
Efficient biomass transformations catalyzed by graphene-like nanoporous carbons functionalized with strong acid ionic liquids and sulfonic groups†
Fujian
Liu
ab,
Weiping
Kong
a,
Liang
Wang
b,
Xianfeng
Yi
c,
Iman
Noshadi
d,
Anmin
Zheng
*c and
Chenze
Qi
*a
aKey Laboratory of Alternative Technologies for Fine Chemicals Process of Zhejiang Province, Department of Chemistry, Shaoxing University, Shaoxing, 312000, China. E-mail: qichenze@usx.edu.cn
bDepartment of Chemistry, Zhejiang University (XiXi Campus), Hangzhou, 310028, China
cWuhan Center for Magnetic Resonance, State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences, Wuhan 430071, China. E-mail: zhenganm@wipm.ac.cn
dPolymer Program, Institute of Materials Science and Department of Chemistry, University of Connecticut, Storrs, CT 06269, USA
First published on 12th September 2014
Abstract
Strong acid ionic liquids and sulfonic group bifunctional graphene-like nanoporous carbons (GNC-SO3H-ILs) have been synthesized by treating nitrogen containing graphene-like nanoporous carbons (GNCs) with 1,3-propanesultone, ion exchanging with HSO3CF3 or H2SO4. Introducing nitrogen is important for grafting strong acid ionic liquids and sulfonic group in GNCs, which were synthesized from carbonization of a mixture of dicyandiamide or melamine and glucose. GNC-SO3H-ILs possess abundant nanopores, nanosheet structure, good dispersion and controlled acidity. By themselves, they are capable of enhancing the fast diffusion of reactants and products, while increasing the exposure degree of acidic sites in GNC-SO3H-ILs throughout various reactions. The above characteristics resulted in their much improved catalytic activity in biomass transformations such as the production of biodiesel and depolymerization of crystalline cellulose into sugars, which was even comparable to those of homogeneous ionic liquid and mineral acids.
1. Introduction
Great effort has been made to develop efficient and cost effective ways to transform biomass into biofuels, due to the low cost, excellent renewability and environmental friendliness of biomass when compared with conventional fossil energy.1–9 Biomass may therefore be the most important feedstock for replacing conventional fossil energy in the future. Generally, biofuels were typically produced by two routes: (i) towards transesterification to biodiesel and (ii) depolymerization of crystalline cellulose into sugars and fine chemicals.1,2,7,10–15 Notably, biodiesel production was focused on employing low cost and low quality plant oil or waste oil as the feedstocks, typically held under green and mild conditions.16 Alternatively, the depolymerization of cellulose was mostly held under severe conditions.7,9 Surprisingly, crystalline cellulose easily transformed into sugars in the presence of alkylmethylimidazolium ionic liquid solvents because of their unique ability to break down the crystalline cellulose into soluble polymer chains. However, the high cost of ionic liquids and the reaction mixtures’ acute recyclability create a demand for new catalysts with extremely high effectiveness. Researchers have discovered that acids, bases or enzyme catalysts were active when producing biomass transformations.1,2,7,10–16 Acid catalysts were highly efficient and low in cost for biomass transformation, which could catalyze the transformation of low-quality and low cost biomass into biofuels under rather mild conditions. The replacement of mineral acids with solid acids in terms of biomass transformation has received considerable attention regarding green and sustainable chemistry.10–19Among various heterogeneous acid catalysts, carbon based solid acids showed controlled wettability, good thermal and chemical stabilities and superior catalytic activity throughout various reactions, combining the advantages of the solid acids with both organic and inorganic frameworks.3,20–24 Graphene based nanomaterials act as novel carbon materials. Materials scientists, chemists and physicist recognize that these materials possess excellent thermal and mechanical stabilities, unique nanosheet structure, controlled wettability and very good dispersion in various systems.25–30 These are the ideal candidates for solid acidic catalysts or catalyst supports because they are capable of largely reducing the limitation of mass transfer, increase the exposure degree of active sites and enhance the recyclability of the catalysts in various reactions.31–33 Due to the frequency of graphene nanomaterial layers sticking together, it is imperative to create pillars between the layers, maximizing the surface area and accessibility during various reactions.31–33 Still, the synthesis of graphene based nanomaterials with large BET surface areas and abundant nanopores through facile and cost-effective routes remains a challenge.34–38 Abundant nanopores in graphene-based nanomaterials must be created to increase the degree of active sites, which contribute importantly to their application during heterogeneous catalysis.31
We report here the successful synthesis of novel acid ionic liquids and sulfonic group bifunctional graphene-like nanoporous carbons (GNC-SO3H-ILs), which were synthesized by treating nitrogen containing graphene-like nanoporous carbons (GNCs) with 1,3-propanesultone. The anions were exchanged with strong acids of H2SO4 or HSO3CF3. The introduction of nitrogen atoms into GNCs forms strong acid ionic liquids and sulfonic groups through the quaternary ammonization reaction. The GNC supports could be easily synthesized using a one-step carbonization mixture. This mixture should consist of dicyandiamide/melamine and glucose, using no additional templates (as shown in Scheme 1). The resulting bifunctional carbon based solid acids showed controlled acidity, unique nanosheet structure, abundant nanopores and good stability. Interestingly, GNC-SO3H-ILs showed significantly improved catalytic activity and good recyclability in biomass transformation, including transesterification to biodiesel and depolymerization of crystalline cellulose into sugars and 5-hydroymethylfurfural (HMF). This process was better than those of acidic resin of Amberlyst 15, sulfonic group functionalized mesoporous SBA-15, and heteropolyacid of H3PW12O40, which was even comparable to those of homogeneous acid ionic liquids and mineral acids. The preparation of GNC-SO3H-ILs fosters a facile and cost-effective method to synthesize highly efficient graphene based solid acids for biomass transformation, which is crucial for their various applications when exploring biofuel through green and sustainable processes.
