
Silver tungstate: a single-component bifunctional catalyst for carboxylation of terminal alkynes with CO2 in ambient conditions†
Chun-Xiang
Guo
,
Bing
Yu
,
Jia-Ning
Xie
and
Liang-Nian
He
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin, 300071, People's Republic of China. E-mail: heln@nankai.edu.cn; Fax: (+86) 22-23503878; Tel: (+86) 22-23503878
First published on 16th September 2014
Abstract
Silver tungstate was successfully developed as a bifunctional catalyst for the ligand-free carboxylation of various terminal alkynes with electron-withdrawing or electron-donating groups under atmospheric pressure of carbon dioxide (CO2) at room temperature. In this protocol, dual activation – i.e., the terminal alkyne activated by silver, and CO2 activation by the tungstate anion – was verified using nuclear magnetic resonance spectroscopy, and means that this reaction can be run under ambient conditions. Notably, this protocol can be applied to the preparation of a phenylacrylate derivative by a cascade reaction using phenylacetylene, CO2 and benzylamine as starting materials.
1. Introduction
The design of efficient methods to conveniently form carbon–carbon bonds is exciting and challenging.1 Recently, because of the advantages of being nontoxic, renewable, abundant and economical, carbon dioxide (CO2) is considered more and more to be an ideal C1 synthon for production of value-added chemicals in both academic and pharmaceutical laboratories.2 However, compared with traditional C1 sources, (e.g., carbon monoxide and phosgene), CO2 is in its highest oxidation state and the molecule is thermodynamically stable and kinetically inert. In this respect, high-energy active reagents, low-energy targets, powerful catalysts, extra energy or high CO2 pressure are usually required for successful CO2 incorporation.3 In particular, utilization of CO2 under mild conditions will inevitably rely on its activation. For example, transition metal mediated CO2 fixation and conversion has been successfully applied to the synthesis of natural products and biologically active molecules. Although significant achievements have been accomplished, chemical conversion of CO2 at atmospheric pressure and room temperature remains a great challenge.4 In this context, bifunctional catalysts able to simultaneously activate both CO2 and the substrate are promising candidates for highly efficient CO2 chemical conversion under milder conditions.5Carboxylic acids, especially unsaturated ones such as acrylic acid, are important starting materials/intermediates in organic synthesis and are popular skeletons in a wide number of natural products, agrochemicals, or pharmaceutically relevant compounds such as Lipitor®, Blopress, Prandin®, Vancomycin, and so on.6 Traditional synthesis of carboxylic acids commonly employs strong oxidants or toxic cyanides via cumbersome operations. In this context, convenient approaches to preparing carboxylic acid derivatives with CO2 as a carboxylative reagent under mild conditions have attracted more and more attention. Indeed, a renaissance has recently been witnessed in the carboxylation of organotin,7 organozinc,8 and organoboron9 reagents with CO2. However, the benefits associated with CO2 are more than offset by unavoidably using such expensive organometallic reagents. Therefore, using the insertion of CO2 into active C–H bonds to give the corresponding carboxylic acids has been used.
Since Inoue et al. reported the first carboxylation of terminal alkynes and CO2,10 remarkable progress has been made in the field of the Ag(I)/Cu(I)-catalyzed carboxylation of terminal alkynes and CO2.2e,11 Silver salts have been widely employed as C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple-bond activators in organic synthesis,12 and several important studies about the silver-catalyzed carboxylation of terminal alkynes with CO2 have been reported. The AgI catalytic system was developed by Lu et al. with a relatively high CO2 pressure and the catalytically active species was investigated by density functional theory (DFT) studies.13 Gooßen et al. found that the AgBF4-catalyzed carboxylation reactions could be conducted under atmospheric pressure of CO2 in dimethyl sulfoxide (DMSO) at 50 °C.14 With the poly-NHC-Ag (NHC, N-heterocyclic carbene) catalytic system developed by Zhang et al., the carboxylation of terminal alkynes under ambient CO2 pressure and room temperature was achieved.15 Unfortunately, rigorous reaction conditions, such as the extra addition of ligands or high CO2 pressure, are commonly indispensable to the previously mentioned catalytic reactions.
C triple-bond activators in organic synthesis,12 and several important studies about the silver-catalyzed carboxylation of terminal alkynes with CO2 have been reported. The AgI catalytic system was developed by Lu et al. with a relatively high CO2 pressure and the catalytically active species was investigated by density functional theory (DFT) studies.13 Gooßen et al. found that the AgBF4-catalyzed carboxylation reactions could be conducted under atmospheric pressure of CO2 in dimethyl sulfoxide (DMSO) at 50 °C.14 With the poly-NHC-Ag (NHC, N-heterocyclic carbene) catalytic system developed by Zhang et al., the carboxylation of terminal alkynes under ambient CO2 pressure and room temperature was achieved.15 Unfortunately, rigorous reaction conditions, such as the extra addition of ligands or high CO2 pressure, are commonly indispensable to the previously mentioned catalytic reactions.
