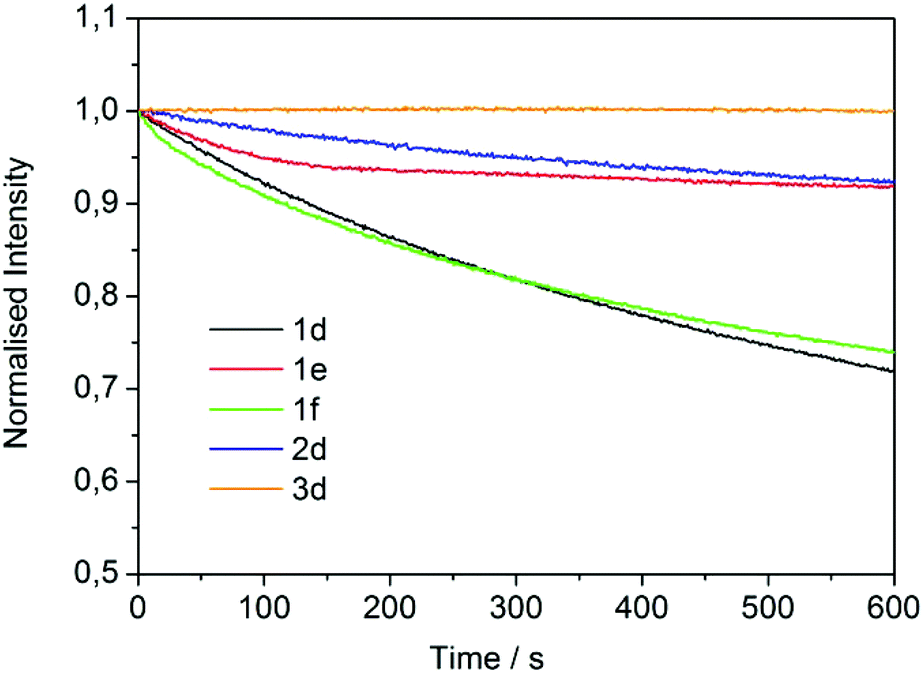 | ||
| Fig. 6 Photostability measurements on neat 1d–f, 2d and 3d liquid samples (excitation 350 nm, emission in the maximum). | ||
Overall, three major observations emerged from the analysis of the luminescent behavior of the investigated ILs: (i) the luminescent properties of the dansyl unit were little affected by the type of ionic liquid in which the chromophore was incorporated. In other words, the presence of an amino acid moiety adjacent to the DNS group did not affect the chromophore optimal properties (Fig. 3); (ii) when in solution, the structure of the ionic liquid was sensitive to changes of solvent polarity. Under such conditions, a luminescence tuning was possible to some extent (Table 5 and Fig. 4); (iii) as demonstrated by the photostability experiment, different structural (anion/cation) compositions of LILs also affected their bulk properties. In perspective, these aspects opened an intriguing way to devise new LILs of interest for material science applications, particularly for luminescent coatings and nanosensors.
Conclusions
A simple and highly reproducible protocol has been implemented for the synthesis of a library of new luminescent ionic liquids (LILs) based on phosphonium and ammonium cationic moieties and dansylated amino acids as counteranions. The originality of the proposed method can be recognized throughout the overall sequence. Each step of the procedure has been optimized to increase the sustainability by maximizing the atom economy, improving the mass index, and favoring the use of low carbon footprint solvents and reagents. In particular, major advantages have been achieved in the following three aspects: (i) the use of a new DMC-based catalytic procedure for the amino acid esterification which allows the replacement of conventional harmful (and stoichiometric) reagents such as thionyl chloride, as well as the formation of contaminated wastes; (ii) the setup of a dansylation step where non-toxic 2-propanol acts as an excellent substitute solvent for CH2Cl2. Under such conditions, isolated yields of desired products have been improved; (iii) the achievement of a metathesis reaction in which methylcarbonate based ILs are key partners to produce highly pure LILs in quantitative yields. This protocol is of particular green appeal since the use of halide salts and the consequent formation of halogenated waste are ruled out in favor of the formation of low boiling point or gaseous by-products (CO2, MeOH, and Et3N).The synthesized LILs are all liquids under standard laboratory conditions and display the typical luminescence behavior of dansyl-based chromophores both in solution and in neat phases. A remarkable photostability, however, has been observed for the glycyl-glycine derivative [P1888][DNS-Gly-Gly]. This compound (3d) may offers a good model for future investigations aimed at the synthesis of other robust LILs for applications in the field of luminescent surface and nanoparticles coatings and nanosensors.
