
Mercury methylation and demethylation in highly contaminated sediments from the Deûle River in Northern France using species-specific enriched stable isotopes
Baghdad
Ouddane
*a,
Mathilde
Monperrus
b,
Milada
Kadlecova
ac,
Mirna
Daye
ad and
David
Amouroux
b
aUniversité, Lille 1, Equipe de Physico-Chimie de l'Environnement, UMR CNRS LASIR 8516, Bat. C8 2° étage, 59655 Villeneuve d'Ascq Cedex, France. E-mail: baghdad.ouddane@univ-lille1.fr; Fax: +33 320434822; Tel: +33 320434481
bUniversité de Pau et des Pays de l'Adour, Laboratoire de Chimie Analytique Bio-Inorganique et Environnement, Institut des Sciences Analytiques et de Physico-chimie pour l'Environnement et les Matériaux, IPREM UMR CNRS 5254, Hélioparc, 2, Avenue Pierre Angot, 64053 Pau Cedex, France
cBrno University of Technology, Institute of Chemistry and Technology of Environmental Protection, Faculty of Chemistry, Purkynova 118, 612 00 Brno, Czech Republic
dLebanese University, Water & Environmental Sciences Laboratory, Tripoli, Lebanon
First published on 20th October 2014
Abstract
The methylation–demethylation processes in sediments of the Deûle River were determined using well-established isotope experiments. For this purpose, species-specific isotopically enriched tracers in the form of inorganic mercury IHg (199Hg) and methylmercury MeHg (Me201Hg) were used to determine Hg dynamics in the Deûle River. Sediment cores were collected at two sampling locations chosen in the most polluted zone of the Deûle River (Northern France) in proximity of a Zn, Pb, Cu, and Ni smelter called “Metaleurop” that had closed in 2003. Site I was chosen in the vicinity of the historic smelter site and site II upstream of the Deûle River. The incubation was realized directly in the sediment cores during the 24 hour experiment under environmental conditions close to the real natural systems (the same temperature, pH, humidity, light/dark conditions, oxygen levels…). The enriched isotopes were injected by needle into different sections of the core. After incubation, the core was sliced and the concentration of Hg species was determined in each section. The highest methylation potentials were found at sediment depths away from the sediment–water-interface. At site I, the methylation potential varied between 0.02–0.9% and at site II between 0.001–0.2%. The demethylation potentials fluctuated between 0.001–60% at site I and between 4–53% at site II. In both sites, negative net methylation potentials were obtained in several sediment depths, representing a net sink for MeHg. The average net methylation potential in site I demonstrated a negative value of 1919 ng g−1 day−1. It seems that in site I the demethylation process predominates methylation. Whereas, in site II, the average net methylation potential was a positive value of 138 ng g−1 day−1, demonstrating the dominance of methylation over demethylation.
Environmental impactMercury reactivity has been investigated in highly contaminated sediments from the Deûle River (Northern France) using species-specific enriched stable isotope tracers. Such experiments can allow the measurements of methylation and demethylation potentials and therefore specify important information about the production and the consumption of different mercury species. Moreover, important biogeochemical factors controlling methylmercury production and degradation in the Deûle River can be closely examined. The study of Hg dynamics in the Deûle River can provide important information essential for the implementation of environmental policies and decontamination strategies in order to control the toxicity and the bioaccumulation of Hg through the food chain. |
1. Introduction
The overall toxicity of mercury (Hg) in aquatic systems is controlled by mercury dynamics of methylation, demethylation and volatilization. Methylmercury (MeHg) is a severe environmental pollutant, because of its potent neurotoxicity and its bioaccumulative character.1 MeHg can enter the food chain first at the base of planktons or benthic fauna and then biomagnifies within the chain to higher trophic levels.2 Understanding the major factors controlling methylation and demethylation is essential in order to control the concentrations and the accumulation of MeHg in sediments. The methylation process is primarily controlled by Sulfate Reducing Bacteria (SRB).3 Whereas demethylation can be abiotically driven by photochemical reduction or biotically mediated via oxidative and reductive pathways.4 Several methods exist for the determination of Hg species transformations. The first Hg transformation studies have used spikes with high concentrations of inorganic mercury (IHg) of natural isotopic composition to determine methylation and demethylation potentials of Hg.5 Later on, radiochemical procedures were used to determine Hg dynamics by spiking with 203Hg.6,7 The concentrations of the Hg radiotracer must be added at least 10 times higher than the ambient mercury concentration. This can influence the microbial community and the selection of resistant strains. Moreover, methylation and demethylation yields cannot be determined simultaneously requiring separate samples. The radiotracer method was improved by the use of high specific activity 203Hg, which allowed low Hg concentrations in the spikes.8Furthermore, developments in the technique have led to significant improvements in the determination of stable isotopes by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). The response of such a technique is based on the precise determination of the Hg isotopic ratio rather than the total metal concentration. Various double-spiking approaches were developed over the last few years based on the addition of enriched mercury species (199Hg and 201MeHg) to the sample in order to simultaneously determine methylation and demethylation rates.9–11 By quantifying all Hg isotopes, methylation and demethylation potentials can be simultaneously determined. Many studies have used this technique coupled to Gas Chromatography (GC) instrumentation, with spikes corresponding to low concentrations of isotopic Hg tracers.10,12,13 The double-spiking methodology was demonstrated to present limitations when employed for the accurate measurement of IHg and MeHg particularly when they are present in a relatively different concentration in the sample.14
Spike addition of Hg isotopes (concentration and type of spiking solution) and incubation times are among the most important parameters influencing MeHg formation and degradation yields.15 Moreover, isotopic tracer experiments permit the simultaneous tracking of both endogenous and exogenous Hg species transformations and the determination of the possible contribution of Hg species to the biogeochemical cycle and to the bioaccumulative process.16 Later on, analytical methods were developed for the determination of Hg isotopic composition in different matrices (fish, sediment, water…) and during different biogeochemical processes (methylation, demethylation and volatilization processes).17,18 Isotopic fractionation of different Hg species was simultaneously determined within the same sample by the hyphenation of Gas Chromatography (GC) with Multicollector-Inductively Coupled Plasma Mass Spectrometry (MC-ICP-MS).19,20
The region of Northern France (Nord Pas-de-Calais) is highly populated and industrialized by metallurgical activities. One of the most important smelters in the region “Metaleurop”, which was localized at the banks of the Deûle River, had perturbed the natural balance of the ecosystem significantly. Metaleurop smelter had refined lead, zinc, copper, antimony, indium, germanium, gold, silver and cadmium. Metaleurop smelter was active for more than a century and was the major contributor of metal pollution to the river. Fortunately, Metaleurop was closed in January 2003; however untreated or not well-refined ore wastes are disposed in the former smelter location, certainly contributing to continuous metal pollution to the soil and to the surrounding river system. Hg pollution in the site is found as a by-product in sulfide-rich ore. Consequently, high quantities of various metals including Hg are easily eroded and leached to the water body by surficial runoffs of the contaminated sites. A recent study on Hg contamination in the Deûle River had shown important Hg pollution in sediments near the Metaleurop site with a mean THg of 10 μg g−1, a mean MeHg of 4.5 ng g−1 and a mean% MeHg of 0.05.21 Consequently, the investigation of Hg methylation and demethylation in the Deûle River is substantial to discover the extent and factors affecting net methylation potentials. The study of Hg dynamics in the Deûle River can provide important information essential for the implementation of environmental policies and decontamination strategies in order to control the toxicity and the bioaccumulation of Hg through the food chain.
The major objectives of this work were: (1) to investigate Hg reactivity in highly contaminated sediments from the Deûle River (Northern France) using species-specific enriched stable isotope tracers, in two sampling locations near the ancient sites of Metaleurop smelter: site I (downstream the river) and site II (upstream the river), and (2) to evaluate important biogeochemical factors controlling methylmercury production and degradation in the Deûle River. Such experiments can allow the measurements of methylation and demethylation potentials and therefore provide important information about the production and the consumption of different Hg species.