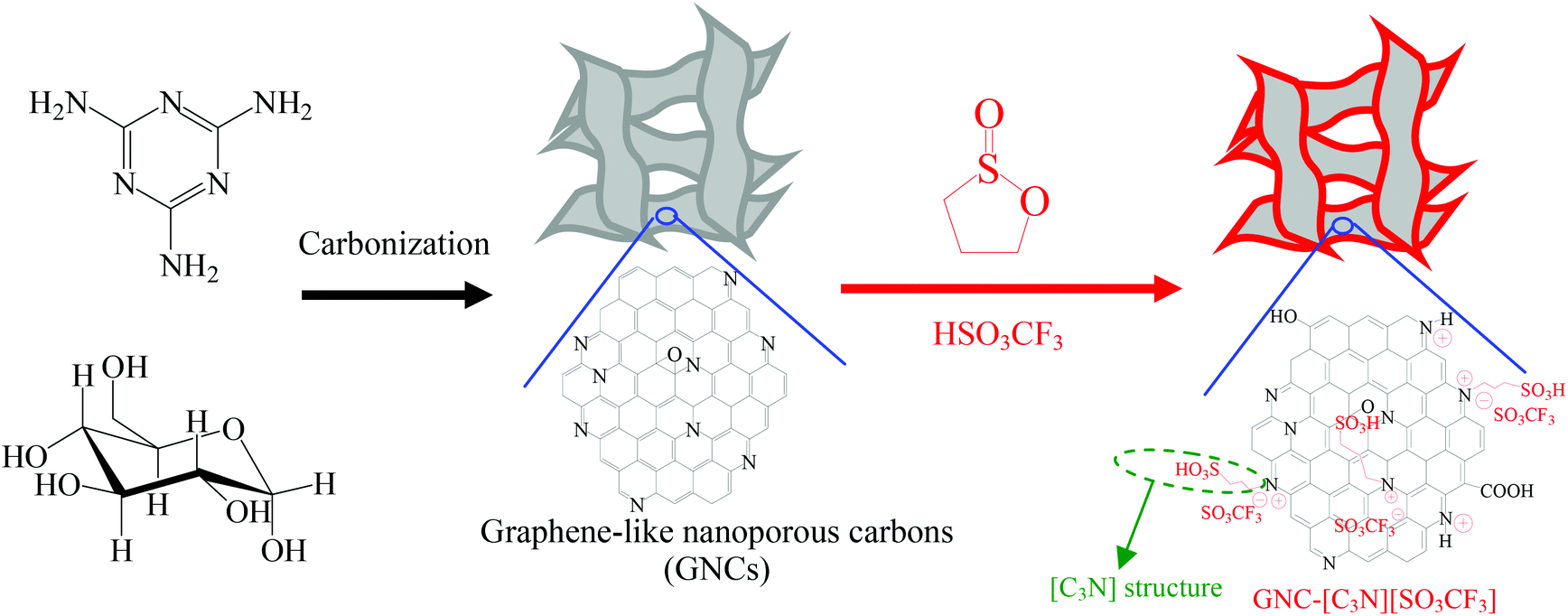 | ||
| Scheme 1 The procedures for synthesizing GNC-[C3N][SO3CF3]. | ||
2. Experimental details
2.1 Preparation of catalysts
2.2 Catalytic reactions
Transesterification of sunflower oil with methanol was executed as follows: 1.0 g of sunflower oil (1.16 mmol) was added into a flask equipped with a condenser and a magnetic stirrer. The temperature was rapidly increased to 65 °C. Then, 3.5 mL of methanol (86.3 mmol) and 0.15 g of catalyst were quickly added under vigorous stirring. The reaction occurred for 18 h. The molar ratio of sunflower oil to methanol was 1/75 and the mass ratio of catalyst to tripalmitin was 0.1. The products were mainly methyl palmitate (C16:0), methyl stearate (C18:0), methyl oleate (C18:1), methyl linoleate (C18:2), and methyl heptacosane (C27:0). The quantification of these products was analyzed by using an Agilent GC/MS instrument (Agilent 6890N/5975I) with a programmable split/splitless injector.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 rpm for 5 min to remove catalysts and unreacted cellulose, giving the reaction mixture. Various acid catalysts such as Amberlyst 15, HCl and [C3vim][SO3CF3] were also used for catalyzing depolymerization of Avicel, which used the same number of acidic sites as in GNC-[C3N][SO3CF3].
800 rpm for 5 min to remove catalysts and unreacted cellulose, giving the reaction mixture. Various acid catalysts such as Amberlyst 15, HCl and [C3vim][SO3CF3] were also used for catalyzing depolymerization of Avicel, which used the same number of acidic sites as in GNC-[C3N][SO3CF3].
Meanwhile, the isolated cellulose was thoroughly washed with water, and recovered through centrifugation. The amount of cellulose isolated was determined by weight.
The concentrations of glucose and cellobiose in the reaction mixture were measured using the Shimadzu LC-20A HPLC system based on the standard curve method, which was equipped with a SCR-101N column using extra-pure water during the mobile phase at a flow rate of 0.5 mL min−1. The column's temperature was set to 55 °C. In the meantime, a refraction index was used for the detection of sugars in the water. The concentration of 5-hydroxymethylfurfural (HMF) was also measured using a Shimadzu LC-20A HPLC system based on the standard curve method. It was equipped with a CAPCELL PAK C18 column using methanol and water (methanol–water = 80:20) during the mobile phase, at a flow rate of 0.7 mL min−1, and the column's temperature was set to 50 °C. At the same time, an ultraviolet detector with the wavelength number at 254 nm was used for the detection of HMF in the reaction mixture.