Experimental
Materials and methods
All reagents and solvents were of reagent grade (>99%), purchased from commercial sources (Sigma Aldrich, Fluka, TCI and Carlo Erba) and used as received. Dimethyloctylamine (95%, Sigma Aldrich) was distilled under reduced pressure (Teb = 27–28 °C, p = 120 mbar) and stored over KOH prior to use.TLC were performed on silica gel plates (4 × 6.7 cm) coated with a fluorescent indicator, λabs = 254 nm. Flash Column Chromatography (FCC) separations were performed on silica gel (60 mesh). FTIR spectra were recorded on both KBr pellets. NMR spectra were recorded on a spectrometer operating at 400 and 101 MHz for 1H and 13C nuclei, respectively. Alternatively, a spectrometer operating at 50 and 81 MHz frequencies for 13C and 31P nuclei, respectively, was also used. [P1888][H3COCO2] and [P1444][H3COCO2] 1H and 13C NMR spectra were recorded on neat phases: a coaxial capillary containing DMSO-d6 was used for internal reference. Unless otherwise specified, other NMR spectra were recorded in deuterated solvents. ESI-MS spectra were recorded in FIA (Flow Injection Analysis) mode, at a 0.05 ml min−1 flow (eluent: acetonitrile).
UV/visible absorption spectra were recorded on a Perkin-Elmer Lambda 25 spectrometer. Photoluminescent emission measurements of the LILs diluted in different solvents were performed on a steady state Perkin-Elmer LS55 fluorimeter and on a combined fluorescence lifetime and steady state spectrometer (Edinburgh Instruments FLS920). Steady state emission spectra were obtained with a Xenon arc lamp as the excitation source and were corrected for detector sensitivity. Quantum yields were calculated against a [Ru(bpy)3]Cl2 standard solution. Emission lifetime measurements were performed using the time-correlated single-photon counting technique, with an EPL400 pulsed diode laser (405 nm, pulse width 95 ps) as the excitation source (time resolution ca. 1 ns). Samples were made up in water, acetonitrile, dichloromethane or toluene at concentrations adjusted to be optically dilute (Amax ∼ 0.1, c ∼ 1 × 10−5 M) and measurements performed in 1 cm pathlength quartz cuvettes.
Optical measurements on the non-diluted matrix as thin films between two quartz slides, including photoluminescence emission (PL) and excitation (PLE) measurements, stability under illumination and emission lifetime measurements were collected with a Horiba JobinYvon Fluorolog3 instrument. A 450 W Xenon lamp coupled to a double grating Czerny-Turner monochromator was used as excitation source, while a iHR320 single grating monochromator coupled to a R928 Hamamatsu PMT allowed the analysis and detection of the emitted photons. Emission lifetime measurements were performed using the time correlated single photon counting technique, with a NanoLED-03 pulsed diode (373 nm, pulse width 1.3 ns) as excitation source (time resolution ca. 1 ns). DSC measurements were performed on a Universal Analysis 2000 (TA instruments). TGA analysis was performed on a Pyris 1 thermogravimetric analyser (Perkin-Elmer), heating at 5 °C min−1.
