Aryl hydrocarbon receptor potency of chlorinated parabens in the aquatic environment†
Received
19th February 2015
, Accepted 20th March 2015
First published on 23rd March 2015
Abstract
Parabens are widely used as preservatives in personal care products. Parabens and their chlorinated by-products are continuously released into aquatic environments where aryl hydrocarbon receptor (AhR) potency was observed. Chlorinated parabens have structural similarities to hydroxylated polychlorinated biphenyls (PCBs, which also possess AhR potency) in the number of chlorine atoms and hydroxyl groups in the aromatic ring. The aryl hydrocarbon receptor potency of 24 compounds, including chlorinated and non-chlorinated parabens, degradation products of non-parabens, and possible degradation products of chlorinated parabens, was measured using a yeast (YCM3) reporter gene and cytochrome P450 (CYP)1, ethoxyresorufin O-deethylase (EROD) assays. We specifically focused on the AhR potency of parabens in water samples from swimming pools and from rivers receiving effluents from sewage treatment plants (STPs). The yeast assay revealed 16 compounds exhibiting AhR potency, with their potency relative to the AhR ligand β-naphthoflavone (BNF) ranging from 5 × 10−5 to 9 × 10−2. In human hepatoma cell line HepG2, four parabens caused significant induction of EROD activity (p < 0.05); contents of isobutyl paraben (iBP) and dichlorinated isopropyl paraben (Cl2iPP) in each treatment group were 2.6- and 1.8-fold greater, respectively, than that of the vehicle control at 100 μM. The occurrences of all compounds in swimming pool samples and of 20 compounds in river samples were analysed using gas chromatography-mass spectrometry, which revealed total concentrations of 2.7–1100 ng L−1 and 83–4600 ng L−1, respectively. Water samples exhibited AhR potency; their BNF equivalents ranged from 4.0 to 960 pM in pool water and from 25 to 3500 pM in river water. The contribution ratio of active compounds accounted for up to 6.3% (pool water) and 0.94% (river water) of the observed potency of water samples. This study provides the first confirmation of AhR-mediated toxicity of parabens using in vitro assays.
Water impact
Activation of the aryl hydrocarbon receptor (AhR) induces a broad spectrum of biological responses such as induction of the cytochrome P450 (CYPs) system, disruption of normal hormone signalling pathways, reproductive toxicity, immunotoxicity and mutagenicity. Parabens are widely used as preservatives in pharmaceuticals and personal care products (PPCPs). Recently, parabens and their chlorinated by-products have been identified as pollutants in many aquatic environments where AhR potencies have been observed. However, little is known about the AhR potencies of parabens and chlorinated parabens. Identifying the undesirable effects of parabens, which are regularly used in PPCPs, is also important for water management. This study provides the first confirmation of the water impact via AhR toxicity of parabens.
|
Introduction
Parabens are widely used as preservatives in pharmaceuticals and personal care products (PPCPs) because they exhibit a broad spectrum of antimicrobial activity, are stable over a wide pH range, and are sufficiently soluble in water.1 Parabens are a group of lower alkyl (C1–C4) or benzyl esters of 4-hydroxybenzoic acid. Methyl- and propylparabens are the most commonly used parabens in numerous cosmetics, and the most widely used preservative system consists of a combination of these compounds.2 The European Community Directive's standards for paraben use permit a maximum concentration of 0.4% (w/w) for single esters and a total maximum concentration of 0.8% (w/w). In Japan, parabens are permitted to be used at concentrations of up to 1% (w/w) in all cosmetic products.
Due to their wide use, parabens are continuously released into aquatic environments via domestic wastewater; concentrations ranging from 3.7 ng L−1 to 85 ng L−1 have been detected in river water receiving effluents from sewage treatment plants.3,4 Compounds including parabens that contain phenolic hydroxyl groups exhibit favorable chlorination kinetics; thus, parabens are easily chlorinated by chlorine in tap water and by sodium hypochlorite, a bleaching agent.5,6 Several chlorinated parabens have been found in aquatic environments. Occurrences of dichlorinated derivatives of methyl- and propylparabens were reported in raw wastewater.7 Chlorinated derivatives of methyl-, propyl-, and benzylparabens have been identified in water samples from public swimming pools at levels up to 25 ng L−1.8 More recently, chlorinated parabens (up to 90 ng L−1) were found in raw wastewater9 and river water (up to 28 ng L−1).4
The most frequently used parabens, methyl- and propylparabens, are considered to be non-toxic to animals via oral or parenteral routes.1,10 However, several studies have demonstrated that parabens exhibit endocrine-disrupting properties to mammals, including in vitro studies on estrogenic activity employing the recombinant yeast estrogen screen transfected with a human estrogen receptor α (hERα) gene and expression plasmids,11 and in vivo studies using rat12 or mouse uterotrophic assays.13 These reports reveal that parabens exert varying degrees of estrogenic activity, with potency ranging from 10−7- to 10−4-fold compared to the activity of 17β-estradiol. Effects of chlorinated paraben by-products on estrogenic activity have also been assessed. Chlorination masked the apparent estrogenic activity of the parent compounds in yeast two-hybrid assays that incorporated hERα or ERα from the freshwater fish medaka (Oryzias latipes).14
Regarding the biological activity of halogenated organic contaminants, groups of chlorinated aromatic hydrocarbons, such as TCDDs and PCBs, interact with the aryl hydrocarbon receptor (AhR), which is a ligand-dependent transcription factor.15 Some hydroxylated PCBs16–18 and phytochemicals19,20 have been shown to act as xenobiotic and natural ligand-like compounds (e.g., polyphenols and indole-3-carbinol, respectively). In addition, it has been suggested that indigo and indirubin are endogenous AhR agonists present in human urine.21 Activation of the AhR by PCB-like compounds induces a broad spectrum of biological responses, including induction of the cytochrome P450 (CYPs) system (particularly CYP1A1); disruption of normal hormone signaling pathways; neuroendocrine, developmental, and reproductive toxicity; immunotoxicity; and mutagenicity.22–24 Following ligand binding, the cytosolic ligand–AhR complex translocates to the nucleus and dimerizes with AhR nuclear translocator (ARNT) protein. The AhR–ARNT complex binds to dioxin-responsive elements (DRE, specific DNA sequences upstream of AhR-responsive genes such as CYP1A1/2) and increases gene transcription rates.25 Therefore, the binding of environmental xenobiotics to AhR is considered an indication of possible toxicity and has recently received considerable attention. In addition, activation of AhR by xenobiotics modulates a number of endocrine systems, perhaps most notably by interfering with estrogen signaling.26 The molecular basis for this inhibitory crosstalk is unclear and may be due to a combination of several proposed mechanisms.27,28
AhR potency has been detected in sewage treatment plant (STP) effluents,29,30 in river water from urban and industrial areas.31 Parabens and chlorinated parabens have also been identified as pollutants in aquatic environments where AhR potency was observed. Chlorinated parabens have structural similarities to hydroxylated PCBs (which also possess AhR potency) in the number of chlorine atoms and hydroxyl groups in the aromatic ring. Mono-hydroxylated PCBs with one and two chlorine atoms exhibit positive responses,16 suggesting that all chlorinated parabens might have AhR potency. In the present study, we aimed to measure the AhR potency of 24 paraben compounds, including parabens and their degradation products and chlorinated parabens and their possible degradation products, and to investigate the contribution of chlorinated parabens to the total AhR potency in water samples. For degradation products, parabens have been shown to form 4-hydroxybenzoic acid (HBA) via aerobic transformation by microbes.32 Further, structurally chlorinated parabens can be hydrolysed to chlorinated hydroxybenzoic acids (ClHBA and Cl2HBA), the secondary product. AhR potency was evaluated using a recombinant yeast reporter gene assay incorporating the human AhR and ARNT and AhR-dependent induction of CYP1A, ethoxyresorufin O-deethylase (EROD) activity in human hepatoma cell line HepG2. Samples were collected from swimming pool water and rivers receiving STP effluents. We used gas chromatography-mass spectrometry (GC-MS) to quantify parabens in the SPE extracts.
Experimental
Yeast assay for aryl hydrocarbon receptor potency
The transcriptional agonist potency of compounds to AhR was measured by a reporter assay using the recombinant yeast YCM3 strain.33 The YCM3 cells were transfected with an expression plasmid, pBEVY-GL-AHR/ARNT, which coexpresses human AhR and ARNT genes, and with a lacZ reporter plasmid, pTXRE5-Z, which carries the response element for the AhR complex, XRE5. The assay procedure was performed as described by Miller,33 with minor modification. Yeast cells were preincubated for 18 h at 30 °C under shaking conditions in synthetic glucose medium (2% glucose, lacking tryptophan). A solution (1.25 μL) of the real sample or test compound in DMSO was mixed with 250 μL of synthetic galactose medium (2% galactose, lacking tryptophan) in the first row of a 96-well microplate. The test solution was diluted 5-fold serially from rows 1 to 7 with the synthetic galactose medium, and the volume was adjusted to 200 μL. Five microliters of yeast cell suspension was then added to each well (including wells in row 8, which served as the controls), and the mixture was incubated at 30 °C for 18 h. After incubation, cell density was determined by reading the absorbance at 600 nm (A600) using a microplate reader (Multiskan GO; Thermo Fisher Scientific KK, Yokohama, Japan). Each cell suspension (10 μL) was transferred into a new microplate and digested by addition of 50 μL of Z buffer containing 1.5 mg mL−1 zymolyase 20 T, and incubated at 30 °C for 1 h. Then, 50 μL of 0.5 M phosphate buffer (pH 7.0) containing 0.5 mg mL−1 chlorophenol red-β-galactopyranoside (CPRG) was added, and the samples were reacted at 30 °C for 10 min; the colorimetric reaction was stopped by addition of 90 μL of 1 M Na2CO3. β-galactosidase activity was assessed by measuring absorbance at 578 nm (A578) and calculated as follows:
| β-galactosidase activity = (1000 × A578)/[A600 × 0.01(cell suspension added, mL) × 10(reaction time, min)]. |
BNF was used as a positive control. AhR potency in the yeast assay was recorded as the 25% relative effective concentration (REC25, the concentration of the test compounds showing 25% of the maximal response to BNF). The dose–response curve of each compound was derived using logistic regression analysis with the following equation:
| Y = bottom + (top–bottom)/(1 + Ks/Xslope) |
where
X is the sample concentration (nM) or sample concentration ratio,
Y is the response (from bottom to top, with a sigmoid shape), Ks is the substrate constant, and slope is the Hill coefficient. Ks and slope were determined by using the Solver function in Microsoft Excel 2010. The sum of the percent contribution of all active compounds to the observed AhR potency of individual water samples was defined as the contribution of total parabens. This analysis excluded active compounds with concentrations below the LOQ. When water samples or individual compounds could not reach the maximal effect within the concentration range tested, we determined the REC25 from two response points closest to the highest dose.
Ethoxyresorufin O-deethylase assay
Human hepatoma HepG2 cells were purchased from American Type Culture Collection (Manassas, VA) and maintained in Dulbecco's modified Eagle's medium (Wako Pure Chemical Industries, Ltd., Osaka, Japan) supplemented with 10% fetal bovine serum, penicillin (100 U mL−1), and streptomycin (100 μg mL−1) at 37 °C under a humidified atmosphere of 95% air and 5% CO2. EROD activity was used as a marker for CYP1A1 activity in HepG2 cells. The cells were seeded, and after 24 h of incubation, they were treated with 50 or 100 μM of each compound, 2 μM BaP as a positive control, or DMSO as a vehicle control, and incubated for 24 h. After incubation, the medium was removed, and the medium containing 7-ethoxyresorufin (2.5 μM) and dicumarol (10 μM) was added to each well. After incubation for 20 min at 37 °C, fluorometric (Ex: 530 nm, Em: 590 nm) endpoint analysis for the conversion of ethoxyresorufin to resorufin was conducted using a fluorescence microplate reader (Thermo Fisher Scientific Inc., Worcester, MA). Cell survival was calculated using Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan), in accordance with the manufacturer's protocol. DMSO concentration in the assay mixture was 0.2%, and we confirmed, in a preliminary study, that DMSO at this concentration did not substantially affect the viability of HepG2 cells as well as EROD activity.
Sample collection and characterization
Water samples were collected in August and December 2012 and May and August 2013 from 17 public swimming pools (17 grab samples) and river sites (20 grab samples) receiving effluents from 20 sewage treatment plants in Shizuoka Prefecture, in the central Pacific region of Japan (Fig. S1 in the ESI†). Water (3 L) was collected in glass bottles with stoppers, quenched with 100 mg of Na2S2O3, transported to the laboratory on ice in coolers, and stored at 4 °C until analysis (within 2 days). The sample characteristics are provided in Tables S1 and S2 (ESI†).
Solid-phase extraction of water samples
Samples (1 L) were spiked with 20 μL of methanolic solution containing 100 μg mL−1 of deuterated bisphenol A as a surrogate standard for chemical analysis. Samples were then adjusted to pH 2 with 2 M hydrochloric acid and passed through a vacuum manifold onto a conditioned styrene polymer cartridge (230 mg of InertSep mini RP-1; GL Science, Inc., Tokyo, Japan), which had been rinsed with 10 mL of methanol followed by 10 ml of deionized water under reduced pressure. After drying for 30 min, the solution was eluted with 10 mL of acetonitrile. The acetonitrile eluent was collected in a test tube and evaporated using a nitrogen stream for bioassay or GC-MS analysis.
Materials and chemical analysis
Chlorinated parabens were synthesized by chlorination using sulfuryl chloride.34 The other reagents and solvents were of analytical reagent grade purchased from commercial suppliers and were not purified further. The molecular structures of the standards used are shown in Fig. 1. Extracted samples were reconstituted into 850 μL of ethyl acetate and 150 μL of BSTFA. The solutions were allowed to stand for 1 h at room temperature to complete trimethylsilylation. After the reaction was complete, 500 μL of samples were mixed with 5 μL of ethyl acetate containing 100 μg mL−1 of an internal standard (pyrene-d10) in an autosampler vial, and examined using GC-MS with a mass-selective detector (HP 5973 N Series; Agilent Technologies, Palo Alto, CA) on an HP-5 fused-silica capillary column (J&W Scientific, Folsom, CA), according to our previously described method.4 Compounds were identified by comparing the GC retention times and mass fragmentograms of the selected ions in the samples with those of authentic standards (Fig. 1). These compounds were quantified by correlating the ratio of their peak areas with that of the internal standard using a calibration curve created with the standard solution. The limit of detection (LOD) and limit of quantification (LOQ) were calculated by multiplying the standard deviation of river water spiked with the lowest calibration concentration of each compound (n = 5 samples per compound) by 3 and 10, respectively. During sample analysis, the LOD ranged from 0.63 to 4.2 ng L−1 and the LOQ ranged from 2.1 to 14 ng L−1. The recovery yield for each compound varied between 92% and 104%, and the repeatability (relative standard deviation) of the results was within 11%. Details on quality assurance and control are presented in Table S3 (ESI†).
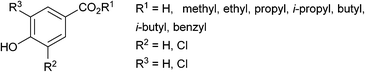 |
| | Fig. 1 Molecular structures of target compounds. | |
Statistical analysis
Data are expressed as mean values ± standard deviation (SD) from a minimum of three assays performed in duplicate. Statistical differences in HepG2 EROD activity were assessed by analyses of variance (ANOVA) followed by Dunnett's multiple comparison test.
Results and discussion
AhR potency of chlorinated parabens
Table 1 presents the REC25 values for AhR potency of chlorinated parabens, their parent compounds, and their primary degradation products, measured using the yeast assay. The dose–response relationships for 24 compounds in the yeast assay are shown in Fig. 2. Sixteen compounds exhibited AhR potency, with their relative potency to BNF (RP) ranging from 5 × 10−5 to 9 × 10−2. The RP values of the active compounds increased progressively with increasing hydrophobicity of the ester group for each compound of parabens and mono- and dichlorinated parabens. All of the monochlorinated parabens also exhibited greater AhR potency than their corresponding parent compounds, a behavior analogous to that of monohalogenated aza-PAHs observed using a yeast assay.35 However, further chlorination of parabens reduced or eliminated their AhR potency. Previous studies using rat hepatoma cell (H4IIE-luc) assay have shown a structure–activity relationship for AhR potency of ClPAHs.36 The AhR potency increased with the number of chlorine substitutions in low-molecular-weight ClPAHs and decreased with the increasing number of chlorine substitutions on the aromatic rings in high-molecular-weight ClPAHs. Hence, the effect of chlorine substitution on AhR potency in parabens was analogous to that of the chlorination of high-molecular-weight PAHs. When parabens underwent electrophilic substitution, most substitutions did not occur at the meta position but at the ortho position of the hydroxyl group. Substitutions, such as those in tri- or tetra-chlorinated parabens, could not occur in the aquatic environment. However, further studies are required to evaluate the AhR potency of highly chlorinated parabens and to elucidate the quantitative structure–activity relationship. Most active compounds yielded a typical dose-dependent response curve. In particular, the responses of 4 monochlorinated parabens (ClPP, CliBP, ClEP, and CliPP) were greater than that of BNF at the maximum concentration (Fig. 2). Some dichlorinated parabens (Cl2iPP, Cl2BP, and Cl2iBP) showed a marked decline in response at higher concentrations (>104 nM) due to their toxicity to the yeast cells. HBA, ClHBA, and Cl2HBA, which are primary degradation products of parabens, did not elicit significant responses at the concentration range tested in this study (<105 nM). Thus, hydrolysis of the ester group in paraben molecules appears to eliminate the AhR potency of these compounds.
Table 1 AhR potency of compounds in a yeast assay (mean ± standard deviation, n = 3)
|
|
Structurea |
REC25b (nM) |
RPc |
| Abbr. |
R
1
|
R
2
|
R
3
|
|
|
|
See Fig. 1.
Concentration of the compound showing 25% of the maximum BNF signal.
Relative potency to BNF.
Not active is defined as compounds showing <25% concentration of the maximum BNF signal.
The value determined from two response points at the high dose.
|
| MP |
Methyl |
H |
H |
Not actived |
|
| EP |
Ethyl |
H |
H |
Not active |
|
| PP |
Propyl |
H |
H |
46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 2500 000 ± 2500 |
5 × 10−5 |
| iPP |
Isopropyl |
H |
H |
Not active |
|
| BP |
Butyl |
H |
H |
25000 ± 4800 |
1 × 10−4 |
| iBP |
Isobutyl |
H |
H |
5900 ± 240 |
4 × 10−4 |
| BnP |
Benzyl |
H |
H |
2600 ± 360 |
1 × 10−3 |
| HBA |
H |
H |
H |
Not active |
|
| ClMP |
Methyl |
Cl |
H |
14000 ± 1000 |
2 × 10−4 |
| ClEP |
Ethyl |
Cl |
H |
5600 ± 670 |
4 × 10−4 |
| ClPP |
Propyl |
Cl |
H |
670 ± 160 |
4 × 10−3 |
| CliPP |
Isopropyl |
Cl |
H |
2100 ± 310 |
1 × 10−3 |
| ClBP |
Butyl |
Cl |
H |
110 ± 14 |
2 × 10−2 |
| CliBP |
Isobutyl |
Cl |
H |
97 ± 48 |
3 × 10−2 |
| ClBnP |
Benzyl |
Cl |
H |
29 ± 11 |
9 × 10−2 |
| ClHBA |
H |
Cl |
H |
Not active |
|
| Cl2MP |
Methyl |
Cl |
Cl |
13000 ± 2700 |
2 × 10−4 |
| Cl2EP |
Ethyl |
Cl |
Cl |
9900 ± 2000e |
3 × 10−4 |
| Cl2PP |
Propyl |
Cl |
Cl |
1300 ± 83e |
2 × 10−3 |
| Cl2iPP |
Isopropyl |
Cl |
Cl |
Not active |
|
| Cl2BP |
Butyl |
Cl |
Cl |
380 ± 160 |
7 × 10−3 |
| Cl2iBP |
Isobutyl |
Cl |
Cl |
Not active |
|
| Cl2BnP |
Benzyl |
Cl |
Cl |
290 ± 110 |
9 × 10−3 |
| Cl2HBA |
H |
Cl |
H |
Not active |
|
| BNF |
|
|
|
2.5 ± 0.24 |
1 |
 |
| | Fig. 2 Dose–response curves of the AhR potency of compounds in a yeast assay. Each point represents the mean ± standard deviation (n = 3). | |
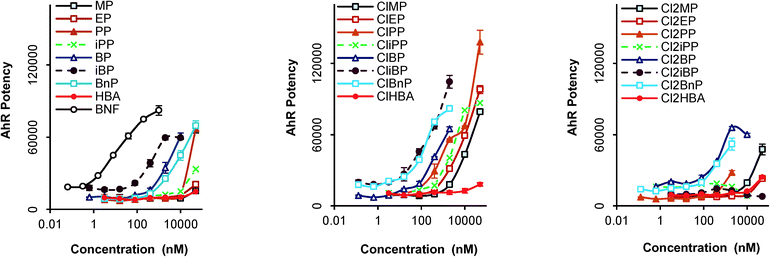
Fig. 3 presents the EROD activity of 24 compounds in HepG2 cells at 100 μM and 150 μM. The tested concentration range did not exhibit cytotoxicity (defined as <80% viability of control cells) except for ClBnP at 100 μM (cell viability = 74%). The CYP1A-dependent EROD activity of four parabens was significantly enhanced compared to that of the vehicle control (p < 0.05). Isobutyl paraben (iBP) caused an increase in EROD activity that was 2.6-fold higher than that of the vehicle control at 100 μM. Increases in EROD activity caused by iBP were similar at 100 and 150 μM, presumably because maximal CYP1A induction occurred at or below 100 μM. The limited data in Fig. 3 also indicates that the EROD activity of parabens with branched side chains is higher than that of parabens with linear side chains (e.g. Cl2iPP vs. Cl2PP and Cl2iBP vs. Cl2BP). The reporter gene and EROD assays demonstrated the specific AhR-mediated toxicity of Cl2iPP, Cl2iBP, iBP, and BP. This observation is analogous to those made with TCDDs, PAHs, and the phytocompound indirubin.37,38 AhR potency of 16 compounds was observed in the yeast assay, but 12 compounds were less potent inducers of EROD activity in HepG2 cells. One of the reasons for this is that parabens may be metabolized in HepG2 cells to be AhR inactive. With regard to the metabolizing potential of HepG2 cells, SULT1A1 and NQO1, which belong to the phase II xenobiotic metabolizing enzyme, are expressed in HepG2.39 Therefore the results indicate that a system using a recombinant yeast is likely to be more effective for bioassay-based monitoring of parabens and their derivatives.
 |
| | Fig. 3 Relative intensity of paraben-induced cytochrome P450 (CYP) activity in HepG2 cells. CYP activity was determined in intact cells by ethoxyresorufin-O-deethylase (EROD) assay. HepG2 cells were stimulated with 100 or 150 μM parabens or 2 μM BaP for 24 hours. Each column represents the mean ± standard deviation (n = 3). *P < 0.05 and **P < 0.01 compared with the vehicle control (DMSO). | |
Chlorinated parabens and AhR agonist potency in water samples
The mean concentrations of the compounds identified in each water sample are summarized in Table 2, and the concentrations of individual compounds in each sample are shown in Tables S4 and S5 (ESI†). All 24 compounds were found in swimming pool water, and 20 of the 24 were found in river water, with total concentrations ranging from 2.7 to 1100 ng L−1 in pool water and from 83 to 4600 ng L−1 in river water. Compounds with methyl or propyl side chains were the most frequently detected and the most abundant analytes in both types of water samples. The concentrations of the different parabens in water reflect their wider use in cosmetic formulations. Dichlorinated parabens accounted for an average of 44% of total parabens (∑parabens), excluding degradation products (HBA, ClHBA, and Cl2HBA), in each sample; monochlorinated derivatives accounted for an average of 5% of ∑parabens. The observed levels of ∑parabens were, on average, 4.1-fold higher than those reported in our previous study, in which fewer compounds were found (4 compounds).8 On the other hand, observed concentrations and patterns of compounds in river water were comparable to those in effluents from wastewater treatment plants (WWTPs) in Spain9 and river water from STP discharge areas in Japan.4 Correlations between concentrations of ∑parabens and the population served by STPs were poor overall (R2 = 0.082). Sewage and domestic wastes are the primary sources of PPCPs in aquatic environments.40 Their concentrations vary depending on several factors, including the type and combination of treatment used, dilution and mixing effects, and distance of sampling locations up- and downstream of STP outfalls.41 These complex factors may have contributed to the observed paraben concentrations reported here. The dose–response relationships for water samples in the yeast assay are shown in Fig. S3 (ESI†). AhR potency was observed in 11 of 17 swimming pool samples; their BNF equivalents ranged from 4.0 to 960 pM with a geometric mean of 93 pM (Table 2). This study is the first to confirm the presence of AhR agonists in swimming pool water. Although active compounds were detected in pool water at sites 5 and 9, AhR potency was not observed in these samples, which might be explained by the presence of AhR antagonists in pool water. At present, several AhR antagonists have been developed and distributed for use in sunscreen products. It has been suggested that AhR activation may play a role in photocarcinogenesis and photoaging.42 Thus, prevention of sunlight-induced AhR activation may help to reduce skin damage. All water samples from the 20 river sites exhibited AhR potency; their BNF equivalents ranged from 25 to 3500 pM with a geometric mean of 130 pM (Table 2). This geometric mean value was equal to the reported potency of WWTP effluents in Australia (16–279 ng L−1; 59–1000 pM as the BNF equivalent)30 and water collected from urban and industrialized rivers in Japan (102–472 pM as the BNF equivalent).32 The high potencies were observed at sites 11, 13, and 17 (Table S5 in the ESI†). Sites 11, 13, and 17 were located in the textile-industry region in Shizuoka Prefecture.43 Recently, AhR agonists were detected in dye wastewater from the same region in Japan and were identified as dyes or dye derivatives.44 AhR agonists originating from industrial effluents, such as discharge from paper mills and textile-dyeing factories, may contribute to the particularly high potencies of these compounds observed in river water. Moreover, many studies have described various naturally occurring dietary substances that may directly activate the AhR-signaling pathway, as described in a review by Denison and Nagy.45 These natural products, such as flavonoids and indoles, have also been found in STP effluents.46,47 Wastewater contains numerous chemical compounds that could include large quantities of well-known or unknown AhR agonists.
Table 2 Concentration and AhR potency of the swimming pool and river water samples
|
|
Paraben concentration (ng L−1) |
AhR potency |
| Sample no. |
∑non-Cl |
∑mono-Cl |
∑di-Cl |
∑(non-, mono-, and di-Cl) |
BNF eq.a (pM) |
SD |
|
β-naphthoflavone equivalents.
Limits of quantification. Compounds were detected but below the LOQ.
Not detected.
The value determined from two response points at the high dose.
|
| Pool 1 |
900 |
68 |
91 |
1100 |
79 |
9.2 |
| 2 |
260 |
LOQb |
44 |
300 |
8.0 |
3.0 |
| 3 |
280 |
36 |
61 |
370 |
4.0 |
0.8 |
| 4 |
270 |
30 |
56 |
360 |
33d |
14 |
| 5 |
8.1 |
LOQ |
LOQ |
8.1 |
Not active |
|
| 6 |
8.0 |
—c |
LOQ |
8.0 |
84d |
13 |
| 7 |
LOQ |
— |
— |
LOQ |
960 |
150 |
| 8 |
LOQ |
— |
— |
LOQ |
Not active |
|
| 9 |
5.9 |
LOQ |
LOQ |
5.9 |
Not active |
|
| 10 |
10 |
100 |
110 |
220 |
160d |
0.40 |
| 11 |
LOQ |
LOQ |
18 |
18 |
220d |
81 |
| 12 |
LOQ |
LOQ |
LOQ |
LOQ |
Not active |
|
| 13 |
250 |
180 |
18 |
450 |
670 |
6.5 |
| 14 |
3.9 |
LOQ |
15 |
18 |
410 |
1.4 |
| 15 |
5.0 |
LOQ |
LOQ |
5.0 |
70 |
5.4 |
| 16 |
2.7 |
— |
LOQ |
2.7 |
Not active |
|
| 17 |
LOQ |
— |
— |
LOQ |
Not active |
|
|
|
|
|
|
|
|
|
| River 1 |
82 |
LOQ |
13 |
95 |
25d |
8.6 |
| 2 |
180 |
36 |
66 |
280 |
73 |
18 |
| 3 |
100 |
LOQ |
28 |
130 |
60 |
5.6 |
| 4 |
160 |
22 |
31 |
210 |
334 |
63 |
| 5 |
150 |
45 |
4400 |
4600 |
180 |
97 |
| 6 |
86 |
20 |
430 |
530 |
44 |
23 |
| 7 |
58 |
LOQ |
220 |
280 |
100 |
26 |
| 8 |
76 |
LOQ |
190 |
270 |
160 |
76 |
| 9 |
120 |
LOQ |
460 |
580 |
81 |
79 |
| 10 |
44 |
LOQ |
210 |
250 |
90 |
7.7 |
| 11 |
61 |
12 |
220 |
290 |
730 |
650 |
| 12 |
12 |
— |
93 |
110 |
90 |
7.6 |
| 13 |
62 |
LOQ |
110 |
170 |
3500 |
2300 |
| 14 |
42 |
LOQ |
71 |
110 |
130 |
83 |
| 15 |
82 |
17 |
130 |
230 |
170 |
11 |
| 16 |
77 |
— |
150 |
220 |
46 |
8.1 |
| 17 |
61 |
LOQ |
73 |
130 |
1400 |
76 |
| 18 |
64 |
— |
54 |
120 |
120 |
7.4 |
| 19 |
44 |
LOQ |
47 |
91 |
41 |
6.0 |
| 20 |
40 |
LOQ |
43 |
83 |
37 |
2.4 |
Implication for risk assessment of parabens affecting AhR potency
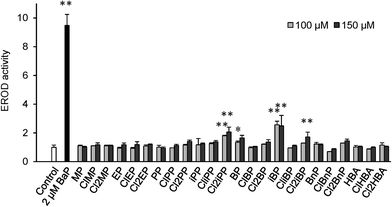
To evaluate the contribution of the active compounds to the observed AhR potency of the water samples, data obtained from the chemical analysis were converted to BNF equivalents by multiplying the concentrations of the components by their corresponding RA values (Fig. 4). Parabens and chlorinated parabens accounted for up to 6.3% (pool water) and 0.94% (river water) of the observed AhR potency of extracts from the water samples. Cl2PP had the highest BNF equivalent among the active components, followed by Cl2EP and Cl2MP. The current data obtained using a yeast AhR assay suggest that parabens and their chlorinated by-products are not likely to produce biological effects at environmentally relevant concentrations in aquatic environment.
 |
| | Fig. 4 The percentage contribution of active compounds to the observed AhR potency of water samples. | |
Conclusions
In this study, AhR potency of chlorinated parabens, including the parent compounds, was revealed for the first time using a yeast assay and human hepatoma cell line HepG2. The AhR mediates most of the toxic responses of halogenated organic contaminants including TCDDs and PCBs. These halogenated contaminants are also persistent and bioaccumulative. Although the metabolic characteristics of chlorinated parabens have not been reported to date, these compounds may be more persistent and difficult to metabolize than their parent parabens. Sustained exposure to low levels of chlorinated parabens may thus result in issues related to PCB-like toxicity. This study should signal concern about a novel aspect of parabens and chlorinated parabens in aquatic environments. Further studies attempt to confirm PCB-like toxicity of chlorinated paraben.
Abbreviations
| BaP |
benzo[a]pyrene
|
| BNF |
β-naphthoflavone
|
| BSTFA |
N,O-bis(trimethylsilyl)trifluoroacetamide
|
| ClPAHs |
chlorinated polycyclic aromatic hydrocarbons
|
| DMSO |
dimethylsulfoxide
|
| NQO1 |
NADP-quinone reductase 1
|
| PCBs |
polychlorinated biphenyls
|
| SULT |
sulfotransferase
|
| TCDD |
2,3,7,8-tetrachlorodibenzo-p-dioxin
|
Acknowledgements
This work was supported by the Grant-in-Aid for Scientific Research (C) no. 26410195 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
We thank the staff of the Public Health Center, City of Shizuoka, for supplying the swimming pool water samples.
Notes and references
- M. G. Soni, I. G. Carabin and G. A. Burdock, Food Chem. Toxicol., 2005, 43, 985–1015, DOI:10.1016/j.fct.2005.01.020.
- D. C. Steinberg, Cosmet. Toiletries, 2010, 125, 46–51 Search PubMed.
- T. Benijts, W. Lambert and D. L. Andre, Anal. Chem., 2004, 76, 704–711, DOI:10.1021/ac035062x.
- M. Terasaki, Y. Takemura and M. Makino, Environ. Chem. Lett., 2012, 10, 401–406, DOI:10.1007/s10311-012-0367-1.
- H. Gallard and U. Gunten, Environ. Sci. Technol., 2002, 36, 884–890, DOI:10.1021/es010076a.
- P. Westerhoff, Y. Yoon, S. Snyder and E. Wert, Environ. Sci. Technol., 2005, 39, 6649–6663, DOI:10.1021/es0484799.
- P. Canosa, I. Rodríguez, E. Rubí, N. Negreira and R. Cela, Anal. Chim. Acta, 2006, 575, 106–113, DOI:10.1016/j.aca.2006.05.068.
- M. Terasaki and M. Makino, Int. J. Environ. Anal. Chem., 2008, 88, 911–922, DOI:10.1080/03067310802272663.
- I. González-Marinõ, J. B. Quintana, I. Rodríguez and R. Cela, Water Res., 2011, 45, 6770–6780, DOI:10.1016/j.watres.2011.10.027.
- R. Golden, J. Gandy and G. Vollmer, Crit. Rev. Toxicol., 2005, 35, 435–458, DOI:10.1080/10408440490920104.
- D. Miller, B. B. Wheals, N. Beresford and J. P. Sumpter, Environ. Health Perspect., 2001, 109, 133–138, DOI:10.1289/ehp.01109133.
- E. J. Routledge, J. Parker, J. Odum, J. Ashby and J. P. Sumpter, Toxicol. Appl. Pharmacol., 1998, 153, 12–19, DOI:10.1006/taap.1998.8544.
- P. D. Darbre, J. R. Byford, L. E. Shaw, R. A. Horton, G. S. Pope and M. J. Sauer, J. Appl. Toxicol., 2002, 22, 219–226, DOI:10.1002/jat.860.
- M. Terasaki, R. Kamata, F. Shiraishi and M. Makino, Environ. Toxicol. Chem., 2009, 28, 204–208, DOI:10.1897/08-225.1.
- L. P. Nguyen and C. A. Bradfield, Chem. Res. Toxicol., 2008, 21, 102–116, DOI:10.1021/tx7001965.
- R. Kamata, F. Shiraishi, D. Nakajima, H. Takigami and H. Shiraishi, Toxicol. In Vitro, 2009, 23, 736–743, DOI:10.1016/j.tiv.2009.03.004.
- M. Machala, L. Bláha, H. J. Lehmler, M. Plísková, Z. Májková, P. Kapplová, I. Sovadinová, J. Vondrácek, T. Malmberg and L. W. Robertson, Chem. Res. Toxicol., 2004, 17, 340–347, DOI:10.1021/tx030034v.
- J. Vondrácek, M. Machala, V. Bryja, K. Chramostová, P. Krcmár, C. Dietrich, A. Hampl and A. Kozubík, Toxicol. Sci., 2005, 83, 53–63, DOI:10.1093/toxsci/kfi009.
- L. F. Bjeldanes, J. Y. Kim, K. R. Grose, J. C. Bartholomew and C. A. Bradfield, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9543–9547, DOI:10.1073/pnas.88.21.9543.
- C. Gouédard, R. Barouki and Y. Morel, Mol. Cell. Biol., 2004, 24, 5209–5222, DOI:10.1128/MCB.24.12.5209-5222.2004.
- J. Adachi, Y. Mori, S. Matsui, H. Takigami, J. Fujino, H. Kitagawa, C. A. Miller, T. Kato, K. Saeki and T. Matsuda, J. Biol. Chem., 2001, 276, 31475–31478, DOI:10.1074/jbc.C100238200.
- A. Poland, E. Glover and A. S. Kende, J. Biol. Chem., 1976, 251, 4936–4946 CAS.
- J. P. Giesy and K. Kannan, Crit. Rev. Toxicol., 1998, 28, 511–569, DOI:10.1080/10408449891344263.
- S. H. Safe, Toxicol. Lett., 2001, 120, 1–7, DOI:10.1016/S0378-4274(01)00301-0.
- O. Hankinson, Annu. Rev. Pharmacol. Toxicol., 1995, 35, 307–340, DOI:10.1146/annurev.pa.35.040195.001515.
- S. H. Safe, Environ. Health Perspect., 1995, 103, 346–351 CrossRef CAS.
- S. H. Safe and M. Wormke, Chem. Res. Toxicol., 2003, 16, 807–816, DOI:10.1021/tx034036r.
- F. Ohtake, A. Baba, I. Takada, M. Okada, K. Iwasaki, H. Miki, S. Takahashi, A. Kouzmenko, K. Nohara, T. Chiba, Y. Fujii-Kuriyama and S. Kato, Nature, 2007, 446, 562–566, DOI:10.1038/nature05683.
- M. Allinson, F. Shiraishi, S. A. Salzman and G. Allinson, Arch. Environ. Contam. Toxicol., 2011, 61, 539–546, DOI:10.1007/s00244-011-9665-z.
- K. O. Kusk, T. Kruger, M. Long, C. Taxvig, A. E. Lykkesfeldt, H. Frederiksen, A. M. Andersson, H. R. Andersen, K. M. Hansen, C. Nellemann and E. C. Bonefeld-Jorgensen, Environ. Toxicol. Chem., 2011, 30, 413–426, DOI:10.1002/etc.385.
- M. Kawanishi, M. Kondo, K. Shiizaki, W. L. Chu, Y. Terasoma and T. Yagi, Environ. Sci. Technol., 2008, 42, 6897–6902, DOI:10.1021/es801464z.
- N. Valkova, F. Lépine, L. Valeanu, M. Dupont, L. Labrie, J. G. Bisaillon, R. Beaudet, F. Shareck and R. Villemur, Appl. Environ. Microbiol., 2001, 67, 2404–2409, DOI:10.1128/AEM.67.6.2404-2409.2001.
- C. A. Miller, Toxicol. Appl. Pharmacol., 1999, 160, 297–303, DOI:10.1006/taap.1999.8769.
- M. Terasaki and M. Makino, J. Health Sci., 2009, 55, 631–635, DOI:10.1248/jhs.55.631.
- K. Saeki, T. Matsuda, T. Kato, K. Yamada, T. Mizutani, S. Matsui, K. Fukuhara and N. Miyata, Biol. Pharm. Bull., 2003, 26, 448–452, DOI:10.1248/bpb.26.448.
- Y. Horii, J. S. Khim, E. B. Higley, J. P. Giesy, T. Ohura and K. Kannan, Environ. Sci. Technol., 2009, 43, 2159–2165, DOI:10.1021/es8030402.
- M. Abdelrahim, R. Smith 3rd and S. Safe, Mol. Pharmacol., 2003, 63, 1373–1381, DOI:10.1124/mol.63.6.1373.
- J. Adachi, Y. Mori, S. Matsui and T. Matsuda, Toxicol. Sci., 2004, 80, 161–169, DOI:10.1093/toxsci/kfh129.
- S. Wilkening, F. Stahl and A. Bader, Drug Metab. Dispos., 2003, 31, 1035–1042 CrossRef CAS PubMed.
-
C. G. Daughton, in Pharmaceuticals and personal care products in the environment: overarching issues and overview, ed. C. G. Daughton and T. L. Jones-Lepp, American Chmiecal Society, Washington, D.C., 2001, ch 1, pp. 2–38 Search PubMed.
- G. L. Brun, M. Bernier, R. Losier, K. Doe, P. Jackman and H. B. Lee, Environ. Toxicol. Chem., 2006, 25, 2163–2176, DOI:10.1897/05-426R.1.
- J. Krutmann, A. Morita and J. H. Chung, J. Invest. Dermatol., 2012, 132, 976–984, DOI:10.1038/jid.2011.394.
- T. Watanabe, H. Ohba, M. Asanoma, T. Hasei, T. Takamura, Y. Terao, T. Shiozawa, T. Hirayama, K. Wakabayashi and H. Nukaya, Mutat. Res., 2006, 609, 137–145, DOI:10.1016/j.mrgentox.2006.06.033.
- P. H. Chou, S. Matsui, K. Misaki and T. Matsuda, Environ. Sci. Technol., 2007, 41, 652–657, DOI:10.1021/es061500g.
- M. S. Denison and S. R. Nagy, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 309–334, DOI:10.1146/annurev.pharmtox.43.100901.135828.
- A. Laganà, A. Bacaloni, I. D. Leva, A. Faberi, G. Fago and A. Marino, Anal. Chim. Acta, 2004, 501, 79–88, DOI:10.1016/j.aca.2003.09.020.
- J. Hu, H. Chang, L. Wang, S. Wu, B. Shao, J. Zhou and Y. Zhao, Environ. Sci. Technol., 2008, 42, 8339–8344, DOI:10.1021/es801038y.
Footnote |
| † Electronic supplementary information (ESI) available: Details of sample collection, characterization, quality control and assurance, concentrations of individual compounds, and AhR dose–response curves for water samples. See DOI: 10.1039/c5ew00047e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 



