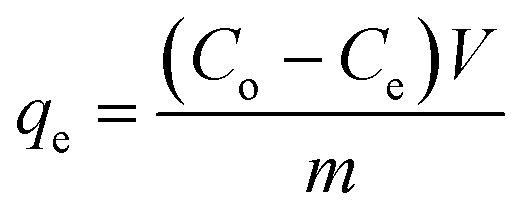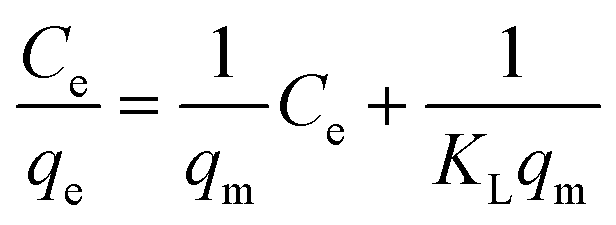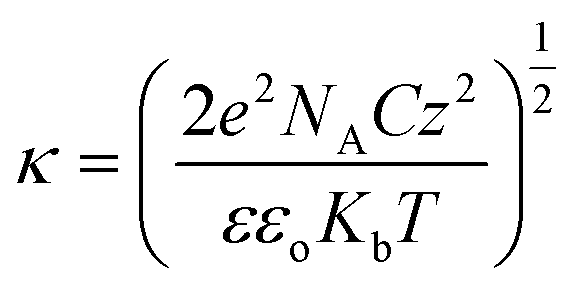Graphene in the Fe3O4 nano-composite switching the negative influence of humic acid coating into an enhancing effect in the removal of arsenic from water†
Received
5th August 2014
, Accepted 19th September 2014
First published on 5th November 2014
Abstract
The humic acid coating was influenced by graphene in the Fe3O4 nano-composite, which turned out to be an enhancing effect in the removal of arsenic from water. Earlier studies have shown the negative role of humic acid (ubiquitous in all natural water systems) in the removal of arsenic from natural water resources. Herein we present for the first time the positive influence of humic acid (HA) on graphene–Fe3O4 nano-composites for the removal of arsenic present in water. The Fe3O4 nanoparticles were assembled on graphene oxide (GO) sheets through the co-precipitation process and concurrently the humic acid coating was brought out. After coating with humic acid, the composite has shown almost double the removal efficiency of As(III) and As(V) at neutral pH.
Water impact
Many countries are at risk of arsenic contamination in ground water. Immense efforts are being done from a nanotechnology standpoint to tackle this conundrum. However, the most critical aspect in solving this problem in the presence of humic acid (ubiquitous in all natural water systems) has been overlooked by most researchers. The negative influence of humic acid for arsenic removal is well documented. This information gap is embarked upon to view into the subject anew. The graphene in our nano-composite could reverse the role of humic acid from negative towards positive. The present study would open a new dimension in the practical utilization of nanotechnology in water research for arsenic removal.
|
Introduction
Exposure of drinking water to inorganic arsenic leads to large-scale cases of poisoning in Argentina, Chile, Bangladesh, Taiwan, USA, Canada and Japan.1 Among them, the biggest arsenic calamity in Bangladesh has been risking the lives of more than 65 million people.2 In recent years a great deal of interest has been developed in nanomaterials for the effective treatment of arsenic in natural ground water.3 However, studies have found that removal of arsenic by nanoparticles in the presence of humic acid has a negative role.4,5 Humic acid represents the major component of humic substances which are produced by the biodegradation of organic matter and are ubiquitous in all natural water systems.6 Therefore, humic acid in turn poses a big hurdle for practical use of nanomaterials for the effective removal of arsenic from the natural water system due to its strong complexation ability.7–10 This inhibitory effect of humic acid in the arsenic removal process therefore causes an extra amount of materials and energy for the pre-treatment.11 This demands extensive research in advancing novel nanomaterials to tackle this conundrum posed by humic acid for the effective removal of arsenic from the ground water system. Although zero-valent iron nanoparticles have been proven to be effective in the arsenic removal process, these systems failed to enhance the adsorption of arsenic in the presence of humic acid.12 Several studies have been conducted to investigate the negative influence of humic acid in water treatment for toxic metals. For example, a binary system composed of zero-valent iron and activated carbon performed with negligible effect in the removal of arsenic in the presence of humic acid.13 The Fe3O4 nanoparticles coated with a polysaccharide were observed with a negative effect in the presence of humic acid in the adsorption of As(V).14 Another Fe–Mn binary oxide has been tested in the presence of humic acid but no significant effect was observed.15 Maghemite coated with Fe–Mn binary oxide was also found to have insufficient influence with humic acid in the removal of arsenite.16 Modification of Fe3O4 with cetyltrimethylammonium bromide (CTAB) greatly enhanced As(V) adsorption capacity but the uptake decreased in the presence of humic acid.17 Moreover, in recent years graphene with its exceptional properties has emerged as the material of choice for clean water technologies.18,19 The water proofing and surface charging properties of graphene sheets can be tailored for the faster separation of ions from water.20 Single-layer graphene has been created with nano-pores in order to improve the water permeation properties for the application of desalination.19 Creating precisely defined pores of the required size on large graphene sheets for separating pollutants is in progress.21 Furthermore, graphene and its composites have been used in the treatment of arsenic.22,23 However, we did not find any study on the role of humic acid in the presence of graphene and its composites. This motivated us to investigate the effect of humic acid on the efficiency of these graphene-based composites in arsenic removal from ground water. For this we chose the graphene–Fe3O4 nano-composite as a model system to conduct our studies. Interestingly, we found that the graphene–Fe3O4 nano-composite sheathed with humic acid (HA) reverses the role of HA to positive for the removal of arsenic from ground water. Our observation is contradictory to the general belief that the association of any system with humic acid reduces the adsorption of arsenic from water.
