
Retracted Article: Reductive immobilization of uranium by PAAM–FeS/Fe3O4 magnetic composites†
Dadong
Shao
a,
Xiangxue
Wang
a,
Jiaxing
Li
*a,
Yongshun
Huang
a,
Xuemei
Ren
a,
Guangshun
Hou
b and
Xiangke
Wang
ac
aInstitute of Plasma Physics, Chinese Academy of Sciences, P.O. Box 1126, 230031 Hefei, PR China. E-mail: lijx@ipp.ac.cn; Fax: +86 551 65591310; Tel: +86 551 65592788
bSchool of Surveying and Land Information Engineering, Henan Polytechnic University, PR China
cSchool for Radiological and interdisciplinary Sciences, Soochow University, PR China
First published on 21st November 2014
Abstract
Ternary magnetic composites of poly(acrylamide)-stabilized FeS/Fe3O4 (denoted as PAAM–FeS/Fe3O4) were in situ synthesized by chemical coprecipitation of FeS/Fe3O4 in the presence of acrylamide (AAM), followed by dielectric barrier discharge (DBD) plasma-induced polymerization of AAM on the FeS/Fe3O4 surface. The modified PAAM improved greatly the stabilization and dispersion properties of PAAM–FeS/Fe3O4 in aqueous solutions. This as-prepared composite was applied as an adsorbent for the preconcentration of U(VI) cations from aqueous solutions. The maximum enrichment capacity of U(VI) on this adsorbent at pH 5.0 and 20 °C was calculated to be 311 mg g−1. The presence of other cations had no obvious influence on the sorption of U(VI) on PAAM–FeS/Fe3O4, suggesting that this absorbent can remove U(VI) with high selectivity. According to the results of XPS spectra analysis, both FeS and amide groups on PAAM–FeS/Fe3O4 surfaces have strong chemical affinities to U(VI), and FeS can also reduce U(VI) to U(IV) effectively. The coagulation of magnetic U-laden PAAM–FeS/Fe3O4 can be easily separated and recovered from aqueous solution by simple magnet separation. This paper highlights the potential application of PAAM–FeS/Fe3O4 composites as suitable materials for the preconcentration and extraction of trace U(VI) ions from aqueous solutions in environmental radioactive pollution control and peaceful utilization of nuclear energy, such as the extraction of trace U(VI) ions from seawater.
Water impactIn this work, PAAM–FeS/Fe3O4 composites were in situ synthesized by the chemical coprecipitation–dielectric barrier discharge plasma technique and used as adsorbents for the preconcentration of U(VI) from aqueous solutions. PAAM improved greatly the stabilization properties and adsorption capability of PAAM–FeS/Fe3O4 in aqueous solutions. The coagulation of PAAM–FeS/Fe3O4 after adsorption of U(VI) can be easily separated and recovered from aqueous solution by simple magnet separation. The results show that PAAM–FeS/Fe3O4 is a suitable material for the preconcentration and extraction of trace U(VI) ions from aqueous solutions in environmental radioactive pollution control and peaceful utilization of nuclear energy. |
Introduction
With the fast development of nuclear techniques, large quantities of radioactive contaminants were released into the environment under certain circumstances during the past decades,1,2 which resulted in serious environmental radioactive pollution. Uranium, one fundamental material of nuclear energy, is one common and important radioactive contaminant in soils, sediments, and groundwater throughout the world.3 The immobilization and enrichment of uranium from radioactive wastewater is an important issue in the environmental friendly utilization of nuclear energy.The environmental risk and diffusion of uranium is strongly affected by its redox state.1,4–6 The reduction of soluble U(VI) to insoluble U(IV) solid phases is widely considered as an effective method for the in situ immobilization/stabilization of uranium contaminants.6,7 The reductive immobilization and enrichment of U(VI) with a variety of minerals and synthesized materials, such as sulfate-reducing bacteria,3,7 FeS,1,4,5,8 and FeS2,2,6,9 have been extensively studied. Among them, FeS can effectively reduce U(VI) to U(IV) and inhibit U(IV) re-oxidation under varied environmental conditions and is recognized as an important material for uranium contamination remediation. However, the easy aggregation of FeS particles in aqueous solution greatly restricts their application.10–12 It is desirable to modify the surface of FeS to enhance its stabilization and dispersion properties in aqueous solutions. Surface modification of FeS/Fe3O4 by hydrophilic materials, such as itaconic acid,10 agarose,11 carboxymethyl cellulose (CMC),12,13etc., is a feasible approach to enhance its stabilization and dispersion properties in aqueous solutions. Due to its excellent hydrophilic properties and complexation ability, poly(acrylamide) (PAAM) has been widely introduced to modify various materials for the enrichment and separation of radionuclides (such as U(VI) and Th(IV)) and metal ions (such as Pb(II), Cu(II), Cd(II), and Hg(II)) from aqueous solutions.14–16 The modified PAAM could provide more available sites for the selective extraction of U(VI) from aqueous solutions.
The effective separation technique for U-laden FeS-based composites from aqueous solution is also critical for the application of FeS in uranium contamination cleanup. Magnetic materials, such as nano- or micro-sized Fe3O4, have attracted great interest in various fields because they can be easily separated from aqueous solution by utilizing a simple magnetic field.17 Based on the reasons mentioned above, one can see that PAAM–FeS/Fe3O4 composites would be attractive materials in uranium pollution management because of the excellent enrichment capacity of PAAM, the reduction of U(VI) to U(IV) by FeS, and the magnetic properties of Fe3O4 for magnetic separation on a large scale.
In this paper, the magnetic composites of PAAM-stabilized FeS/Fe3O4 (denoted as PAAM–FeS/Fe3O4) were in situ synthesized by chemical coprecipitation of FeS/Fe3O4 in the presence of AAM, followed by DBD plasma-induced polymerization of AAM on the FeS/Fe3O4 surface. The as-synthesized PAAM–FeS/Fe3O4 was applied as an adsorbent to selectively enrich and separate U(VI) from aqueous solutions under a variety of operation conditions. The results showed that PAAM–FeS/Fe3O4 presented exceptional performance in the selective enrichment and separation of trace U(VI) ions from aqueous solutions, and the surface-adsorbed U(VI) was also partly reduced to insoluble U(IV). This paper highlighted the applicability of PAAM–FeS/Fe3O4 composites for the enrichment and reduction of U(VI) ions from aqueous solutions in uranium contamination cleanup.
