
Microbial electrolysis cell accelerates phosphate remobilisation from iron phosphate contained in sewage sludge
Fabian
Fischer
*,
Géraldine
Zufferey
,
Marc
Sugnaux
and
Manuel
Happe
Institute of Life Technologies, University of Applied Sciences and Arts Western Switzerland, HES-SO Valais-Wallis, Route du Rawyl 64, CH-1950 Sion 2, Switzerland. E-mail: fabian.fischer@hevs.ch
First published on 20th October 2014
Abstract
Phosphate was remobilised from iron phosphate contained in digested sewage sludge using a bio-electric cell. A significant acceleration above former results was caused by strongly basic catholytes. For these experiments a dual chambered microbial electrolysis cell with a small cathode (40 mL) and an 80 times larger anode (2.5 L) was equipped with a platinum sputtered reticulated vitreous carbon cathode. Various applied voltages (0.2–6.0 V) generated moderate to strongly basic catholytes using artificial waste water with pH close to neutral. Phosphate from iron phosphate contained in digested sewage sludge was remobilised most effectively at pH ∼13 with up to 95% yield. Beside minor electrochemical reduction, hydroxyl substitution was the dominating remobilisation mechanism. Particle–fluid kinetics using the “shrinking core” model allowed us to determine the reaction controlling step. Reaction rates changed with temperature (15–40 °C) and an activation energy of Ea = 55 kJ mol−1 was found. These analyses indicated chemical and physical reaction control, which is of interest for future scale-up work. Phosphate remobilisation rates increased significantly, yields doubled and recovered PO43− concentrations increased four times using a task specific bio-electric system. The result is a sustainable process for decentralized phosphate mining and a green chemical base generator useful also for many other sustainable processing needs.
Environmental impactPhosphate extraction from daylight mines will peak in 2030 and then decline in the following 50 years to a much lower level. Phosphate contained in waste is probably the most important concentrated phosphate source in the future. Since recently microbial fuel cells are proposed for phosphate recycling as they combine conceptually well with waste water treatment and nutriment extraction. In microbial fuel cells and other bioelectric systems Na+, K+ and other cations migrate from the anode to the cathode. This cation migration allows sustainable chemical base generation, which remobilises phosphate from insoluble iron-phosphate comprised in digested sewage sludge. This approach is also useful for other industrial processes where chemical base is needed. The integration of this bio-electric system in waste water treatment plants is proposed to mine phosphorus with energy and chemicals contained in waste water. |
Introduction
Low cost phosphorus mining enabled large scale phosphate use in industry and food production over the last hundred years. In the second half of the 19th century, lakes, rivers and sea shores eutrophicated due to increasing phosphate use.1 Since then, legislation enforces phosphate removal from waste water. For this purpose, iron chloride (FeCl3) is applied as an efficient ortho-phosphate removal reagent.2 In state of the art waste water treatment plants, more than 93% of phosphate is precipitated as insoluble iron phosphate (FePO4). No other method is considered more convenient and efficient to use in small to large scale waste water treatment plants.3 The resulting FePO4 precipitate is a significant constituent of digested sewage sludge (2–3% elemental phosphorus). Unfortunately, sewage sludge contains toxic metals and other contaminants and therefore a direct use as a fertilizer is forbidden in agricultural production, nor is it appreciated by the food industry.4 In consequence digested sewage sludge is incinerated and ashes deposited in landfills.5It is an unpleasant truth that phosphate will deplete in 50–80 years and its availability from daylight mines will peak in ∼2030.6 As a consequence of reduced phosphate availability, there is a risk of agricultural production declining, hunger rising and the world population shrinking. Resource shortage effects are non-linear and were predicted by Hardin as “The tragedy of the commons”.7 A way to counteract the looming phosphate shortage is to develop efficient recycling or other extraction processes.8
Phosphorus is an abundant element in the earth's crust but highly dispersed in minerals.9 Current phosphate use in agriculture, households and industry leads to a total loss to the environment. Once washed away into river, lake and sea sediments, phosphate enters a geological cycle for millions of years. Thus, today's mined phosphate is from magmatic apatite,10 phosphorite sediments,11 and from more recent organogenic guano deposits of sea birds12 and bats.13 Guano accumulation is faster than the geological cycle, but still takes thousands of years to form relevant deposits. A much faster phosphate accumulation system is human iron phosphate production from waste water treatment including related processes. This technology is a starting point for sustainable phosphate recycling as proposed in this paper.
There are 30 methods to recover phosphate from digested sewage sludge and its ashes.3 None of these methods ensure economic plant operation. Current phosphate prices are low. In addition, increasing phosphorus recycling in industrial countries will keep prices steady at current levels, but, will rise at some point rapidly.14 An idea how briskly prices double was experienced in 2008 due to uncertainties in financial markets15. High fertilizer prices are problematic for sustainable food production. Therefore, recycling systems need to be low-cost to ensure food production at an affordable price. A smooth transition from today's phosphate rock mining to tomorrow's recycling regime is a great challenge.
An emerging phosphorus recycling method is based on the microbial fuel cell (MFC) technology using microbial power and chemicals contained in municipal waste water. Bio-electrical systems are ideal technology to become integrated in waste water treatment plants.16 In preceding work, microbial fuel cell (MFC) power enabled phosphate extraction from digested sewage sludge up to 82% or 600 mg L−1.17 The remobilised phosphate can be transformed into different fertilizers such as superphosphate, triple superphosphate, ammonium phosphate and less traded struvite.18 The MFC approach was also investigated using swine manure in a single chambered MFC, where phosphate was deposited as struvite in the vicinity of an air cathode.19 Microbial electrolysis cell conditions equally caused struvite deposition on hydrogen releasing cathodes.20 Also combined nitrogen and phosphate recovery was reported using microbial fuel cell power.21 There are also non-bioelectric processes such as electrodynamic recovery from packed beds22 and electrochemical phosphate isolation from nanofiltration concentrates.23
The microbial fuel cell enabled phosphate remobilisation from insoluble FePO4 is potentially quantitative but slow. To improve reaction rates, an understanding of the remobilisation mechanism is important to scale-up the process. Earlier results showed that electrochemical reduction occurred under microbial fuel cell conditions, as iron content increased in iron-phosphate particles.17 Another observation concerned a pH increase up to ∼9 in the abiotic cathode using a dual chambered microbial fuel cell. This is based on the fact that iron and oxygen are reduced in the cathode and a base (OH−) is formed, which stays there because alkali cations Mn+ migrate into the cathode. Therefore, a basic iron-phosphate substitution mechanism is a second reaction mechanism.