Characterization of products
The 1H, 13C, and 31P NMR characterization of trimethyl octylammonium methylcarbonate ([N1118][H3COCO2]), amino acids methyl esters (1a and 2a), dansylated amino acid esters (1b, 2b, and 3b), compounds 1′c, 2′c, and 3c, and LILs 1d–f, 2d, and 3d is available in the ESI† section.Synthesis of methylcarbonate based ILs
Methylcarbonate based ILs, including tributylmethylphosphonium methylcarbonate [P1444][H3COCO2] and methyltrioctylphosphonium methylcarbonate [P1888][H3COCO2] were prepared according to a procedure previously reported by us.17a These ILs were obtained in quantitative yields (>99%) and high purity (>98%). The same procedure was also used to prepare trimethyloctylammonium methylcarbonate [N1118][H3COCO2], the synthesis of which was never previously described in the literature. In particular, this new methylcarbonate ILs was obtained by the following method: a mixture of freshly distilled dimethyloctylamine (49 mmol, 10 mL), dimethyl carbonate (DMC, 392 mmol, 33 mL), and methanol (20 mL) were charged stainless steel autoclave (V = 120 mL), equipped with a pressure gauge and a thermocouple for temperature control. Three freeze–pump–thaw cycles were performed for efficient water and gas removal from the reaction mixture. The autoclave was finally purged with nitrogen and heated at 120 °C for 24 h, under magnetic stirring. Then, the autoclave was allowed to cool to rt and it was vented. The reaction mixture was concentrated by rotary evaporation and further vacuum treatment at 80 mbar. [N1118][H3COCO2] was obtained as a white powder (12 g, 48.50 mmol; >99%).Synthesis of amino acids methyl esters (compounds 1a and 2a)
A conventional SOCl2-based methodology was used for initial experiments. Accordingly, the selected amino acid (L-phenylalanine or L-tryptophan: 25.0 mmol) was suspended in methanol (75 mL). The mixture was kept under vigorous stirring at 0 °C, while excess thionyl chloride (4.4 mL, 60 mmol, 2.5 equiv.) was added dropwise to the alcohol suspension. Once the addition was complete, a pale yellow homogeneous solution was obtained. This was heated up at 65 °C (reflux temperature) for 2 h. The formation of the amino acid methyl ester was confirmed by TLC (eluent: toluene–acetone 3:1 v/v). The mixture was evaporated to dryness and the resulting methyl ester hydrochloric salt was dissolved in the minimum amount necessary of MilliQ water. pH of the aqueous solution was adjusted to 9 by dropwise addition of aq. NH3 (33%, v/v). Methyl ester of tryptophan and phenylalanine (compounds 1a and 2a, respectively) were isolated by extraction with diethyl ether (4 × 15 mL) and dichloromethane (2 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated by rotary evaporation. After an additional drying cycle under vacuum at 80 mbar, 1a was isolated in 87% yield (3.82 g, 21.0 mmol), while 2a was isolated in 75% yield (4.09 g, 19.0 mmol).
Esterification reactions were also carried out by a DMC-based sustainable methodology. Accordingly, at rt, H2SO4 (96%; 750 μl; 1.2 equiv.) was added dropwise to a mixture of the selected amino acid (L-Phe or L-Trp: 10 mmol) and dimethylcarbonate (DMC: 5 mL). The resulting sticky suspension was kept under vigorous stirring and heated at 90 °C for 12 h, until the completion of the reaction was confirmed by TLC (eluent: toluene–acetone 3:1 v/v). The final reaction mixture was concentrated by rotary evaporation. Then, the residue (methyl ester hydrogensulphate salt) was dissolved in the minimum volume of MilliQ water, and pH was adjusted to 9 by dropwise addition of NaOH 1 M. The product was extracted with diethyl ether (3 × 15 mL for phenylalanine derivative; 10 × 10 mL for tryptophan derivative). The combined organic layers were dried over Na2SO4, filtered, and concentrated by rotary evaporation. After an additional drying cycle under vacuum at 80 mbar, methyl ester of tryptophan was isolated in 90% yield (1a: 3.95 g, 21.7 mmol), while methyl ester of phenylalanine was isolated in 93% yield (2a: 5.07 g, 23.6 mmol).
Preparation of dansylated amino acid esters (compounds 1b, 2b, and 3b)
Methyl esters 1a and 2a, and commercial glycyl-glycine ethyl ester hydrochloride (3a) were used as reactants. A conventional methodology was used for initial experiments. Accordingly, the chosen ester (1a, 2a, or 3a: 5.50 mmol), and triethylamine (5.77 mmol, 1.05 equiv. for 1a and 2a; 12.10 mmol, 2.10 equiv. for 3a), were dissolved in CH2Cl2 (30 mL). A solution of dansyl chloride (DNS-Cl, 6.05 mmol, 1.05 equiv.) in CH2Cl2 (50 mL) was added dropwise in 1 h. The mixture was then kept under vigorous stirring at rt. The reaction progress was monitored by TLC (eluent: toluene–acetone 3:1 v/v): the complete disappearance of the reactant (C-protected amino acid/peptide) was observed after 12 h. The reaction mixture was dried by rotary evaporation, and the corresponding dansylated product was purified by FCC (eluant: toluene–acetone 4:1 v/v). Methyl (2-dansylamido-3-indolyl)propanoate (1b) was isolated as a bright yellow powder in 99% yield (2.46 g, 5.48 mmol). Methyl (2-dansylamido-3-phenyl)propanoate (2b) was isolated as a green yellow solid in 75% yield (1.79 g, 4.34 mmol). Ethyl (2-dansylamido-3-oxo)amidoetanoate (3b) was isolated as a white solid in 93% yield (2.01 g, 5.11 mmol).