Experimental
Synthesis of PAAM–FeS/Fe3O4 composites
PAAM–FeS/Fe3O4 composites were in situ synthesized by chemical coprecipitation of FeS/Fe3O4 in the presence of AAM, followed by DBD plasma-induced polymerization of AAM on the FeS/Fe3O4 surface. NaOH and Na2S were used as precipitants for Fe3O4 and FeS, respectively. Briefly, under argon and continuous mechanical stirring conditions, the solution containing NaOH (20.0 g L−1) and Na2S (0–12.5 g L−1) was added dropwise into a suspension containing FeCl2·4H2O (1.7 g L−1), FeCl3·6H2O (4.6 g L−1), and AAM (0–2.0 g L−1) to achieve pH ~7.0. After aging for 1 h, the argon DBD plasma technique was used to induce the polymerization of AAM on the FeS/Fe3O4 surface. The DBD plasma discharging power, time, and argon flow rate were 50 W, 1 h, and 10 mL min−1, respectively. The product was centrifuged and rinsed repeatedly with degassed Milli-Q water and dried at 50 °C for 24 h in a vacuum drier to obtain PAAM–FeS/Fe3O4 composites. In order to compare the sorption capacities of PAAM–FeS/Fe3O4 composites, Fe3O4 and PAAM/Fe3O4 were synthesized by the same method.Characterization
The physiochemical properties of PAAM–FeS/Fe3O4 were measured and evaluated by scanning electron microscopy (SEM), transmission electron microscopy (TEM) – element distribution mapping, a vibrating sample magnetometer (VSM), Fourier transform infrared (FT-IR) spectroscopy, UV-vis spectroscopy, powder X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) in detail. SEM images were obtained with a JSM 6320F FE-SEM (JEOL). TEM image and element distribution mapping investigations were carried out using a JEOL JEM 2010 microscope. VSM curves were obtained using a Model 155 VSM at room temperature in the measurement range of 0 to ±25 kOe. FT-IR spectroscopy measurements were performed using a Perkin-Elmer 100 spectrometer in KBr pellets under environmental conditions. UV-vis spectroscopy analysis was performed using a Shimadzu 2550 UV-vis spectrophotometer. The XRD pattern of PAAM–FeS/Fe3O4 was obtained using a Rigaku D/max 2550 X-ray diffractometer with Cu Kα radiation (λ = 0.15406 nm) at room temperature. XPS spectroscopy measurements were performed with an ESCALab220i XL surface microanalysis system (VG Scientific) equipped with an Al Kα (hν = 1486.6 eV) source at a chamber pressure of 3 × 10−9 mbar. The surface charging effects were corrected with the C 1s peak at 284.4 eV as a reference.Batch enrichment
The enrichment properties of radionuclides (U(VI), 90-Sr(II), and 137-Cs(I)) and metal ions (Pb(II), Cu(II), and Ni(II)) on Fe3O4, PAAM/Fe3O4, and PAAM–FeS/Fe3O4 were studied at 20 ± 1 °C by using the batch technique. The bulk suspensions of adsorbent and NaCl were pre-equilibrated for 24 h, then the solution containing radionuclides or metal ions was added, and the pH of the suspensions was adjusted to the desired values. The experiments were carried out under argon protection by shaking for 48 h to achieve sorption equilibration. The solid and liquid phases were separated by centrifugation at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min (Beckman Coulter 64R). The concentration of U(VI) in the supernatant was analyzed by the arsenazo III spectrophotometric method at 650 nm. The concentrations of 90-Sr(II) and 137-Cs(I) were analyzed by liquid scintillation counting (Packard 3100 TR/AB liquid scintillation analyzer, Perkin-Elmer) with the scintillation cocktail (Ultima Gold AB™, Packard). The concentrations of Pb(II), Cu(II), and Ni(II) were determined by atomic absorption spectrometry. All the experimental data were the average of triplicate determinations, and the data relative errors were <5%.
000 rpm for 30 min (Beckman Coulter 64R). The concentration of U(VI) in the supernatant was analyzed by the arsenazo III spectrophotometric method at 650 nm. The concentrations of 90-Sr(II) and 137-Cs(I) were analyzed by liquid scintillation counting (Packard 3100 TR/AB liquid scintillation analyzer, Perkin-Elmer) with the scintillation cocktail (Ultima Gold AB™, Packard). The concentrations of Pb(II), Cu(II), and Ni(II) were determined by atomic absorption spectrometry. All the experimental data were the average of triplicate determinations, and the data relative errors were <5%.
Recovery of uranium
The recovery of uranium from U-laden PAAM–FeS/Fe3O4 was investigated by dispersing 20 mg of U-laden PAAM–FeS/Fe3O4 in a beaker containing 50 mL of HNO3 solution with different HNO3 concentrations. The reduced U(IV) can be re-oxidized to U(VI) effectively under oxidizing conditions,3,10 which would dissolve in acidic solutions. After being exposed to air and mechanical stirring for 3 h, the suspension was centrifuged and analyzed under the same conditions as in the enrichment experiments. The residual materials in the 0.10 mol L−1 HNO3 solution were washed repeatedly with Milli-Q water, dried at 50 °C for 24 h under argon protection, and denoted as recovered PAAM–FeS/Fe3O4 for XPS and FT-IR analysis.Results and discussion
PAAM–FeS/Fe3O4 characterization
SEM, TEM and element distribution mapping were employed to investigate the morphology, microstructure and element distribution of PAAM–FeS/Fe3O4. As depicted in Fig. 1A and B, PAAM–FeS/Fe3O4 nanoparticles are highly dispersed and spherical-like in shape with a diameter of 10–20 nm, which is much smaller than that of bare and CMC-stabilized FeS particles (38.5 ± 5.4 nm) measured by TEM.12,13 It reveals that AAM can effectively prevent the aggregation of FeS/Fe3O4 particles in aqueous solution. Compared with aggregated bare FeS, stabilized FeS particles present higher specific surface area, which is due to their sorption capacity and affinity toward contaminants.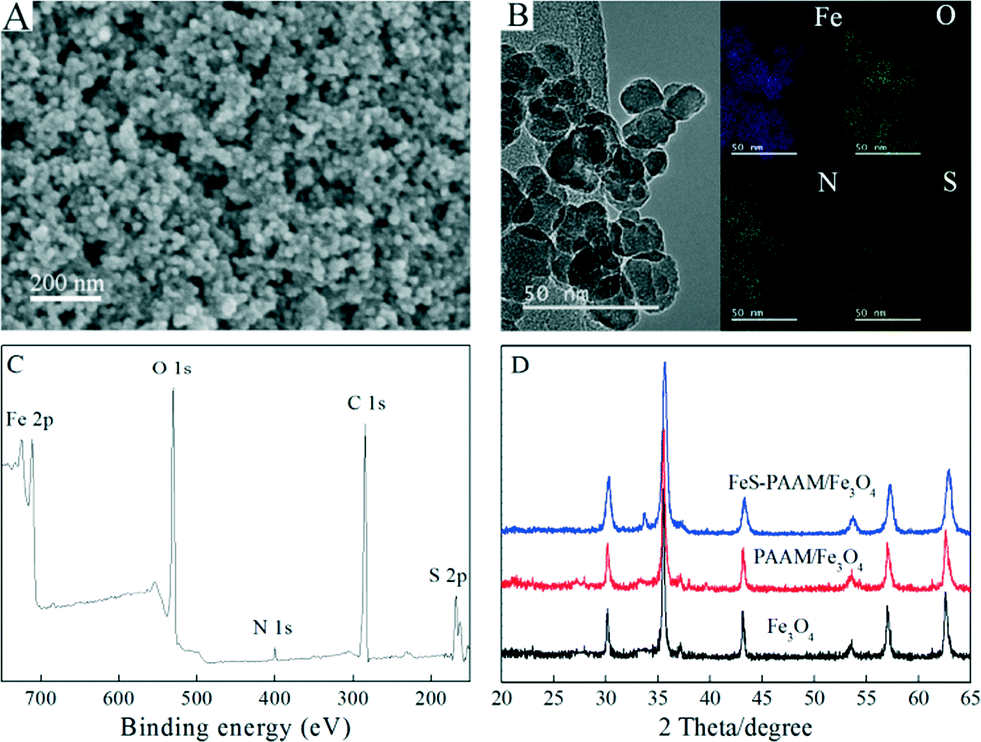 | ||
| Fig. 1 Characterization of PAAM–FeS/Fe3O4. SEM image (A), TEM image and the related element distribution mapping (B), XPS survey spectrum (C), and XRD pattern (D) of PAAM–FeS/Fe3O4. | ||
The Fe, O, N, and S element mapping (Fig. 1B) demonstrates that nitrogen and sulfur are uniformly dispersed. This indicates the homogeneous distribution of PAAM and FeS on the surfaces of PAAM–FeS/Fe3O4.
