
Spatial and temporal variability of incidental nanoparticles in indoor workplaces: impact on the characterization of point source exposures
Jianjun
Niu
a,
Pat E.
Rasmussen
*ab,
Robert
Magee
c and
Gregory
Nilsson
c
aEnvironmental Health Science and Research Bureau, HECSB, Health Canada, Ottawa, Ontario K1A 0K9, Canada. E-mail: Pat.Rasmussen@hc-sc.gc.ca; Fax: +1(613)952-8133; Tel: +1(613)941-9868
bEarth Sciences Department, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada
cNational Research Council of Canada, Construction, 1200 Montreal Road, Ottawa, Ontario K1A 0R6, Canada
First published on 29th October 2014
Abstract
This study deployed a suite of direct-reading instruments in six locations inside one building to characterize variability of the background aerosol, including incidental nanoparticles (NP), over a six month period. The instrument suite consisted of a portable Condensation Particle Counter (CPC) and a Scanning Mobility Particle Sizer (SMPS) for assessing particle number concentrations and size distributions in the nano-scale range; an Aerodynamic Particle Sizer (APS) for assessing micron-scale particle number concentrations and size distributions; plus a desktop Aerosol Monitor (DustTrak DRX) and a Diffusion Charger (DC2000CE) for assessing total particle mass and surface area concentrations respectively. In terms of number concentration, NPs (<100 nm) were the dominant particles observed in the background aerosol, contributing up to 53–93% of the total particle number concentrations. The particle size distributions were bimodal with maxima around 19–79 nm and 50–136 nm, respectively, depending on workplace locations. The average detected background particle number, surface area and total mass concentrations were below 7.1 × 103 # cm−3, 22.9 μm2 cm−3 and 33.5 μg m−3, respectively in spring samples and below 1.8 × 103 # cm−3, 10.1 μm2 cm−3 and 12.0 μg m−3, respectively in winter samples. A point source study using an older model laser printer as the emission source indicated that NPs emitted from the investigated printer were distinguishable from background. However, more recent low emitting printers are likely to be indistinguishable from background, and chemical characterization (e.g. VOCs, metals) would be required to help identify emission sources.
Environmental impactsAn important component of assessing exposure to engineered nanoparticles is characterization of background. Quantification of engineered nanomaterial exposure can be very challenging due to temporal and spatial variability of incidental nanoparticles. In this study particle size distribution, particle count, and particle surface area are monitored in six locations of an indoor workplace environment, where incidental nanoparticles are found to be the largest contributor to the background aerosol. Strong temporal trends, i.e. a clear increase or decrease in background nanoparticles, are observed in several locations, demonstrating the importance of including time series data in an exposure assessment. The study concludes that the most appropriate measurement strategy is situation specific, and should include multiple direct-reading instruments to cover all the relevant measurands. |
1. Introduction
An important but challenging aspect of characterizing nanoparticle (NP) exposures in the workplace is the necessity to quantify background or “incidental” NPs.1–3 The term nanoparticle originally referred to engineered nanoparticles but is now defined as any particle less than 100 nm in three dimensions,4 which includes ultrafine particles (UFPs) that originate from multiple incidental sources in indoor environments including office electronic equipment, cosmetics, food and packaging, clothing and textiles, heating and cooking5–8 and infiltration from outdoor sources such as combustion and vehicle emissions.7,9,10 Appropriate characterization of background NPs is a critical step to avoid misidentification of exposure sources11–13 but currently there is limited guidance for measurement and characterization of background NPs and for differentiating background from engineered NP exposure in the workplace.14–16 Background NP concentrations may vary widely depending on the unique conditions of the workplace,17,18 and thus it may be difficult or impractical to correct for background using a simple subtraction approach. Background correction to identify point source contributions of NP becomes quite complex if the investigated source contributes relatively small elevations in particle counts while the background particle count is relatively high.18There is also a lack of international consensus about which measurement parameters (solubility, size, surface area, morphology, composition, degree of agglomeration/aggregation, surface modifications or reactivity, number concentration, and/or mass) provide the most reliable exposure metrics.2,17,19,20 Animal studies have indicated that the toxicity of inhaled NPs or UFPs is more closely associated with particle surface area and particle number concentrations than with the particle mass concentration when comparing aerosols having different particle size distributions.21–23 However, mass concentration measurements are still considered valuable and necessary in many situations.3 For this reason, it has been recommended that several different exposure metrics should be captured (e.g., particle number concentration, surface area concentration, and mass concentration) which requires the use of multiple instruments operated simultaneously.17–20,24–29 If source identification is required, NPs may need to be sampled for off-line chemical and physical characterization using techniques such as inductively coupled plasma-mass spectrometry (ICP-MS), energy dispersive X-ray fluorescence (ED-XRF), atomic force microscopy (AFM), electron microscopy (EM), and X-ray diffraction (XRD).9,13,30–33
The present paper is organized into two parts: a background study and a point source study. The main focus of the background study is the investigation of spatial and temporal variability of indoor background aerosols using key parameters: particle number concentrations, total surface area and mass concentrations, and particle number and mass size distributions. This study was carried out at different locations and times in a single federal workplace building as a preliminary characterization of incidental (background) nano-sized particles. The point source study focuses on assessing the capacity of the instrument suite to measure and distinguish a known point source of NP emissions (compared to background) in a workplace environment. Laser printers, which are a very common point source of NP emissions,5,17,34–36 were used in this study to assess the analytical ability to discriminate these point sources from background levels in each of the six investigated locations. In addition to NPs, the fine and coarse particle size ranges (100–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nm) were also monitored using both research grade instruments and analyzers traditionally used by occupational hygienists for characterization of aerosol trends.
000 nm) were also monitored using both research grade instruments and analyzers traditionally used by occupational hygienists for characterization of aerosol trends.
2. Experimental
2.1. Background study
The suite of instruments listed in Table 1 was set up on carts for simultaneous direct monitoring of the key parameters. A limitation of the background study was the availability of only one complete suite of instruments, due to the total combined cost of these instruments. Therefore the six locations could not be monitored simultaneously as it was necessary to move the instrument suite consecutively from one location to another. Continuous monitoring was conducted for a 3–4 day period in each location, from which a 24 h subset was extracted based on time stamps (midnight to midnight) to allow comparisons of 24 h averages and standard deviations. The criteria for selection of the 24 h subsets were (1) presence of a complete data set for all instruments, and (2) minimal human activity during monitoring to avoid disrupting the suite of instruments. Apart from the single operator of the instrument suite, there were no other occupants present in the four investigated rooms, but human activity was not controlled in the corridor (20–50 passers-by during the day/2–4 at night) or the receiving/delivery area (5–15 deliveries per day/none at night). The 24 h subsets were further divided to obtain 12 h daytime averages (6:30 am to 6:30 pm) and 12 h nighttime averages (6:30 pm to 6:30 am).