In this work a supplemental voltage is applied to a dual chambered microbial fuel cell to improve phosphate remobilisation rates (Fig. 1). This transforms the MFC into a microbial electrolysis cell. Particle-fluid kinetics is elaborated to identify the reaction resistance in the phosphate remobilisation process.
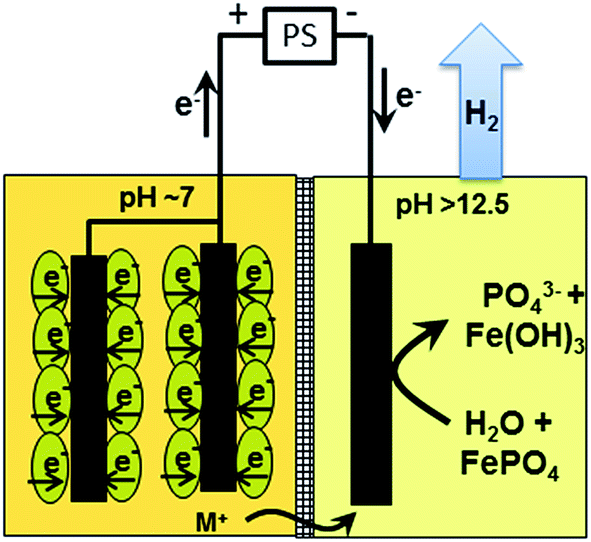 | ||
| Fig. 1 Microbial electrolysis conditions (Process C) caused hydrogen evolution in the abiotic cathode and the formation of a strong chemical base (OH−), which substituted insoluble iron phosphate (FePO4) and liberated ortho-phosphate (PO43−). The pH in the bio-anode was buffered and remained neutral. | ||
Experimental
Material
Escherichia coli K12 DSM 498 was obtained from the “Deutsche Sammlung für Mikroorganismen und Zellkulturen” (DSMZ) [Braunschweig, Germany]. Yeast extract was purchased from Lonza Ltd. [Visp, Switzerland], and enzymatic hydrolysate of soybean meal was purchased from Becton Dickinson AG [Allschwil, Switzerland]. KH2PO4, NaHCO3, NaH2PO4, Na2HPO4 and glucose were Sigma-Aldrich products [Buchs, Switzerland]. Methylene blue was from Fluka [Buchs, Switzerland]. Potassium hexacyanoferrate, K3Fe(CN)6, was obtained from Acros Organics [Geel, Belgium]. Ferric phosphate hydrate FePO4·H2O as the reference compound was a Riedel-de-Haen product [Seelze, Germany]. Nafion™ proton exchange membranes (PEM) and carbon felt electrodes were obtained from NCBE [Reading, United Kingdom]. Reticulated vitreous carbon foam (RVC), 3% density and 100 ppi, was purchased from the ERG aerospace corporation [Emeryville, CA, USA]. Digested sewage sludge was from the waste water treatment plant “Châteauneuf” of the city of Sion Switzerland.Microbial fuel/electrolysis cell construction
The dual chambered bio-electric cell was constructed to serve as microbial fuel cell as well as microbial electrolysis cell. It consisted of a 2.5 L anode and a 40 mL remobilisation cathode made from polyvinylchloride (PVC)17 (an important size difference between anode and cathode is strongly recommended). The remobilisation cathode was separated by a Nafion™ proton exchange membrane from the anode. The anode was made from six carbon fibre tissue electrodes of 1150 mm2. The cathode contained an electrode made from platinum sputtered reticulated vitreous carbon 25 × 30 × 5 mm and was equipped with a mechanical stirrer with 60 rpm. The top of the cathode cavity contained a small opening for hydrogen exhaust, liquid handling and sludge particle insertion.Anodic cultivation
Artificial waste water was made from Escherichia coli K12, which was pre-cultivated in a medium consisting of 1% yeast extract, 1% enzymatic soybean hydrolysate and 2% D-glucose. Overnight, the mixture was incubated in a 300 mL flask at 30 °C and 180 rpm and 50 mL added to the MFC anode. The cultivation medium in the anode contained 5 g L−1 yeast extract, 8.2 g L−1 KH2PO4, 10 g L−1 NaHCO3, 20 g L−1 glucose and 0.2 mM methylene blue.24 The microbial growth and readiness to provide power were monitored by an integrated MFC-sensor.25 The process was started when the electromotive force of this sensor reached 0.7–0.9 V. The anodic cultivation was not protected from contamination and biofilms were reused repeatedly. To verify results and trends closely, new cultivations with new anodes were employed. In these cases, the anode chamber was cleaned with anti-microbial solution and ethanol and the cultivation medium was sterilized at 120 °C.Generation of basic catholytes
In the MEC cathode with a platinized RVC electrode, 30 mL demineralized water was added, and a power supply Voltcraft output 0–30 V/0–2.5 A was connected between anode and cathode. Current flux was calculated from potentiometric data recorded by a multimetre over an 11 Ω resistance. Various applied voltages between 0.2 and 6.0 V generated basic solutions reaching high pH values within 24 hours.Sewage sludge particles
Dewatered digested sewage sludge was lyophilised and milled with a Zentrifugenmühle (Retsch) equipped with an 80 μm sieve. Before milling the dry sewage sludge was cooled with liquid nitrogen to prevent overheating in the mill. Obtained particles were analysed with a Mastersizer 2000 from Malvern Instruments [Worcestershire, England] and elemental composition analysed by a Varian 720-ES ICP-Optical Emission Spectrometer.Phosphate remobilisation from FePO4 comprised in sewage sludge
Once the pH of the catholyte reached a steady value, 0.2–1.2 g of dried sewage sludge particles (23 μm) were added to the cathode (30 mL). The resulting suspension was mechanically stirred at 60 rpm throughout the remobilisation process. In defined intervals (one to 24 hours) 1.5 mL catholyte samples were centrifuged at 10’000 g for 20 minutes at 20 °C (HighCen Zentrifuge Hettich). Then the supernatant solution was decanted off and analysed for phosphor and iron content by ICP-OES.Kinetics analysis
A semi-artificial reaction system was analysed outside of the microbial electrolysis cell as a well controllable setup. The basic catholyte was replaced by a 0.05 M NaOH solution and adjusted to pH 12.6 by hydrochloric acid. 30 mL of the artificial catholyte was added into a 100 mL flask, covered and stirred at 100 rpm at room temperature (24 °C) and atmospheric pressure. 0.2 g dry sewage sludge particles were added and samples of 1.5 mL were taken in defined intervals and centrifuged at 9300 rpm at 20 °C per 30 min. The same procedure was applied at 15 °C and 40 °C to determine the activation energy Ea. Also the pH was varied between 12 and 13 by adapting the method. All experiments were realized in triplicate.Elemental analysis of catholytes