In this paper, we would like to report on the use of a simple bifunctional silver tungstate (Ag2WO4) catalyst for the direct carboxylation of terminal alkynes without utilization of any ligand or additive. The synergistic effect of the silver cation being able to activate the C–C triple bond and the tungstate anion able to activate the CO2 molecule allows the reaction to perform smoothly at room temperature and under the atmospheric pressure of CO2 without the extra addition of ligand, as shown in Scheme 1.
 | ||
| Scheme 1 Metal-catalyzed carboxylation of terminal alkynes with CO2. | ||
2. Results and discussion
In a prototype experiment designed to test the validity of the concept outlined previously (Scheme 1), 5 mol% of Ag2WO4 was used as a catalyst for the carboxylation of phenylacetylene 1a in the presence of n-butyl iodide and caesium carbonate (Cs2CO3), with atmospheric pressure of CO2 in dimethylformamide (DMF) at room temperature. The desired product, butyl 3-phenylpropiolate 2a was obtained at a 71% yield as a colorless oil (entry 1, Table 1). As shown in Table S1 (see ESI†), it was found that CO2, Cs2CO3 and silver(I) catalyst are prerequisites for performing the carboxylation. In addition, 2.5 mol% Ag2WO4 was found to be enough for the reaction to run smoothly (entries 1–3). Furthermore, the presence of the ligand had a negative effect on the catalyst activity. For example, the yield decreased to 46% and 66% in the presence of triphenyl phosphine (Ph3P) and 1,4-diazabicyclo[2.2.2]octane (DABCO), respectively (entries 4 and 5). A high catalyst loading and the addition of extra ligand resulted in a lower yield, which might be tentatively be ascribed to the decarboxylative activity of the Ag(I) salt.16|
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Additive (mol%) | Yieldb (%) |
| a Reaction conditions: phenylacetylene (0.0511 g, 0.5 mmol), catalyst (molar percentage to 1a), Cs2CO3 (0.1955 g, 0.6 mmol), nBuI (0.1104 g, 0.6 mmol), DMF (3 mL), CO2 (99.999%, balloon), room temperature, 12 h. b Determined by gas chromatography (GC) with biphenyl as the internal standard. | |||
| 1 | Ag2WO4 (5) | — | 71 |
| 2 | Ag2WO4 (2.5) | — | 76 |
| 3 | Ag2WO4 (1) | — | 46 |
| 4 | Ag2WO4 (2.5) | Ph3P (3) | 46 |
| 5 | Ag2WO4 (2.5) | DABCO (3) | 66 |
| 6 | AgCl (5) | — | 45 |
| 7 | AgBr (5) | — | 43 |
| 8 | AgI (5) | — | 52 |
| 9 | AgOAc (5) | — | 26 |
| 10 | Ag2CO3 (2.5) | — | 27 |
| 11 | AgOTf (5) | — | 42 |
| 12 | AgBF4 (5) | — | 22 |
| 13 | AgO2CCF3 (5) | — | 26 |
| 14 | Ag2O (2.5) | — | 4 |
| 15 | AgNO3 (5) | — | 54 |
| 16 | Na2WO4 (2.5) | — | <1 |
| 17 | AgNO3 (5) | Na2WO4 (2.5) | 72 |
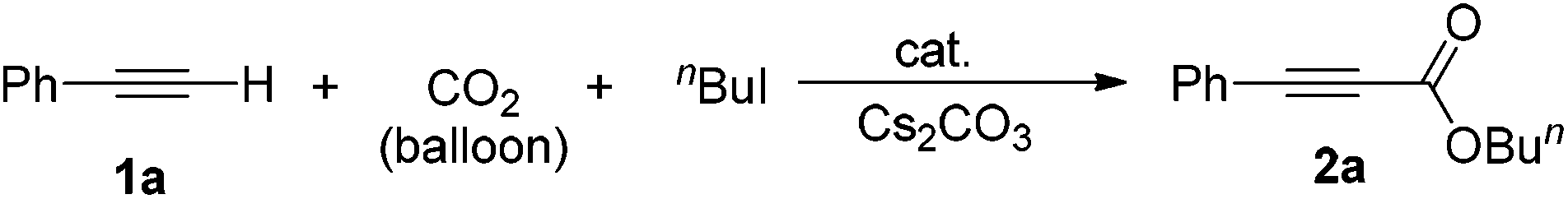
Subsequently, several silver salts were investigated. Most of the tested silver compounds exhibited poor to moderate activity without any additive at room temperature (entries 6–14). Interestingly, when Na2WO4 was added to the AgNO3-catalyzed system, the yield of 2a dramatically improved from 54% to 72%. However, Na2WO4 itself showed no catalytic activity under the reaction conditions (entries 15–17). This implied that the tungstate anion could efficiently activate CO2. As a result, Ag2WO4 was determined to be the most active silver catalyst for the carboxylation of terminal alkynes with CO2.