The successful formation of PAAM–FeS/Fe3O4 was also confirmed by XPS analysis. Fig. 1C shows the XPS survey spectrum of PAAM–FeS/Fe3O4. The peaks at ~168, ~285 and ~400 eV are related to S 2p, C 1s, and N 1s, which confirm the successful introduction of FeS and PAAM onto Fe3O4 surfaces.
Fig. 1D presents the XRD patterns of Fe3O4, PAAM/Fe3O4, and PAAM–FeS/Fe3O4. The XRD patterns of Fe3O4 and PAAM/Fe3O4 were similar to each other, and the characteristic XRD peaks at 2θ values of 30.3°, 35.6°, 37.3°, 43.3°, 53.7°, 57.2°, and 62.8° can be assigned to the (220), (311), (222), (400), (422), (511) and (440) planes of Fe3O4, respectively (JCPDS card no. 19-0629). Due to the fact that FeS shows the characteristic XRD peaks at similar 2θ values of 30.0°, 33.7°, 43.2°, 53.2°, and 56.9°,18,19 the XRD pattern of PAAM–FeS/Fe3O4 presents a new characteristic XRD peak at the 2θ value of 33.7° and has a much broader peak width at other positions. The average crystallite size of PAAM–FeS/Fe3O4 was calculated to be ~15.8 nm by using the Debye–Scherrer formula (D = 0.9λ/(β cosθ), where D is the crystallite size, λ is the wavelength of X-ray, β is the value of full width at half maximum and θ is the Bragg angle).
The stabilization and dispersion of Fe3O4, FeS/Fe3O4, and PAAM–FeS/Fe3O4 in aqueous solution is straightly demonstrated in Fig. 2A. After allowing to stand for 4 min, most Fe3O4 and FeS/Fe3O4 particles aggregated and precipitated at the bottom of the bottle. Moreover, the visible color change in the FeS/Fe3O4 suspension from black to black yellow indicated that part of FeS/Fe3O4 could be oxidized by dissolved oxygen in solution.20,21 On the other hand, due to the abundant hydrophilic amide groups in the molecular chains of PAAM, PAAM–FeS/Fe3O4 composites dispersed well in aqueous solution and no precipitate was formed even after allowing to stand for 6 months, which was further confirmed by UV-vis analysis (Fig. 2B). Meanwhile, the color of PAAM–FeS/Fe3O4 in aqueous solution was kept as gray black during the measurements, which demonstrated the excellent physical and chemical stability of PAAM–FeS/Fe3O4 in aqueous solution. Black yellow coagulation of U-laden PAAM–FeS/Fe3O4 could be observed when U(VI) solutions were added (Fig. 2A), which can be explained by the deposition of insoluble U(IV) on the oxidized PAAM–FeS/Fe3O4 surface. The coagulation and precipitation of U-laden FeS in aqueous solutions was consistent with the previous reports.1,5,6,22 Bi et al.1 found loose and porous U(IV) precipitates on FeS surfaces by using TEM images. The coagulation can be easily separated from the supernatant, which results in the separation and recovery of U(VI) from aqueous solutions by PAAM–FeS/Fe3O4 composites.
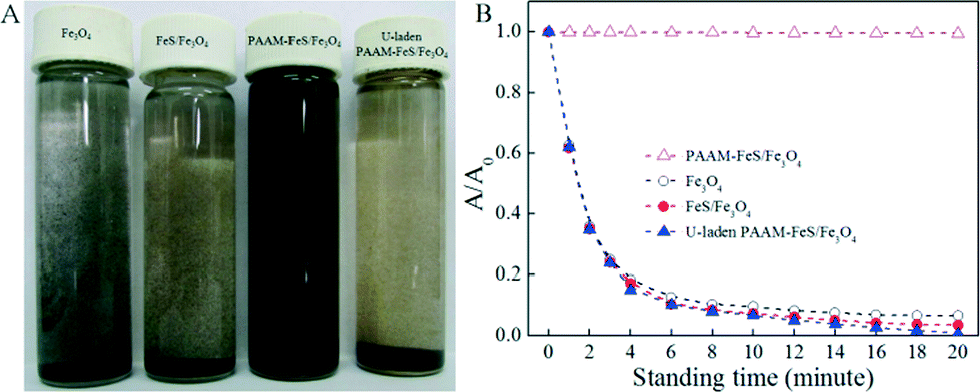 | ||
| Fig. 2 Photographs of dispersion (A) after allowing to stand for 20 minutes and UV-vis absorbance at 380 nm as a function of standing time (B) of Fe3O4, FeS/Fe3O4, raw and U-laden PAAM–FeS/Fe3O4. | ||
The magnetization properties of PAAM–FeS/Fe3O4 were also studied (Fig. SI-1†). The saturation magnetization of PAAM–FeS/Fe3O4 was detected to be 41.9 emu g−1 at room temperature and at a magnetic field of ±25 kOe. As observed from Fig. SI-1,† the PAAM–FeS/Fe3O4 composites could be attracted quickly by using a simple permanent magnet, and the clear solution can be easily removed by using a pipet or decantation. This result proved the excellent magnetic properties of PAAM–FeS/Fe3O4 composites, which can be used as magnetic adsorbents to enrich and preconcentrate U(VI) ions from aqueous solutions, followed by easy separation from aqueous solutions.