N-dansylation reactions were carried out also by a more sustainable methodology
Accordingly, the chosen aminoacid/peptide ester (1a or 3a: 25 mmol) was dissolved (1a) or suspended (3a) in 2-propanol (1 mL) and treated with Et3N (0.23 mmol, 1.00 equiv.). A solution of DNS-Cl (0.25 mmol, 1 equiv.) in 2-propanol (2 mL) was then added dropwise to the mixture. The reaction progress was monitored by TLC (eluent: toluene–acetone 3:1 v/v). In both cases, the conversion of reactant esters was quantitative after 2 h. The crude mixture was concentrated by rotary evaporation and the product was purified by FCC (eluent: toluene–acetone 4:1 v/v). Methyl (2-dansylamido-3-indolyl)propanoate (1b) was isolated as a bright yellow powder in 93% yield (0.11 g, 0.23 mmol), while ethyl (2-dansylamido-3-oxo)amidoetanoate (3b) was isolated as a white powder in 96% yield (0.9 g, 0.24 mmol). Due to solubility issues, the same procedure was not effective for compound 2b (phenylalanine derivative). Spectral characterizations were in agreement with those above reported.
Dansylated amino acid esters hydrolysis (compounds 1′c, 2′c, and 3c)
Ester 1b or 2b (2.00 mmol) was dissolved in THF (30 mL). To this solution, aq. LiOH (0.2 M; 2.0 equiv.) was added dropwise at 0 °C, under vigorous stirring, in 30 min. A cloudy pale yellow solution was obtained. The mixture was kept under stirring at rt. The reaction progress was monitored by TLC (eluent: toluene–acetone 3:1 v/v). The hydrolysis was complete in 12 h. Then, pH was adjusted to ≈5 upon dropwise addition of HCl 3 M. The reaction mixture was extracted by ethyl acetate (4 × 10 mL). The combined organic layers were washed with MilliQ water (2 × 10 mL), dried over Na2SO4, and concentrated by both rotary evaporation and vacuum at 0.8 mbar. Both products (plausibly 2-dansylamido-3-indolylpropanoic acid and 2-dansylamido-3-phenylpropanoic acid: compounds 1c and 2c, respectively, of Scheme 3) were highly deliquescent solids that were used as such for a subsequent reaction with Et3N. In particular, the dansylated amino acid 1c or 2c was dissolved in a H2O–acetone (1:1 v/v) mixture and set to react with Et3N (1.10 equiv.). The mixture was kept under vigorous stirring for 30 min. Then, extractions with CH2Cl2 (3 × 10 mL) followed. The combined organic layers were dried over Na2SO4, and concentrated by rotatory evaporation and vacuum at 0.8 mbar. Triethylammonium (2-dansylamido-3-indolyl)propanoate (1′c) was isolated in 83% yield (0.36 g, 0.83 mmol); while, triethylammonium(2-dansylamido-3-phenyl)propanoate (2′c) was isolated in 90% yield (0.36 g, 0.90 mmol). Both compounds 1′c and 2′c were white easy-to-handle solids.
A similar, though simpler, procedure was used for ester 3b. Compound 3b (2.00 mmol) was dissolved in THF (20 mL). To this solution, aq. LiOH (0.34 M; 3.0 equiv.) was added dropwise at 0 °C, under vigorous stirring, in 30 min. The reaction progress was monitored by TLC (eluent: toluene–acetone 3:1 v/v). The hydrolysis was complete in 2 h. Then, pH was adjusted to ≈5 upon dropwise addition of HCl 3 M. The reaction mixture was extracted by ethyl acetate (6 × 10 mL). The combined organic layers were washed with MilliQ water (2 × 10 mL), dried over Na2SO4, and concentrated by both rotary evaporation and vacuum at 0.8 mbar. Pure 3-dansylamido-2-ossoaminoetanoic acid (3c) was recovered in 61% yield (0.47 g, 1.27 mmol).