ICP-OES spectroscopy was used to determine phosphor and iron content. The supernatant cathode solutions were diluted with 2% HNO3, for phosphorus 10 times, and iron dosage 100 times, respectively. Also catholytes containing Na+, K+, Ca2+ and others were analysed with this method.Results and discussion
Phosphate remobilisation
Strongly basic abiotic catholytes accelerated phosphate remobilisation from sewage sludge significantly above the state of the art17 (Table 1, entry 24 and 25). Sludge particles were added into the cathode and a voltage was applied; however, foam formation rendered this approach irreproducible with the given cathode architecture. Therefore, a step by step protocol was developed. In this improved process, the cathode was turned basic first, and then digested sewage sludge particles (23 μm) were added for the remobilisation step. Good to highest remobilisation yields (95%) (Table 1, entry 22) resulted from strong basic catholytes (pH > 12.3). The high reaction rate was verified by using pure FePO4 as the model system, yielding 85% in just one hour (Table 1, entry 20). All in all, in comparison to former microbial fuel cell conditions,17 the reaction rate increased significantly and yields doubled.| Entry | Bio-electric system | pH | Remobilisation timec | Dry sludge (FePO4) | Sludge mixture | Remobilized PO43− | Yield of PO43− |
|---|---|---|---|---|---|---|---|
| [—] | [min] | [g (mg)] | [g L−1] | [mg L−1] | [%] | ||
| a Pure FePO4 particles as the model compound. b Semi-artificial experiment realized outside of the microbial electrolysis cell in a flask using 0.05 M NaOH instead of basic catholyte and heated to 40 °C. c Remobilisation time after pH was reached. Preparation time for the chemical base: one day (Fig. 2A). d In former work the same MEC reactor operated as the microbial fuel cell.17 | |||||||
| 1 | Microbial electrolysis cell | 11.9 | 30 | 0.2 | 8.4 | 87 | 11.7 |
| 2 | Microbial electrolysis cell | 12.7 | 120 | 0.2 (17.7) | 9.3 | 352 | 42.8 |
| 3 | Microbial electrolysis cell | 13.3 | 90 | 0.3 (26.6) | 10.4 | 430 | 46.6 |
| 4 | Microbial electrolysis cell | 13.5 | 90 | 0.2 (17.7) | 6.3 | 265 | 47.9 |
| 5 | Microbial electrolysis cell | 13.5 | 240 | 0.8 (71) | 26.7 | 1228 | 51.9 |
| 6 | Microbial electrolysis cell | 13 | 240 | 0.3 (26.6) | 10.7 | 499 | 52.5 |
| 7 | Microbial electrolysis cell | 13.1 | 600 | 0.5 (44.4) | 25 | 1632 | 52.9 |
| 8 | Microbial electrolysis cell | 12.8 | 240 | 0.1 (8.9) | 3.6 | 175 | 55.1 |
| 9 | Microbial electrolysis cell | 13.6 | 360 | 0.8 (71) | 13.6 | 1728 | 56 |
| 10 | Microbial electrolysis cell | 12.8 | 75 | 0.2 (17.7) | 9.5 | 478 | 56.8 |
| 11 | Microbial electrolysis cell | 13.3 | 360 | 0.2 (17.7) | 10 | 511 | 57.5 |
| 12 | Microbial electrolysis cell | 13.8 | 360 | 1.2 (106.5) | 42.9 | 2190 | 57.6 |
| 13 | Microbial electrolysis cell | 13.8 | 360 | 0.2 (17.7) | 10 | 534 | 60.1 |
| 14 | Microbial electrolysis cell | 12.8 | 1380 | 0.8 (71) | 36.4 | 2231 | 69.1 |
| 15 | Microbial electrolysis cell | 12.6 | 1440 | 0.8 (71) | 26.7 | 1730 | 73 |
| 16 | Microbial electrolysis cell | 12.8 | 1380 | 0.2 (17.7) | 8 | 540 | 76.1 |
| 17 | Microbial electrolysis cell | 13.4 | 600 | 0.3 (26.6) | 13.4 | 1693 | 76.3 |
| 18 | Microbial electrolysis cell | 13.5 | 240 | 0.2 (17.7) | 6.7 | 474 | 80.2 |
| 19 | Microbial electrolysis cell | 12.6 | 1440 | 0.2 (17.7) | 6.7 | 486 | 82.3 |
| 20 | Microbial electrolysis cell | 12.5 | 60 | 0.0 (200)a | 0 | 2567 | 85 |
| 21 | Microbial electrolysis cell | 13.8 | 60 | 0.2 (17.7) | 6.7 | 549 | 91.5 |
| 22 | Microbial electrolysis cell | 13.3 | 60 | 0.2 (17.7) | 6.7 | 570 | 95 |
| 23 | Chemical base | 12.6b | 1200 | 0.2 (17.7) | 6.7 | 580 | 98 |
| 24d | Microbial fuel cell17 | 7–8.5 | 3 × 104 | 0.2 (30) | 6.7 | 11.2 | 82 |
| 25d | Microbial fuel cell17 | 7–8.5 | 2.7 × 104 | 1.9 (270) | 67.9 | 196 | 14.6 |
The basic abiotic catholyte was a non-buffered system and remobilisation rates depended on the quantity of added FePO4 particles (Table 1). Therefore, the power supply was not switched off during the remobilisation process to compensate an eventual pH drop and to use electrical reduction of Fe3+ as the second mechanism (Table 2). The solubility product of FePO4 is Ks = 1.3 × 10−22, which is as such low and is the reason that phosphate can be removed from waste water.26 The presence of specific ortho-phosphate forms (H3PO4, H2PO4−, HPO42− and PO43−) in contrast depends also on the protonation stage and shows that pH needs to be controlled.27 Under applied strong basic conditions, the remobilised phosphate was entirely deprotonated and a high pH was needed to avoid precipitation with iron to ensure solubility. Basicity and remobilisation rates correlated closely as shown by changing the pH gradually between 12 and 13. A pH of at least >12.3 was needed to enter into an effective remobilisation regime. From graphical interpretation, the effective pH was around 12.8 (Fig. 2B). A secondary product was Fe(OH)3, whose solubility product28Ks = 6.3 × 10−38 is rather low and enabled a separation of the iron cations from supernatant phosphate solutions (0.5–3%).
| Phosphate remobilisation under microbial fuel cell conditions | ||
|---|---|---|
| Bio-anode | Process | Abiotic cathode |
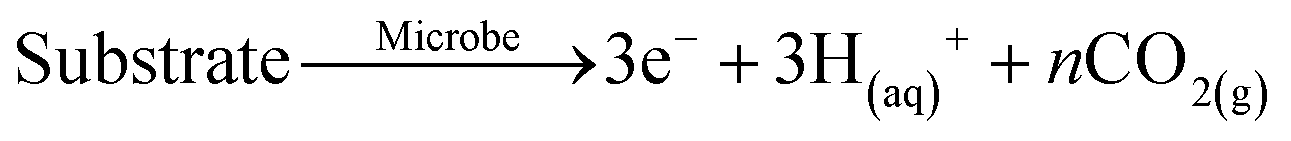
|
A |
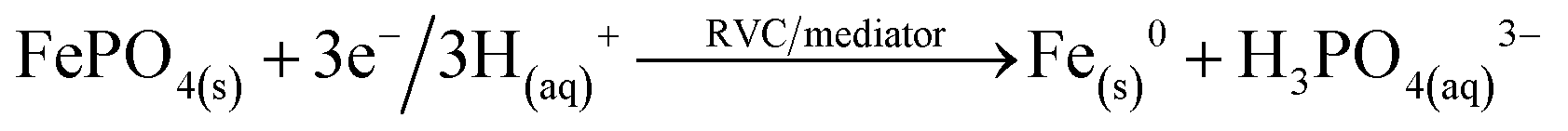
|
| B |
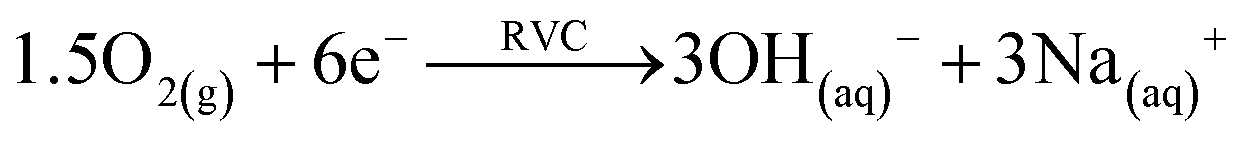
|
|
| FePO4(s) + 3OH(aq)− → PO4(aq)3− + 3Fe(OH−)3(s) | ||
| Phosphate remobilisation under microbial electrolysis cell conditions | ||
|---|---|---|
| Bio-anode | Process | Abiotic cathode |
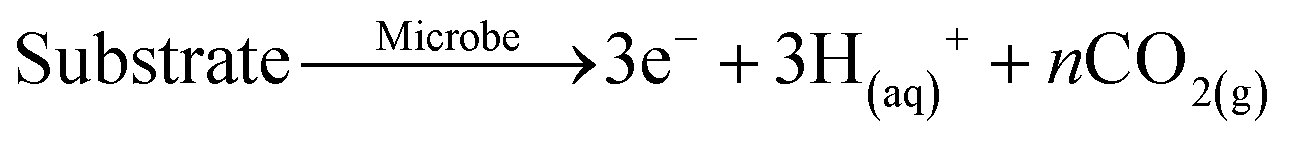
|
C |
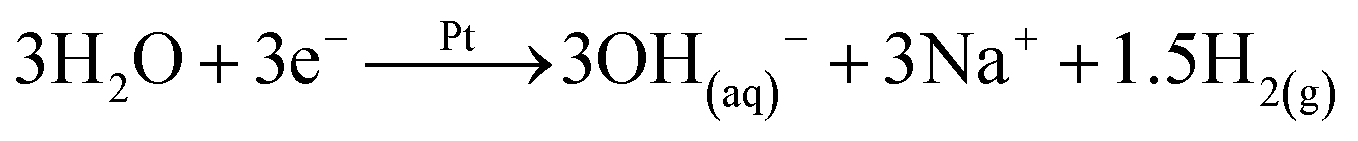
|
| FePO4(s) + 3OH(aq)− → PO4(aq)3− + 3Fe(OH−)3(s) | ||
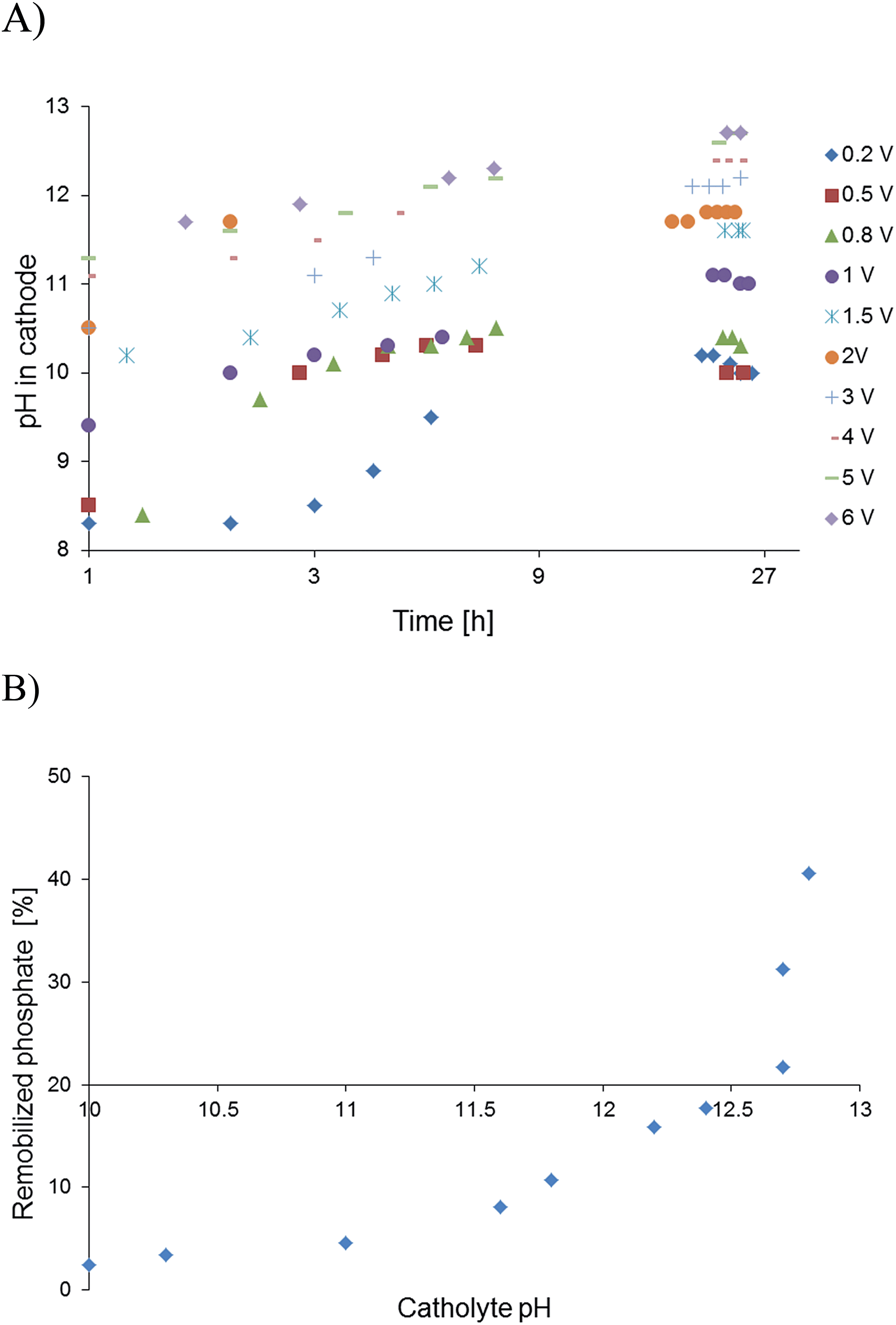 | ||
| Fig. 2 (A) pH evolution in 24 hours by microbial electrolysis related to applied voltages. (B) pH and phosphate remobilisation efficiency from iron phosphate in digested sewage sludge after one hour at ambient temperature. | ||
The best recovery of 95% was registered after 1 hour and is superior to microbial fuel cell conditions under which 82.3% was obtained in 21 days.17 This represents a significant acceleration of the process. Beside enhanced remobilisation rates, also PO43− concentrations increased from 600 mg L−1 to 2.23 g L−1. The preparation of basic catholytes required in general 24 hours and therefore the acceleration of the whole process including preparation corresponded to a time gain of a factor 20. Overall, the second generation phosphate remobilisation process developed here was much faster than that under pure microbial fuel cell conditions and became quantitative.
Microbial electrolysis to generate a strong base
Microbial electrolysis turned abiotic catholytes strongly basic using a platinized reticulated vitreous carbon (RVC) cathode. A final pH of 10 was possible by only 0.2 V of applied voltage, which corresponds to microbial electrolysis conditions just above the threshold voltage of 0.12 V.29 To increase the basicity further up to pH 11, an applied voltage of 1.0 V was needed. Next higher pH values of 12 and 13 were achieved with applied voltages between 3.0 and 5.0 V (Fig. 2A). Cation (Mn+) migration into cathodes with bioelectric systems is widely observed and increases the pH in the cathode. This is a major problem to scale-up microbial fuel cells for current production.30 Conversely, in the phosphate remobilisation investigated here this unwanted cation migration is an advantage and accelerated green chemical base formation strongly31 (Fig. 1). In a dual chambered microbial electrolysis cell the electron flux into the cathode is ideally accompanied by an equal quantity of protons that migrate through the proton exchange membrane and then combine on a catalytic surface with hydrogen. However, proton supply was highly insufficient, as good quantities of Na+, K+ and Ca2+ cations migrated from their place into the cathode according to ICP-OES analysis. As mentioned previously, the alkali cation migration was significant as bio-anodic protons were buffered at neutral pH in the cultivation medium. Sodium cations migrated faster than larger calcium or potassium ions (Fig. 3). An electron transfer efficiency analysis showed that 20–80% of alkali cations migrated per delivered electron, and the registered loss was presumably proton migration and also hydroxide crossover from the cathode into the anode compartment. It has to be mentioned at this point in terms of repeatability that the used microbial fuel cell was strongly asymmetric. The abiotic cathode was 80 times smaller than the bio-anode. This batch type base generator is an alternative to adjust anode and cathode fluxes.31 The results show that an important industrial chemical base can be produced in a sustainable manner employing a bio-electric batch reactor.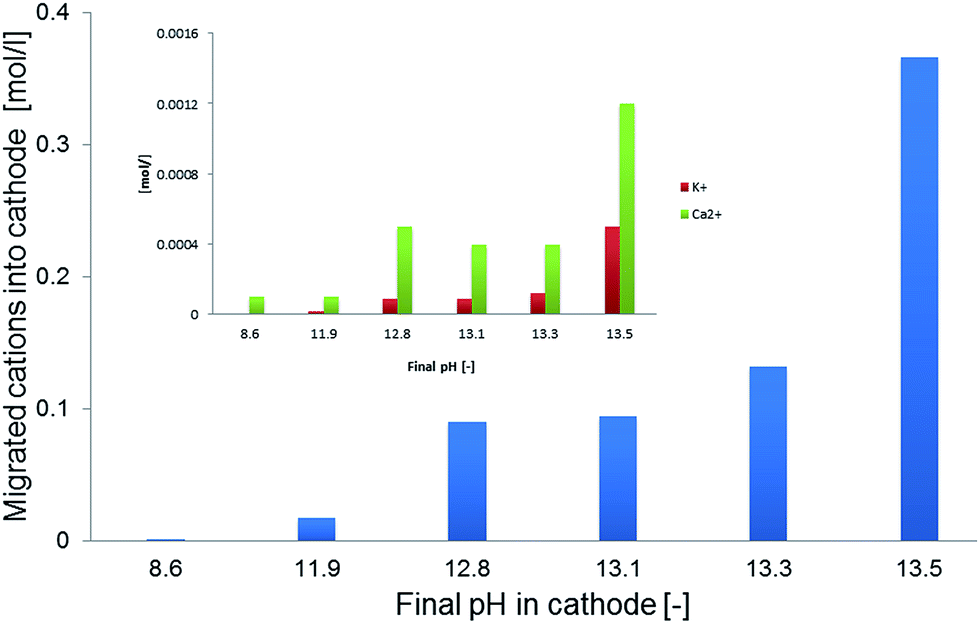 | ||
| Fig. 3 Migrated Na+ cations (blue) from the bioanode into the abiotic catholyte in correlation with achieved pH values. The small inserted figure shows final K+ and Ca2+ concentrations. | ||
Fluid-particle kinetics in phosphate remobilisation
The non-biological phosphate remobilisation followed fluid-particle kinetics and a theoretical model was searched to find the rate determining step. The microbial electrolysis cell based remobilisation was fast and quantitative that allowed to design a semi-artificial kinetic experiment. The phosphate remobilisation from sewage sludge particles was adjusted to a 24 hour experiment with close to quantitative FePO4 conversion. A ∼0.05 M NaOH solution replaced the catholyte and was set to a pH of 12.6 and 6.67 g L−1 digested sewage sludge particles were added. Three fluid-particle kinetic models were considered: diffusion through an ash/sludge layer, diffusion through a liquid film, and chemical reaction control32 (Fig. 4A). Instead of ash as the model theory suggests, here sewage sludge particles were used. They were considered to be a reasonable match to compare the semi-artificial process and identify the reaction resistance in phosphate remobilisation. The base (OH−) was thought to enter the sewage sludge matrix and to substitute FePO4 and release ortho-phosphate. The resistance of the reaction was found to be caused by the phosphate substitution and the solid FePO4 core shrinked toward zero over time. The release of ortho-phosphate from the particle was well possible at 12.6 pH. The side product Fe(OH)3 precipitated most likely in the particle due to its low solubility28Ks = 6.3 × 10−38. A comparison of the experimental results with the aforementioned models showed a mix of chemical and physical reaction control (Fig. 4B). The degree of chemical reaction control was verified by determining the activation energy Ea = 55 kJ mol−1 using the Arrhenius equation and assuming first order reaction kinetics to calculate reaction constants kobs (Table 3). The activation energy was above a purely diffusion controlled process where an Ea = 8–25 kJ mol−1 is expected, but below an entire chemical reaction control because the activation energy was below 80 kJ mol−1. Therefore, physical and chemical reaction control was involved. The physical control increased with time as visible in Fig. 4B. Beside the kinetic particle fluid analysis, the pH was of great importance and changing from 12 to 13 influenced reaction rates very much (Fig. 2B). And in this respect the remobilisation was controlled in the first place by the pH and then other factors, such as diffusion and temperature influenced the reaction rate. Overall, the kinetic analysis allowed us to determine important reaction parameters and facilitated the following mechanistic discussion.| Temperature [°C] | Initial rate constants kobs [s−1] |