2. Methods and materials
2.1. Reagents and standards
Mercury standard solution of 199Hg(II) (91.71%) and Me201Hg (96.5%) solution was used in this study. The Hg standard enriched isotopes were purchased from ISc Science (Oveido, Spain). Stock standard solutions of 199IHg and Me201Hg (1000 mg L−1) were prepared by dissolving mercury chloride in 1% HNO3 and methylmercury chloride in methanol. Working standard solutions were prepared fresh daily by appropriate dilution of stock standard solution in 1% HCl and stored in the dark at 4 °C. A 0.1 M acetic acid–acetate buffer solution (pH 4.7) (CH3COONa: Merck Darmstadt, Germany; CH3COOH: Scharlau Extrapur) was prepared. A solution of 1% tetraethyl borate prepared for ethylation reactions (98%, Stream Chemicals, Newburyport, USA). All reagents were of analytical grade. Ultrapure water was obtained from Milli-Q system (Quantum EX, Millipore, USA).2.2. Field sampling
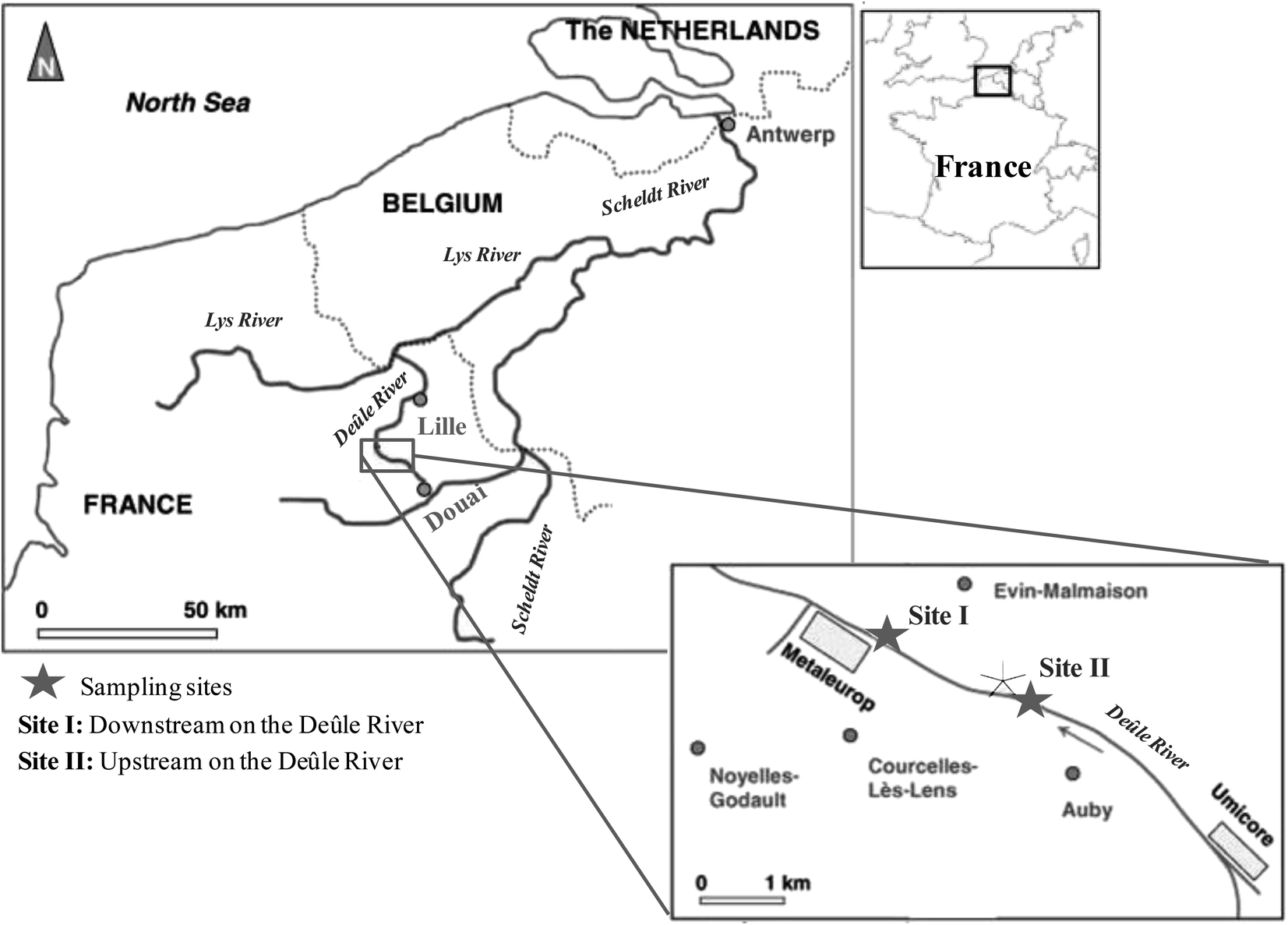 | ||
| Fig. 1 Locations of sampling sites along the Deûle River. Site I: downstream of the Deûle River and site II: upstream of the Deûle River. | ||
2.3. Determination of methylation and demethylation potentials
Hg methylation and demethylation potentials were determined from a single analysis by spiking the same sample with mercury enriched isotopic tracers (199Hg(II), Me201Hg). Sediment samples were spiked with aqueous solutions of isotope enriched 199Hg(NO3)2 (91.97%) to 10–40% of THg and Me201HgCH3COO (96.49%) to 0.1% of THg concentration. Duplicates in analysis were made for each sediment layer. For accuracy in measurements, each spiked sample is divided into two parts. One part is immediately frozen at −20 °C representing t0. The other part of the sample is tightly sealed in an argon atmosphere and incubated for 24 hours in darkness and at bottom water temperature (10 °C). After 24 hours, the incubation was stopped by adding high purity HCl (1% v/v) and the sample was frozen at −20 °C, representing t2. t0 represents the theoretical initial concentration of 199Hg(II) and Me201Hg and t2 represents the recovered concentrations of 199Hg(II) and Me201Hg after various transformations. The determination of the concentrations of 199Hg(II) and Me201Hg at t0 and t2 allows calculation of methylation and demethylation potentials. Three isotopes should be measured to determine the amount of methylated and demethylated mercury, Me199Hg representing the newly produced Me199Hg from 199Hg, 201Hg representing the demethylation of Me201Hg, and 200Hg representing the changes in ambient 200Hg. Methylation and demethylation potentials were calculated by isotope reverse dilution calculations.15The potential of Hg formation and degradation potentials were deducted based on the initial concentrations of Hg isotopically enriched species (t0) and on the newly formed and degraded Hg isotopes after 24 hours of incubation time (t2). The methylation potential is calculated from the amount of newly formed Me199Hg found after incubation in a system containing 199Hg according to eqn (1). The demethylation potential is determined based on the decrease of the added Me201Hg found after incubation according to eqn (2). The net methylation potential is calculated according to eqn (3).