Furthermore, the presence of a base could strongly affect the carboxylation (entries 1–4, Table 2). Similarly to previous research,17 Cs2CO3 could play an essential role in the carboxylation reaction. Other inorganic bases, such as K2CO3, KOH, CsF, t-BuOLi, CsOAc, and NaNH2 (Table S1†), gave unsatisfactory yields under identical conditions. In addition, several organic bases were also examined. Among them (Table S1†), only 1,5,7-triazabicyclo[4.4.0]dec-1-ene (TBD) was effective (entry 4), suggesting that the reaction goes through the TBD-CO2 intermediate pathway.18
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base (eq.) | n BuI (eq.) | Solvent | Time (h) | Yieldb (%) |
| a Unless otherwise stated, the reactions were carried out with 0.5 mmol 1a in 3 mL solvent in the presence of base and nBuI, at room temperature. b GC yield with biphenyl as the internal standard. | |||||
| 1 | Cs2CO3 (1.2) | 1.2 | DMF | 12 | 76 |
| 2 | K2CO3 (1.2) | 1.2 | DMF | 12 | 5 |
| 3 | t BuOLi (1.2) | 1.2 | DMF | 12 | 2 |
| 4 | TBD (1.2) | 1.2 | DMF | 12 | 21 |
| 5 | Cs2CO3 (1.2) | 1.2 | PC | 12 | <1 |
| 6 | Cs2CO3 (1.2) | 1.2 | DMC | 12 | <1 |
| 7 | Cs2CO3 (1.2) | 1.2 | DEC | 12 | <1 |
| 8 | Cs2CO3 (1.2) | 1.2 | THF | 12 | <1 |
| 9 | Cs2CO3 (1.2) | 1.2 | 1,4-Dioxane | 12 | <1 |
| 10 | Cs2CO3 (1.2) | 1.2 | DMSO | 12 | <1 |
| 11 | Cs2CO3 (1.2) | 1.2 | MeCN | 12 | 25 |
| 12 | Cs2CO3 (1.2) | 1.2 | DMF | 12 | 81 |
| 13 | Cs2CO3 (1.2) | 1.2 | DMF | 12 | 82 |
| 14 | Cs2CO3 (1.2) | 1.5 | DMF | 12 | 76 |
| 15 | Cs2CO3 (1.2) | 2.0 | DMF | 12 | 78 |
| 16 | Cs2CO3 (1.2) | 1.2 | DMF | 18 | 91 |
| 17 | Cs2CO3 (1.2) | 1.2 | DMF | 24 | >99 |
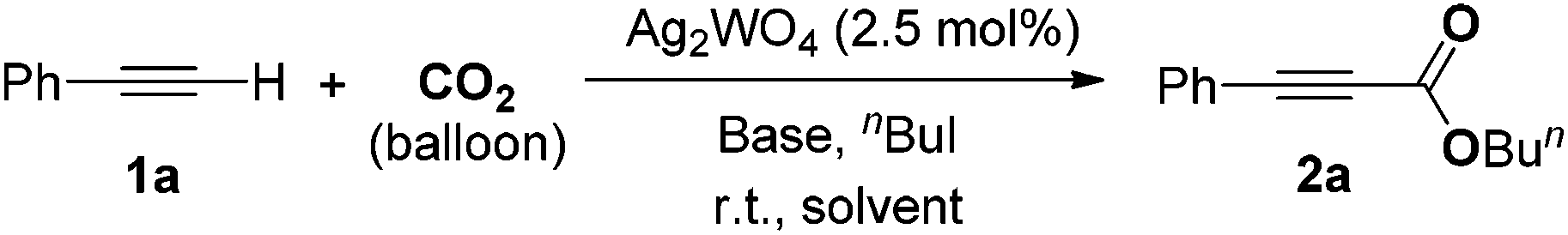
To prove the origin of the carboxylate, an isotope labeling experiment was performed. The carboxylation of 1a was conducted using labeled 13CO2, and the 13C-labeled product 2ac was obtained as shown in Fig. 1. This result clearly demonstrated that CO2 was the carboxyl source of this transformation.
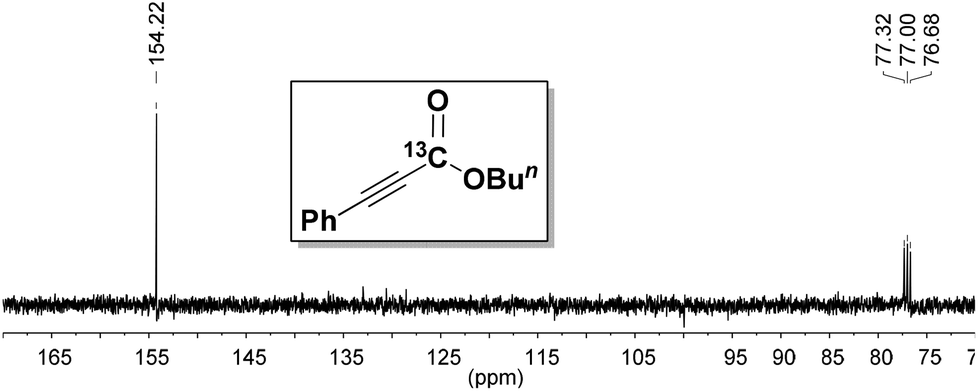 | ||
| Fig. 1 Proton-nuclear magnetic resonance (13C-NMR) spectrum of 13Ccarbonyl-labeled product 2ac. | ||
Type of solvent is another important influencing factor for this transformation. Organic carbonates – e.g., propylene carbonate (PC), dimethyl carbonate (DMC), or diethyl carbonate (DEC) – were found to be inefficient (entries 5–7, Table 2).17 Other conventional solvents did not work at all (entries 8–10) or gave poor yield (entry 11) compared with DMF (entry 1). Therefore, DMF was chosen as the reaction medium for further investigations. On the other hand, an excess amount of Cs2CO3 and n-BuI hardly affected the reaction (entries 12–15). Further prolonging the reaction time to 24 h gave 2a in quantitative yield (entries 1, 16, 17).