Luminescent ionic liquid (LILs) synthesis
ESI-MS (FIA, CH3CN): 385 ([P1888]+); 436 ([C23H22N3O4S]−.
IR (KBr): 3435 (b), 2960, 2920, 2850 (m), 1640 (b), 1455 (sh), 1265 (sh), 1105 (b), 805 (sh). Anal. Calcd for C48H76N3O4PS·2H2O: C, 67.2; H, 9.4; N, 4.9. Found: C, 67.2; H, 9.4; N, 4.9.
ESI-MS (FIA, CH3CN): 217 ([P1444]+); 436 ([C23H22N3O4S]−.
IR (KBr): 3430 (b), 2970, 2920, 2860 (w), 1640 (b), 1385 (sh), 1260 (sh), 1105, 1030 (b), 810 (sh). Anal. Calcd for C36H52N3O4PS: C, 66.1; H, 8.0; N, 6.4. Found: C, 66.0; H, 8.0; N, 6.52.
ESI-MS (FIA, CH3CN): 172 ([N1118]+); 436 ([C23H22N3O4S]−.
IR (KBr): 3455 (b), 2955 (m), 2960, 2930, 2860 (w), 1620 (b), 1145 (sh), 1090 (m), 795 (sh). Anal. Calcd for C34H48N4O4S·3H2O: C, 61.6; H, 8.2; N, 8.4. Found: C, 62.4; H, 9.1; N, 8.3.
ESI-MS (FIA, CH3CN): 385 ([P1888]+); 397 ([C21H21N2O4S]−).
IR (KBr): 3440 (b), 2955, 2935, 2860 (sh), 1745, 1640 (b), 1460 (sh), 111500 (sh), 1090 (b), 790 (sh). Anal. Calcd for C46H75N2O4PS·3H2O: C, 66.9; H, 10.0; N, 3.2. Found: C, 66.75; H, 10.3; N, 3.6.
IR (KBr): 3440 (w), 2955 (m), 2920 (m), 2850 (sh), 1635 (b), 1265 (sh), 1105, 1025 (m), 795 (sh). Anal. Calcd for C41H72N3O5PS·2H2O: C, 62.6; H, 9.7; N, 5.4. Found: C, 63.1; H, 11.0; N, 5.2.
Acknowledgements
GF acknowledges the Department of Molecular Sciences and Nanosystems (DSMN) of Ca’ Foscari University of Venice for co-funding her post-doctoral research fellowship. Prof. Vittorio Lucchini and Dr Marco Bortoluzzi are also acknowledged for helpful discussion.Notes and references
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965–967 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- (a) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef PubMed; (b) H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS PubMed; (c) Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC; (d) M. D. Joshi and J. L. Anderson, RSC Adv., 2012, 2, 5470–5484 RSC.
- (a) P. Domínguez de María, Angew. Chem., Int. Ed., 2008, 47, 6960–6968 CrossRef PubMed; (b) V. Lucchini, M. Noè, M. Selva, M. Fabris and A. Perosa, Chem. Commun., 2012, 48, 5178–5180 RSC; (c) D. E. Siyutkin, A. S. Kucherenko and S. G. Zlotin, in Comprehensive Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2013, pp. 617–650 Search PubMed.
- (a) E. F. Borra, O. Seddiki, R. Angel, D. Eisenstein, P. Hickson, K. R. Seddon and S. P. Worden, Nature, 2007, 447, 979–981 CrossRef CAS PubMed; (b) L. D. Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef; (c) J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780–1804 RSC; (d) C. Vollmer and C. Janiak, Coord. Chem. Rev., 2011, 255, 2039–2057 CrossRef CAS PubMed; (e) M. Zahmakran and S. Ozkar, Nanoscale, 2011, 3, 3462–3481 RSC.
- (a) D. R. MacFarlane, M. Forsyth, P. C. Howlett, J. M. Pringle, J. Sun, G. Annat, W. Neil and E. I. Izgorodina, Acc. Chem. Res., 2007, 40, 1165–1173 CrossRef CAS PubMed; (b) M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed; (c) D. S. Silvester, Analyst, 2011, 136, 4871–4882 RSC; (d) U. A. Rana, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Electrochim. Acta, 2012, 84, 213–222 CrossRef CAS PubMed; (e) S. H. Kim, K. Hong, W. Xie, K. H. Lee, S. Zhang, T. P. Lodge and C. D. Frisbie, Adv. Mater., 2013, 25, 1822–1846 CrossRef CAS PubMed; (f) M. J. Park, I. Choi, J. Hong and O. Kim, J. Appl. Polym. Sci., 2013, 129, 2363–2376 CrossRef CAS.