|---|---|
| 15 | 7 × 10−6 ± 2 × 10−6 |
| 25 | 1.7 × 10−5 ± 6 × 10−6 |
| 40 | 4.5 × 10−5 ± 7 × 10−6 |
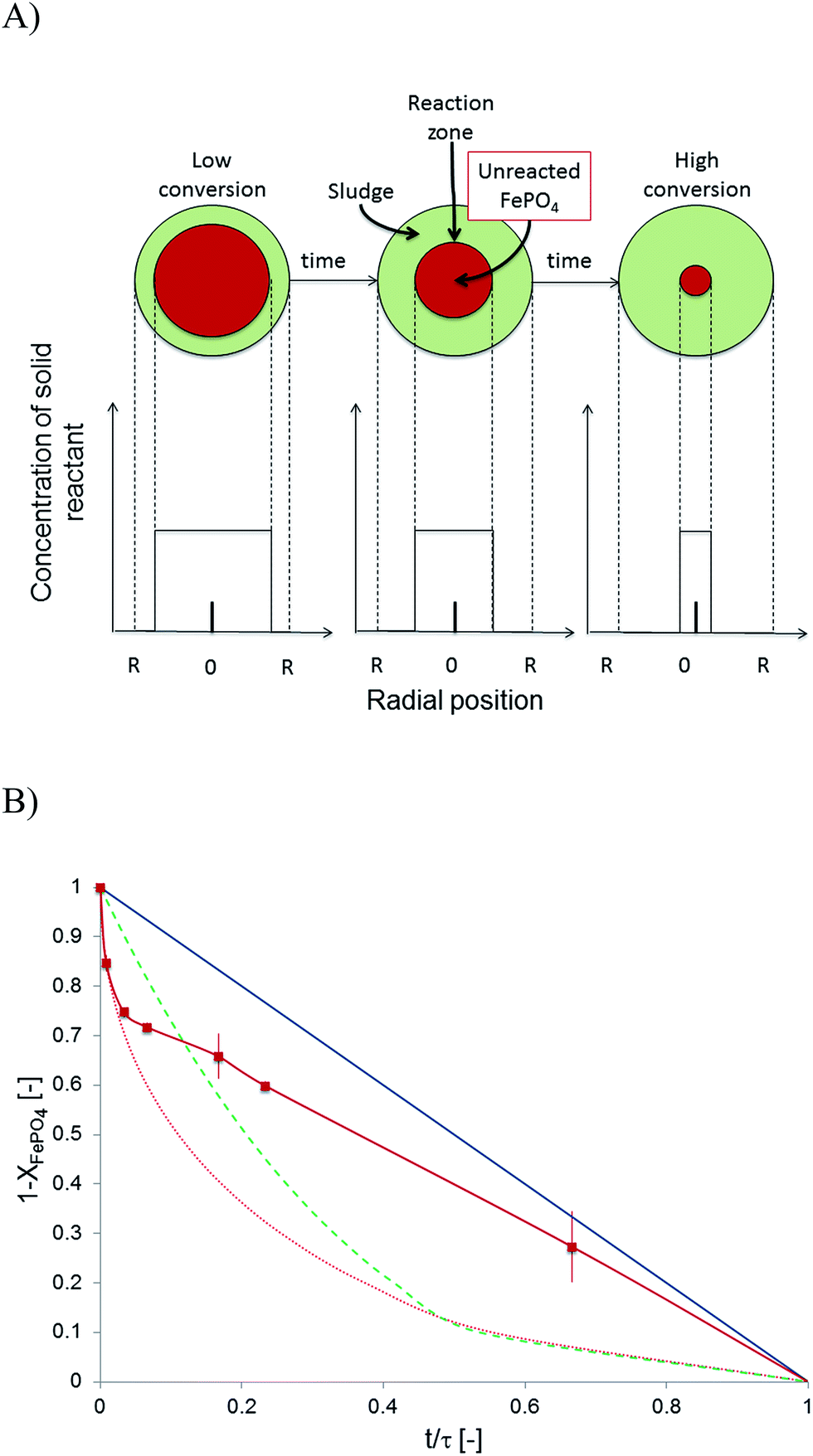 | ||
Fig. 4 (A) Particle shrinking core kinetics adapted to digested sewage sludge to determine the rate controlling step in phosphate remobilisation.30 The solid FePO4 core in sewage sludge particles shrinked with time. (B) Particle fluid kinetics models compared to experimental ( ) data from a semi-artificial remobilisation system at pH 12.6 at 25 °C with 6.67 g L−1 of sewage sludge particles (Process C,Table 2). The model predictions are: diffusion through a liquid film (blue), chemical reaction (green), and diffusion through an ash layer (red). ) data from a semi-artificial remobilisation system at pH 12.6 at 25 °C with 6.67 g L−1 of sewage sludge particles (Process C,Table 2). The model predictions are: diffusion through a liquid film (blue), chemical reaction (green), and diffusion through an ash layer (red). | ||
Remobilisation mechanisms
An unresolved question in former phosphate remobilisation work using a microbial fuel cell was the reaction mechanism (Table 2). Electrochemical reduction was proposed from qualitative analysis but also a substitution mechanism was not excluded. The hypothesis was that electrochemical reduction of FePO4 liberates ortho-phosphate in a slow reaction17 and a vivianite type compound is formed. But it remained unclear why yields varied. Using pure FePO4·H2O particles (∼11 μm) in a model remobilisation indicated iron accumulation in particles by electron reduction. The alternative substitution mechanism works because OH− is present that originates from iron and oxygen reduction in the cathode. A pH increase up to 9 was observed with MFC conditions (Process B, Table 2) and assumed to be high enough that a slow substitution mechanism competes with the equally slow reduction mechanism that is expected under pure microbial fuel cell conditions (Process A).33 Hydroxyl anions substituted iron phosphate and ortho-phosphate was liberated into the supernatant solution while the side product Fe(OH)3 precipitated (Processes B and C, Table 2). This substitution mechanism removes iron and possibly also toxic cations from reaction mixtures, which is a welcomed purification effect. Alternatively, cations could be galvanized on the cathode. The substitution mechanism was confirmed by using commercially available chemical base (NaOH) that equally remobilised phosphate and this under non-reductive conditions (Table 1, entry 23).34 The base propelled mechanism is therefore also possible under microbial fuel cell conditions (Process A, Table 2) with moderate basic catholytes (pH ∼9).17 In other terms, a higher pH increases the solubility of FePO4 and the substitution