
(−)-Epicatechin prevents alterations in the metabolism of superoxide anion and nitric oxide in the hearts of L-NAME-treated rats
Barbara
Piotrkowski†
,
Valeria
Calabró†
,
Mónica
Galleano
and
César G.
Fraga
Physical Chemistry and Institute of Molecular Biochemistry and Medicine, University of Buenos Aires (IBIMOL) – CONICET, School of Pharmacy and Biochemistry, University of Buenos Aires, Argentina. E-mail: barbara@ffyb.uba.ar; Fax: +(54-11)4508-3646, ext. 102; Tel: +(54-11)4964-8245, ext. 105
First published on 31st October 2014
Abstract
The aim of this work was to evaluate the effects of (−)-epicatechin administration in the heart of a rat model with reduced NO production that follows a short-term treatment with L-NAME. Sprague-Dawley rats were treated for 4 d with L-NAME in the absence or presence of (−)-epicatechin in the diet. The redox status in cardiac tissue was improved by (−)-epicatechin administration. L-NAME treatment induced a decrease in NO synthase activity (−62%, p < 0.05) and an increase in NADPH-dependent superoxide anion production (+300%, p < 0.05) that were totally prevented by (−)-epicatechin administration. These effects of (−)-epicatechin were associated with a higher endothelial NO synthase phosphorylation at an activation site and a reduced expression of the regulatory subunit, p47phox, suggesting the involvement of posttranslational mechanisms in (−)-epicatechin action. Thus, the (−)-epicatechin treatment would restore NO steady state levels in vivo through effects on both, its synthesis and degradation via the reaction with superoxide anion. The fact that (−)-epicatechin is commonly present in human diet makes this compound a reasonable explanation for the positive cardiovascular effects of a high consumption of fruits and vegetables.
Introduction
Epidemiological data show positive associations between high consumption of fruits and vegetables and improvements in health parameters.1–3 Numerous studies reveal that hypertension and cardiovascular disease incidence is diminished in people who regularly consume diets rich in fruits and vegetables, especially the ones rich in certain flavanols, e.g. cocoa, tea, and grapes.4–6 Thus, changes in blood pressure (BP) and vascular function were observed, associated with consumption of cocoa or chocolate,7,8 wine9,10 and tea,11,12 among others.These epidemiological and clinical studies support the idea that the benefits may be causally related to the presence of certain polyphenols, mainly flavonoids, in edible plants.13,14 (−)-Epicatechin (EC) is a flavonoid highly represented in human diet as it is present in many foods rich in this type of polyphenol.15 Studies in humans and laboratory animals showed that EC administered pure or as part of foods like chocolate or wine modifies the vascular function, resulting in an antihypertensive effect.16–20 Moreover, EC administered to long-term Nω-nitro-L-arginine methyl ester (L-NAME)-treated or deoxycorticosterone acetate (DOCA)-salt hypertensive rats prevented cardiac hypertrophy associated with increased BP,21 and the development of hypertension,22 respectively.
Nitric oxide (NO) plays an important role in modulating tissue function, including vascular and heart function.23 The NO steady state level depends both on its synthesis and degradation. The superoxide anion, mostly produced in cardiovascular tissue by NADPH oxidases (NOX),24,25 reacts with NO, modulating its bioavailability. NOX activity may undergo regulation by angiotensin II, thrombin, TNF-α, among many other agents and conditions related to the modulation of cardiovascular function.26–29 In endothelial cells in culture, it was reported that EC may cause an increase in NO bioavailability, either as a result of an increase in NO generation30 or a decrease in superoxide anion production.31 Thus, it is thought that the EC effects on BP might be associated with the modulation of vascular levels of NO through modifications in superoxide anion production.
So the aim of this work was to evaluate the effects of EC administration in the heart of a rat model of reduced NO production that follows a short-term treatment with L-NAME.
Materials and methods
Materials and chemicals
(−)-Epicatechin, Nω-nitro-L-arginine methyl ester (L-NAME), superoxide dismutase (SOD), glutathione reductase, N,N′-dimethyl-9,9′-biacridinium dinitrate (lucigenin) diphenyleneiodonium (DPI), tempol, Nω-nitro-L-arginine (LNNA), nicotinamide adenine dinucleotide (NADH), nicotinamide adenine dinucleotide phosphate (NADPH), ethylenediaminetetraacetic acid (EDTA), bovine serum albumin (BSA), and epinephrine were purchased from Sigma-Aldrich (St. Louis, MO). eNOS, iNOS, p47phox, and actin antibodies were acquired from Santa Cruz Biotechnology (Dallas, TX) and p-eNOS from Cell Signaling (Danvers, MA). All other reagents were of the highest purity commercially available.Animals and treatments
Male Sprague-Dawley rats (150 g) were randomly divided into 3 experimental groups (8 rats per group) that during 4 d received: (i) tap water and a basal diet (control); (ii) 360 mg l−1L-NAME in tap water and a basal diet (LN); and (iii) 360 mg l−1L-NAME in tap water and a basal diet added with 4 g kg−1 EC (LN-EC). L-NAME was administered at a dose within the range generally used,32,33 but the length of the treatment was shorter than the intervals normally used (4 d vs. 4–12 weeks) in order to evaluate the modifications produced during the early stages of hypertension development. The dose of EC was selected based on previous studies from our laboratory that were confirmatory of a BP-lowering effect of EC.34,35 Nevertheless it can be considered to be high as compared to a median dietary human consumption; the intention is to study potential effects under acute conditions.The rats had free access to beverages and food and were kept under 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light–dark cycles. Body weight and systolic BP, evaluated by tail plethysmography (Polygraph Grass Instrumental, Quincy, MA), were determined at the beginning and at the end of the study. At day 4, the rats were euthanized with CO2, blood was collected by cardiac puncture, and the heart was excised for further determinations. A portion of the left ventricle was used for NOS activity and glutathione determinations, while the other biochemical analyses were performed in the rest of the heart. All the procedures were carried out according to protocols for animal use approved by the School of Pharmacy (UBA) Animal Ethics Committee.
:12 h light–dark cycles. Body weight and systolic BP, evaluated by tail plethysmography (Polygraph Grass Instrumental, Quincy, MA), were determined at the beginning and at the end of the study. At day 4, the rats were euthanized with CO2, blood was collected by cardiac puncture, and the heart was excised for further determinations. A portion of the left ventricle was used for NOS activity and glutathione determinations, while the other biochemical analyses were performed in the rest of the heart. All the procedures were carried out according to protocols for animal use approved by the School of Pharmacy (UBA) Animal Ethics Committee.
Heart tissue preparations
The tissue was minced and homogenized at 4 °C in phosphate buffer, pH 7.4. After centrifugation of these homogenates at 600g for 10 min, the pellets, containing nuclei and cellular debris, were discarded. A fraction of the supernatant was separated to be used for most of the determinations. For superoxide anion production, the remaining supernatant was further centrifuged at 10000g for 10 min to obtain a fraction free of mitochondria and peroxisomes. The protein content was assayed according to Lowry, using BSA as the standard.
Matrix metalloproteases-9 activity (MMP-9)
MMP-9 activity was evaluated by zymography according to Shimokawa et al. (2002).36Thiobarbituric acid reactive substances
The malondialdehyde (MDA) content in the heart was evaluated as thiobarbituric acid reactive substances (TBARS) with the use of a fluorescence method with modifications.37 Butylhydroxytoluene (4% w/v in ethanol) was used to prevent non-physiological TBARS formation during the sample processing. Results are expressed as pmoles of MDA per mg of protein. The MDA standard was prepared from 1,1,3,3,-tetramethoxypropane.Reduced and oxidized glutathione
A portion of approximately 100 mg of heart tissue was added with 1.0 M HClO4 containing 2 mM EDTA. After 20 min of centrifugation at 12000g, supernatants were filtered through 0.22 μm membranes, diluted to 1:10 (v/v) with the mobile phase, and subjected to HPLC separation using an LC-18-DB column (25 cm × 4.6 mm, 5 μm particle size). Reduced (GSH) and oxidized (GSSG) glutathione were eluted with 20 mM sodium phosphate (pH 2.7) and detected electrochemically at 0.800 V.38
Nitric oxide synthase (NOS) activity
The NOS activity in the heart was measured using [14C]-L-arginine as the substrate in tissue slices prepared as reported.39 Specific NOS activity was assessed as the formation of [14C]-L-citrulline in the presence of 10−4 M L-NAME. Values were expressed as pmoles of [14C]-L-citrulline min−1 per g wet weight.Superoxide anion production
Superoxide anion production was determined in homogenates free of mitochondria by following the chemiluminescence emission of the samples added with 25 μM lucigenin.24 The reaction was monitored for 7 min and controls with SOD, DPI, tempol, and LNNA were performed to confirm superoxide anion production and that NOX was the main source producing this species. Superoxide anion production was calculated as the SOD-inhibitable fraction of lucigenin. Results are expressed as μmoles of superoxide min−1 per mg of protein.Western blot
Proteins were prepared as reported40 and were separated on SDS polyacrylamide gels and then transferred to PVDF membranes by liquid electroblotting (MiniProtean 3, Bio-Rad) in a transfer solution containing 25 mM Tris, 190 mM glycine, and 20% (v/v) methanol, pH 8.3, at 110 V for 90 min. After transfer, membranes were blocked by incubating with phosphate buffered saline (PBS) containing 5% (w/v) dry low-fat milk for 2 h at room temperature. Antibodies were incubated overnight at 4 °C. Peroxidase-conjugated secondary antibodies were incubated for 90 min at room temperature. To reveal the bound secondary antibody, a chemiluminescence-enhanced kit and autoradiography were used. The intensity of immunospecific bands was quantified using ImageJ software (NIH, Bethesda, MD). To normalize samples for protein loading, antibodies against β-actin were used.Glutathione peroxidase (GPx) activity
GPx activity was determined in the presence of glutathione reductase by following the oxidation of NADPH at 340 nm. Results are expressed as nmoles of NADPH min−1 per mg of protein.41Superoxide dismutase (SOD) activity
SOD activity was determined through the inhibition of epinephrine autooxidation at 480 nm.42 One unit of SOD was defined as the amount of enzyme necessary to cause a 50% inhibition of epinephrine autooxidation. Results are expressed as units of SOD per mg of protein.42Catalase activity
Catalase activity was evaluated by following the reduction of H2O2 at 240 nm and calculating the pseudo-first order constant of the reaction.43 The catalase content was expressed as pmoles of catalase per mg of protein.Statistical analysis
Values in the text, table, and figures are expressed as means ± SEM. Statistical analyses were performed by one-way ANOVA using Graphpad Prism 4 software (GraphPad Software Inc., San Diego, California, USA) to establish the significance of between-group differences. p values <0.05 were considered significant.Results
Table 1 shows the body and heart weight, food and beverage intake, systolic BP, and heart matrix metalloprotease activity of rats from control, LN, and LN-EC groups. At the end of the 4 d study period, body and heart weight, heart/body weight, and food and beverages showed no significant differences among the groups. At day 4, systolic BP in LN was significantly higher than in control. Co-administered EC prevented such an increase (p > 0.05 vs. LN; Table 1), in accordance with a previous observation in which EC exerted antihypertensive effects in rats with previously established hypertension.44| Control | LN | LN-EC | |
|---|---|---|---|
| a p < 0.05 vs. control and LN-EC. | |||
| Initial body weight (g) | 171 ± 13 | 175 ± 10 | 180 ± 13 |
| Final body weight (g) | 202 ± 11 | 209 ± 9 | 204 ± 13 |
| Heart weight (g) | 0.96 ± 0.07 | 0.95 ± 0.06 | 0.84 ± 0.06 |
| Food intake (mg per rat per d) | 22.0 ± 0.5 | 22.0 ± 0.9 | 21.9 ± 0.7 |
| Beverage intake (ml per rat per d) | 29 ± 5 | 26 ± 3 | 30 ± 2 |
| Systolic blood pressure (mmHg) | 127 ± 5 | 159 ± 7* | 140 ± 5 |
| Heart/total weight ratio (10−3) | 4.7 ± 0.2 | 4.5 ± 0.2 | 4.1 ± 0.1 |
| Matrix metalloprotease activity (A.U.) | 1.07 ± 0.06 | 1.30 ± 0.09 | 1.20 ± 0.03 |
Matrix metalloprotease-9 (MMP-9) activity was determined to be an indicator of cardiac hypertrophy and remodeling. There were no significant differences among the studied groups (Table 1), so even though animals were hypertensive it can be speculated that the study period was too short to find any evidence of hypertrophy in the heart, as is also indicated by the absence of heart weight changes.
To evaluate the redox status in cardiac tissue, we evaluated the TBARS content and glutathione oxidation as the ratio of GSSG/GSH2. The TBARS content (1.9 ± 0.3 pmoles MDA per mg of protein) showed no significant differences among the three groups (Fig. 1A). The GSSG/GSH2 ratio was 37% lower in the LN-EC group compared to the LN group (Fig. 1B).
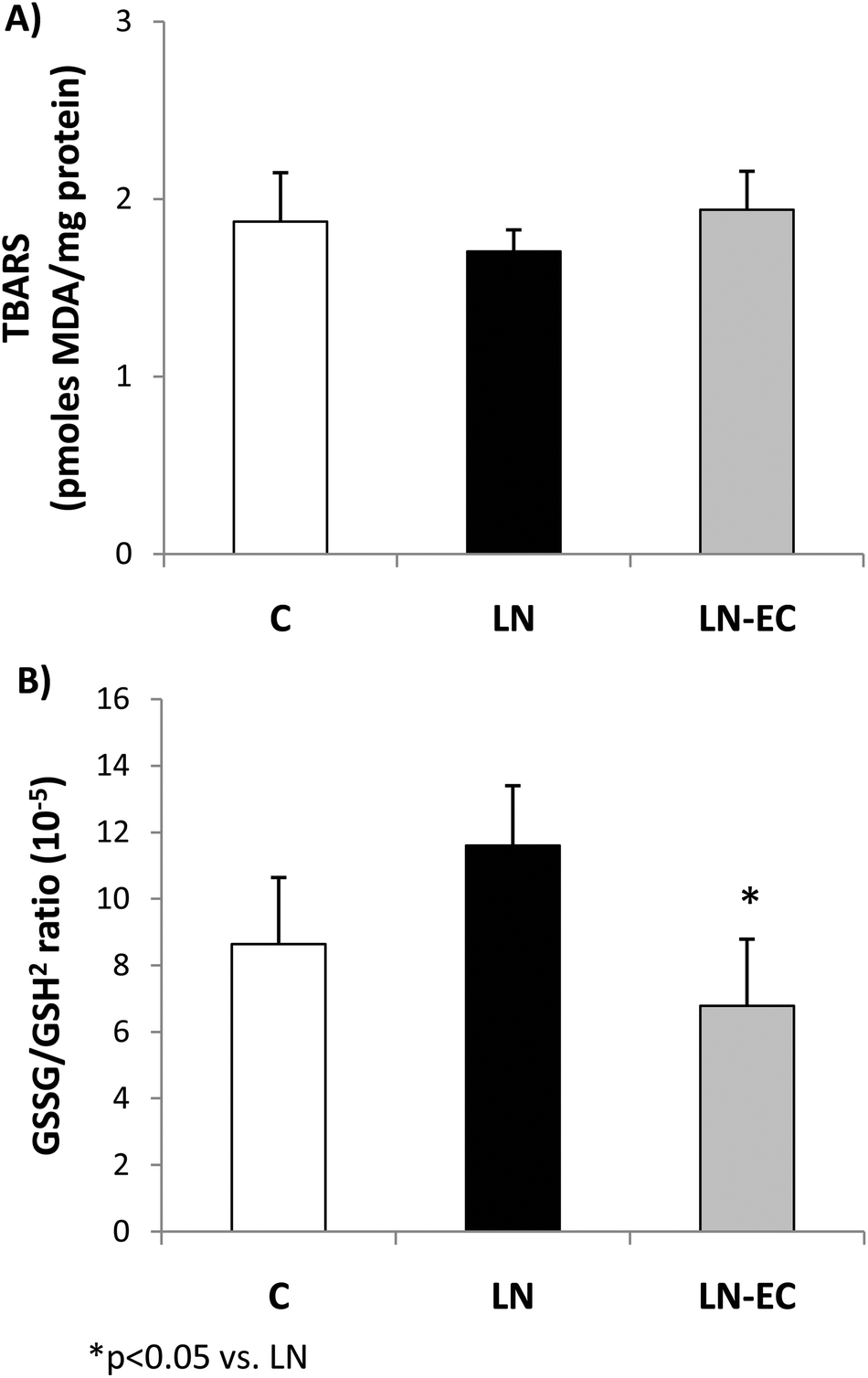 | ||
| Fig. 1 Effects of dietary EC on oxidative stress markers. (A) TBARS content and (B) GSSG/GSH2 ratio in the heart of control rats treated with L-NAME and those treated with L-NAME and EC for 4 d. Values are expressed as means ± SEM. *p < 0.05 with respect to the LN group. | ||
As NO plays a key role in the vascular function, NOS was evaluated in the heart. EC administration prevented the decrease in NOS activity observed in LN (64 ± 12 vs. 168 ± 6 pmoles [14C]-L-citrulline min−1 per g wet weight, p < 0.05) (Fig. 2A). What is more, EC administration maintained the levels of phosphorylation of eNOS protein within control values (Fig. 2B). The expression of iNOS protein in the heart was not affected by the treatments (data not shown).
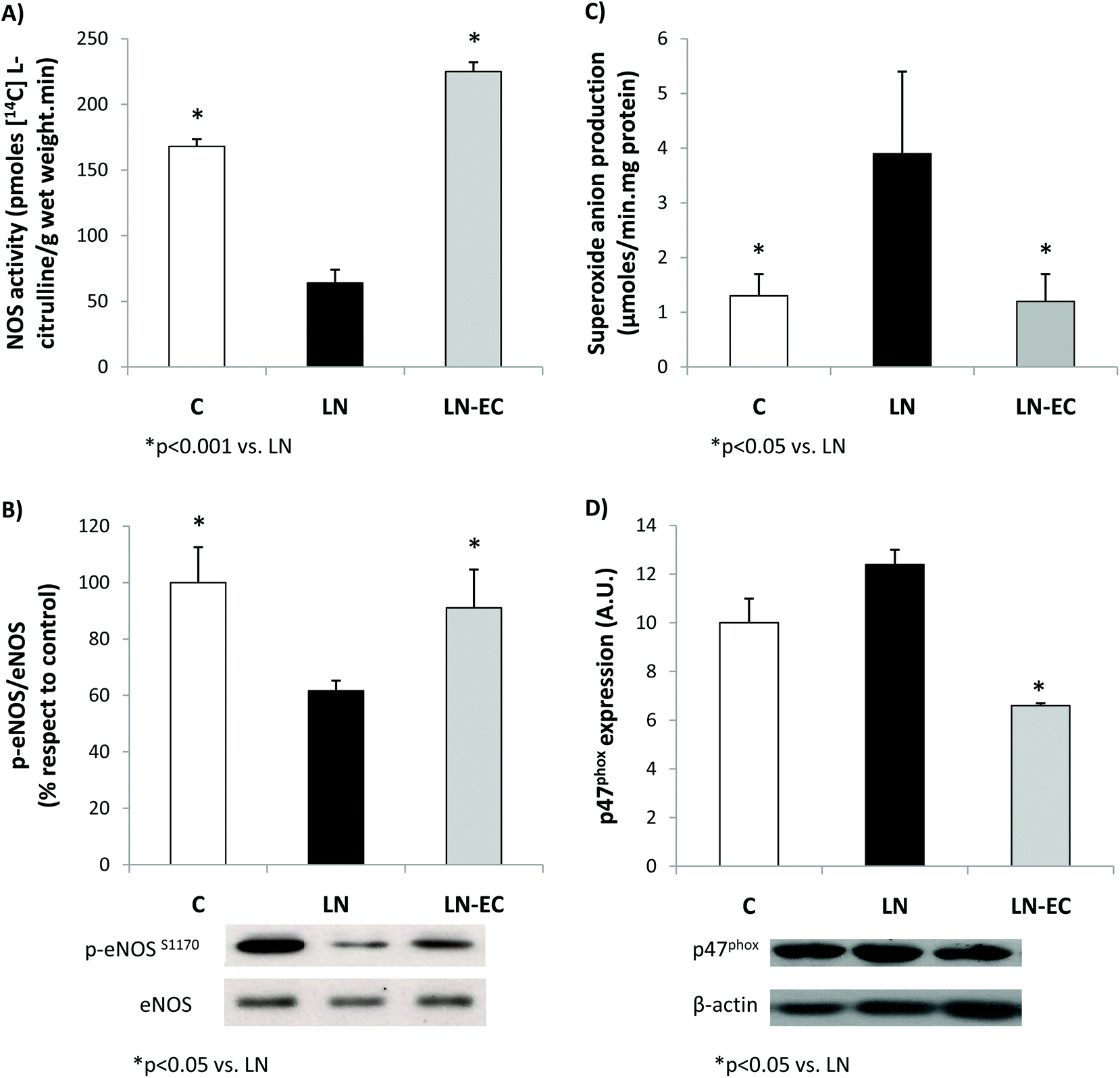 | ||
| Fig. 2 Effects of dietary EC on (A) NOS activity, (B) eNOS phosphorylation levels, (C) superoxide anion production, and (D) p47phox expression in the heart of control rats treated with L-NAME and those treated with L-NAME and EC for 4 d. Values are expressed as means ± SEM. *p < 0.05 with respect to the LN group; #p < 0.001 with respect to the LN group. | ||
Regarding NOX activity, we evaluated the capacity of the heart to generate superoxide anions in the 3 groups.
EC administration prevented the increase in NOX-dependent superoxide production observed in the LN group (p < 0.05, Fig. 2C). This decrease was accompanied by a lower expression of p47phox (only marginally increased by L-NAME treatment, p = 0.06 LN vs. C), the cytosolic and regulatory subunit of NOX1 and NOX2 (Fig. 2D). Expression of gp91 (the catalytic subunit of NOX2) and NOX4 subunits was similar in the three groups studied (data not shown).
As oxidative stress was increased by L-NAME administration, we studied antioxidant enzyme activities. The increase in GPx activity observed in LN with respect to the control group (p < 0.05, Fig. 3A) was absent in the LN-EC group. SOD activity was lower in LN-EC than in the LN group (p < 0.05, Fig. 3B). Catalase activity showed no significant differences between groups (Fig. 3C).
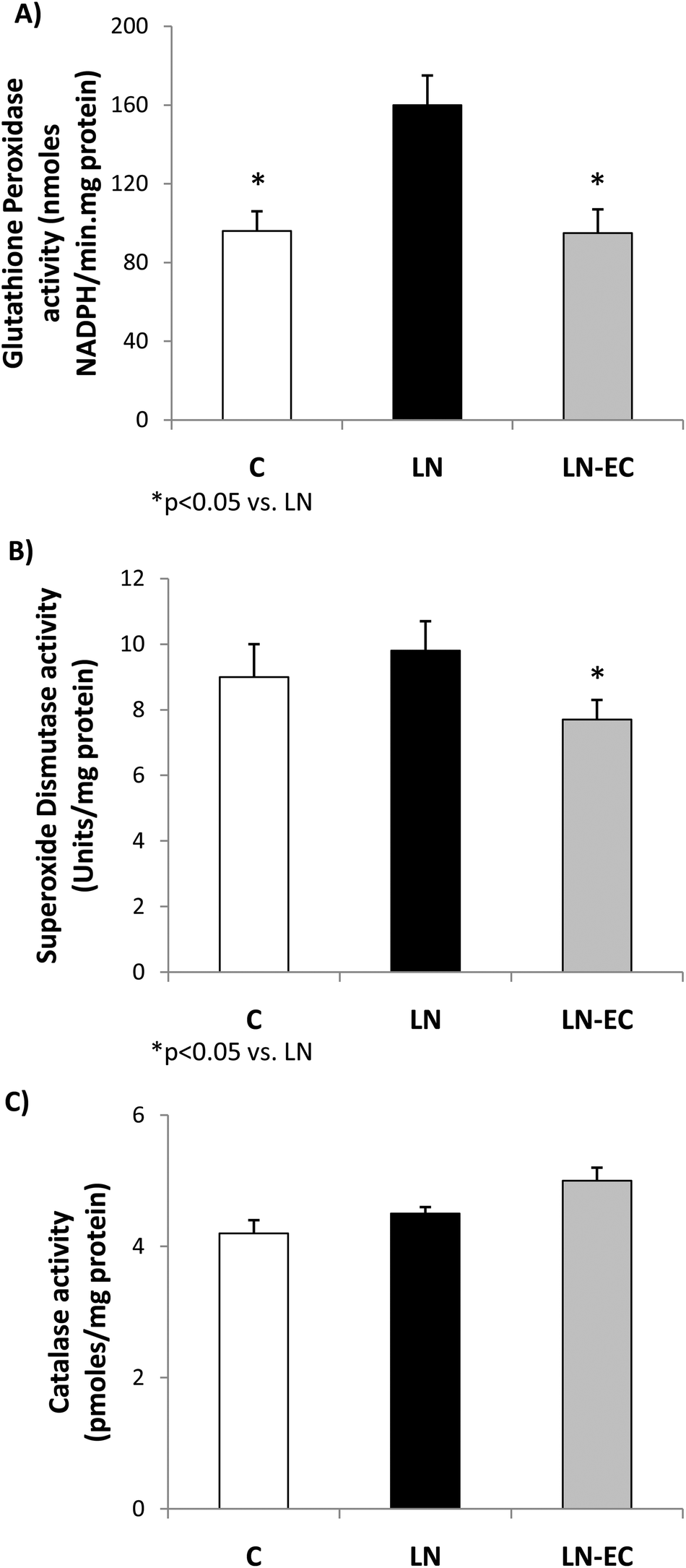 | ||
| Fig. 3 Effects of dietary EC on the activities of (A) glutathione peroxidase, (B) superoxide dismutase, and (C) catalase in the heart of control rats treated with L-NAME and those treated with L-NAME and EC for 4 d. Values are expressed as means ± SEM. *p < 0.05 with respect to the LN group. | ||
Discussion
This work shows that short-term administration of EC prevented changes in oxidative stress and NO metabolism induced by L-NAME in the heart.Besides its vascular effects, in heart cells, NO has been shown to regulate cardiac function, e.g. controlling the coronary vessel tone, thrombogenicity, cardiomyocyte contractility, and mitochondrial respiration.23,32 Molecularly, a major reaction of NO is the posttranslational-like S-nitrosylation of proteins. These modifications may cause changes in cells and heart functions and are highly dependent on the amount of NO bioavailable.45 NO bioavailability is associated with oxidative stress mainly as a result of the reaction between NO and superoxide anions, but also through the actions of both NO and superoxide on NOS and NOX.
Long-term L-NAME administration has been associated with an increase in oxidative stress in several tissues,46–48 including heart.49 Even in this short-term treatment, we observed an increase in oxidative stress in the heart. Oxidative stress was evaluated by two methodologies: the content of MDA in the tissue as TBARS and the glutathione oxidation/reduction status.50 Interestingly, neither L-NAME nor EC affected endogenous TBARS formation but did modify GSSG/GSH2 ratios. One of the reasons why TBARS did not show differences could be given by the fact that glutathione oxidation is an early event taking place after an oxidative insult. At that point, enzymatic defenses prevent the accumulation of lipid oxidation products at a level to be detected as TBARS. Besides, under such mild oxidation conditions, glutathione peroxidase,51,52 which is also enhanced by L-NAME treatment, can cope with the formation of peroxides contributing to the increase of GSSG/GSH2 ratios. EC co-administration to L-NAME-treated rats increased GSH and lowered GSSG contents in cardiac tissue, which led to a significant decrease in the GSSG/GSH2 ratio to control values. These results suggest that EC balances the changes in the tissue redox state due to L-NAME.
As was previously shown, the imbalance in the tissue redox status mediated by L-NAME appears to be related to the increased capacity of the heart to generate superoxide anions as a result of a higher expression and activity of NOX.22,53 EC can directly scavenge superoxide anions or regulate NOX activity.19,54–56 Under our experimental conditions, EC bioavailability is rather low to scavenge radicals at a physiologically significant rate,57 but prevented the increase in the superoxide anion production observed in the LN group. These effects of both L-NAME and EC seem to be mostly associated with the NOX regulation through changes in the expression of the regulatory subunit p47phox, since the catalytic subunits measured were not affected under the present experimental conditions.
The reduced NOS activity observed in the LN group was not present in the LN-EC group. In coincidence with the results in HUVECs showing that EC induced NO production via eNOS activation,30 we observed an increase in the level of eNOS phosphorylation. Integrating our results, it is possible to conclude that the compromise in NOS activity observed after the treatment with L-NAME was absent when EC was administered in the diet. This effect could be due to: (i) an increased activity of NOS and/or (ii) a decrease in NOX activity leading to a decrease in superoxide anion production, which leads to a decreased NO consumption (by its reaction with superoxide anions). Other possibilities of cross-talking between NOS (NO) and NOX (superoxide anion) that should be taken into consideration are: (i) the increased S-nitrosylation of NOX, maintaining the enzyme generating superoxide anions at a low controlled rate;58 and/or (ii) the superoxide anion-mediated uncoupling of NOS, enhancing superoxide anion production in a self-sustained cycle.
SOD, catalase, and GPx activities are responsible for detoxifying superoxide anions and hydrogen peroxide. GPx activity was enhanced after L-NAME treatment, which is in accordance with previous reports.59 This increase could be associated with a priming effect of a higher production of superoxide anions, which in turn generates hydrogen peroxide. As a consequence, the absence of the increase in GPx activity and the decrease in SOD activity after the EC treatment appear to be due to the maintenance of a controlled NOX activity.
The present study is complementary of another investigation in which using the same rat model, EC prevented oxidative stress and the development and maintenance of high BP.34 Integrating these studies, it can be concluded that L-NAME leads to changes in the cell redox status and NO-metabolism that can be prevented by EC administration.
Given the ubiquitous role of NO in maintaining heart physiology and the implication of oxidative stress in several unhealthy conditions, e.g. inflammation and high BP, the effects of EC appear to be highly relevant. The fact that EC is commonly present in human diet makes this compound a reasonable explanation for the positive cardiovascular effects of a high consumption of fruits and vegetables. This information is valuable for designing healthy diets and/or pharmacological interventions based on experimental evidence. In addition, it opens the possibility of studying other compounds chemically related, i.e. flavonoids and polyphenols that can share some of these health benefits.
Acknowledgements
This study was supported by grants from the Universidad de Buenos Aires (UBACyT 20020100300012, 20020120100177 and 20020100100659) and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT BID PICT 2409-2011). BP, MG and CGF are members of CIC, CONICET. VC holds a CONICET fellowship.References
- H. C. Hung, K. J. Joshipura, R. Jiang, F. B. Hu, D. Hunter, S. A. Smith-Warner, G. A. Colditz, B. Rosner, D. Spiegelman and W. C. Willett, J. Natl. Cancer Inst., 2004, 96(21), 1577–1584 CrossRef PubMed.
- B. Buijsse, E. J. Feskens, F. J. Kok and D. Kromhout, Arch. Intern. Med., 2006, 166(4), 411–417 Search PubMed.
- R. Iqbal, S. Anand, S. Ounpuu, S. Islam, X. Zhang, S. Rangarajan, J. Chifamba, A. Al-Hinai, M. Keltai and S. Yusuf, Circulation, 2008, 118(19), 1929–1937 CrossRef CAS PubMed.
- D. Grassi, G. Desideri, S. Necozione, C. Lippi, R. Casale, G. Properzi, J. B. Blumberg and C. Ferri, J. Nutr., 2008, 138(9), 1671–1676 CAS.
- L. I. Mennen, D. Sapinho, A. de Bree, N. Arnault, S. Bertrais, P. Galan and S. Hercberg, J. Nutr., 2004, 134(4), 923–926 CAS.
- D. Taubert, R. Roesen, C. Lehmann, N. Jung and E. Schomig, JAMA, J. Am. Med. Assoc., 2007, 298(1), 49–60 CrossRef CAS PubMed.
- C. G. Fraga, L. Actis-Goretta, J. I. Ottaviani, F. Carrasquedo, S. B. Lotito, S. Lazarus, H. H. Schmitz and C. L. Keen, Clin. Dev. Immunol., 2005, 12(1), 11–17 CrossRef CAS.
- L. Hooper, C. Kay, A. Abdelhamid, P. A. Kroon, J. S. Cohn, E. B. Rimm and A. Cassidy, Am. J. Clin. Nutr., 2012, 95(3), 740–751 CrossRef CAS PubMed.
- I. Bernatova, O. Pechanova, P. Babal, S. Kysela, S. Stvrtina and R. Andriantsitohaina, Am. J. Physiol. Heart Circ. Physiol., 2002, 282(3), H942–H948 CAS.
- D. Juric, P. Wojciechowski, D. K. Das and T. Netticadan, Am. J. Physiol. Heart Circ. Physiol., 2007, 292(5), H2138–H2143 CrossRef CAS PubMed.
- J. M. Hodgson, I. B. Puddey, V. Burke, L. J. Beilin and N. Jordan, J. Hypertens., 1999, 17(4), 457–463 CrossRef CAS PubMed.
- J. M. Hodgson, I. B. Puddey, V. Burke, G. F. Watts and L. J. Beilin, Clin. Sci., 2002, 102(2), 195–201 CrossRef.
- L. Bravo, Nutr. Rev., 1998, 56(11), 317–333 CrossRef CAS PubMed.
- M. L. McCullough, J. J. Peterson, R. Patel, P. F. Jacques, R. Shah and J. T. Dwyer, Am. J. Clin. Nutr., 2012, 95(2), 454–464 CrossRef CAS PubMed.
- A. Vogiatzoglou, A. A. Mulligan, R. N. Luben, M. A. Lentjes, C. Heiss, M. Kelm, M. W. Merx, J. P. Spencer, H. Schroeter and G. G. Kuhnle, Br. J. Nutr., 2014, 111(8), 1463–1473 CrossRef CAS PubMed.
- E. N. Frankel, J. Kanner, J. B. German, E. Parks and J. E. Kinsella, Lancet, 1993, 341(8843), 454–457 CrossRef CAS.
- E. N. Frankel, A. L. Waterhouse and J. E. Kinsella, Lancet, 1993, 341(8852), 1103–1104 CrossRef CAS.
- G. G. Mackenzie, F. Carrasquedo, J. M. Delfino, C. L. Keen, C. G. Fraga and P. I. Oteiza, FASEB J., 2004, 18(1), 167–169 CAS.
- M. Sarr, M. Chataigneau, S. Martins, C. Schott, J. El Bedoui, M. H. Oak, B. Muller, T. Chataigneau and V. B. Schini-Kerth, Cardiovasc. Res., 2006, 71(4), 794–802 CrossRef CAS PubMed.
- C. G. Fraga, IUBMB Life, 2007, 59(4–5), 308–315 CrossRef CAS PubMed.
- M. Gomez-Guzman, R. Jimenez, M. Sanchez, M. Romero, F. O'Valle, R. Lopez-Sepulveda, A. M. Quintela, P. Galindo, M. J. Zarzuelo, E. Bailon, E. Delpon, F. Perez-Vizcaino and J. Duarte, Br. J. Nutr., 2011, 106(9), 1337–1348 CrossRef CAS PubMed.
- M. Gomez-Guzman, R. Jimenez, M. Sanchez, M. J. Zarzuelo, P. Galindo, A. M. Quintela, R. Lopez-Sepulveda, M. Romero, J. Tamargo, F. Vargas, F. Perez-Vizcaino and J. Duarte, Free Radicals Biol. Med., 2012, 52(1), 70–79 CrossRef CAS PubMed.
- P. B. Massion, O. Feron, C. Dessy and J. L. Balligand, Circ. Res., 2003, 93(5), 388–398 CrossRef CAS PubMed.
- K. K. Griendling, C. A. Minieri, J. D. Ollerenshaw and R. W. Alexander, Circ. Res., 1994, 74(6), 1141–1148 CrossRef CAS.
- K. K. Griendling and M. Ushio-Fukai, Regul. Pept., 2000, 91(1–3), 21–27 CrossRef CAS.
- G. W. De Keulenaer, R. W. Alexander, M. Ushio-Fukai, N. Ishizaka and K. K. Griendling, Biochem. J., 1998, 329(Pt 3), 653–657 CAS.
- C. Patterson, J. Ruef, N. R. Madamanchi, P. Barry-Lane, Z. Hu, C. Horaist, C. A. Ballinger, A. R. Brasier, C. Bode and M. S. Runge, J. Biol. Chem., 1999, 274(28), 19814–19822 CrossRef CAS PubMed.
- J. M. Li, A. M. Mullen, S. Yun, F. Wientjes, G. Y. Brouns, A. J. Thrasher and A. M. Shah, Circ. Res., 2002, 90(2), 143–150 CrossRef CAS.
- B. Lassegue and R. E. Clempus, Am. J. Physiol. Regul. Integr. Comp. Physiol., 2003, 285(2), R277–R297 CAS.
- I. Ramirez-Sanchez, L. Maya, G. Ceballos and F. Villarreal, Hypertension, 2010, 55(6), 1398–1405 CrossRef CAS PubMed.
- Y. Steffen, T. Schewe and H. Sies, Biochem. Biophys. Res. Commun., 2007, 359(3), 828–833 CrossRef CAS PubMed.
- J. F. Arnal, L. Warin and J. B. Michel, J. Clin. Invest., 1992, 90(2), 647–652 CrossRef CAS PubMed.
- M. Kashiwagi, M. Shinozaki, H. Hirakata, K. Tamaki, T. Hirano, M. Tokumoto, H. Goto, S. Okuda and M. Fujishima, J. Am. Soc. Nephrol., 2000, 11(4), 616–624 CAS.
- M. C. Litterio, G. Jaggers, G. Sagdicoglu Celep, A. M. Adamo, M. A. Costa, P. I. Oteiza, C. G. Fraga and M. Galleano, Free Radicals Biol. Med., 2012, 53(10), 1894–1902 CrossRef CAS PubMed.
- M. Galleano, I. Bernatova, A. Puzserova, P. Balis, N. Sestakova, O. Pechanova and C. G. Fraga, IUBMB Life, 2013, 65(8), 710–715 CrossRef CAS PubMed.
- K. Shimokawa Ki, M. Katayama, Y. Matsuda, H. Takahashi, I. Hara, H. Sato and S. Kaneko, Mol. Hum. Reprod., 2002, 8(1), 32–36 CrossRef.
- L. Actis-Goretta, G. G. Mackenzie, P. I. Oteiza and C. G. Fraga, Ann. N. Y. Acad. Sci., 2002, 957, 279–283 CrossRef CAS PubMed.
- A. Rodriguez-Ariza, F. Toribio and J. Lopez-Barea, J. Chromatogr., B: Biomed. Appl., 1994, 656(2), 311–318 CrossRef CAS.
- M. A. Costa, A. Loria, R. Elesgaray, A. M. Balaszczuk and C. Arranz, J. Hypertens., 2004, 22(8), 1561–1569 CrossRef CAS.
- A. Bettaieb, M. A. Vazquez Prieto, C. Rodriguez Lanzi, R. M. Miatello, F. G. Haj, C. G. Fraga and P. I. Oteiza, Free Radicals Biol. Med., 2014, 72, 247–256 CrossRef CAS PubMed.
- W. A. Gunzler, H. Kremers and L. Flohe, Z. Klin. Chem. Klin. Biochem., 1974, 12(10), 444–448 CAS.
- H. P. Misra and I. Fridovich, J. Biol. Chem., 1972, 247(10), 3170–3175 CAS.
- H. Aebi, Methods Enzymol., 1984, 105, 121–126 CAS.
- C. G. Fraga, M. C. Litterio, P. D. Prince, V. Calabro, B. Piotrkowski and M. Galleano, J. Clin. Biochem. Nutr., 2011, 48(1), 63–67 CrossRef CAS PubMed.
- E. Murphy, M. Kohr, J. Sun, T. Nguyen and C. Steenbergen, J. Mol. Cell Cardiol., 2012, 52(3), 568–577 CrossRef CAS PubMed.
- A. M. Cardoso, C. C. Martins, S. Fiorin Fda, R. Schmatz, F. H. Abdalla, J. Gutierres, D. Zanini, A. M. Fiorenza, N. Stefanello, J. D. Serres, F. Carvalho, V. P. Castro, C. M. Mazzanti, L. F. Royes, A. Bello-Klein, J. F. Goularte, V. M. Morsch, M. D. Bagatini and M. R. Schetinger, Cell Biochem. Funct., 2013, 31(2), 136–151 CrossRef CAS PubMed.
- M. Saravanakumar and B. Raja, Eur. J. Pharmacol., 2011, 671(1–3), 87–94 CrossRef CAS PubMed.
- Z. Selamoglu Talas, Cell Biochem. Funct., 2014, 32(2), 150–154 CrossRef CAS PubMed.
- K. Husain and S. R. Hazelrigg, Biochim. Biophys. Acta, 2002, 1587(1), 75–82 CrossRef CAS.
- A. Meister, Strategies for increasing cellular glutathione, in Biothiols in Health and Disease, ed. L. Packer and E. Cadenas, Marcel Dekker Inc., New York, NY, 1995. pp. 165–188 Search PubMed.
- L. Flohe, Biochim. Biophys. Acta, 2013, 1830(5), 3139–3142 CrossRef CAS PubMed.
- F. Ursini and M. Maiorino, Int. J. Androl., 2005, 28(1), 61–62 CrossRef CAS PubMed ; author reply 63–64.
- T. M. Paravicini, S. Chrissobolis, G. R. Drummond and C. G. Sobey, Stroke, 2004, 35(2), 584–589 CrossRef CAS PubMed.
- R. Jimenez, R. Lopez-Sepulveda, M. Kadmiri, M. Romero, R. Vera, M. Sanchez, F. Vargas, F. O'Valle, A. Zarzuelo, M. Duenas, C. Santos-Buelga and J. Duarte, Free Radicals Biol. Med., 2007, 43(3), 462–473 CrossRef CAS PubMed.
- R. Lopez-Sepulveda, R. Jimenez, M. Romero, M. J. Zarzuelo, M. Sanchez, M. Gomez-Guzman, F. Vargas, F. O'Valle, A. Zarzuelo, F. Perez-Vizcaino and J. Duarte, Hypertension, 2008, 51(4), 1088–1095 CrossRef CAS PubMed.
- M. Galleano, S. V. Verstraeten, P. I. Oteiza and C. G. Fraga, Arch. Biochem. Biophys., 2010, 501(1), 23–30 CrossRef CAS PubMed.
- C. G. Fraga, M. Galleano, S. V. Verstraeten and P. I. Oteiza, Mol. Aspects Med., 2010, 31(6), 435–445 CrossRef CAS PubMed.
- J. Qian, F. Chen, Y. Kovalenkov, D. Pandey, M. A. Moseley, M. W. Foster, S. M. Black, R. C. Venema, D. W. Stepp and D. J. Fulton, Free Radicals Biol. Med., 2012, 52(9), 1806–1819 CrossRef CAS PubMed.
- J. Sainz, R. Wangensteen, I. Rodriguez Gomez, J. M. Moreno, V. Chamorro, A. Osuna, P. Bueno and F. Vargas, Am. J. Hypertens., 2005, 18(6), 871–877 CrossRef CAS PubMed.
Footnote |
| † Both authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2015 |