3. Results and discussion
3.1 Structural characterization
Fig. 1A shows XRD patterns of GNC and GNC-[C3N][SO3CF3]. Notably, GNC exhibits two broad peaks centered at 26.1 and 43.2°. These are associated with the graphite structure, which was extremely close to the peaks of graphene based nanomaterials with a nanosheet structure, reported previously.31,37 This result indicated that graphene-like structures with several layers was formed in the GNC. After grafting the strong acid ionic liquids and the sulfonic group onto the network of GNC, the sample of GNC-[C3N][SO3CF3] was given, which showed almost no change in XRD patterns compared to the GNC support. The above results indicated that the graphene-like nanosheet structure was well maintained in GNC-[C3N][SO3CF3].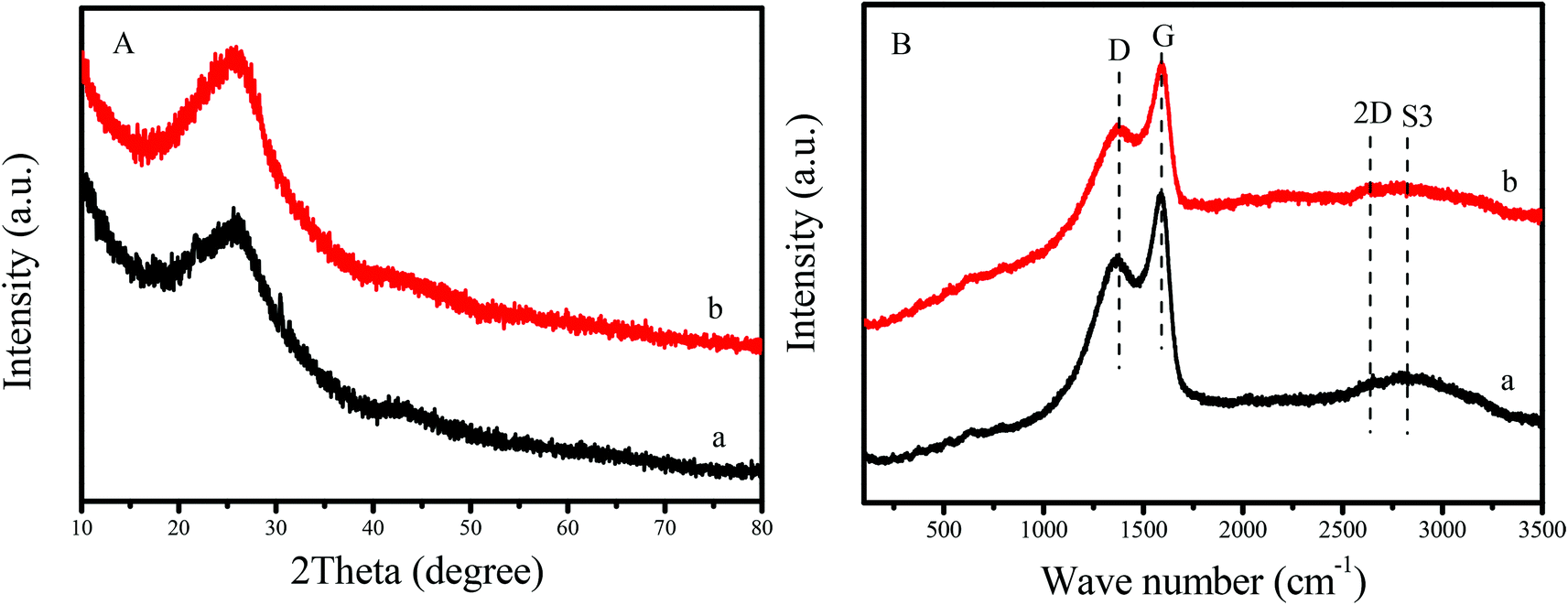 | ||
| Fig. 1 (A) XRD patterns and (B) Raman spectra of (a) GNC and (b) GNC-[C3N][SO3CF3]. | ||
In addition, the graphene-like structures of GNC and GNC-[C3N][SO3CF3] have also been confirmed by Raman spectra. It is well known that Raman spectroscopy is a powerful approach for determining the properties of graphene based nanomaterials such as the number and orientation of layers, the quality and types of edges and the effects of perturbations involving the structure strain, doping, disorder and functional groups.40,41Fig. 1B shows the Raman spectra of GNC and GNC-[C3N][SO3CF3]. It was noted that a series of peaks at approximately 1378, 1591, 2639 and 2822 cm−1 were clearly observed in these samples, which could be attributed to the signals of D, G, 2D and S3 bands.26 The broadened D-band indicated the presence of sp3 carbon atoms with defects and disorders at the edges and the boundaries of the graphene domains. The sharp G-band indicated the presence of sp2 carbon atoms in a graphitic 2D hexagonal lattice, confirming the formation of graphitic carbon, in good agreement with XRD results. Compared with GNC, GNC-[C3N][SO3CF3] showed a decreased intensity of the Raman spectrum, which could be attributed to the introduction of acidic groups into the sample. It is also noteworthy that the position and shape of each peak in the Raman spectrum does not shift after the introduction of acidic groups into GNC. This indicates that the graphene-like nanosheet structure was well maintained in GNC-[C3N][SO3CF3], in good agreement with XRD results.
Table 1 presents the parameters of various acid catalysts. GNC-[C3N][SO3CF3] showed the content of acidic sites at 1.25 mmol g−1, lower than those of various acid catalysts including SBA-15-SO3H (1.98 mmol g−1), H3PW12O40 (3.5 mmol g−1), Amberlyst 15 (4.3 mmol g−1) and H2SO4 (10.2 mmol g−1). Alternatively, the BET surface area of GNC-[C3N][SO3CF3] (184 m2 g−1, Fig. S1†) was higher than that of acidic resin of Amberlyst 15 (45 m2 g−1). Compared to the GNC support (945 m2 g−1), the decreased surface area in GNC-[C3N][SO3CF3] should result from the grafting of acid active sites, which largely increases the weight of the GNC network. Previously, similar results have also been reported.39
| Samples | Acid contentsa (mmol g−1) | N contenta (mmol g−1) | S BET (m2 g−1) |
V
pb (cm3 g−1) |
D
pc (nm) |
|---|---|---|---|---|---|
| a The acid contents were measured by CHNS elemental analysis. b Pore volumes (Vp) were estimated at relative pressure p/p0 ≈ 0.98. c Average pore diameters were estimated from the BJH model. d GNC-[C3N][SO3CF3] after being recycled five times in transesterification of sunflower oil with methanol. e The acid contents of H3PW12O40, [C3vim][SO3CF3] and H2SO4 were calculated from the molecular formula. | |||||
| GNC | — | 1.82 | 945 | 3.1 | 3.8 & 33.7 |
| GNC-[C3N][SO3CF3] | 1.25 | 1.57 | 184 | 0.59 | 3.6 & 31.2 |
| GNC-[C3N][SO3CF3]d | 1.22 | 1.62 | 173 | 0.55 | 3.4 & 30.2 |
| SBA-15-SO3H | 1.98 | — | 814 | 1.4 | 7.5 |
| H3PW12O40e |
3.50 | — | 4.2 | — | — |
| Amberlyst 15 | 4.30 | — | 45 | 0.31 | 40 |
| [C3vim][SO3CF3]e | 2.96 | — | — | — | — |
| H2SO4e |
10.20 | — | — | — | — |
In order to obtain the relationship between the catalyst structure and reactive activity, the surface morphology of the GNC-[C3N][SO3CF3] was analyzed in detail by the multiple image techniques (TEM, HR-TEM and AFM images). Typically, TEM images indicate a typical nanosheet structure with three dimensional network clusters in GNC-[C3N][SO3CF3], exhibiting abundant nanoporous and hollow structures (Fig. 2a, b and S2†). Furthermore, highly-resolved TEM images were also used to confirm the graphene structure with crumpled and entangled character (Fig. 2c–f and S1†), exhibiting several nanocrystallites with well-defined lattice planes. When the XRD and Raman aforementioned were combined with TEM and HR-TEM, the uniform graphitized networks were determined. In the meantime, the AFM image of GNC-[C3N][SO3CF3] further affirms its nanosheet structure (Fig. 2g). The thickness of layers ranged from 1.7 to 3.5 nm, indicating the stick had several graphene layers in GNC-[C3N][SO3CF3]. These results confirmed that the graphene-like nanoporous carbon network was formed in GNC-[C3N][SO3CF3].