- (a) L. C. Branco and F. Pina, Chem. Commun., 2009, 6204–6206 RSC; (b) T. Torimoto, T. Tsuda, K.-I. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS PubMed; (c) S. Zhang, S. Liu, Q. Zhang and Y. Deng, Chem. Commun., 2011, 47, 6641–6643 RSC; (d) V. F. Curto, C. Fay, S. Coyle, R. Byrne, C. O'Toole, C. Barry, S. Hughes, N. Moyna, D. Diamond and F. Benito-Lopez, Sens. Actuators, B, 2012, 171–172, 1327–1334 CrossRef CAS PubMed; (e) P. G. Jessop, S. M. Mercer and D. J. Heldebrant, Energy Environ. Sci., 2012, 5, 7240–7253 RSC; (f) G. Li, F. Song, D. Wu, J. Lan, X. Liu, J. Wu, S. Yang, D. Xiao and J. You, Adv. Funct. Mater., 2014, 24, 747–753 CrossRef CAS; (g) H. Maeda, in Intelligent Stimuli-Responsive Materials: From Well-Defined Nanostructures to Applications, ed. Q. Li, John Wiley & Sons Inc., Hoboken, 2013, pp. 56–140 Search PubMed.
- (a) S. S. Babu, J. Aimi, H. Ozawa, N. Shirahata, A. Saeki, S. Seki, A. Ajayaghosh, H. Möhwald and T. Nakanishi, Angew. Chem., Int. Ed., 2012, 51, 3391–3395 CrossRef PubMed; (b) S. S. Babu and T. Nakanishi, Chem. Commun., 2013, 49, 9373–9382 RSC; (c) C. C. Weber, A. F. Masters and T. Maschmeyer, Green Chem., 2013, 15, 2655–2679 RSC.
- (a) K. Binnemans, Chem. Rev., 2007, 107, 2592–2614 CrossRef CAS PubMed; (b) J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387–410 RSC.
- J. Avó, L. Cunha-Silva, J. C. Lima and A. Jorge Parola, Org. Lett., 2014, 16, 2582–2585 CrossRef PubMed.
- J.-F. Huang, H. Luo, C. Liang, I. W. Sun, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2005, 127, 12784–12785 CrossRef CAS PubMed.
- S. Hernández-Ainsa, J. Barberá, M. Marcos and J. L. Serrano, Macromolecules, 2011, 45, 1006–1015 CrossRef.
- (a) S. Wakizono, K. Yamamoto and J.-I. Kadokawa, J. Photochem. Photobiol., A, 2011, 222, 283–287 CrossRef CAS PubMed; (b) S. Wakizono, K. Yamamoto and J.-I. Kadokawa, J. Mater. Chem., 2012, 22, 10619–10624 RSC.
- (a) J.-H. Olivier, F. Camerel, J. Barberá, P. Retailleau and R. Ziessel, Chem. – Eur. J., 2009, 15, 8163–8174 CrossRef CAS PubMed; (b) Q. Zhang, B. Yang, S. Zhang, S. Liu and Y. Deng, J. Mater. Chem., 2011, 21, 16335–16338 RSC.
- (a) T. S. Jo, W. L. McCurdy, O. Tanthmanatham, T. K. Kim, H. Han, P. K. Bhowmik, B. Heinrich and B. Donnio, J. Mol. Struct., 2012, 1019, 174–182 CrossRef CAS PubMed; (b) K. Tanabe, Y. Suzui, M. Hasegawa and T. Kato, J. Am. Chem. Soc., 2012, 134, 5652–5661 CrossRef CAS PubMed.
- E. Westphal, D. H. d. Silva, F. Molin and H. Gallardo, RSC Adv., 2013, 3, 6442–6454 RSC.
