New insights into the origin of the visible light photocatalytic activity of Fe(III) porphyrin surface anchored TiO2
Received
25th July 2014
, Accepted 3rd December 2014
First published on 3rd December 2014
Abstract
In order to utilize visible light more effectively in photocatalytic reactions, the surfaces of TiO2 nanoparticles are sensitized by Hemin molecules (H–TiO2) and the catalyst is characterized by various analytical techniques like powder X-ray diffraction (PXRD), Fourier transform infrared spectroscopy (FTIR), UV-Visible absorption spectroscopy, X-ray photoelectron spectroscopy (XPS), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) with an energy-dispersive X-ray (EDX) technique, BET surface area measurements and thermogravimetric analysis (TGA). The results strongly confirm the chemisorption of Hemin molecules on the TiO2 surface through O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O–Ti bonds. The photocatalytic activity of H–TiO2 was investigated by the degradation of 4-nitrophenol as a model compound in an aqueous solution under solar light irradiation with the assistance of an appropriate amount of a sacrificial electron donor. The enhanced activity of H–TiO2 confirms the sensitization process. Intermediate products were identified by HPLC analysis and a possible degradation reaction mechanism was proposed. The development of this porphyrin-based photocatalyst provides an alternative approach in harnessing visible solar light and shows promise for waste water treatment in future industrial applications.
C–O–Ti bonds. The photocatalytic activity of H–TiO2 was investigated by the degradation of 4-nitrophenol as a model compound in an aqueous solution under solar light irradiation with the assistance of an appropriate amount of a sacrificial electron donor. The enhanced activity of H–TiO2 confirms the sensitization process. Intermediate products were identified by HPLC analysis and a possible degradation reaction mechanism was proposed. The development of this porphyrin-based photocatalyst provides an alternative approach in harnessing visible solar light and shows promise for waste water treatment in future industrial applications.
Water impact
TiO2 mediated semiconductor photocatalysis is an efficient wastewater treatment method for the chemical utilization of solar energy. TiO2 is a most promising photocatalyst, however it is only active under the UV fraction of solar light. Hence to extend its response to the visible region, the TiO2 surface is modified with an Fe(III)–porphyrin (Hemin) moiety (H–TiO2). Hemin has the ability to trap the solar energy and pass it on from one molecule to another until the conditions are favorable for the sensitization reaction because of an extensive delocalized π electron system bonded to TiO2 through OC–O–Ti bonds, benefiting both the adsorption and channeling electron transfer processes. H–TiO2 is very active and sustains the complete mineralization of pollutants under solar light for several repetitive cycles with an excellent reproducibility and great industrial applicability.
|
1. Introduction
Water pollution by the presence of toxic and non-biodegradable organic materials, especially in parts per million (ppm) concentrations in drinking water, is a serious threat to living species. The enormous diversity of pollutants with different chemical structures and compositions excludes the possibility of using a universal treatment method and thus requires the development of specific treatment techniques to address the problem. Recently, advanced oxidation processes (AOPs) for wastewater treatment have attracted more attention due to the generation of highly reactive radicals in a sufficient quantity for effective water purification. Among the AOP technologies, titanium dioxide (TiO2) mediated photocatalysis has gained a growing acceptance as an effective technique in treating wastewater containing persistent organic pollutants because of its stability, nontoxicity, hydrophilicity, cheap availability and its capability for the complete mineralization of pollutants under ambient temperature and pressure. However, TiO2 has two major drawbacks, i) its relatively large band gap (3.2 eV for anatase) requires an excitation wavelength that falls in the UV region, which hampers the exploitation of solar light in photocatalytic reactions, and ii) the high rate of recombination of photogenerated charge carriers, which unfortunately reduces the efficiency of the photocatalytic process.1,2 Hence, attempts have been made to modify TiO2 using various strategies like coupling with a narrow band gap semiconductor, doping of metal/nonmetal ions, codoping with two or more foreign ions and surface modifications with either organic dyes or with metal complexes, surface fluorination/sulfation/phosphation, noble metal deposition, and photosensitization to overcome the drawbacks of electron–hole recombination and also to extend the response to the visible region.1–5 The photosensitization of TiO2 by dyes, ruthenium polypyridyl complexes, porphyrins, squaraines and natural dyes, etc., has shown to be a better technique to achieve excellent visible light harvesting with higher efficiency compared with other methods.6 Usually the inorganic metal complexes which act as TiO2 photosensitizers have a transition metal ion with inorganic or organic ligands. These ligands are coordinatively bound to the central metal ion and the complex in turn is covalently linked to the TiO2 surface via various organic (e.g. hydroxyl, carboxyl, amino and other groups)/inorganic (e.g. CN−, F−, PO43−) anchoring groups which act as a bridge between the titanium surface and the metal complex. Among the complexes, porphyrins (including metal-free porphyrins, metalloporphyrins and supramolecular porphyrins) are recognized as the most promising sensitizers to harvest visible light, since they are structural analogues of chlorophyll in plant photosynthesis.7 Porphyrins show very strong absorption in the visible region due to an extensive system of delocalized π electrons and they also have excellent photophysical properties like high quantum yield for intersystem crossing with small singlet–triplet splitting energy and long triplet state life time. Additionally, the photophysical properties of porphyrins can be easily tuned by the incorporation of transition metal ions like Fe into the central cavity, resulting in a stronger and broader photoresponse in the visible region.8–10 Furthermore, studies show that metal complexes of porphyrins are highly photostable when adsorbed on the surface of TiO2.11–13 Porphyrin–TiO2 photocatalysts are highly effective and can be considered as a hybrid of organic and inorganic counterparts which show real promise in water purification.7,9 Recently, we reported the superior photocatalytic activity of Hemin anchored TiO2 (H–TiO2) under UV light.14 Extending our research work in this context, we have now tested the photocatalytic activity of H–TiO2 under solar light irradiation. The model compound chosen for photocatalytic degradation is 4-nitrophenol (4-NP), which is known to be a very stable and refractory pollutant mainly found in paper mill and dye-industry wastewater.
2. Experimental section
2.1. Materials
Titanium(IV) chloride (TiCl4 ≥ 99.9%), Hemin, acetonitrile and methanol (HPLC grade) were obtained from Merck Chemicals Limited. 4-Nitrophenol, dimethyl sulfoxide and triethanolamine were obtained from S D Fine-Chemicals Limited. Double distilled water was used throughout the experiment.