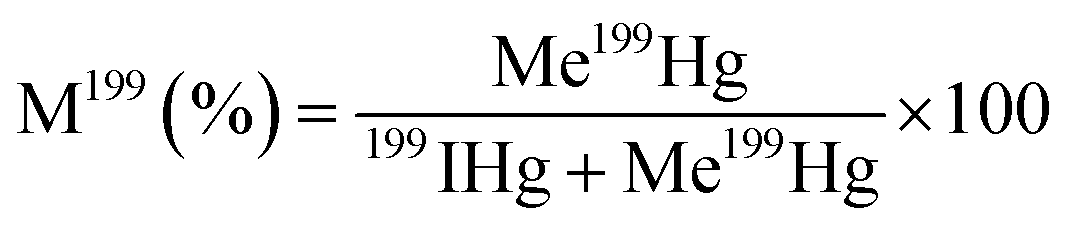 | (1) |
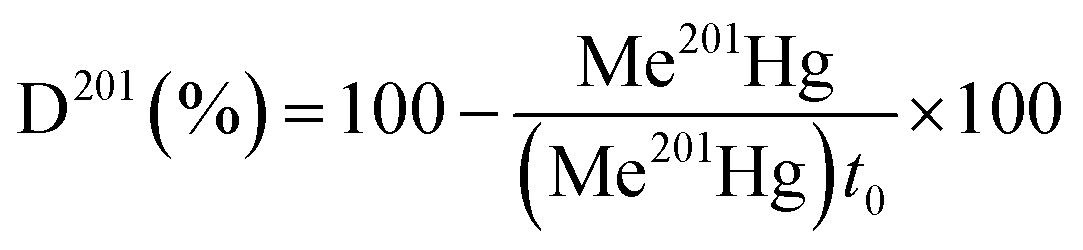 | (2) |
| Net methylation (ng per g per day) = (methylation potential (per day) × [IHg]ambient (ng g−1)) − (demethylation potential (per day) × [MeHg]ambient (ng g−1)) | (3) |
The relative standard deviation of the recovered 199Hg(II) spike was 3% and of the formed Me199Hg was 21%. The relative standard deviation of the recovered Me201Hg spike was 21% and of the formed 201Hg(II) was 5%. Normally, 199Hg(II) spike recovery should be less than 199Hg(II) theoretically spiked for methylation to occur and Me201Hg spike recovery should be less than Me201Hg theoretically spiked for demethylation to occur. However, higher recoveries of 199Hg(II) and Me201Hg than the theoretical spike were observed in some sediment depth. This higher recovery of Hg isotopic tracers can be explained by a redistribution of Hg species by bioirrigation driven by benthic organisms found in the core, substantially affecting the fluxes of the sediment–water interface.
2.4. Determination of isotopically enriched mercury species in sediments
The determination of mercury species was performed according to the procedure described by Monperrus et al. (2008).14 The samples were analyzed by Gas Chromatography (Trace GC, Thermo Element) coupled to Inductively Coupled Plasma Mass Spectrometry (ICPMS X7, Thermo Element). The gas chromatography was equipped with a capillary column MXT-1 (crossbond 100% dimethylplysiloxane 30 m, id 0.53 mm and 1 mm coating). The absolute detection limit based on three standard deviations of the digestion blank was 0.02 ng g−1 and 0.05 ng g−1 for MeHg and Hg(II), respectively. Precision was based on the relative standard deviation of five replicates of certified reference materials (IAEA 405-estuarine sediment and BCR 580-estuarine sediment) and was 2.1% for MeHg and 1.8% for Hg(II). The accuracy of the analytical results was checked by analyzing international certified materials of IAEA 405 (MeHg 5.5 ± 0.5 ng g−1; IHg: 808.7 ± 40.1 ng g−1) and BCR 580 (MeHg: 75.5 ± 3.7 ng g−1; IHg 131.9 ± 3 μg g−1). Speciation analysis of IAEA 405 and BCR 580 gave 5.8 ± 0.4 ng g−1 and 83.5 ± 4 ng g−1 for MeHg concentrations, respectively and the IHg concentration of 800 ± 140 ng g−1 and 131 ± 3 μg g−1, respectively. Therefore, there were no statistically significant differences with obtained experimental and certified values.2.5. Analysis of ambient mercury species
Total mercury analysis in dry sediment samples (THg) was performed without any pre-treatment by using an atomic absorption spectrometer Advanced Mercury Analyzer; model AMA 254 (Altec Ltd, Czech Republic). The detection limit for AMA 254 based on 100 mg of sample was 0.1 μg kg−1. The relative standard deviation of six replicate measurements was 1.2%. For precision objectives, certified reference materials were analyzed including IAEA 405 (0.750 ± 0.06 μg g−1), IAEA-433 (0.155 ± 0.01 μg g−1) and IAEA-158 (0.121 ± 0.013 μg g−1). The obtained values of THg in IAEA 405 (estuarine sediment), IAEA 433 (marine sediment), and IAEA 158 (marine sediment) were 0.81 ± 0.003 μg g−1, 0.168 ± 0.017 μg g−1, and 0.132 ± 0.014 μg g−1, respectively. Therefore, there were no statistically significant differences with obtained experimental and certified values. MeHg in sediments was determined as proposed by Kadlecova et al.,22 by aqueous ethylation followed by Headspace (HS) injection, Gas Chromatography separation (Clarus 500, PerkinElmer, USA), and detection by Atomic Fluorescence Spectroscopy (Tekran, Model 2600 CVAFS Mercury Analysis System, USA). The detection limit of HS-GC-CVAFS was 1.12 ng kg−1. The accuracy was examined by international reference materials (IAEA-405, IAEA-433). The obtained values of certified reference materials (IAEA-405: 5.85 ± 0.45 ng Hg g−1, IAEA-433: 0.18 ± 0.09 ng Hg g−1) presented no statistically significant differences with the certified values (IAEA-405: 5.49 ± 0.53 ng Hg g−1, IAEA-433: 0.17 ± 0.07 ng Hg g−1). Precision is given by the relative standard deviation of 5 replicate analyses and was determined to be 4.8%.2.6. Sulfur analysis
The inorganic sulfides in the sediment are generally grouped into two main categories: unstable sulfides that are freshly precipitated (AVS: Acid Volatile Sulfides) and the more stable sulfides, mainly pyrite and elemental sulfur (CRS: Chromium Reducible Sulfur). The reduced sulfur species is determined by sequential extraction as described previously by Canfield et al.23 Manipulations are performed in a glove bag under an inert atmosphere (N2). About 1 g of sediment is reacted with 40 mL of 6 mol L−1 hydrochloric acid, then the volatilized sulfide in the form of H2S is purged by N2 flow and trapped in 20 mL of a basic solution ([NaOH] 1 mol L−1, [EDTA] 1 mol L−1). CRS (Chromium Reducible Sulfides) sequentially follows AVS, and is extracted by adding about 40 mL of 1 mol L−1 chromium II solution, produced in the column of Jones and HCl solution. Again the volatile sulfur is trapped in 20 mL of a basic solution ([NaOH] 1 mol L−1 [EDTA] 1 mol L−1). The sulfide concentration is then determined for each trap by potentiometry using an automatic titrator (Metrohm, model 736 GP Titrino). The titration is carried out with a cadmium solution of 8.9 × 10−3 mol L−1, a calomel reference electrode (Hg/Hg2Cl2, [KCl] = 3 mol L−1) and a measuring electrode specific to sulfide ions (Orion). The accuracy of the two methods is determined to be <8%. The lower limit of determination of 1 g of sediment was 20 μg g−1 of S.3. Results
3.1. Ambient Hg species distribution
Mean values of ambient IHg and MeHg were calculated from duplicate cores and the relative standard deviation was 3–5% and 20–23%, respectively for the two sites. Mercury speciation analyses were conducted for both sites and the ambient concentrations of mercury in sediment are shown in Fig. 2. At site I, the sediments of the Deûle River have shown statistically significant lower concentration of THg between 5330–11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 660 ng g−1 than site II of values between 8510–19604 ng g−1 (Fig. 2a and d). Similarly, natural IHg at site II showed statistically significant higher values of 13517 ± 1327 ng g−1 than site I of 9292 ± 296 ng g−1 (Fig. 2b and e). Conversely, site I demonstrated statistically significant higher mean concentration of MeHg 210 ± 49 ng g−1 as compared to site II of a mean concentration of MeHg 75 ± 15 ng g−1 (Fig. 2b and e). The average value of methylmercury percentage (% MeHg/THg) is 2% for site I and 0.6% for site II. For site I, IHg depth profiles fluctuated between 5323–11658 ng g−1. While MeHg varied between 64–1007 ng g−1 with maximal concentrations occurring deeper at 15 cm in the sediment, slightly decreasing at higher depth values (Fig. 2b). As for site II, IHg concentrations were almost homogeneous until 15 cm sediment depth with values ranging 8507–1100 ng g−1, doubling at depth profiles from 20 to 30 cm to 19604 ng g−1 (Fig. 2e). Whereas, MeHg concentrations varied between 35–208 ng g−1, where maximal concentrations were found at sediment surface of 3 cm, then the MeHg profile decreased significantly with depth and remained constant (Fig. 2e).
660 ng g−1 than site II of values between 8510–19604 ng g−1 (Fig. 2a and d). Similarly, natural IHg at site II showed statistically significant higher values of 13517 ± 1327 ng g−1 than site I of 9292 ± 296 ng g−1 (Fig. 2b and e). Conversely, site I demonstrated statistically significant higher mean concentration of MeHg 210 ± 49 ng g−1 as compared to site II of a mean concentration of MeHg 75 ± 15 ng g−1 (Fig. 2b and e). The average value of methylmercury percentage (% MeHg/THg) is 2% for site I and 0.6% for site II. For site I, IHg depth profiles fluctuated between 5323–11658 ng g−1. While MeHg varied between 64–1007 ng g−1 with maximal concentrations occurring deeper at 15 cm in the sediment, slightly decreasing at higher depth values (Fig. 2b). As for site II, IHg concentrations were almost homogeneous until 15 cm sediment depth with values ranging 8507–1100 ng g−1, doubling at depth profiles from 20 to 30 cm to 19604 ng g−1 (Fig. 2e). Whereas, MeHg concentrations varied between 35–208 ng g−1, where maximal concentrations were found at sediment surface of 3 cm, then the MeHg profile decreased significantly with depth and remained constant (Fig. 2e).
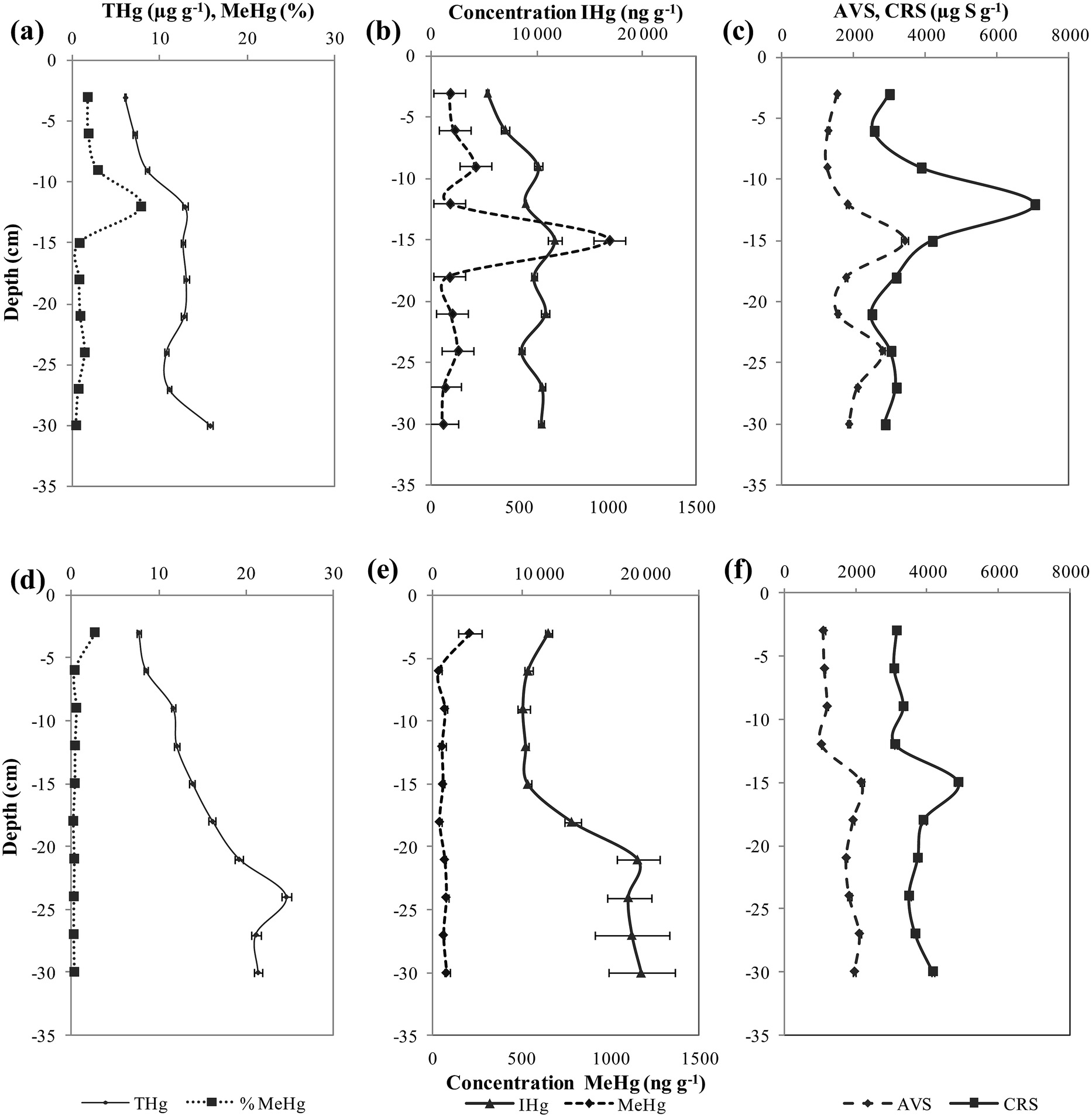 | ||
| Fig. 2 Vertical depth distribution of Hg species (IHg, MeHg) and sulfides (AVS, CRS) in sediment cores of site I (a–c) and II (d–f). | ||
In this study, an insignificant correlation between THg and MeHg was observed for site I while in site II, good association was found (r2 = 0.7, p < 0.01, n = 8). When MeHg contents in both sites were normalized to THg, good associations were detected between % MeHg and THg in site I (r2 = 0.6, p < 0.01, n = 9), with a statistically insignificant correlation observed in site II.
In site I, a relationship was remarked between % MeHg and CRS (r2 = 0.8, p < 0.001, n = 10) with an insignificant relationship with AVS. Whereas in site II, an opposite trend was determined with a statistically good relationship between % MeHg and AVS (r2 = 0.4, p < 0.05, n = 9) and a lack of correlation with CRS.
3.2. Methylation and demethylation yields
The measurement of formed Me199Hg and 201Hg(II) during the 24 hour incubation period permits the calculation of methylation and demethylation potentials. The precision of methylation and demethylation potentials calculated from standard deviation between two cores is calculated to be 0.02–0.05% and 6–10%, respectively for both sites.At site I, the methylation potential varied between 0.02 and 0.9% with an average value of 0.2 ± 0.03% (Fig. 3a). The maximal methylation potential of 0.9% occurred at a sediment depth of 12 cm corresponding to maximal concentrations of AVS and CRS (Fig. 2c). At site II, less significant methylation potentials are observed fluctuating between 0.001 and 0.19% with an average value of 0.1 ± 0.02% (Fig. 3c). When methylation potentials are plotted against ambient % MeHg, significant correlations were found for site I (r2 = 0.8, p < 0.001, n = 10) (Fig. 4a). However, irrelevant relationships were observed between methylation potentials and ambient % MeHg for site II. Likewise in site I, a correlation was found between in situ MeHg and the amount of Me199Hg produced (r2 = 0.8, p < 0.001, n = 10) (Fig. 4b) with the lack of relationship between these two parameters in site II. This demonstrates that site I is more active with respect to Hg transformations than site II.