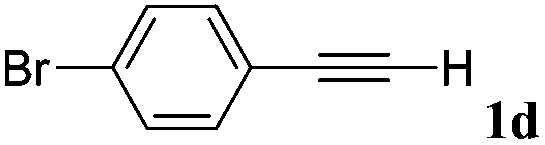
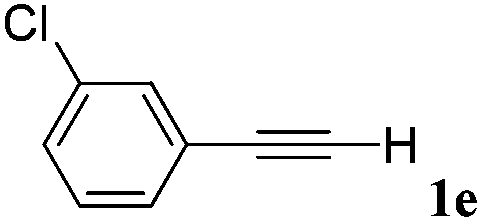
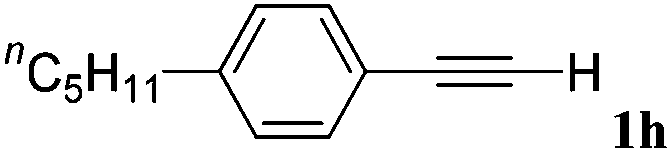
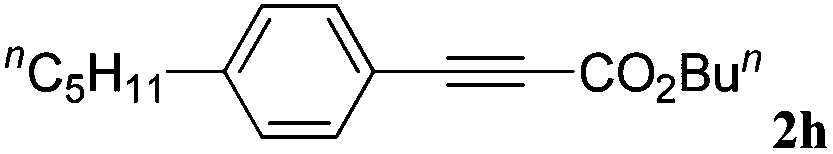
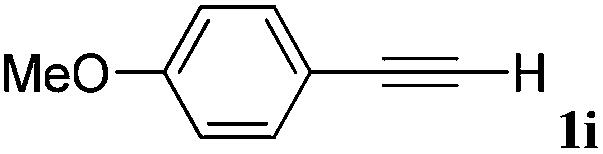
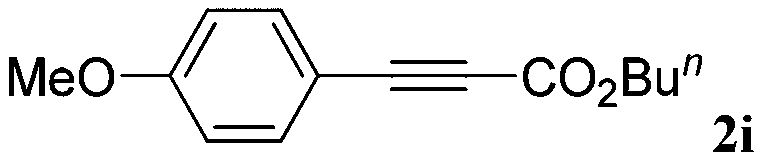
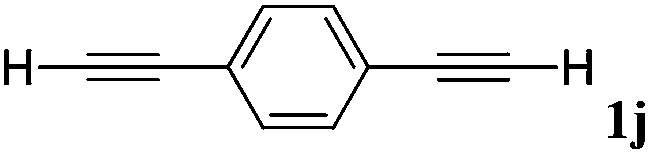
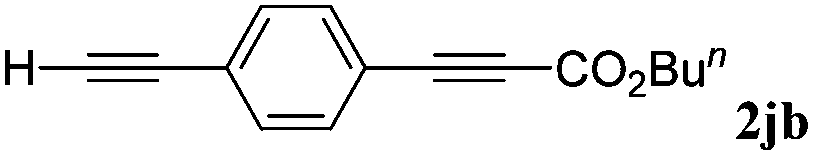
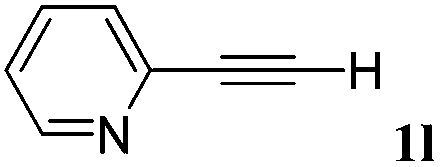
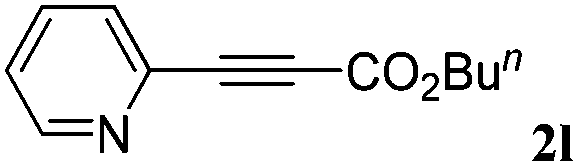
With the optimized reaction conditions to hand, we evaluated the scope of the substrate in this reaction. All the products were characterized by conventional spectroscopic methods and by comparison with the data available in the literature.17,19 The reaction conditions were broadly adapted to a series of terminal alkynes 1a–1n and the corresponding alkyl 2-alkynoates 2a–2n were obtained in good to excellent yields, as summarized in Table 3. Both electron-donating and electron-withdrawing group substituted phenylacetylenes were successfully carboxylated with this method (entries 2–9). For 1,4-diethynylbenzene, biscarboxylative product 2ja was formed as the major product with 64% yield and 24% yield of the monocarboxylative product 2jb, which was also detected (entry 10). Aliphatic alkynes (entry 11) and heteroaromatic acetylenes (entries 12–15) were also found to be suitable substrates. In agreement with the DFT calculation, 3-ethynylpyridine showed lower activity when compared with 2-ethynylpyridine (entry 12 vs. 13), indicating that the acidity of the alkyne proton strongly impacts on the activity.20
|
|
||||
|---|---|---|---|---|
| Entry | RCCH |
R1X | Product | Yieldb (%) |
| a Reaction conditions: Substrate (1.0 mmol), Ag2WO4 (0.0116 g, 0.025 mmol), Cs2CO3 (0.3910 g, 1.2 mmol; 2.4 mmol for entry 10), alkyl halide (1.2 mmol; 2.4 mmol for entry 10), DMF (3 mL; 5 mL for entry 10), CO2 (99.999%, balloon), 25 °C, 24 h. b Isolated yield. | ||||
| 1 |
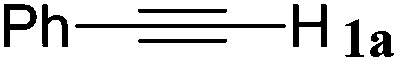
|
n BuI |
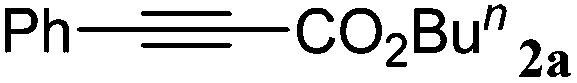
|
96 |
| 2 |
|
n BuI |
|
80 |
| 3 |
|
n BuI |
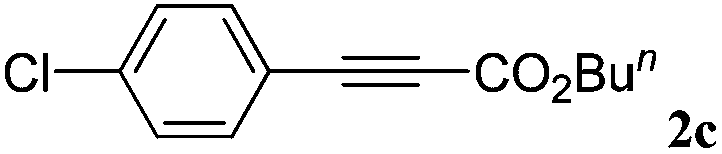
|
82 |
| 4 |
|
n BuI |
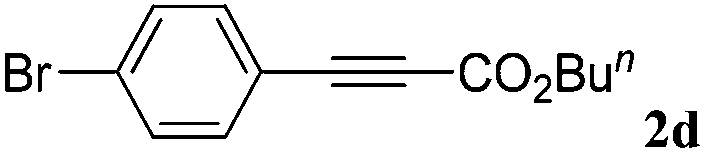
|
81 |
| 5 |
|
n BuI |
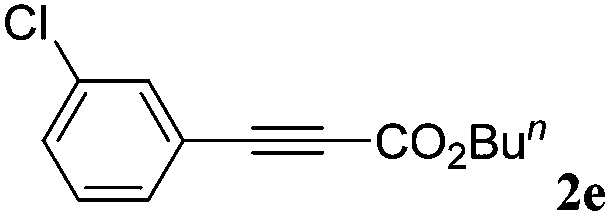
|
84 |
| 6 |
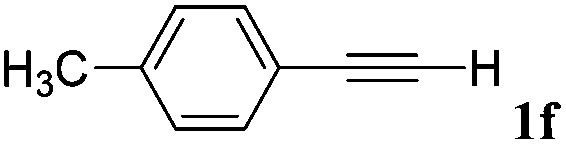
|
n BuI |
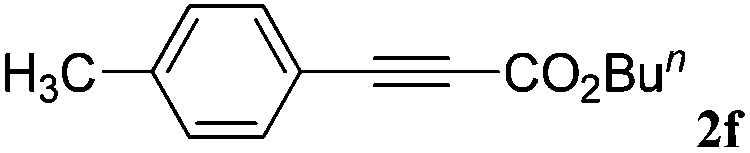
|
80 |
| 7 |
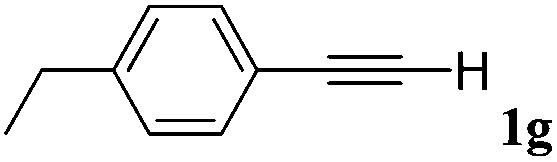
|
n BuI |
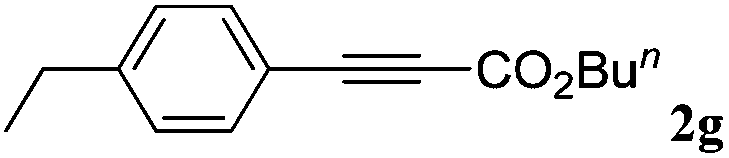
|
75 |
| 8 |
|
n BuI |
|
68 |
| 9 |
|
n BuI |
|
91 |
| 10 |
|
n BuI |

|
64 |
|
24 | |||
| 11 |
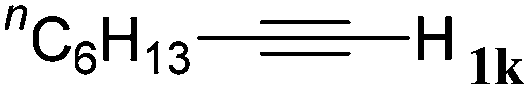
|
n BuI |
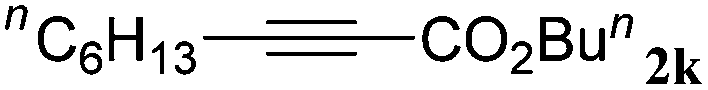
|
60 |
| 12 |
|
n BuI |
|
84 |
| 13 |
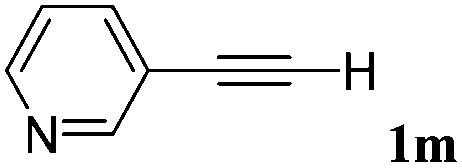
|
n BuI |
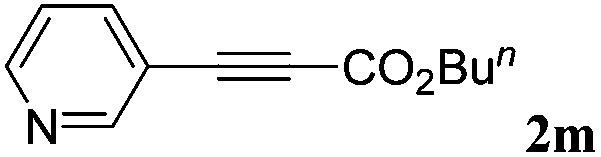
|
70 |
| 14 |
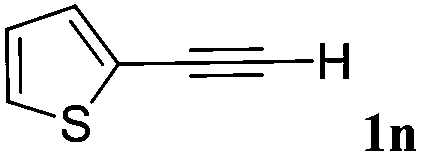
|
n BuI |

|
90 |
| 15 |
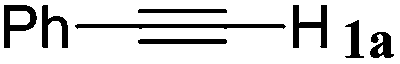
|
n BuBr |
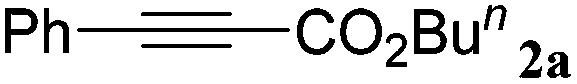
|
92 |
| 16 |
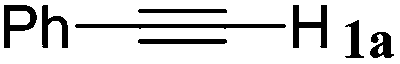
|
EtBr |
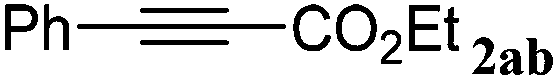
|
92 |
| 17 |
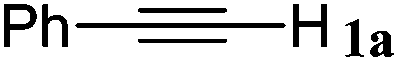
|
n BuCl |
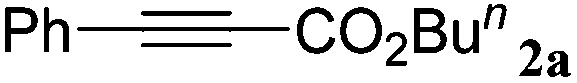
|
59 |
The reactivity of the alkylating reagent was also examined. Bromoalkanes such as n-BuBr and EtBr gave 2a in excellent yields (entries 15 and 16). Even n-BuCl with poor activity21 also worked well in this protocol to give 2a with a 59% yield (entry 17).
To explore the reaction mechanism, the interactions of WO42− with CO2 were examined by NMR spectroscopy. In the 13C-NMR spectrum, a new signal appeared at δ = 157.8 ppm when CO2 was absorbed by a suspension of deuterated DMSO (DMSO-d6) and Ag2WO4 (Fig. 2), indicating that the interaction of the tungstate anion with CO2 {Ag+2[WO4]2−-CO2} leads to the activation of the CO2 molecule.