Enrichment capability of PAAM–FeS/Fe3O4 composites
The enrichment capability of PAAM–FeS/Fe3O4 is critical for its potential application. The effect of AAM and Na2S concentration used in the experiments on the enrichment capability of PAAM–FeS/Fe3O4 for U(VI) was investigated. Firstly, we studied the effect of AAM concentration on PAAM/Fe3O4 enrichment capability for U(VI). As illustrated in Fig. 3A and C, the enrichment of U(VI) on PAAM/Fe3O4 was much higher than that of U(VI) on Fe3O4. The PAAM/Fe3O4 composites prepared with 1.0 g L−1 AAM exhibited the highest enrichment ability for aqueous U(VI) under the experimental conditions. Therefore, 1.0 g L−1 AAM was selected to prepare the PAAM–FeS/Fe3O4 composites in this study. The effect of Na2S concentration on the sorption capability of PAAM–FeS/Fe3O4 for U(VI) was also investigated with the highest enrichment capability at 10.0 g L−1 Na2S (Fig. 3B and D). Together, 1.0 g L−1 AAM and 10.0 g L−1 Na2S solutions were used to prepare PAAM–FeS/Fe3O4 in this study. | ||
| Fig. 3 Effect of AAM (A, C) and Na2S (B, D) concentrations used in the experiment on the enrichment capability of PAAM–FeS/Fe3O4 for U(VI), pH-dependent enrichment of U(VI) on PAAM–FeS/Fe3O4 and the distribution of 50.0 mg L−1 U(VI) species in aqueous solution at different pH values (E), and the effect of contact time on U(VI) enrichment on PAAM–FeS/Fe3O4 (F). T = 20 ± 1 °C, m/V = 0.10 g L−1, C[NaCl] = 0.10 mol L−1. Fig. 3A–D: contact time: 48 h, pH = 5.0 ±0.1. Fig. 3E: contact time: 48 h, C[U(VI)](initial) = 50.0 mg L−1. Fig. 3F: C[U(VI)](initial) = 50.0 mg L−1, pH = 5.0 ±0.1. | ||
The experimental data were fitted with the classical Langmuir model (Cs = b × Csmax× Ce/(1 + b × Ce), Csmax is the maximum enrichment capacity, b is the Langmuir constant) and the Freundlich model (Cs = K × Ce1/n, K and 1/n are the constants indicative of enrichment capacity and enrichment intensity, respectively), which are the two most popular models applied in the reductive enrichment of multi-valent metal ions and radionuclides, such as U(VI),6 Hg(II),12 Se(IV) and Se(VI).23 Herein, the correlation coefficients (R2) (Table 1) indicated that the experimental data could be better described by the Langmuir model than by the Freundlich model. The applicability of the Langmuir model suggested that PAAM/Fe3O4 and PAAM–FeS/Fe3O4 surfaces were uniform and homogeneous for U(VI) sorption. The maximum enrichment capacity of U(VI) on PAAM–FeS/Fe3O4 at pH 5.0 and 20 °C was calculated to be 311 mg g−1 by the Langmuir model, which was much higher than that of U(VI) on Fe3O4 (90 mg g−1) and on PAAM/Fe3O4 (121 mg g−1) and comparable to today's nanomaterials under similar experimental conditions (see Table SI-1†). This observation highlights the potential application of PAAM–FeS/Fe3O4 as a suitable material for the high sorption of U(VI) from aqueous solutions in radioactive pollution cleanup.
| AAM (g L−1) | Na2S (g L−1) | Langmuir model | Freundlich model | ||||
|---|---|---|---|---|---|---|---|
| C smax (mg g−1) | b (L mg−1) | R 2 | K (mg g−1) | 1/n | R 2 | ||
| Fe3O4 | |||||||
| — | — | 89.5 | 0.152 | 0.995 | 27.7 | 0.272 | 0.919 |
| PAAM/Fe3O4 | |||||||
| 0.25 | — | 100 | 0.184 | 0.998 | 35.0 | 0.245 | 0.905 |
| 0.50 | 113 | 0.162 | 0.995 | 36.2 | 0.265 | 0.910 | |
| 1.0 | 121 | 0.157 | 0.996 | 37.1 | 0.279 | 0.913 | |
| 2.0 | 112 | 0.118 | 0.996 | 29.4 | 0.302 | 0.923 | |
| PAAM–FeS/Fe3O4 | |||||||
| 1.0 | 2.5 | 231 | 0.0358 | 0.993 | 22.9 | 0.469 | 0.955 |
| 5.0 | 249 | 0.0461 | 0.996 | 31.5 | 0.431 | 0.965 | |
| 7.5 | 290 | 0.0778 | 0.995 | 55.5 | 0.361 | 0.950 | |
| 10.0 | 311 | 0.0251 | 0.997 | 83.0 | 0.301 | 0.927 | |
| 12.5 | 251 | 0.0576 | 0.996 | 39.7 | 0.391 | 0.973 | |
Solution pH is an important factor that affects U(VI) sorption. The effect of pH on U(VI) sorption on PAAM–FeS/Fe3O4 was investigated by the batch technique in 0.10 mol L−1 NaCl solutions. As illustrated in Fig. 3E, the sorption of U(VI) on the PAAM–FeS/Fe3O4 surface increased with increasing solution pH. At pH ~7.0, PAAM–FeS/Fe3O4 can effectively remove ~95% U(VI) from aqueous solutions under experimental conditions. It is widely accepted that the protonation–deprotonation reactions of functional groups on the adsorbent surface and the aqueous U(VI) species are fairly dependent on solution pH.14,24 The relative distribution of U(VI) species (50.0 mg L−1) in aqueous solution was calculated from the hydrolysis constants of U(VI)24 and is also shown in Fig. 3E. The distribution of U(VI) species in aqueous solution was dependent on pH values, and the dominating U(VI) species were positively charged under the experimental conditions. U(VI) mainly existed as free uranyl ions (UO22+) at pH <4.5 and mainly existed as U(VI) hydrolysis complexes (such as UO2OH+) and U(VI) multinuclear hydroxide complexes (such as (UO2)2(OH)22+, (UO2)3(OH)5+, and (UO2)4(OH)7+) at pH >4.5 in aqueous solutions. Moreover, the enrichment capacity of FeS was also affected by solution pH because the hydroxyl and bisulfide functional groups on FeS surfaces would undergo different protonation–deprotonation reactions as solution pH changes.25 Similar phenomena were confirmed by other studies, such as those on U(VI),5,9,26 Se(IV) and Se(VI),23 and Hg(II),12 and organic contaminants.25
The effect of contact time on the removal of U(VI) ions from aqueous solutions on PAAM–FeS/Fe3O4 was studied to evaluate the required operation time of PAAM–FeS/Fe3O4 in real possible applications. As can be seen from Fig. 3F, the sorption of U(VI) on PAAM–FeS/Fe3O4 increased quickly within 1 h and then maintained the high level with further increasing contact time. Hua and Deng5 also found that the uptake of U(VI) from aqueous solution by amorphous FeS was quick and the sorption equilibrium was achieved within 1 h. The experimental data were analyzed by using pseudo-first-order kinetic models (ln(qe − qt) = lnqe − k1t, where qe and qt are the maximum and the experimental enrichment capacity, respectively; K1 (h−1) is the rate constant of enrichment) and pseudo-second-order kinetic models (t/Cs = 1/(2K′·C′s) + t/C′s, where C′s (mg g−1) is the enrichment capacity; K′ (g mg−1 h−1) is the rate constant of enrichment). The parameters for pseudo-first-order were calculated to be qe = 224.7 mg g−1, K1 = 9.07 h−1, and R2 = 0.725, and the parameters for pseudo-second-order were calculated to be C′s = 225.1 mg g−1, K′ = 0.199 g mg−1 h−1, and R2 = 0.999. The pseudo-second-order kinetic model fitted the experimental data better than the pseudo-first-order model. In the reaction of U(VI) with iron sulfide materials, the enrichment behavior was widely considered as a complex process of surface sorption of U(VI), partial reduction of U(VI) to U(IV), and precipitation of U(IV)/U(VI).6,9 The principle mechanism of U(VI) sorption on PAAM–FeS/Fe3O4 was further evidenced by XPS characterization.