 | ||
| Fig. 2 (a, b) TEM images, (c–f) high-resolved TEM images, and (g) AFM image and corresponding thickness analysis of GNC-[C3N][SO3CF3]. | ||
Fig. 3 shows the X-ray photoelectron spectroscopy (XPS) spectra of GNC-[C3N][SO3CF3], which exhibits the signals of S2p (168.8 eV), C1s (284.8 eV), N1s (402.1 eV), O1s (532.4 eV) and F1s (686.8 eV). Notably, the broad C1s spectrum could be fitted and deconvoluted into four peaks, centered at approximately 284.8, 286.1, 287.0 and 292.3 eV, which should be assigned to C–C and C–N in the GNC network, and C–O and C–F in the strongly acidic ionic liquids and sulfonic group.8 In the meantime, the N1s spectrum could be fitted and deconvoluted into two peaks, centered at approximately 398.4, 400.4 and 401.8 eV, which should be assigned to the C–N, N+–C3 ionic liquid group and the protonated structure of GNC-[N+H][SO3CF3] in GNC-[C3N][SO3CF3].8 Similarly, the O1s associated with O–S (531.9 eV) and O–SCF3 (533.1 eV) could also be observed.8 The above results confirm that strongly acidic ionic liquids and sulfonic groups have been successfully grafted onto the network of GNCs, in good agreement with FT-IR and elemental analysis results (Fig. S3† & Table 1).
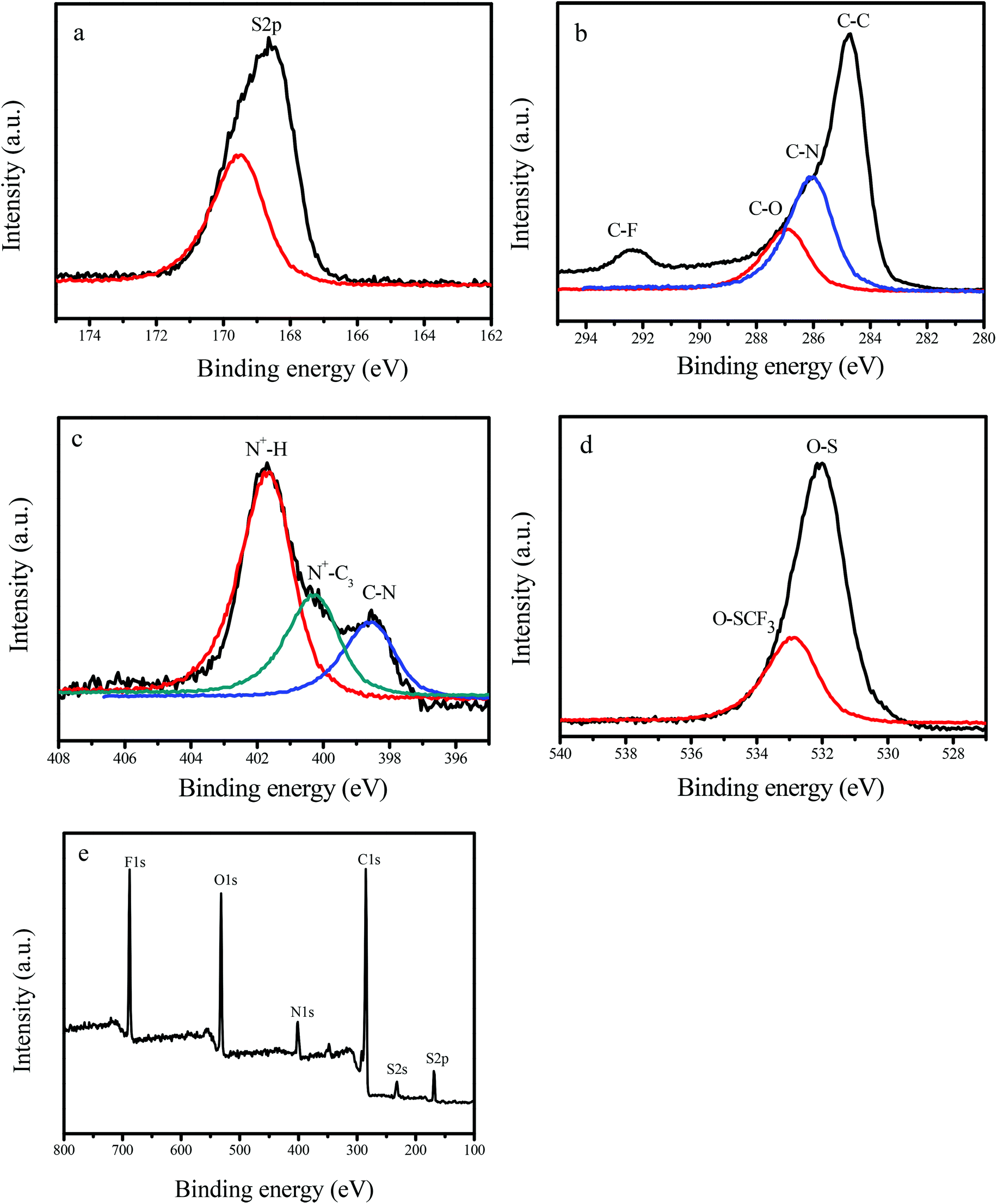 | ||
| Fig. 3 XPS spectra of (a) S2p, (b) C1s, (c) N1s, (d) O1s and (e) survey of GNC-[C3N][SO3CF3]. | ||
Furthermore, the acidity properties of GNC-[C3N][SO3CF3] were also characterized by the 31P NMR probe techniques involving the adsorbed trimethylphosphine (TMP) and trimethylphosphine oxygen (TMPO). It is noteworthy that the 31P NMR probe technique is a sensitive and reliable technique to determine the acid type (Brønsted or Lewis acid) and acid strength of solid acid catalysts.42–44 As shown in Fig. 4a, using TMP as a probe molecule, the 31P resonances at −3.2 ppm were assigned to the protonated adducts of [(CH3)3P-H]+, and attributed to the reaction of TMP with the Brønsted acidic protons. In contrast, almost no resonances were observed in the range of −20 to −60 ppm due to the interaction with Lewis acid sites.42,43 Therefore, it is indicative that no Lewis acid was formed over GNC-[C3N][SO3CF3]. As aforementioned in the Introduction, solid-state 31P MAS NMR of adsorbed TMPO is an efficient technique for acidity characterization of solid acid catalysts such as zeolites, sulfated mesoporous metal oxides and heteropolyacids.42,43 Interestingly, GNC-[C3N][SO3CF3] showed multiple 31P resonance peaks at approximately 45, 48, 51.2, 52.8, 54.5, 56.6 and 63.1 ppm, indicating the presence of various Brønsted acid sites with different acid strengths in GNC-[C3N][SO3CF3] (Fig. 4b). The weakly acidic sites (45 & 48 ppm) may be attributed to the presence of the carboxylic or hydroxyl group in GNC-[C3N][SO3CF3]. While the relatively strongly acidic sites (51.2, 52.8, 54.5, 56.6 and 63.1 ppm) should be attributed to the strong acid ionic liquids, sulfonic group and protonated structure of GNC-[N+H][SO3CF3] located in the outer and inner surfaces of GNC-[C3N][SO3CF3]. The GNC-[N+H][SO3CF3] structure should be formed from protonation of the N atom in GNC by HSO3CF3. It is noteworthy that the acidity of the –SO3H group is stronger than GNC-[N+H][SO3CF3], and the –SO3H group has a better catalytic role for its activity. The strongly acidic sites in GNC-[C3N][SO3CF3] showed a similar acid strength to that of HY zeolites (55 and 65 ppm) and bifunctional carbon–silica nano composited solid acid (40–70 ppm).44,45
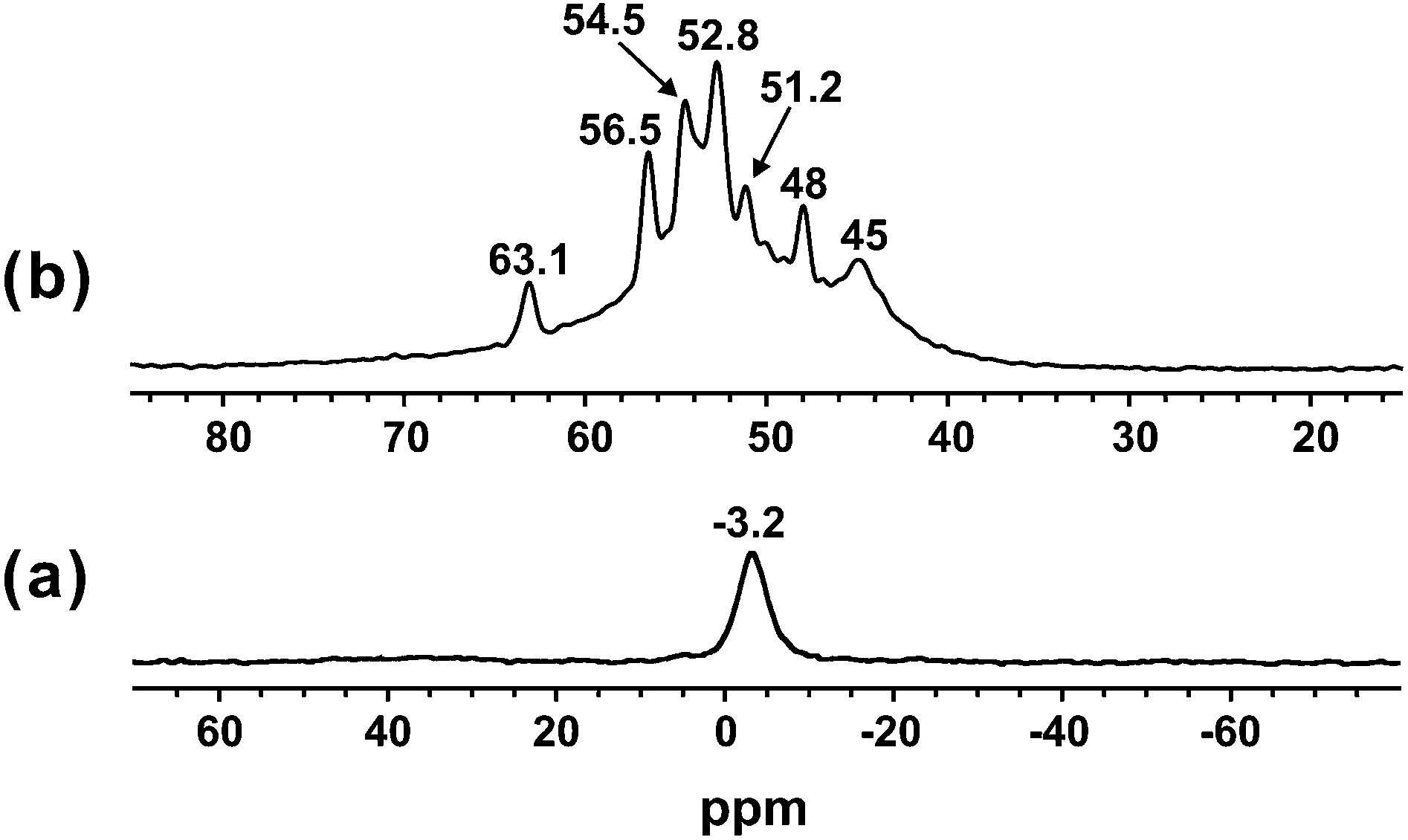 | ||
| Fig. 4 Room temperature 31P MAS NMR spectra of adsorbed (a) TMP acquired with proton decoupling, and (b) TMPO of GNC-[C3N][SO3CF3]. | ||
3.2 Catalytic reactions
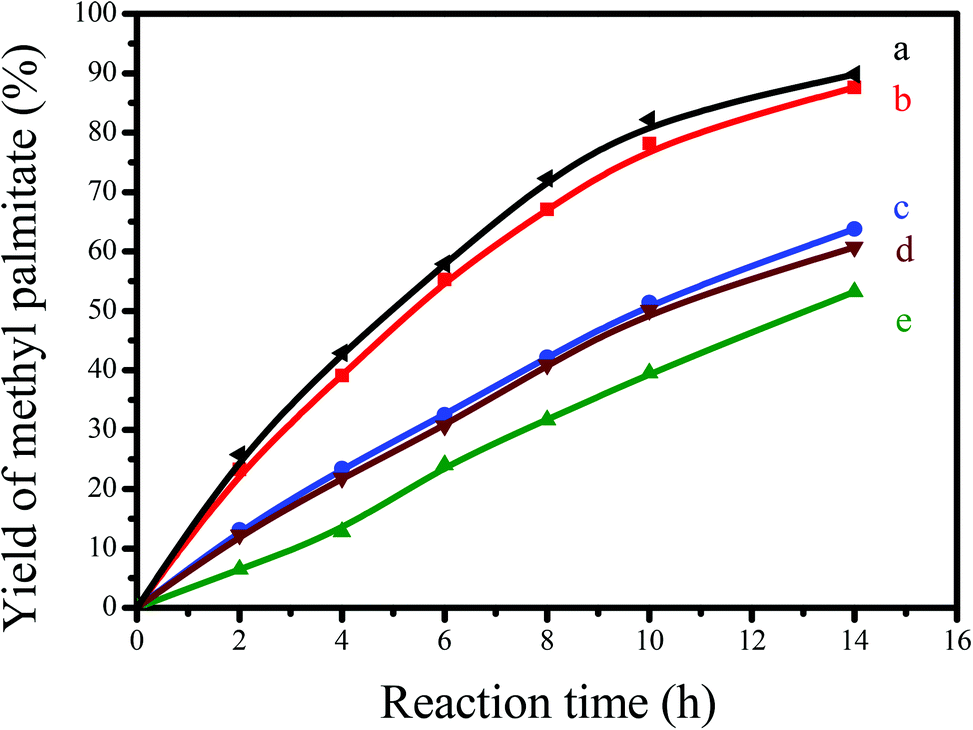 | ||
| Fig. 5 Dependences of catalytic activities on the time in transesterification of tripalmitin with methanol catalyzed by (a) [C3vim][SO3CF3] (the same number of acidic sites as that of GNC-[C3N][SO3CF3]), (b) GNC-[C3N][SO3CF3], (c) H3PW12O40, (d) SBA-15-SO3H and (e) Amberlyst 15. Reaction conditions: 0.84 g tripalmitin; 2.47 mL methanol; 0.1 g catalyst; reaction temperature, 65 °C; reaction time, 14 h. | ||