- (a) M. Fabris, V. Lucchini, M. Noè, A. Perosa and M. Selva, Chem. – Eur. J., 2009, 15, 12273–12282 CrossRef CAS PubMed; (b) M. Selva, M. Fabris, V. Lucchini, A. Perosa and M. Noè, Org. Biomol. Chem., 2010, 8, 5187–5198 RSC; (c) M. Fabris, M. Noè, A. Perosa, M. Selva and R. Ballini, J. Org. Chem., 2012, 77, 1805–1811 CrossRef CAS PubMed; (d) M. Selva, M. Noè, A. Perosa and M. Gottardo, Org. Biomol. Chem., 2012, 10, 6569–6578 RSC; (e) M. Selva, A. Caretto, M. Noè and A. Perosa, Org. Biomol. Chem., 2014, 12(24), 4143–4155 RSC.
- (a) S. Silvestrini, P. Riello, I. Freris, D. Cristofori, F. Enrichi and A. Benedetti, J. Nanopart. Res., 2010, 12, 993–1002 CrossRef; (b) I. Freris, P. Riello, F. Enrichi, D. Cristofori and A. Benedetti, Opt. Mater., 2011, 33, 1745–1752 CrossRef CAS PubMed; (c) A. Dhahri, K. Horchani-Naifer, A. Benedetti, F. Enrichi and M. Ferid, Opt. Mater., 2012, 34, 1742–1746 CrossRef CAS PubMed; (d) R. Marin, G. Sponchia, E. Zucchetta, P. Riello, F. Enrichi, G. De Portu and A. Benedetti, J. Am. Ceram. Soc., 2013, 96, 2628–2635 CrossRef CAS PubMed.
- (a) C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927–934 RSC; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. Bartzatt, J. Biochem. Biophys. Methods, 2001, 47, 189–195 CrossRef CAS.
- C. Gros and B. Labouesse, FEBS J., 1969, 7, 463–470 CrossRef CAS PubMed.
- A. J. Brouwer, S. J. E. Mulders and R. M. J. Liskamp, Eur. J. Org. Chem., 2001, 1903–1915 CrossRef CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- (a) W. H. J. Boesten and D. Heemskerk, EU Pat., EP1931619, 2008 Search PubMed; (b) M. J. Earle, M. Noè, A. Perosa and K. R. Seddon, RSC Adv., 2014, 4, 1204–1211 RSC.
- J. L. Ferguson, J. D. Holbrey, S. Ng, N. V. Plechkova, K. R. Seddon, A. A. Tomaszowska and D. F. Wassell, Pure Appl. Chem., 2012, 84, 723–744 CAS.
- Green Chemistry Metrics: Measuring and Monitoring Sustainable Processes, ed. A. Lapkin and D. J. C. Constable, John Wiley & Sons Ltd., Chichester (UK), 2008 Search PubMed.
- E. Brasola, F. Mancin, E. Rampazzo, P. Tecilla and U. Tonellato, Chem. Commun., 2003, 3026–3027 RSC.
- D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596–600 RSC.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Hoboken, 6th edn, 2007 Search PubMed.
- (a) G. H. Tao, L. He, N. Sun and Y. Kou, Chem. Commun., 2005, 3562–3564 RSC; (b) Q.-P. Liu, X.-D. Hou, N. Li and M.-H. Zong, Green Chem., 2012, 14, 304–307 RSC.
- Y.-H. Li, L.-M. Chan, L. Tyer, R. T. Moody, C. M. Himel and D. M. Hercules, J. Am. Chem. Soc., 1975, 97, 3118–3126 CrossRef CAS.
- L. Ding, Y. Fang, L. Jiang, L. Gao and X. Yin, Thin Solid Films, 2005, 478, 318–325 CrossRef CAS PubMed.
- K. M. Johansson, E. I. Izgorodina, M. Forsyth, D. R. MacFarlane and K. R. Seddon, Phys. Chem. Chem. Phys., 2008, 10, 2972–2978 RSC.
- S. Chen, S. Zhang, X. Liu, J. Wang, J. Wang, K. Dong, J. Suna and B. Xu, Phys. Chem. Chem. Phys., 2014, 16, 5893–5906 RSC.
- (a) M. Kogiso, M. Masuda and T. Shimizu, Supramol. Chem., 1998, 9, 183–189 CrossRef CAS; (b) A. Abo-Riziq, L. Grace, B. Crews, M. P. Callahan, T. van Mourik and M. S. de Vries, J. Phys. Chem. A, 2011, 115, 6077–6087 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 31P NMR, ESI and IR spectra of LILS 1d–f, 2d, and 3d. See DOI: 10.1039/c4gc01198h |
| This journal is © The Royal Society of Chemistry 2015 |