| Instrument | Particulate analysis capability | |
|---|---|---|
| Aerasense NanoTracer | • Particle counter for size range 10–300 nm; monitors total particle number concentration; units # cm−3 | |
| (Philips Electronics, Eindhoven, The Netherlands) | ||
| TSI Model 3936 scanning mobility particle sizer (SMPS) (TSI Inc., St. Paul, MN, USA), including: | • Monitors particle number size distributions; units dN/dlogDp (# cm−3) | |
| • A model 3080 electrostatic classifier | • 64 channels | |
| • A model 3081 long differential mobility analyzer (DMA) | • size range 10.6–495.8 nm | |
| • A model 3786 ultrafine water-based CPC | ||
| TSI Model 3321 aerodynamic particle sizer (APS) | • Monitors particle number size disturbutions; units dN/dlogDp (# cm−3) | |
| (TSI Inc., St. Paul, MN, USA) | • 32 channels | |
| • size range 500 nm to 20 μm | ||
| EcoChem DC2000CE diffusion charger (DC) | • Measures particle surface area concentration (units μm2 cm−3) | |
| (EcoChem Analytics, League City, TX, USA) | ||
| TSI DustTrak DRX Model 8533 | • Monitors total particle mass (units μg m−3), corresponding to PM1, PM2.5, respirable (RESP = PM4), PM10 and total PM (equal to PM15) | |
| (TSI Inc. St. Paul, MN, USA) | ||
Measurements were taken twice in each location: once in the winter (November 2011 to January 2012) and once in the spring (April to May 2012). Although the resulting 24 h data sets cannot be considered representative of an entire season, sampling over a six-month period does provide an indication of long-term variations that may need to be considered in a future monitoring campaign, related to changes in ventilation, infiltration, and heating/air conditioning associated with changes in season.
s for the NanoTracer and 1 s for the TSI 3786 CPC.
To prepare for the present study, all instrument units were either sent to the manufacturers for calibration or calibrated on-site according to manufacturers' recommended procedures. An instrument comparison study was also performed in the full-scale chamber (see 2.2.2) using sodium chloride aerosol37 to investigate consistency amongst the various particle counters before initiation of monitoring. Strong correlations were confirmed (e.g., R2 ≥ 0.94 for NanoTracer vs. TSI 3787 CPC and NanoTracer vs. TSI P-Trak CPC37).
| Location | Dates sampled | Area (m2) | Description |
|---|---|---|---|
| a Rm – room; Cor – corridor; R/D – receiving and delivering area. | |||
| Rm1 | May 08–10, Dec 03–07 | 22.0 | Clean laboratory (HEPA system in ceiling) with benches and a sink lining walls |
| Rm2 | May 10–16, Nov 26–29 | 8.7 | Regular laboratory (without HEPA) with laminar fumehood |
| Cor | Apr 26–30, Nov 29 to Dec 03 | 160.4 | A corridor (88.1 m long and 1.82 m wide) |
| Rm3 | Apr 23–26, Dec 07–11 | 12.7 | Combined office/laboratory environment |
| Rm4 | May 03–07, Jan 08–13 | 16.9 | A storage room containing floor-to-ceiling shelving |
| R/D | Apr 30 to May 03, Jan 02–04 | 90.5 | A receiving and delivering area with loading dock and service entrance |
2.2. Point source study
3. Results and discussion
3.1. Background study
| Day (12 h) | Night (12 h) | 24 h | ||||
|---|---|---|---|---|---|---|
| Avea | Stdevb | Ave | Stdev | Ave | Stdev | |
| a Average values. b Standard deviation. | ||||||
| Number concentration (# cm−3) | 2.4 × 103 | 5.1 × 102 | 2.0 × 103 | 4.8 × 102 | 2.2 × 103 | 5.1 × 102 |
| Total surface area concentration (μm2 cm−3) | 10.1 | 3.9 | 8.6 | 3.7 | 9.5 | 3.9 |
| Total mass concentration (μg m−3) | 8.5 | 2.3 | 4.4 | 1.1 | 6.5 | 2.1 |
Typical particle number concentrations (10–300 nm) for the ground floor of the test building are in the range of 2.0–2.4 × 103 # cm−3 based on an overall average of all six locations (Table 3). These are comparable to reported background NP values for other indoor environments (2.4 × 103 # cm−3)40 and office workplaces (1.5–7.5 × 103 # cm−3),5 and are low compared to NP exposures from cooking (∼5 × 103 to 2 × 105 # cm−3), second-hand smoking (∼1 × 104 # cm−3), and average outdoor ambient background concentrations (8 × 103 # cm−3).40
Background total surface area concentrations during the day and night in the six locations averaged 10.1 μm2 cm−3 and 8.6 μm2 cm−3 respectively (Table 3). Again, these values are low compared to outdoor ambient particle surface areas reported for an urban background site (50–70 μm2 cm−3) and an outdoor site near a freeway (100–150 μm2 cm−3),41 and much lower than values reported in a residential kitchen during cooking activities (135–2000 μm2 cm−3), inside an automotive engine plant (250–2000 μm2 cm−3)23 and in a pizzeria (200–20000 μm2 cm−3).6
Background measurements of total PM mass concentrations averaged 8.5 μg m−3 for the six locations (Table 3). The corresponding PM1 and PM2.5 averages were 5.5 μg m−3 and 5.6 μg m−3, respectively (not shown in Table 3), which were 10–20 times lower than PM1 or PM2.5 levels reported for indoor environments with cooking sources.6,42 In summary, daytime averages exceeded nighttime averages for all metrics, notably total PM which decreased to 4.4 μg m−3 at night (Table 3).
| Location | Time | First peak | Second peak | ||
|---|---|---|---|---|---|
| Peak location (nm) | Peak height (dN/dlogDp, # cm−3) | Peak location (nm) | Peak height (dN/dlogDp, # cm−3) | ||
| a Bold number indicates the modes with larger peak height values. | |||||
| Rm1 | Day | — | — | — | — |
| Night | — | — | — | — | |
| Rm2 | Day | 27 | 1.5 × 103 | 98 | 1.4 × 103 |
| Night | 19 | 2.7 × 102 | 106 | 1.2 × 103 | |
| Cor | Day | 79 | 3.7 × 103 | 106 | 3.7 × 103 |
| Night | 74 | 3.4 × 103 | 106 | 3.7 × 103 | |
| Rm3 | Day | 29 | 1.2 × 103 | 136 | 5.0 × 102 |
| Night | 39 | 7.1 × 102 | 136 | 5.0 × 102 | |
| Rm4 | Day | 33 | 3.7 × 102 | 109 | 6.8 × 102 |
| Night | 33 | 2.3 × 103 | 109 | 5.9 × 102 | |
| R/D | Day | 30 | 8.9 × 103 | 71 | 5.8 × 103 |
| Night | 21 | 1.8 × 103 | 50 | 5.6 × 103 | |
| Location/time | Nano particles | Fine particles | Coarse particles | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10–20 nm | 20–50 nm | 50–100 nm | 100–500 nm | 500–1000 nm | 1000–5000 nm | 5000–10000 nm |
10000–20000 nm |
||||||||||
| C (# cm−3) | % | C (# cm−3) | % | C (# cm−3) | % | C (# cm−3) | % | C (# cm−3) | % | C (# cm−3) | % | C (# cm−3) | % | C (# cm−3) | % | ||
| a C – particle number concentration; Rm – room; R/D – receiving and delivering area; % – percentage of the total particle number concentration. The bold number indicates the main particle size distribution sections. | |||||||||||||||||
| Rm1 | Day | 7.6 × 10−2 | 4.4 | 5.2 × 10−1 | 30.1 | 1.0 × 100 | 58.5 | 8.6 × 10−2 | 5.0 | 2.9 × 10−2 | 1.7 | 5.1 × 10−3 | 0.3 | 0 | 0.0 | 0 | 0.0 |
| Night | 4.0 × 10 −1 | 39.4 | 2.6 × 10−1 | 25.6 | 2.5 × 10−1 | 24.6 | 8.1 × 10−2 | 8.0 | 1.4 × 10−2 | 1.4 | 1.0 × 10−2 | 1.0 | 0 | 0.0 | 0 | 0.0 | |
| Rm2 | Day | 9.2 × 101 | 8.3 | 4.7 × 102 | 42.3 | 3.3 × 102 | 29.7 | 1.1 × 102 | 10.3 | 1.0 × 102 | 9.2 | 2.9 × 100 | 0.2 | 7.5 × 10−2 | 0.0 | 0 | 0.0 |
| Night | 9.9 × 101 | 9.2 | 3.4 × 10 2 | 31.2 | 3.1 × 102 | 28.4 | 2.2 × 102 | 20.8 | 1.1 × 102 | 10.3 | 1.1 × 100 | 0.1 | 4.8 × 10−3 | 0.0 | 0 | 0.0 | |
| Cor | Day | 2.5 × 101 | 1.9 | 2.2 × 102 | 16.7 | 4.5 × 102 | 34.2 | 5.0 × 102 | 38.1 | 1.2 × 102 | 9.0 | 8.8 × 10−1 | 0.1 | 5.0 × 10−3 | 0.0 | 0 | 0.0 |
| Night | 1.7 × 101 | 1.7 | 1.2 × 102 | 12.0 | 3.9 × 102 | 39.2 | 3.9 × 102 | 39.2 | 7.8 × 101 | 7.8 | 6.8 × 10−1 | 0.1 | 2.5 × 10−3 | 0.0 | 0 | 0.0 | |
| Rm3 | Day | 5.3 × 102 | 23.2 | 1.1 × 103 | 49.5 | 2.5 × 102 | 11.0 | 2.7 × 102 | 11.7 | 9.5 × 101 | 4.2 | 9.5 × 100 | 0.4 | 0 | 0.0 | 0 | 0.0 |
| Night | 1.2 × 102 | 4.0 | 9.1 × 102 | 29.2 | 7.0 × 102 | 22.7 | 8.9 × 102 | 28.5 | 4.7 × 102 | 15.0 | 2.0 × 100 | 0.6 | 8.1 × 10−3 | 0.0 | 0 | 0.0 | |
| Rm4 | Day | 5.0 × 102 | 13.1 | 1.5 × 103 | 39.7 | 1.0 × 103 | 27.2 | 6.3 × 102 | 16.6 | 1.3 × 102 | 3.4 | 1.6 × 100 | 0.0 | 1.8 × 10−2 | 0.0 | 0 | 0.0 |
| Night | 6.7 × 101 | 1.9 | 1.1 × 103 | 29.9 | 1.3 × 103 | 37.3 | 9.0 × 102 | 25.3 | 2.0 × 102 | 5.6 | 1.0 × 100 | 0.0 | 1.5 × 10−3 | 0.0 | 0 | 0.0 | |
| R/D | Day | 8.1 × 102 | 8.1 | 4.2 × 103 | 41.9 | 2.4 × 103 | 24.2 | 1.6 × 103 | 17.6 | 7.9 × 102 | 7.9 | 3.1 × 101 | 0.3 | 7.8 × 10−1 | 0.0 | 5.9 × 10−3 | 0.0 |
| Night | 1.7 × 102 | 3.2 | 2.1 × 103 | 40.0 | 1.9 × 103 | 36.2 | 6.2 × 102 | 12.0 | 3.9 × 102 | 7.5 | 5.4 × 101 | 1.1 | 2.8 × 10−2 | 0.0 | 0 | 0.0 | |
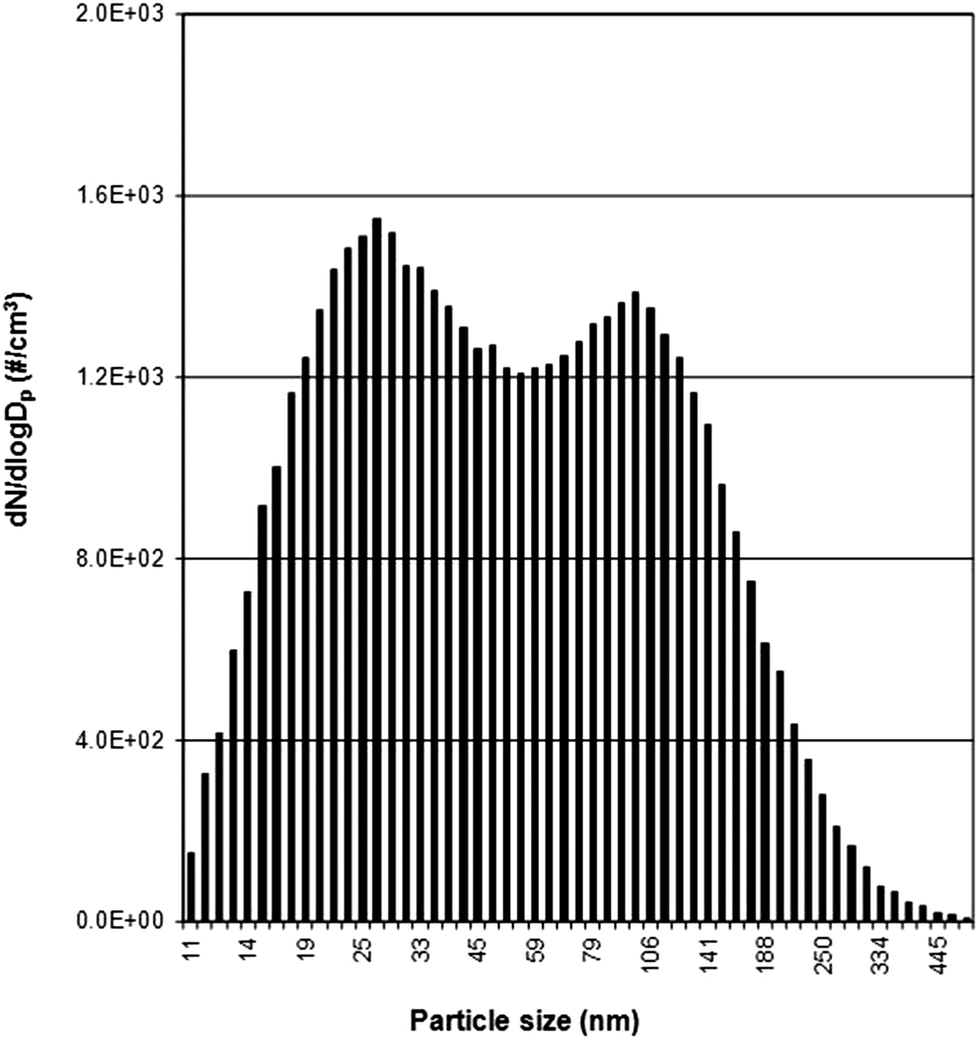 | ||
| Fig. 1 Example of bimodal particle size distribution of the background signal monitored using TSI SMPS (Rm2 winter sample is shown here; see Table 4 for other locations). | ||
Table 4 summarizes the bimodal peak locations and peak heights in the different locations, and shows that the bimodal maxima vary from day to night, with daytime peak values being generally higher than night time peak values. Higher values during the day are related to increased human activity and equipment usage during working hours, consistent with the well-documented “personal cloud” effect.45Table 4 also indicates large spatial variability, particularly in the first peak of the bimodal background signal which varies by more than an order of magnitude from Rm4 (3700 at 33 nm) to the R/D location (89000 at 30 nm) during the day. The origin of the bimodal size distributions could be partly explained by the nucleation of new particles leading to the first peak in the size distribution. Ageing processes then lead to the second peak, which is the accumulation mode. This might be the reason for the much higher peak at 30 nm in the R/D area (e.g. due to vehicle emissions).