2.2. Preparation of TiO2 and Hemin anchored TiO2
Anatase TiO2 was prepared by a sol–gel method through the hydrolysis of TiCl4 as mentioned in earlier literature.15,16 Accurately weighed TiO2 (1 g) was immersed for a day in a 5 μM Hemin solution made up of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of dimethyl sulfoxide and acetonitrile (DMSO/CH3CN) at pH 4 and then centrifuged to remove the solvent. The resulting Hemin anchored TiO2 (H–TiO2) sample was dried at room temperature until the solvent volatilized completely.
:1 mixture of dimethyl sulfoxide and acetonitrile (DMSO/CH3CN) at pH 4 and then centrifuged to remove the solvent. The resulting Hemin anchored TiO2 (H–TiO2) sample was dried at room temperature until the solvent volatilized completely.
2.3. Characterization methods
The powder X-ray diffraction (PXRD) patterns were obtained for the samples using a Philips powder diffractometer PW/1050/70/76. The diffraction patterns were recorded at room temperature using Cu Kα radiation with a Ni filter in the 2θ range 20°–80° at a scan rate of 2° per min. To study the light absorption characteristics of the prepared photocatalyst, the absorption spectra were recorded using a Shimadzu-UV 3101 PC UV-VIS-NIR UV-Visible spectrophotometer in the range of 200–800 nm. The baseline correction was done using a calibrated sample of BaSO4. FTIR spectra were obtained using a NICOLLET IMPACT 400 D FTIR spectrometer. The X-ray photoelectron spectroscopy (XPS) measurements were carried out using an AXIS ULTRA from AXIS 165, integrated with a Kratos patented magnetic immersion lens along with a charge neutralization system and spherical mirror analyzer. The XPS experiments were calibrated according to the binding energy of C 1s (284.6 eV). The morphological features were studied using an ULTRA 55 field emission scanning electron microscope (FESEM) and a TECNAIF-30 transmission electron microscope (TEM) with energy-dispersive X-ray (EDX) techniques. The specific surface area and pore volume of the powders were measured with a Nova Quanta Chrome Corporation multipoint BET Digisorb 2006 instrument system using the dynamic Brunner–Emmet–Teller (BET) method in which N2 gas was adsorbed at 77 K. Thermogravimetric analysis (TGA) was carried out using a TA instrument along with a model Q 600 SDT analyzer in the temperature range of 25–1000 °C under a nitrogen atmosphere with a heating rate of 20 °C min−1. The qualitative identification of the initial substrate and photocatalytic degradation reaction intermediates was carried out by high performance liquid chromatography (HPLC) using a Shimadzu 2010HTC instrument equipped with a Perkin-Elmer C 18 column. A mixture of water–methanol (50:50 v/v) was used as an eluant with a flow rate of 1 mL min−1. The products were identified by comparison with the respective standards. The detection was performed using a UV absorption wavelength at 280 nm. The temperature of the column was kept around 45 °C throughout the analysis. The injection volume of the samples was 20 μL.
2.4. Photocatalytic degradation procedure
The photocatalytic activity of TiO2 and H–TiO2 were evaluated by the photocatalytic decomposition of 4-NP in an aqueous suspension under solar light illumination. In a typical experiment 100 mg of catalyst powder (H–TiO2/TiO2) was suspended in 250 ml of 10 ppm 4-NP along with an appropriate amount of triethanolamine (TEOA) solution. The catalyst suspension was dispersed for 5 minutes in an ultrasonic bath and then the system was kept under solar illumination with continuous stirring (100 rpm) using a magnetic stirrer. All the experiments were performed with continuous bubbling of air through the solution to saturate the catalyst surface with oxygen during the photooxidation process. Solar light irradiation was carried out from 11 a.m. to 2 p.m. during the summer season in Bangalore, India. The latitude and longitude are 12.58 N and 77.38 E, respectively. The average sunlight intensity was found to be around 1200 W m−2. The intensity of the solar light was concentrated by using a convex lens and the reaction mixture was exposed to this concentrated sunlight. To compare the photocatalytic activity of all the catalysts, the experiments were conducted simultaneously to avoid errors arising due to fluctuations in solar intensity. The filtrates were collected at different time intervals and analyzed by UV-visible spectroscopy. The activity of H–TiO2 was compared with various reaction systems like i) TiO2 ii) TiO2/TEOA and iii) H–TiO2/TEOA. The contribution of direct photolysis was checked by performing a blank experiment containing only the pollutant in the absence of photocatalyst under solar light illumination.
3. Results and discussion
3.1. PXRD studies
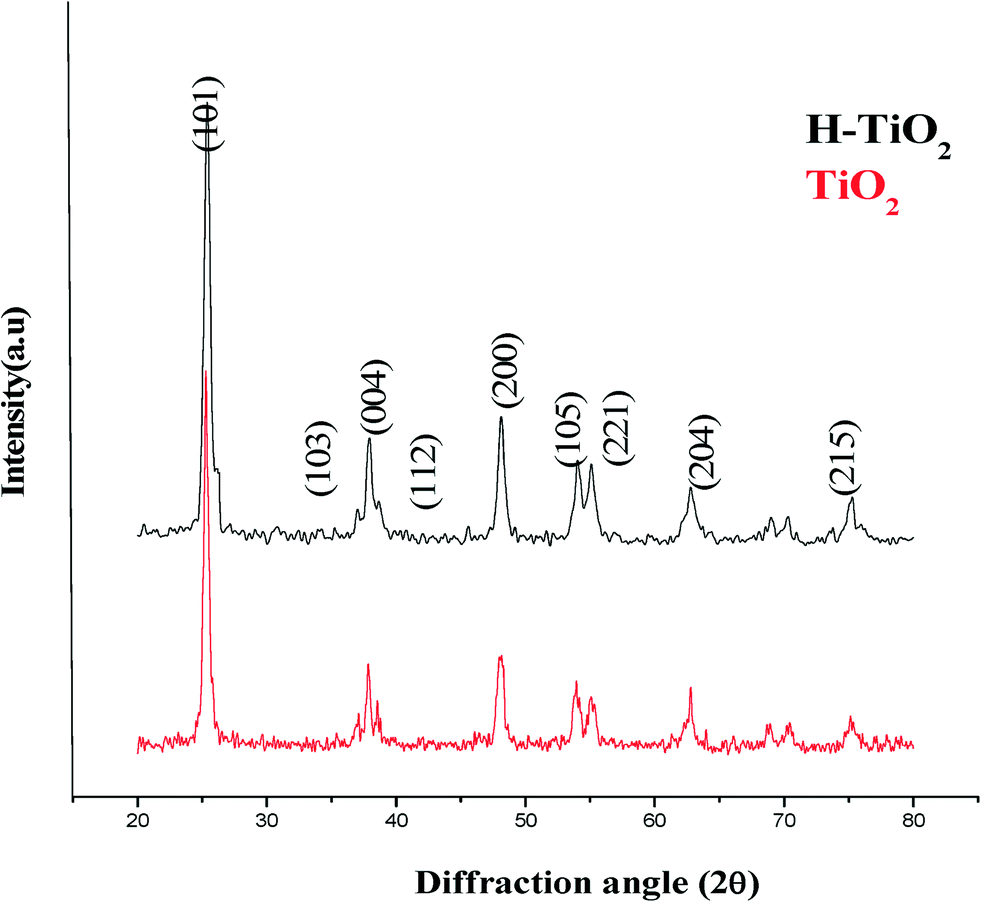
Fig. 1 shows the PXRD patterns of TiO2 and H–TiO2. Both the samples show peaks at 2θ values of 25.3° (101), 38° (112), 48° (200), 54° (105), 55° (211), 62° (204) and 68° (116) corresponding to the anatase phase of TiO2. The numbers in parentheses represent hkl values. From the PXRD patterns, it can be concluded that the structure of Hemin anchored TiO2 remains unchanged compared with pristine TiO2, indicating that the process of Hemin anchoring did not alter the characteristic anatase crystal structure of TiO2.