rate. The lowest solubility of FePO4 is around pH 5.5 and is one of the reasons why FeCl3 and similar reagents are applied to remove phosphate from waste water.35From above observations basic and electrochemical reduction mechanisms are possible. The electron reduction process is very much limited by low electron density that is difficult to enhance. Conversely, high base concentrations are more easily established. In the present investigation, the working potential was increased by a power supply initially from 0.3 to 0.5 V. It was assumed that also the electron reduction process would increase electron density and enhance reduction rates; however, higher voltage caused hydrogen formation. This effect was facilitated by using platinised RVC electrodes. It is noteworthy that microbial electrolysis starts at a low applied voltage of 0.13 V using Platin cathodes.29 Hydrogen formation caused a substantial pH increase in the catholyte as OH− was formed. The pH increased up to 10 on applying 0.2 V only. The basic remobilisation mechanism was then used extensively by producing strong bases at elevated voltages. Overall, there are two remobilisation mechanisms that should be used – if possible combined – to achieve efficient phosphate remobilisation.
Conclusions
Quantitative abiotic phosphate remobilisation from iron phosphate was significantly faster using a microbial electrolysis cell than microbial fuel cell conditions. Applied voltages between 0.2 and 6.0 V generated a strong chemical base in a sustainable manner. The application of the base in the remobilisation of ortho-phosphate from iron phosphate comprised in abiotic digested sewage sludge was optimized to become fast and quantitative.Five processes were involved in phosphate remobilisation: electron supply by microbes, hydrogen evolution, cation migration, phosphate substitution, and iron precipitation. The focus was set on the phosphate substitution reaction in the abiotic cathode. The substitution was accelerated as much as possible by increasing the pH in the non-biologic catholyte, while the pH in the bio-anode remained unchanged. The base in the cathode was either a result of oxygen intake from air or more efficiently produced by microbial electrolysis. Fluid-particle kinetics showed that the remobilisation was chemically and physically controlled, but dominated by the pH as the solubility products of FePO4, PO43− and Fe(OH)3 need to be considered too. The kinetics was also influenced by temperature variation. The related activation energy was 55 kJ mol−1, and both analyses confirmed a mix of chemical and physical reaction control. By using the novel extraction approach reported here phosphate remobilisation rates enhanced significantly, yields doubled and obtained free PO43− concentrations quadrupled. In addition, the remobilisation process is considered as an add-on procedure for waste water treatment plants where phosphate is removed with iron chloride or related reagents. In conclusion, four products result from the investigated bio-electric system: phosphate, hydrogen, a green chemical base, and certainly purified water.