Under the effective immobilization efficiency uncertainties, the lesser the amount of PAAM–FeS/Fe3O4 used, the lower the cost. Thus, the effect of PAAM–FeS/Fe3O4 content on the enrichment of U(VI) was studied and the results are shown in Fig. SI-2.† It is worth noting that a very small amount of PAAM–FeS/Fe3O4 (such as 0.10 g L−1) was needed to extract U(VI) from aqueous solutions, and the distribution coefficient (Kd) values reached the 104 mL g−1 level. Thus, PAAM–FeS/Fe3O4 presents high enrichment capacity in the immobilization and separation of U(VI) from large volumes of aqueous solutions.
Selectivity is a critical factor that affects the extraction of U(VI) from aqueous solutions due to the possible competitive sorption of other radionuclides. Herein 137-Cs(I), 90-Sr(II), Pb(II), Cu(II), and Ni(II) ions were selected as coexisting radionuclides and metal ions. According to the results of selective experiments (Fig. 4), it is observed that PAAM–FeS/Fe3O4 presents an excellent selectivity for U(VI) under the experimental conditions. The experimental data were also simulated by Langmuir and Freundlich models, and the relative fitting results are listed in Table 2. The Langmuir model matched the experimental data much better than the Freundlich model. This observation further confirmed that PAAM–FeS/Fe3O4 is a promising adsorbent for the selective removal and extraction of U(VI) ions from aqueous solutions.
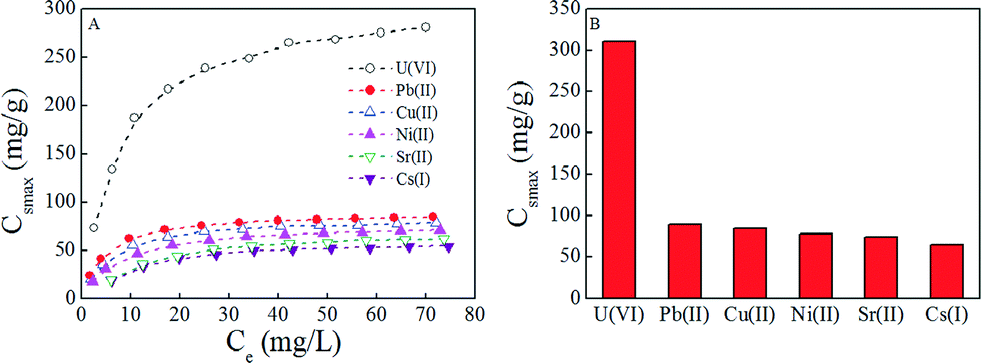 | ||
| Fig. 4 Comparison of the enrichment performance of PAAM–FeS/Fe3O4 for radionuclides and metal ions (A) and the related Csmax values calculated from the Langmuir model (B). Contact time: 48 h, T = 20 ±1 °C, m/V = 0.10 g L−1, pH = 5.0 ±0.1, C[NaCl] = 0.10 mol L−1. | ||
| Ions | Langmuir model | Freundlich model | ||||
|---|---|---|---|---|---|---|
| C smax (mg g−1) | b (L mg−1) | R 2 | K (mg g−1) | 1/n | R 2 | |
| U(VI) | 311 | 0.0251 | 0.997 | 83.0 | 0.301 | 0.927 |
| Pb(II) | 89.7 | 0.226 | 0.999 | 32.1 | 0.243 | 0.901 |
| Cu(II) | 85.0 | 0.170 | 0.998 | 26.5 | 0.270 | 0.902 |
| Ni(II) | 78.2 | 0.132 | 0.999 | 21.0 | 0.299 | 0.934 |
| Sr(II) | 74.5 | 0.0710 | 0.990 | 14.9 | 0.347 | 0.916 |
| Cs(I) | 64.5 | 0.0836 | 0.983 | 15.0 | 0.314 | 0.892 |
Enrichment mechanism
In order to get a better understanding of the interaction mechanism between U(VI) and PAAM–FeS/Fe3O4, the U-laden and recovered PAAM–FeS/Fe3O4 samples were characterized by the XPS technique. The XPS spectra of raw, U-laden, and recovered PAAM–FeS/Fe3O4 are shown in Fig. 5. In the U-laden PAAM–FeS/Fe3O4 sample, the occurrence of U 4f in U-laden PAAM–FeS/Fe3O4 is in accordance with the apparent changes in the N 1s, O 1s, and S 2p spectra, which suggested that the enrichment of U(VI) on PAAM–FeS/Fe3O4 was relevant to FeS and amide groups.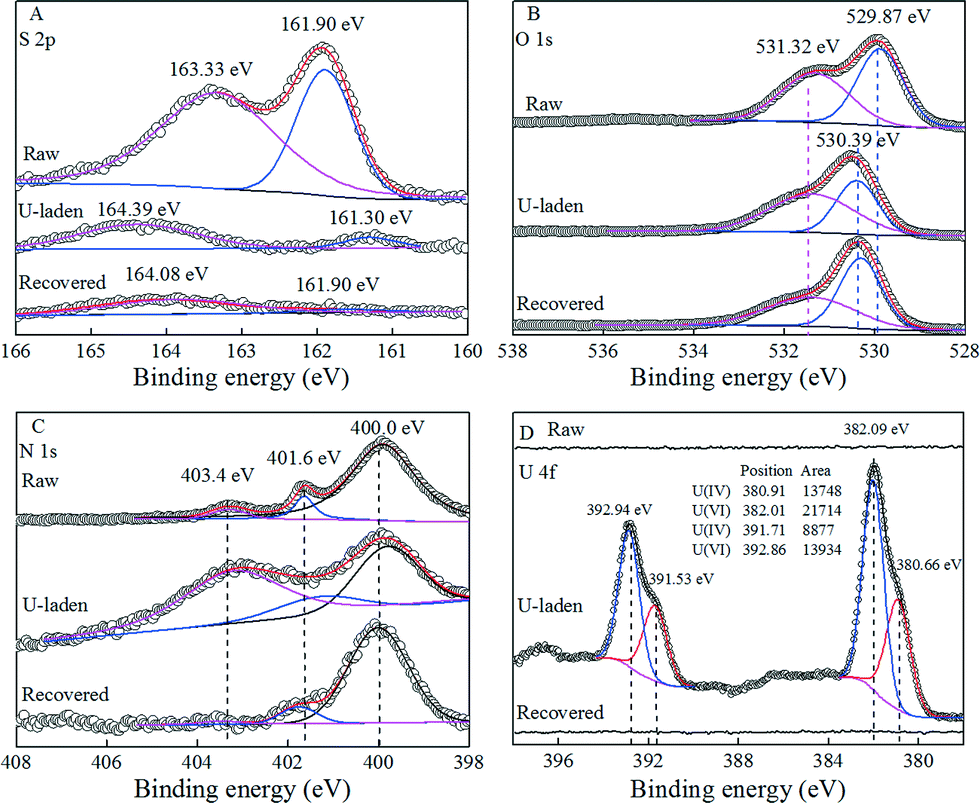 | ||
| Fig. 5 XPS S 2p spectra (A), O 1s spectra (B), N 1s spectra (C), and U 4f (D) spectra of raw, U-laden, and recovered PAAM–FeS/Fe3O4. | ||
As can be seen from the S 2p spectra in Fig. 5A, the raw PAAM–FeS/Fe3O4 showed the typical S 2p XPS peaks related to FeS at 161.90 and 163.33 eV.5,27 After the sorption of U(VI), U-laden PAAM–FeS/Fe3O4 also showed a new peak centered at ~161.30 eV, which was related to the formation of polysulfides (S2+x2−).1,5,9,28 Researchers1,5,6,9,26 have reported this observation that the reaction of U(VI) with FeSx resulted in the formation of U(IV) species and a variety of various sulfur species, including S2+x2−, dissolved S2O32−, dissolved S2O42−, etc. The formed S2+x2− could further react with U(VI) and showed a typical peak centered at ~164.39 eV.28 The conversion of insoluble FeS to dissolved sulfur species also significantly decreased the relative peak intensities and signal-to-noise ratios of S 2p in the XPS spectrum of U-laden PAAM–FeS/Fe3O4. Bargar et al.4 found that the distribution of uranium on the FeS surface was just associated with the sulfur-containing materials on the scale of the whole sediment due to the reaction of U(VI) with FeS. After rinsing with 0.1 mol L−1 HNO3, the peak centered at 164.39 eV related to S2+x2− shifted to 164.08 eV,29 which was due to the removal of U(IV)/U(VI) from the surface of PAAM–FeS/Fe3O4. The regenerated PAAM–FeS/Fe3O4 also showed a weak S 2p XPS peak related to FeS at 161.90 eV, indicating that the quantitative recovery of uranium from the surfaces of PAAM–FeS/Fe3O4 can be achieved by elution with 0.1 mol L−1 HNO3.
The O 1s spectra of raw, U-laden, and recovered PAAM–FeS/Fe3O4 samples (Fig. 5B) can be deconvoluted into the O2− anions in the bulk structure of Fe3O4 and the surface bond oxygen29,30 on the PAAM–FeS/Fe3O4 surface. The significantly increasing content of chemical bond oxygen in the U-laden PAAM–FeS/Fe3O4 sample confirmed the enrichment of U(IV)/U(VI)30 on the PAAM–FeS/Fe3O4 surface (Table 3). Moreover, after enrichment of U(VI), the peak related to the O2− anions in the bulk structure of Fe3O4 increased from ~529.9 eV29,30 to ~530.4 eV, which is similar to the report by Scott et al.30 that the O 1s spectrum of Fe3O4 showed a ~0.4 eV shift to a higher binding energy after enriching with U(VI). They explained the shift by the electron transfer between U(VI) and structural Fe(II) on the Fe3O4 surface, which resulted in the oxidation of Fe(II) to Fe(III) and the reduction of U(VI) to U(IV).
| Peak | BEa (eV) | FWHMb (eV) | % | |
|---|---|---|---|---|
| a Binding energy. b Full width at half maximum. | ||||
| Raw PAAM–FeS/Fe3O4 | ||||
| O 1s | Fe3O4 | 529.87 | 1.19 | 52.2 |
| Bridging OH | 531.32 | 1.76 | 47.8 | |
| N 1s | –NH2 | 399.90 | 1.70 | 87.1 |
| –N–O | 401.64 | 0.52 | 8.49 | |
| –NH2+ | 403.30 | 0.90 | 4.45 | |
| U-laden PAAM–FeS/Fe3O4 | ||||
| O 1s | Fe3O4 | 530.39 | 1.08 | 40.7 |
| Bridging OH | 531.38 | 2.22 | 59.3 | |
| N 1s | –NH2 | 400.10 | 1.66 | 33.1 |
| –N–O | 401.60 | 2.17 | 12.7 | |
| –NH2+ | 403.50 | 2.61 | 54.2 | |
| Recovered PAAM–FeS/Fe3O4 | ||||
| O 1s | Fe3O4 | 530.31 | 1.08 | 54.0 |
| Bridging OH | 531.41 | 2.33 | 46.0 | |
| N 1s | –NH2 | 400.00 | 1.55 | 87.7 |
| –N–O | 401.76 | 0.95 | 9.44 | |
| –NH2+ | 403.50 | 1.00 | 2.90 | |
The high-resolution scans for the N 1s spectra (Fig. 5C and Table 3) of raw, U-laden, and recovered PAAM–FeS/Fe3O4 can be deconvoluted into two peaks at 400.0 ± 0.1 and 401.7 ± 0.1 eV, corresponding to –NH2 and –N–O, respectively.31,32 The U-laden PAAM–FeS/Fe3O4 presents a new peak at 403.4 ± 0.1 eV corresponding to the complex of protonated amide groups and U(VI), which was ascribed to one of the uranyl oxo-oxygen atoms by Sather et al.33 from the protonation of amide groups (denoted as –NH2+). After the recovery of U(IV)/U(VI) from the surface of PAAM–FeS/Fe3O4, the relative content of –NH2+ decreased significantly and can be neglected. The results of XPS spectrum analysis indicated that amide groups and FeS on the surface of PAAM–FeS/Fe3O4 were attributed to the enrichment of U(VI).