 |
| | Fig. 1 PXRD patterns of TiO2 and H–TiO2. | |
3.2. UV–Vis absorption spectroscopy
Fig. 2 shows the absorption spectra of bare TiO2 and H–TiO2. The absorption spectrum of TiO2 includes a single broad intense absorption band around 400 nm due to the process of charge-transfer from the valence band (chiefly formed by 2p orbitals of the oxygen atoms) to the conduction band (formed by 3d t2g orbitals of the Ti4+ cations).17–19 In the case of H–TiO2, the observed red shift could be assigned to delocalized π-electrons in the porphyrin ring and was also due to the interaction of the carboxylate groups of Hemin with the –OH groups on the TiO2 surface. The strong Soret band of porphyrin appears at 480 nm due to the transition from a1u(π) to eg*(π) as expected and a less intense Q-band appears around 500–600 nm corresponding to a2u(π) to eg*(π) transition (as shown in the inset of Fig. 2) which confirms the chemisorption of Hemin molecules on the TiO2 surface.20
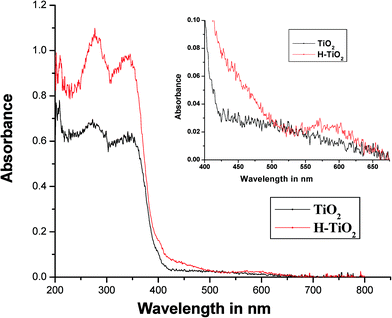 |
| | Fig. 2 UV-Visible absorption spectra of TiO2 and H–TiO2. | |
3.3. FTIR spectra
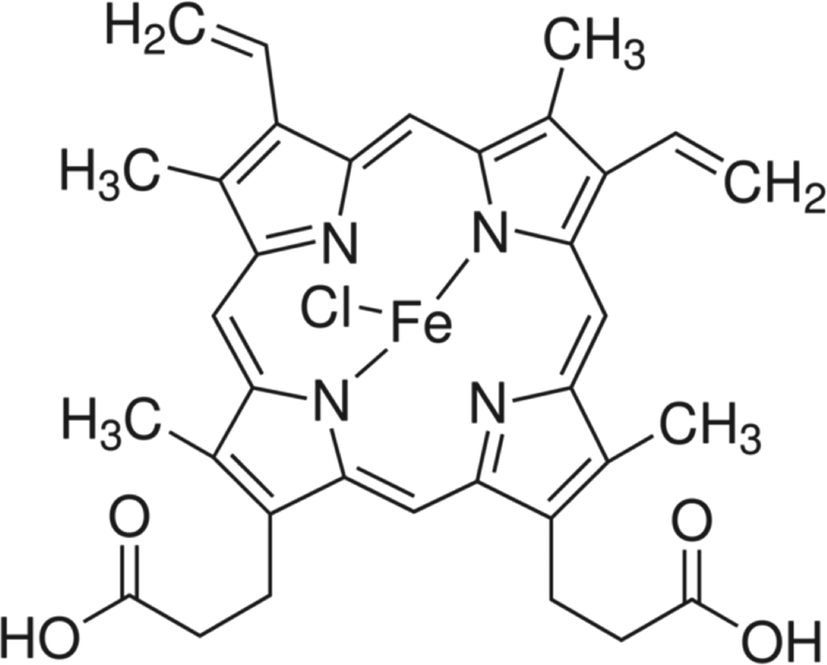
The vibrational spectra of the system can be better understood considering the structure of Hemin. It consists of a protoporphyrin (IX) ring system with the Fe(III) metal ion at the cavity centre along with a co-ordinated chloride ion as shown in the Scheme 1. The Fe(III) center is square pyramidal in geometry, due to the high spin penta-coordination of ligands. The porphyrin ring has several side chains, such as carboxyethyl, vinyl, and methyl groups.21
 |
| | Scheme 1 | |
Fig. 3 shows the FTIR spectra of TiO2 and H–TiO2. The spectra display two characteristic broad bands centered at 3480 (not shown in the figure) and 1640 cm−1 which can be assigned to the stretching and bending vibration modes of adsorbed water molecules, and alternatively they can also be assigned to Ti–OH vibration. The band centered at 735 cm−1 is associated with the stretching vibration mode of bridged Ti–O bonds. The