Acknowledgements
This work was supported by the Swiss Federal Office for the Environment (FOEN), WWTP Worblental, Lonza Ltd., WWTP Sion, fenaco, Saia-Burgess Controls AG and HES-SO Valais.References
- V. H. Smith, G. D. Tilman and J. C. Nekola, Environ. Pollut., 1999, 100, 179–196 CrossRef CAS.
- S. K. Gupta, H. Häni and P. W. Schindler, Z. Pflanzenernaehr. Bodenkd., 1979, 142, 705–718 CrossRef CAS.
- H. Ludwig 2009: Rückgewinnung von Phosphor aus der Abwassereinigung. Eine Bestandesaufnahme. Umwelt-Wissen Nr. 0929. Bundesamt für Umwelt, Bern. 196 S.
- D. Fytili and A. Zabaniotou, Renewable Sustainable Energy Rev., 2008, 12, 116–140 CrossRef CAS.
- M. Horttanainen, J. Kaikko, R. Bergman, M. Pasila-Lehtinen and J. Nerg, Appl. Therm. Eng., 2010, 30, 110–118 CrossRef CAS.
- D. Cordell, J.-O. Drangert and S. White, Global Environ. Chang., 2009, 19, 292–305 CrossRef.
- G. Hardin, Science, 1968, 162, 1243–1248 CrossRef CAS.
- S. R. Carpenter and E. M. Bennett, Environ. Res. Lett., 2011, 6, 014009 CrossRef.
- J. V. Smith, Nature, 1981, 289, 762–765 CrossRef CAS.
- D. London, Am. Mineral., 1992, 77, 126–145 CAS.
- C. R. Glenn, K. B. Follmi, S. R. Riggs, G. N. Baturin, K. A. Grimm, J. Trappe, A. M. Abed, C. Galli-Olivier, R. E. Garrison, A. V. Ilyin, C. Jehl, V. Rohrlich, R. M. Y. Sadaqah, M. Schidlowski, R. E. Sheldon and H. Siegmund, Eclogae Geol. Helv., 1994, 87, 747–788 Search PubMed.
- R. J. M. Crawford and J. Jahncke, S. Afr. J. Mar. Sci., 1999, 21, 145–156 CrossRef.
- J. A. Harris, Science, 1970, 169, 1342–1343 CAS.
- H.-P. Weikard and D. Seyhan, Ecol. Econ., 2009, 68, 1749–1755 CrossRef.
- S. A. Mueller, J. E. Anderson and T. J. Wallington, Biomass Bioenergy, 2011, 35, 1623–1632 CrossRef.
- P. T. Kelly and Z. He, Bioresour. Technol., 2014, 153, 351–360 CrossRef CAS.
- F. Fischer, C. Bastian, M. Happe, E. Mabillard and N. Schmidt, Bioresour. Technol., 2011, 102, 5824–5830 CrossRef CAS.
- X. Hao, C. Wang, M. C. M. van Loosdrecht and Y. Hu, Environ. Sci. Technol., 2013, 47, 4965–4966 CrossRef CAS.
- O. Ichihashi and K. Hirooka, Bioresour. Technol., 2012, 114, 303–307 CrossRef CAS.
- R. D. Cusick and B. E. Logan, Bioresour. Technol., 2012, 107, 110–115 CrossRef CAS.
- Q. Tao, J. Luo, J. Zhou, S. Zhou, G. Liu and R. Zhang, Bioresour. Technol., 2014, 164, 402–407 CrossRef CAS.
- G. Sturm, H. Weigand, C. Marb, W. Weiß and B. Huwe, J. Appl. Electrochem., 2010, 40, 1069–1078 CrossRef CAS PubMed.
- C. Kappel, K. Yasadi, H. Temmink, S. J. Metz, A. J. B. Kemperman, K. Nijmeijer, A. Zwijnenburg, G.-J. Witkamp and H. H. M. Rijnaarts, Sep. Purif. Technol., 2013, 120, 437–444 CrossRef CAS PubMed.
- M. Sugnaux, S. Mermoud, A. Ferreira da Costa, M. Happe and F. Fischer, Bioresour. Technol., 2013, 148, 567–573 CrossRef CAS PubMed.
- M.-F. Favre, D. Carrard, R. Ducommun and F. Fischer, J. Ind. Microbiol. Biotechnol., 2009, 36, 1307–1314 CrossRef CAS PubMed.
- D. E. C. Corbridge, Outline of its Chemistry, Biochemistry and Uses, Elsevier Science, New York, 5th edn, 1995, p. 143 Search PubMed.
- Y. Bashan, A. A. Kamnev and L. E. de-Bashan, Biol. Fertil. Soils, 2013, 49, 465–479 CrossRef CAS.
- J. W. Hill, R. H. Petrucci, T. W. McCreary and S. S. Perry, Chimie des solutions (ERPi), 2e édn, 1988 Search PubMed.
- B. E. Logan and K. Rabaey, Science, 2012, 337, 686–690 CrossRef CAS PubMed; H. Liu, H. Hu, J. Chignell and Y. Fan, Biofuels, 2010, 1, 129–142 CrossRef.
- K. J. Chae, M. Choi, F. F. Ajayi, W. Park, I. S. Chang and I. S. Kim, Energy Fuels, 2008, 22, 169–176 CrossRef CAS.
- K. Rabaey, S. Bützer, S. Brown, J. Keller and R. A. Rozendal, Environ. Sci. Technol., 2010, 44, 4315–4321 CrossRef CAS PubMed.
- O. Levenspiel, Chemical Reaction Engineering, John Wiley & Sons, New York, 3rd edn, 1998 Search PubMed.
- R. A. Rozendal, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5206–5211 CrossRef CAS.
- A. Sano, M. Kanomata, H. Inoue, N. Sugiura, K.-Q. Xu and Y. In, Chemosphere, 2012, 89, 1243–1247 CrossRef CAS PubMed.
- R. Sedlak, Phosphorus and N. Removal from Municipal Wastewater; Principles and Practise, Lewis Publishers, 2nd edn, ISBN 0-87371-683-3, 1991 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |