
A diet with lactosucrose supplementation ameliorates trinitrobenzene sulfonic acid-induced colitis in rats†
Yan
Zhou
a,
Zheng
Ruan
*a,
Xiaoli
Zhou
a,
Xiaoliu
Huang
a,
Hua
Li
b,
Ling
Wang
b,
Cui
Zhang
a,
Shiqiang
Liu
a,
Zeyuan
Deng
a,
Guoyao
Wu
c and
Yulong
Yin
ad
aState Key Laboratory of Food Science and Technology, Nanchang University, Nanchang 330047, China. E-mail: ruanzheng@ncu.edu.cn; Fax: +86-791-88304402
bThe Second Affiliated Hospital, Nanchang University, Nanchang 330006, China
cDepartment of Animal Science, Faculty of Nutrition, Texas A&M University, College Station, TX 77843-2471, USA
dInstitute of Subtropical Agriculture, Chinese Academy of Sciences, Changsha 410125, China
First published on 15th October 2014
Abstract
Chronic intestinal inflammation contributes to an increased risk of colon cancer. Lactosucrose (LS), a kind of functional trisaccharide, can modulate immunity and promote microbe growth. The aim of this study was to investigate the effect of LS on 2,4,6-trinitrobenzene sulfonic acid (TNBS) induced colitis in rats. Rats were randomly divided into four treatment groups: the normal group, TNBS group, LS group, and salicylazosulfapyridine (SASP) group for five weeks. LS supplementation ameliorated TNBS-induced colitis. LS supplementation increased IL-10 production and suppressed the secretion of IL-12 in the colon, as compared to the TNBS group. LS decreased the production of TLR-2 protein and nuclear NF-κB p65 protein, as well as mRNA levels, as compared with colitic rats. These results indicate that chronic feeding of LS inhibited TNBS-induced chronic inflammation. LS has potential nutraceutical intervention to combat colitis.
1. Introduction
Inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), is a chronically recurrent disorder of the intestine.1 IBD involves the dysregulation of the intestinal immune response. Immune and non-immune cells within the intestinal mucosa recognize conserved microbial structures through so-called pattern recognition receptors (PRRs), including toll-like receptors (TLRs).2 Moreover, TLR-mediated cellular responses can lead to chronic inflammation, which in turn contributes to the development of IBD.3 Polymorphisms in TLR-1, -2, -4 and -9 loci have been associated with IBD in humans.4–6Nuclear factor-kappa B (NF-κB) may serve as the main mediator of TLR signaling in IBD.7 It is a key regulator of the inducible expression of numerous genes involved in immune and inflammatory responses in the gut.8,9 Increased NF-κB activation has been detected in the intestinal lamina propria of patients with CD or murine TNBS colitis models.10,11 Colonic mucosal biopsies as well as lamina propria mononuclear cells from IBD patients display an increased presence of NF-κB p65 in the nucleus in comparison with mucosa samples from healthy individuals.12 Neurath et al. were able to prevent and even to abrogate established TNBS-induced colitis in mice by administrating antisense oligonucleotides directed against NF-κB p65.13 Growing evidence in the association of NF-κB activation and IBD suggests that modulation of the NF-κB signaling pathway is the main target for the anti-inflammatory treatment of IBD.
Activation of NF-κB then upregulates the expression of numerous proinflammatory cytokines, such as interleukin-6 (IL-6) and IL-12, involved in intestinal inflammation. IL-6 is a pleiotropic cytokine. An elevation in serum IL-6 levels during remission in CD patients was found to be a clinically relevant parameter for predicting inflammatory activity14 as well as for corresponding with a high frequency of disease relapse.15 TNBS-induced colitis can be abrogated with antibodies specific for IL-12,16 in which excessive production of IL-12 seems to be the crucial underlying genetic abnormality in susceptible mice.17 However, IL-10 is an anti-inflammatory cytokine that inhibits both antigen presentation and subsequent release of proinflammatory cytokines, thereby attenuating mucosal inflammation. IL-10 can inhibit the effector functions of activated macrophages and monocytes in vitro and downregulate IL-12,18 and inactivation of IL-10 in mice results in an increased production of IL-12.19 IL-10−/− Mice spontaneously develop colitis in a bacteria-containing environment but are healthy under germ-free conditions.20
Although the etiology of IBD is not well understood, the promoting effects of functional oligosaccharides on intestinal health have been attracted increasing attention. Fructooligosaccharide attenuates the production of pro-inflammatory cytokines.21 Fruit intake is negatively associated with the risk of IBD and can reduce the risk factors of colon cancer.22 So far, a number of experimental findings have suggested that intestinal metabolites (short-chain fatty acids) and beneficial bacterium in gut lumen contribute to the remission of IBD. For example, butyric acid attenuates intestinal inflammation in murine colitis.23 In addition, it is different for luminal and mucosa-associated microflora between IBD patients and healthy controls.24
Several animal models have been developed to investigate the pathology of IBD. One of these models is 2,4,6-trinitrobenzene sulfonic acid (TNBS) induced colitis in which rodents receiving TNBS develop an acute and chronic transmural colitis resembling colitis.25 The TNBS/ethanol induced colitis model has been very useful in studying many important aspects of gut inflammation, including cytokine secretion patterns, cell adhesion, and immunotherapy.26
Lactosucrose (4G-β-D-galactosylsucrose, LS) is an indigestible trisaccharide, which is composed of galactose, glucose, and fructose. It was approved as a commercial prebiotic by the Japan Health Department 20 years previously and has been prevalent until now. Several studies have shown that LS confers anti-inflammatory and anti-allergic effects.27–30 Hino et al. reported that LS could stimulate overproduction of transforming growth factor-β1 (TGF-β1) in Peyer's patches.28 LS was found to inhibit immunoglobulin E production and increase the secretive yield of IL-10 in ovalbumin/alum-immunized mice.29 In addition, LS has been shown to promote the growth of Bifidobacteria in patients with chronic inflammatory bowel disease or in a healthy human.30,31 Although a number of studies have investigated the effects of LS on inflammation, the anti-inflammatory effect and mechanisms of LS have not been studied thoroughly.
In our previous study, we found that LS increased short chain fatty acid (SCFA) production, promoted Bifidobacterium and Lactobacillus, and decreased the number of Escherichia in TNBS-induced colonic inflammation.32 The main objective of the present work is to investigate the effects of LS on TNBS-induced colitic rats and its effect on TLR-2, TLR-4 and nuclear NF-κB p65 expression in colitis.
2. Materials and methods
2.1 Chemicals
TNBS (0.5% w/v) was purchased from Sigma-Aldrich Chemical Co. (MO, USA). Rabbit polyclonal NF-κB p65, TLR-2, and Lamin B1 antibodies were purchased from Santa Cruz Biotechnology (CA, USA). NE-PER nuclear and cytoplasmic extraction reagents and enhanced chemiluminescence were purchased from Thermo Scientific (MA, USA). Peroxidase conjugated anti-rabbit immunoglobulin G, IL-12, IL-10, and IL-6 ELISA kits were purchased from Boster Biological Technology (Wuhan, China). Alanine transaminase (ALT) and aspartate transaminase (AST) were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Blood clinical chemistry reagents for measurement of alkaline phosphatase (ALP), total protein (TP), albumin (ALB), urea nitrogen (BUN), low-density lipoprotein (LDL) and cholesterol (CHO) were from Leadman Biochemistry (Beijing, China). Trizol was purchased from Invitrogen (NY, USA). The Reverse Transcription System Kit was purchased from TransGen Biotech (Beijing, China).2.2 Preparation of lactosucrose
LS was catalytically synthesized from sucrose and lactose solution by β-fructofuranosidase. β-Fructofuranosidase was derived from Arthrobacter sp.10138 in our laboratory. In this enzyme-catalyzed reaction (pH 7.0 and 30 °C), the solution contained 550 mmol L−1 sucrose, 550 mmol L−1 lactose, and β-fructofuranosidase (250 U mL−1). After a 24 h period, the whole solution was freeze-dried under vacuum, and LS was obtained. The concentration of saccharides were then analyzed by high performance liquid chromatography (HPLC) 1200 from Agilent (USA) with an evaporative light scattering detector (ELSD) model 300S (Softa, America) according to Ruan et al.33 The chromatographic column is a Kromasil 100-5NH2 column (250 mm × 4.6 mm). The mobile phase consisted of acetonitrile–water (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 v/v) with a flow rate of 1.0 mL min−1, and the temperature of the column oven was 25 °C. The drift tube and evaporator temperatures were set at 70 °C and 30 °C, respectively.
:25 v/v) with a flow rate of 1.0 mL min−1, and the temperature of the column oven was 25 °C. The drift tube and evaporator temperatures were set at 70 °C and 30 °C, respectively.
2.3 Animals care
Female Sprague-Dawley rats weighing 200 ± 20 g were obtained from SIPPR-BK Experimental Animal (Shanghai, China). All animals were housed under specific pathogen-free conditions, a temperature-controlled atmosphere (25 ± 1 °C at a 50% relative humidity) and under a 12 h light/12 h dark cycle. The rats had free access to a standard diet (GB14924.1-2001, Table 1) and sterile drinking water at all times. This study was carried out in Jiangxi Province Center for Disease Control and Prevention (Nanchang, China), and performed in accordance with the Chinese guidelines for the Laboratory Animals Care.| Ingredients | Content (%) | Chemical composition | Content |
|---|---|---|---|
|
a The vitamin–mineral premix provided (per kilogram feed): vitamin A, 14000 IU; vitamin D3, 1500 IU; vitamin E, 5 mg; vitamin K, 5 mg; thiamine, 13 mg; riboflavin, 12 mg; pyridoxine, 12 mg; vitamin B12, 0.022 mg; niacin 60 mg; pantothenic acid, 24 mg; biotin, 0.2 mg; folic acid, 6 mg; choline chloride, 350 mg; Fe (as iron sulfate), 120 mg; Cu (as copper oxide), 10 mg; Mn (as manganous oxide), 75 mg; Zn (as zinc oxide), 30 mg; I (as ethylenediamine dihydroiodide), 0.5 mg; and Se (as sodium selenite), 0.2 mg.
|
|||
| Wheat | 14 | Digestible energy (MJ kg−1) | 3.40 |
| Corn | 43 | Crude protein (%) | 21.0 |
| Soybean meal | 24 | Crude fat (%) | 4.5 |
| Full fat soybean extruded | 8 | Calcium (%) | 1.0 |
| Soybean oil | 1.4 | Total phosphate (%) | 0.7 |
| Whey powder | 3 | ||
| Fish meal | 3.2 | Sodium (%) | 0.3 |
| Limestone | 1.3 | Met + Cys (%) | 0.78 |
| Dicalcium phosphate | 1.1 | Lys (%) | 1.35 |
| Vitamin–mineral premixa | 1.0 | Thr (%) | 0.88 |
The rats with intestinal inflammation induced by TNBS were assigned randomly into one of the three treatments: the TNBS group (receiving physiological saline), the LS group (intragastric administration, 250 mg kg−1 body weight every day), and the SASP group (intragastric administration receiving salicylazosulfapyridine (SASP), 250 mg kg−1 body weight every day) as a positive control. SASP is commonly used in clinical therapeutics for IBD patients. Normal rats that had not been exposed to TNBS were given physiological saline (the normal group). There were 6 rats in each group. All rats in each group were treated for 35 days, and then sacrificed with ether anesthesia to obtain tissue samples.
| Score | Weight loss (% of initial wt) | Blood in feces | Stool consistency |
|---|---|---|---|
| 0 | <1 | Negative | Normal pelletets |
| 1 | 1–4.99 | Occult blood+ | Slightly loose feces |
| 2 | 5–10 | Occult blood ++ | Loose feces |
| 3 | >10 | Gross blood | Watery diarrhea |
2.4 Histological grading of colitis
Histological examination was performed as previously described.36 The colon tissues were fixed in 10% (v/v) buffered formalin phosphate, embedded in paraffin, and cut into 5 μm sections. Then the sections were stained with hematoxylin and eosin, and assessed under light microscopy. The degree of inflammation was graded semi-quantitatively from 0 to 4 (0, no signs of inflammation; 1, very low level of inflammation; 2, low level of leukocyte infiltration; 3, high level of leukocyte infiltration, high vascular density, and thickening of the colon wall; and 4, transmural infiltration, loss of goblet cells, high vascular density, and thickening of the colon wall). Grading was performed in a blinded fashion.2.5 Assay of blood clinical chemistry
When rats were sacrificed, fresh blood samples were obtained and collected in tubes containing heparin. The blood samples were centrifuged at 3500 r min−1 and the supernatant fluid (plasma) was obtained. All samples were stored at −20 °C until analysis.Hematological parameters ALP, BUN, LDL, TP, CHO, and ALB were determined using a biochemistry analyzer (Beckman, USA). ALT and AST were determined by spectrophotometry using commercial kits.
2.6 Assay of secretory production of IL-12, IL-6 and IL-10 in the colon
The production of IL-12, IL-6 and IL-10 in the colon was measured using an enzyme-linked immunosorbent assay (ELISA). The sections of the proximal colon (100 mg) were taken from inflamed areas and homogenized in physiological saline (10 mL). The solution was centrifuged at 3500 r min−1 for 15 min. The supernatant was obtained for the analysis of cytokines. These cytokines were measured with ELISA kits (Boster Biological Technology, Wuhan, China) according to the manufacturer's protocol. The ELISA microplate was read using an ELISA reader (Dynatech Laboratories, USA) with an absorbance maximum at 450 nm.2.7 Immunohistochemistry
Immunohistochemistry was performed as previously described.37 Samples from the colon were fixed in 4% paraformaldehyde in phosphate buffer solution and dehydrated with ethanol. Then those samples were embedded in paraffin. Sections (5 μm thick) were prepared and exposed to immunoperoxidase. Endogenous peroxidase was cleared by treatment with 3% H2O2 phosphate buffered saline (PBS) for 20 min at room temperature. After washing with 0.05 mol L−1 PBS (pH 7.4), slides were blocked with 5% bovine serum albumin (BSA) in PBS for 20 min at room temperature to prevent non-specific protein binding. Then, the slides were sequentially exposed to the primary antibody TLR-2 (Santa Cruz Biotechnology, CA, USA, 1:200) and NF-κB p65 (Santa Cruz Biotechnology, CA, USA, 1:80) in PBS and incubated overnight at 4 °C. After rinsing, the slides were incubated with a secondary antibody conjugated to biotinylated goat peroxidase and 0.1% (v/v) diaminobenzidine substrate. Slides processed without a primary antibody served as the negative control. There are four slides for every sample (rat) and there are four observations for a slide. The optical densities of TLR-2 and NF-κB p65 proteins in the colonic epithelial cells were determined by densitometric scanning using the Motic Med System 6.0 Software.
2.8 Western blot analysis of NF-κB p65 in nucleus
The activation of NF-κB in the colon was assessed by western blot. The nuclear protein in the colon tissue was extracted by a commercial kit (Thermo Scientific, MA, USA) according to the manufacturer's instructions. The protein content was estimated by the Coomassie brilliant blue method. Proteins (100 μg) were separated with 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene difluoride (PVDF) membrane (2 h, 200 mA). The membranes were blocked in 5% BSA for 1 h at room temperature. The different proteins were visualized using NF-κB p65 (1:1000) and Lamin B1 (1:500) primary antibodies and developed using enhanced chemiluminescence (Thermo Scientific, MA, USA). The image of the blot was acquired with Molecular Imager Gel Doc XR (Bio Rad). The optical density of bands was calculated and analyzed by means of Image J (NIH, Bethesda, MD, USA).
2.9 Quantitative analysis of gene expression with fluorescence quantitative polymerase chain reaction
Real-time fluorescence quantitative polymerase chain reaction (PCR) analysis was used to measure the gene expression of TLR-2, TLR-4, and NF-κB. Colonic tissues from the normal, TNBS, LS, and SASP groups were immediately frozen in liquid nitrogen for RNA extraction. Total RNA was isolated from tissues using the Trizol reagent (Invitrogen, America) method as described by the manufacturer. cDNA synthesis was carried out using the Reverse Transcription System Kit (Takara, Dalian, China), according to the manufacturer's instructions. Fluorescence quantitative PCR was carried out using SYBR Premix Ex Taq (Takara, Japan) on an ABI 7900HT PCR instrument (Applied Biosystems, USA). The experimental conditions were: denaturation for 30 s at 95 °C, annealing for 5 s at 95 °C, and extension for 30 s at 60 °C, for 40 cycles. Rat primers were designed using Primer 3 and synthesized by Sangon Biotech Co., Ltd (Table 3) (Shanghai, China). The relative mRNA levels of the genes were calculated using the 2−ΔΔCt formula38 and the rat GAPDH as the housekeeping gene.| Primers | Product (bp) | Genebank accession no. | ||
|---|---|---|---|---|
| TLR-2 | Forward primer | 5′-AGAGGGAAATCGTGCGTGAC-3′ | 137 | NM_198769.2 |
| Reverse primer | 5′-CCATACCCAGGAAGGAAGGCT-3′ | |||
| TLR-4 | Forward primer | 5′-AGAATGAGGACTGGGTGA-3′ | 323 | NM_019178 |
| Reverse primer | 5′-GCTAAGAAGGCGAGTACAACCT-3′ | |||
| NF-κB p65 | Forward primer | 5′-GCACTGTGAGGACGGCATA-3′ | 96 | AF 079314.1 |
| Reverse primer | 5′-CGTGAAGTATTCCCAGGTTTG-3′ | |||
| GAPDH | Forward primer | 5′-GGCAAGTTCAACGGCACAG-3′ | 142 | NM_017008.3 |
| Reverse primer | 5′-CGCCAGTAGACTCCACGACAT-3′ |
2.10 Statistical analysis
All values in the figures and text are expressed as mean ± standard deviation (SD). Differences between the means of individual groups were determined by one-way analysis of variance with the Duncan multiple range test. P value <0.05 were considered statistically significant. The statistical software package SPSS 11.5 (SPSS Institute, Illinois, USA) was used for these analyses.3. Results
3.1 Production and analysis of LS
In the present work, LS production was carried out using a reaction mixture of β-fructofuranosidase, sucrose and lactose (1:1) in an incubator at 30 °C for 24 h. In this reaction, the fructosyl moiety of sucrose is transferred to lactose by fructofuranosidase, thus forming LS. The concentration of oligosaccharide formation during a course of the periods was analyzed by HPLC (Fig. 1). The maximum amount of LS formation was observed as 55.04% (w/w) in which the concentration of lactose was remarked as 22.81% (w/w), sucrose as 19.04% (w/w), glucose as 1.86% (w/w), and fructose as 1.25% (w/w) respectively.
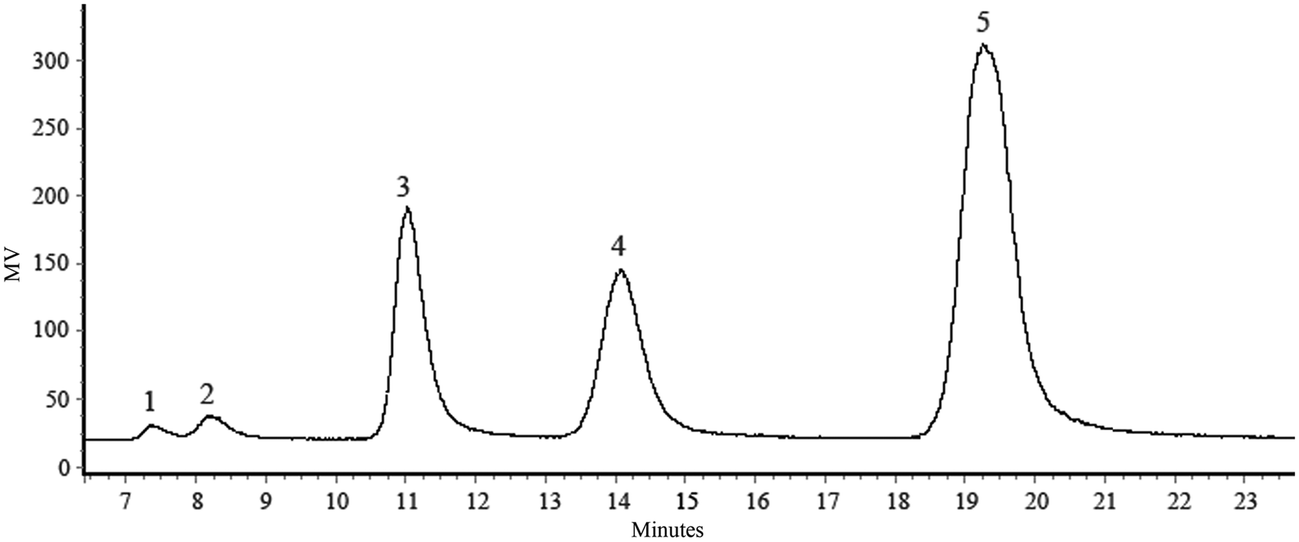 | ||
| Fig. 1 HPLC chromatogram of LS formation. HPLC chromatogram of LS: (1) fructose; (2) glucose; (3) sucrose; (4) lactose; (5) LS. | ||
3.2 LS improves colitis clinical symptoms and colon damage in rats
Rats exposed to TNBS developed symptoms of colitis as assessed by DAI. After LS treatment for 14 days, the DAI was significantly reduced compared to TNBS control rats. After SASP supplementation for 7 days, the DAI in the SASP group was significantly decreased, as compared with the TNBS control group. Compared to the normal group, DAI in the LS group or the SASP group showed no significant difference after 14 days. There were no significant changes among all treatment groups after 28 days (Fig. 2A).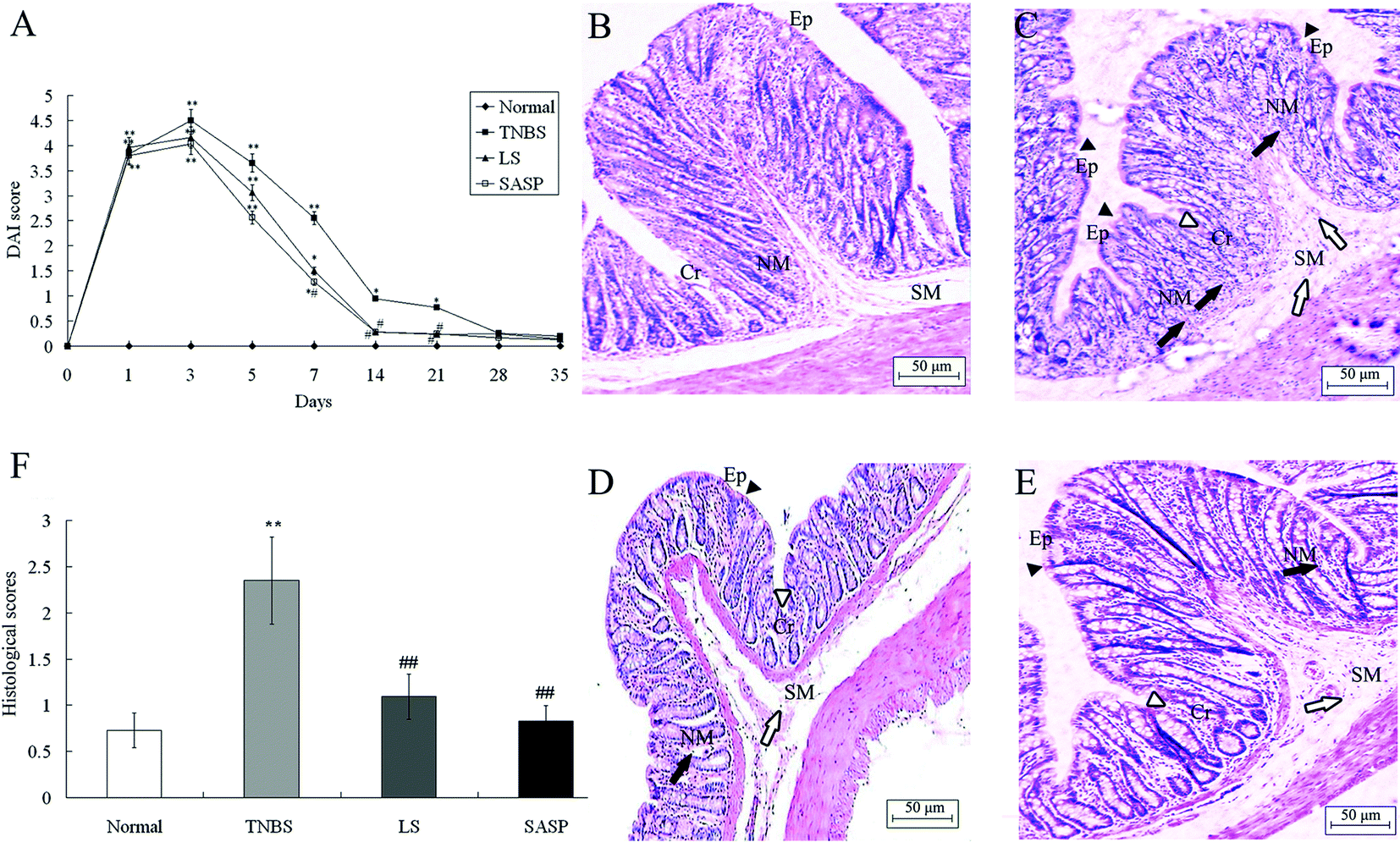 | ||
| Fig. 2 Administration of LS attenuates the development of TNBS induced colitis in rats. (A) The disease activity index (DAI) indicated the grade of intestinal inflammation. Histological findings of colon samples stained with hematoxylin and eosin. (B) Normal control rats. Micrograph showed normal mucosa (NM), submucosa (SM), epithelium (Ep) and crypt (Cr). (C) TNBS induced colitic rats. Micrograph showed surface epithelial (arrow-heads) and crypt (hollow arrow-heads) loss, severe infiltration of inflammatory cells in NM (arrows) and SM area (hollow arrows). (D) LS-treated and (E) SASP-treated rats with colitis induced by TNBS, micrograph showed that integrated surface epithelium (arrow-heads) and normal crypt (hollow arrow-heads), the infiltration of inflammatory cells was mild in NM (arrows) and SM area (hollow arrows). (F) The values mean a grading scale for histological assessment of inflammation in colitis. Values are expressed as mean ± S.D., n = 6. *Mean values were significantly different compared with normal rats (*P < 0.05; **P < 0.01); #mean values were significantly different compared with TNBS rats (#P < 0.05; ##P < 0.01), n = 6. | ||
Histological analysis of the colon (Fig. 2B–E) showed that there are differences in the intestinal microstructures between 4 groups. In the TNBS group, there was a change in the colonic architecture such as crypt and surface epithelial cells as well as massive transluminal infiltration of inflammatory cells. In contrast, infiltration of inflammatory cells was mild in the submucosal area in animals fed with the LS or SASP diets. Histological scores of inflammatory groups were significantly increased after TNBS enema compared to normal animals (Fig. 2F). Dietary LS or SASP supplementation significantly mitigated the intestinal injury compared to the TNBS group, and no significant difference was found between LS and SASP groups. Hence, the LS treated rat had a recovery effect for inflammation.
3.3 Effects of LS supplementation on blood chemistry in colitic rats
TNBS significantly increased the activities of AST (P < 0.05) and ALT (P < 0.05) as well as concentrations of TP (P < 0.05) and CHO (P < 0.05) in serum, but decreased ALP (P < 0.05) activity, as compared with normal rats (Table 4). Compared to the normal group, the content of TP (P < 0.05), ALB (P < 0.05), and CHO (P < 0.05) in the LS or the SASP group was increased. The activities of AST (P < 0.05) and LDL (P < 0.05) content in the SASP group were increased, as compared with normal rats.| Group | Normal | TNBS | LS | SASP |
|---|---|---|---|---|
| a Values are expressed as mean ± S.D., n = 6. *Mean values were significantly different compared with normal rats (*P < 0.05); #mean values were significantly different compared with TNBS rats (#P < 0.05). AST, aspartate aminotransferase; ALT, alanine transaminase; ALP, alkaline phosphatase; TP, total protein; ALB, albumin; BUN, urea nitrogen; LDL, low-density lipoprotein; CHO, cholesterol. | ||||
| AST (U L−1) | 31.8 ± 3.1 | 42.2 ± 6.4* | 32.2 ± 3.3# | 38.7 ± 3.2* |
| ALT (U L−1) | 11.5 ± 1.4 | 19.8 ± 5.9* | 15.0 ± 2.8 | 16.3 ± 3.5 |
| ALP (U L−1) | 62.6 ± 21.8 | 41.0 ± 15.4* | 67.6 ± 17.7# | 73.7 ± 15.0# |
| TP (g L−1) | 58.9 ± 6.6 | 65.4 ± 3.3* | 67.6 ± 4.9* | 65.3 ± 1.9* |
| ALB (g L−1) | 28.6 ± 2.7 | 30.8 ± 1.9 | 31.4 ± 2.8* | 31.4 ± 1.1* |
| BUN (mmol L−1) | 5.90 ± 0.88 | 6.48 ± 0.44 | 5.99 ± 1.10 | 6.78 ± 1.18 |
| LDL (mmol L−1) | 0.36 ± 0.09 | 0.43 ± 0.07 | 0.46 ± 0.09 | 0.48 ± 0.06* |
| CHO (mmol L−1) | 0.97 ± 0.17 | 1.25 ± 0.17* | 1.27 ± 0.45* | 1.02 ± 0.35* |
ALP is a membrane-bound enzyme that can dephosphorylate multiplex phosphate substrates.39 ALP removes the phosphate group of lipopolysaccharide (LPS) to generate a monophage lipid A which is much less toxic than LPS.40 AST activity was significantly decreased (P < 0.05) and ALP activity was increased (P < 0.05) in the LS group compared to the TNBS group. No significant differences of AST activity and ALP activity were found in the LS group compared to the normal group. In the SASP group, the activity of ALP was increased compared to the TNBS group colitic rats (P < 0.05), and there was no significant difference between the SASP group and the normal group (Table 4). LS and SASP showed a similar effect on blood chemistry.
3.4 Effect of LS supplementation on IL-12, IL-6 and IL-10 secretion
IL-12 and IL-6 are known as proinflammatory cytokines. IL-10 is a well-known anti-inflammatory cytokine.41 In this study, TNBS-induced colitis was characterized by a significantly increased levels of IL-12 (P < 0.01) and IL-6 (P < 0.05), and a significantly decreased level of IL-10 (P < 0.05) compared to normal rats. Following administration of LS, the level of IL-12 was decreased (P < 0.01) (Fig. 3A), and the level of IL-10 (P < 0.05) was increased, compared to TNBS group rats (Fig. 3C). The level of IL-6 also tended to decrease in LS-treated rats (P > 0.05), compared to the TNBS group (Fig. 3B). SASP supplementation decreased the level of IL-12 (P < 0.01), and increased the IL-10 level (P < 0.05), as compared with the TNBS group. When compared with normal rats, the content of IL-12, IL-10, and IL-6 in the LS or the SASP group showed no significant difference. The effect of LS on cytokine modulation was similar to SASP. These results suggest that the anti-inflammatory effect of LS may be involved in the down-regulation of the IL-12 level and up-regulation of the IL-10 level.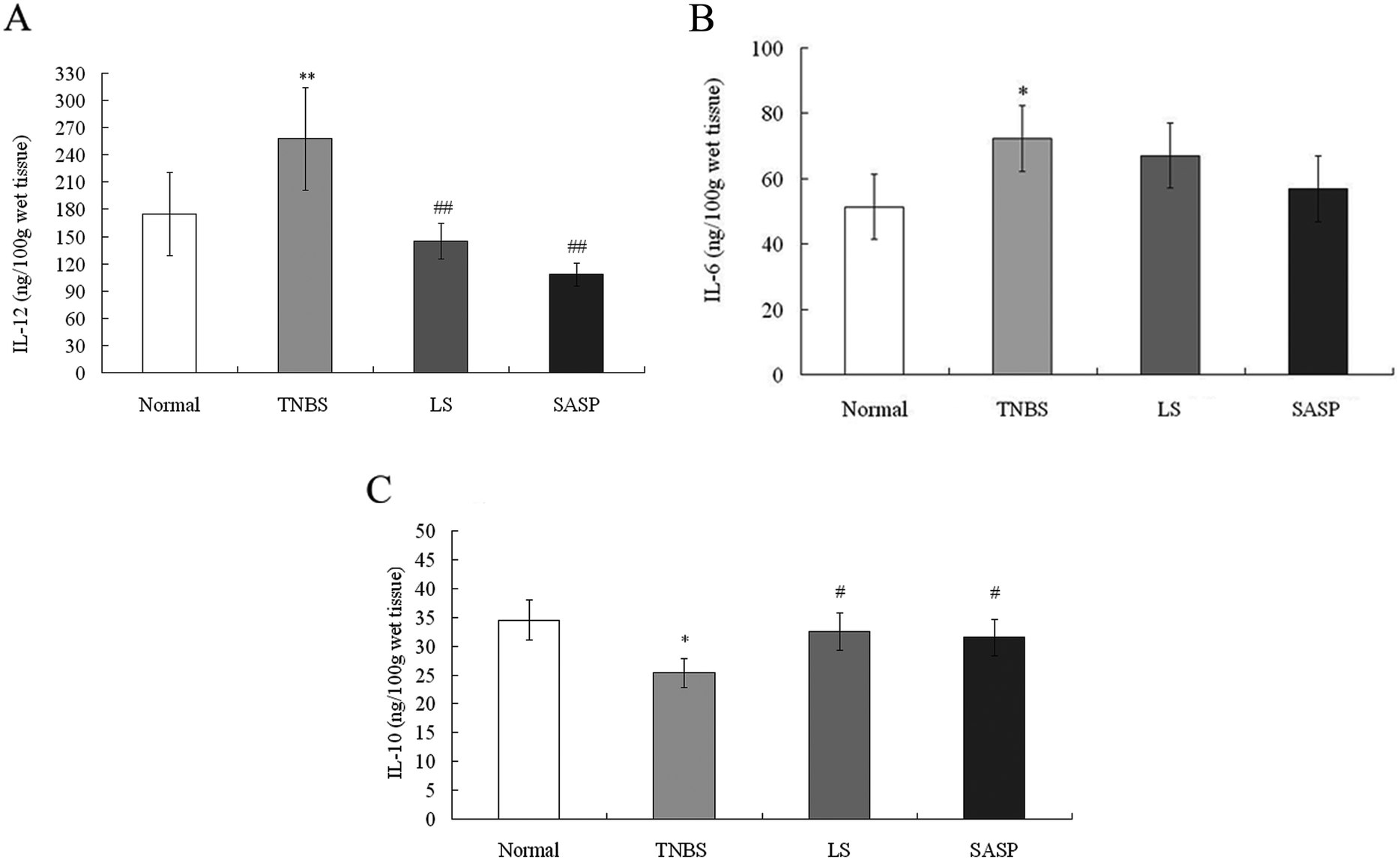 | ||
| Fig. 3 Effects of LS on inflammatory cytokines IL-12 (A), IL-6 (B), and IL-10 (C) in colon of colitic rats. The contents of IL-12 (A), IL-6 (B), and IL-10 (C) were measured using ELISA. Values are expressed as mean ± S.D., n = 6. *Mean values were significantly different compared with normal rats (*P < 0.05; **P < 0.01); #mean values were significantly different compared with TNBS rats (#P < 0.05; ##P < 0.01), n = 6. | ||
3.5 Modulatory effect of LS on the TLR-2, TLR-4 and NF-κB mRNA levels
Gut bacteria influence TLR expression.42 LS supplementation modulated cecum and colonic bacteria in our previous study. The mRNA levels for TLR-2, TLR-4 and NF-κB p65 were then determined. Compared to normal rats, significant increases in mRNA abundances of these proteins in the inflammatory TNBS group was observed after administration of TNBS (Fig. 4). The expression of TLR-2 and NF-κB p65 mRNA in the LS group was significantly down-regulated compared to the inflammatory TNBS group (P < 0.05), and showed no significant difference compared to the normal group. TLR-4 mRNA was reduced by LS supplementation compared to the colitic rat (P > 0.05). SASP supplementation decreased the expression of TLR-2 (P < 0.01) and TLR-4 (P < 0.05) mRNA, as compared with colitic rats, and showed no significant difference compared to normal rats (Fig. 4). The mRNA levels for TLR-2, TLR-4 and NF-κB p65 in the LS group had no significant difference compared to SASP rats.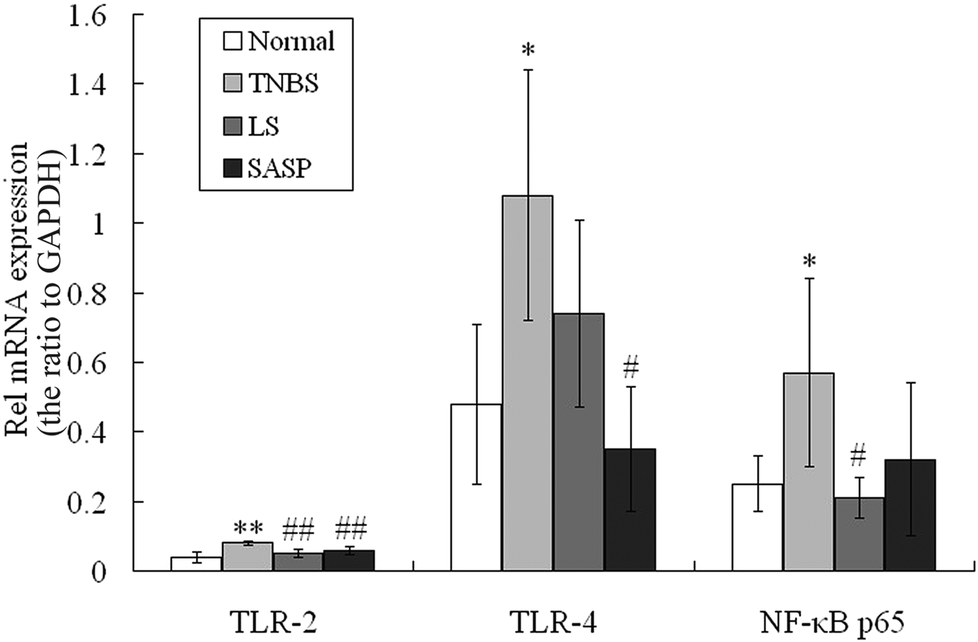 | ||
| Fig. 4 Effects of LS on mRNA expression of TLR and NF-κB. Total RNA was extracted from colon tissues for quantification by real-time fluorescent quantitative PCR. The relative mRNA expression of the target genes was calculated using the 2−ΔΔCt formula and were normalized using GAPDH as the housekeeping gene. Values are expressed as mean ± S.D., n = 6. *Mean values were significantly different compared with normal rats (*P < 0.05; **P < 0.01); #mean values were significantly different compared with TNBS rats (#P < 0.05; ##P < 0.01), n = 6. | ||
3.6 Inhibitory effect of LS supplementation on TLR-2 and NF-κB p65 protein levels in TNBS-induced colitis
Next, the levels of TLR-2 and NF-κB p65 proteins were determined by immunohistochemistry (Fig. 5). The immunohistochemistry of TLR-2 in the colon is shown in Fig. 5A–5D, and the NF-κB p65 is shown in Fig. 5F–5I. Arrows indicate the positive cells. The results showed that the TNBS treatment caused a significant increase in TLR-2 (71.7 ± 20.0 to 583.2 ± 61.0) (Fig. 5E) and NF-κB p65 (97.6 ± 19.1 to 667.9 ± 109.9) (Fig. 5J). TLR-2 and NF-κB p65 protein abundance in the LS group or the SASP group was significantly decreased compared to the TNBS group (inflammatory rats). Compared to the normal group, the levels of TLR-2 and NF-κB p65 in the LS or the SASP group showed no significant difference.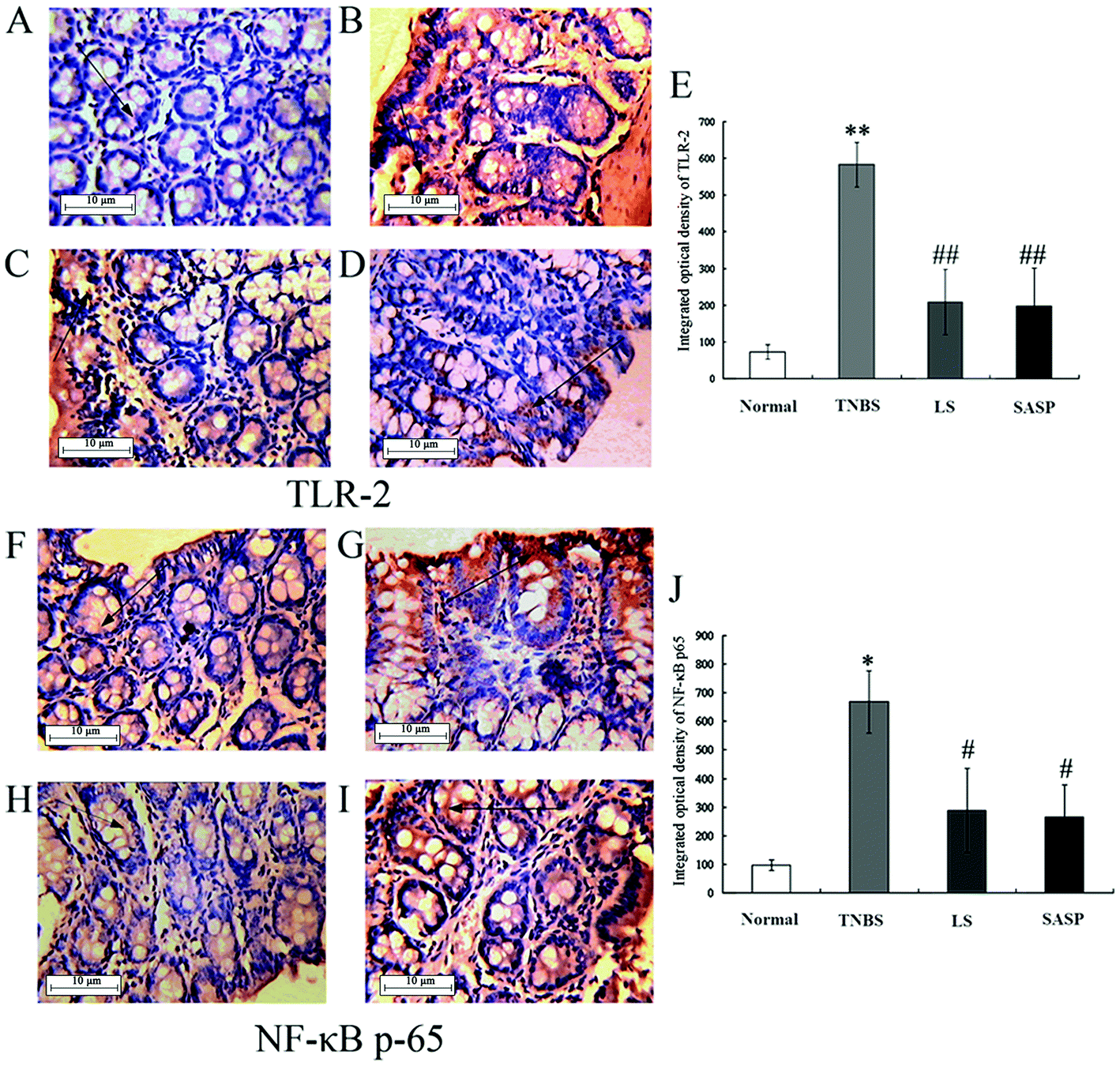 | ||
| Fig. 5 Representative images for the effect of LS on TLR-2 and NF-κB p65 proteins in the colon. TLR-2 and NF-κB p65 proteins in the colonic tissue were measured by immunohistochemistry methods. Immunohistochemistry of TLR-2 expression in the colon: (A) normal control rats, (B) TNBS induced colitic rats, (C) LS-treated rats with colitis induced by TNBS, (D) SASP-treated rats with colitis induced by TNBS, (E) integrated optical density of TLR-2. Immunohistochemistry of NF-κB p65 expression in the colon: (F) normal control rats, (G) TNBS induced colitic rats, (H) LS-treated rats with colitis induced by TNBS, (I) SASP-treated rats with colitis induced by TNBS. (J) Integrated optical density of NF-κB p65. TLR-2 and NF-κB p65 positive cells (arrows) were stained in brown. Values are expressed as mean ± S.D., n = 6. *Mean values were significantly different compared with normal rats (*P < 0.05; **P < 0.01); #Mean values were significantly different compared with TNBS rats (#P < 0.05; ##P < 0.01), n = 6. | ||
The expression of NF-κB p65 in the nucleus was determined by western blot. NF-κB was an important downstream molecule of TLR activation. When NF-κB was activated, it was transported into the nucleus and stimulated various pro-cytokines production. In this study, the expression of NF-κB p65 in the nucleus of intestinal cells in TNBS group rats was increased (P < 0.01), as compared with the normal group. Colitic rats supplemented with LS (P < 0.01) or SASP (P < 0.01) decreased the expression of nuclear NF-κB, as compared with colitic rats, and showed no significant difference compared to normal rats (Fig. 6). The anti-inflammatory effect of LS or SASP may correlate with the inhibition of NF-κB activation, and the effect of LS on the inhibition of NF-κB activation is equivalent to SASP.
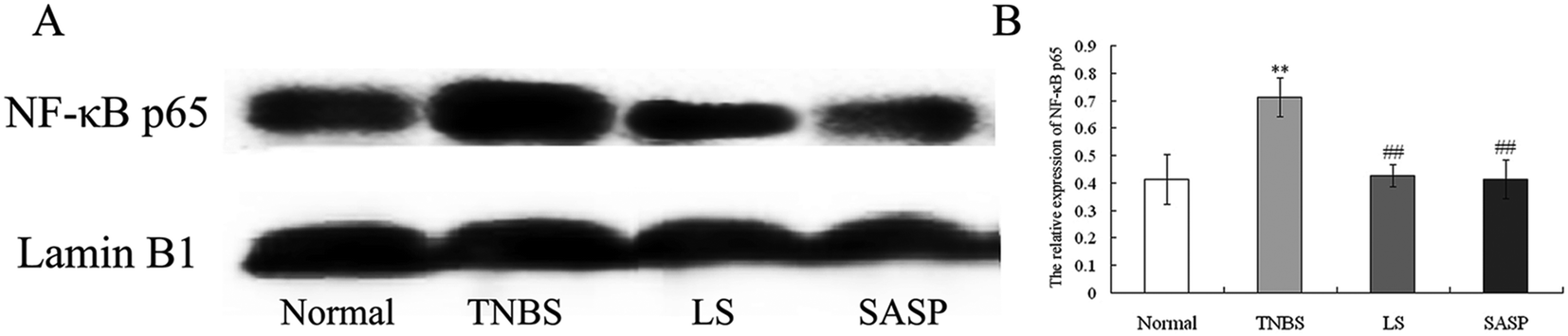 | ||
| Fig. 6 Effect of LS on the expression of nuclear NF-κB p65 in the colon of colitic rats. (A) Western analysis of NF-κB p65 and Lamin B1 expression in the nucleus of the colon. (B) Relative density of NF-κB p65, the density of NF-κB p65 band was normalized to that of Lamin B1. Values were expressed as the mean ± S.D.; n = 3 per group. *Mean values were significantly different compared to normal rats (**P < 0.01); #mean values were significantly different compared with TNBS rats (##P < 0.01). | ||
4. Discussion
The incidence of IBD is on the rise not only in developed countries but also in developing countries, such as China.43 Thus, it is imperative to increase the repertoire of methods available for inhibition of intestinal inflammation. Recent studies have shown that dietary compounds cause remission of IBD by their anti-inflammatory effect and modification of microbiota composition.44–46 Prebiotics, including inulin and fructooligosaccharide, may play an important role in controlling inflammatory disease.47,48 Oligosaccharides have not been extensively studied, but they may offer ideal treatment or co-treatment options due to their capacity to reduce intestinal inflammation. Here, we demonstrated that LS supplementation successfully ameliorates colitis. The mitigatory effect of LS on colitis was found to be similar to that of SASP. Thus, functional oligosaccharides, including LS and fructooligosaccharide, may serve as novel alternative options for the treatment or remission in IBD.In recent years, many studies have demonstrated that development of intestinal inflammation is partly attributed to TLR activation.3,49 Among this family of receptors, TLR-2 and TLR-4 in colitis have been reported in numerous articles and the development of IBD has been paid much attention.5,50–53 TLR-2−/−, TLR-4−/− and TLR-2/-4−/− mice displayed reduced macroscopic signs of acute colitis and the amelioration of inflammation.54 In TNBS-induced murine colitis, the levels of TLR-2 and TLR-4 were increased.55,56 In conclusion, TLR-2 and TLR-4 play an important role in colitis.
TLR-2 and TLR-4 are regulated by intestinal bacteria. TLR-2 recognizes lipoteichoic acid, peptidoglycan and related cell-wall glycopolymers in Gram-positive bacteria and probiotics (including Lactobacilli and Bifidobacteria). TLR-4 recognizes LPS, which is a bacterial cell wall component of Gram-positive bacteria (including Escherichia). Escherichia stimulate TLR-4 activation.57 Lee et al.58 reported that Lactobacillus suntoryeus HY7801 inhibited TLR-4 linked NF-κB activation in TNBS-induced colitic mice. Lactobacillus protects intestinal epithelium from radiation injury in a TLR-2/cyclo-oxygenase-2-dependent manner.59 Oral administration of Bifidobacterium bifidum activated TLR-2 in the intestinal epithelium necrotizing enterocolitis rat.60 In our previous study, LS supplementation in TNBS-induced colonic inflammation increased the levels of Bifidobacterium and Lactobacillus, and decreased the number of Escherichia.32 Therefore, in this study, the expression of TLR-2 and TLR-4 was determined to explore the effect of LS on TNBS-induced rat colitis.
In the present study, LS supplementation in colitic rats significantly decreased the levels of TLR-2 mRNA and protein compared to colitic rats (P < 0.05). The expression of TLR-4 mRNA was decreased compared to inflammatory rats (P > 0.05). These results indicated that LS alleviated colitis associated with down-regulated TLR-2 expression rather than TLR-4. When TLR-2 was activated by its ligands, the production of pro-inflammatory cytokines (such as IL-12) was increased and the production of anti-inflammatory cytokines (such as IL-10) was decreased, suggesting induced inflammation.61,62 It was reported that TLR-2 polymorphisms and protein were up-regulated in IBD patients and animal models with colitis.51,52,55 Inhibited or down-regulated TLR-2 expression may alleviate colonic inflammation. TLR-2 monoclonal antibody supplementation in dextran sulfate sodium-induced colitic mice decreased colonic inflammation.63 Following granulocyte and monocyte adsorption apheresis treatment, TLR-2 cells in the colorectal mucosa decreased.64
The TLRs related signaling pathway might be influenced by microbes and short chain fatty acids. Intestinal microbes alone or combined with a short chain fatty acid, such as butyric acid, impacted TLR-2 activation.65–67 Caco-2 cells cultured with yeast increased the level of TLR-2 mRNA, while Caco-2 cells incubated with butyrate (10 mmol L−1) and yeast decreased the level of TLR-2 mRNA.65 Mirmonsef et al.66 reported that the TLR-2 ligand could enhance IL-8 and TNF-α production in blood mononuclear cells and neutrophils pretreated with low levels of SCFA (0.02–2 mmol L−1), while the TLR-4 ligand did not have such an effect. Kovarik et al.67 suggested that higher concentrations of n-butyrate (0.06–1 mmol L−1) inhibited the release of IL-12/23p40 from blood mononuclear cells (obtained from IBD patients) after activation via TLR-2 agonists, but not TLR-4 engagement. According to several reported papers with vitro experiments, SCFA play a dual-directional regulation in the TLR-2 signaling pathway. In our previous study, the numbers of Lactobacilli and Bifidobacteria, as well as the production of SCFA in LS-supplemented rats were increased, and the numbers of Escherichia were decreased, compared to the inflammatory group.32 In this study, after LS administration, the TLR-2 level decreased, and the decreased TLR-2 expression was related to the increase of Lactobacilli and Bifidobacteria. Short chain fatty acids need further research.
LS possesses an anti-inflammatory effect by up-regulating the IL-10 level, and down-regulating IL-12 and NF-κB levels in the colon. IL-10 is an important anti-inflammatory cytokine for preventing intestinal inflammation. It is evidenced by the spontaneous colitis in IL-10−/− mice.68 Taniguchi et al.29 reported that LS increased the production of IL-10 in ovalbumin/alum-immunized mice. NF-κB can be activated by TLRs, such as TLR-2 and TLR-4, and then stimulates the production of various pro-inflammation cytokines (IL-12 and IL-6 etc.).69 Inhibition of NF-κB is a possible way for combating inflammation. B. longum HY8004 and curcumin have been reported to attenuate colitis via inhibition of NF-κB activation.57,70 In our studies, a diet supplemented with LS or SASP in colitic rats inhibited IL-12 production and nuclear NF-κB p65 expression, and increased the IL-10 production in the colon.
5. Conclusion
LS has an inhibitory effect on intestinal inflammation induced by TNBS. LS exerts similar effects on the ameliorating colitis that is shown by the SASP widely used in the treatment of IBD. LS can increase the production of anti-inflammatory cytokine IL-10 and decrease the production of IL-12, while inhibiting TLR-2 and NF-κB expression at the mRNA and protein levels, and inhibits NF-κB activation. LS may be used as a functional food for IBD patients.Author contributions
Z. Ruan was in charge of the whole project and was involved in the designing of the study and revising the paper. Y. Zhou conducted the animal trial and wrote the part of the paper. H. Li, L. Wang, S. Liu assisted with tissue collection and chemical analyses; X. Zhou, and C. Zhang assisted with RT-PCR; X. Huang assisted with H&E staining and immunohistochemistry. Z. Deng, G. Wu, and Y. Yin assisted with the discussion.Acknowledgements
This research was financially supported by National Natural Science Foundation of China (grant no. 31001014) and the Research Foundation (SKLF-TS-201108 and SKLF-TS-200817) and the Open Project Program (SKLF-KF-201005 and SKLF-KF-201216) of State Key Laboratory of Food Science and Technology, Nanchang University.References
- A. Kaser, S. Zeissig and R. S. Blumberg, Annu. Rev. Immunol., 2010, 28, 573–621 CrossRef CAS PubMed.
- S. Rakoff-Nahoum, J. Paglino, F. Eslami-Varzaneh, S. Edberg and R. Medzhitov, Cell, 2004, 118, 229–241 CrossRef CAS PubMed.
- M. T. Abreu, M. Fukata and M. Arditi, J. Immunol., 2005, 174, 4453–4460 CrossRef CAS.
- P. L. De Jager, D. Franchimont, A. Waliszewska, A. Bitton, A. Cohen, D. Langelier, J. Belaiche, S. Vermeire, L. Farwell, A. Goris, C. Libioulle, N. Jani, T. Dassopoulos, G. P. Bromfield, B. Dubois, J. H. Cho, S. R. Brant, R. H. Duerr, H. Yang, J. I. Rotter, M. S. Silverberg, A. H. Steinhart, M. J. Daly, D. K. Podolsky, E. Louis, D. A. Hafler and J. D. Rioux, Genes Immun., 2007, 8, 387–397 CrossRef CAS PubMed.
- M. Pierik, S. Joossens, K. Van Steen, N. Van Schuerbeek, R. Vlietinck, P. Rutgeerts and S. Vermeire, Inflammatory Bowel Dis., 2006, 12, 1–8 CrossRef.
- H. P. Torok, J. Glas, L. Tonenchi, G. Bruennler, M. Folwaczny and C. Folwaczny, Gastroenterology, 2004, 127, 365–366 CrossRef PubMed.
- I. Atreya, R. Atreya and M. F. Neurath, J. Intern. Med., 2008, 263, 591–596 CrossRef CAS PubMed.
- M. F. Neurath, C. Becker and K. Barbulescu, Gut, 1998, 43, 856–860 CrossRef CAS PubMed.
- A. J. Schottelius and A. S. Baldwin Jr., Int. J. Colorectal Dis., 1999, 14, 18–28 CrossRef CAS.
- G. Rogler, K. Brand, D. Vogl, S. Page, R. Hofmeister, T. Andus, R. Knuechel, P. A. Baeuerle, J. Schölmerich and V. Gross, Gastroenterology, 1998, 115, 357–369 CrossRef CAS.
- I. A. Lee, Y. J. Park, H. K. Yeo, M. J. Han and D. H. Kim, J. Agric. Food Chem., 2010, 58, 10929–10934 CrossRef CAS PubMed.
- S. Schreiber, S. Nikolaus and J. Hampe, Gut, 1998, 42, 477–484 CrossRef CAS PubMed.
- M. F. Neurath, S. Pettersson, K. H. Meyer zum Buschenfelde and W. Strober, Nat. Med., 1996, 2, 998–1004 CrossRef CAS PubMed.
- W. Reinisch, C. Gasche, W. Tillinger, J. Wyatt, C. Lichtenberger, M. Willheim, C. Dejaco, T. Waldhör, S. Bakos, H. Vogelsang, A. Gangl and H. Lochs, Am. J. Gastroenterol., 1999, 94, 2156–2164 CrossRef CAS PubMed.
- C. Van Kemseke, J. Belaiche and E. Louis, Int. J. Colorectal Dis., 2000, 15, 206–210 CrossRef CAS.
- M. F. Neurath, I. Fuss, B. L. Kelsall, E. Stuber and W. Strober, J. Exp. Med., 1995, 182, 1281–1290 CrossRef CAS.
- G. Bouma, A. Kaushiva and W. Strober, Gastroenterology, 2002, 123, 554–565 CrossRef CAS PubMed.
- D. F. Fiorentino, M. W. Bond and T. R. Mosmann, J. Exp. Med., 1989, 170, 2081–2095 CrossRef CAS.
- R. Kuhn, J. Lohler, D. Rennick, K. Rajewsky and W. Muller, Cell, 1993, 75, 263–274 CrossRef CAS.
- T. Karrasch, J. S. Kim, M. Muhlbauer, S. T. Magness and C. Jobin, J. Immunol., 2007, 178, 6522–6532 CrossRef CAS.
- S. L. Yeh, T. C. Wu, S. T. Chan, M. J. Hong and H. L. Chen, Eur. J. Nutr., 2013, 53, 449–456 CrossRef PubMed.
- J. K. Hou, B. Abraham and H. El-Serag, Am. J. Gastroenterol., 2011, 106, 563–573 CrossRef CAS PubMed.
- T. Mishiro, R. Kusunoki, A. Otani, M. M. U. Ansary, M. Tongu, N. Harashima, T. Yamada, S. Sato, Y. Amano, K. Itoh, S. Ishihara and Y. Kinoshita, Lab. Invest., 2013, 93, 834–843 CrossRef CAS PubMed.
- H. Sokol, P. Seksik, L. Rigottier-Gois, C. Lay, P. Lepage, I. Podglajen, P. Marteau and J. Doré, Inflammatory Bowel Dis., 2006, 12, 106–111 CrossRef PubMed.
- J. L. Wallace, T. Le, L. Carter, C. B. Appleyard and P. L. Beck, J. Pharmacol. Toxicol., 1995, 33, 237–239 CrossRef CAS.
- S. Wirtz and M. F. Neurath, Int. J. Colorectal Dis., 2000, 15, 144–160 CrossRef CAS.
- K. Honda, T. Matsumoto, F. Kuroki, M. Iida, M. Oka and I. Sawatani, Scand. J. Gastroenterol., 1999, 34, 264–269 CAS.
- K. Hino, M. Kurose, T. Sakurai, S. I. Inoue, K. Oku, H. Chaen and S. Fukuda, J. Appl. Glycosci., 2007, 54, 169–172 CrossRef CAS.
- Y. Taniguchi, A. Mizote, K. Kohno, K. Iwaki, K. Oku, H. Chaen and S. Fukuda, Biosci., Biotechnol., Biochem., 2007, 71, 2766–2773 CrossRef CAS PubMed.
- F. Teramoto, K. Rokutan, Y. Kawakami, Y. Fujimura, J. Uchida, K. Oku, M. Oka and M. Yoneyama, J. Gastroenterol., 1996, 31, 33–39 CrossRef CAS.
- T. Ohkusa, Y. Ozaki and S. Tokoi, Gastroenterology, 1993, 104, A757–757S Search PubMed.
- X. Zhou, Z. Ruan, X. Huang, Y. Zhou, S. Liu and Y. Yin, Food Sci. Biotechnol., 2014, 23, 1–7 CrossRef PubMed.
- Z. Ruan, S. Liu, Z. Cui, D. Su, X. Wu, Z. Dai, C. Luo, C. Liao and Y. Yin, J. Food Agric. Environ., 2012, 10, 176–181 CAS.
- Z. Ruan, Y. Lv, X. Fu, Q. He, Z. Deng, W. Liu, X. Wu, G. Wu, X. Wu and Y. Yin, Amino Acids, 2013, 45, 877–887 CrossRef CAS PubMed.
- S. Melgar, A. Karlsson and E. Michaëlsson, Am. J. Physiol.: Gastrointest. Liver Physiol., 2005, 288, G1328–G1338 CrossRef CAS PubMed.
- B. R. MacPherson and C. J. Pfeiffer, Digestion, 1978, 17, 135–150 CAS.
- Y. M. Kim, R. Romero, T. Chaiworapongsa, J. Espinoza, G. Mor and C. J. Kim, Histopathology, 2006, 49, 506–514 CrossRef CAS PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- K. Poelstra, W. W. Bakker, P. A. Klok, J. A. Kamps, M. J. Hardonk and D. K. Meijer, Am. J. Pathol., 1997, 151, 1163–1169 CAS.
- H. Bentala, W. R. Verweij, A. Huizinga-Van der Vlag, A. M. van Loenen-Weemaes, D. K. Meijer and K. Poelstra, Shock, 2002, 18, 561–566 CrossRef PubMed.
- M. G. Neuman, Transl. Res., 2007, 149, 173–186 CrossRef CAS PubMed.
- S. Rakoff-Nahoum, J. Paglino, F. Eslami-Varzaneh, S. Edberg and R. Medzhitov, Cell, 2004, 118, 229–241 CrossRef CAS PubMed.
- APDW2004 Chinese IBD Working Group, J. Gastroenterol. Hepatol., 2006, 21, 1009–1015 CrossRef PubMed.
- M. H. Pan, C. S. Lai, S. Dushenkov and C. T. Ho, J. Agric. Food Chem., 2009, 57, 4467–4477 CrossRef CAS PubMed.
- M. Lee, J. Kovacs-Nolan, T. Archbold, M. Z. Fan, L. R. Juneja, T. Okubo and Y. Mine, J. Funct. Foods, 2009, 1, 161–169 CrossRef CAS PubMed.
- T. Wakuda, K. Azuma, H. Saimoto, S. Ifuku, M. Morimoto, I. Arifuku, M. Asaka, T. Tsuka, T. Imagawa, Y. Okamoto, S. Osaki and S. Minami, J. Funct. Foods, 2013, 5, 516–523 CrossRef CAS PubMed.
- H. M. Celine, L. Leenen and A. Dieleman, J. Nutr., 2007, 137, 2572S–2575S Search PubMed.
- A. R. Lomax and P. C. Calder, Br. J. Nutr., 2009, 101, 633–658 CrossRef CAS PubMed.
- K. O. Arseneau, H. Tamagawa, T. T. Pizarro and F. Cominelli, Curr. Gastroenterol. Rep., 2007, 9, 508–512 CrossRef PubMed.
- E. Cario and D. K. Podolsky, Infect. Immun., 2000, 68, 7010–7017 CrossRef CAS.
- B. Szebeni, G. Veres, A. Dezsofi, K. Rusai, A. Vannay, M. Mraz, E. Majorova and A. Arato, Clin. Exp. Immunol., 2008, 151, 34–41 CrossRef CAS PubMed.
- L. Frolova, P. Drastich, P. Rossmann, K. Klimesova and H. Tlaskalova-Hogenova, J. Histochem. Cytochem., 2008, 56, 267–274 CrossRef CAS PubMed.
- F. Wang, T. Tahara, T. Arisawa, T. Shibata, M. Nakamura, H. Fujita, M. Iwata, Y. Kamiya, M. Nagasaka, K. Takahama, M. Watanabe, I. Hirata and H. Nakano, J. Gastroenterol. Hepatol., 2007, 22, 925–929 CrossRef CAS PubMed.
- M. M. Heimesaat, A. Fischer, B. Siegmund, A. Kupz, J. Niebergall, D. Fuchs, H. K. Jahn, M. Freudenberg, C. Loddenkemper, A. Batra, H. A. Lehr, O. Liesenfeld, M. Blaut, U. B. Göbel, R. R. Schumann and S. Bereswill, PLoS One, 2007, 2, e662 Search PubMed.
- A. Arranz, C. Abad, Y. Juarranz, M. Torroba, F. Rosignoli, J. Leceta, R. P. Rosignoli and C. Martinez, Anal. N. Y. Acad. Sci., 2006, 1070, 129–134 CrossRef CAS PubMed.
- Y. Zhao, S. Zhang, L. Jiang, J. Jiang and H. Liu, J. Gastroenterol. Hepatol., 2009, 24, 1775–1780 CrossRef PubMed.
- R. I. Tapping, S. Akashi, K. Miyake, P. J. Godowski and P. S. Tobias, J. Immunol., 2000, 165, 5780–5787 CrossRef CAS.
- I. A. Lee, E. A. Bae, J. H. Lee, H. Lee, Y. T. Ahn, C. S. Huh and D. H. Kim, Inflammation Res., 2010, 59, 359–368 CrossRef CAS PubMed.
- J. H. Lee, B. Lee, H. S. Lee, E. A. Bae, H. Lee, Y. T. Ahn, K. S. Lim, C. S. Huh and D. H. Kim, Int. J. Colorectal Dis., 2009, 24, 231–237 CrossRef PubMed.
- L. Khailova, S. K. M. Patrick, K. M. Arganbright, M. D. Halpern, T. Kinouchi and B. Dvorak, Am. J. Physiol.: Gastrointest. Liver Physiol., 2010, 299, G1118–G1127 CrossRef CAS PubMed.
- T. U. Sybille, S. M. Kiertscher, M. T. Ochoa, D. A. Michael, V. Norgard, K. Miyake, P. Godowski, J. M. D. Roth and R. L. Modlin, J. Immunol., 2000, 165, 3804–3810 CrossRef.
- H. L. Jin, L. Kumar, C. Mathias, D. Zurakowski, H. Oettgen, L. Gorelik and R. Geha, J. Allergy Clin. Immunol., 2009, 123, 875–882 CrossRef CAS PubMed.
- L. Dong, J. Li, Y. Liu, W. Yue and X. Luo, J. Gastroenterol. Hepatol., 2012, 27, 110–119 CrossRef CAS PubMed.
- V. Muratov, A. K. Ulfgren, M. Engström, K. Elvin, O. Winqvist, R. Löfberg and J. Lundahl, J. Gastroenterol., 2008, 43, 277–282 CrossRef PubMed.
- S. Saegusa, M. Totsuka, S. Kaminogawa and T. Hosoi, FEMS Immunol. Med. Microbiol., 2004, 41, 227–235 CrossRef CAS PubMed.
- P. Mirmonsef, M. R. Zariffard, D. Gilbert, H. Makinde, A. L. Landay and G. T. Spear, Am. J. Reprod. Immunol., 2012, 67, 391–400 CrossRef CAS PubMed.
- J. J. Kovarik, W. Tillinger, J. Hofer, M. A. Hölzl, H. Heinzl, M. D. Saemann and G. J. Zlabinger, Eur. J. Clin. Invest., 2011, 41, 291–298 CrossRef CAS PubMed.
- R. Kühn, J. Löhler, D. Rennick, K. Rajewsky and W. Müller, Cell, 1993, 75, 263–274 CrossRef.
- R. G. Baker, M. S. Hayden and S. Ghosh, Cell Metab., 2011, 13, 11–22 CrossRef CAS PubMed.
- M. S. Baliga, N. Joseph, M. V. Venkataranganna, A. Saxena, V. Ponemone and R. Fayad, Food Funct., 2012, 3, 1109–1117 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4fo00381k |
| This journal is © The Royal Society of Chemistry 2015 |