spectrum of H–TiO2 shows a highly intense band at 1020 cm−1 due to the C–O stretching vibration and a split peak around 1430–1400 cm−1 corresponding to CO vibrations of surface bound carboxylic acid (to TiO2) and hydrogen bonded carboxylic acid, respectively. The weak intensity peak observed at 1310 cm−1 is due to the C–O stretching modes of adsorbed Hemin molecules. This implies the chemisorption of Hemin molecules on the surface of TiO2 through the –COOH functional group. The OH group on the TiO2 surface deprotonates the carboxylic acid group of Hemin with the elimination of a water molecule. This interaction increases the adherence of the Hemin molecule on the TiO2 surface through a OC–O–Ti bond.22
 |
| | Fig. 3 FTIR spectra of TiO2 and H–TiO2. | |
3.4 X-ray photoelectron spectroscopic (XPS) studies
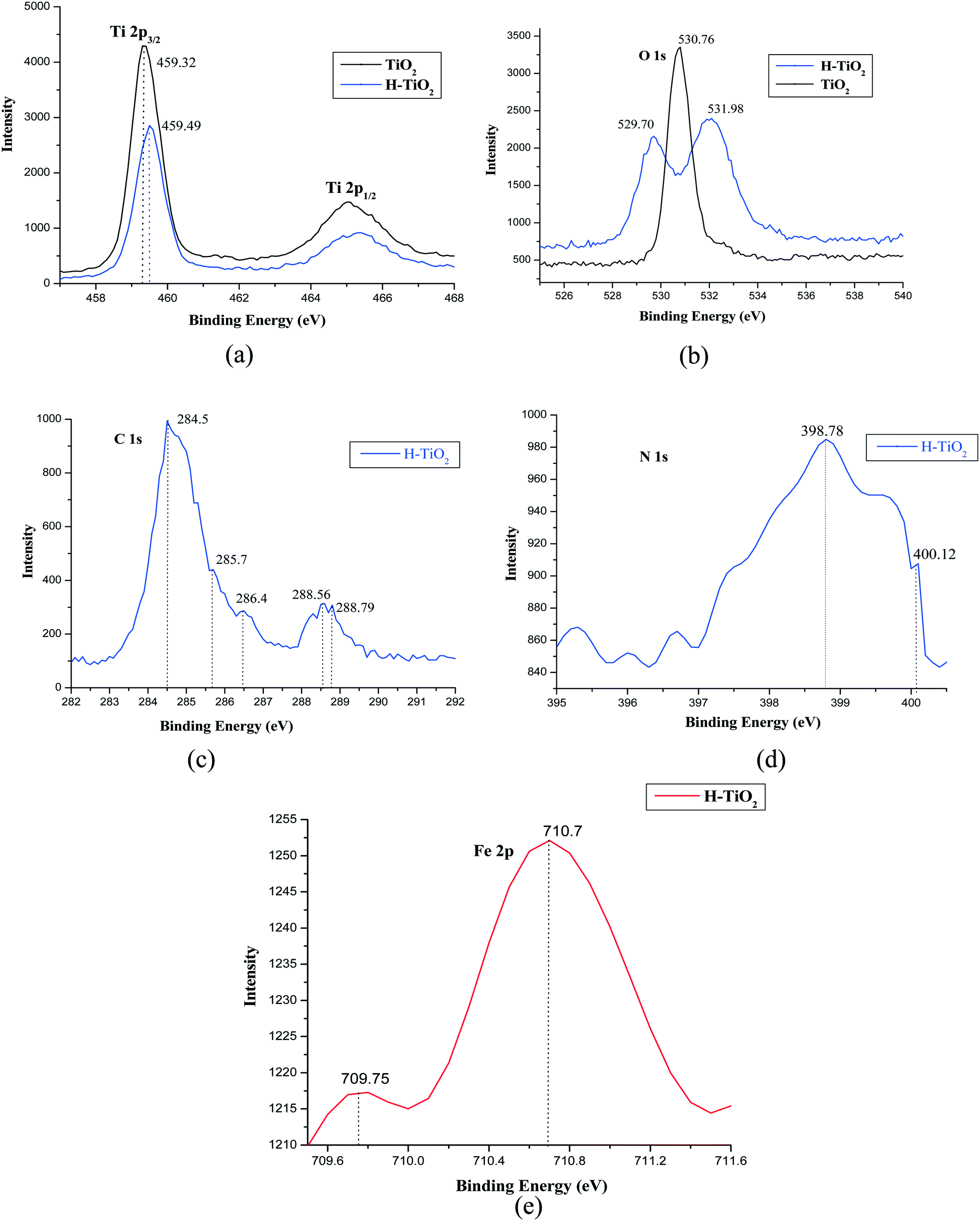
The chemical composition of H–TiO2 was analyzed by the XPS technique, which is a highly surface selective process used to explore surface composition and local chemical environments. The survey scan of H–TiO2 only showed traceable signals of Fe 2p (710 eV), C 1s (284 eV) and N 1s (400 eV) peaks, which gives evidence for the surface modification of TiO2 with Hemin. These peaks were absent for pristine TiO2.
In order to distinguish the surface changes, XPS spectra of the Ti 2p and O 1s regions (Fig. 4a and b) of pure TiO2 and H–TiO2 were recorded. The Ti (2p3/2) binding energy values of TiO2 and H–TiO2 are found to be 459.32 and 459.49 eV, respectively. The peak of H–TiO2 is shifted by 0.17 eV compared to pure TiO2. This suggests that the Hemin is chemisorbed on the TiO2 surface through the carbonyl group and it substitutes the H atom of TiO2 in Ti–O–H bonds, finally forming Ti–O–CO bonds. The carbonyl group is electron withdrawing in nature, and it withdraws electrons from the O atom, resulting in the decrease of electron cloud density on the Ti atom, thereby increasing the valence state of the Ti atom leading to a slight increase in the binding energy of the Ti (2p3/2).
 |
| | Fig. 4 (a) Ti 2p XPS spectra of TiO2 and H–TiO2, (b) O 1s XPS spectra of TiO2 and H–TiO2 (c) C 1s XPS spectra of H–TiO2, (d) N 1s XPS spectra of H–TiO2, (e) Fe 2p XPS spectra of H–TiO2. | |
The O 1s XPS spectra of TiO2 consists of a single peak located at 530 eV which is related to the oxygen (O–Ti–O) in the crystal lattice. H–TiO2 consists of two peaks, implying the presence of O in more than one chemical state. The peak at 531.9 eV is related to oxygen bonded to Ti metal as Ti–O and the other peak at ~529.7 eV can be assigned to the oxygen bonded to carbon as Ti–O–C in the case of H–TiO2.23
The anchoring of Hemin on the TiO2 surface through chemisorption is further substantiated by the presence of the C 1s (284 eV), N 1s (398.8 eV) and Fe 2p (710.7 eV) bands. Fig. 4c shows the C 1s region and the five peaks observed in the spectra imply the presence of five different binding energies (BE) for the carbon in H–TiO2. The carbon may be involved in different bonds of the following types: C–C (284.5 eV), C–N (285.7 eV), C–O (286.4 eV), CO (288.5 eV), O–CO (288.7 eV).24 In the N 1s spectrum (Fig. 4d) a single band at 398.8 eV can be ascribed to the four chemically equivalent N atoms which are bound to the central iron atom in the porphyrin ring. The XPS spectra of Fe 2p (Fig. 4e) show a binding energy peak at 710.7 eV, which indicates the presence of iron in the +3 oxidation state. According to the literature, the binding energy of pure Hemin was found to be 712.5 eV for Fe 2p and 401.8 eV for N 1s, whereas in the case of H–TiO2 a pronounced shift in binding energy by ~1.8 eV for Fe 2p and ~3 eV for N 1s suggests the strong interaction of Hemin with TiO2.25
3.5 Morphology of TiO2 and H–TiO2
The surface morphology of TiO2 and H–TiO2 was studied by the FESEM technique as shown in Fig. 5a and b. The FESEM micrographs show that the samples consist of nanoparticles of varying size and shape. The particle size varies from 19.69 to 29.49 nm and 20.69 to 26.57 nm, with an average of 21.19 and 23.023 nm for TiO2 and H–TiO2, respectively. TiO2 shows a flat platelet like structure with a smooth surface, whereas H–TiO2 has a rough and coarse surface due to the surface modification. Particles in H–TiO2 are agglomerated due to the interlinkage of TiO2 nanoparticles through the –COOH functional groups of Hemin, indicating the strong chemisorption of Hemin molecules on the TiO2 surface.
 |
| | Fig. 5 Field emission scanning electron microscope images of a) TiO2 and b) H–TiO2. | |
Fig. 6a and b show typical TEM images of TiO2 and H–TiO2. TEM permits easy differentiation of Hemin and TiO2 with lighter and darker shades. The Hemin molecules are highly dispersed on the surface of the TiO2 particles with a distinguishable grain boundary. EDX analysis confirmed the presence of only O and Ti elements in TiO2 (Fig. 6c) whereas Fe, N, C, O and Ti elements were present in H–TiO2 with the weight percentages shown in Fig. 6d. These are derived from the Hemin which is present on the surface of the TiO2, while only O and Ti elements exist in the bare TiO2.
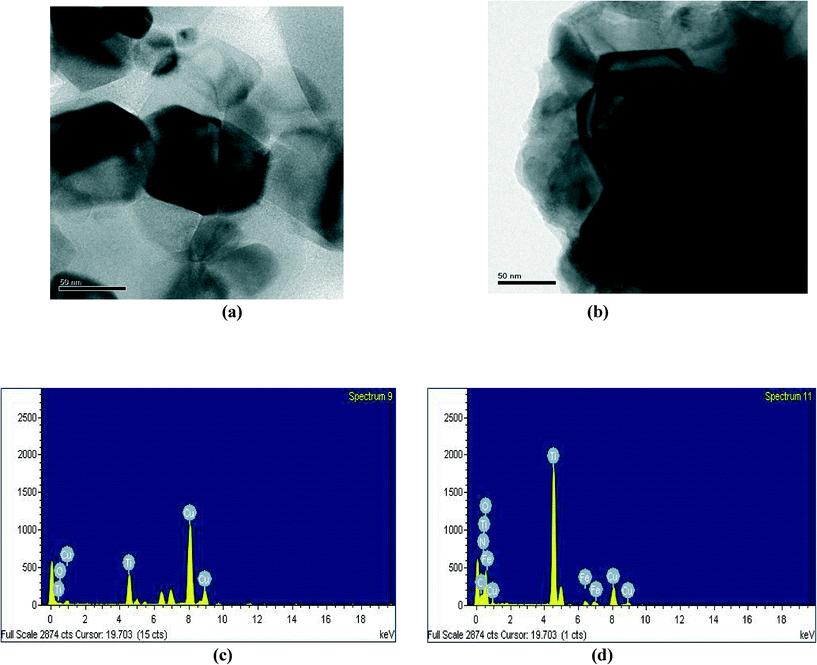 |
| | Fig. 6 TEM images of (a) TiO2 and (b) H–TiO2, and EDX spectra of (c) TiO2, (d) H–TiO2. | |
3.6. BET nitrogen adsorption–desorption isotherms
It is widely believed that the efficiency of photocatalytic reactions is much dependent on the specific surface area of the photocatalyst. The specific surface area and porosity of the as-prepared samples were investigated using nitrogen adsorption and desorption isotherms as shown in Fig. 7. The isotherms were identified as type IV with H2 hysteresis loops, implying the existence of cylindrical-shaped and slit-shaped pores, which suggests that the samples were mesoporous according to the IUPAC classification.26 The specific surface area, pore diameter and pore volume of the samples were calculated from the N2 isotherms and the values are listed in Table 1. It is observed that there was a decrease in the average pore diameter and an increase in the specific surface area when the TiO2 surface was modified with Hemin and these results are also in good agreement with the data obtained by Niu et al.27
 |
| | Fig. 7 The N2 adsorption–desorption isotherms with an inset showing the BJH pore size distribution curves of TiO2 and H–TiO2. | |
Table 1 BET data of the TiO2 and H–TiO2 samples
| Photocatalyst |
BET surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore diameter in nm |
| TiO2 |
26.98 |
1.03 |
9.49 |
| H–TiO2 |
74.81 |
0.435 |
8.77 |
The plots of pore volume versus pore diameter of both samples are shown in the inset of Fig. 7. The pore diameter values are determined by the Barrett–Joyner–Halenda (BJH) method. The mesopore diameter of the samples was found to be 9.49 and 8.77 nm for TiO2 and H–TiO2, respectively.
3.7. Thermogravimetric analysis (TGA)
TGA of TiO2 and H–TiO2 samples are shown in Fig. 8. The thermograms show a weight loss of 1.7% and 2.6% for TiO2 and H–TiO2, respectively. The weight loss in TiO2 is due to the surface adsorbed water molecules and also due to surface bound hydroxyl groups. The excess weight loss observed in the case of H–TiO2 is due to the burning off of organic residues found in the porphyrin group of Hemin.
 |
| | Fig. 8 Thermograms of a) TiO2 and b) H–TiO2. | |
4. Photocatalytic degradation studies
4.1. Solar light driven photocatalytic activity
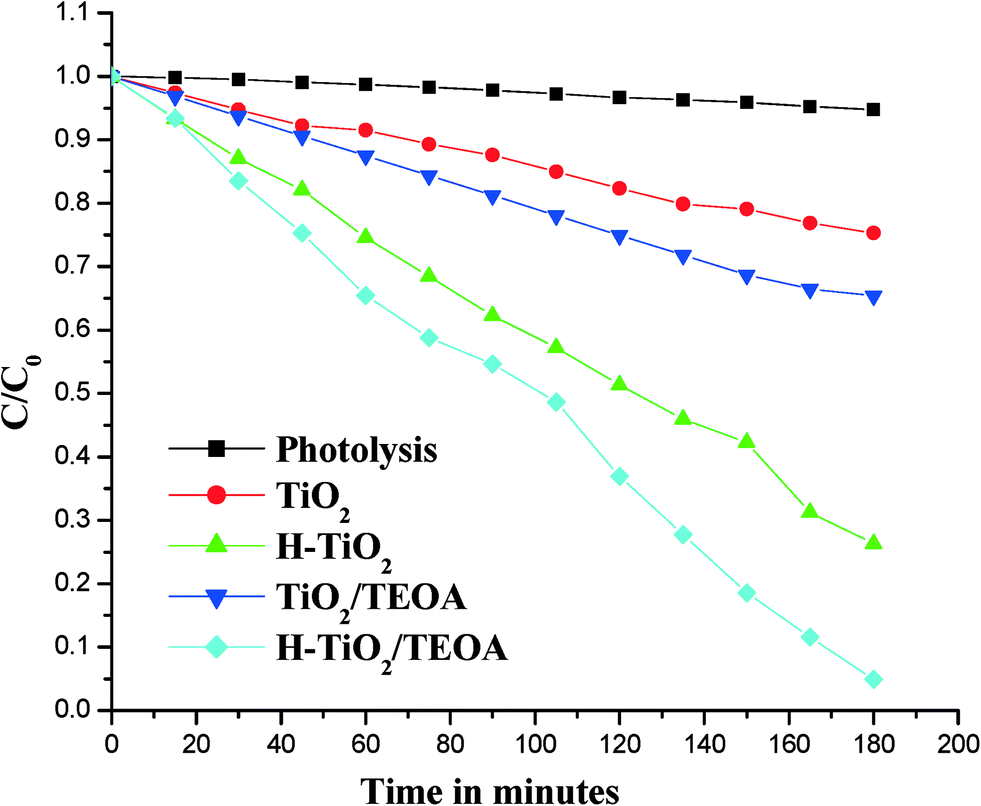
The photocatalytic activity of TiO2 and H–TiO2 was evaluated by the degradation of 4-NP in an aqueous system under solar light irradiation. In order to obtain insights into the degradation reaction mechanism, several experiments were planned in the presence and absence of a sacrificial electron donor, TEOA, and these include: i) direct photolysis, ii) bare TiO2, iii) TiO2 with TEOA, iv) H–TiO2 and v) H–TiO2 with TEOA. These were performed and the results were compared with one another as shown in Fig. 9.
 |
| | Fig. 9 Plots of C/C0versus time under solar light illumination for various systems. | |
The porphyrin moiety has the ability to absorb blue and red light corresponding to the Soret and Q-bands, respectively. These transitions make the Hemin molecule an efficient photosensitizer under sunlight. In the photosensitization process, the porphyrin moiety gets excited instead of TiO2 by absorbing the photons of wavelengths corresponding to the visible region. A ground state Hemin molecule [H] absorbs the visible light and this results in the excited singlet state 1[H]*. Strong intramolecular perturbations can be expected within the Hemin molecule since the excited energy can be taken up by the iron metal ion which is chelated to an organic porphyrin molecule and hence the creation of a triplet state is more probable. Through a process of intersystem crossing these excited singlet states relax to the lowest excited triplet state 3[H]*. The lifetime of this triplet state is in the time regime of milliseconds, which is quite a lot longer than the nanosecond time regime of the excited singlet state.7
| |  | (1) |
Due to the longer lifetime of the triplet state, it gives enough time for the transfer of electrons from the excited Hemin molecule to the TiO2 conduction band (CB), this transfer is also favored due to the potential gradient created between the energy levels. The possibility of electron transfer from both 1[H]* and 3[H]* excited states cannot be ruled out since the CB edge of TiO2 is below these excited states, however the latter transition is more probable due to the longer lifetime. This transition is facilitated through the channels of OC–O–Ti bonds, which results in the oxidation of the Hemin molecule [H+–TiO2]. The electron from the TiO2 CB is further trapped by adsorbed O2 molecules on the surface of TiO2 to produce reactive oxygen species like hydroxyl and superoxide radicals which oxidatively degrade the 4-NP molecule as shown below:6–13,20,27–32
| | | 1 or 3[H]* → [H]+ + e− | (2) |
| | | H–TiO2 + e− → H–TiO2(e−CB) | (3) |
| | | H–TiO2(e−CB) + O2 → H+–TiO2 + O2−˙ | (4) |
| | | H2O2 + ecb− → OH− + OH˙ | (7) |
| | | O2−˙ + NO2–PhOH → → → Mineralized products | (8) |
| | | OH˙ + NO2–PhOH → → → Complete mineralization | (9) |
Alternatively, the Kautsky mechanism suggests the formation of singlet oxygen [1O2] by the energy transfer from the excited triplet state sensitizer 3[H]* to molecular oxygen upon collision with each other.7 At an ordinary temperature in an air saturated aqueous solution the concentration of oxygen is around 10−4 to 10−5 M, and hence the possibility of the formation of singlet oxygen [1O2] is high. Thus formed singlet oxygen [1O2] can also cause the oxidation of 4-NP molecules. The oxidized Hemin [H+–TiO2] can interact with either the pollutant or water or with a sacrificial electron donor (TEOA) molecule to regenerate the Hemin molecule. The excited sensitizer on the surface of TiO2 gives rise to a series of reactions producing reactive intermediates for effective degradation.
| | | H+–TiO2 + NO2–PhOH → H–TiO2 + NO2–PhOH+ | (10) |
| | | H+–TiO2 + TEOA → H–TiO2 + TEOA+ | (11) |
| | | H+–TiO2 + H2O → H–TiO2 + H+ + OH˙ | (12) |
4.2. Influence of TEOA concentration on the rate of degradation
A sacrificial electron donor is usually used in a photosensitization reaction. The role of this sacrificial electron donor is to facilitate the reduction of the excited photosensitizer, in other words it regenerates the photosensitizer. In such reactions TEOA is frequently used as a sacrificial electron donor (E0[TEOA/TEOA+] = 0.82 V). The addition of TEOA enhances the rate of regeneration of Hemin by quenching the oxidized form.33,34 TEOA is a simple molecule containing functional groups such as tertiary amine and triol. The reductive quenching of the activated Hemin by TEOA generates a nitrogen-centered radical cation. Abstraction of a proton by the nitrogen-centered radical cation from another TEOA molecule yields carbon-centered radicals as given by Reithmeier et al. in their reaction scheme.34 These carbon-centered radicals are strong reducing agents that are capable of donating a second electron, thereby regenerating the Hemin molecule. Alternatively H+–TiO2 can also be reduced by water molecules or even by the pollutant 4-NP molecules which can act as electron donors.
The plots of C/C0versus time for H–TiO2 at different concentrations of TEOA are depicted in Fig. 10. The photodegradation efficiency increases with an increase in the concentration of TEOA up to 0.3 M and then the efficiency decreases slowly. This suggests that an optimum concentration of 0.3 M TEOA would be needed for efficient degradation. At lower concentrations of TEOA, H+–TiO2 is reduced to H–TiO2 by abstracting an electron from the TEOA molecule (eqn (11)). The regenerated Hemin can then undergo excitation again by absorbing visible light, which means it can undergo a further series of reactions. By this cyclic process the efficiency of 4-NP degradation is enhanced. However an excess of TEOA could reduce the degradation efficiency by 25%. This retardation in the degradation rate can be attributed to the domination of the reaction process shown in the eqn (11), as simultaneously the rate of production of hydroxyl radicals (eqn (10)) and oxidative degradation of 4-NP (eqn (12)) decreases.
 |
| | Fig. 10 Plots of C/C0versus time at different concentrations of TEOA. | |
4.3. Stability of the photocatalysts
The stability and reuse of photocatalysts is very essential for their practical application in water purification. In order to evaluate the stability of H–TiO2, recycling experiments were carried out by employing similar experimental conditions (250 ml of 10 ppm 4-NP solution, 100 mg of H–TiO2, 0.3 M of TEOA solution). After each experiment the photocatalyst was separated from the solution, washed, and air dried for the next run. The absence of structural modifications was confirmed by FTIR analysis. The spectrum reveals the characteristic stretching modes of the porphyrin ring and carboxylate group which confirms the adsorption of Hemin to the TiO2 surface, and it is found that the characteristic structure is maintained for three successive cycles.
4.4. 4-NP mineralization mechanism
The photocatalytic degradation of 4-NP as a function of irradiation time was followed using HPLC analysis. The percentage concentration of the major degradation intermediates formed at various time intervals and their disappearance is shown in Fig. 11.
 |
| | Fig. 11 Plots of the % concentration of reaction intermediates with respect to time. | |
HPLC analysis of the solutions at different irradiation time intervals reveals the presence of several initial reaction intermediates like hydroquinone, 1,4-benzoquinone and a small amount of catechol. The photodegradation of the 4-NP seems to occur in two steps: a rapid step destroying the aromatic group, followed by a much slower oxidation of the aliphatic chains. The first stage of the photodegradation implies the addition of a hydroxyl radical to the benzene ring with the formation of a hydroxylated radical, more specifically a dihydroxycyclohexadienyl (DHCHD) radical. This radical may further take up different reaction pathways according to the nature of the reaction medium.35–37 In the oxidative medium this radical can undergo hydroxylation reactions. The identification of intermediates during the degradation process by HPLC analysis shows that degradation proceeds through the elimination of the nitro group followed by hydroxylation. Hydroquinone and 1,4-benzoquinone were identified as the major degradation intermediate products, these results are in agreement with previous studies.38,39 In the second reaction pathway the nitrous acid is eliminated by the attack of photogenerated hydroxyl radicals on 4-NP to yield 1,4-benzosemiquinone as an intermediate, which subsequently disproportionate into hydroquinone and 1,4-benzoquinone.40 The possibility of hydroquinone formation also occurs by an ipso attack of the hydroxyl radical with simultaneous elimination of the nitro group, since the nitro group in nitro aromatics is always a very good leaving group.37,38,41 Furthermore, the degradation reaction proceeds with the attack of hydroxyl radicals on the intermediate products, mainly leading to cleavage of the benzene ring and also ends in the formation of oxygenated aliphatic compounds. Successive oxidation of aliphatic products ends in the formation of CO2 and H2O as shown in Scheme 2.
 |
| | Scheme 2 | |
5. Conclusions
A visible light active photocatalyst has been successfully developed by the surface modification of TiO2 with Hemin (chloro(protoporhyinato)iron(III)). Hemin is chemisorbed on the surface of TiO2 through OC–O–Ti bonds. These bonds not only enhance the extent of adsorption, but can also act as electron transfer channels facilitating the electron transfer process. Hemin is an exceptionally efficient photosensitizer because of its ability to trap solar energy and pass it on to TiO2 molecules. The presence of oxygen in the photocatalytic reaction medium favors the formation of reactive singlet oxygen in addition to enhancing the transition process of singlet to triplet. The presence of a sacrificial electron donor like TEOA increases the efficiency of the degradation process by regenerating the neutral Hemin molecule from its oxidized form. A general scheme of the degradation was proposed with the quantitative identification of reaction intermediates like hydroquinone, benzoquinone and a small amount of catechol by HPLC analysis.
Acknowledgements
The authors acknowledge the financial assistance from Department of Science and Technology (DST-IDP & DST-SERC) and University Grants Commission (UGC), government of India.
References
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS PubMed.
- M. V. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2013, 14, 13–28 CrossRef CAS PubMed.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol., C, 2013, 15, 1–20 CrossRef CAS PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. Oshea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS PubMed.
- L. G. Devi and R. Kavitha, Appl. Catal., B, 2013, 140–141, 559–587 CrossRef CAS PubMed.
- D. Chen, D. Yang, J. Geng, J. Zhu and Z. Jiang, Appl. Surf. Sci., 2008, 255, 2879–2884 CrossRef CAS PubMed.
- C. Wang, J. Li, G. Mele, G.-M. Yang, F.-X. Zhang, L. Palmisano and G. Vasapollo, Appl. Catal., B, 2007, 76, 218–226 CrossRef CAS PubMed.
- S. Afzal, W. A. Daoud and S. J. Langford, ACS Appl. Mater. Interfaces, 2013, 5, 4753–4759 CAS.
- W.-J. Suna, J. Li, G.-P. Yao, F.-X. Zhang and J.-L. Wang, Appl. Surf. Sci., 2011, 258, 940–945 CrossRef PubMed.
- M.-Y. Duan, J. Li, M. Li, Z.-Q. Zhang and C. Wang, Appl. Surf. Sci., 2012, 258, 5499–5504 CrossRef CAS PubMed.
- X.-F. Lü, J. Li, C. Wang, M.-Y. Duan, Y. Luo, G.-P. Yao and J.-L. Wang, Appl. Surf. Sci., 2010, 257, 795–801 CrossRef PubMed.
- C. A. Paez, S. D. Lambert, D. Poelman, J.-P. Pirard and B. Heinrichs, Appl. Catal., B, 2011, 106, 220–227 CAS.
- S. Afzal, W. A. Daoud and S. J. Langford, Appl. Surf. Sci., 2013, 275, 36–42 CrossRef CAS PubMed.
- L. G. Devi and M. L. ArunaKumari, Appl. Surf. Sci., 2013, 276, 521–528 CrossRef CAS PubMed.
- L. G. Devi and G. M. Krishnaiah, J. Photochem. Photobiol., A, 1999, 121, 141–145 CrossRef.
- L. G. Devi, N. Kottam and S. G. Kumar, J. Phys. Chem. C, 2009, 113, 15593–15601 CAS.
- K. Nagaveni, M. S. Hegde and G. Madras, J. Phys. Chem. B, 2004, 108, 20204–20212 CrossRef CAS.
- N. Venkatachalam, M. Palanichamy and V. Murugesan, J. Mol. Catal. A: Chem., 2007, 273, 177–185 CrossRef CAS PubMed.
- R. Rahimi, E. H. Fard, S. Saadati and M. Rabbani, J. Sol-Gel Sci. Technol., 2012, 62, 351–357 CrossRef CAS.
- H. Huang, X. Gu, J. Zhou, K. Ji, H. Liu and Y. Feng, Catal. Commun., 2009, 11, 58–61 CrossRef CAS PubMed.
- R. T. Tom and T. P. Pradeep, Langmuir, 2005, 21, 11896–11902 CrossRef CAS PubMed.
- J. Desilvetro, M. Graetzel, L. Kavan and J. Moser, J. Am. Chem. Soc., 1985, 107, 2988–2990 CrossRef CAS and K. S Finnie, J. R. Bartlett and J. L. Woolfrey, Langmuir, 1998, 14, 2744–2749 CrossRef.
- H. Wang, D. Zhou, Z. Wu, J. Wan, X. Zheng, L. Yu and D. L. Phillips, Mater. Res. Bull., 2014, 57, 311–319 CrossRef CAS PubMed.
- Y. Guo, L. Deng, J. Li, S. Guo, E. Wang and S. Dong, ACS Nano, 2011, 5, 1282–1290 CrossRef CAS PubMed.
- Z.-X. Liang, H.-Y. Song and S.-J. Lia, J. Phys. Chem. C, 2011, 115, 2604–2610 CAS.
- G. Guo, B. Yu, P. Yu and X. Chen, Talanta, 2009, 79, 570–575 CrossRef CAS PubMed.
- J. Niu, B. Yao, Y. Chen, C. Peng, X. Yu, J. Zhang and G. Bai, Appl. Surf. Sci., 2013, 271, 39–44 CrossRef CAS PubMed.
- W.-J. Sun, J. Li, G.-P. Yao, M. Jiang and F.-X. Zhang, Catal. Commun., 2011, 16, 90–93 CrossRef CAS PubMed.
- R. Słota, G. Dyrda, K. Szczegot, G. Mele and I. Pio, Photochem. Photobiol. Sci., 2011, 10, 361–366 Search PubMed.
- P. Moro, M. P. Donzello, C. Ercolani, F. Monacelli and G. Moretti, J. Photochem. Photobiol., A, 2011, 220, 77–83 CrossRef CAS PubMed.
- G.-P. Yao, J. Li, Y. Luo and W.-J. Sun, J. Mol. Catal. A: Chem., 2012, 361–362, 29–35 CrossRef CAS PubMed.
- G. Mele, R. D. Sole, G. Vasapollo, E. García-López, L. Palmisano and M. Schiavello, J. Catal., 2003, 217, 334–342 CrossRef CAS.
- P. Chowdhury, J. Moreira, H. Gomaa and A. K. Ray, Ind. Eng. Chem. Res., 2012, 51, 4523–4532 CrossRef CAS.
- X. Zhang, Z. Jin, Y. Li, S. Li and G. Lu, J. Power Sources, 2007, 166, 74–79 CrossRef CAS PubMed and R. Reithmeier, C. Bruckmeier and B. Rieger, Catalysts, 2012, 2, 544–571 CrossRef PubMed.
- M. K. Eberhardt, J. Phys. Chem., 1974, 78, 1795–1797 CrossRef CAS.
- R. Chen and J. J. Pignatello, Environ. Sci. Technol., 1997, 31, 2399–2406 CrossRef CAS.
- M. A. Outran, J. Peiroten, P. Chartin and A. J. Acher, Environ. Sci. Technol., 2000, 34, 3474–3479 CrossRef.
- M. S. Dieckmann and K. A. Gray, Water Res., 1996, 30, 1169–1183 CrossRef CAS.
- A. Di Paola, V. Augugliaro, L. Palmisano, G. Pantaleo and E. Savinov, J. Photochem. Photobiol., A, 2003, 155, 207–214 CrossRef CAS.
- C. Suarez, F. Louys, K. Günther and K. Eiben, Tetrahedron Lett., 1970, 8, 575–578 CrossRef.
- M. I. O. Ishag and P. G. N. Moseley, Tetrahedron, 1977, 33, 3141–3144 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.