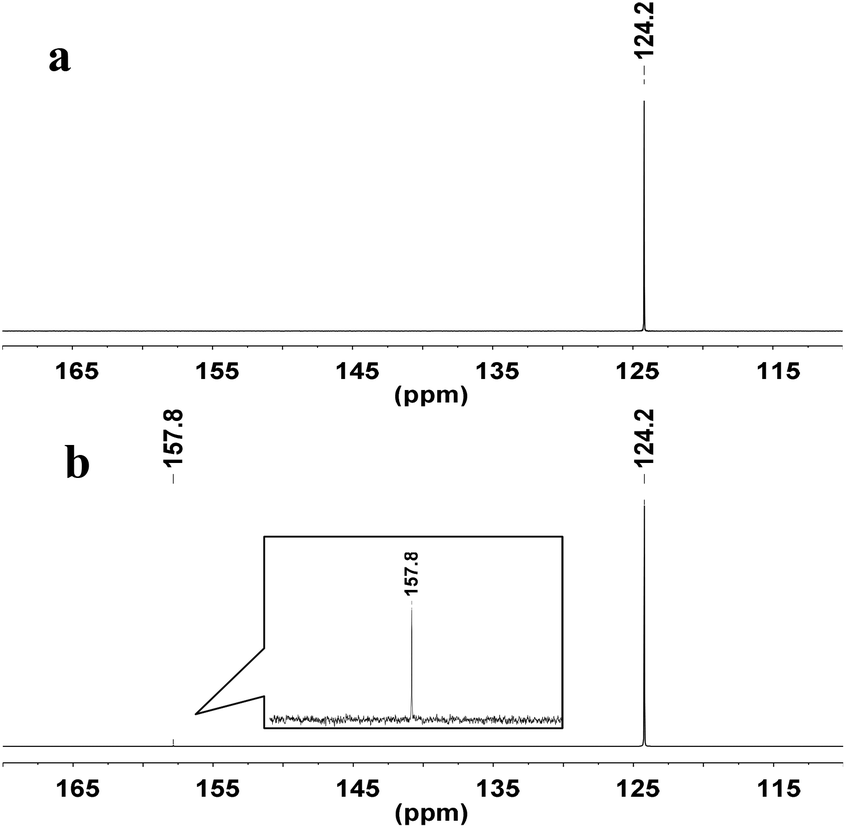 | ||
| Fig. 2 13C-NMR study: (a) DMSO-d6 under atmospheric CO2 (1 atm, 25 °C); (b) Ag2WO4 in DMSO-d6 under atmospheric CO2 (1 atm, 25 °C). | ||
A catalytic cycle for the single-component bifunctional carboxylation catalytic system is proposed, as delineated in Scheme 2. On the basis of the coordination of silver with a C–C triple bond, phenylacetylene 1a is converted to silver acetylide II with the aid of a base, and CO2 is simultaneously activated by tungstate to form WO42−-CO2 adduct III. Subsequently, insertion of III into the sp-hybridized Ag–carbon bond forms the silver phenylpropiolate IV with release of WO42−. Finally, IV is esterified in situ by using n-BuI to produce the ester 2a.
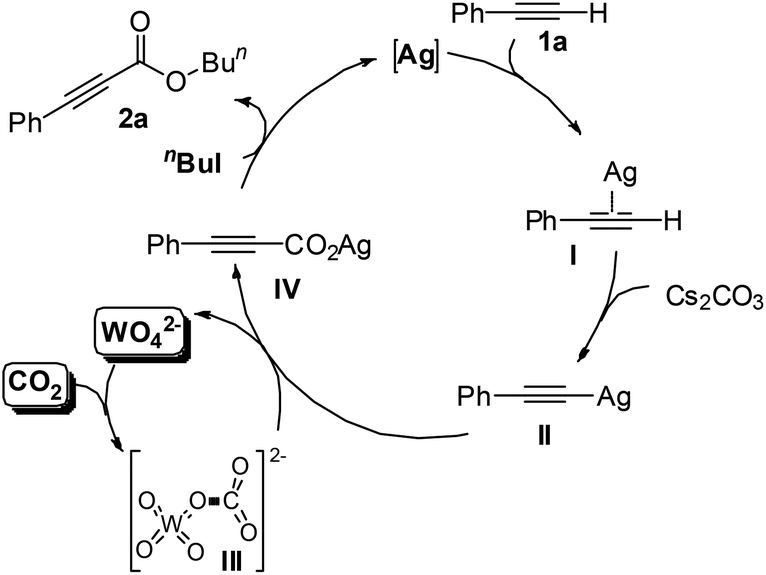 | ||
| Scheme 2 The dual activation pathway for the present Ag2WO4-catalyzed carboxylation of terminal alkynes with CO2. | ||
The effectiveness of the Ag2WO4-catalyzed carboxylation of terminal alkynes and CO2 provides a convenient approach for the synthesis of carboxylic acid derivatives and various useful chemicals under the very mild reaction conditions. For example, β-enaminoesters, widely used in organic syntheses, were traditionally prepared from β-ketoesters.22 Here, we have successfully prepared such a β-enaminoester through the carboxylation and hydroamination sequence from a terminal alkyne in one pot as demonstrated in Scheme 3. The Ag(I)-catalyzed carboxylation of 1a with CO2 gave 2-alkynoate 2ab, which further reacted with benzylamine, resulting in the formation of ethyl (Z)-3-(benzylamino)-3-phenylacrylate 3a in 80% isolated yield. The configuration of the enamino double bond was identified by NMR spectroscopy and compared with those previously reported.23
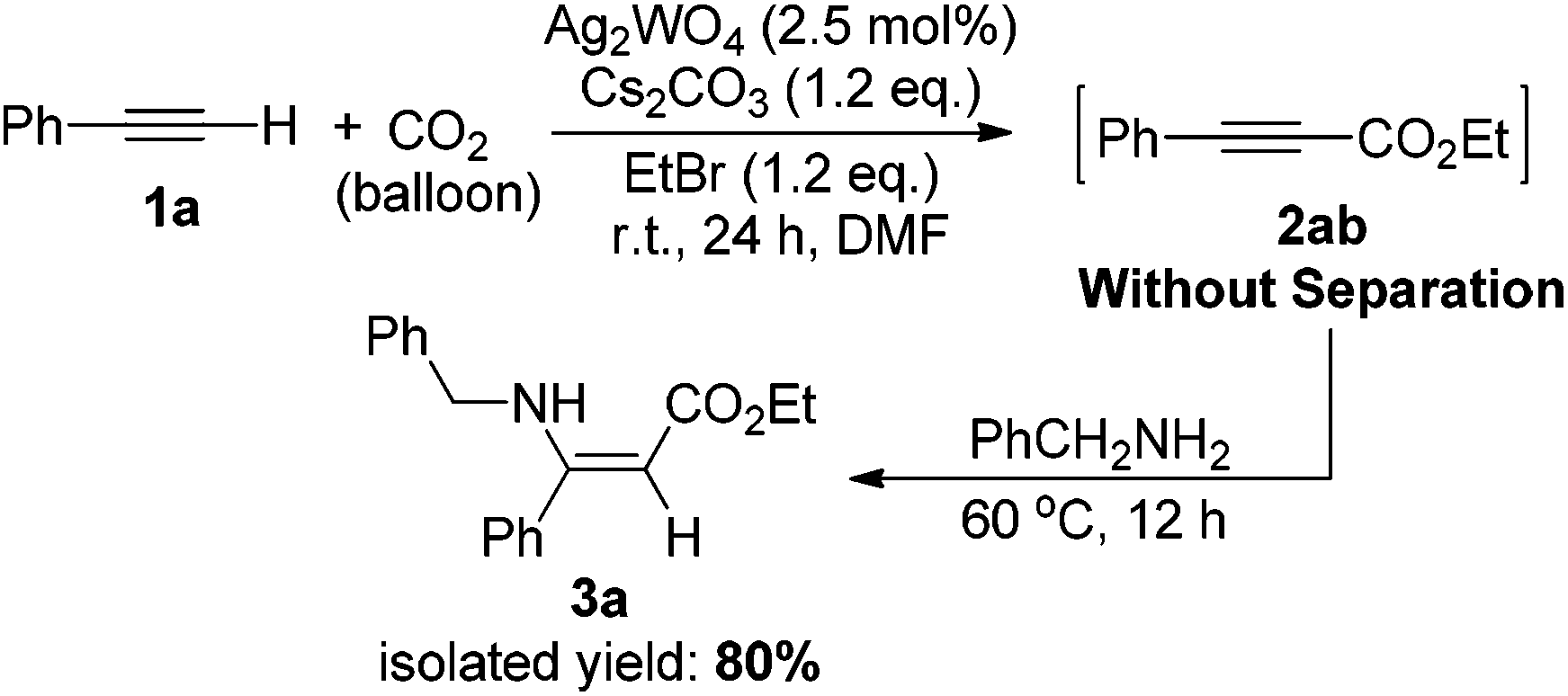 | ||
| Scheme 3 One-pot synthesis of ethyl (Z)-3-(benzylamino)-3-phenylacrylate from phenylacetylene, benzylamine and CO2. | ||
3. Conclusions
In summary, we have developed a convenient ligand-free Ag(I)-based bifunctional catalytic system, i.e., silver tungstate, which can simultaneously activate both CO2 and a C–C triple bond for the direct carboxylation of terminal alkynes with atmospheric CO2 at room temperature, as confirmed using 13CO2. This protocol also features a halogen-free catalyst and a simple process with low energy consumption and avoidance of complicated preparation of the catalyst. The origin of the carboxylate was corroborated by the isotope labeling experiment. Dual activation of the alkyne substrate by silver(I) and CO2 by tungstate could account for such carboxylation working under extremely mild conditions. Furthermore, the utility of this catalytic system was demonstrated by the convenient synthesis of β-enaminoester in a one-pot process.Such findings in this study encourage us to further develop cost-effective, green and efficient bifunctional catalysts for the chemical fixation of atmospheric CO2 under mild reaction conditions.