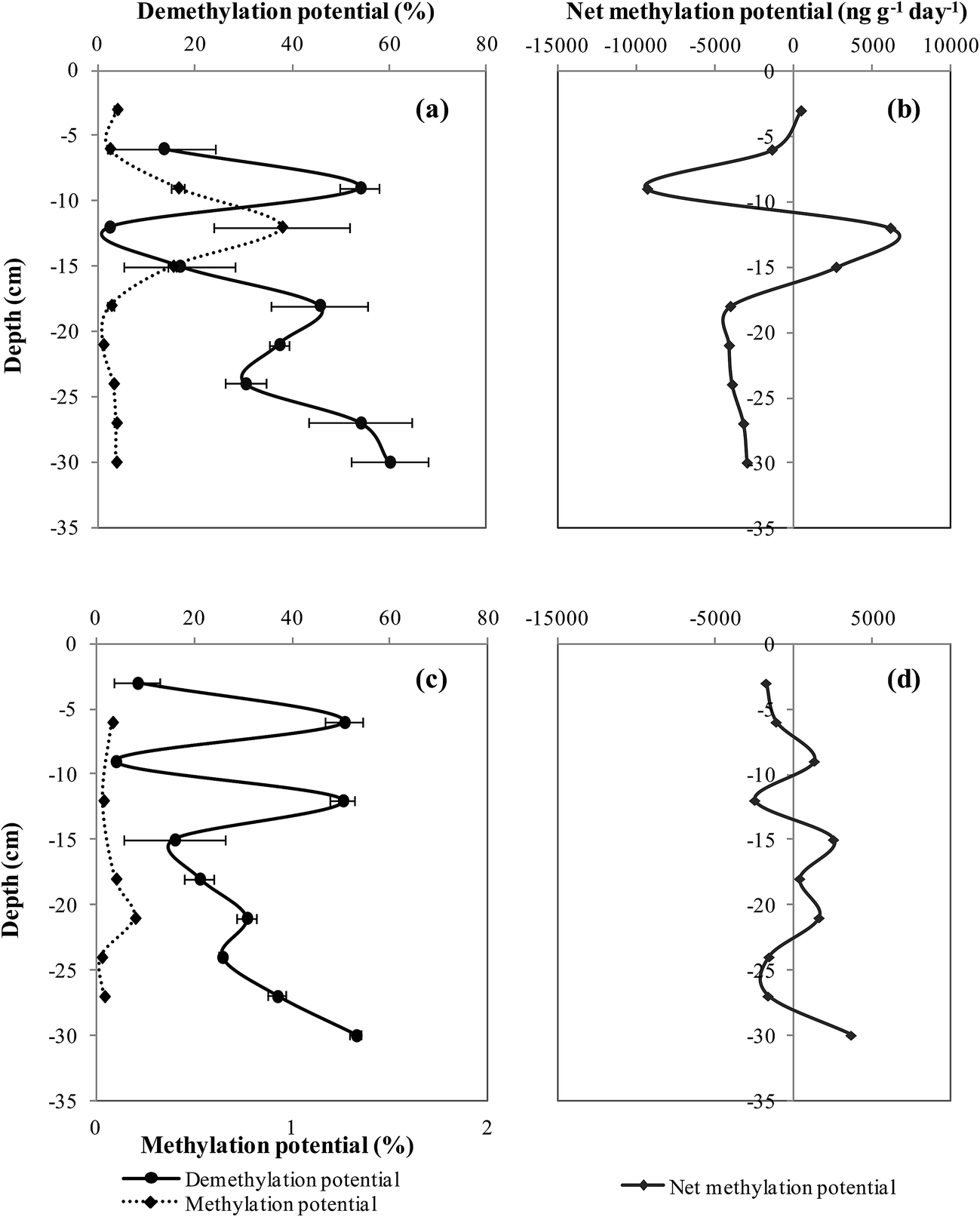 | ||
| Fig. 3 Vertical depth distribution of Hg methylation, demethylation and net methylation potentials in site I (a and b) and site II (c and d). | ||
 | ||
| Fig. 4 Relationships between (a) methylation potential and % MeHg in site I, (b) % MeHg and Me199Hg in site I, and (c) CRS and methylation potential in site II and II. | ||
When methylation potentials were plotted against AVS, insignificant correlations were found for both sites. Whereas, positive correlations of the methylation potential with CRS were found for site I (r2 = 0.96, p < 0.001, n = 10) and site II (r2 = 0.6, p < 0.001, n = 10) (Fig. 4c). Therefore, sulfides particularly CRS control Hg methylation processes.
In addition to methylation, MeHg concentrations can also be affected by the simultaneous process of MeHg demethylation. Demethylation may significantly affect the increase of MeHg concentrations. The demethylation potential vertical profiles in both sites have followed variable profiles. In site I, the demethylation potential varied between 0.001–60% with an average value of 31 ± 8% (Fig. 3a). The high value of the demethylation potential for site I was found around 10 cm sediment depth and higher values of 60% were observed deeper than 18 cm sediment depth. At site II, the demethylation potential fluctuated between 4–53% with an average value of 29.6 ± 6% (Fig. 3c). Maximal values of demethylation of 50% occurred at sub-surface sediment around 6 cm and 12 cm sediment depths. Sediments deeper than 15 cm had an increasing trend of demethylation. Demethylation potentials and % MeHg profiles followed opposite trends in both sites in which demethylation maximal values corresponded to % MeHg minimal values and vice versa. Good associations were established between demethylation potentials and the concentration of THg for site I (r2 = 0.4, p < 0.05, n = 8) and site II (r2 = 0.6, p < 0.05, n = 8). However, when the demethylation potential was correlated with IHg, remarkable associations were found for site I (r2 = 0.8, p < 0.001, n = 9) and site II (r2 = 0.7, p < 0.01, n = 9). The strong correlation observed between demethylation potentials and IHg suggests that the same biogeochemical parameters control IHg availability and demethylation processes.
4. Discussion
4.1. Long-term methylation controls
The high values of MeHg and THg concentrations encountered in the Deûle River are in accordance with other estuaries affected by point source contamination such as chlor-alkali industry in the Lavaca bay24 and the Gulf of Trieste affected by the old Idrijca mine.25,26 Moreover, Hg contents in the Deûle River sediments were comparable to other riverine systems such as the downstream of the Wuli River in China affected by a chlor-alkali plant,27 the Thur River in France affected by a chlor-alkali plant28 and the Nura River in Kazakhstan influenced by an acetaldehyde plant.29 The higher MeHg concentration in site I than site II demonstrates the more reactivity of sediments of site I and the bioavailability of its Hg pool for methylation. The overall partitioning of mercury with different phases, organic matter, sulfides and Fe/Mn oxyhydroxides under oxic and anoxic conditions, respectively controls the concentration of Hg species in the environment and their subsequent transformations.30The % MeHg in sites I and II is found to be similar to the Passamaquoddy Bay of 0.5–1.2%,31 of 1% in the Chesapeake Bay,32 of 0.7% at the bay of Fundy,33 of 0.6% at the Lavaca Bay,24 of 0.02–0.4% at the Tagus estuary,34 of 1.5% in the Idrijca River35 and of 0.12–2.5% in Adour estuary.36 Other estuaries have presented lower % MeHg i.e. at Loire of 0.075–0.13%,37 at Gironde of 0.2% (ref. 38) and at the Seine estuary of 0.2–0.4%.39
The methylation zone at site I is found at deeper zones in contrast to previous studies of near sediment-surface methylation zones (Fig. 2a and b).33,40,41 Higher MeHg concentrations in deeper sediments suggest MeHg burial of historic pollution and its preservation under anoxic conditions. In contrast, site II is characterized by high MeHg concentrations at the water–sediment interface which suggests that methylation is limited to the surface and the microbial activity is the overriding controller of MeHg concentrations (Fig. 2d and e). The prevalence of the methylation zone in site II at the water–sediment interface is similar to other studies.4,13
Previous studies showed significant correlations between THg and MeHg31,39,42,43 and others demonstrated a lack of dependence between THg and MeHg in different ecosystems.44–46 These confusing differences between the studies might be ascribed to intrinsic site characteristics and conditions affecting the bioavailability of mercury. The good association found between % MeHg and THg in site I and the lack of correlation in site II are in agreement with the results of the previous study on the Deûle River where an important relationship was determined for the site downstream the river with irrelevant association determined upstream the river.21 In site I, THg is one of the driving factors of long-term methylation as shown in other studies.41,47 In contrast to site I, the lack of dependency of % MeHg on THg in site II, signifies the potential role of other factors than THg in the prevailing methylation process.
Factors controlling Hg methylation can be grouped into those affecting the bioavailability of Hg (sulfide and organic matter) and those influencing SRB activity (sulfate, organic matter, and temperature). It seems that factors affecting the speciation of mercury have more influential effects on MeHg formation, in which the concentration of dissolved neutral mercury sulfide HgS0 is the major controller of the methylation process. In site I, a remarkable relationship was observed between % MeHg and CRS with an insignificant relationship with AVS (freshly precipitated sulfide). Whereas in site II, an opposite trend was determined with a good relationship between % MeHg and AVS and a lack of correlation with CRS. This is in accordance with the previous study on the Deûle River,21 which demonstrated that site I, the most proximal to the former smelter site, is characterized by the accumulation of metal sulfide minerals (ZnS, CdS…) discharged by the old smelter. This leads to the accumulation of sulfides and their subsequent pyritization during past decades of intensive methylation. This can definitely illustrate the interdependence between % MeHg and CRS which signifies an old event of methylation in site I and the association present between % MeHg and AVS in site II signifying a more recent event of methylation.
4.2. Biogeochemical controls on methylation potentials
The average values of methylation potentials (site I: 0.2% and site II: 0.1%) and demethylation potentials (site I: 31% and site II: 29.6%) are in agreement with values previously reported in Hg-polluted environments.4,26,38Maximum methylation potentials are observed in different depth ranges other than the known methylation zone, i.e. sediment–water interface. This observation is in agreement with other studies.30,33,38 These variations are explained by changes in bacterial activity leading to zonal stratification of SRB genera as a function of depth. Methylation potentials are depth dependent relative on different depth gradients of electron–donor acceptors.48 The maximal methylation depth zone can be altered by environmental variables including bioturbation and Fe(III)/Mn(III)/(IV) reduction that can shift the methylation depth zone to greater depth gradients.
Another explanation for the non-occurrence of the methylation zone near the surface–sediment interface where SRB tends to thrive is the possibility of mercury methylation by other bacterial genera. Iron reducing bacteria (FeRB) have been demonstrated to methylate mercury in nature.49,50 It is shown that the bacterial community composition changes with seasons, with the methylation seizure by SRB during summer and autumn and the overriding control of FeRB of Hg methylation during winter.25 However, at both sites, the highest methylation potentials occurred at the same depth profiles as sulfides (Fig. 2c and f and 3a and c) with good associations established between % MeHg, methylation potentials and sulfides. Therefore, there is a relationship between Hg methylation and sulfides conquering the bioavailability of HgS0. The depth profiles of the methylation potential in both sites are consistent with ambient MeHg and sulfide concentration profiles. It is little confusing whether to consider the results obtained representative for the behavior of the ambient IHg and MeHg or to consider it as a reflection for the methylation and demethylation potentials of sediment obtained under artificial conditions. It seems that the long-term methylation zone differs from the short-term methylation zone in site II. Short-term methylation as determined in the laboratory does not necessarily reflect long-term MeHg build-up in sediments. The significant correlations found between methylation potentials and % MeHg for site I (Fig. 4a) are similar to those established in Ore River estuary,46 Hudson River,51 Patuxent River33 and Florida Everglades.52 Site II was characterized by the absence of significant relationships between methylation potentials and % MeHg as observed in the Bay of Fundy.33 These differences can be explained by the fact that the added Hg is more readily available with the possibility of complex formation of ambient Hg with organic and inorganic ligands. Therefore, ambient mercury in site I is more bioavailable for SRB than site II and isotopic methylation potentials in sediments of site I may reflect the real behavior of ambient Hg.
A significant correlation in site I was found between in situ MeHg and the amount of Me199Hg produced (Fig. 4b) similar to previous studies.31,51 Conversely, in site II, an insignificant correlation was observed between MeHg and Me199Hg. Accordingly, it is assumed that site I is more reactive, and that Hg pool is more available for various biochemical transformations. It is verified by the presence of a relationship between ambient MeHg and Me199Hg, methylation potential and % MeHg and the complete absence of these associations in site II. This further signifies that site I is an Hg reactor showing higher methylation potentials and % MeHg with a total reflection of the isotopic Hg transformations to the real in situ methylation/demethylation processes.
The average methylation potentials are higher for site I than site II with 0.2 and 0.1%, respectively. As a result, the % MeHg differs largely between the two sites. At site I, the average % MeHg is 2% which is 3 times higher than site II of 0.6%. This is attributed to a higher sulfide content (AVS) at site I (1933 μg S g−1) than site II (1613 μg S g−1) and a less IHg concentration in site I (9292 ng g−1) than site II (13517 ng g−1). Thereby, the site is conferred by higher reactivity of sediments towards Hg transformations. Mercury availability for the Hg-methylating microbes is controlled by organic matter, sulfur speciation, microbial activity and the partitioning of mercury between the solid and the dissolved phase. These interdependent factors regulate the MeHg formation in marine sediments.53,54 The insignificant correlations found in both sites between methylation potentials and AVS with an important relationship between methylation potential and CRS (Fig. 4c) suggest the predominance of CRS over AVS on Hg bioavailability. This can be elucidated by the effect of the discharges of mineral sulfides by the old smelter “Metaleurop” contributing to extremely high levels of sulfides (CRS) in both sites. Sulfide levels and organic matter contents are the most important biogeochemical controls affecting mercury methylation.46 In the studied sites, CRS seems to control Hg methylation.
4.3. Biogeochemical controls on demethylation potentials
Demethylation can be abiotically (photochemical or chemical reactions) or biotically mediated. The photochemical degradation of MeHg is the most dominant process of the demethylation mechanism at surface water.55 Other abiotic processes are probable including abiotic MeHg degradation in the presence of reducible Fe(III) citrate.56 The bacterial demethylation mechanism includes two pathways: reduction producing Hg° and oxidation generating Hg(II) and CO2.57,58 Reductive methylation is shown to prevail under contaminated oxic and anoxic conditions57 whereas oxidative demethylation seems to predominate in unpolluted anoxic sediments.58 Ambient MeHg is equivalent to the relative rate of methylation and demethylation processes. Since both sites of the Deûle River have prominent MeHg and THg characteristics of polluted sites, this can certainly trigger the bacterial Mer-system for MeHg degradation and eventual Hg volatilization from the system following a reductive demethylation pathway. In contaminated sites with high THg, demethylation is induced by Mer bacterial enzymes degrading MeHg and reducing Hg(II) into Hg° with ultimate elimination of Hg from the system.59 The vertical profiles of demethylation potentials in both sites are not even but rather variable rates occur within the sediment depth. Thus, there exists a clear dependency between the microbial community composition of the sediment and MeHg degradation. This excludes the abiotic processes and confirms the possible biotic degradation of mercury in these sites.Moreover, when demethylation potentials were plotted against CRS and AVS, no correlations were found. Thereby, sulfides have no control on the demethylation process. Hines et al.25 reported a lack of dependency between SO42− reduction rates and demethylation potentials in winter season and suggested that Hg-resistant bacteria are more implicated in the MeHg reductive degradation. These observations imply that SRB are not important in MeHg degradation in the Deûle River sediments. Therefore, it is noteworthy to investigate bacterial species other than SBR for Hg degradation. In anoxic environments, methanogens and sulfidogens60 are implicated in Hg demethylation. Iron reducing bacteria (FeRB) could be influential demethylators in winter with the over control of SRB in summer season.25 Moreover, MeHg degradation pathways can change from the oxidative process in warm seasons to reductive pathways in winter season.25 It is suggested that FeRB could predominate over SRB for this particular winter season, the time of the sampling campaign of the studied sediment cores. This indicates that Hg-resistant bacteria of iron reducing genera can actively degrade MeHg in a reductive demethylation pathway. Reductive demethylation is demonstrated to be more evident in high Hg concentration systems such as calcine samples as opposed to the oxidative pathway which dominates in the moderately Hg contaminated environment.61 Although, the reductive degradation pathway is a characteristic of Hg polluted environments, it is not necessary to be extremely Hg contaminated since important relationships were exhibited between Gram negative merA genes and the concentration of THg.62 In the Deûle River sediments, strong correlations between demethylation potentials and IHg were observed in both sites, as found in a previous study.58 The IHg pool is shown to be the main trigger of mer-mediated reductive demethylation of MeHg in the Deûle River.
4.4. Net methylation potential
The determination of the net methylation potential is important to identify the dominating process (methylation, demethylation) in a specific sediment depth. In both sites, negative net methylation potentials were obtained in several sediment depths, representing a net sink for MeHg (Fig. 3b and d). Net methylation in site I showed positive values near surface sediment at 3 cm (502 ng g−1 day−1) and higher values at greater depths of 6180 ng g−1 day−1 and 2736 ng g−1 day−1 at 12 and 15 cm, respectively (Fig. 3b). The average net methylation potential in this site I demonstrated a negative value of 1919 ng g−1 day−1. It seems that in site I the demethylation process predominates methylation. The negative methylation potential at site I is due to high demethylation potentials provoked by high Hg concentrations.In site II, net methylation has occurred deeper at 22 cm sediment depth and more frequently at 8, 15, and 22 cm sediment depth than site I (Fig. 3d). The average net methylation potential in site II, is a positive value of 138 ng g−1 day−1, showing the over control of methylation on Hg transformation dynamics. Still, higher values of % MeHg and methylation potential occur in site I, the most proximal to the former site of the Metaleurop smelter. This demonstrates the high reactivity of site I with a potential high accumulation of MeHg corresponding to a long-term MeHg build-up.
In contaminated Hg ecosystems, the high concentration of Hg can trigger the demethylation process at increased rates as observed at site I; the most proximal location to the Hg contamination source. Site II is situated 200 m away from the historic smelter site and upstream the Deûle River, consequently it is considered to be less influenced by Hg contamination than site I. The positive net methylation potential in site II is clear evidence that in less contaminated Hg environments methylation is enhanced. High methylation potentials and MeHg concentrations can be found in pristine environments.63,64 Likewise, high methylation potentials of 22% were observed in the uncontaminated sediments of the Carson River.65
5. Conclusion
High average methylation and demethylation potentials of 0.15% and 30%, respectively are observed in the Deûle River, a characteristic of the Hg-contaminated system. Results have shown that the site, the most proximal to the former smelter (i.e. site I), had higher methylation potentials and % MeHg values than upstream the Deûle River (i.e. site II). Site I is considered as a Hg powerhouse with a bioavailable Hg pool for various transformations. On the other hand, site II was less bioreactive, relative to the scavenging of Hg with potent binding phases e.g. sulfides. Thus, short-term methylation as measured by enriched Hg isotopes may reflect long-term methylation in site I, as opposed to site II where short-term methylation does not reflect the ambient Hg transformations. In both sites, methylation seems to be controlled by sulfate reducing bacteria. The overall MeHg degradation in the Deûle River sediments appears to be controlled by Hg-resistant bacteria FeRB rather than with SRB following a reductive demethylation pathway. Further examinations are deemed to be indispensable to further verify the dominant MeHg degradation pathway in the Deûle River. Average negative net methylation potentials were found to dominate site I, the most proximal to the pollution source while positive values were determined for site II the less affected site by Hg pollution. In highly polluted sites with direct impact on the source of Hg contamination i.e. site I, various processes of Hg transformations are highly stimulated particularly the demethylation mechanism. The presence of high concentration of Hg and sulfides in site I makes it highly active in methylating Hg in the presence of favorable conditions of sulfides and in demethylating Hg in the presence of IHg stimulant of mer genes of the microbial communities. It seems that the environment in the Deûle River is undergoing an auto-recovery with its extraordinary capacity of recycling and eliminating mercury from the aquatic ecosystem to other environmental compartments.Overall, this study is the first assessment of mercury transformation in the Deûle River sediments using isotopically labeled mercury species incubations. Despite the fact that Metaleurop smelter had been closed since 2003, high MeHg concentrations and methylation potentials were found and the bioactive sediments of the Deûle River continue to transform Hg species. Further investigation on methylation potentials of the microbial communities involved and the seasonal variation of mercury dynamics are needed to be explored.
Acknowledgements
This work was financially supported by the Region “Nord Pas-de-Calais” and the “Agence de l'Eau Artois-Picardie”. The authors would like to thank Michel Arold (Agence de l'Eau Artois-Picardie), Christine Grare and Romain Descamps for the samplings and all the technical support and assistance. The help for GC-ICP-MS analysis in LCABIE (UMR 5254) in Université de Pau et des Pays de l'adour was greatly appreciated.References
- D. Mergler, H. A. Anderson, L. H. M. Chan, K. R. Mahaffey, M. Murray, M. Sakamoto and A. H. Stern, Ambio, 2007, 36, 3–11 CrossRef CAS.
- R. A. Lavoie, T. D. Jardine, M. M. Chumchal, K. A. Kidd and L. M. Campbell, Environ. Sci. Technol., 2013, 47, 13385–13394 CrossRef CAS PubMed.
- R. C. Rodriguez Martin Doimeadios, E. Tessier, D. Amouroux, R. Guyoneaud, R. Duran, P. Caumette and O. F. X. Donard, Mar. Chem., 2004, 90, 107–123 CrossRef CAS PubMed.
- M. Monperrus, E. Tessier, D. Point, K. Vidimova, D. Amouroux, R. Guyoneaud, A. Leynaert, J. Grall, L. Chauvaud, G. Thouzeauc and O. F. X. Donard, Estuarine, Coastal Shelf Sci., 2007, 72, 485–496 CrossRef CAS PubMed.
- S. Jensen and A. Jernelov, Nature, 1969, 223, 753–754 CrossRef CAS.
- J. W. Rudd, A. Furutani and M. A. Turner, Appl. Environ. Microbiol., 1980, 40, 777–782 CAS.
- E. T. Korthals and M. R. Winfrey, Appl. Environ. Microbiol., 1987, 53, 2397 CAS.
- C. C. Gilmour and G. S. Riedel, Water, Air, Soil Pollut., 1995, 80, 747–756 CrossRef CAS.
- H. Hintelmann and R. D. Evans, Fresenius' J. Anal. Chem., 1997, 358, 378–385 CrossRef CAS.
- H. Hintelmann, K. Keppel-Jones and R. D. Evans, Environ. Toxicol. Chem., 2000, 19, 2204–2211 CrossRef CAS.
- G. M. M. Rahman and H. M. S. Kingston, Anal. Chem., 2004, 76, 3548–3555 CrossRef CAS PubMed.
- H. Hintelmann, R. Douglas Evans and J. Y. Villeneuve, J. Anal. At. Spectrom., 1995, 10, 619–624 RSC.
- L. Lambertsson, E. Lundberg, M. Nilsson and W. Frech, J. Anal. At. Spectrom., 2001, 16, 1296–1301 RSC.
- M. Monperrus, P. Rodriguez Gonzalez, D. Amouroux, J. I. G. Alonso and O. F. X. Donard, Anal. Bioanal. Chem., 2008, 390, 655–666 CrossRef CAS PubMed.
- P. Rodriguez-Gonzalez, S. Bouchet, M. Monperrus, E. Tessier and D. Amouroux, Environ. Sci. Pollut. Res., 2013, 20, 1269–1280 CrossRef CAS PubMed.
- S. Bouchet, D. Amouroux, P. Rodriguez-Gonzalez, E. Tessier, M. Monperrus, G. Thouseau, J. Clavier, E. Amice, J. Deborde, S. Bujan, J. Grall and P. Anschutz, Biogeochemistry, 2013, 114, 341–358 CrossRef CAS.
- V. Perrot, V. N. Epov, M. V. Pastukhov, V. I. Grebenshchikova, C. Zouiten, J. E. Sonke, S. Husted, O. F. X. Donard and D. Amouroux, Environ. Sci. Technol., 2010, 44, 8030–8037 CrossRef CAS PubMed.
- P. Rodriguez-Gonzalez, V. N. Epov, R. Bridou, E. Tessier, E. Guyoneaud, M. Monperrus and D. Amouroux, Environ. Sci. Technol., 2009, 43, 9183–9188 CrossRef CAS PubMed.
- V. N. Epov, P. Rodriguez-Gonzalez, J. E. Sonke, E. Tessier, D. Amouroux, L. M. Bourgoin and O. F. X. Donard, Anal. Chem., 2008, 80, 3530–3538 CrossRef CAS PubMed.
- V. N. Epov, S. Berail, M. Jimenez-Moreno, V. Perrot, C. Pecheyran, D. Amouroux and O. F. X. Donard, Anal. Chem., 2010, 82, 5652–5662 CrossRef CAS PubMed.
- M. Daye, M. Kadlecova and B. Ouddane, Environ. Sci. Pollut. Res., 2014 DOI:10.1007/s11356-014-3528-x.
- M. Kadlecova, M. Daye and B. Ouddane, Anal. Lett., 2014, 47, 1–10 CrossRef.
- D. E. Canfield, R. Raiswell, J. T. Westrich, C. M. Reaves and R. A. Berner, Chem. Geol., 1986, 54, 149–155 CrossRef CAS.
- N. S. Bloom, G. A. Gill, S. Cappellino, C. Dobbos, L. Mcshea, C. Driscoll, R. Manson and J. Rudd, Environ. Sci. Technol., 1999, 33, 1–13 CrossRef.
- M. E. Hines, E. N. Poitras, S. Covelli, J. Faganeli, A. Emili, S. Zizek and M. Horvat, Estuarine, Coastal Shelf Sci., 2012, 113, 85–95 CrossRef CAS PubMed.
- M. E. Hines, J. Faganeli, I. Adatto and M. Horvat, Appl. Geochem., 2006, 21, 1924–1939 CrossRef CAS PubMed.
- S. Wang, D. Xing, Y. Jia, B. Li and K. Wang, Appl. Geochem., 2012, 27, 343–351 CrossRef CAS PubMed.
- C. Hissler and J. L. Probst, Sci. Total Environ., 2006, 361, 163–178 CrossRef CAS PubMed.
- S. M. Ullrich, M. A. Ilyushchenko, G. A. Uskov and T. W. Tanton, Appl. Geochem., 2007, 22, 2706–2734 CrossRef CAS PubMed.
- K. A. Merritt and A. Amirbahman, Limnol. Oceanogr., 2008, 53, 1064–1075 CrossRef CAS.
- E. M. Sunderland, F. A. P. C. Gobas, B. A. Branfireun and A. Heyes, Mar. Chem., 2006, 102, 111–123 CrossRef CAS PubMed.
- R. P. Manson and A. L. Lawrence, Environ. Toxicol. Chem., 1999, 18, 2438–2447 Search PubMed.
- A. Heyes, R. P. Mason, E. H. Kim and E. Sunderland, Mar. Chem., 2006, 102, 134–147 CrossRef CAS PubMed.
- J. Canario, C. Vale and M. Caetano, Mar. Pollut. Bull., 2005, 50, 1142–1145 CrossRef CAS PubMed.
- D. Kocman, T. Kanduc, N. Ogrinc and M. Horvat, Biogeochemistry, 2011, 104, 183–201 CrossRef CAS.
- T. Stoichev, D. Amouroux, J. C. Wasserman, D. Point, A. De Diego, G. Bareille and O. F. X. Donard, Estuarine, Coastal Shelf Sci., 2004, 59, 5111 CrossRef PubMed.
- M. Coquery, D. Cossa and J. Sanjuan, M. Chem., 1997, 58, 213–227.
- J. Schafer, S. Castelle, G. Blanc, A. Dabrin, M. Masson, L. Lanceleur and C. Bossy, Estuarine, Coastal Shelf Sci., 2010, 90, 80–92 CrossRef PubMed.
- B. Ouddane, N. Mikac, A. B. Cundy, L. Quillet and J. C. Fischer, Appl. Geochem., 2008, 23, 618–631 CrossRef CAS PubMed.
- C. R. Hammerschmidt and W. F. Fitzgerald, Environ. Sci. Technol., 2004, 38, 1487–1495 CrossRef CAS.
- C. R. Hammerschmidt and W. F. Fitzgerald, Geochim. Cosmochim. Acta, 2006, 70, 918–930 CrossRef CAS PubMed.
- M. Kadlecova, B. Ouddane and H. Docekalova, J. Environ. Monit., 2012, 14, 961–967 RSC.
- V. Rimondi, J. E. Gray, P. Costagliola, O. Vaselli and P. Lattanzi, Sci. Total Environ., 2012, 414, 318–327 CrossRef CAS PubMed.
- M. E. Hines, M. Horvat, J. Faganeli, J. C. Bonzongo, T. Barkay, E. B. Major, K. J. Scott, E. A. Bailey, J. J. Warwick and W. B. Lyons, Environ. Res., 2000, 83, 129–139 CrossRef CAS PubMed.
- E. M. Sunderland, F. A. P. C. Gobas, A. Heyes, B. A. Branfireun, A. K. Bayer, R. E. Cranston and M. B. Parsons, Mar. Chem., 2004, 90, 91–105 CrossRef CAS PubMed.
- L. Lambertsson and M. Nilsson, Environ. Sci. Technol., 2006, 40, 1822–1829 CrossRef CAS.
- J. K. King, S. Saunders, R. F. Lee and R. A. Jahnke, Environ. Toxicol. Chem., 1999, 18, 1362–1369 CrossRef CAS.
- R. Devereux, M. R. Winfrey, J. Winfrey and D. A. Stahl, FEMS Microbiol. Ecol., 1996, 20, 23–31 CrossRef CAS PubMed.
- E. J. Kerin, C. C. Gilmour, E. Roden, M. T. Suzuki, J. D. Coates and R. P. Mason, Appl. Environ. Microbiol., 2006, 72, 7919–7921 CrossRef CAS PubMed.
- C. P. J. Mitchell and C. C. Gilmour, J. Geophys. Res., 2008, 113, G00C04 CrossRef.
- A. Heyes, C. Miller and R. P. Manson, Mar. Chem., 2003, 90, 75–89 CrossRef PubMed.
- C. C. Gilmour, G. S. Riedel, M. C. Ederington, J. T. Bell, J. M. Benoit, G. A. Gill and M. C. Stordal, Biogeochemistry, 1998, 40, 327–345 CrossRef CAS.
- T. A. Hollweg, C. C. Gilmour and R. P. Mason, Mar. Chem., 2009, 114, 86–101 CrossRef CAS PubMed.
- S. Han, P. Narasingarao, A. Obraztsova, J. Gieskes, A. C. Hartmann, B. M. Tebo, E. E. Allen and D. D. Deheyn, Environ. Sci. Technol., 2010, 44, 3752–3757 CrossRef CAS PubMed.
- N. J. O'Driscoll, D. R. S. Lean, L. L. Loseto, R. Carignan and S. D. Siciliano, Environ. Sci. Technol., 2004, 38, 2664–2672 CrossRef.
- T. Peretyazhko, L. Charlet and M. Grimaldi, Eur. J. Soil Sci., 2006, 57, 190–199 CrossRef CAS PubMed.
- M. C. Marvin-DiPasquale, J. L. Agee, C. McGowan, R. Oremland, M. Thomas, D. Krabbenhoft and C. C. Gilmour, Environ. Sci. Technol., 2000, 34, 4908–4916 CrossRef CAS.
- T. Barkay, S. M. Miller and A. O. Summers, FEMS Microbiol. Rev., 2003, 27, 355–384 CrossRef CAS.
- J. K. Schaefer, J. Yagi, J. R. reinfelder, T. Cardona, K. M. Ellickson, S. Tel-Or and T. Barkay, Environ. Sci. Technol., 2004, 38, 4304–4311 CrossRef CAS.
- R. S. Oremland, C. W. Culbertson and M. R. Winfrey, Appl. Environ. Microbiol., 1991, 57, 130–137 CAS.
- J. E. Gray, M. E. Hines, P. L. Higueras, I. Adatto and B. K. Lasorsa, Environ. Sci. Technol., 2004, 38, 4285–4292 CrossRef CAS.
- J. Ramond, T. Berthe, R. Lafite, J. Deloffre, B. Ouddane and F. Petit, Mar. Pollut. Bull., 2008, 56, 1168–1176 CrossRef CAS PubMed.
- L. Lambertsson and M. Nilsson, Environ. Sci. Technol., 2006, 40, 1822–1829 CrossRef CAS.
- Z. Kwokal, S. Franciskovic-Bilinski, H. Bilinski and M. Branica, Mar. Pollut. Bull., 2002, 44, 1152–1169 CrossRef CAS.
- R. S. Oremland, L. G. Miller, P. Dowdle, T. Connell and T. Barkay, Appl. Environ. Microbiol., 1995, 61, 2745–2753 CAS.
| This journal is © The Royal Society of Chemistry 2015 |