Apart from tripalmitin, GNC-[C3N][SO3CF3] also showed good catalytic activity for the transformation of plant oils such as sunflower oil into biodiesel. Table 2 presents catalytic data on transesterification of sunflower oil with methanol for the production of biodiesel over various catalysts. Interestingly, after 18 h of reaction, the yields of biodiesel including C16:0, C18:0, C18:1, C18:2 and C27:0 catalyzed by GNC-[C3N][SO3CF3] were almost as high as inside the pure acidic ionic liquid of [C3vim][SO3CF3] (89.1, 94.8, 95.2, 93.1, and 86.7% in homogeneous vs. 88.5, 93.6, 94.7, 92.3 and 81.4% in heterogeneous), which was evencomparable to that of H2SO4 (87.4, 94.1, 95.9, 94.4 and 82.1%). Therefore, it is clear that the high catalytic reactivity has been well retained. As presented in Table 2, the activity of GNC-[C3N][SO3CF3] was much higher than those of heteropolyacid of H3PW12O40 (58.9, 59.2, 54.3, 60.1 and 50.9%), Amberlyst 15 (54.8, 50.2, 48.6, 50.5 and 26.4%) and SBA-15-SO3H (60.9, 56.5, 55.4, 52.6 and 45.7%). The excellent catalytic activity found in GNC-[C3N][SO3CF3] for biodiesel production could be attributed to its controlled acidity, good dispersion, abundant and hierarchical nanoporosity, and unique nanosheet structure. By themselves, they are capable of enhancing the fast diffusion of reactants and products, increasing the exposure degree of catalytically active sites, in processes of various reactions. Compared to GNC-[C3N][SO3CF3], the low BET surface areas of H3PW12O40 and Amberlyst 15 resulted in their low exposure degrees and easy poison of active sites, further leading to their low catalytic activities. Although SBA-15-SO3H has large BET surface areas, its functional group could be easily occluded in the silica wall.46 Moreover, the accessibility of reactants to catalytically active sites located into the mesopores of SBA-15-SO3H is usually difficult in comparison to two dimensional nanosheet structures of GNC-[C3N][SO3CF3].27 These factors resulted in a lower catalytic activity of SBA-15-SO3H. The above results prove that GNC-[C3N][SO3CF3] could be used as a highly efficient solid acid for biodiesel production.
| Catalysts | Conversion of esters (%) | ||||
|---|---|---|---|---|---|
| Methyl palmitate (C16:0) | Methyl stearate (C18:0) | Methyl oleate (C18:1) | Methyl linoleate (C18:2) | Methyl heptacosane (C27:0) | |
| a 1.0 g sunflower oil; 2.47 mL methanol; 0.15 g catalyst; reaction temperature, 65 °C; reaction time, 18 h. The catalytic activity of the catalysts was characterized quantitatively by the conversion of fatty acid methyl esters (FAME, Y %) which was calculated as follows: Yield = (MD/MT) × 100%, where MD and MT are the numbers of moles of each FAME produced and expected, respectively. In this section, MT is the number of moles of FAME catalyzed by 0.2 g of H2SO4 for 24 h, which was performed at 65 °C with the same content of feedstock as that of GNC-[C3N][SO3CF3]. b Imidazole based acidic ionic liquid synthesized using the same procedure as that of GNC-[C3N][SO3CF3]. c The same number of acidic sites as that of GNC-[C3N][SO3CF3]. d The 5th cycle in transesterification of sunflower oil with methanol. | |||||
| [C3vim][SO3CF3]b | 89.1 | 94.8 | 95.2 | 93.1 | 86.7 |
| H2SO4c |
87.4 | 94.1 | 95.9 | 94.4 | 82.1 |
| GNC-[C3N][SO3CF3] | 88.5 | 93.6 | 94.7 | 92.3 | 81.4 |
| GNC-[C3N][SO3CF3]d | 84.3 | 89.1 | 92.3 | 90.7 | 78.3 |
| H3PW12O40 | 58.9 | 59.2 | 54.3 | 60.1 | 50.9 |
| Amberlyst 15 | 54.8 | 50.2 | 48.6 | 50.5 | 26.4 |
| SBA-15-SO3H | 60.9 | 56.5 | 55.4 | 52.6 | 45.7 |
More importantly, GNC-[C3N][SO3CF3] showed very good recyclability for catalyzing biodiesel production. For example, in the reaction of sunflower oil with methanol, even after being recycled five times, the yields of biodiesel catalyzed by GNC-[C3N][SO3CF3] were still up to 86.2, 90.8, 92.1, 89.4 and 79.5% (Table 2), comparable to fresh GNC-[C3N][SO3CF3] (88.5, 93.6, 94.7, 92.3 and 81.4%, Table 2).
Fig. 6 shows the dependences of catalytic activities on the time in depolymerization of Avicel catalyzed by GNC-[C3N][SO3CF3], Amberlyst 15, HCl and [C3vim][SO3CF3]. Notably, GNC-[C3N][SO3CF3] showed significantly better catalytic activity than that of Amberlyst 15, one of the most efficient commercial acidic resins, comparable to those of homogeneous HCl and [C3vim][SO3CF3] (the same number of active sites as in GNC-[C3N][SO3CF3]). For example, the yields of total reducing sugars, mono- and disaccharides catalyzed by GNC-[C3N][SO3CF3] was 94.6% after 5 h of incubation, higher than those of Amberlyst 15 (40.5%), HCl (87.1%) and [C3vim][SO3CF3] (84.9%). Similar results could also be obtained from HPLC data (as shown in Table 3); the yields of the main products including glucose (44.1%), cellobiose (32.7%) and HMF (11.6%) catalyzed by GNC-[C3N][SO3CF3] were much higher than that of Amberlyst 15 (14.3, 17.5 and 3.2%), which were comparable to those of homogeneous HCl (39.8, 20.4 and 10.7%) and [C3vim][SO3CF3] (26.3, 34.5 and 10.1%). Presumably, the drastic enhancement of the catalytic effectiveness found in GNC-[C3N][SO3CF3] during biomass transformation was attributed to the synergistic effects from the abundant nanopores, unique nanosheet structure, good dispersion, controlled acidity and good network stability of the catalyst.
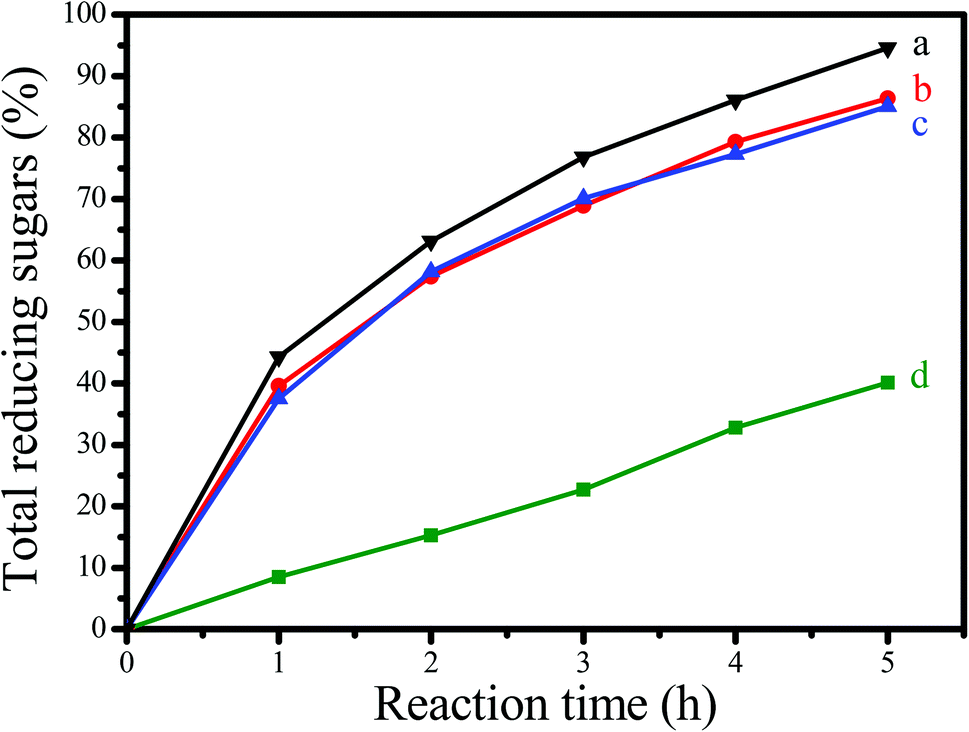 | ||
| Fig. 6 Dependences of catalytic activities on the time in depolymerization of Avicel into sugars catalyzed by (a) GNC-[C3N][SO3CF3], (b) HCl, (c) [C3vim][SO3CF3] and (d) Amberlyst 15 (b, c, d used the same number of acidic sites as those in GNC-[C3N][SO3CF3]). Reaction conditions: 0.1 g, Avicel; 1.5 g [C4mim]Cl ionic liquid, and 0.5 mL DMSO mixed solvents; 20 mg catalyst; reaction temperature, 100 °C; reaction time, 5 h. The total reducing sugars were monitored using the 3,5-dinitrosalicylic acid (DNS) reagent. | ||
| Run | Samples | Glucose yieldb (%) | Cellobiose yieldb (%) | HMF yieldb (%) | TRSc (%) |
|---|---|---|---|---|---|
| a 0.1 g Avicel; 1.5 g [C4mim]Cl ionic liquid, and 0.5 mL DMSO mixed solvents; 20 mg catalyst; reaction temperature, 100 °C; reaction time, 5 h. b Measured by the HPLC method. c Measured by the DNS method. d The same number of acidic sites as in GNC-[C3N][SO3CF3]. | |||||
| 1 | Amberlyst 15c | 14.3 | 17.5 | 3.2 | 40.5 |
| 2 | HCld | 39.8 | 20.4 | 10.7 | 87.1 |
| 3 | [C3vim][SO3CF3]d | 26.3 | 34.5 | 10.1 | 84.9 |
| 4 | GNC-[C3N][SO3CF3] | 44.1 | 32.7 | 11.6 | 94.6 |
The preparation of GNC-[C3N][SO3CF3] will open an effective method for synthesizing graphene-like nanoporous carbon based solid acids with excellent catalytic activity and a long life for biomass transformation, which will be potentially important for the wide applications of graphene based nanomaterials in the areas of biofuel exploitation in the near future.
4. Conclusion
Efficient graphene-like nanoporous carbon based solid acids (GNC-SO3H-ILs) were successfully synthesized and tested for their effectiveness in biomass transformation. GNC-SO3H-ILs showed controlled acidity, abundant nanopores, unique nanosheet structure and good dispersion in various reaction media, resulting in their excellent catalytic efficiency for biodiesel production and depolymerization of crystalline cellulose into biofuel precursors. The preparation of GNC-SO3H-ILs will offer a facile and cost-effective way to synthesize nanoporous graphene based solid strong acids, which will be very important for their wide applications in the areas of biofuel production through green and sustainable processes.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21203122, 21473244 and 21173255), the Foundation of Science, Key Sci-Tech Innovation Team Project of Zhejiang Province (2010R50014-20), Postdoctoral Foundation of China (2012M520062, Bsh1201018), the Foundation of State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences (T151303) and State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University (2013-12).References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- M. Toda, A. Takagaki, M. Okamura, J. N. Kondo, S. Hayashi, K. Domen and M. Hara, Nature, 2005, 438, 178 CrossRef CAS PubMed.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. Dumesic, Nature, 2007, 447, 982 CrossRef PubMed.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef CAS PubMed.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef PubMed.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096 CrossRef CAS PubMed.
- F. J. Liu, L. Wang, Q. Sun, L. F. Zhu, X. J. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948 CrossRef CAS PubMed.
- F. J. Liu, R. K. Kamat, I. Noshadi, D. Peck, R. S. Parnas, A. Zheng, C. Qi and Y. Lin, Chem. Commun., 2013, 49, 8456 RSC.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS PubMed.
- F. J. Liu, A. M. Zheng, I. Noshadi and F.-S. Xiao, Appl. Catal., B, 2013, 136–137, 193 CrossRef CAS PubMed.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516 CrossRef CAS PubMed.
- L. Wang, H. Wang, F. J. Liu, A. Zheng, J. Zhang, Q. Sun, J. P. Lewis, L. Zhu, X. Meng and F.-S. Xiao, ChemSusChem, 2014, 7, 402 CrossRef CAS PubMed.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047 CrossRef CAS PubMed.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS PubMed.
- I. Noshadi, B. Kanjilal, S. C. Du, G. M. Bollas, S. L. Suib, A. Provatas, F. J. Liu and R. S. Parnas, Appl. Energy, 2014, 129, 112 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS PubMed.
- F. Su and Y. H. Guo, Green Chem., 2014, 16, 2934 RSC.
- H. L. Cai, C. Z. Li, A. Q. Wang, G. L. Xu and T. Zhang, Appl. Catal., B, 2012, 123–124, 333 CrossRef CAS PubMed.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787 CrossRef CAS PubMed.
- M. Hara, T. Yoshida, A. Takagaki, T. Takata, J. N. Kondo, S. Hayashi and K. Domen, Angew. Chem., Int. Ed., 2004, 43, 2955 CrossRef CAS PubMed.
- R. Xing, Y. M. Liu, Y. Wang, L. Chen, H. H. Wu, Y. W. Jiang, M. Y. He and P. Wu, Microporous Mesoporous Mater., 2007, 105, 41 CrossRef CAS PubMed.
- Z. X. Ding, X. F. Chen, M. Antonietti and X. C. Wang, ChemSusChem, 2011, 4, 274 CAS.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- K. S. Noveselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef PubMed.
- M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher Jr. and T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067 CrossRef CAS PubMed.
- S. B. Yang, X. L. Feng, X. C. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339 CrossRef CAS PubMed.
- X.-H. Li, J.-S. Chen, X. C. Wang, M. E. Schuster, R. Schlögl and M. Antonietti, ChemSusChem, 2012, 5, 642 CrossRef CAS PubMed.
- F. J. Liu, J. Sun, L. F. Zhu, X. J. Meng, C. Z. Qi and F.-S. Xiao, J. Mater. Chem., 2012, 22, 5495 RSC.
- P. P. Upare, J.-W. Yoon, M. Y. Kim, H.-Y. Kang, D. W. Hwang, Y. K. Hwang, H. H. Kung and J.-S. Chang, Green Chem., 2013, 15, 2935 RSC.
- H. L. Wang, T. S. Deng, Y. X. Wang, X. J. Cui, Y. Q. Qi, X. D. Mu, X. L. Hou and Y. L. Zhu, Green Chem., 2013, 15, 2379 RSC.
- J.-S. Lee, S.-I. Kim, J.-C. Yoon and J.-H. Jang, ACS Nano, 2013, 7, 6047 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. S. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS PubMed.
- X.-H. Li, S. Kurasch, U. Kaiser and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 9689 CrossRef CAS PubMed.
- Z. G. Xiong, L. L. Zhang and X. S. Zhao, Chem. – Eur. J., 2011, 17, 2428 CrossRef CAS PubMed.
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- A. Zheng, S. J. Huang, S. B. Liu and F. Deng, Phys. Chem. Chem. Phys., 2011, 13, 14889 RSC.
- Y. Chu, Z. Yu, A. Zheng, H. Fang, H. Zhang, S. J. Huang, S. B. Liu and F. Deng, J. Phys. Chem. C, 2011, 115, 7660 CAS.
- M. D. Karra, K. J. Sutovich and K. T. Mueller, J. Am. Chem. Soc., 2002, 124, 902 CrossRef CAS PubMed.
- F. de Clippel, M. Dusselier, R. V. Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089 CrossRef CAS PubMed.
- T. Yokoi, H. Yoshitake, T. Yamada, Y. Kubota and T. Tatsumi, J. Mater. Chem., 2006, 16, 1125 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01052c |
| This journal is © The Royal Society of Chemistry 2015 |