4. Experimental details
The general procedure for the carboxylation of terminal alkynes is described here.The terminal alkyne (1.0 mmol), Ag2WO4 (0.0116 g, 0.025 mmol), Cs2CO3 (0.3910 g, 1.2 mmol), nBuI (0.2208 g, 1.2 mmol) and DMF (3 mL) were added in a 10 mL Schlenk flask. The flask was capped and sealed. Then a gas-exchanging process was conducted using the “freeze–pump–thaw” method. The reaction mixture was stirred at 25 °C for 24 h under an atmosphere of CO2 (99.999%, balloon). After the reaction, water was added to the mixture, which was extracted with acetic ether five times. The combined organic layer was washed with saturated aqueous NaCl and then dried over MgSO4. Then the solvent was removed under reduced pressure to get a crude product and the residue was purified by column chromatography (silica gel, petroleum ether–EtOAc) to give the desired product. The products were further identified by NMR and mass spectroscopy (see the ESI†), which are consistent with those reported in the literature and were in good agreement with the assigned structures. 2a. Colorless oil; 1H-NMR (400 MHz, deuterated chloroform (CDCl3)) δ (ppm): 7.58–7.57 (d, J = 4.0 Hz, 2H), 7.45–7.34 (m, 3H), 4.23 (t, J = 6.7 Hz, 2H), 1.73–1.66 (m, 2H), 1.48–1.38 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ (ppm): 154.1, 132.9, 130.5, 128.5, 119.6, 86.0, 80.7, 65.9, 30.4, 19.0, 13.6; EI-MS, m/z (%): 129.15 (100), 201.20 (11.72) [M+].
Acknowledgements
We are grateful to the National Natural Sciences Foundation of China (no. 21172125, 21121002, 21472103), Specialized Research Fund for the Doctoral Program of Higher Education (20130031110013), MOE Innovation Team (IRT13022) of China for financial support.Notes and references
- W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC.
- Chosen reviews for C–C bond formation with CO2 as C1-synthon see: (a) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (b) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed; (c) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC; (d) B. Yu, Z.-F. Diao, C.-X. Guo and L.-N. He, J. CO2 Utilization, 2013, 1, 60–68 CrossRef CAS; (e) F. Manjolinho, M. Arndt, K. Gooßen and L. J. Gooßen, ACS Catal., 2012, 2, 2014–2021 CrossRef CAS; (f) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395–3403 RSC; (g) A. Ueno, Y. Kayaki and T. Ikariya, Organometallics, 2014, 33, 4479–4485 CrossRef CAS.
- K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC.
- Reviews and books for the chemical transformation of CO2: (a) Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 RSC; (b) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC; (c) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (d) L.-N. He, Carbon Dioxide Chemistry, Science Press, Beijing, 2013 Search PubMed; (e) W. J. Yoo, T. V. Nguyen and S. Kobayashi, Angew. Chem., Int. Ed., 2014, 53, 10213–10217 CrossRef CAS PubMed.