Table 5 provides additional information about NP size distributions at different locations. Size distributions in the fine (100–1000 nm) and coarse (1–20 μm) particle ranges were also recorded to complete the background size spectra. Data provided include the daytime and night time averages for three nano-specific ranges (10–20 nm, 20–50 nm and 50–100 nm), two fine size ranges (100–500 nm, 500–1000 nm), and three coarse size ranges (1000–5000 nm, 5000–10000 nm and 10000–20000 nm). Note that the NPs (sum of the three nano-specific ranges) contribute 53–93% of the total indoor background particle number concentrations, while very fine (100–500 nm) and fine particle ranges (500–1000 nm) make smaller contributions: 5–39% and 1.4–15%, respectively (Table 5). Minor numbers of coarse particles from 1000–5000 nm were detected in indoor room environments, contributing <0.05–0.4% and <0.05–1.1% for day and night time, respectively. Particles larger than 5000 nm were negligible (<0.01%; Table 5). With respect to day-to-night variations, NPs (<100 nm) contribute 61–89% during the day and 50–76% at night, respectively. Thus it can be concluded that particles in the nano size range dominate the indoor background aerosol when particle number concentrations are used as the metric.
| Mass (mg m−3) | Day (12 h) | Night (12 h) | 24 h | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Avea | Stdevb | %c | Ave | Stdev | % | Ave | Stdev | % | |
| a Average values. b Standard deviation. c Percentages, calculated using PM10 as the total. | |||||||||
| <PM1 | 5.55 | 1.13 | 78.4 | 4.21 | 0.90 | 95.7 | 4.88 | 1.26 | 85.5 |
| PM1 → PM2.5 | 0.08 | 0.02 | 1.1 | 0.04 | 0.01 | 0.9 | 0.06 | 0.02 | 1.0 |
| PM2.5 → RESP | 0.21 | 0.05 | 3.0 | 0.05 | 0.01 | 1.1 | 0.14 | 0.03 | 2.5 |
| RESP → PM10 | 1.24 | 0.39 | 17.5 | 0.10 | 0.03 | 2.3 | 0.63 | 0.19 | 11.0 |
Due to the limitations of the direct-reading mass measurement instruments, the precise contribution of NPs (<100 nm) to the total background mass concentration is unknown. Previous work using filter-based gravimetric methods in Rm2 showed that, of the total particle mass (16 μg m−3), fine and ultrafine particles (10–560 nm) contributed 73% and coarse particles (1–10 μm) contributed 27%.17,25,46
| Location | Number (# cm−3) | Surface area (μm2 cm−3) | Mass (μg m−3) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Meana | stdev | Peak | Meana | stdev | PM1a | stdev | PM2.5a | stdev | PM10a | stdev | |
| a 24 h average concentration; stdev – standard deviation; Rm – room; Cor – corridor and R/D – receiving and delivering area. | |||||||||||
| Rm1 | 2.6 × 102 | 1.7 × 102 | 6.4 × 102 | 2.1 | 1.9 | 0.02 | 0.13 | 0.02 | 0.13 | 0.02 | 0.13 |
| Rm2 | 8.8 × 102 | 2.6 × 102 | 1.6 × 103 | 5.1 | 2.6 | 2.75 | 0.62 | 2.82 | 0.64 | 3.09 | 0.78 |
| Cor | 8.8 × 102 | 2.9 × 102 | 2.1 × 103 | 3.3 | 2.2 | 1.61 | 0.91 | 1.66 | 0.98 | 2.18 | 1.87 |
| Rm3 | 2.2 × 103 | 8.0 × 102 | 4.6 × 103 | 9.9 | 4.5 | 4.45 | 1.03 | 4.46 | 1.04 | 4.47 | 1.05 |
| Rm4 | 3.2 × 103 | 6.0 × 102 | 5.5 × 103 | 17.7 | 4.6 | 7.89 | 1.73 | 7.92 | 1.74 | 7.96 | 1.76 |
| R/D | 6.3 × 103 | 1.7 × 103 | 1.4 × 104 | 20.1 | 6.6 | 11.68 | 6.92 | 11.95 | 7.13 | 14.20 | 11.01 |
Variations of particle number concentrations in spring sampling versus winter sampling are shown in Fig. 2a for daytime and Fig. 2b for night. Although day and night trends are similar, number concentrations at night are generally lower than daytime concentrations, especially in spring at locations R/D and Rm3. R/D is a busy location during the day (Fig. 2a) with frequent human activities that increase infiltration of outdoor air (receiving/delivering from a sliding garage door, and entry and exit of personnel from a regular entrance door). The corresponding night time value for R/D is much lower (Fig. 2b) when such activities are at minimum. The difference between day and night in this area is a good example of the influence of human activities and outdoor air infiltration on indoor NP background values.
 | ||
| Fig. 2 Comparison of spatial variations of the background particle number concentrations using NanoTracer between spring (■) and winter (○) for day (a) and night (b). Error bars = 1 std dev for 12 h. | ||
Fig. 2 shows that particle number concentrations in spring were higher than in winter (spring > winter) for Rm3, Rm4 and R/D. However, the reverse trend (spring ≤ winter) was observed for Rm1, Rm2 and Cor (Fig. 2). Researchers have reported5,40 that indoor particle number concentrations are usually directly related to outdoor particle number concentration, and that outdoor concentrations during winter are generally lower.47 This reported seasonal trend (spring > winter) is most reflected by Rm3, Rm4 and R/D as indicated in Fig. 2, suggesting that these areas are most influenced by outdoor air. The observation of a reverse trend (spring ≤ winter) for Rm1, Rm2 and Cor reflects the influence of environmental controls in the two positive pressure labs (Rm1 and Rm2) and in the corridor area receiving the lab air (Cor). Particle number concentrations at these locations are more strongly influenced by the air handling systems within the building, and in the case of Rm1 (HEPA system in ceiling) diurnal and seasonal change is minimal.
Fig. 3 and 4 illustrate temporal and spatial variations in surface area and mass, respectively, monitored simultaneously with particle number (Fig. 2) at the same six locations. Similar to Fig. 2, surface area values are 3–7 times lower in winter than in spring for Rm3, Rm4 and R/D, with values for R/D as high as 22.9 μm2 cm−3 during the day and 17.2 μm2 cm−3 at night. Fig. 4 shows that spatial and seasonal variations of PM1 (Fig. 4a and b) and total PM (Fig. 4c and d) bear similarities to the trends shown in Fig. 2 and 3.
 | ||
| Fig. 3 Comparison of spatial variations of the background active surface areas using DC2000CE between spring (■) and winter (○) for day (a) and night (b). | ||
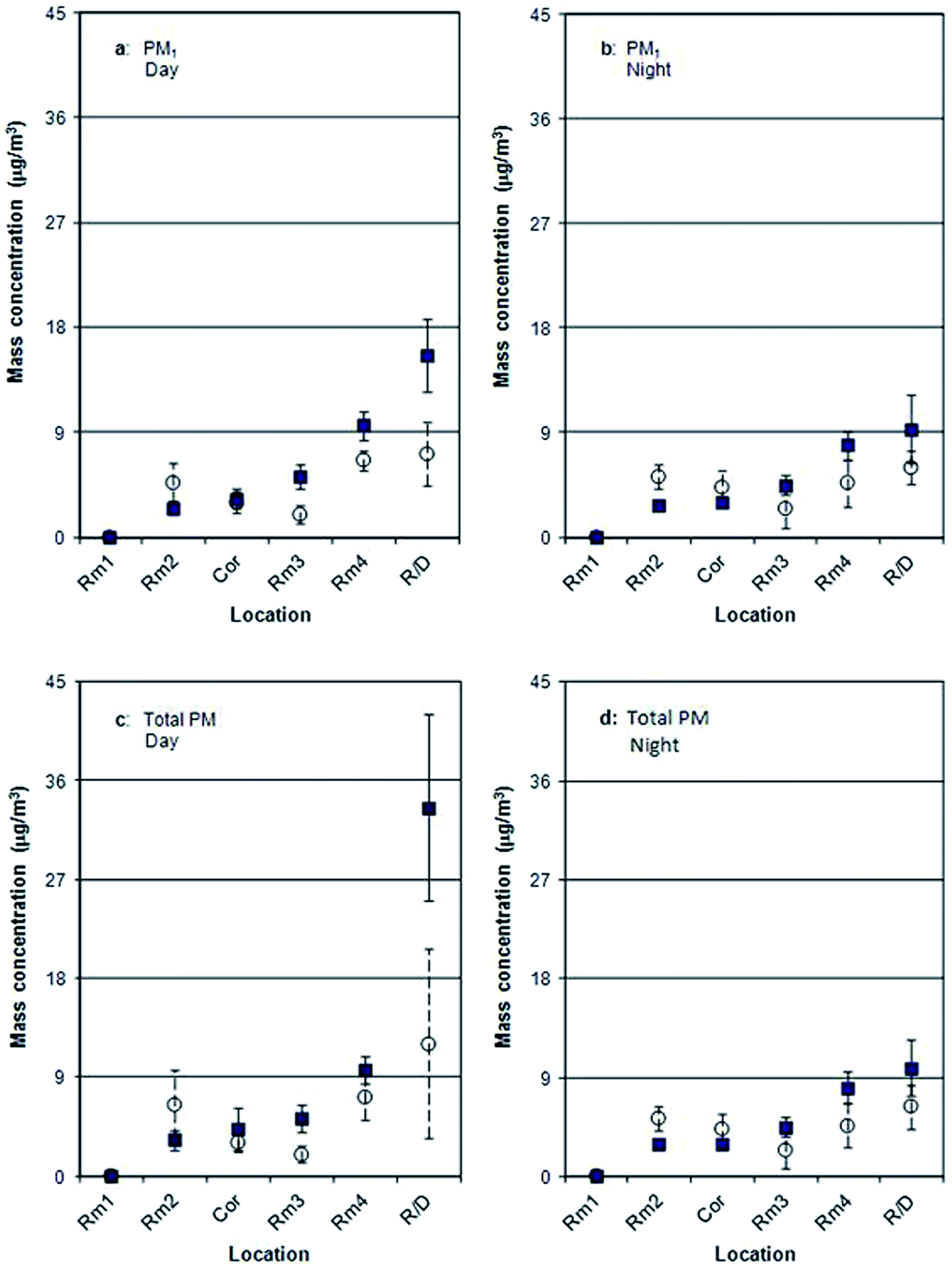 | ||
| Fig. 4 Comparison of spatial variations of the background PM1 (a and b) and the total mass (c and d) concentrations using DustTrak between spring (■) and winter (○) for day (a and c) and night (b and d). | ||
These results indicate that average values and standard deviations of background NP concentrations in the study building are dependent upon such factors as diurnal and seasonal variations, proximity to building entranceways such as the receiving area and loading dock, the degree of environmental controls in different workplace locations (e.g. laboratories versus office areas), and human activities. It is important to understand these background characteristics in order to select an appropriate exposure assessment approach that will be able to distinguish releases of engineered nano-objects from background. As indicated in CSA (2012) guidance,15 such indoor environmental factors need to be considered when making background corrections in exposure assessments.
Based on observations for background number, surface area and mass (Fig. 2–4), the lowest and least variable values were observed in Rm1, which can be attributed to the greater environmental controls in this room. The evidence of time trends observed in other locations (e.g. the increase from winter to spring in Rm3 and Rm4) indicates that an evaluation based only on mean and standard deviation is not sufficient and that time series data should be provided as well. Average and standard deviation values for particle number concentrations (e.g., Tables 3 and 7) do not provide adequate information about temporal trends in variability. Fig. 5 provides an example of a time series over a 24 h period in Rm2, a location that was characterized in Fig. 2 as being one of the least-variable locations. Despite being a positive pressure lab with environmental controls, Rm2 displays large diurnal variability and even successive midnight values show a three-fold variation (from 900 to 300 # cm−3; Fig. 5). Surface area and mass (total PM) monitored simultaneously in the same location also displayed large diurnal variability (from 3 to 37 μm2 cm−3 and from 8 to 25 μg m−3, respectively; not shown here). No occupants used the room during the monitoring process, and there were no other identifiable causes for the variations. Such complicated and unpredictable temporal variations indicate the value of collecting time series data to characterize background nanoparticles in exposure assessments.
 | ||
| Fig. 5 Diurnal variations of background particle number concentrations recorded using NanoTracer in Rm2 (spring sample; see Fig. 2 for other locations and seasons). | ||
3.2. Point source study: distinguishing point source emissions (printer example) from background
Fig. 6 shows the particle number size distribution of emissions during continuous printing by a laser printer in the full scale chamber. The recorded particle size distribution ranges from 40–280 nm (mainly within 45–180 nm) with modes at 79 nm and 109 nm, respectively. Note that Fig. 6 represents only a “snapshot” of the size distribution which continuously changes due to particle coagulation and loss processes. Also, based on previous work,35 it should be noted that the printer model employed in this study is classified as a high emitting printer and that the magnitude of printer emissions varies according to make and model. Therefore these results cannot be generalized to other printer emissions.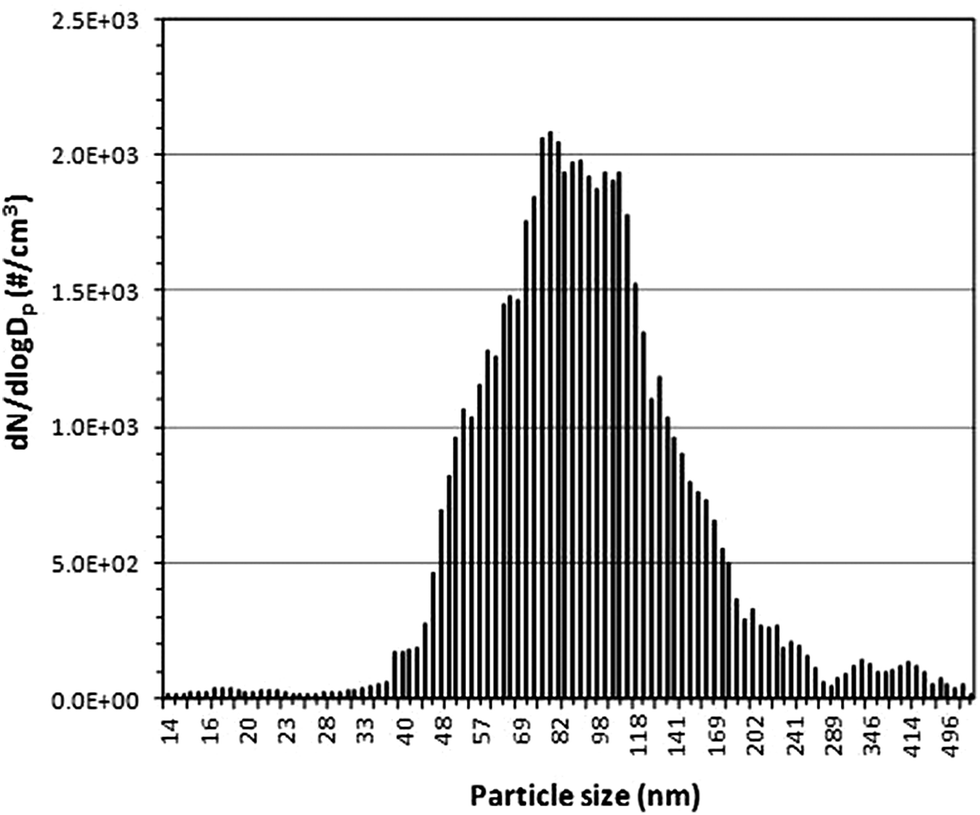 | ||
| Fig. 6 Particle size distribution of emissions from the HP LaserJet model 4200 dtn during continuous printing recorded in the full scale chamber using TSI SMPS. | ||
Fig. 7 shows the time profile for emissions from printing a single page using the same laser printer, quantified using the NanoTracer. (It was not possible to capture the particle size distribution of a single print emission due to the longer response time of the SMPS used in this study.) The NanoTracer data in Fig. 7 were collected in the chamber where the background particle count averaged 209 # cm−3 (compared to the much higher background values in the real-world example shown in Fig. 5; 1000–2000 # cm−3 during daytime).
 | ||
| Fig. 7 Particle number concentrations emitted from a HP LaserJet model 4200 dtn printer for a single printing recorded in the full scale chamber using NanoTracer in fast scan mode (data logging every 3 s). Background average is 209 # cm−3. | ||
Fig. 7 indicates that, inside the chamber, the peak number concentration is about 400 times higher than the chamber background, showing clear distinguishability between the tested printer emissions and background signals. Had the single-page printer emission experiment (Fig. 7) been conducted in the non-HEPA laboratory background environment shown in Fig. 5, the emission peak to background peak (e-peak/b-peak) ratio would be 54, large enough to distinguish source emissions from the background. Actually, the e-peak/b-peak ratios are at least one to two orders of magnitude in five out of the six tested locations, i.e. 134×, 54×, 41×, 19×, and 16× for Rm1, Rm2, Cor, Rm3, and Rm4, respectively (referring to the peak values listed in Table 7). The e-peak/b-peak ratio is about 6× in the R/D location. Thus, even taking into account the complex temporal variations, it would be possible to distinguish this printer emission from the background at each of the six locations due to the large e-peak/b-peak ratios.
4. Conclusions
This study provided information about the variability of the background aerosol that will assist in designing real-time measurement strategies (e.g., duration and frequency of monitoring, monitoring locations) for future assessments of exposure to airborne engineered NPs. It shows that, depending on location in the building, time series data may be required to characterize diurnal variations and longer-term trends (such as seasonal variations) in addition to shorter-term average and standard deviation values.Simultaneous monitoring of particle size, number, mass and surface area using multiple direct-reading instruments (e.g., SMPS, CPC, APS, DC and mass monitors) provides information about the full size range of background aerosols (nano- to micron-scale) and is a recommended strategy for monitoring both NP background and point source exposures.
The particle number size distribution results show that incidental NPs are a major contributor to the background, accounting for 53–93% of the total number concentrations. The size distributions at the tested workplaces were bimodal with the first mode located within the 21–79 nm range and second mode within the 70–136 nm range, depending on location.
The point source study indicates some limitations of the existing direct-reading instrumentation. First, the response time of the SMPS is too slow to capture rapid particle size distribution changes in NP emissions, such as laser printer emissions when printing a single page. The time resolution of particle counters (e.g. CPC and NanoTracer) is sufficient to monitor rapid changes in total particle number concentrations (as shown in Fig. 7). Therefore, both types of instruments are recommended for emission monitoring as they provide complementary information. Second, the direct-reading instruments used in this study do not have the capacity to distinguish amongst different types of particles and therefore cannot directly identify emission sources. In this study, an older model laser printer emitted NPs at peak concentrations 6 to 134 times background in the studied locations and thus were distinguishable on the basis of number concentration. However, lower emissions from more recent printers are likely to be indistinguishable, requiring off-line chemical analysis and/or electron microscopy to characterize particle components as necessary to identify sources.
Acknowledgements
We gratefully acknowledge the assistance of F. Muhammad, C. Levesque and L. Lavigne-Brunette. We are grateful for constructive comments and suggestions from two anonymous reviewers, and for helpful reviews of an early draft version by Drs I. Jayawardene and L. Avramescu. This project was funded by Health Canada's Chemicals Management Plan, Clean Air Regulatory Agenda, and Environmental Health Sciences Research Bureau.References
- D. Brouwer, M. Berges, M. A. Virji, W. Fransman, D. Bello, L. Hodson, S. Gabriel and E. Tielemans, Harmonization of measurement strategies for exposure to manufactured nano-objects; report of a workshop, Ann. Occup. Hyg., 2012, 56, 1–9 CrossRef PubMed.
- T. A. J. Kuhlbusch, C. Asbach, H. Fissan, D. Göhler and M. Stintz, Nanoparticle exposure at nanotechnology workplaces: A review, Part. Fibre Toxicol., 2011, 8, 1–18 CrossRef PubMed , http://www.particleandfibretoxicology.com/content/8/1/22.
- M. Methner, L. Hodson and C. Geraci, Nanoparticle emission assessment technique (NEAT) for the identification and measurement of potential inhalation exposure to engineered nanomaterials – Part A, J. Occup. Environ. Hyg., 2009, 7, 127–132 CrossRef PubMed.
- ISO (International Organization for Standardization), Nanotechnologies: terminology and definitions for nano-objects—nanoparticle, nanofibre and nanoplate, ISO/TS 27687, Geneva, Switzerland, 2008 Search PubMed.
- P. McGarry, L. Morawska, R. Jayaratne, M. Falk, Q. Tran and H. Wang, Exposure to particles from laser printers operating within office workplaces, Environ. Sci. Technol., 2011, 45, 6444–6452 CrossRef CAS PubMed.
- G. Buonanno, L. Morawska, L. Stabile and A. Viola, Exposure to particle number, surface area and PM concentrations in pizzerias, Atmos. Environ., 2010, 44, 3963–3969 CrossRef CAS PubMed.
- L. A. Wallace, F. Wang, C. Howard-Reed and A. Persily, Contribution of gas and electric stoves to residential ultrafine particle concentrations between 2 nm and 64 nm: size distributions and emissions and coagulation rates, Environ. Sci. Technol., 2008, 42, 8641–8647 CrossRef CAS.
- B. Nowack and T. D. Bucheli, Occurrence, behavior and effects of nanoparticles in the environment, Environ. Pollut., 2007, 150, 5–22 CrossRef CAS PubMed.
- J. Niu, P. E. Rasmussen, N. M. Hassan and R. Vincent, Concentration distribution and bioaccessibility of trace elements in nano and fine urban airborne particulate matter: influence of particle size, Water, Air, Soil Pollut., 2010, 213, 211–225 CrossRef CAS.
- T. A. J. Kuhlbusch, S. Neumann and H. Fissan, Number size distribution, mass concentration, and particle composition of PM1, PM2.5, and PM10 in bag filling areas of carbon black production, J. Occup. Environ. Hyg., 2004, 1, 660–671 CrossRef CAS PubMed.
- NIOSH, Approaches to Safe Nanotechnology: Managing the Health and Safety Concerns Associated with Engineered Nanomaterials, DHHS (NIOSH) Publication No. 2009–125, March 2009.
- D. H. Brouwer, J. H. J. Gijsbers and M. W. M. Lurvink, Personal exposure to ultrafine particles in the workplace: exploring sampling techniques and strategies, Ann. Occup. Hyg., 2004, 48, 439–453 CrossRef CAS PubMed.
- M. Methner, L. Hodson, A. Dames and C. Geraci, Nanoparticle emission assessment technique (NEAT) for the identification and measurement of potential inhalation exposure to engineered nanomaterials – Part B: results from 12 field studies, J. Occup. Environ. Hyg., 2009, 7, 163–176 CrossRef PubMed.
- C. Asbach, T. A. J. Kuhlbusch, H. Kaminski, B. Stahlmecke, S. Plitzko, U. Götz, M. Voetz, H. J. Kiesling and D. Dahmann, NanoGEM Standard Operation Procedures for assessing exposure to nanomaterials, following a tiered approach, 2012, http://www.nanogem.de/cms/nanogem/upload/Veroeffentlichungen/nanoGEM_SOPs_Tiered_Approach.pdf Search PubMed.
- CSA Group, Nanotechnologies – exposure control program for engineered nanomaterials in occupational settings, Z12885-12, Sept. 2012 Search PubMed.
- OECD, Emission assessment for the identification of sources and release of airborne manufactured nanomaterials in the workplace: compilation of existing guidance ENV/JM/MONO 16, 2009 Search PubMed.
- J. Niu, P. E. Rasmussen and F. Muhammad, Characterization of airborne nanoparticles using a combination of in situ direct-reading and filter-based mass measurement strategies, 7th International Conference on the Environmental Effects of Nanoparticles and Nanomaterials, P15, Banff, Alberta, Sept. 10–12, 2012 Search PubMed.
- M. M. Dahm, D. E. Evans, M. K. Schubauer-Berigan, M. E. Birch and J. A. Deddens, Occupational exposure assessment in carbon nanotube and nanofiber primary and secondary manufacturers: mobile direct-reading sampling, Ann. Occup. Hyg., 2013, 57, 328–344 CrossRef CAS PubMed.
- S. Y. Paik, D. M. Zalk and P. Swuste, Application of a pilot control banding tool for risk level assessment and control of nanoparticle exposures, Ann. Occup. Hyg., 2008, 52, 419–428 CrossRef CAS PubMed.
- A. D. Maynard and R. J. Aitken, Assessing exposure to airborne nanomaterials: Current abilities and future requirements, Nanotoxicology, 2007, 1, 26–41 CrossRef CAS.
- R. Duffin, C. L. Tran, A. Clouter, D. M. Brown, W. MacNee, V. Stone and K. Donaldson, The importance of surface area and specific reactivity in the acute pulmonary inflammatory response to particles, Ann. Occup. Hyg., 2002, 46, 242–245 CrossRef.
- G. Oberdörster, J. Ferin and B. E. Lehnert, Correlation Between Particle-Size, In-Vivo Particle Persistence, and Lung Injury, Environ. Health Perspect., 1994, 102, 173–179 CrossRef.
- W. A. Heitbrink, D. E. Evans, B. K. Ku, A. D. Maynard, T. J. Slavin and T. M. Peters, Relationships among particle number, surface area, and respirable mass concentrations in automotive engine manufacturing, J. Occup. Environ. Hyg., 2008, 6, 19–31 CrossRef PubMed.
- J. Niu, P. E. Rasmussen, F. Muhammad, C. Levesque and M. Chénier, From micro to nano: Instrumentation for measurement of airborne particles in the workplace, 2011 Health Canada Science Forum, J-1.44, Ottawa, Ontario, Nov. 7–8, 2011 Search PubMed.
- J. Niu, P. E. Rasmussen and B. G. Fraser, Characterization of nanoparticles in the indoor environment: Collection strategy, size distributions and element signatures, 2010 Health Canada Science Forum, J-3.18, Ottawa, Ontario, Nov. 1–2, 2010 Search PubMed.
- G. Ramachandran, D. Paulsen, W. Watts and D. Kittelson, Mass, surface area and number metrics in diesel occupational exposure assessment, J. Environ. Monit., 2005, 7, 728–735 RSC.
- F. E. Pfefferkorn, D. Bello, G. Haddad, J.-Y. Park, M. Powell, J. Mccarthy, K. L. Bunker, A. Fehrenbacher, Y. Jeon, M. A. Virji, G. Gruetzmacher and M. D. Hoover, Characterization of exposures to airborne nanoscale particles during friction stir welding of aluminum, Ann. Occup. Hyg., 2010, 54, 486–503 CrossRef CAS PubMed.
- D. E. Evans, W. A. Heitbrink, T. J. Slavin and T. M. Peters, Ultrafine and respirable particles in an automotive grey iron foundry, Ann. Occup. Hyg., 2008, 52, 9–21 CrossRef PubMed.
- W. A. Heitbrink, D. E. Evans, T. M. Peters and T. J. Slavin, The characterization and mapping of very fine particles in an engine machining and assembly facility, J. Occup. Environ. Hyg., 2007, 4, 341–351 CrossRef CAS PubMed.
- A. Elder, R. Gelein, V. Silva, T. Feikert, L. Opanashuk, J. Carter, R. Potter, A. Maynard, Y. Ito, J. Finkelstein and G. Oberdörster, Translocation of inhaled ultrafine manganese oxide particles to the central nervous system, Environ. Health Perspect., 2006, 114, 1172–1178 CrossRef CAS.
- A. Wiebel, R. Bouchet, F. Boulch and P. Knauth, The big problem of particle size: a comparison of methods for determination of particle size in nanocrystalline anatase powers, Chem. Mater., 2005, 17, 2378–2385 CrossRef.
- P. E. Rasmussen, I. Jayawardene, D. Gardner, M. Chénier, L. Christine and J. Niu, Metal impurities provide useful tracers for identifying exposures to airborne single-wall carbon nanotubes released from work-related processes, J. Phys.: Conf. Ser.,, 2013, 429, 012007, DOI:10.1088/1742-6596/429/1/012007.
- J. Niu, P. E. Rasmussen and M. Chénier, Ultrasonic dissolution for ICP-MS determination of trace elements in lightly loaded airborne PM filters, Int. J. Environ. Anal. Chem., 2013, 93, 661–678 CrossRef CAS.
- H. Destaillats, R. L. Maddalena, B. C. Singer, A. T. Hodgson and T. E. McKone, Indoor pollutants emitted by office equipment: a review of reported data and information needs, Atmos. Environ., 2008, 42, 1371–1388 CrossRef CAS PubMed.
- C. He, L. Morawska and L. Taplin, Particle emission characteristics of office printers, Environ. Sci. Technol., 2007, 41, 6039–6045 CrossRef CAS.
- J. Niu, P. E. Rasmussen and B. G. Fraser, Exposure to nanoparticles and associated metals in the office environment: concentration distributions and laser printer emissions, HECSB Science, Policy and Regulatory Symposium-2010, Health Canada, Ottawa, Ontario, Canada, March 23, 2010 Search PubMed.
- J. Niu, P. E. Rasmussen, R. Magee and G. Nilsson, Monitoring strategy for characterization of airborne nanoparticles, Proceedings of NanoTech-Advanced Materials and Applications (NanoTech 2014), Washington DC, USA, June 15–18, 2014, vol. 1, no. 488, 495–496 Search PubMed.
- National Research Council Canada, Material Emissions Laboratory – Full-Scale Test Chamber, http://www.nrc-cnrc.gc.ca/eng/solutions/facilities/material_emissions.html.
- BAM Federal Institute for Materials Research and Testing, Germany. Achromatic test chart with area coverage 5% (900 kByte, 504 pages), Emissions from office equipment with print functions according to RAL-UZ122:2006-04, http://www.ps.bam.de/RALUZ122/RALA00.PDF.
- L. Wallace and W. Ott, Personal exposure to ultrafine particles, J. Exposure Sci. Environ. Epidemiol., 2011, 21, 20–30 CrossRef CAS PubMed.
- L. Ntziachristos, A. Polidori, H. Phuleria, M. D. Geller and C. Sioutas, Application of a diffusion charger for the measurement of particle surface, Aerosol Sci. Technol., 2007, 41, 571–580 CrossRef CAS.
- G. Buonanno, L. Morawska and L. Stabile, Particle emission factors during cooking activities, Atmos. Environ., 2009, 43, 3235–3242 CrossRef CAS PubMed.
- Y. Zhu, W. C. Hinds, M. Krudysz, T. Kuhn, J. Froines and C. Sioutas, Penetration of freeway ultrafine particles into indoor environments, Aerosol Sci., 2005, 36, 303–322 CrossRef CAS PubMed.
- T. Tritscher, M. Beeston, A. F. Zerrath, S. Elzey, T. J. Krinke, E. Filimundi and O. F. Bischof, NanoScan SMPS – A novel, portable nanoparticle sizing and counting instrument, J. Phys.: Conf. Ser., 2013, 429, 012061, DOI:10.1088/1742-6596/429/1/012061.
- USEPA, Review of the national ambient air quality standards for particulate matter: Policy Assessment of Scientific and Technique Information, EPA-452/R-05-005a, December 2005, ch. 2.7, 2-68–2-69 Search PubMed.
- P. E. Rasmussen, J. Niu and H. D. Gardner, Characterization of airborne nanoparticles using filter-based methods: metals and gravimetric analysis, Tri-National Workshop on Standards for Nanotechnology, National Research Council, Ottawa, Canada, Feb 3–4, 2010. http://download.isiglobal.ca/inms/enter.html Search PubMed.
- C. O. Stanier, A. Y. Khlystov and S. N. Pandis, Ambient aerosol size distributions and number concentrations measured during the Pittsburgh air quality study (PAQS), Atmos. Environ., 2004, 38, 3275–3284 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |