
Phosphorus recovery from urine and anaerobic digester filtrate: comparison of adsorption–precipitation with direct precipitation†
Jeremy A.
O'Neal
and
Treavor H.
Boyer
 *
*
Department of Environmental Engineering Sciences, Engineering School of Sustainable Infrastructure & Environment (ESSIE), University of Florida, P.O. Box 116450, Gainesville, Florida 32611-6450, USA. E-mail: thboyer@ufl.edu; Tel: +1 352 846 3351
First published on 21st April 2015
Abstract
Hybrid anion exchange resin containing hydrous ferric oxide (HAIX-Fe) was used in column tests to remove phosphate (PO4) from fresh urine, hydrolyzed urine, and anaerobic digester filtrate, and subsequently recover PO4 as struvite (MgNH4PO4·6H2O) or potassium struvite (MgKPO4·6H2O) via precipitation in the spent regenerant. The recovery of PO4 using the two-step adsorption–precipitation process was compared with direct precipitation in urine and anaerobic digester filtrate considering chemical requirements for precipitation and mineral purity. Following the saturation of HAIX-Fe resin with PO4 from urine and anaerobic digester filtrate, up to 95% of the PO4 was desorbed using caustic brine during the regeneration phase. The spent regenerants were more concentrated in PO4 than the original urine and anaerobic digester filtrate due to recycling of the regeneration solution. Precipitation in the spent regenerants and original wastewaters (urine and filtrate) yielded 96.7–99.8% PO4 recovery as struvite or potassium struvite. Direct precipitation in fresh urine and hydrolyzed urine was more efficient than precipitation in the corresponding spent regenerants based on lower chemical requirements. Precipitation in the spent regenerant from HAIX-Fe resin saturated with PO4 from anaerobic digester filtrate produced a higher purity mineral than direct precipitation in the anaerobic digester filtrate.
Water impact statementPhosphorus (P) is a pollutant, finite resource, and critical element in agriculture. As such, there is growing interest in P recovery from waste streams for beneficial use as fertilizer. The key findings of this research are new experimental data on (1) P adsorption from urine and anaerobic digester filtrate to resin under continuous-flow operation and corresponding regeneration efficiency, and (2) chemical requirements for and mineral purity of P precipitation in the spent regenerants from resin adsorption/desorption and original waste waters. The results of this research are generalizable to resource recovery from domestic and industrial waste streams, and are significant because they advance source-separated and decentralized approaches to wastewater management that aim to increase recovery of water, nutrients, and energy. |
Introduction
Recovery of phosphorus (P) from various types of wastewater has been proposed as a more sustainable alternative to phosphate rock mining due to concerns about the finite supply of phosphate rock, economic scarcity of phosphate, and the critical need for P in agriculture and food production.1–4 Moreover, recovery of P from wastewater is attractive because wastewater treatment plants serve as a point source of P to receiving waters,5 which can harm aquatic ecosystems. In addition, excess P in wastewater can cause operational problems at wastewater treatment plants during sludge handling due to mineral scaling in pipes and pumps.6 Thus, P recovery from the most concentrated waste streams that comprise domestic wastewater, such as human urine and anaerobic digester sidestreams,7,8 would provide an alternative to phosphate rock fertilizers while having the important benefits of decreasing nutrient loading to receiving waters and reducing operational problems caused by mineral scaling at wastewater treatment plants.Phosphorus recovery by precipitation is a well-established process whereby the minerals struvite (MgNH4PO4·6H2O),7,9–12 and to a lesser extent potassium struvite (MgKPO4·6H2O),13,14 have been investigated for various types of wastewater. For example, previous researchers have achieved 95% P recovery by precipitation of either struvite or potassium struvite in human urine,14 and 94% P recovery by precipitation of struvite in anaerobic digester sidestreams.7 In addition, struvite has the potential to be used as an effective slow-release fertilizer based on plant growth studies.15 One of the major challenges to P recovery by precipitation is achieving a high purity product. For example, the presence of calcium during struvite precipitation can decrease mineral purity by co-precipitating calcium carbonate and calcium phosphate minerals with struvite.16,17 The extent to which co-precipitation is a problem depends of the final use of the struvite product. In some cases, potential contamination of struvite due to heavy metals18,19 and pathogens20 is possible, making the final product less desirable as fertilizer.
As an alternative to precipitation, adsorption processes can effectively and selectively remove phosphate (PO4) from many types of wastewater.21–23 For instance, hybrid anion exchange (HAIX) or ligand exchange resins combine polymer anion exchange resin with transition metals (e.g., Zr(IV),24 Cu(II),25 and Fe(III)26) thereby enabling selective removal of PO4 over competing anions such as sulfate, chloride, and bicarbonate.27 This study used HAIX resin infused with hydrous ferric oxide (HFO); hereafter HAIX-Fe. The predominant mechanism of removal of PO4 by HAIX-Fe resin is ligand exchange,26,28 whereby the HFO immobilized in the resin serves as the Lewis acid which forms an inner-sphere complex with the Lewis base, PO4.29 HAIX-Fe resin has been used to remove PO4 from synthetic and real secondary effluent (0.26–4.7 mg P L−1),26–28,30 wastewater-derived reverse osmosis concentrate (12 mg P L−1),31 synthetic (324 mg P L−1) and real (472 mg P L−1) sludge liquor,32 and synthetic human urine (460–620 mg P L−1).21,33 The previous studies show greater removal of PO4 by HAIX-Fe resin than anion exchange resins or granular ferric hydroxide, and selective removal of PO4 by HAIX-Fe resin in the presence of competing anions. Upon saturation of HAIX-Fe resin with PO4, regeneration of HAIX-Fe resin using caustic brine can desorb >90% of the adsorbed PO4.26–28,32 The drawback of PO4 removal by adsorption is that an additional step is needed to recover the PO4 in a useable form such as for fertilizer. For instance, PO4 recovery has been demonstrated by adding magnesium and ammonia to the spent regenerant produced by HAIX-Fe regeneration thereby precipitating struvite.28
The gap in the literature that motivated this research was the absence of data comparing PO4 recovery by precipitation in waste streams (i.e., direct precipitation) with precipitation in the spent regenerant following regeneration of HAIX-Fe resin saturated with PO4 (i.e., two-step adsorption–precipitation). Furthermore, there is only one previous study on struvite precipitation in the spent regenerant from HAIX-Fe resin28 and no data on potassium struvite precipitation in spent regenerant. As such, there is not sufficient data available to compare the adsorption–precipitation process with direct precipitation in terms of chemical requirements for precipitation or purity of the final precipitated solid. In addition, there is limited data available on the continuous-flow column operation of HAIX-Fe resin to remove PO4 from wastewater high in PO4, total dissolved solids (TDS), and organics,32 and as such, it is not known how continuous-flow operation of HAIX-Fe resin will perform for source-separated urine. Accordingly, the goal of this research was to compare the two-step adsorption–precipitation process with direct precipitation for P recovery from source-separated human urine and anaerobic digester filtrate. The specific objectives of this research were to (1) evaluate PO4 adsorption from urine and filtrate to virgin and regenerated HAIX-Fe resin under continuous-flow column operation; (2) evaluate the regeneration efficiency of HAIX-Fe resin saturated with PO4 using fresh and recycled caustic brine; (3) quantify the chemical requirements for PO4 precipitation as struvite or potassium struvite in the spent regenerants and wastewaters through chemical addition and batch precipitation; and (4) evaluate the P recovery and mineral purity achieved by batch precipitation in the spent regenerants and wastewaters. Source-separated urine and anaerobic digester filtrate were selected for this research because both have high PO4 concentrations. In addition, there is increasing interest in urine diversion and anaerobic digestion as part of source-separated and decentralized approaches to wastewater management to increase recovery of water, nutrients, and energy.34,35
Methods
Wastewaters
Three types of wastewater were used in this study: synthetic fresh urine, synthetic hydrolyzed urine, and real anaerobic digester filtrate (Table 1). Human urine represents wastewater high in PO4 and TDS, whereas anaerobic digester filtrate represents wastewater high in PO4, TDS, and organics. The composition of synthetic urine was based on previous studies.14,36,37 Synthetic urine was used to ensure that the composition of urine was consistent throughout the study. Previous studies have shown similar precipitation behavior in synthetic and real urine,36 and comparable adsorption results in synthetic urine38 and real urine.39 The composition of synthetic fresh urine assumed that no urea hydrolysis had occurred, whereas the synthetic hydrolyzed urine assumed complete urea hydrolysis with spontaneous precipitation of struvite and hydroxyapatite to eliminate the magnesium and calcium, respectively, and reduce the PO4 concentration.40,41 Theoretically, the ammonia would also be very slightly reduced by struvite precipitation. Potassium struvite does not precipitate during urea hydrolysis because it is not supersaturated. Therefore, the potassium concentration, as well as the concentrations of sodium, chloride, and sulfate, remained approximately constant in fresh and hydrolyzed urine.| Chemicala | Fresh urineb | Hydrolyzed urineb | Anaerobic digester filtratec |
|---|---|---|---|
| a All concentrations mmol L−1, except pH and phosphate distribution (unitless) and ionic strength (mol L−1). b Prepared using NH4Cl, NH4HCO3, urea, NaCl, NaH2PO4, Na2SO4, KCl, CaCl2·2H2O, and MgCl2·6H2O in deionized water based on previous studies.14,36,37 Concentrations are calculated not measured, except pH measured. c Real filtrate spiked with NaH2PO4; measured concentrations. d Not added, assumed 0. e From O'Neal and Boyer calculated using Visual MINTEQ.21 f Measured as total nitrogen. Not measured (nm). | |||
| Urea (as N) | 500 | —d | nm |
| Total ammonia | —d | 500 | 134f |
| Cl− | 100 | 100 | 20 |
| Total phosphate | 20 (619 mg P L−1) | 13.6 (421 mg P L−1) | 2.5f (77 mg P L−1) |
| H2PO4−/total PO43− | 0.940e | 0.0078e | 0.284e |
| HPO42−/total PO43− | 0.0596e | 0.991e | 0.716e |
| SO42− | 15 | 15 | 2.6 |
| Total carbonate | —d | 250 | nm |
| Na+ | 94 | 104 | 14 |
| K+ | 40 | 40 | 1.9 |
| Ca2+ | 4 | —d | 4.3 |
| Mg2+ | 4 | —d | 2 |
| DOC | —d | —d | 7.0 |
| pH | 5.8 | 9.3 | 7.0 |
| Ionic strength | 0.145e | 0.476e | 0.158e |
The anaerobic digester filtrate was collected from the belt press filtrate of anaerobically digested sludge from the Howard F. Curren Advanced Wastewater Treatment Plant in Tampa, FL, USA. The anaerobic digesters are operated at approximately 37 °C with a typical solids residence time of 15–25 d and a combination of mixed sludge and waste activated sludge. The anaerobic digester filtrate was filtered in series through filters with pore sizes of 5, 3, and 0.45 μm (Millipore). The composition of the anaerobic digester filtrate given in Table 1 is presented to allow for comparison with the synthetic urine used in this work. More detailed analysis of anaerobic digestion liquors can be found in the literature.42,43 In particular, the nature of the dissolved organics could affect the adsorption and precipitation processes investigated in this work but was outside the scope of this project. Because the Curren plant does not practice enhanced biological P removal, the anaerobic digester filtrate was spiked with NaH2PO4 to increase the PO4 concentration to approximately 80 mg P L−1 to allow for comparison with previous data on anaerobic digestion sidestreams.21 In addition, the rationale for using synthetic urine and real filtrate was because the composition of urine is well documented in the literature, whereas there is not a standard recipe for anaerobic digester filtrate.
HAIX-Fe adsorption column tests and regeneration
The HAIX-Fe resin used in this work was a commercially available product (trade names PhosXnp or LayneRT, SolmeteX, Northborough, MA). Phosphate adsorption to HAIX-Fe resin using fresh urine, hydrolyzed urine, and anaerobic digester filtrate was investigated using continuous-flow column tests. The columns (Omnifit) were glass with a 10 mm inner diameter. All column tests were conducted using 8 mL of HAIX-Fe resin, which was defined as one bed volume (BV). The column tests were operated up-flow by pumping wastewater (Masterflex peristaltic pump) at a flow rate of 2.5 mL min−1, which yielded an empty bed contact time (EBCT) of 3.2 min and a superficial linear velocity (SLV) of 3.1 cm min−1. All column tests were performed at laboratory temperature of 24 °C. Effluent samples were collected at predetermined times and analyzed for pH and PO4 concentration. Phosphate breakthrough was defined as C/C0 = 0.1 and saturation was defined as C/C0 = 1. An initial column experiment was performed in triplicate using fresh HAIX-Fe resin and fresh urine to ensure reproducible results (see Fig. S1 in ESI†). In subsequent experiments HAIX-Fe resin was used to treat each wastewater three times as follows: first with virgin HAIX-Fe resin, second with once-used HAIX-Fe resin that was regenerated with fresh regeneration solution, and third with twice-used HAIX-Fe resin that was regenerated with once-used regeneration solution. All column tests were run until HAIX-Fe resin was saturated with PO4, at which point the test was stopped and the resin was regenerated as described in the next paragraph. The column containing HAIX-Fe resin was rinsed with 10 BVs of DI water between the adsorption and regeneration tests. The mass of PO4 adsorbed to HAIX-Fe resin was calculated by taking the difference in influent and effluent PO4 concentrations for each sample multiplied by the volume of wastewater treated per sample. The total mass of PO4 adsorbed to HAIX-Fe resin was the summation of PO4 adsorbed for all samples. The capacity (or column loading) of HAIX-Fe resin for PO4 was calculated by dividing the total mass of PO4 adsorbed by the mass of resin contained in the column.Regeneration was performed in the same column as the adsorption tests using a solution containing 2.5% (m/m) NaCl and 2% (m/m) NaOH, pH 13.7, based on previous work.28 Effluent samples were collected at predetermined times and analyzed for pH and PO4 concentration. An initial regeneration test was performed in triplicate using HAIX-Fe resin that was previously saturated with PO4 from column tests using fresh urine (see Fig. S2 in ESI†). After the initial regeneration test, the flow rate was decreased to 0.8 mL min−1 for all subsequent regeneration tests, which corresponded to an EBCT of 10 min and a SLV of 1.0 cm min−1. The HAIX-Fe resin was regenerated after each column adsorption test described in the previous paragraph. This resulted in three regeneration tests for each wastewater: first with fresh regeneration solution, second with recycled (i.e., once-used) regeneration solution, and third with recycled (i.e., twice-used) regeneration solution. No additional NaCl or NaOH was added to the recycled regeneration solution. The twice-used regeneration solution is referred to as spent regenerant, and was used in the precipitation experiments. The total mass of PO4 desorbed from HAIX-Fe resin during regeneration was calculated by combining all effluent samples into a single sample, measuring the PO4 concentration, and multiplying by the total volume of regeneration solution. The regeneration efficiency was calculated as the amount of PO4 desorbed divided by the amount adsorbed.
Precipitation experiments
Precipitation experiments were conducted using each wastewater and spent regenerant, and either struvite or potassium struvite was precipitated. Potassium struvite was precipitated in fresh urine and spent regenerant from HAIX-Fe resin saturated with PO4 from fresh urine and hydrolyzed urine. Struvite was precipitated in hydrolyzed urine, anaerobic digester filtrate, and spent regenerant from HAIX-Fe resin saturated with PO4 from hydrolyzed urine and anaerobic digester filtrate. Potassium struvite precipitation was conducted by adding the appropriate chemicals (MgCl2·6H2O and KCl) such that the solution contained a K![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg:P molar ratio of at least 1.5:1.5:1. (Fresh urine had a K:P molar ratio of >1.5:1.) Struvite precipitation was conducted by adding the appropriate chemicals (NH4Cl and MgCl2·6H2O) such that the solution contained a Mg:N:P molar ratio of at least 1.5:1.5:1. (Hydrolyzed urine and anaerobic digester filtrate had an N:P model ratio of >1.5:1.) All precipitation experiments were conducted at ambient laboratory temperature of 24 °C. Precipitation was performed in 125 mL amber bottles containing 50 mL of solution. The pH was adjusted to 9.3 using 1 M NaOH or 12.1 M HCl to create favorable conditions for precipitation and avoid precipitation of Mg(OH)2 at high pH. Samples were mixed on an Innova 2000 Platform Shaker at 200 rpm for 2 h. Precipitation experiments using wastewater were conducted in triplicate; precipitation experiments using spent regenerant were conducted singly because of the limited volume of solution. After the precipitation experiment was complete the solution was filtered through 1.5 μm glass fiber filter (Whatman 934-AH RTU), the solid precipitate was dried at 24 °C in a desiccator for a minimum of 48 h and weighed. The PO4 concentration in solution was measured before and after precipitation to determine the P recovery by precipitation. The percent P by mass of the solid precipitate was calculated using the measured mass of solid and the difference in aqueous PO4 concentrations. X-ray diffraction (XRD) was used to identify the crystal structure of the solid precipitate.
:Mg:P molar ratio of at least 1.5:1.5:1. (Fresh urine had a K:P molar ratio of >1.5:1.) Struvite precipitation was conducted by adding the appropriate chemicals (NH4Cl and MgCl2·6H2O) such that the solution contained a Mg:N:P molar ratio of at least 1.5:1.5:1. (Hydrolyzed urine and anaerobic digester filtrate had an N:P model ratio of >1.5:1.) All precipitation experiments were conducted at ambient laboratory temperature of 24 °C. Precipitation was performed in 125 mL amber bottles containing 50 mL of solution. The pH was adjusted to 9.3 using 1 M NaOH or 12.1 M HCl to create favorable conditions for precipitation and avoid precipitation of Mg(OH)2 at high pH. Samples were mixed on an Innova 2000 Platform Shaker at 200 rpm for 2 h. Precipitation experiments using wastewater were conducted in triplicate; precipitation experiments using spent regenerant were conducted singly because of the limited volume of solution. After the precipitation experiment was complete the solution was filtered through 1.5 μm glass fiber filter (Whatman 934-AH RTU), the solid precipitate was dried at 24 °C in a desiccator for a minimum of 48 h and weighed. The PO4 concentration in solution was measured before and after precipitation to determine the P recovery by precipitation. The percent P by mass of the solid precipitate was calculated using the measured mass of solid and the difference in aqueous PO4 concentrations. X-ray diffraction (XRD) was used to identify the crystal structure of the solid precipitate.
Chemical equilibrium calculations
Visual MINTEQ (ver. 3.0) was used to calculate the concentration of dissolved species, ion activity product (IAP), ionic strength, and saturation index (SI) of relevant solids in synthetic urine and spent regenerants used in the precipitation experiments.44 Two different calculation approaches were used: supersaturated solids not allowed to precipitate and struvite or potassium struvite allowed to precipitate. The former approach was used to identify all possible solid phases that were supersaturated. The later approach was used to calculate the extent of PO4 removal by precipitation of struvite or potassium struvite. The input values were taken from Table 1 or the initial composition of the regeneration solution, and were augmented by the addition of chemicals required for precipitation as described later in Table 3. The SI was calculated as log(IAP/Ks0) where Ks0 is the solubility product and SI > 0 indicates supersaturated conditions. The SI for anaerobic digester filtrate was not calculated because of incomplete information on its composition especially related to organics. Activity corrections were made using the Davies approximation with B = 0.3 instead of 0.2,29 which was shown previously to be the most appropriate approximation to the activity coefficient in urine.36 The Ks0 for potassium struvite (10−10.62) was added to the database.45 The Ks0 in the database for struvite (10−13.26) was consistent with published data and was not changed.36| Column test | Phosphate adsorption/desorption | Fresh urine | Hydrolyzed urine | ADF |
|---|---|---|---|---|
| Anaerobic digester filtrate (ADF).a Adsorbed/(volume HAIX-Fe resin × density HAIX-Fe resin).b Freundlich parameters from O'Neal and Boyer.21c Fouled = adsorbed − desorbed.d Total = adsorbed + fouled.e Total/(volume HAIX-Fe resin × density HAIX-Fe resin).f Synthetic anaerobic digester supernatant. | ||||
| First use | Adsorbed, mg P | 31.9 | 20.7 | 19.9 |
| Column loading,a mg P g−1 | 10.2 | 6.7 | 6.4 | |
| Freundlich capacity,b mg P g−1 | 10.1 | 5.7 | 5.1f | |
| Desorbed, mg P | 30.2 | 17.4 | 18.9 | |
| Fouled,c mg P | 1.7 | 3.4 | 1.0 | |
| Regeneration efficiency | 94.7% | 83.8% | 95.0% | |
| Second use | Adsorbed, mg P | 30.0 | 19.4 | 20.0 |
| Total,d mg P | 31.7 | 22.8 | 21.0 | |
| Column loading,e mg P g−1 | 10.2 | 7.3 | 6.7 | |
| Desorbed, mg P | 15.9 | 15.3 | 14.4 | |
| Fouled,c mg P | 15.8 | 7.4 | 6.6 | |
| Regeneration efficiency | 50.1% | 67.3% | 68.6% | |
| Third use | Adsorbed, mg P | 25.9 | 15.9 | 17.3 |
| Total,d mg P | 41.7 | 23.3 | 23.9 | |
| Column loading,e mg P g−1 | 13.4 | 7.5 | 7.7 | |
| Desorbed, mg P | 7.2 | 10.1 | 12.1 | |
| Fouled,c mg P | 34.5 | 13.2 | 11.8 | |
| Regeneration efficiency | 17.3% | 43.4% | 50.7% | |
| Direct precipitation | Precipitation in spent regenerant | ||||||
|---|---|---|---|---|---|---|---|
| FU | HU | ADF | FU | HU | HU | ADF | |
| Fresh urine (FU). Hydrolyzed urine (HU). Anaerobic digester filtrate (ADF).a Total species.b NaOH/HCl added to reach pH 9.3.c Magnesium added to achieve Mg:P molar ratio of 1.5:1.d Potassium added to achieve K:P molar ratio of 1.5:1.e Ammonia added to achieve N:P molar ratio of 1.5:1.f Calculated from difference in aqueous concentrations.g Potassium struvite (MPP; MgKPO4·6H2O); struvite (MAP; MgNH4PO4·6H2O); halite (H). |
|||||||
| Initial PO43−,a mg P L−1 | 674 | 487 | 78.2 | 783 | 652 | 652 | 693 |
| Initial pH | 5.8 | 9.3 | 7.0 | 12.8 | 12.8 | 12.8 | 13.0 |
| 1 M NaOH added,b mL L−1 | 28 | — | 36 | — | — | — | — |
| 12.1 M HCl added,b mL L−1 | — | — | — | 54 | 54 | 52 | 52 |
| MgCl2·6H2O added,c g g−1 P | 8 | 8.8 | 4.5 | 10 | 8.6 | 8.4 | 8.3 |
| KCl added,d g g−1 P | — | — | — | 3.7 | 3.1 | — | — |
| NH4Cl added,e g g−1 P | — | — | — | — | — | 2.2 | 2.2 |
| Final PO43−,a mg P L−1 | 15.0 | 1.37 | 0.510 | 16.9 | 21.4 | 5.26 | 1.18 |
| P removed,f % | 97.8% | 99.7% | 99.3% | 97.8% | 96.7% | 99.2% | 99.8% |
| P recovered,f g L−1 | 0.66 | 0.49 | 0.078 | 0.77 | 0.63 | 0.65 | 0.69 |
| Solids collected, g L−1 | 5.3 | 3.7 | 1.1 | 6.2 | 5.2 | 5.1 | 6.0 |
| P in solid, % (m/m) | 12.5 | 13.2 | 7.1 | 12.4 | 12.1 | 12.7 | 11.5 |
| Mineral identifiedg | MPP | MAP | MAP | MPP, H | MPP, H | MAP | MAP, H |
Analytical methods
All stock solutions and synthetic urine were prepared using DI water and ACS reagent grade purity chemicals. Phosphate (as P) was measured following Standard Method 4500-P using a Hitachi U-2900 spectrophotometer at 880 nm and a 1 cm quartz cuvette.46 Standard calibration checks and matrix spikes were performed to assess the accuracy of measurements; the relative difference between measured and known concentrations was <5% for standard calibration checks and matrix spike recoveries were 94–106%. The pH of each sample was measured with an Accumet AB 15 pH meter and Accumet combination pH/temperature electrode (Fisher Scientific), which was calibrated before use with pH 4, 7, and 10 buffer solutions (Fisher Scientific). Dissolved organic carbon (DOC) was measured using a Shimadazu TOC-VCPH total organic carbon analyzer equipped with an ASI-V autosampler.47 Inorganic anions and cations, excluding PO4, were measured using a Dionex ICS-3000 ion chromatograph as described elsewhere.48 X-ray diffraction of solid precipitates was performed with a computer-controlled Rigaku Ultima IV X-ray powder diffractometer with a stepping motor and graphite crystal monochrometer as described elsewhere.49Results and discussion
Phosphate adsorption column tests and regeneration
Fig. 1 shows the effluent concentration of PO4 divided by the initial concentration of PO4 as a function of the number of BVs treated by HAIX-Fe resin. The trends for increasing number of BVs of wastewater treated by HAIX-Fe resin until breakthrough (i.e., C/C0 = 0.1) were hydrolyzed urine < fresh urine ≪ anaerobic digester filtrate and third use < second use ≤ first use (see Table S1 in ESI†). The trends for increasing number of BVs of wastewater treated by HAIX-Fe resin until saturation (i.e., C/C0 = 1) were fresh urine < hydrolyzed urine ≪ anaerobic digester filtrate with no clear trend related to first, second, or third use (see Table S1 in ESI†). The results in Fig. 1, especially for anaerobic digester filtrate, share the greatest similarity with previous results on HAIX-Fe resin treatment of real and synthetic sludge liquor (472 and 324 mg P L−1, respectively) in which saturation of HAIX-Fe resin using real and synthetic sludge liquor occurred at 31 and 120 BVs, respectively.32 All other previous column studies using HAIX-Fe resin has been conducted using wastewater with low PO4 concentration (≤12 mg P L−1) and low ionic strength (<1 × 10−3 M). Therefore, the results in Fig. 1 add new data to the limited data available on the continuous flow operation of HAIX-Fe resin in high PO4 concentration (>400 mg P L−1) and high ionic strength (>1 × 10−1 M) wastewater.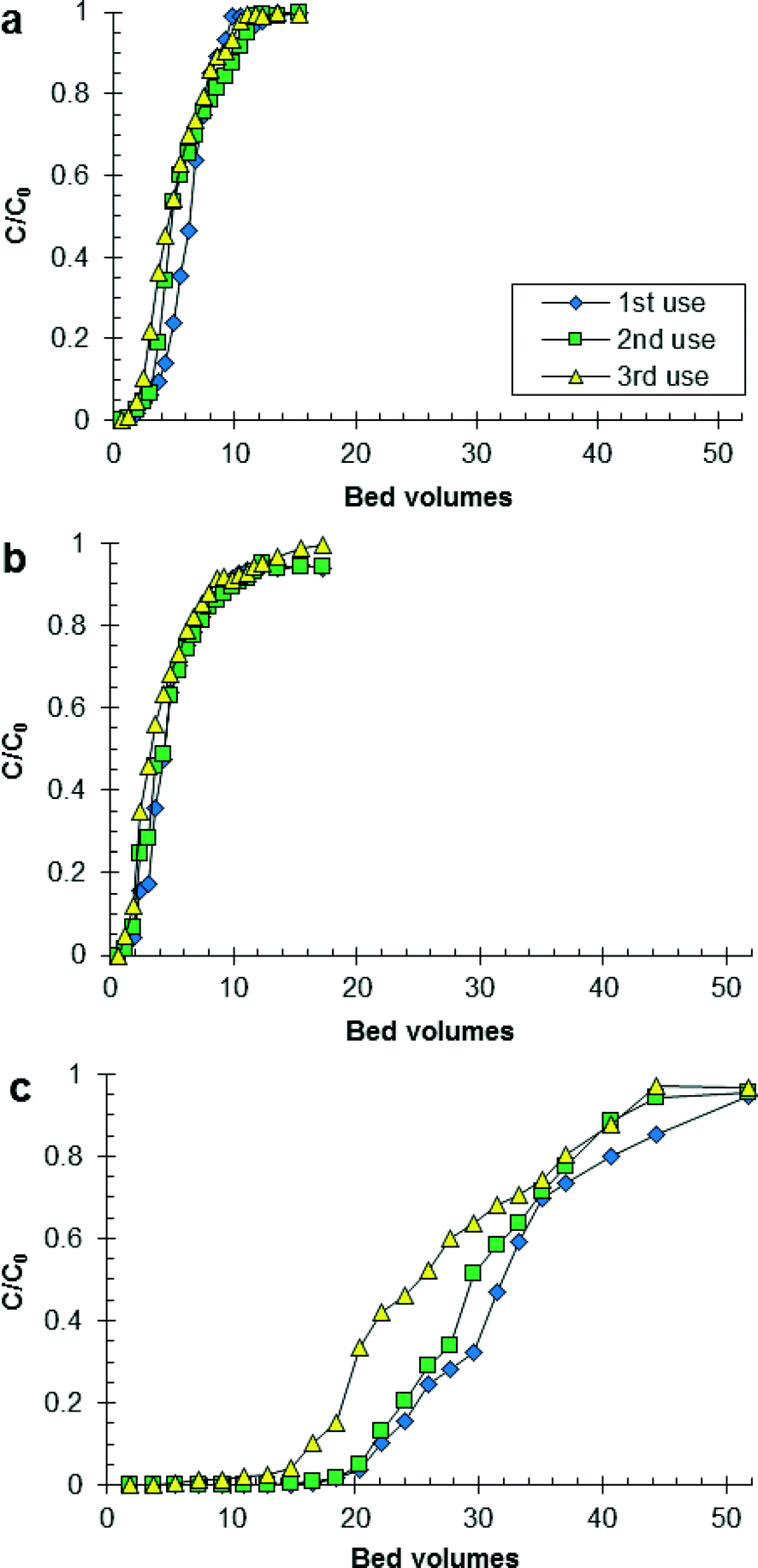 | ||
| Fig. 1 Phosphate adsorption to HAIX-Fe resin under continuous-flow column operation: (a) synthetic fresh urine (C0 = 672 mg P L−1, pH = 5.8), (b) synthetic hydrolyzed urine (C0 = 491 mg P L−1, pH = 9.3), and (c) real anaerobic digester filtrate (C0 = 77 mg P L−1, pH = 7.0). Column operating conditions: BV = 8 mL; EBCT = 3.2 min; SLV = 3.1 cm min−1. | ||
The pH and ionic strength of the wastewaters appears to explain some of the trends for number of BVs treated by HAIX-Fe resin until breakthrough and saturation (see Fig. S3 in ESI†). For instance, the lower number of BVs treated by HAIX-Fe resin in hydrolyzed urine than the other wastewaters was due to the higher pH and higher ionic strength of hydrolyzed urine than the other wastewaters. As the pH of the solution increases, the speciation of the resin-phase HFO shifts from positively charged (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) FeOH2+) to neutral (FeOH) to negatively charged (FeO−), and the speciation of PO4 shifts from monovalent to divalent (i.e., H2PO4− ↔ HPO42− + H+, pKa = 7.20, I = 0 M).50 Moreover, the charged HFO species become more prevalent as the ionic strength increases.50 Hence, it is speculated that in hydrolyzed urine unfavorable electrostatic interactions occur between HFO and PO4, whereas in fresh urine and anaerobic digester filtrate more favorable electrostatic interactions occur between HFO and PO4. This is supported by previous research in which the maximum PO4 adsorption to HAIX-Fe resin in secondary wastewater effluent was at pH 7–7.5.26,27
FeOH2+) to neutral (FeOH) to negatively charged (FeO−), and the speciation of PO4 shifts from monovalent to divalent (i.e., H2PO4− ↔ HPO42− + H+, pKa = 7.20, I = 0 M).50 Moreover, the charged HFO species become more prevalent as the ionic strength increases.50 Hence, it is speculated that in hydrolyzed urine unfavorable electrostatic interactions occur between HFO and PO4, whereas in fresh urine and anaerobic digester filtrate more favorable electrostatic interactions occur between HFO and PO4. This is supported by previous research in which the maximum PO4 adsorption to HAIX-Fe resin in secondary wastewater effluent was at pH 7–7.5.26,27
Fig. 2 shows the effluent concentration of PO4 as a function of the BVs of regeneration solution that passed through the HAIX-Fe resin bed. Phosphate desorption from HAIX-Fe resin showed very similar trends among fresh urine, hydrolyzed urine, and anaerobic digester filtrate, e.g., regeneration was complete within 5 BVs for the first, second, and third regeneration. This similarity was because the composition of the regeneration solution was initially the same for all three wastewaters. The concentration of PO4 in the regeneration solution increased with repeated use of the regeneration solution (see Table S2 in ESI†), and this increase was most substantial for anaerobic digester filtrate. The concentration of PO4 in the final, combined regeneration solution was 263 mg P L−1 after the first regeneration, 502 mg P L−1 after the second regeneration, and 693 mg P L−1 after the third regeneration, which corresponded to 9.0× higher PO4 concentration in spent regenerant than initial anaerobic digester filtrate. Fresh urine and hydrolyzed urine had 1.2× and 1.3× higher PO4 concentration in spent regenerant than initial urine. Although not measured in this work, HAIX-Fe resin is able to bind sulfate to the strong-base anion exchange sites in the resin. For context, the phosphate–sulfate separation factor for HAIX-Fe resin was previously determined to be 46.26 Although HAIX-Fe resin has a higher selectivity for PO4 than sulfate, given the high concentration of sulfate in urine, HAIX-Fe resin likely accumulated sulfate and thereby the regeneration solution likely accumulated sulfate to some extent. The sulfate concentration in the regeneration solution was not measured, which is a limitation of the experimental results. Desorption of PO4 and sulfate from HAIX-Fe resin during regeneration was reported by Sengupta and Pandit.28 As a result, the accumulation of sulfate in the regeneration solution is expected to lead to decreased regeneration efficiency and possibly decrease the purity of the recovered PO4 solid.
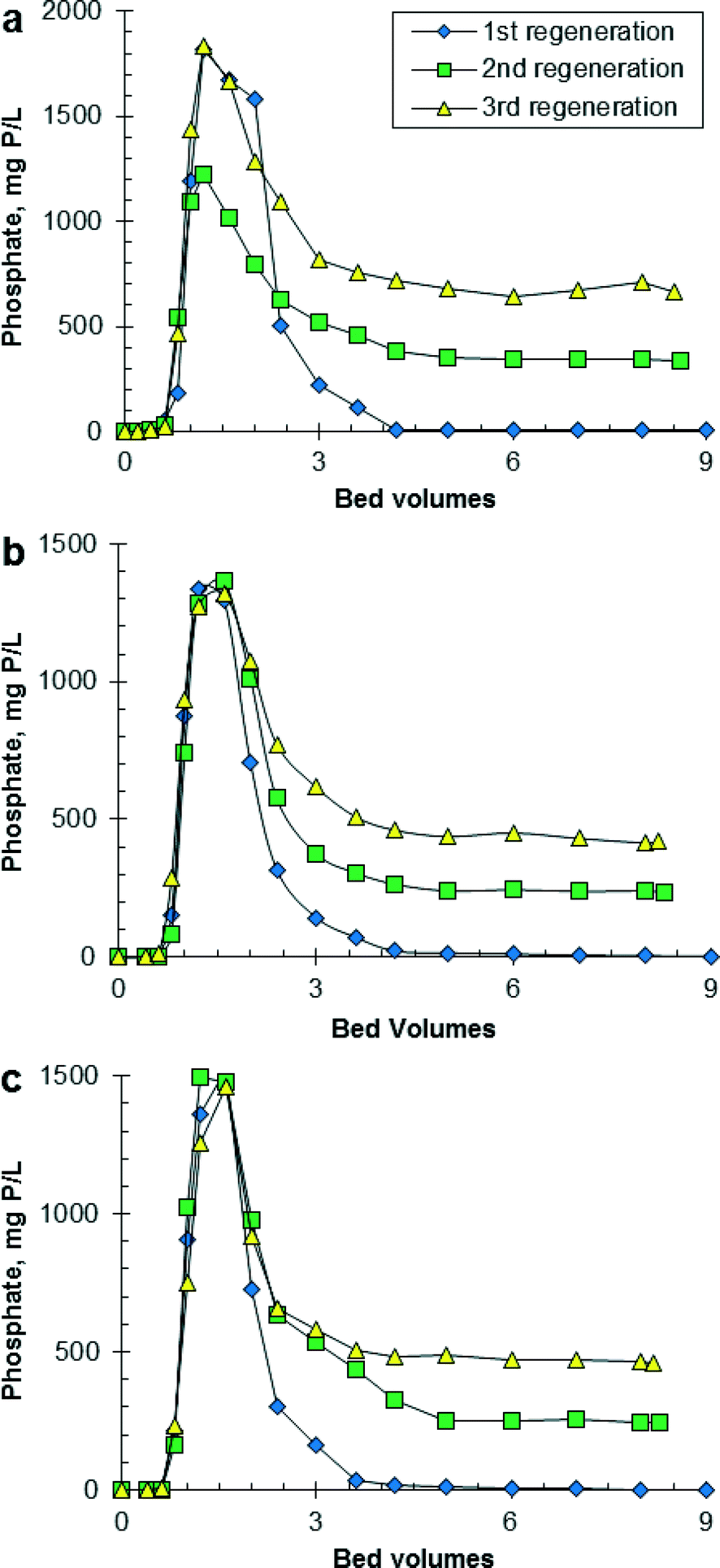 | ||
| Fig. 2 Phosphate desorption from HAIX-Fe resin during column regeneration corresponding to adsorption tests using (a) synthetic fresh urine, (b) synthetic hydrolyzed urine, and (c) real anaerobic digester filtrate. Column regeneration conditions: 2.5% NaCl + 2% NaOH regeneration solution; BV = 8 mL; EBCT = 10 min; SLV = 1.0 cm min−1. | ||
It is possible that even higher PO4 concentrations could be achieved in the spent regenerant if the regeneration volume was reduced from 9 to 5 BVs. However, the accumulation of PO4 in the regeneration solution has the potential to decrease the regeneration efficiency as illustrated in Table 2. The regeneration efficiency decreased from 84–95% for the first regeneration to 17–51% for the third regeneration. It is not known why the first regeneration of HAIX-Fe resin saturated with PO4 from hydrolyzed urine was 11 percentage points lower than the corresponding regeneration for fresh urine and anaerobic digester filtrate. Based on reported data (i.e., 90–98% regeneration efficiency of HAIX-Fe resin saturated with PO4 from a variety of wastewaters excluding urine),28,31,32 95% regeneration efficiency for the first regeneration is a more likely value than 84% regeneration efficiency. Overall, with each adsorption–regeneration cycle an increasing amount of PO4 was retained (i.e., fouled) on HAIX-Fe resin, which resulted in a lower amount of PO4 adsorbed in the subsequent adsorption step. HAIX-Fe resin saturated with PO4 from fresh urine exhibited greater fouling than HAIX-Fe resin saturated with PO4 from hydrolyzed urine or anaerobic digester filtrate because of the higher PO4 concentration in the recycled regeneration solution. Fig. S4 in ESI† shows that the regeneration efficiency decreased linearly with increasing PO4 concentration in the regeneration solution. As an alternative approach, Sengupta and Pandit were able to achieve 93% regeneration efficiency using recycled regeneration solution by precipitating PO4 in the regeneration solution and then adding 1.5% NaOH to compensate for the loss of hydroxide during regeneration.28
The maximum PO4 loading on HAIX-Fe resin during the column tests was achieved using fresh urine, and increased from 10.2 mg P g−1 (first use, adsorbed) to 13.4 mg P g−1 (third use, total). The maximum PO4 loading observed in this study was less than the maximum PO4 loading reported by Sengupta and Pandit (23 mg P g−1),28 but greater than the PO4 loading reported by Martin et al. (7.7 mg P g−1).30 The calculated PO4 loading on HAIX-Fe resin using the Freundlich isotherm model (and model parameters corresponding to fresh urine, hydrolyzed urine, and anaerobic digester supernatant21) was in relatively good agreement (1–21% relative difference) with the experimental PO4 loading on HAIX-Fe resin from the column tests (Table 2) and better than the Langmuir isotherm model (results not shown). Thus, batch equilibrium experiments and Freundlich isotherm modeling can predict the capacity of HAIX-Fe resin for PO4 under continuous-flow column operation, and following the adsorption step, fresh regeneration solution can desorb up to 95% of PO4 making it available for P recovery via precipitation.
Chemical requirements for phosphate precipitation
All chemical additions required for precipitation of struvite and potassium struvite in wastewaters and spent regenerants are listed in Table 3. Fresh urine and anaerobic digester filtrate required the addition of strong base to increase the pH to 9.3. The anaerobic digester filtrate required more NaOH than fresh urine because it had a higher buffer intensity than fresh urine due to the higher concentration of ammonia, carbonate, and DOC. Hydrolyzed urine did not require pH adjustment due to the increase in pH that occurs during urea hydrolysis, which yields a final pH of approximately 9.41All wastewaters required the addition of magnesium to achieve an Mg:P molar ratio of 1.5:1. The anaerobic digester filtrate required the least amount of magnesium because the initial Mg:P molar ratio was 0.8:1 due to the initial magnesium present, whereas the hydrolyzed urine required the greatest amount of magnesium because magnesium was not initially present. No additional chemicals were required because fresh urine contained a stoichiometric excess of potassium for potassium struvite precipitation, and hydrolyzed urine and anaerobic digester filtrate contained a stoichiometric excess of ammonia for struvite precipitation. The chemical additions described herein are specific to the waste streams investigated and could vary for other wastewaters.
Although the comparison of saturation indexes for various solid phases does not allow for the determination of the least soluble phase, it is a useful approach for defining the universe of solid phases that could precipitate. Chemical equilibrium calculations (not allowing solid phases to precipitate) showed that potassium struvite, as well as nine other calcium and magnesium phosphate solid phases, were supersaturated in fresh urine at an Mg:K:P molar ratio of 1.5:2:1 and pH 9.3 (see Table S3 in ESI†). The presence of calcium in fresh urine increased the number of solid phases that were supersaturated. In hydrolyzed urine at an Mg:N:P molar ratio of 1.5:37:1 and pH 9.3, chemical equilibrium calculations showed that struvite, potassium struvite, magnesium carbonate, and magnesium phosphate solid phases were supersaturated (see Table S4 in ESI†). Of the supersaturated species, the solids that precipitate will depend on the nucleation kinetics. For example, previous precipitation studies investigating the NH4H2PO4–CaCl2–MgCl2–H2O system showed that brushite (CaHPO4·2H2O), monetite (CaHPO4), amorphous calcium phosphate (Ca3(PO4)2·xH2O), newberyite (MgHPO4·3H2O), and struvite precipitated first, and that brushite, monetite, newberyite, and struvite were present as the final phases depending on concentration and pH.51,52 The results in Tables S3 and S4† show that struvite had a higher SI in hydrolyzed urine (SI = 3.97) than potassium struvite in fresh urine (SI = 1.02). This was because struvite has a lower Ks0 than potassium struvite,45 and the IAP of struvite in hydrolyzed urine was greater than the IAP of potassium struvite in fresh urine. Previous data are not available to compare the nucleation kinetics of struvite with potassium struvite; however, previous results for struvite show that crystallization induction time decreases with increasing supersaturation.53 The amount and purity of the solids collected during the precipitation experiments, and the potential for co-precipitation of multiple solid phases are discussed in the following subsections.
The precipitation conditions in the spent regenerants were designed to be similar to the conditions in urine and filtrate, i.e., same pH and same Mg:P molar ratio. All of the spent regenerants required similar additions of strong acid to decrease the pH from 13 to 9.3 to avoid the precipitation of magnesium hydroxide at pH 13. The large amount of HCl required by the spent regenerants was due to the high buffer intensity of hydroxide at pH 13. The spent regenerants required the addition of two other chemicals: magnesium and potassium for precipitation of potassium struvite, or magnesium and ammonia for precipitation of struvite. The amount of magnesium, potassium, and ammonia required for precipitation was determined by the PO4 concentration because none of these chemicals were initially present in the regeneration solution.
In the spent regenerant corresponding to fresh urine (Mg:K:P = 1.5:1.5:1 and pH 9.3), chemical equilibrium calculations showed that potassium struvite, Mg3(PO4)2, and newberyite (MgHPO4·3H2O) were supersaturated (see Table S5 in ESI†). The SI for potassium struvite was lower in the spent regenerant (SI = 0.8) than in fresh urine (SI = 1.02) because the IAP was smaller in the spent regenerant than fresh urine. Because of the absence of calcium in the spent regenerant, only one solid phase had a higher SI than potassium struvite in the spent regenerant (i.e., Mg3(PO4)2), whereas six other solid phases had a higher SI than potassium struvite in fresh urine (see Table S3 in ESI†). In the spent regenerant corresponding to hydrolyzed urine (Mg:N:P = 1.5:1.5:1 and pH 9.3), chemical equilibrium calculations showed that struvite, Mg3(PO4)2, and newberyite were supersaturated (see Table S6 in ESI†). Ma et al. investigated struvite precipitation in MgCl2–(NH4)2HPO4–NaCl–H2O system, which contained the same components as spent regenerant corresponding to hydrolyzed urine, and calculated that struvite (SI = 3.05), Mg3(PO4)2 (SI = 3.98), and newberyite (SI = 0.74) were supersaturated at pH 9.12 The SI for struvite was lower in the spent regenerant (SI = 3.08) than hydrolyzed urine (SI = 3.97) because the IAP was smaller in the spent regenerant than hydrolyzed urine. Struvite had the second highest SI in the spent regenerant and hydrolyzed urine; however, the solid with the highest SI changed from Mg5(CO3)4(OH)2·4H2O in hydrolyzed urine to Mg3(PO4)2 in spent regenerant. In comparing the PO4 precipitation potential in urine and spent regenerant, fewer solid phases were supersaturated in the spent regenerant than urine.
Phosphate recovery by precipitation
The results on PO4 recovery by direct precipitation and adsorption–precipitation are listed in Table 3. Struvite precipitation in hydrolyzed urine and anaerobic digester filtrate resulted in >99% PO4 removal from solution, and chemical equilibrium calculations (setting struvite allowed to precipitate) predicted that essentially 100% of the PO4 would precipitate as struvite in hydrolyzed urine. Additionally, the experimental results agree with previous literature, e.g., 99% PO4 removal by precipitation of struvite in synthetic urine14 and 95% PO4 removal by precipitation of struvite in real anaerobic digester supernatant.54Potassium struvite precipitation in fresh urine resulted in 98% PO4 removal from solution. However, chemical equilibrium calculations (setting potassium struvite allowed to precipitate) predicted that only 75% of the PO4 would precipitate as potassium struvite in fresh urine. Previous reports on PO4 removal by potassium struvite precipitation differ, e.g., approximately 75% PO4 removal by precipitation of potassium struvite in synthetic urine14 and 98% PO4 removal by precipitation of potassium struvite and magnesium sodium phosphate in synthetic urine.55 Thus, the high PO4 removal observed for potassium struvite precipitation in this work is likely due to co-precipitation of potassium struvite and other phosphate-containing solid phases as discussed later.
Struvite precipitation in the spent regenerants corresponding to hydrolyzed urine and anaerobic digester filtrate resulted in >99% PO4 removal from solution. Additionally, chemical equilibrium calculations (setting struvite allowed to precipitate) showed >99% PO4 precipitation as struvite in spent regenerant. Previous reports show 99.9% PO4 removal by struvite precipitation in synthetic spent regenerant31 and >90% PO4 removal by struvite precipitation in real spent regenerant.28 Hence, the experimental results, chemical equilibrium calculations, and previous literature all show high PO4 removal by struvite precipitation in spent regenerant.
Potassium struvite precipitation in the spent regenerants corresponding to fresh urine and hydrolyzed urine showed 97–98% PO4 removal from solution. However, chemical equilibrium calculations (setting potassium struvite allowed to precipitate) predicted only 55% PO4 precipitation as potassium struvite in spent regenerant. The experimental results and chemical equilibrium calculations suggest that additional solid phases precipitated with potassium struvite, which is discussed in the next subsection. There are no published data on potassium struvite precipitation in spent regenerant to compare with, so the results of this work fill a gap in the literature on potassium struvite precipitation and illustrate the need for additional experimental work.
Identity and purity of precipitated solids
The percent P in the precipitated solid gives an indication of its mineral identity and purity, e.g., pure struvite is 12.62% (m/m) P and pure potassium struvite is 11.62% (m/m) P. Based on percent P, the solid precipitated in the spent regenerant corresponding to hydrolyzed urine showed the best agreement with pure struvite followed by the solid precipitated in hydrolyzed urine (see Table 3). The slightly higher P content in the solid precipitated in hydrolyzed urine than pure struvite suggests the presence of struvite and other magnesium phosphate solid phases. For example, newberyite (17.77% (m/m) P), bobierrite (Mg3(PO4)2·8H2O, 15.22% (m/m) P), and trimagnesium phosphate (Mg3(PO4)2·22H2O, 9.40% (m/m) P) are known to precipitate with struvite in solutions resembling urine.12,45,52 Additionally, previous reports show that the percent P of struvite precipitated in urine can vary from 7.2–18%.56 Based on XRD results, the solid precipitated in hydrolyzed urine matched well with the major d-spacings and relative intensities of pure struvite, whereas the solid precipitated in the spent regenerant corresponding to hydrolyzed urine matched the major d-spacings for pure struvite but the relative intensities deviated from pure struvite possibly due to orientation effects (see Fig. S4 in ESI†).49,57The solid precipitated in fresh urine and the spent regenerants corresponding to fresh urine and hydrolyzed urine showed higher P content than pure potassium struvite (see Table 3). This suggests that other calcium and magnesium phosphate solid phases precipitated with potassium struvite. For example, brushite (CaHPO4·2H2O, 18.00% (m/m) P), monetite (CaHPO4, 22.77% (m/m) P), and hydroxyapatite (Ca5(PO4)3OH), 18.50% (m/m) P) are known to precipitate in solutions resembling urine.51,52 Additionally, trimagnesium phosphates (Mg3(PO4)2·8H2O, 15.22% (m/m) P; Mg3(PO4)2·22H2O, 9.40% (m/m) P) and magnesium sodium phosphate (MgNaPO4·7H2O, 11.54% (m/m) P) are known to precipitate with potassium struvite.45,55 Furthermore, previous work investigating the co-precipitation of potassium struvite and magnesium sodium phosphate in synthetic urine reported 98% PO4 removal,55 which is in excellent agreement with 97–98% PO4 removal by potassium struvite precipitation in this work. Based on XRD results, the solid precipitated in fresh urine and spent regenerants corresponding to fresh and hydrolyzed urine matched relatively well with the major d-spacings and relative intensity of pure potassium struvite (see Fig. S6 in ESI†).57 In addition, the XRD results suggested the presence of halite in the solid collected from the spent regenerants even though the chemical equilibrium calculations showed halite was undersaturated. The XRD results were not quantitative in terms providing the amount of halite.
The solid precipitated in the anaerobic digester filtrate contained substantially lower P content than pure struvite (i.e., 5.5 percentage points lower), and the solid precipitated in the spent regenerant corresponding to anaerobic digester filtrate contained 1.1 percentage points lower P than pure struvite. The solid formed by direct precipitation in anaerobic digester filtrate was likely a combination of struvite, amorphous calcium phosphate (based on the Mg:Ca molar ratio of 1:2.1 in the filtrate16), and other unknown solids phases. Based on previous reports, the percent P of struvite precipitated in anaerobic digester sidestreams can vary from low to close match with pure struvite.7,58 The XRD results for the solid precipitated in anaerobic digester filtrate matched most of the major d-spacings for pure struvite but the relative intensities deviated from pure struvite (see Fig. S5 in ESI†).57 The XRD results for the solid precipitated in the spent regenerant corresponding to anaerobic digester filtrate showed a better match with the major d-spacings and relative intensities for pure struvite than the solid from direct precipitation. The XRD results for the solid precipitated in the spent regenerant corresponding to anaerobic digester filtrate also showed the presence of halite, which could account for the lower percent P than the solid precipitated in the spent regenerant corresponding to hydrolyzed urine in which halite was not detected.
Environmental implications
Phosphate adsorption to HAIX-Fe resin and desorption using caustic brine was more effective for anaerobic digester filtrate than fresh and hydrolyzed urine. Considering the waste streams investigated, the order of increasing chemical requirements for precipitation of struvite and potassium struvite was hydrolyzed urine < fresh urine ≈ anaerobic digester filtrate < spent regenerants. Based on the experimentally determined P content of the precipitated solids, the purity of struvite and potassium struvite precipitated in the spent regenerants was equal to or higher than the purity of the same mineral precipitated in urine and filtrate. Thus, direct precipitation of struvite in hydrolyzed urine is more efficient in terms of lower chemical requirements and similar mineral purity than adsorption–precipitation. However, adsorption–precipitation has potential benefits for PO4 recovery from anaerobic digester filtrate and fresh urine as described below. Although outside the scope of this work, two possible scenarios in which PO4 recovery via adsorption–precipitation could be more favorable than direct precipitation in source-separated urine are (1) to minimize contamination of the final mineral product19,20 and (2) to improve process automation and control.11,59 For example, the well-defined composition of the spent regenerant could allow for measurements such as pH, conductivity, and turbidity to control the precipitation process, whereas this type of monitoring and automation is challenging in urine because of its varying composition. Based on the data generated in this work, these scenarios are speculation; however, the scenarios represent the next logical step in the continuum of research on PO4 recovery by adsorption–precipitation.The adsorption–precipitation process is considered favorable for PO4 recovery as struvite from anaerobic digester filtrate considering that direct precipitation produced a lower purity solid in this study and previous studies. If, however, the goal is to remove PO4 to prevent fouling due to precipitation during sludge handling processes at the wastewater treatment plant then direct precipitation would be favored over adsorption–precipitation due to lower chemical addition requirements. For example, Lew et al. found that struvite precipitation of belt press filtrate from an anaerobic digester can be a solution to prevent clogging due to precipitation in pumps and pipes during sludge dewatering processes, but produces precipitates that could not be used for land application in agriculture due to impurities such as metals and low nutrient content.58
The adsorption–precipitation process also has the potential to be more favorable than direct precipitation for PO4 recovery as potassium struvite from fresh urine, especially if potassium struvite precipitation can be achieved at pH 13. Struvite precipitation is much less favorable at pH > 11 than pH 9–10 because of the shift in ammonia from NH4+ to NH3, and the subsequent decrease in the activity of NH4+ and the corresponding decrease in the IAP and SI of struvite.12 In contrast to struvite, Xu et al. showed nearly 100% PO4 recovery as potassium struvite over the pH range 10–12.51.55 Hence, if potassium struvite could be precipitated in spent regenerant with minimal pH adjustment, it would substantially decrease chemical requirements for the adsorption–precipitation process. To make the spent regenerant more conducive to potassium struvite precipitation, the regeneration solution could be prepared using KCl in place of NaCl so that magnesium would be the only external chemical needed.
Acknowledgements
This publication is based upon work supported by NSF CAREER grant number CBET-1150790. Any opinions, findings, conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the National Science Foundation. The authors thank Willie Harris (University of Florida) for XRD analysis and Daniel Yeh (University of South Florida) for assistance obtaining anaerobic digester filtrate. This manuscript was improved by the comments of two anonymous reviewers.References
- J. S. Guest, S. J. Skerlos, J. L. Barnard, M. B. Beck, G. T. Daigger, H. Hilger, S. J. Jackson, K. Karvazy, L. Kelly, L. Macpherson, J. R. Mihelcic, A. Promanik, L. Raskin, M. C. M. Van Loosdrecht, D. Yeh and N. G. Love, A New Planning and Design Paradigm to Achieve Sustainable Resource Recovery from Wastewater, Environ. Sci. Technol., 2009, 43(16), 6126–6130 CrossRef CAS.
- D. P. Van Vuuren, A. F. Bouwman and A. H. W. Beusen, Phosphorus demand for the 1970–2100 period: A scenario analysis of resource depletion, NATO ASI Ser., Ser. I, 2010, 20(3), 428–439 Search PubMed.
- D. Cordell, J.-O. Drangert and S. White, The story of phosphorus: Global food security and food for thought, NATO ASI Ser., Ser. I, 2009, 19(2), 292–305 Search PubMed.
- R. W. Scholz, A. E. Ulrich, M. Eilittä and A. Roy, Sustainable use of phosphorus: A finite resource, Sci. Total Environ., 2013, 461–462(0), 799–803 CrossRef CAS PubMed.
- R. O. Carey and K. W. Migliaccio, Contribution of Wastewater Treatment Plant Effluents to Nutrient Dynamics in Aquatic Systems: A Review, Environ. Manage., 2009, 44(2), 205–217 CrossRef PubMed.
- J. D. Doyle, K. Oldring, J. Churchley and S. A. Parsons, Struvite formation and the fouling propensity of different materials, Water Res., 2002, 36(16), 3971–3978 CrossRef CAS.
- E. V. Munch and K. Barr, Controlled struvite crystallisation for removing phosphorus from anaerobic digester sidestreams, Water Res., 2001, 35(1), 151–159 CrossRef CAS.
- J. A. Wilsenach and M. C. M. van Loosdrecht, Integration of processes to treat wastewater and source-separated urine, J. Environ. Eng., 2006, 132(3), 331–341 CrossRef CAS.
- L. E. de-Bashan and Y. Bashan, Recent advances in removing phosphorus from wastewater and its future use as fertilizer (1997–2003), Water Res., 2004, 38(19), 4222–4246 CrossRef CAS PubMed.
- K. S. Le Corre, E. Valsami-Jones, P. Hobbs and S. A. Parsons, Phosphorus Recovery from Wastewater by Struvite Crystallization: A Review, Crit. Rev. Environ. Sci. Technol., 2009, 39(6), 433–477 CrossRef CAS.
- B. Etter, E. Tilley, R. Khadka and K. M. Udert, Low-cost struvite production using source-separated urine in Nepal, Water Res., 2011, 45(2), 852–862 CrossRef CAS PubMed.
- N. Ma, A. A. Rouff and B. L. Phillips, A 31P NMR and TG/DSC-FTIR Investigation of the Influence of Initial pH on Phosphorus Recovery as Struvite, ACS Sustainable Chem. Eng., 2014, 2(4), 816–822 CrossRef CAS.
- K. Xu, C. Wang, X. Wang and Y. Qian, Laboratory experiments on simultaneous removal of K and P from synthetic and real urine for nutrient recycle by crystallization of magnesium–potassium–phosphate–hexahydrate in a draft tube and baffle reactor, Chemosphere, 2012, 88(2), 219–223 CrossRef CAS PubMed.
- J. A. Wilsenach, C. A. H. Schuurbiers and M. C. M. van Loosdrecht, Phosphate and potassium recovery from source separated urine through struvite precipitation, Water Res., 2007, 41(2), 458–466 CrossRef CAS PubMed.
- A. E. Johnston and I. R. Richards, Effectiveness of different precipitated phosphates as phosphorus sources for plants, Soil Use Manage., 2003, 19(1), 45–49 CrossRef PubMed.
- K. S. Le Corre, E. Valsami-Jones, P. Hobbs and S. A. Parsons, Impact of calcium on struvite crystal size, shape and purity, J. Cryst. Growth, 2005, 283(3–4), 514–522 CrossRef CAS PubMed.
- X. D. Hao, C. C. Wang, L. Lan and M. C. M. van Loosdrecht, Struvite formation, analytical methods and effects of pH and Ca2+, Water Sci. Technol., 2008, 58(8), 1687–1692 CrossRef PubMed.
- S. R. Sakthivel, E. Tilley and K. M. Udert, Wood ash as a magnesium source for phosphorus recovery from source-separated urine, Sci. Total Environ., 2012, 419(0), 68–75 CrossRef CAS PubMed.
- A. A. Rouff, Sorption of Chromium with Struvite During Phosphorus Recovery, Environ. Sci. Technol., 2012, 46(22), 12493–12501 CrossRef CAS PubMed.
- L. Decrey, K. M. Udert, E. Tilley, B. M. Pecson and T. Kohn, Fate of the pathogen indicators phage ΦX174 and Ascaris suum eggs during the production of struvite fertilizer from source-separated urine, Water Res., 2011, 45(16), 4960–4972 CrossRef CAS PubMed.
- J. A. O'Neal and T. H. Boyer, Phosphate recovery using hybrid anion exchange: Applications to source-separated urine and combined wastewater streams, Water Res., 2013, 47(14), 5003–5017 CrossRef PubMed.
- M. R. Awual and A. Jyo, Assessing of phosphorus removal by polymeric anion exchangers, Desalination, 2011, 281(0), 111–117 CrossRef CAS PubMed.
- P. L. Sibrell, G. A. Montgomery, K. L. Ritenour and T. W. Tucker, Removal of phosphorus from agricultural wastewaters using adsorption media prepared from acid mine drainage sludge, Water Res., 2009, 43(8), 2240–2250 CrossRef CAS PubMed.
- X. P. Zhu and A. Jyo, Column-mode phosphate removal by a novel highly selective adsorbent, Water Res., 2005, 39(11), 2301–2308 CrossRef CAS PubMed.
- D. Zhao and A. K. SenGupta, Ligand Separation with a Copper(II)-Loaded Polymeric Ligand Exchanger, Ind. Eng. Chem. Res., 2000, 39, 455–462 CrossRef CAS.
- L. M. Blaney, S. Cinar and A. K. SenGupta, Hybrid anion exchanger for trace phosphate removal from water and wastewater, Water Res., 2007, 41(7), 1603–1613 CrossRef CAS PubMed.
- B. J. Pan, J. Wu, B. C. Pan, L. Lv, W. M. Zhang, L. L. Xiao, X. S. Wang, X. C. Tao and S. R. Zheng, Development of polymer-based nanosized hydrated ferric oxides (HFOs) for enhanced phosphate removal from waste effluents, Water Res., 2009, 43(17), 4421–4429 CrossRef CAS PubMed.
- S. Sengupta and A. Pandit, Selective removal of phosphorus from wastewater combined with its recovery as a solid-phase fertilizer, Water Res., 2011, 45(11), 3318–3330 CrossRef CAS PubMed.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, John Wiley & Sons, Inc., New York, 1996 Search PubMed.
- B. D. Martin, S. A. Parsons and B. Jefferson, Removal and recovery of phosphate from municipal wastewaters using a polymeric anion exchanger bound with hydrated ferric oxide nanoparticles, Water Sci. Technol., 2009, 60(10), 2637–2645 CrossRef CAS PubMed.
- M. Kumar, M. Badruzzaman, S. Adham and J. Oppenheimer, Beneficial phosphate recovery from reverse osmosis (RO) concentrate of an integrated membrane system using polymeric ligand exchanger (PLE), Water Res., 2007, 41(10), 2211–2219 CrossRef CAS PubMed.
- A. Bottini and L. Rizzo, Phosphorus Recovery from Urban Wastewater Treatment Plant Sludge Liquor by Ion Exchange, Sep. Sci. Technol., 2012, 47(4), 613–620 CrossRef CAS.
- A. Sendrowski and T. H. Boyer, Phosphate removal from urine using hybrid anion exchange resin, Desalination, 2013, 322(0), 104–112 CrossRef CAS PubMed.
- T. Tervahauta, T. Hoang, L. Hernández, G. Zeeman and C. Buisman, Prospects of Source-Separation-Based Sanitation Concepts: A Model-Based Study, Water, 2013, 5(3), 1006–1035 CrossRef CAS PubMed.
- R. Rajagopal, J. W. Lim, Y. Mao, C.-L. Chen and J.-Y. Wang, Anaerobic co-digestion of source segregated brown water (feces-without-urine) and food waste: For Singapore context, Sci. Total Environ., 2013, 443(0), 877–886 CrossRef CAS PubMed.
- M. Ronteltap, M. Maurer and W. Gujer, Struvite precipitation thermodynamics in source-separated urine, Water Res., 2007, 41(5), 977–984 CrossRef CAS PubMed.
- K. M. Udert and M. Wächter, Complete nutrient recovery from source-separated urine by nitrification and distillation, Water Res., 2012, 46(2), 453–464 CrossRef CAS PubMed.
- B.-B. Lind, Z. Ban and S. Bydén, Nutrient recovery from human urine by struvite crystallization with ammonia adsorption on zeolite and wollastonite, Bioresour. Technol., 2000, 73(2), 169–174 CrossRef CAS.
- B. Beler-Baykal, N. P. Kocaturk, A. D. Allar and B. Sari, The effect of initial loading on the removal of ammonium and potassium from source-separated human urine via clinoptilolite, Water Sci. Technol., 2009, 60(10), 2515–2520 CrossRef CAS PubMed.
- H. L. Mobley and R. P. Hausinger, Microbial ureases: significance, regulation, and molecular characterization, Microbiol. Rev., 1989, 53(1), 85–108 CAS.
- K. M. Udert, T. A. Larsen, M. Biebow and W. Gujer, Urea hydrolysis and precipitation dynamics in a urine-collecting system, Water Res., 2003, 37(11), 2571–2582 CrossRef CAS.
- L.-L. Wei, Q.-L. Zhao, K. Hu, D.-J. Lee, C.-M. Xie and J.-Q. Jiang, Extracellular biological organic matters in sewage sludge during mesophilic digestion at reduced hydraulic retention time, Water Res., 2011, 45(3), 1472–1480 CrossRef CAS PubMed.
- P. Battistoni, A. De Angelis, P. Pavan, M. Prisciandaro and F. Cecchi, Phosphorus removal from a real anaerobic supernatant by struvite crystallization, Water Res., 2001, 35(9), 2167–2178 CrossRef CAS.
- J. P. Gustafsson, Visual MINTEQ, version 3.0.http://www2.lwr.kth.se/English/OurSoftware/vminteq/ (accessed 24 June 2013) Search PubMed.
- A. W. Taylor, A. W. Frazier and E. L. Gurney, Solubility products of magnesium ammonium and magnesium potassium phosphates, Trans. Faraday Soc., 1963, 59(0), 1580–1584 RSC.
- A. D. Eaton, L. S. Clesceri, E. W. Rice and A. E. Greenberg, Standard Methods for the Examination of Water and Wastewater, APHA, AWWA, WEF, 21st edn, 2005 Search PubMed.
- J. N. Apell and T. H. Boyer, Combined ion exchange treatment for removal of dissolved organic matter and hardness, Water Res., 2010, 44(8), 2419–2430 CrossRef CAS PubMed.
- K. Indarawis and T. H. Boyer, Alkaline Earth Metal Cation Exchange: Effect of Mobile Counterion and Dissolved Organic Matter, Environ. Sci. Technol., 2012, 46(8), 4591–4598 CrossRef CAS PubMed.
- W. G. Harris and G. N. White, X-ray diffraction techniques for soil mineral identification. In Methods of Soil Analysis: Part 5 – Mineralogical Methods, ed. A. L. Ulery and L. R. Drees, Soil Science Society of America, Madison, WI, 2008, pp. 81–115 Search PubMed.
- F. M. M. Morel and J. G. Hering, Principles and Applications of Aquatic Chemistry, John Wiley & Sons, New York, 1993 Search PubMed.
- F. Abbona, H. E. Lundager Madsen and R. Boistelle, The final phases of calcium and magnesium phosphates precipitated from solutions of high to medium concentration, J. Cryst. Growth, 1988, 89(4), 592–602 CrossRef CAS.
- F. Abbona, H. E. L. Madsen and R. Boistelle, The initial phases of calcium and magnesium phosphates precipitated from solutions of high to medium concentrations, J. Cryst. Growth, 1986, 74(3), 581–590 CrossRef CAS.
- N. C. Bouropoulos and P. G. Koutsoukos, Spontaneous precipitation of struvite from aqueous solutions, J. Cryst. Growth, 2000, 213(3–4), 381–388 CrossRef CAS.
- L. Pastor, D. Mangin, J. Ferrer and A. Seco, Struvite formation from the supernatants of an anaerobic digestion pilot plant, Bioresour. Technol., 2010, 101(1), 118–125 CrossRef CAS PubMed.
- K. Xu, C. Wang, H. Liu and Y. Qian, Simultaneous removal of phosphorus and potassium from synthetic urine through the precipitation of magnesium potassium phosphate hexahydrate, Chemosphere, 2011, 84(2), 207–212 CrossRef CAS PubMed.
- S. Antonini, M. A. Arias, T. Eichert and J. Clemens, Greenhouse evaluation and environmental impact assessment of different urine-derived struvite fertilizers as phosphorus sources for plants, Chemosphere, 2012, 89(10), 1202–1210 CrossRef CAS PubMed.
- H. Yang, H. J. Sun and R. T. Downs, Hazenite, KNaMg2(PO4)2·14H2O, a new biologically related phosphate mineral, from Mono Lake, California, U.S.A., Am. Mineral., 2011, 96(4), 675–681 CrossRef CAS.
- B. Lew, S. Phalah and M. Rebhum, Controlled struvite precipitation from belt press filtrate of anaerobic digester in a CSTR, Environ. Prog. Sustainable Energy, 2011, 30(4), 640–647 CrossRef CAS PubMed.
- A. Triger, J.-S. Pic and C. Cabassud, Determination of struvite crystallization mechanisms in urine using turbidity measurement, Water Res., 2012, 46(18), 6084–6094 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and tables as referenced in the text. This material is available via the internet. See DOI: 10.1039/c5ew00009b |
| This journal is © The Royal Society of Chemistry 2015 |