- Ultilization of bifounctional catalysts for CO2 fixation and conversion: (a) T. Kimura, K. Kamata and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 6700–6703 CrossRef CAS PubMed; (b) Q.-W. Song, B. Yu, X.-D. Li, R. Ma, Z.-F. Diao, R.-G. Li, W. Li and L.-N. He, Green Chem., 2014, 16, 1633–1638 RSC; (c) Y. Zhao, B. Yu, Z. Yang, H. Zhang, L. Hao, X. Gao and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 5922–5925 CrossRef CAS PubMed.
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef PubMed.
- (a) M. Shi and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 5057–5058 CrossRef CAS; (b) R. Johansson and O. F. Wendt, Dalton Trans., 2007, 488–492 RSC.
- (a) H. Ochiai, M. Jang, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2681–2683 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826–7827 CrossRef CAS PubMed; (c) K. Kobayashi and Y. Kondo, Org. Lett., 2009, 11, 2035–2037 CrossRef CAS PubMed.
- Carboxylation of organoboron regents: (a) X. Zhang, W.-Z. Zhang, L.-L. Shi, C.-X. Guo, L.-L. Zhang and X.-B. Lu, Chem. Commun., 2012, 48, 6292–6294 RSC; (b) K. Ukai, M. Aoki, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706–8707 CrossRef CAS PubMed; (c) J. Takaya, S. Tadami, K. Ukai and N. Iwasawa, Org. Lett., 2008, 10, 2697–2700 CrossRef CAS PubMed; (d) T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792–5795 CrossRef CAS PubMed; (e) Y. Makida, E. Marelli, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2014, 50, 8010–8013 RSC.
- Y. Fukue, S. Oi and Y. Inoue, J. Chem. Soc., Chem. Commun., 1994, 2091–2091 RSC.
- (a) S. H. Kim, K. H. Kim and S. H. Hong, Angew. Chem., Int. Ed., 2014, 53, 771–774 CrossRef CAS PubMed.
- Samples for using silver to active CC bond:
(a) C. He, S. Guo, J. Ke, J. Hao, H. Xu, H. Chen and A. Lei, J. Am. Chem. Soc., 2012, 134, 5766–5769 CrossRef CAS PubMed;
(b) F. Li, J. Nie, L. Sun, Y. Zheng and J. A. Ma, Angew. Chem., Int. Ed., 2013, 52, 6255–6258 CrossRef CAS PubMed;
(c) U. Halbes-Letinois, J. M. Weibel and P. Pale, Chem. Soc. Rev., 2007, 36, 759–769 RSC;
(d) J. Liu, Z. Fang, Q. Zhang, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2013, 52, 6953–6957 CrossRef CAS PubMed;
(e) M. Gao, C. He, H. Chen, R. Bai, B. Cheng and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 6958–6961 CrossRef CAS PubMed;
(f) X. Yao and C.-J. Li, Org. Lett., 2005, 7, 4395–4398 CrossRef CAS PubMed.
- For AgI-catalyzed carboxylation of terminal alkynes: (a) X. Zhang, W.-Z. Zhang, L.-L. Shi, C. Zhu, J.-L. Jiang and X.-B. Lu, Tetrahedron, 2012, 68, 9085–9089 CrossRef CAS; (b) X. Zhang, W.-Z. Zhang, X. Ren, L.-L. Zhang and X.-B. Lu, Org. Lett., 2011, 13, 2402–2405 CrossRef CAS PubMed; (c) C. Liu, Y. Luo, W. Zhang, J. Qu and X. Lu, Organometallics, 2014, 33, 2984–2989 CrossRef CAS.
- M. Arndt, E. Risto, T. Krause and L. J. Gooßen, ChemCatChem, 2012, 4, 484–487 CrossRef CAS.
- D. Yu, M. X. Tan and Y. Zhang, Adv. Synth. Catal., 2012, 354, 969–974 CrossRef CAS.
- (a) L. J. Gooßen, N. Rodríguez, C. Linder, P. P. Lange and A. Fromm, ChemCatChem, 2010, 2, 430–442 CrossRef; (b) C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132, 14006–14008 CrossRef CAS PubMed.
- B. Yu, Z.-F. Diao, C.-X. Guo, C.-L. Zhong, L.-N. He, Y.-N. Zhao, Q.-W. Song, A.-H. Liu and J.-Q. Wang, Green Chem., 2013, 15, 2401–2407 RSC.
- X. Wang, Y. N. Lim, C. Lee, H.-Y. Jang and B. Y. Lee, Eur. J. Org. Chem., 2013, 1867–1871 CrossRef CAS.
- K. Inamoto, N. Asano, K. Kobayashi, M. Yonemoto and Y. Kondo, Org. Biomol. Chem., 2012, 10, 1514–1516 CAS.
- The density functional theory calculation [B3LYP/6-31+G(d) level] indicated that the negative charge at terminal carbon of 2-ethynylpyridine is −0.405 and that of 3-ethynylpyridine was −0.312.
- K. Inamoto, N. Asano, K. Kobayashi, M. Yonemoto and Y. Kondo, Org. Biomol. Chem., 2012, 10, 1514–1516 CAS.
- For some applications of β-enaminoesters: (a) R. A. Bunce, B. Nammalwar, K. K. Gnanasekaran and N. R. Cain, Tetrahedron, 2014, 70, 838–844 CrossRef CAS; (b) M. Inman and C. J. Moody, J. Org. Chem., 2010, 75, 6023–6026 CrossRef CAS PubMed.
- H. Cao, X. Wang, H. Jiang, Q. Zhu, M. Zhang and H. Liu, Chem. – Eur. J., 2008, 14, 11623–11633 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization products and copies of the NMR spectra. See DOI: 10.1039/c4gc01638f |
| This journal is © The Royal Society of Chemistry 2015 |