Materials and methods
Chemicals
The starting reagents for the preparation of graphene oxides and magnetite particles were all purchased from Sigma-Aldrich and were used as received. Sulphuric acid, phosphoric acid, hydrochloric acid and ammonia solution were obtained from Merck.
Preparation of graphene oxide
Natural graphite powder was exfoliated to form graphene oxide using a modified Hummers method.24 In a typical process, 360 mL of concentrated sulphuric acid and 48 mL of concentrated phosphoric acid were mixed in a round-bottom flask for 10 min, and 3 g of graphite platelets was added to the homogeneous mixture under vigorous stirring to obtain a dark-coloured suspension with the graphite platelets dispersed uniformly. Subsequently, 18 g of KMnO4 was added slowly into the above suspension. The mixture was stirred vigorously for 12 h at 50 °C. A reddish brown viscous mixture was obtained, indicating the completion of oxidation reaction. This mixture was cooled to room temperature, and then 400 mL of deionized water with 3 mL of H2O2 (30%) was poured slowly and a uniform suspension was obtained. Afterwards, the suspension was centrifuged and washed with a total of 1000 mL of water and 200 mL of 30% HCl. The solid materials were collected after centrifugation and dispersed again in 1000 mL of water. After recovering the solids from the solution through centrifugation, the solid was then dispersed in 200 mL of ether. The resulting suspension was then filtered over a PTFE membrane with a 0.45 μm pore size. The graphene oxide obtained was vacuum-dried overnight at room temperature.
Immobilization of Fe3O4 nanoparticles on graphene sheets and coating with humic acid
The growth of magnetite (Fe3O4) nanoparticles on graphene sheets was carried out through the co-precipitation method. The iron oxide precipitation techniques were based on previous reports.25–27 Vacuum-dried graphene oxide (55.8 mg) was dispersed in a 100 mL solution of well-dissolved 6.1 g of FeCl3·6H2O and 4.2 g of FeSO4·7H2O. The resulting solution was heated to 90 °C with continual stirring in order to get a homogenous dispersion. 10 mL of NH4OH (25%) solution was rapidly poured into the dispersion. A black precipitate suddenly appeared and was heated at a steady temperature of 90 °C. After 1 h of heating, ascorbic acid (1 g) was introduced for the reduction of graphene sheets. After cooling, the product was filtered, washed several times with water and finally with anhydrous ethanol to remove unreacted ions and then dried in a vacuum at 100 °C for 10 h. The resulting solids were denoted as rGO–Fe. The same procedure was followed for the coating with humic acid but which was introduced into the dispersion simultaneously with ammonia solution. Two separate solutions of 0.1 and 0.2 g of the humic acid sodium salt dissolved in 50 mL of water were used for the coating. The modified products were denoted as rGO–FeHA1 and rGO–FeHA2, respectively.
Characterization techniques
XRD patterns were recorded with a Rigaku D/MAX 2000 PC X-ray diffractometer using Cu Kα radiation (k = 0.154 nm) and operated at 40 kV and 20 mA with a scan rate of 1° min−1 in a 2θ range of 0.5–30°. The FTIR spectra were acquired using Nicolet 380 FTIR spectrometers equipped with a Ge/KBr beam splitter and a dGTS/KBr detector. The collection time was about 1 min (64 scans) for the background and the sample. The spectrometer was purged with dry air. To prepare KBr pellets, about 2 mg of the sample was taken and ground for 1–2 min together with about 200 mg of dried KBr (FT-IR grade, Fluka). The pellets were pressed under vacuum for 4–6 min at 8t pressure to produce transparent discs about 1 mm thick and 13 mm in diameter. The samples were dried before preparation. An empty KBr pellet was used as reference and its spectrum was subtracted from the sample spectrum to suppress the spectral lines caused by KBr impurities and water. Raman spectra were obtained with a Renishaw Raman microscope at an excitation laser wavelength of 514.5 nm. All samples were deposited on glass slides in powder form without any solvent. The reduced graphene oxide was dispersed in acetone using an ultrasonicator, and some pieces were collected on carbon-coated 300-mesh copper grids for TEM observation. The morphology was determined by transmission electron microscopy (TEM) using a Hitachi H-800 transmission electron microscope operating at an accelerating voltage of 200 kV. Surface charge values were generated using a Zetasizer (Malvern Nano ZS).
Batch adsorption experiment
The arsenic adsorption experiments were carried out using the batch equilibration method. For each determination, 10 mg of an air-dried sample was mixed with 50 mL of arsenic solutions with a concentration range of 1, 5 and 10 ppm in different batches. The samples were equilibrated for 24 h at room temperature (23 °C) using a linear shaker in 100 mL scorch bottles at neutral pH. Following equilibration, the samples were centrifuged and the amounts of pollutants in the supernatant solutions were determined by ICP-OES (Spectro Arcos ICP-OES). The concentration of phosphate was kept at 0.001 M to study the effect of coexisting ions.
Results and discussion
Characterization of the Fe3O4 based nano-composite
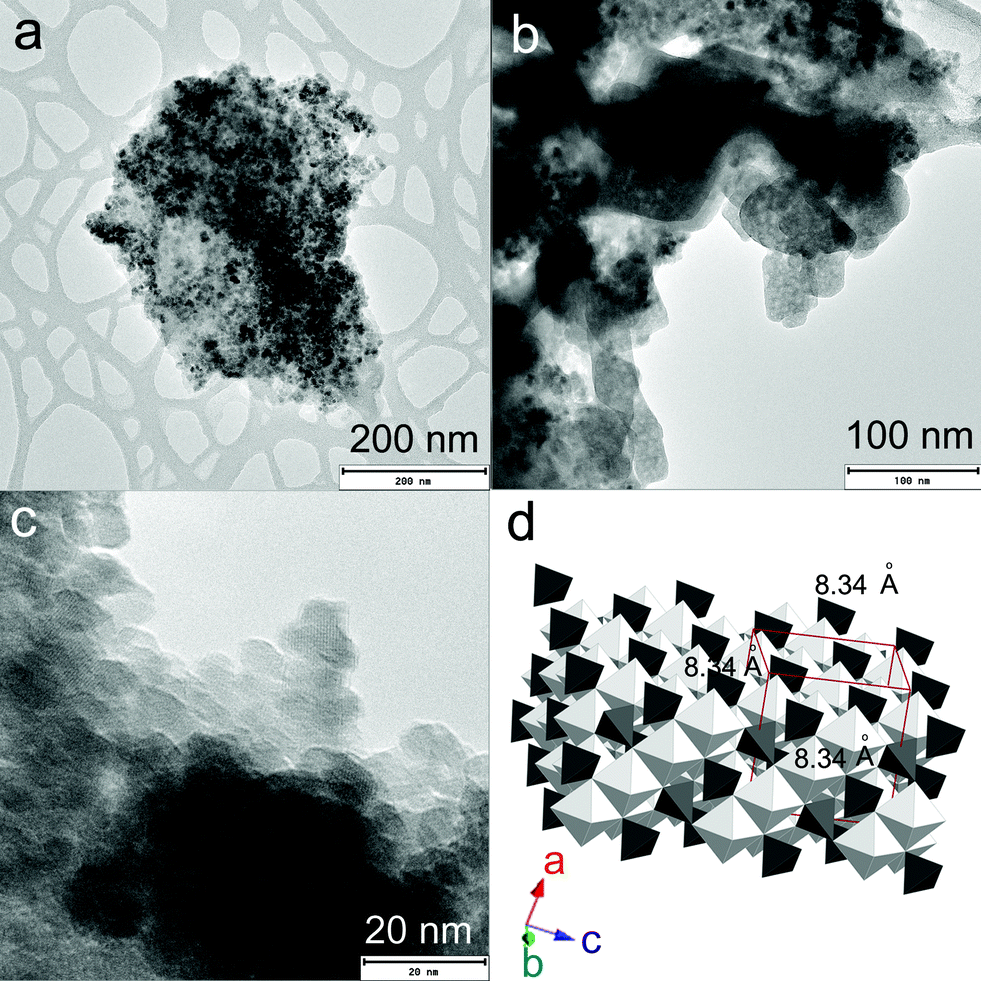
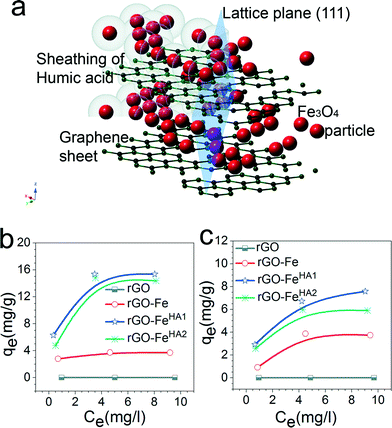
Fig. 1a–c represent the TEM images of rGO–FeHA1. The Fe3O4 nanoparticles acquired an average size distribution of 8 nm and were found assembled along the GO sheets. The graph of the lattice-fringe spectrum of Fe3O4 nanoparticles in rGO–FeHA1 shows a crystallographic spacing (d) of 2.80 Å but a d-spacing of 5.33 Å was also observed in the same crystal in the (112) orientation (ESI†). Fe3O4 is ferromagnetic and both divalent and trivalent irons are present in its structure (Fig. 1d). It has been reported that the Fe3O4 structure includes layers made of octahedral and mixed tetrahedral/octahedral units stacked together. This is the reason we observed two different values of d-spacing.28 Furthermore, it is known that some of the free oxygen from graphene sheets can be involved in the formation of Fe3O4 nanoparticles which provided the strong binding of particles with the GO sheets.29
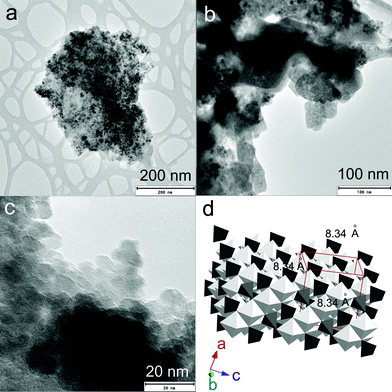 |
| | Fig. 1 (a) TEM image of rGO–FeHA1. (b) TEM of rGO–FeHA1 at higher resolution. (c) Lattice fringes of Fe3O4 nanoparticles are clearly observed on rGO–FeHA1. (d) Schematic presentation of crystallographic structure of Fe3O4. | |
Fig. 2a represents the magnetic interaction of rGO–FeHA1 under an external magnetic field. The association of Fe3O4 with rGO provides an extra edge that the nano-composites can be separated and collected easily by applying an external magnetic field after removing arsenic from the water.
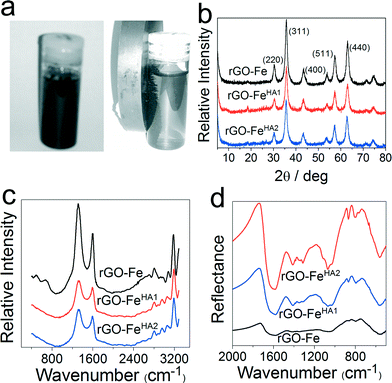 |
| | Fig. 2 (a) Agglomeration of humic acid-coated composites upon the interaction with a permanent magnet; (b) XRD patterns of graphene-based iron composites; (c) and (d) Raman and FTIR spectra of graphene-based composites, respectively. | |
Fig. 2b represents the XRD patterns of rGO–Fe, rGO–FeHA1 and rGO–FeHA2. The refraction peak associated with rGO at a 2θ value of 25° is not pronounced compared to that of the Fe3O4 nanoparticles for all of the samples due to the lack of stacking of graphene sheets as explained by Si et al.30 Furthermore, the XRD pattern confirms that the Fe3O4 nanoparticles exist in cubic structures within the nano-composite. Fig. 2c shows a typical Raman spectrum of the rGO–Fe, rGO–FeHA1 and rGO–FeHA2. Two peaks, at ~1330 and ~1580 cm−1, are observed in the Raman spectrum. These bands are referred to as the so-called D and G-peaks, which are characteristic of sp2 bonded honeycomb structures.31 Moreover, we observed a weak peak at ~600 cm−1 corresponding to Fe3O4 nanoparticles due to low Raman scattering.32Fig. 2d represents the FTIR spectra of rGO–Fe, rGO–FeHA1 and rGO–FeHA2. The band at 1320 cm−1 represents the skeleton of aromatic C–N bonds.33 The band in the 1550 to 1720 cm−1 region is due to the functional groups such as aldehydes, ketones, esters, amide and carboxylic groups.34 A strong absorption band at 1700 cm−1 is significantly due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carboxylic acid and ketone. An absorption band at 1602 cm−1 is ascribed to stretching of carboxylate ions.35,36 The band assigned in the range of 1040–1090 cm−1 is due to the C–O stretching of alcoholic compounds, and that around 500 cm−1 is the deformation of COOH.37 The FTIR spectra clearly demonstrate the coating of humic acid on nano-composites. Fig. 3a depicts schematically the diagram of humic acid coating on Fe3O4-based graphene nano-composites.
O stretching of carboxylic acid and ketone. An absorption band at 1602 cm−1 is ascribed to stretching of carboxylate ions.35,36 The band assigned in the range of 1040–1090 cm−1 is due to the C–O stretching of alcoholic compounds, and that around 500 cm−1 is the deformation of COOH.37 The FTIR spectra clearly demonstrate the coating of humic acid on nano-composites. Fig. 3a depicts schematically the diagram of humic acid coating on Fe3O4-based graphene nano-composites.
 |
| | Fig. 3 (a) Schematic diagram of humic acid coating on iron-based graphene composites; (b) and (c) adsorption isotherms of graphene based composites for arsenate and arsenite, respectively. | |
Adsorption of arsenate and arsenite
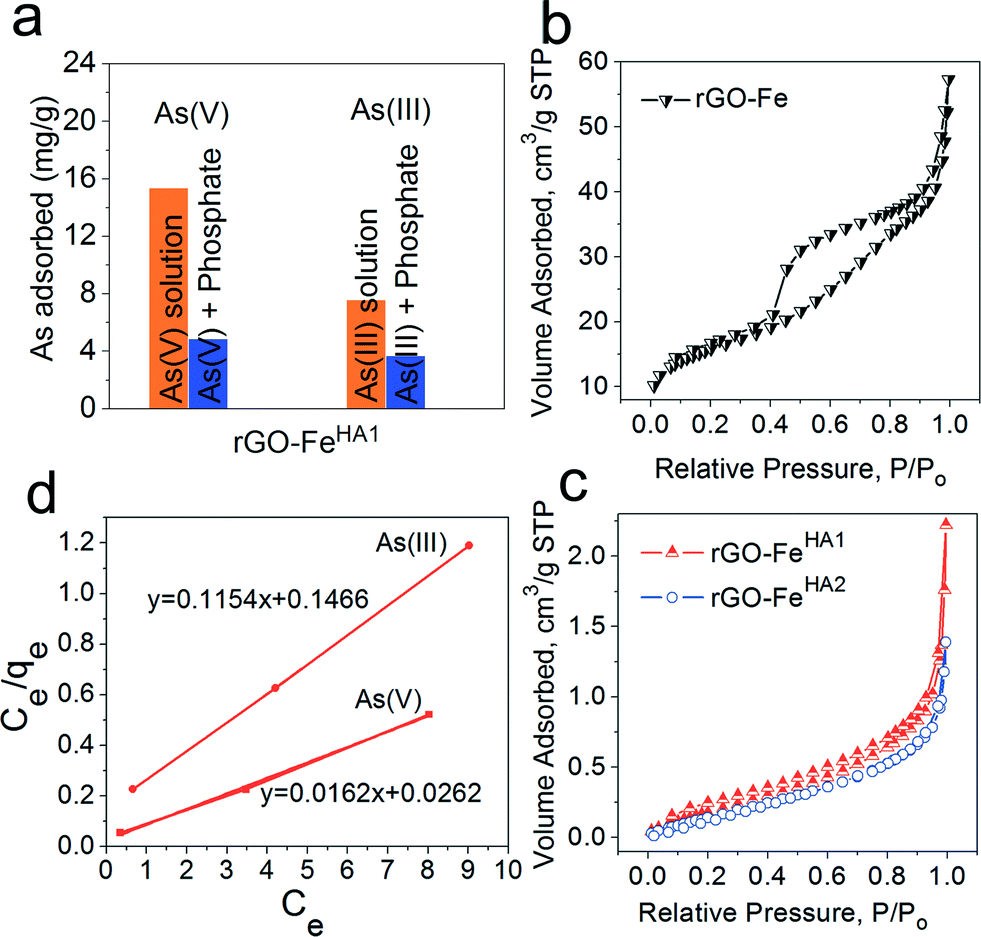
Fig. 3b and c demonstrate that the maximum uptake of arsenate and arsenite observed with rGO–FeHA1 was 16 mg g−1 and 7.5 mg g−1, respectively. This positive observation of humic acid for removal of arsenic in the graphene oxide–Fe3O4 nano-composite materials is not only surprising but also very motivating for the future of nanotechnology in water research. The isotherm curves show L-type adsorption that is well described using the Langmuir equation given below (1)| | 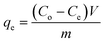 | (1) |
where qe is the amount of arsenic adsorbed onto the composite (mg g−1), Co and Ce are the initial and final concentrations of arsenic in solution at equilibrium (mg L−1), respectively, V is the volume of the solution (L) and m is the mass of the adsorbent (g). The linear form of the equation can be used to analyse the data obtained from the experiment.| | 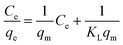 | (2) |
K
L and qm are Langmuir constants representing the adsorption energy and the adsorption capacity, respectively. A linear form of the graph is obtained when Ce/qe is plotted against Ce over a range of arsenic concentrations from the experimental data. The maximum monolayer coverage of arsenic species with the composite represents qm which is obtained from the slope and the enthalpy of adsorption represents KL which is from the intercept of the linear plot (Fig. 5d). The adsorption data for arsenate and arsenite follow the Langmuir behaviour effectively. The maximum monolayer capacity is found to be 8.67 and 61.73 mg g−1 for arsenite and arsenate, respectively. The enthalpy of adsorption (KL) obtained for As(III) is 0.787 L mg−1 and that for As(V) is 0.618 L mg−1.
Mechanism of arsenate and arsenite uptake
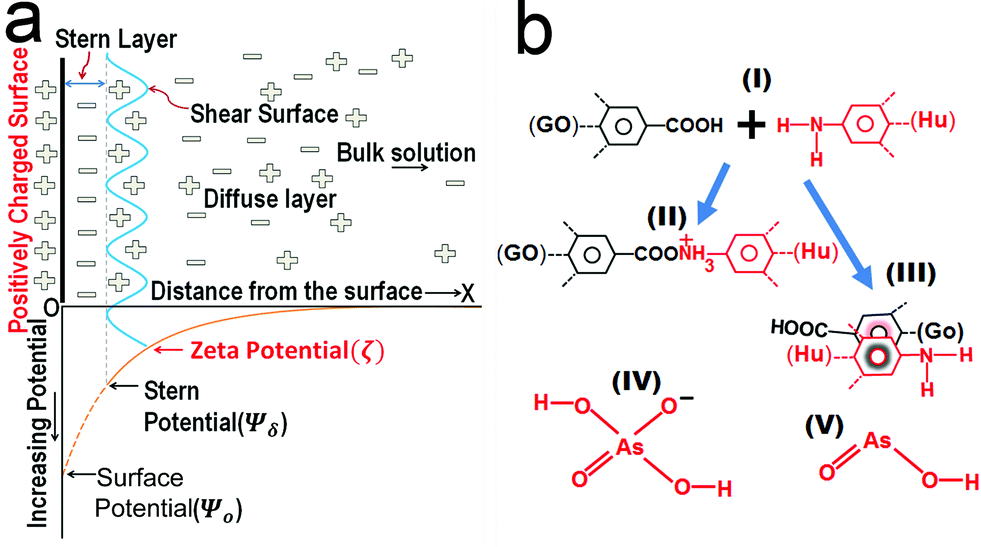
To comprehend this positive role of humic acid towards arsenic, we performed zeta potential measurements on all of the samples, which led us to envision the role of pH and surface charge of humic acid dynamics with GO in the Fe3O4 composite for arsenic removal. The pH-dependent humic acid influence on surface charges is shown in Table 1. In the absence of humic acid coating the surface charge of rGO–Fe was −34.8 mV at pH 7. The humic coating on Fe3O4 nanoparticles adhered on rGO, which not only influence the surface charge but also the diffusion of arsenic towards the interaction sites of iron surfaces. After humic acid coating the distribution of electrical charges around the Fe3O4 particles became different. A model can be developed based on the electrical double layer theory which is shown in Fig. 4a. According to the theory, an immobile Stern layer is created close to the particle surface and an outer layer is distributed with diffusible ions.38,39 When the distance from the surface (x) increases the potential decreases. The potential at the particle surface (Ψo) is impossible to monitor. However, the potential at the shear surface (zeta potential, ζ) can be experimentally measured and is directly proportional to the surface potential.40 The potential at the Stern layer (Ψδ) has a linear relationship with the potential at the surface and the potential exponentially decreases to zero towards the bulk of the solution after the linear drop in the potential from the surface to the Stern layer. This can be expressed as an equation:Here, x is the distance from the particle surface and κ is the Debye parameter:| | 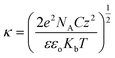 | (4) |
Table 1 The zeta potential values of rGO–Fe, rGO–FeHA1 and rGO–FeHA2 at pH 4, 7 and 10
| pH |
Zeta potential (mV) |
| rGO–Fe |
rGO–FeHA1 |
rGO–FeHA2 |
| 4 |
−14.1 |
−11.3 |
−10.7 |
| 7 |
−34.8 |
−27.8 |
−22.3 |
| 10 |
−28.1 |
−30.4 |
−33.6 |
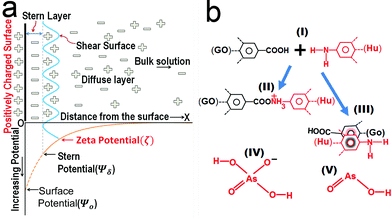 |
| | Fig. 4 (a) Model based on the double layer theory illustrating the interaction of Fe3O4 particles in a solution containing negatively charged arsenic species. (b) The schematic reactions (II) and (III) showing the chemical bonding and π–π interaction of the humic acid–graphene system, respectively. (IV) and (V) are the two major arsenic species at pH 7. | |
Here, e is the protonic charge, NA is Avogadro's constant, C is the concentration of the electrolyte of valence z, ε is the dielectric constant, εo is the permittivity of free space and Kb is the Boltzmann constant. The Debye parameter is proportional to the electrolyte concentration and the potential decreases when the parameter increases. The surface potential (Ψo) is experimentally inaccessible and the Stern potential (Ψδ) is considered almost identical to the zeta potential (ζ) as the shear plane is located at the outer surface of the Stern plane. The measurement of mobility (velocity divided by field strength) of a dilute dispersion of colloidal particles under an electrical field gives zeta potential values. The mobility (μE) is proportional to the zeta potential (ζ) and is known as the Hückel–Onsager equation:
| | 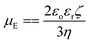 | (5) |
where
η is the shear viscosity of water.
As discussed in the above equation, when the composites were dispersed in water a sheath of negatively charged arsenic builds up along the surface in the Stern layer simultaneously; this led to the formation of a positively charged layer in the shear surface next to the Stern layer. As the process proceeds, the arsenic ions diffuse inside the Fe3O4 particles and establish bonding with them. Therefore, the net negative charge in the Stern layer will decrease but the reduction of charges is maintained through diffusion from the bulk solution. Moreover, humic acid has the ability to form complexes with Fe3+ through the presence of carboxylate and phenolate groups.37 If a bare Fe3O4 nanoparticle is sheathed with a layer of humic acid, the positively charged nature of the particle decreases due to the large number of carbonyl groups present in the humic acid structure. As a result, the number of negatively charged arsenic ions reached in the Stern layer is suppressed due to the repulsion of carbonyl groups. Consequently, humic acid decreases the ability of Fe3O4 nanoparticles to form complexes with arsenic in water. The phenolic and carboxylic groups contribute most of the surface charges which influence the behaviour of humic acid in solution. Similarly, the aromatic and aliphatic moieties inspire the hydrophobic nature of humic acid, whereas when graphene is present in the composite it turns into a different chemistry. Fig. 4b displays the equation which explains the mechanism of interaction between graphene (rGO) and humic acid (Hu). The interaction can be of two types: a chemical bonding interaction (II) and a π–π interaction (III). The humic acid itself behaves as negatively charged poly-anions in slightly acidic to basic solutions and is suitable for the removal of cationic ions from water. Furthermore, humic acid shows a repulsive nature towards other negative species such as anionic arsenic in water at neutral pH. The repulsive interactions of phenolic and carboxylic functional groups are pronounced at pH 7 due to the absence of protonation of functional groups. However, the composite was found with a decrease in negative surface charge at pH 7 after humic acid coating. It is evident that humic acid acts as a π binding stabilizer in the composite which involved π–π interactions with rGO. The resulting reduction in charge would be due to the non-covalent bonding interaction such as lone pair–π and π–π interactions.41,42 Graphene is also capable of establishing chemical bonding with humic acid.43 It has been reported that the amine groups associated with humic acid acquired a more positive charge which would help to attract more arsenic under appropriate pH conditions.44 The interaction of humic acid with rGO enhances the ability of amine groups to attract arsenic efficiently. Moreover, if the thickness of humic coating is beyond an optimum level this reduces the ability of arsenic to diffuse through the layers to interact with Fe3O4 nanoparticles. We found that the layering of humic acid has resettled the surface charges associated with the composites which can be obvious from the changes in zeta potential values (Table 1).
Furthermore, the study was conducted in the presence of PO43− for rGO–FeHA1 with arsenate and arsenite (Fig. 5a). The arsenic uptake capacities decreased in the presence of PO43− which can be explained by the competition effect. The phosphate and arsenic species have almost similar molecular structures which provided similar chemical properties in the adsorption process. We have also investigated the relationship between adsorption and porosity. It is well reported that when there is an adsorbent with high surface area, one can expect an enhancing effect on the adsorption properties through various interacting groups present in internal pores and cavities. On the other hand, with a decrease in surface area, one would expect a negative effect on the adsorption properties due to fewer available interacting groups.45 Therefore, in order to exclude the role of porosity of nano-composites in high uptake of arsenic we performed BET studies on rGO–Fe, rGO–FeHA1 and rGO–FeHA2. The nitrogen adsorption isotherms of rGO–Fe, rGO–FeHA1 and rGO–FeHA2 samples are shown in Fig. 5b–c. The rGO–Fe shows hysteresis between the adsorption and the desorption branches of the nitrogen isotherm, which is classified as type IV according to the IUPAC.45 The phenomenon of the hysteresis loop arises from the capillary condensation of mesopores in the range of 2 to 50 nm. The surface area observed for rGO–Fe was 53 m2 g−1 and the average pore size distribution was 7 nm. The contribution of the surface area is from the iron particles as well as from the graphene slit pores. The width of the hysteresis loop highlights the fact that large pores are interconnected with capillary pores. The desorption branch indicated that the evaporation of the condensed liquid phase in the large cavity cannot escape due to the condensation effect in the capillary pores. Moreover, the surface areas for rGO–FeHA1 and rGO–FeHA2 are 0.9060 and 0.7883 m2 g−1, respectively. The humic acid has clogged the pores associated with the two samples, causing a very low surface area. The isotherms related to rGO–FeHA1 and rGO–FeHA2 indicate a non-porous flat surface or macropores. The curvature of isotherms is the representation of multilayer adsorption of weakly interacting systems. The BET results clearly demonstrate that humic acid coating has completely clogged the internal pores and cavities, which highlight a total decrease in the surface area. It can thus be concluded that the porosity has no role in the high uptake capacity of arsenic.
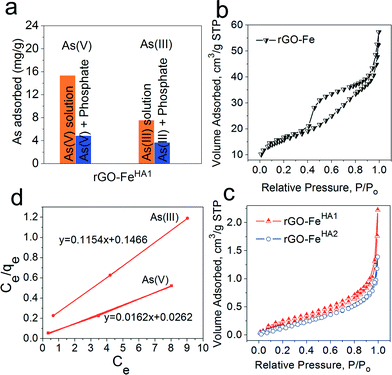 |
| | Fig. 5 (a) Effect of coexisting anion such as PO43− for rGO–FeHA1 with arsenate and arsenite. N2 adsorption/desorption isotherms (b) and (c) for graphene–iron composites and humic-coated graphene–iron composites, respectively. (d) Linear form of the Langmuir adsorption isotherm for rGO–FeHA1. | |
The negative influence of HA on the binding or removal of arsenic using various nanomaterials and composites is well documented. In contrast, we did not find any study which reports the positive influence of HA with any nanomaterial or nano-composite for the removal of arsenic. This work demonstrates that HA coating could significantly modify the mechanism through π–π interactions of aromatic fractions of amine groups with rGO. These interactions of HA with rGO led into an enhancing effect on the adsorption of As(V) and As(III). However, intensive research is needed to understand the mechanism behind this positive effect of HA. The current work opens new doors in graphene research and its interaction with the natural system. We are working to test the efficiency of this nano-composite for arsenic removal from a polluted natural ground water system that in turn is affecting millions of people around the world.
Conclusions
In spite of numerous studies on the negative influence of humic acid on the binding mechanism of arsenic on various iron-based particles, none of them has reported any positive influence with any of the composites. This work demonstrated that humic acid coating could significantly modify the mechanism through π–π interactions of aromatic fractions of amine groups with rGO. Probably the amine fractions of humic acid become more positive due to the association of graphene oxide which would provide an enhancing effect on the adsorption of arsenic. The composites became less negative in their surface charge which enhanced the binding of arsenic at pH 7, which was found better for As(V) than for As(III). This work opens new avenues for arsenic removal in the presence of humic acid, which to date restricts the advancement in nanotechnology water research specifically in removing arsenic from water. We plan to further test our nano-composites from lab to field experiments on arsenic-affected natural ground water systems.
Acknowledgements
Authors acknowledge the University of Johannesburg and NRF-South Africa for funding support.
Notes and references
- A. Mukherjee, M. K. Sengupta, M. A. Hossain, S. Ahamed, B. Das, B. Nayak, D. Lodh, M. M. Rahman and D. Chakraborti, J. Health Popul. Nutr., 2006, 24, 142–163 Search PubMed.
- C. F. Harvey, C. H. Swartz, A. B. M. Badruzzaman, N. Keon-Blute, W. Yu, M. A. Ali, J. Jay, R. Beckie, V. Niedan, D. Brabander, P. M. Oates, K. N. Ashfaque, S. Islam, H. F. Hemond and M. F. Ahmed, Science, 2002, 298, 1602–1606 CrossRef CAS PubMed.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS PubMed.
- M. S. H. Mak, I. M. C. Lo and T. Liu, Water Res., 2011, 45, 6575–6584 CrossRef CAS PubMed.
- A. B. M. Giasuddin, S. R. Kanel and H. Choi, Environ. Sci. Technol., 2007, 41, 2022–2027 CrossRef CAS.
- A. Imai, T. Fukushima, K. Matsushige, Y.-H. Kim and K. Choi, Water Res., 2002, 36, 859–870 CrossRef CAS.
- C. Catrouillet, M. Davranche, A. Dia, M. B.-L. Coz, R. Marsac, O. Pourret and G. Gruau, Chem. Geol., 2014, 372, 109–118 CrossRef CAS PubMed.
- S. Wang, J. Hu, J. Li and Y. Dong, J. Hazard. Mater., 2009, 167, 44–51 CrossRef CAS PubMed.
- T. Wang, W. Liu, L. Xiong, N. Xu and J. Ni, Chem. Eng. J., 2013, 215–216, 366–374 CrossRef CAS PubMed.
- M. S. H. Mak and I. M. C. Lo, Chemosphere, 2011, 84, 234–240 CrossRef CAS PubMed.
- V. Tanboonchuy, N. Grisdanurak and C.-H. Liao, J. Hazard. Mater., 2012, 205–206, 40–46 CrossRef CAS PubMed.
- S. R. Kanel, B. Manning, L. Charlet and H. Choi, Environ. Sci. Technol., 2005, 39, 1291–1298 CrossRef CAS.
- X. Dou, R. Li, B. Zhao and W. Liang, J. Hazard. Mater., 2010, 182, 108–114 CrossRef CAS PubMed.
- Q. Liang, D. Zhao, T. Qian, K. Freeland and Y. Feng, Ind. Eng. Chem. Res., 2011, 51, 2407–2418 CrossRef.
- G. Zhang, J. Qu, H. Liu, R. Liu and R. Wu, Water Res., 2007, 41, 1921–1928 CrossRef CAS PubMed.
- C. Shan and M. Tong, Water Res., 2013, 47, 3411–3421 CrossRef CAS PubMed.
- Y. Jin, F. Liu, M. Tong and Y. Hou, J. Hazard. Mater., 2012, 227–228, 461–468 CrossRef CAS PubMed.
- M. Hu and B. Mi, Environ. Sci. Technol., 2013, 47, 3715–3723 CrossRef CAS PubMed.
- D. Konatham, J. Yu, T. A. Ho and A. Striolo, Langmuir, 2013, 29, 11884–11897 CrossRef CAS PubMed.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- L. Jiang and Z. Fan, Nanoscale, 2014, 6, 1922–1945 RSC.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- K. C. Kemp, H. Seema, M. Saleh, N. H. Le, K. Mahesh, V. Chandra and K. S. Kim, Nanoscale, 2013, 5, 3149–3171 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. Maity and D. C. Agrawal, J. Magn. Magn. Mater., 2007, 308, 46–55 CrossRef CAS PubMed.
- J.-F. Liu, Z.-S. Zhao and G.-B. Jiang, Environ. Sci. Technol., 2008, 42, 6949–6954 CrossRef CAS.
- X. Liu, Z. Ma, J. Xing and H. Liu, J. Magn. Magn. Mater., 2004, 270, 1–6 CrossRef CAS PubMed.
- W. H. Bragg, Nature, 1915, 95, 561 CrossRef.
- K. Zhou, Y. Zhu, X. Yang and C. Li, New J. Chem., 2010, 34, 2950–2955 RSC.
- Y. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792–6797 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2007, 8, 36–41 CrossRef PubMed.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- S. Dheivamalar and V. Silambarasan, Spectrochim. Acta, Part A, 2012, 96, 480–484 CrossRef CAS PubMed.
- O. Celebi, A. Kilikli and H. N. Erten, J. Hazard. Mater., 2009, 168, 695–703 CrossRef CAS PubMed.
- A. G. S. Prado, J. D. Torres, P. C. Martins, J. Pertusatti, L. B. Bolzon and E. A. Faria, J. Hazard. Mater., 2006, 136, 585–588 CrossRef CAS PubMed.
- A. G. S. Prado, B. S. Miranda and J. A. Dias, Colloids Surf., A, 2004, 242, 137–143 CrossRef CAS PubMed.
- C.-F. Lin, S.-H. Liu and O. J. Hao, Water Res., 2001, 35, 2395–2402 CrossRef CAS.
- J. A. Greathouse, S. E. Feller and D. A. McQuarrie, Langmuir, 1994, 10, 2125–2130 CrossRef CAS.
- P. Attard, D. Antelmi and I. Larson, Langmuir, 2000, 16, 1542–1552 CrossRef CAS.
- E. A. Guggenheim, J. Phys. Chem., 1928, 33, 842–849 CrossRef.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- Z. Lu, P. Gamez, I. Mutikainen, U. Turpeinen and J. Reedijk, Cryst. Growth Des., 2007, 7, 1669–1671 CAS.
- T. Hartono, S. Wang, Q. Ma and Z. Zhu, J. Colloid Interface Sci., 2009, 333, 114–119 CrossRef CAS PubMed.
- A. Saada, D. Breeze, C. Crouzet, S. Cornu and P. Baranger, Chemosphere, 2003, 51, 757–763 CrossRef CAS.
- K. S. W. Sing, Colloids Surf., 1989, 38, 113–124 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and lattice fringe spectrum. See DOI: 10.1039/c4ew00034j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.