The immobilized uranium on PAAM–FeS/Fe3O4 is most likely to exist as U(IV)/U(VI) mixture species because the enrichment of U(VI) on the surfaces of PAAM–FeS/Fe3O4 is a combined process of sorption and reductive immobilization. The XPS U 4f of U-laden PAAM–FeS/Fe3O4 was soundly determined and quantitatively resolved into U(IV) (centered at 380.66 and 391.53 eV) and U(VI) (centered at 382.09 and 392.94 eV).5,9,28 Although the formed U(IV) might be partly re-oxidized1,5,6,8 when it was exposed to air, the relative ratio of U(IV) to U(VI) on the PAAM–FeS/Fe3O4 surface (Fig. 5D) was still found to be ~0.63:1. It further confirmed that PAAM–FeS/Fe3O4 could reduce U(VI) to U(IV) on PAAM–FeS/Fe3O4 surfaces. Similarly, Hua and Deng5 reported that U(VI) was firstly adsorbed on the FeS surface and then reduced to U(IV), which resulted in U(IV)/U(VI) mixture species.
The reductive precipitation of U(VI) to U(IV) was also reported as the major reaction mechanism for the enrichment of U(VI) by reductive materials, such as sulfate-reducing biofilms,3 iron sulfide materials,9 and zero-valent iron.34 Compared with that of U-laden PAAM–FeS/Fe3O4, the XPS U 4f of recovered PAAM–FeS/Fe3O4 was undetectable, which indicated that U(VI) and U(VI) were completely rinsed from the surfaces of PAAM–FeS/Fe3O4.
The recovery of uranium from PAAM–FeS/Fe3O4 was also investigated. The adsorbed U(IV) can effectively be re-oxidized to soluble U(VI) under oxidizing conditions.3,10,34,35 Cerrato et al.35 reported that the oxidization and dissolution rates of U(IV) increased with increasing dissolved oxygen and decreasing pH values. The recovery efficiency of U(IV)/U(VI) versus HNO3 concentration is shown in Fig. 6A. It can be seen that 0.1 mol L−1 HNO3was enough for quantitative elution of adsorbed uranium, indicating the promising recovery efficiency of uranium from PAAM–FeS/Fe3O4. The recovery of U(IV)/U(VI) from PAAM–FeS/Fe3O4 was also confirmed by FT-IR spectroscopy analysis.
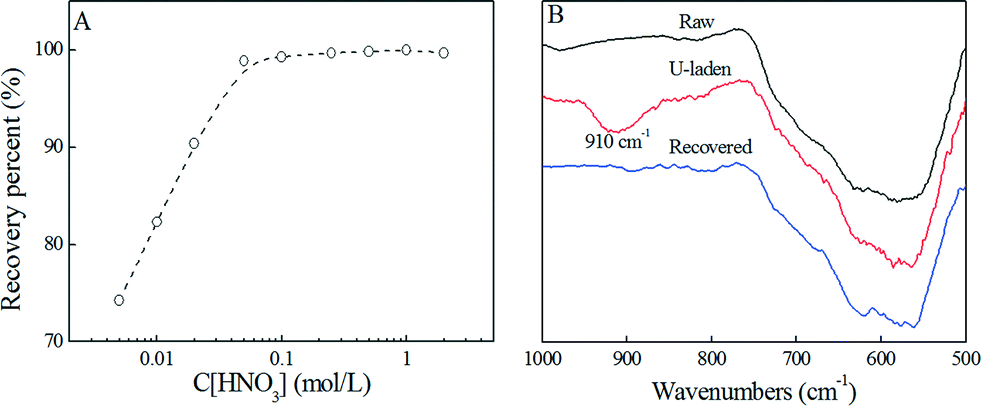 | ||
| Fig. 6 Effect of HNO3 concentration on the recovery of U(VI) from PAAM–FeS/Fe3O4 (A) and the related FT-IR spectra of raw, U-laden, and recovered PAAM–FeS/Fe3O4 (B). | ||
As depicted in the FT-IR spectrum of U-laden PAAM–FeS/Fe3O4 (Fig. 6B), the immobilized uranium on the PAAM–FeS/Fe3O4 surface was evidenced by the broad peak at ~910 cm−1 related to O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO stretching vibration.33,36,37 Compared with the peak of free aqueous U(VI) ions at ~963 cm−1, the significant red shift indicated the strong affinity of uranium to PAAM–FeS/Fe3O4.38 After rinsing with 0.1 mol L−1 HNO3 solution, the peak related to OUO stretching vibration disappeared, which confirmed the quantitative recovery of uranium from the surfaces of PAAM–FeS/Fe3O4 by elution with 0.1 mol L−1 HNO3.
UO stretching vibration.33,36,37 Compared with the peak of free aqueous U(VI) ions at ~963 cm−1, the significant red shift indicated the strong affinity of uranium to PAAM–FeS/Fe3O4.38 After rinsing with 0.1 mol L−1 HNO3 solution, the peak related to OUO stretching vibration disappeared, which confirmed the quantitative recovery of uranium from the surfaces of PAAM–FeS/Fe3O4 by elution with 0.1 mol L−1 HNO3.
Conclusions
In conclusion, PAAM–FeS/Fe3O4 composites were successfully in situ synthesized by chemical coprecipitation of FeS/Fe3O4 in the presence of AAM, followed by DBD plasma-induced polymerization of AAM on the FeS/Fe3O4 surface. The enrichment of U(VI) on PAAM–FeS/Fe3O4 was mainly attributed to the complexation of U(VI) with amide groups and the reductive reaction of U(VI) with FeS to U(IV). Enrichment results indicated that PAAM–FeS/Fe3O4 presented a highly efficient enrichment for U(VI) and can be easily separated and recovered from aqueous solution by a simple magnetic separation method. The results highlighted the potential application of PAAM–FeS/Fe3O4 in the reductive immobilization of U(VI) in nuclear waste management and environmental pollution cleanup.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (21272236, 91326202 and 21225730), the Science Foundation of Institute of Plasma Physics, Chinese Academy of Sciences (DSJJ–13–YY01), and Hefei Center for Physical Science and Technology (2012FXZY005).Notes and references
- Y. Bi, S. P. Hyun, R. K. Kukkadapu and K. F. Hayes, Geochim. Cosmochim. Acta, 2013, 102, 175 CrossRef CAS PubMed.
- F. Dullies, W. Lutze, W. Gong and H. E. Nuttall, Sci. Total Environ., 2010, 408, 6260 CrossRef CAS PubMed.
- H. Beyenal, R. K. Sani, B. M. Peyton, A. C. Dohnalkova, J. E. Amonette and Z. Lewandowski, Environ. Sci. Technol., 2004, 38, 2067 CrossRef CAS.
- J. R. Bargar, K. H. Williams, K. M. Campbell, P. E. Long, J. E. Stubbs, E. I. Suvorova, J. S. Lezama-Pacheco, D. S. Alessi, M. Stylo, S. M. Webb, J. A. Davis, D. E. Giammar, L. Y. Blue and R. Bernier-Latmani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4506 CrossRef CAS.
- B. Hua and B. Deng, Environ. Sci. Technol., 2008, 42, 8703 CrossRef CAS.
- C. Bruggeman and N. Maes, Environ. Sci. Technol., 2010, 44, 4210 CrossRef CAS PubMed.
- J. L. Druhan, M. E. Conrad, K. H. Williams, L. N'Guessan, P. E. Long and S. S. Hubbard, Environ. Sci. Technol., 2008, 42, 7842–7849 CrossRef CAS.
- Y. Bi and K. F. Hayes, Environ. Sci. Technol., 2014, 48, 632 CrossRef CAS PubMed.
- T. B. Scott, O. R. Tort and G. C. Allen, Geochim. Cosmochim. Acta, 2007, 71, 5044 CrossRef CAS PubMed.
- Y. Kalyan, S. Das, A. K. Pandey, G. R. K. Naidu, P. K. Sharma and A. V. R. Reddy, Anal. Methods, 2011, 3, 2017 RSC.
- J. Li, Z. Guo, S. Zhang and X. Wang, Chem. Eng. J., 2011, 172, 892 CrossRef CAS PubMed.
- Y. Gong, Y. Liu, Z. Xiong and D. Zhao, Environ. Sci. Technol., 2014, 48, 3986 CrossRef CAS PubMed.
- Z. Xiong, F. He, D. Zhao and M. O. Barnett, Water Res., 2009, 43, 5171 CrossRef CAS PubMed.
- B. L. Rivas, S. A. Pooley, H. A. Maturana and S. Villegas, Macromol. Chem. Phys., 2001, 202, 443 CrossRef CAS.
- R. Akkaya and U. Ulusoy, J. Hazard. Mater., 2008, 151, 380 CrossRef CAS PubMed.
- U. Ulusoy and R. Akkaya, J. Hazard. Mater., 2009, 163, 98 CrossRef CAS PubMed.
- A. A. Atia, A. M. Donia and H. H. El-Nomany, J. Dispersion Sci. Technol., 2009, 30, 451 CrossRef CAS.
- C. Xu, Y. Zeng, X. Rui, N. Xiao, J. Zhu, W. Zhang, J. Chen, W. Liu, H. Tan, H. H. Hng and Q. Yan, ACS Nano, 2012, 6, 4713 CrossRef CAS PubMed.
- L. Fei, Q. Lin, B. Yuan, G. Chen, P. Xie, Y. Li, Y. Xu, S. Deng, S. Smirnov and H. Luo, ACS Appl. Mater. Interfaces, 2013, 5, 5330 CAS.
- J. M. Senko, J. M. Suflita and L. R. Krumholz, Geomicrobiol. J., 2005, 22, 371 CrossRef CAS.
- I. T. Burke, C. Boothman, J. R. Lloyd, F. R. Livens, J. M. Charnock, J. M. Mcbeth, R. J. G. Mortimer and K. Morris, Environ. Sci. Technol., 2006, 40, 3529 CrossRef CAS.
- S. Y. Lee, M. H. Baik, H. R. Cho, E. C. Jung, J. T. Jeong, J. W. Choi, Y. B. Lee and Y. J. Lee, J. Radioanal. Nucl. Chem., 2013, 296, 1311 CrossRef CAS PubMed.
- D. S. Han, B. Batchelor and A. Abdel-Wahab, J. Hazard. Mater., 2011, 186, 451 CrossRef CAS PubMed.
- D. Shao, Z. Jiang, X. Wang, J. Li and Y. Meng, J. Phys. Chem. B, 2009, 113, 860 CrossRef CAS PubMed.
- E. J. Kim, K. Murugesan, J. H. Kim, P. G. Tratnyek and Y. S. Chang, Ind. Eng. Chem. Res., 2013, 52, 9343 CrossRef CAS.
- M. Yang, A. B. Fidalgo, S. Sundin and M. Jonsson, J. Nucl. Mater., 2013, 434, 38 CrossRef CAS PubMed.
- E. J. Kim, J. H. Kim, A. M. Azad and Y. S. Chang, ACS Appl. Mater. Interfaces, 2011, 3, 1457 CAS.
- Q. Wu, B. V. Yakshinskiy, T. Gouder and T. E. Madey, Catal. Today, 2003, 85, 291 CrossRef CAS.
- P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. Mcintyre, Surf. Interface Anal., 2004, 36, 1564 CrossRef.
- T. B. Scott, G. C. Allen, P. J. Heard and M. G. Randell, Geochim. Cosmochim. Acta, 2005, 69, 5639 CrossRef CAS PubMed.
- Q. Song, L. Ma, J. Liu, C. Bai, J. Geng, H. Wang, B. Li, L. Wang and S. Li, J. Colloid Interface Sci., 2012, 386, 291 CrossRef CAS PubMed.
- H. Nagai, S. Takashima, M. Hiramatsu, M. Hori and T. Goto, J. Appl. Phys., 2002, 91, 2615 CrossRef CAS PubMed.
- A. C. Sather, O. B. Berryman and J. Rebek Jr., J. Am. Chem. Soc., 2010, 132, 13572 CrossRef CAS PubMed.
- R. A. Crane, M. Dickinson, I. C. Popescu and T. B. Scott, Water Res., 2011, 45, 2931 CrossRef CAS PubMed.
- J. M. Cerrato, C. J. Barrows, L. Y. Blue, J. S. Lezama-Pacheco, J. R. Bargar and D. E. Giammar, Environ. Sci. Technol., 2012, 46, 2731 CrossRef CAS PubMed.
- C. R. Preetha, J. M. Gladis, T. P. Rao and G. Venkateswaran, Environ. Sci. Technol., 2006, 40, 3070 CrossRef CAS.
- M. J. Manos and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 16441 CrossRef CAS PubMed.
- D. E. Bryant, D. I. Stewart, T. P. Kee and C. S. Barton, Environ. Sci. Technol., 2003, 37, 4011 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ew00014e |
| This journal is © The Royal Society of Chemistry 2015 |