
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAza-nickelacycle key intermediate in nickel(0)-catalyzed transformation reactions
Masato
Ohashi
a,
Yoichi
Hoshimoto
ab and
Sensuke
Ogoshi
*ac
aDepartment of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: ogoshi@chem.eng.osaka-u.ac.jp
bFrontier Research Base for Global Young Researchers, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
cJST, Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C), Suita, Osaka 565-0871, Japan
First published on 13th April 2015
Abstract
This Perspective provides an overview of the oxidative cyclization reactions of alkynes and imines with nickel(0) to give five-membered aza-nickelacycles. These reactions could be a key step in multicomponent coupling and cycloaddition reactions to afford nitrogen-containing organic compounds.
 Masato Ohashi | Masato Ohashi received his Ph.D. from the Tokyo Institute of Technology under the supervision of Professor Hiroharu Suzuki in 2003. After joining the research group of Kazushi Mashima at Osaka University as a JST post-doctoral fellow, he moved to Aachen in 2006, where he was a Humboldt research fellow with Jun Okuda (RWTH-Aachen). In 2007, he joined the group of S. Ogoshi at Osaka University as an assistant professor. In 2012, he was promoted to associate professor of Osaka University. His current research interests include transition metal-catalyzed transformation reactions of organofluorine compounds as well as their mechanistic investigation. |
 Yoichi Hoshimoto | Yoichi Hoshimoto received his M.Sc. and Ph.D. from Osaka University under the supervision of Professor S. Ogoshi in 2013. He then joined the Frontier Research Base for Global Young Researchers, Osaka University as a tenure-track assistant professor. His recent research interests include homogeneous catalysis with organometallic complexes and Lewis acid–base chemistry. |
 Sensuke Ogoshi | Sensuke Ogoshi studied at Osaka University and received his Ph.D. under the supervision of Professor Shinji Murai in 1993. In that year he joined the faculty at Osaka University as an assistant professor. He was promoted to an associate professor in 1999, and has been a full professor since 2007. In the meantime, he pursued his academic career also at the University of Alberta during 1996–1997, where he joined the research group of Professor Jeffrey M. Stryker (JSPS Fellowships for Research Abroad). His research directs toward the discovery of new transition metal complexes that can act as a key reaction intermediate in new transformation reactions of unsaturated compounds. In addition, he has been focusing on the correlation between the structure and the reactivity of organo-transition-metal complexes. |
1. Introduction
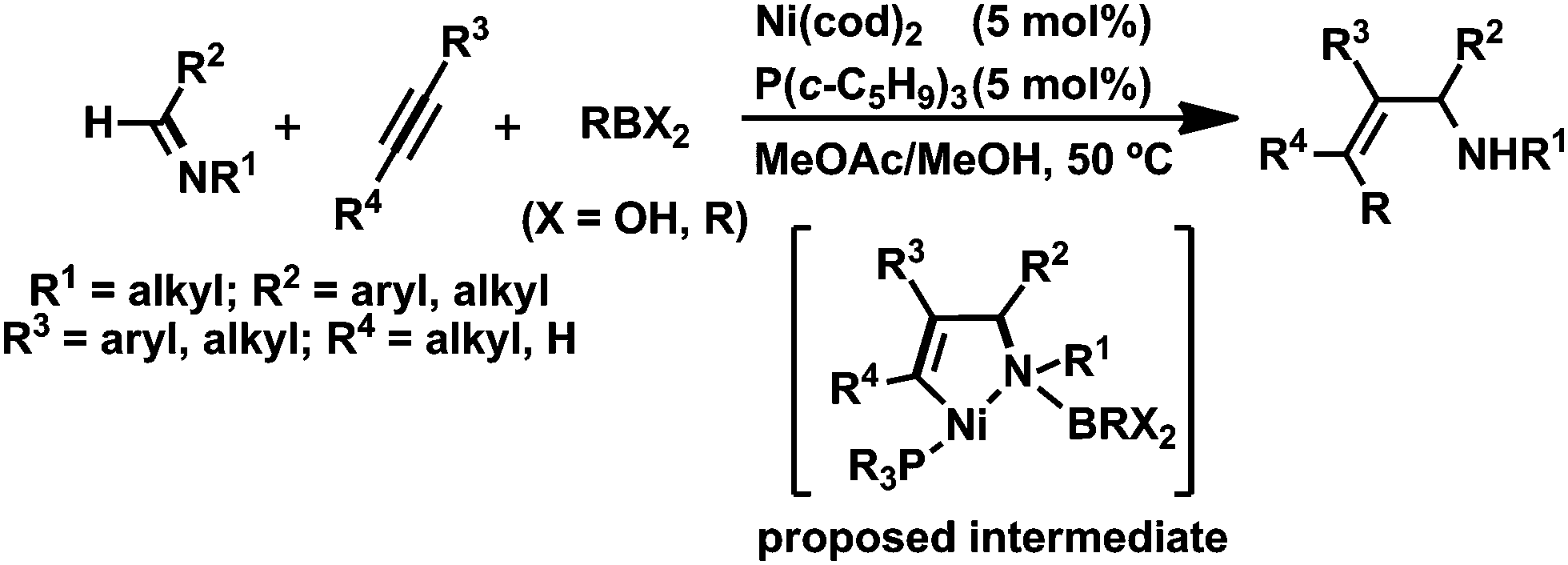
Oxidative cyclization with low-valent transition metals has received considerable attention because the reaction enables the construction of a C–C bond between a variety of unsaturated compounds, and indeed, the resulting five-membered metallacycles are assumed to be key reaction intermediates in transition-metal-catalyzed cycloaddition as well as multicomponent coupling reactions.1 Therefore, the development of efficient methods to generate a variety of metallacycles offers more opportunities to access highly complicated organic compounds. Among transition-metal candidates, nickel has shown great promise as a catalyst, because a number of oxidative cyclization reactions have yielded nickelacycles when using two unsaturated compounds with nickel(0).2,3 In particular, hetero-nickelacycles are assumed to be key intermediates in the nickel-catalyzed multi-component coupling reactions between alkynes and either aldehydes or imines.This Perspective focuses on the preparation of a five-membered aza-nickelacycle generated via the oxidative cyclization of an imine and an alkyne with nickel(0). Such aza-nickelacycles are much rarer than the related oxa-nickelacycles generated via the oxidative cyclization of an aldehyde and an alkyne with nickel(0) because imines are generally weaker electrophiles than aldehydes.2,4 Therefore, it is logical to assume that electron-withdrawing substituents on the nitrogen of the imine are required for oxidative cyclization by promoting back donation from nickel(0) to imines. In addition, the generation of a five-membered aza-nickelacycle is efficiently promoted by chelate coordination of a donor atom on the N-substituent group to a vacant coordination site on the nickel center.3a,b Given this background, in 2007 we achieved the first isolation of a corresponding aza-nickelacycle via the oxidative cyclization of N-sulfonylimine and an alkyne.3e,5 Herein, we discuss three different types of nickel-catalyzed transformation reactions, (a) [2 + 2 + 2] cycloaddition reaction leading to 1,2-dihydropyridines; (b) multi-component coupling or cyclocondensation reactions with alkylmetal reagents to yield allylamine derivatives; and (c) [2 + 2 + 1] carbonylative cycloaddition to give γ-lactams (Scheme 1). These nitrogen-containing products are ubiquitous structural motifs for natural products in small molecules that have biomedical relevance and are among the most versatile synthetic intermediates for use in the synthesis of a wide range of other valuable molecules.6–8
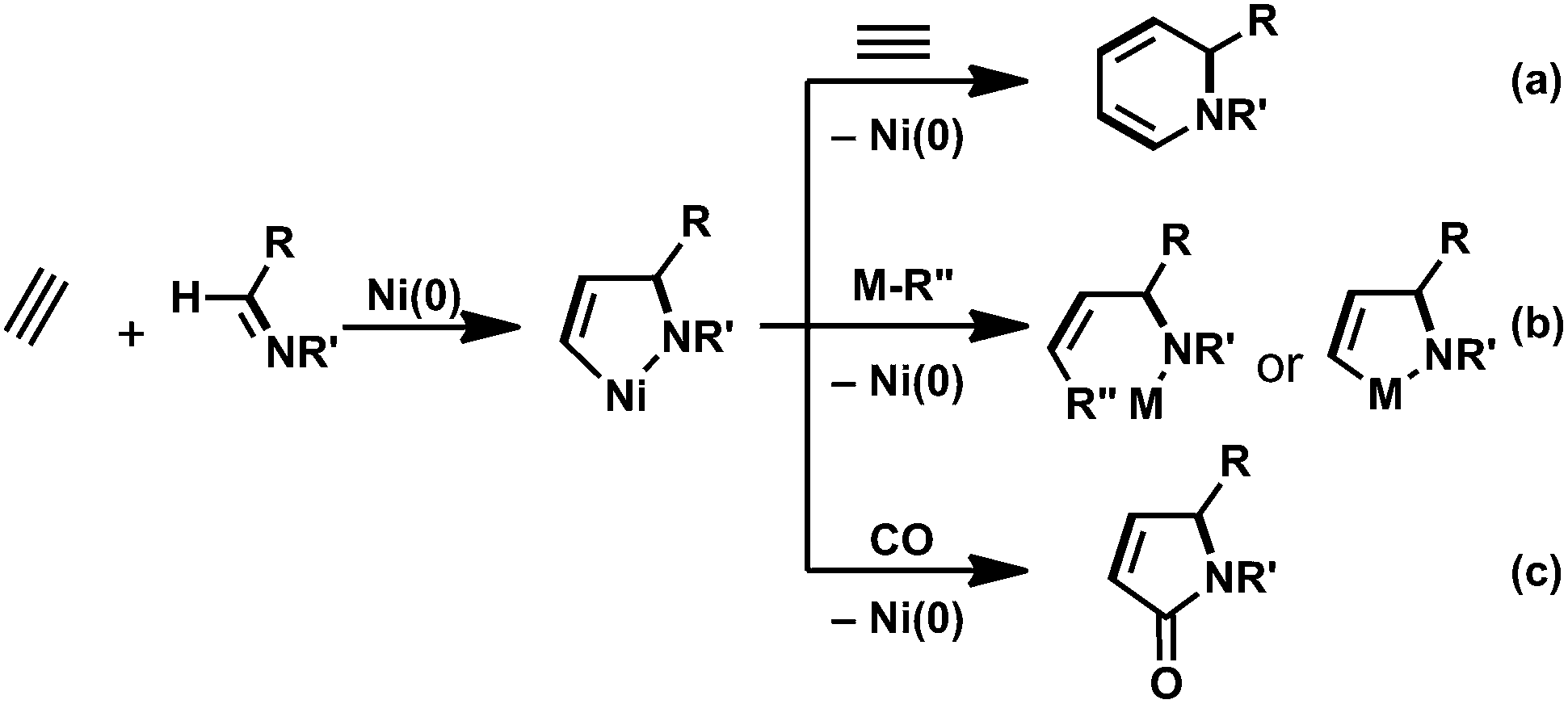 | ||
| Scheme 1 Formation of five-membered aza-nickelacycles generated via oxidative cyclization of an imine and an alkyne, and their key role in the nickel-catalyzed transformation reactions leading to nitrogen-containing products. | ||
2. Generation of five-membered aza-nickelacycles
The reaction of N-(benzenesulfonyl)benzaldimine (1a) with an equimolar amount of diphenylacetylene (2a) in the presence of Ni(cod)2 and PCy3 at room temperature resulted in the quantitative formation of a five-membered nickelacycle (3aa; Scheme 2).3e Treating 3aa with carbon monoxide (5 atm) afforded the corresponding γ-lactam (4aa), which was consistent with the structure of 3aa depicted in Scheme 2. The treatment of 3aa with an additional equimolar amount of 2a gave a seven-membered nickelacycle (5aa) in quantitative yield. The insertion of 2-butyne (2b) into 3aa proceeded much faster (within 10 min) than that of diphenylacetylene, and yielded the corresponding seven-membered nickelacycle (5az).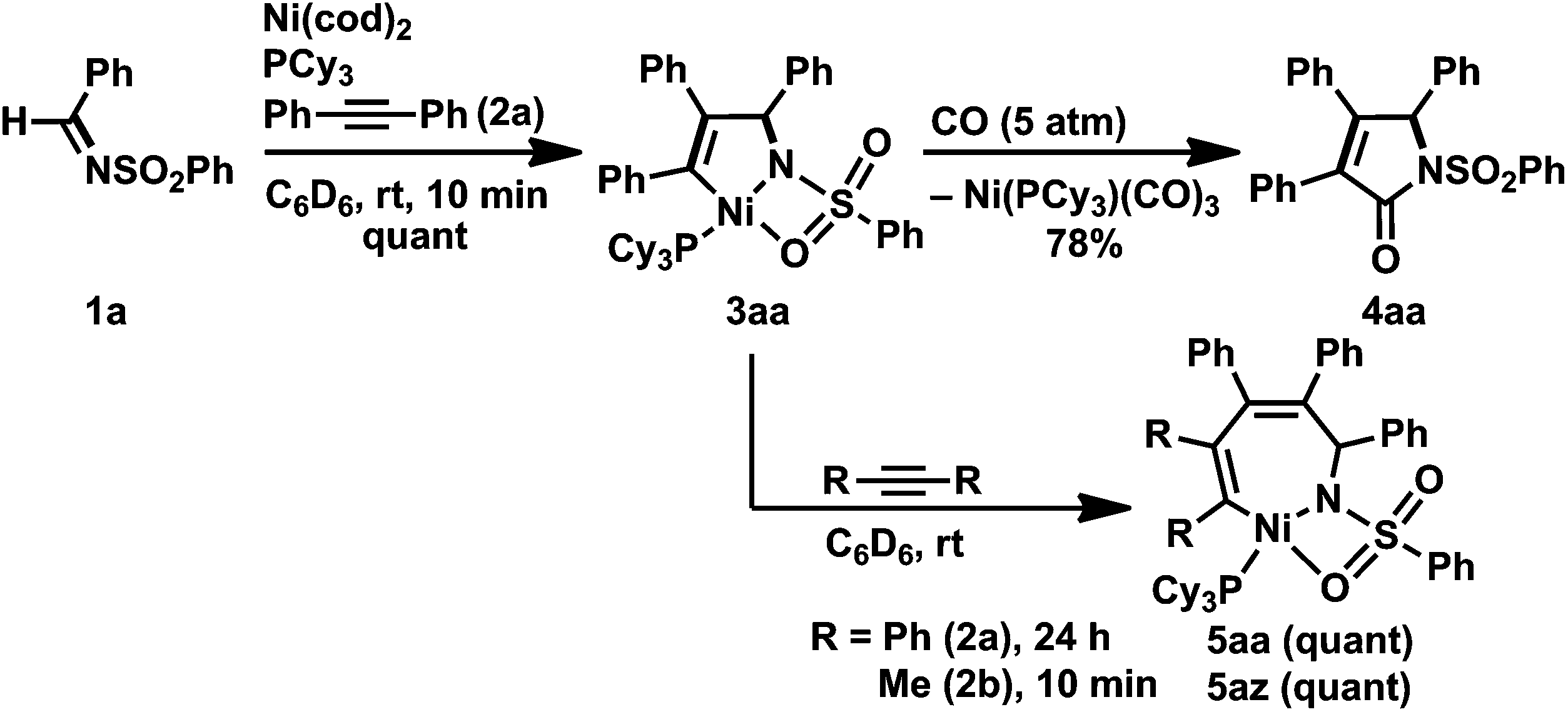 | ||
| Scheme 2 Formation of five-membered aza-nickelacycle 3aa and its reactivity. Yields, determined by 1H NMR spectroscopy, are given. | ||
Such a higher reactivity of 2b explained the formation of an inseparable mixture of a five-membered nickelacycle (3ab), a seven-membered nickelacycle (5ab), and an η2-iminenickel complex (6a) when the reaction of 1a with an equimolar amount of 2b was conducted in the presence of Ni(cod)2 and PCy3 at room temperature for 10 min (Scheme 3).3e Although the five-membered nickelacycle 3ab could not be isolated, the addition of an additional equimolar amount of 2-butyne to this mixture gave 5ab as the sole product in 95% yield. The molecular structure of 5ab was determined by X-ray crystallography (Fig. 1a). The coordination of one of the oxygen atoms in the benzenesulfonyl group to nickel might have played an important role in stabilizing 5ab as an isolable square-planar nickel(II) complex.9 The molecular structure of 6a, in which η2-coordination of the carbon–nitrogen double bond was observed, was also confirmed by X-ray crystallography (Fig. 1b). The N–C1 bond length was 1.405(5) Å, which was obviously elongated compared with a typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond length (ca. 1.27–1.30 Å),10 which was due to a back donation from the nickel(0) center.
N bond length (ca. 1.27–1.30 Å),10 which was due to a back donation from the nickel(0) center.
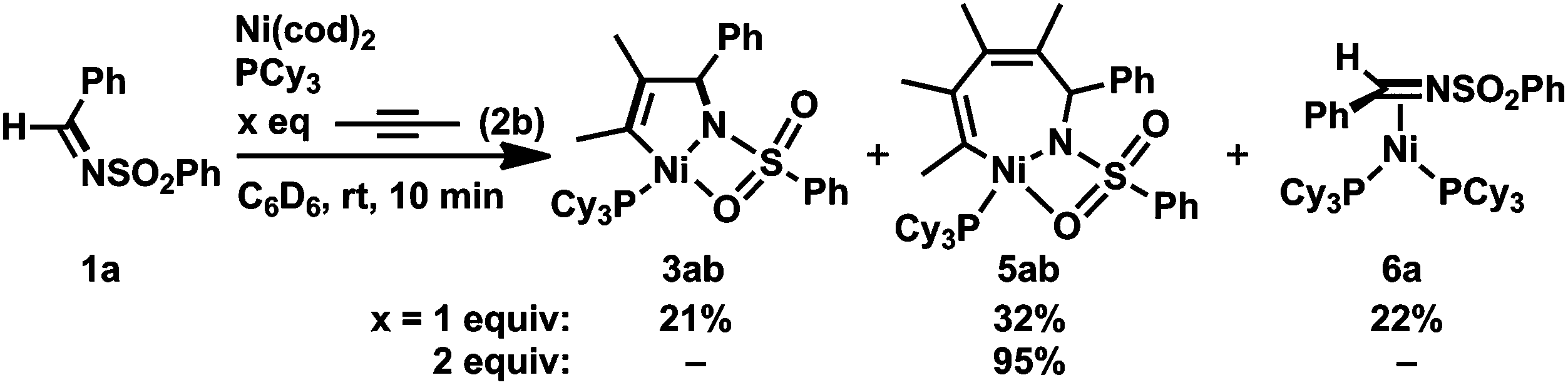 | ||
| Scheme 3 Reaction of 1a with 2b (1 or 2 equiv.) in the presence of Ni(0)/PCy3. Yields, determined by 1H NMR spectroscopy, are given. | ||
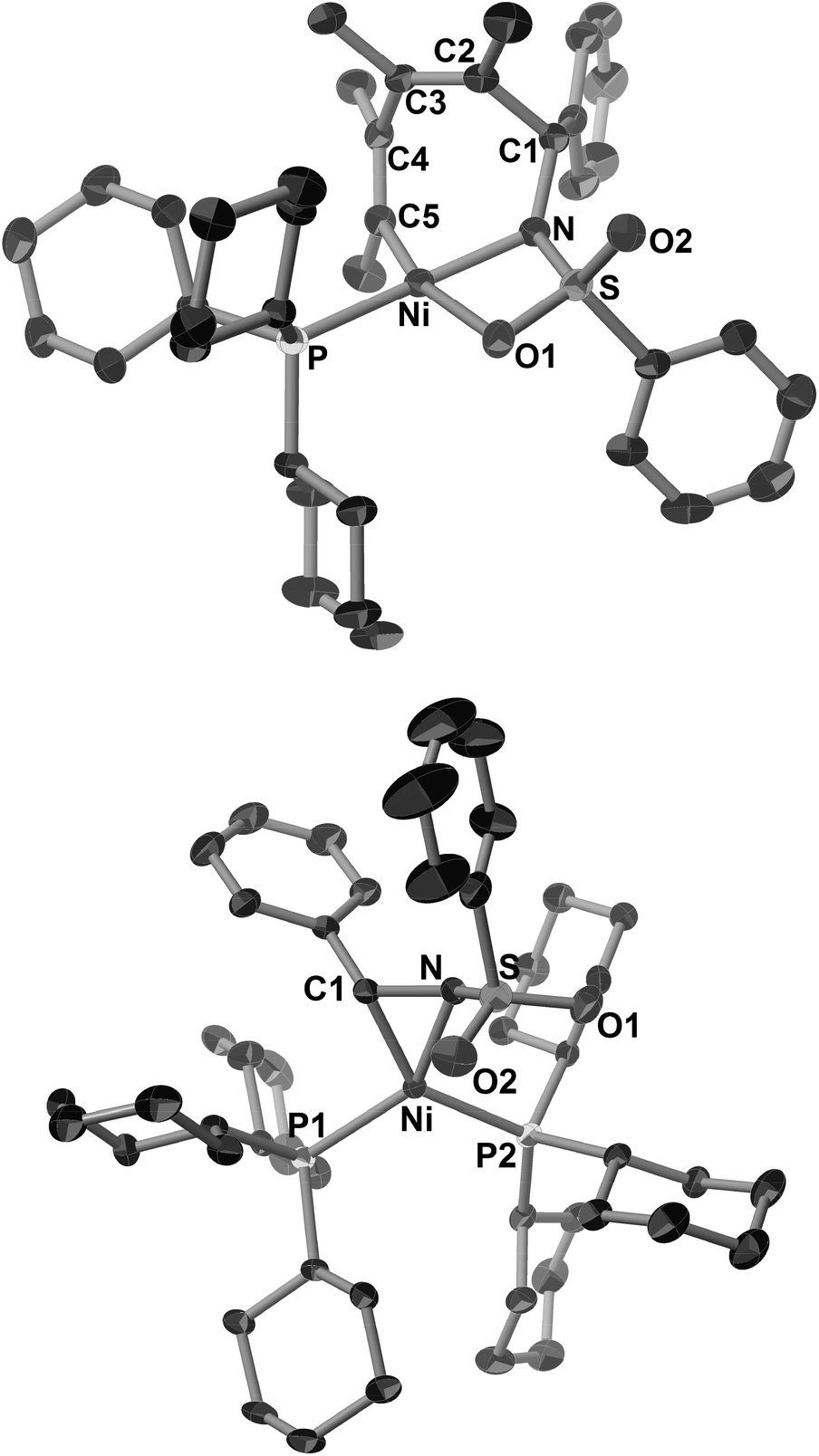 | ||
| Fig. 1 ORTEP drawings of 5ab (a) and 6a (b) with thermal ellipsoids at the 30% probability level. H atoms have been omitted for clarity. | ||
In contrast to N-sulfonyl imine 1a, the reaction of N-benzylidene-P,P-diphenylphosphinic amide (1b) with 2b, Ni(cod)2, and PCy3 (1 equiv. each) was completed in 24 h to afford the corresponding five-membered aza-nickelacycle 3bb in 87% yield with the concomitant formation of an η2-iminenickel complex 6b in 13% yield (Scheme 4).3n An aza-nickelacycle analogue (3ba) was prepared from 1b and diphenylacetylene 2a, and its five-membered framework was unambiguously determined by X-ray crystallography (Fig. 2a). Complex 3ba had a square-planar nickel(II) center with an intramolecular coordination of oxygen to nickel. In addition, the formation of a γ-lactam derivative (4bb) by the carbonylation of 3bb should support the five-membered nickelacycle skeleton of 3bb. The η2-iminenickel complex 6b was isolated in 74% yield by the reaction of 1b with one equivalent of Ni(cod)2 and PCy3 in toluene for 2 h. While NMR analysis revealed that complex 6b existed as a mixture of syn/anti dimeric isomers in solution (syn/anti = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :82, in C6D6, rt), only an anti-isomer (6b-anti) was observed in the crystal lattice, as shown in Fig. 2b. No reaction occurred when an excess amount of 2b was added at room temperature to 6b, indicating that 6b would be highly stabilized through the intramolecular coordination of oxygen to nickel, and thus the simultaneous coordination of N-phosphinyl imine 1b and alkyne 2b might be inhibited.
:82, in C6D6, rt), only an anti-isomer (6b-anti) was observed in the crystal lattice, as shown in Fig. 2b. No reaction occurred when an excess amount of 2b was added at room temperature to 6b, indicating that 6b would be highly stabilized through the intramolecular coordination of oxygen to nickel, and thus the simultaneous coordination of N-phosphinyl imine 1b and alkyne 2b might be inhibited.
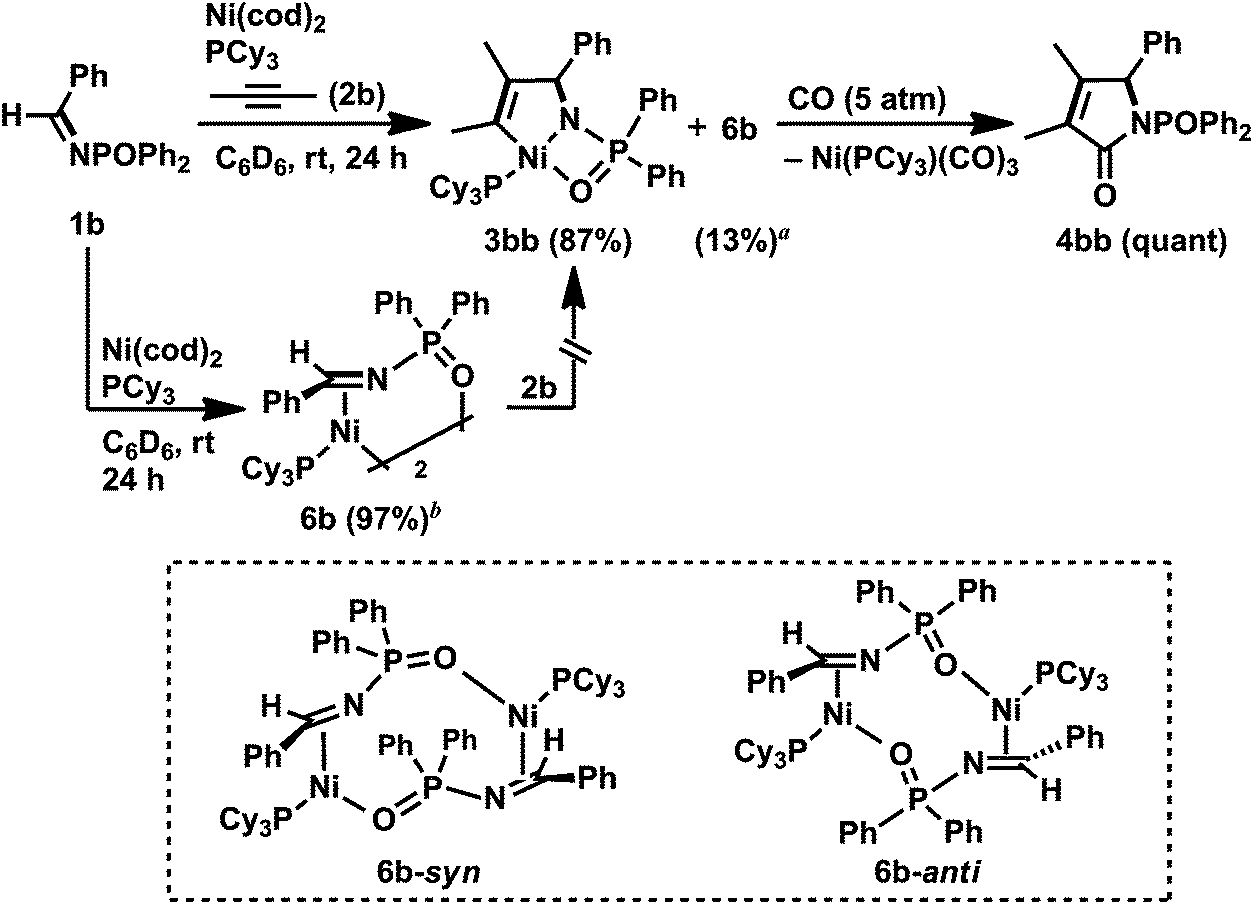 | ||
| Scheme 4 Formation of five-membered aza-nickelacycle 3bb and its reactivity with carbon monoxide. Yields, determined by 1H NMR spectroscopy, are given in parentheses. Diagrams in the dotted frame represent the two isomers of 6b. (a) Syn/anti ratio = 42:58. (b) Syn/anti ratio = 18:82. | ||
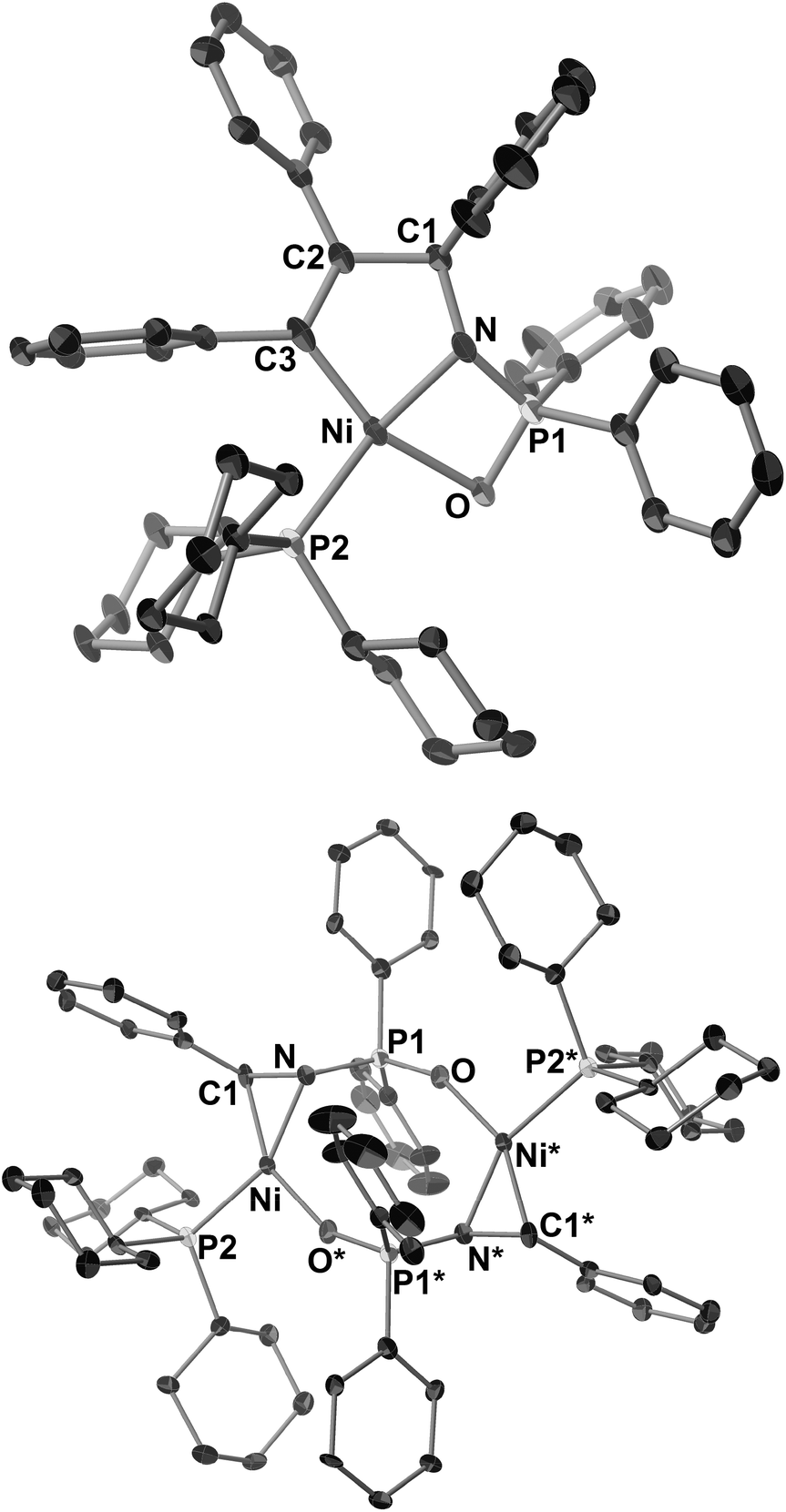 | ||
| Fig. 2 ORTEP drawings of 3ba (a) and 6b-anti (b) with thermal ellipsoids at the 30% probability level. H atoms and solvated molecules in 6b-anti (hexane) have been omitted for clarity. Symmetry transformation used to generate equivalent atoms S* for 6b-anti: −X, 1 − Y, −Z. | ||
Next, we turned our attention to employing NHCs as ligands to investigate whether these stronger electron-donating and more steric-demanding ligands could enhance the formation of aza-nickelacycle compounds via the oxidative cyclization of alkynes and imines without a chelation group. In fact, we demonstrated the preparation of T-shaped 14-electron hetero-nickelacycles bearing a NHC ligand.3g,j The reaction of N-benzylidene-4-trifluoromethyl aniline (1c) or N-benzylidene-2-aminopyridine (1d) with a stoichiometric amount of Ni(cod)2 and IPr (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene) was completed within 10 min to yield the corresponding η2-imine complex, [Ni(IPr)(η2-imine)] (7c and 7d), in 95% and 92% yields, respectively (Scheme 5).3n The molecular structure of 7c was confirmed by X-ray crystallography (Fig. 3a). The C1–N bond length was 1.37(1) Å comparable to those observed in 6a and 6b. It should be mentioned that 7c had a 14-electron nickel(0) center while PCy3-ligated imine complexes 6a and 6b possessed 16-electron centers. Such a structural difference might have been caused by the steric hindrance of IPr; a bulker IPr could stabilize the highly reactive 14-electron nickel(0) complex by cloaking its vacant coordination sites. By contrast, treating sulfonyl imine 1a with Ni(cod)2 and IPr did not afford the corresponding [Ni(IPr)(η2-imine)] complex, and unidentified white precipitates were observed.11
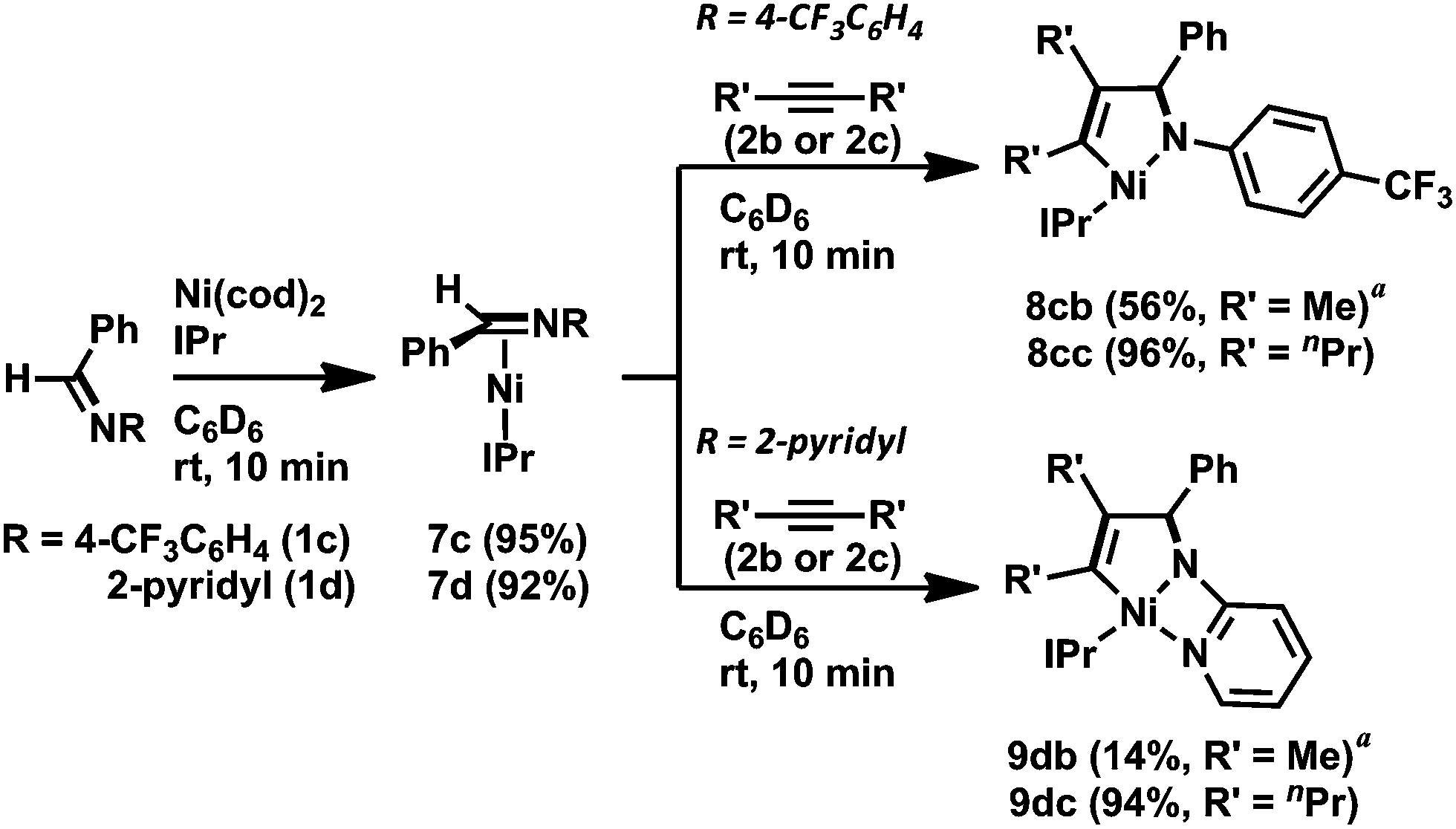 | ||
| Scheme 5 The stoichiometric reactions using N-aryl imines and alkynes with Ni(0)/IPr. Yields were determined by 1H NMR spectroscopy. (a) The reaction was carried out in toluene. Isolated yields after recrystallization are shown. | ||
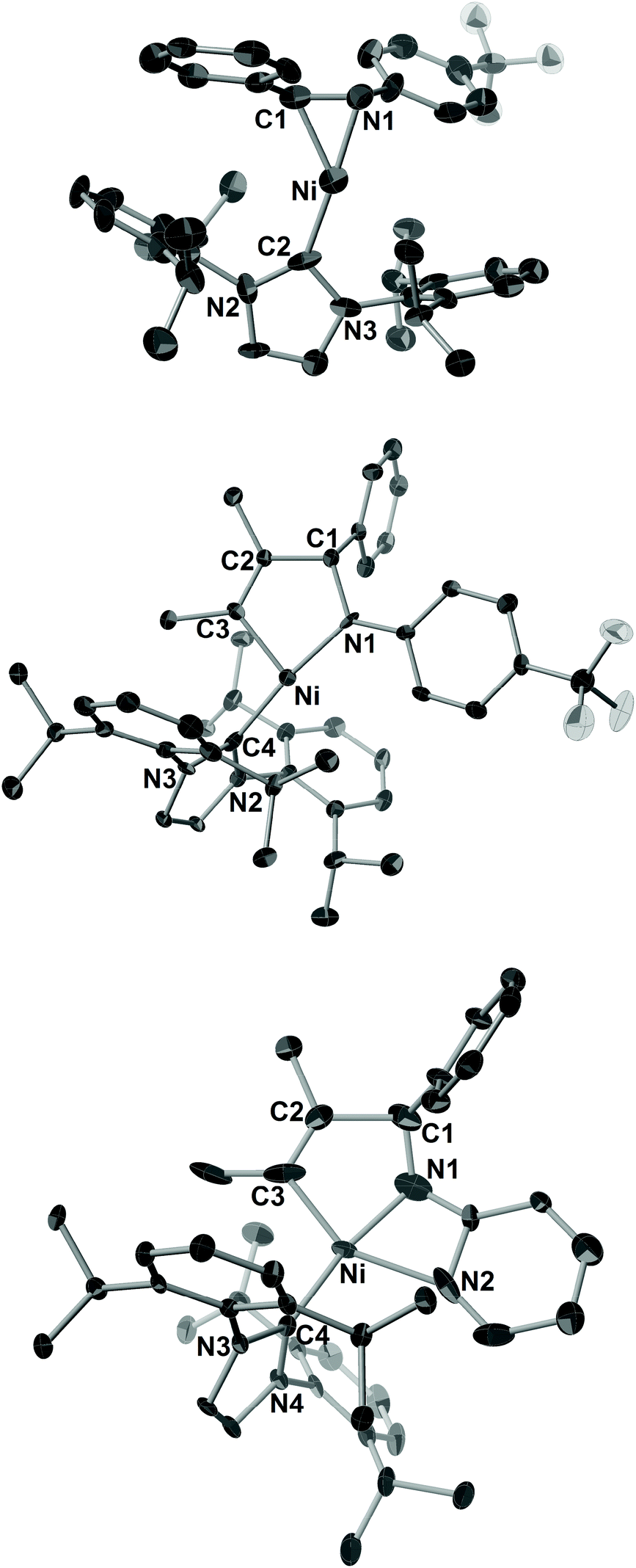 | ||
| Fig. 3 ORTEP drawings of 7c (a), 8cb (b), and 9db (c) with thermal ellipsoids at the 30% probability level. H atoms have been omitted for clarity. | ||
Treatment of 7c with 2b or 4-octyne (2c) in C6D6 at room temperature gave five-membered aza-nickelacycles (8cb and 8cc; Scheme 5).3n An X-ray diffraction study of 8cb demonstrated its T-shaped 14-electron nickel(II) center (Fig. 3b), and the sum of the bond angles around nickel along the C3, N, and C4 was 359.0°, indicating that nickel and these three atoms are on the same plane. A space-filling model of 8cb clearly indicated that such a unique geometry was mostly due to the bulkiness caused by the aryl group on the imine nitrogen atom together with the bulky IPr ligand. On the other hand, the structures of aza-nickelacycles 9db and 9dc, which were prepared by the reaction of 7d with either 2b or 2c, had a planar tetracoordinate nickel(II) center with an intramolecular coordination of the N-pyridine moiety (Fig. 3c).
Yoshikai and co-workers reported that an related aza-nickelacycle similar to 9db was proposed as a reaction intermediate in the [2 + 2 + 2] cycloaddition reaction of two alkynes and an imine bearing a 3-methyl-2-pyridyl group on the nitrogen atom,12 and their DFT calculation of the model compound revealed that intramolecular coordination of the pyridyl ring to the nickel center stabilized the five-membered aza-nickelacycle regardless of the steric strain caused by the four-membered chelation structure (vide infra). It should be mentioned that Jamison and co-workers proposed the related five-membered aza-nickelacycle, generated from the oxidative cyclization of N-methyl imine and an alkyne, as a key intermediate in the nickel-catalyzed three-component coupling reaction of an imine, an alkyne, and BEt3.13
3. Nickel(0)-catalyzed [2 + 2 + 2] cycloaddition reaction of an imine with two alkynes: formation of 1,2-dihydropyridine derivatives
Heating the seven-membered nickelacycles 5aa, 5ab, and 5az at 100 °C promoted a reductive elimination to yield 1,2-dihydropyridine derivatives (10aa, 10ab, 10az), respectively (Scheme 6a).3e The formation of a 1,2-dihydropyridine by reductive elimination suggested that the development of a nickel-catalyzed [2 + 2 + 2] cycloaddition reaction of two alkynes and an imine might be possible. In fact, the intermolecular [2 + 2 + 2] cycloaddition of N-sulfonyl imine 1a and 2b in the presence of catalytic amounts of Ni(cod)2 and PMetBu2 at 100 °C gave the expected 1,2-dihydropyridine 10ab in 87% yield (Table 1, entry 1). 3-Hexyne (2d) and trimethylsilylacetylene (2e) also afforded the corresponding 1,2-dihydropyridines (10ad and 10ae), respectively (entries 5 and 7). The reaction also proceeded catalytically in the presence of PCy3, although PtBu2Me gave better results (entries 2 and 6). In the case of N-phosphinyl imine 1b, the [2 + 2 + 2] cycloaddition reaction with 2b proceeded at 100 °C in the presence of Ni(cod)2 and PCy3 (10 and 20 mol%, respectively), giving the corresponding 1,2-dihydropyridine 10bb in 64% yield (Table 1, entry 8).3n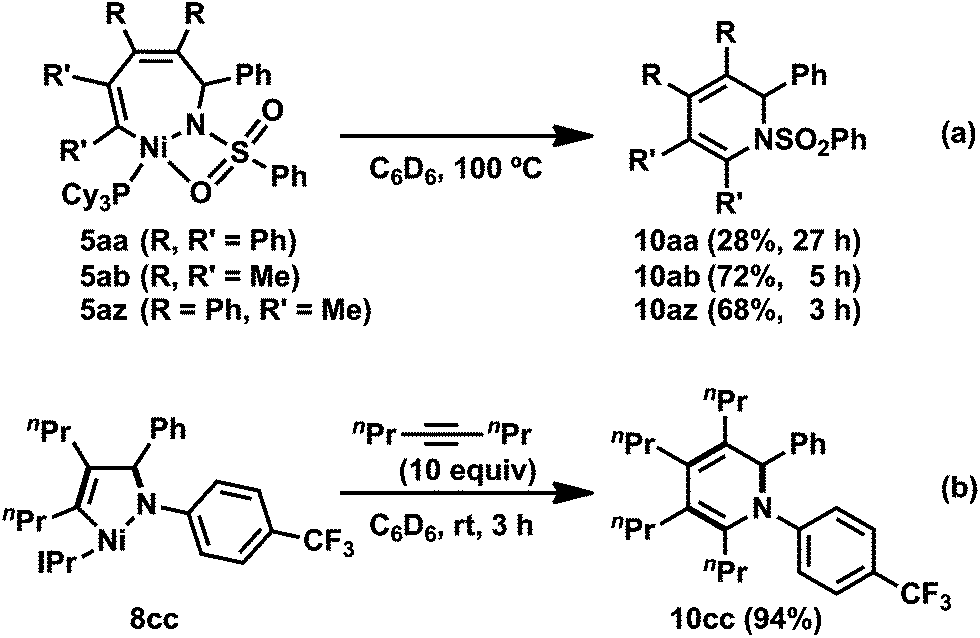 | ||
| Scheme 6 Stoichiometric formation of 1,2-dihydropyridines 10: (a) reductive elimination from seven-membered nickelacycle 5. (b) Reaction of five-membered nickelacycle 8cc with 2c. Yields were determined by 1H NMR spectroscopy. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Imine 1 | Alkyne 2 | Phosphine | Time (h) | Product 10 | Yieldb (%) |
| a General conditions: imines (0.10 mmol), alkynes (0.25 mmol), Ni(cod)2 (0.01 mmol), and phosphine ligand (0.02 mmol) were reacted in C6D6 (0.5 mL) at 100 °C. b Yields, determined by 1H NMR spectroscopy, are given. The values in parentheses are of isolated yield. c Ni(cod)2 (0.04 mmol), PCy3 (0.08 mmol), 1b (0.40 mmol), and 2b (1.20 mmol) were employed in toluene (1.0 mL). | ||||||
| 1 | 1a | 2b (R1, R2 = Me) | PtBu2Me | 48 | 10ab | 87 (50) |
| 2 | PCy3 | 24 | 10ab | 64 | ||
| 3 | PnBu3 | 24 | 10ab | 17 | ||
| 4 | P(o-tolyl)3 | 29 | 10ab | 6 | ||
| 5 | 2d (R1, R2 = Et) | PtBu2Me | 70 | 10ad | 64 (55) | |
| 6 | PCy3 | 24 | 10ad | 26 | ||
| 7 | 2e (R1 = SiMe3, R2 = H) | PtBu2Me | 18 | 10ae | 58 (38) | |
| 8c | 1b | 2b | PCy3 | 3 | 10bb | — (64) |
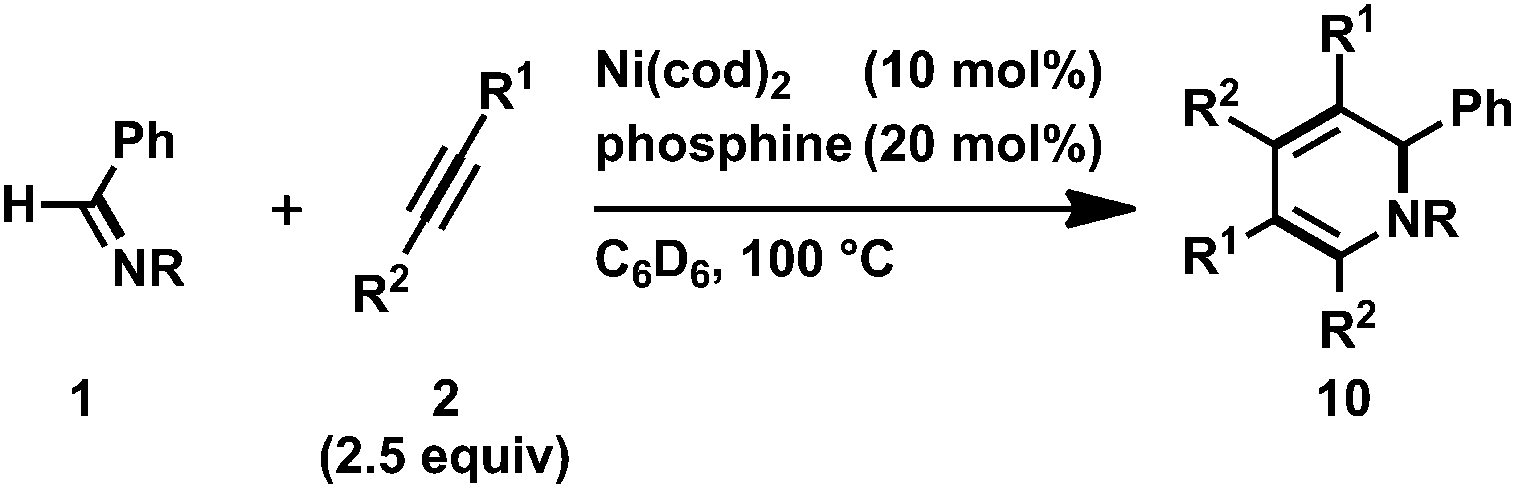
In contrast to the reactivity of the PCy3-ligated five-membered aza-nickelacycle 3aa, 8cc reacted with the second 2c at room temperature to yield 1,2-dihydropyridines (10cc) in 94% yield (Scheme 6b).3n The formation of the corresponding seven-membered aza-nickelacycle could not be observed by 1H NMR spectroscopy. This result might indicate that the rate of reductive elimination from the seven-membered aza-nickelacycle to give 1,2-dihydropyridine was much faster than that of the insertion of the second alkyne into the five-membered aza-nickelacycle. In sharp contrast, complex 9dc did not react with 2c at room temperature due to the suppression of the coordination of 2c by the intramolecular coordination of the N-pyridine.
Ni(0)/IPr-catalyzed [2 + 2 + 2] cycloaddition reactions of N-aryl imines with alkynes were carried out (Scheme 7).3n The reaction of 1c with 2c proceeded efficiently with 5 mol% of Ni(cod)2 and IPr to afford 10cc in 91% yield. In addition, the catalyst loading could be decreased to 2 mol% without a loss of efficiency by utilizing 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene (IMes) as a ligand (10cc; 92% NMR yield, 83% isolated yield). N-Benzylidene-3-(trifluoromethyl)aniline (1e) gave the corresponding 1,2-dihydropyridines 10eb (from 2b) or 10ec (from 2c) in 76 and 86% yields, respectively, by using 2 mol% of Ni(cod)2 and IMes; however, N-benzylidene-2-(trifluoromethyl)aniline (1f) did not afford the product under the same reaction conditions. The present reaction conditions were successfully applied to a simple N-phenyl imine (1g) and gave 10gc in 43% isolated yield in the presence of Ni(cod)2 and IPr (5 mol% each). Furthermore, N-benzylidene-4-fluoroaniline (1h) also reacted with 2c to give 10hc in moderate yield. The use of unsymmetrical alkynes such as 2-hexyne (2f) gave a mixture of four 1,2-dihydropyridines (10ef; total product yield: 79%, ratio of regioisomers: 30:29:21:20) when 1e was used as an imine partner. However, the reaction of imine 1c with 2-methyl-1-hexen-3-yne (2g) at 100 °C for 72 h resulted in the formation of a mixture of two products (10cg and 10cg′, 10cg/10cg′ = 83:17) in a total yield of 58%. This result can be rationalized by the occurrence of the regioselective incorporation of the first alkyne as a result of the formation of the thermodynamically favorable η3-butadienyl structure.3n,14 Thus, the oxidative cyclization of 1c and 2g with Ni(cod)2 in the presence of IPr at room temperature gave aza-nickelacycle 11 in 77% isolated yield (Scheme 8 and Fig. 4).3n Since thermolysis of 11 in C6D6 at 100 °C in the presence of 2g (10 equiv.) led to a regeneration of the starting imine 1c along with the concomitant formation of a mixture of 1,2-dihydropyridines 10cg and 10cg′, trimers of 2g, and unidentified products, this oxidative cyclization was reversible, taking place regioselectively to afford 11. The transition state of the insertion of the second 2g into 11, which proceeded at 100 °C, might also have been stabilized by the assistance of η3-butadienyl coordination, and therefore, 10cg was formed as a major product whereas the regioselectivity of the second insertion was not perfectly controlled at such a higher temperature (100 °C).
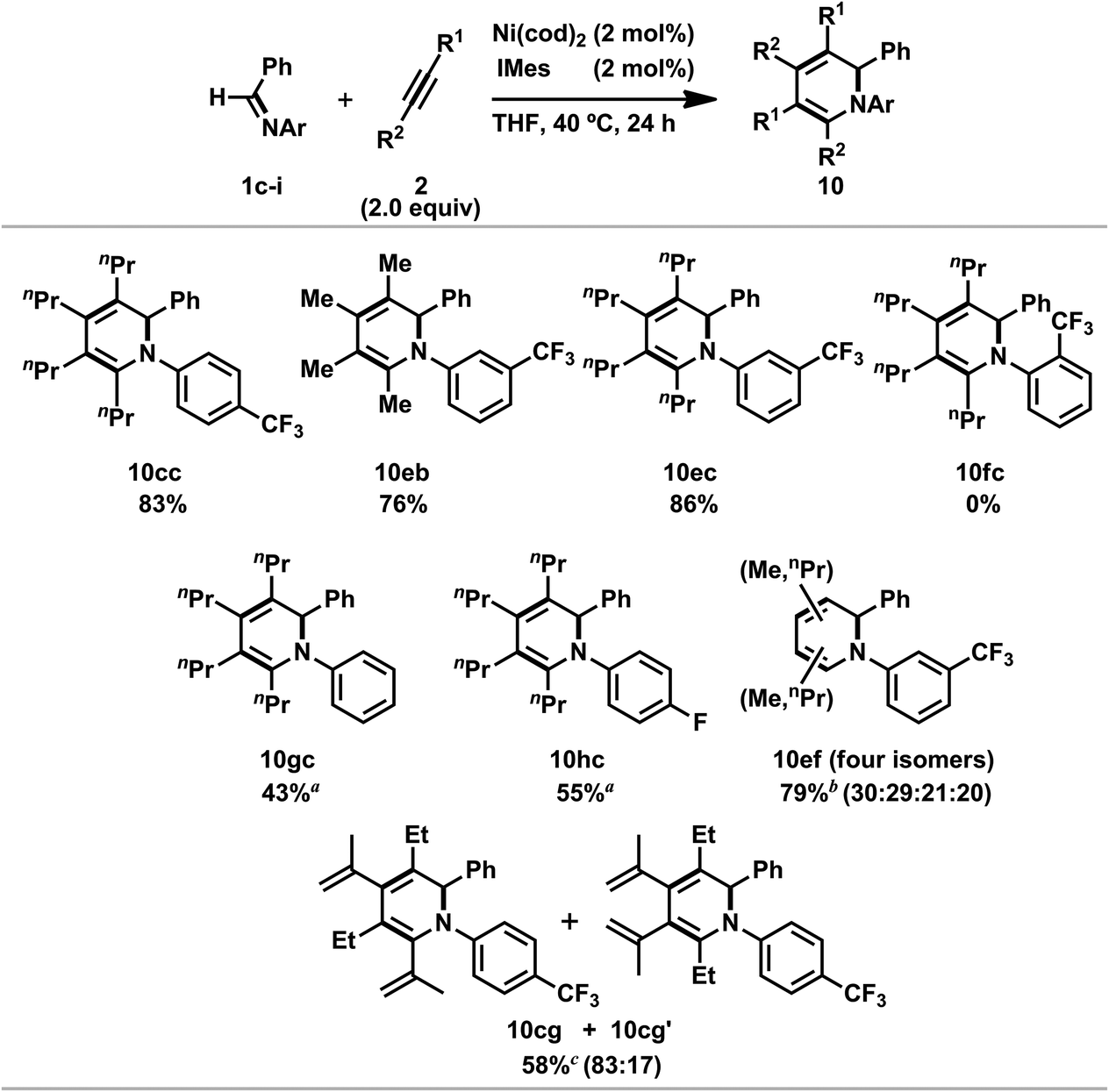 | ||
| Scheme 7 Nickel(0)/NHC-catalyzed [2 + 2 + 2] cycloaddition reaction of N-aryl imines with alkynes. General conditions: imines (1.00 mmol), alkynes (2.00 mmol), and Ni(cod)2/IMes (2 mol% each) were reacted in THF (1.0 mL) at 40 °C for 24 h. Yields of isolated products are given. (a) 5 mol% of Ni(cod)2 and IPr was used. (b) Total yield of the four products after isolation. (c) 10 mol% of Ni(cod)2 and IPr was used in 1,4-dioxane at 100 °C (72 h). Total yield of 10cg and 10cg′ is given. | ||
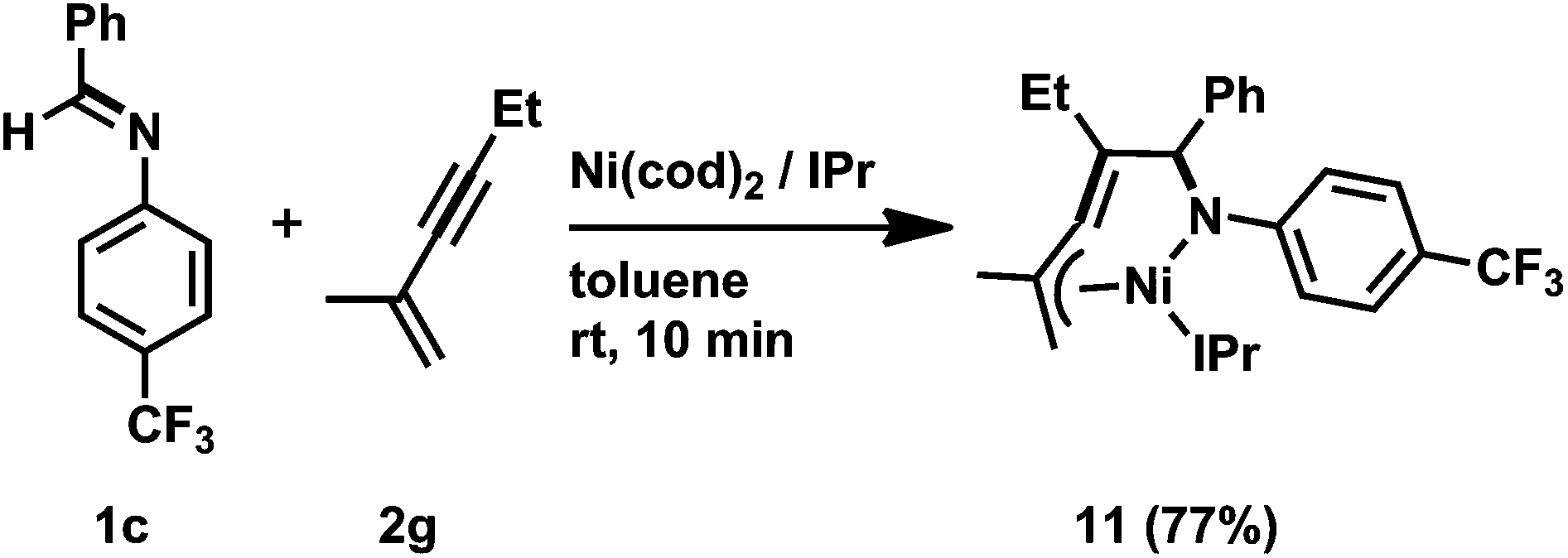 | ||
| Scheme 8 The stoichiometric reaction of 1c and 2g with Ni(0)/IPr. Isolated yield of 11 is given. | ||
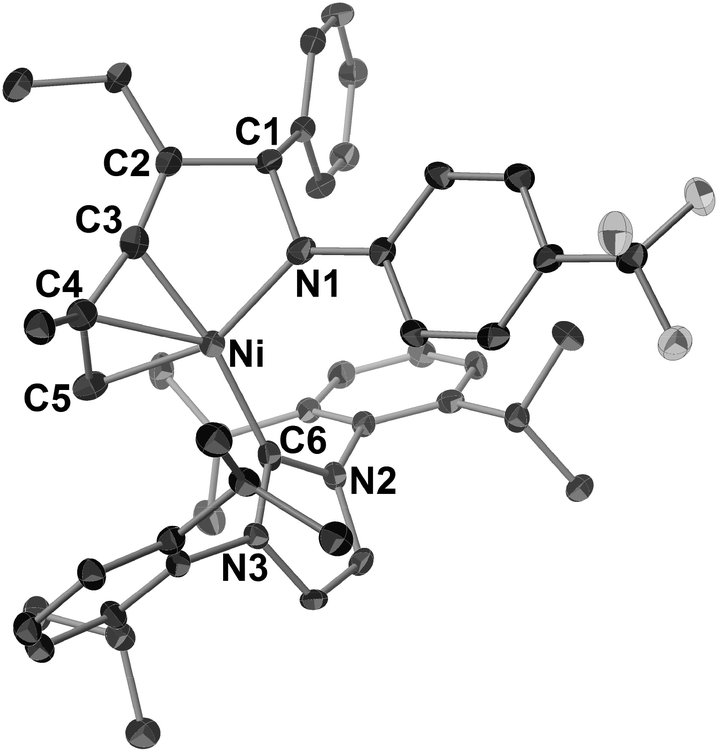 | ||
| Fig. 4 ORTEP drawing of 11 with thermal ellipsoids at the 30% probability level. H atoms have been omitted for clarity. | ||
The nickel(0)-catalyzed [2 + 2 + 2] cycloaddition of imines with alkynes proceeded as follows3e,n: (1) oxidative cyclization of an imine and an alkyne with nickel(0), giving a five-membered aza-nickelacycle; (2) insertion of a second alkyne, forming a seven-membered aza-nickelacycle; and, (3) reductive elimination from the seven-membered aza-nickelacycle, yielding a 1,2-dihydropyridine with the concomitant regeneration of nickel(0). In the reaction using benzenesulfonyl imine 1a, reductive elimination from the seven-membered aza-nickelacycle to give 1,2-dihydropyridine took place at 100 °C whereas the formation of the seven-membered intermediate was observed at room temperature. In addition, Yoshikai also proposed that, based on DFT calculations, reductive elimination would be the rate-limiting step of the reaction with N-pyridyl imines.12 In stark contrast, the reaction rate of the [2 + 2 + 2] cycloaddition of N-aryl imines with alkynes in the presence of a nickel(0)/NHC catalyst might have been determined by the insertion of the second alkyne into the five-membered aza-nickelacycle.3n
As previously described, Yoshikai and co-workers reported a related nickel-catalyzed [2 + 2 + 2] cycloaddition of an aldimine bearing a 3-methyl-2-pyridyl directing group on the nitrogen atom with alkynes to give 1,2-dihydropyridines (Scheme 9).12 Furthermore, rhodium-catalyzed cycloaddition reactions of imines and alkynes or diynes leading to 1,2-dihydropyridine derivatives have been reported;15 however, the initial formation of rhodacyclopentadienes, rather than the corresponding five-membered aza-rhodacycle intermediates, was proposed in these reactions.
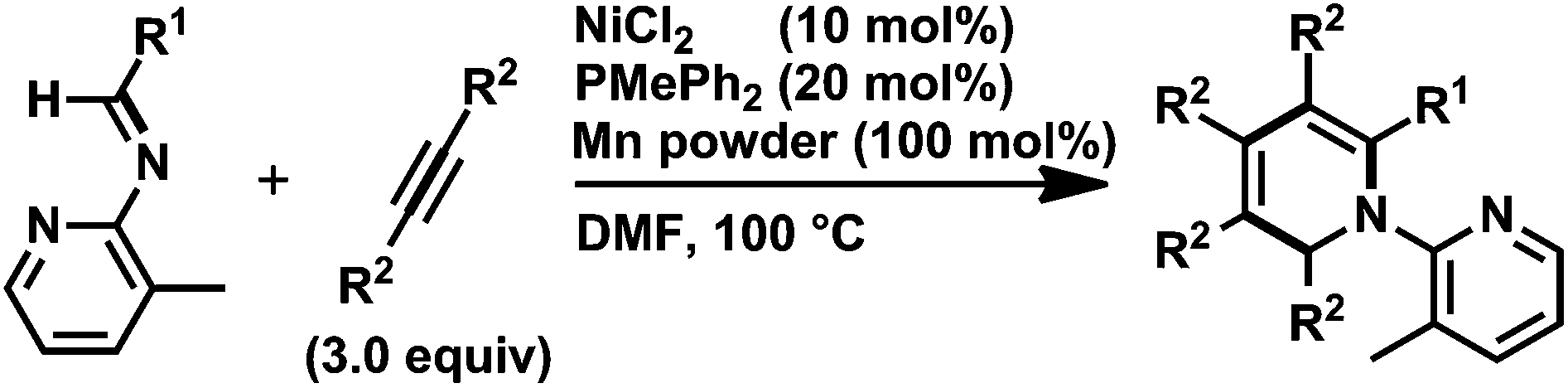 | ||
| Scheme 9 Nickel(0)-catalyzed [2 + 2 + 2] cycloaddition reaction of N-pyridyl imines with alkynes.8 | ||
4. Nickel(0)-catalyzed three-component coupling and cyclocondensation reactions of an imine, an alkyne, and alkylmetal reagents
Next, we investigated the reactivity of the five-membered aza-nickelacycle 3aa toward alkylmetal reagents. First, the reaction of 3aa with ZnMe2 in toluene at room temperature was conducted in the presence of vinyltrimethylsilane, the role of which was to trap the generated nickel(0) species as the known nickel(0) bisalkene complex, (PCy3)Ni(CH2CHTMS)2.3d,i As a result, the expected methylzincamido (12aa) was obtained in 74% isolated yield together with the formation of (PCy3)Ni(CH2CHTMS)2 (Scheme 10a and Fig. 5a).3h This stoichiometric reaction was successfully applied to a catalytic reaction wherein a three-component coupling reaction of 1a, 2a, and ZnMe2 afforded 12aa, and it also proceeded in the presence of catalytic amounts of Ni(cod)2 and PCy3 (10 and 20 mol%, respectively). It should be mentioned that the five-membered aza-nickelacycle 3aa did not react with BEt3 even when heated at 60 °C for 2 h, while 3aa was analogous to the reaction intermediate proposed in Jamison's work.13 This was consistent with the fact that N-tosyl imines cannot participate in the nickel-catalyzed three-component coupling of an alkyne, an imine, and a triethylborane (Scheme 11).13
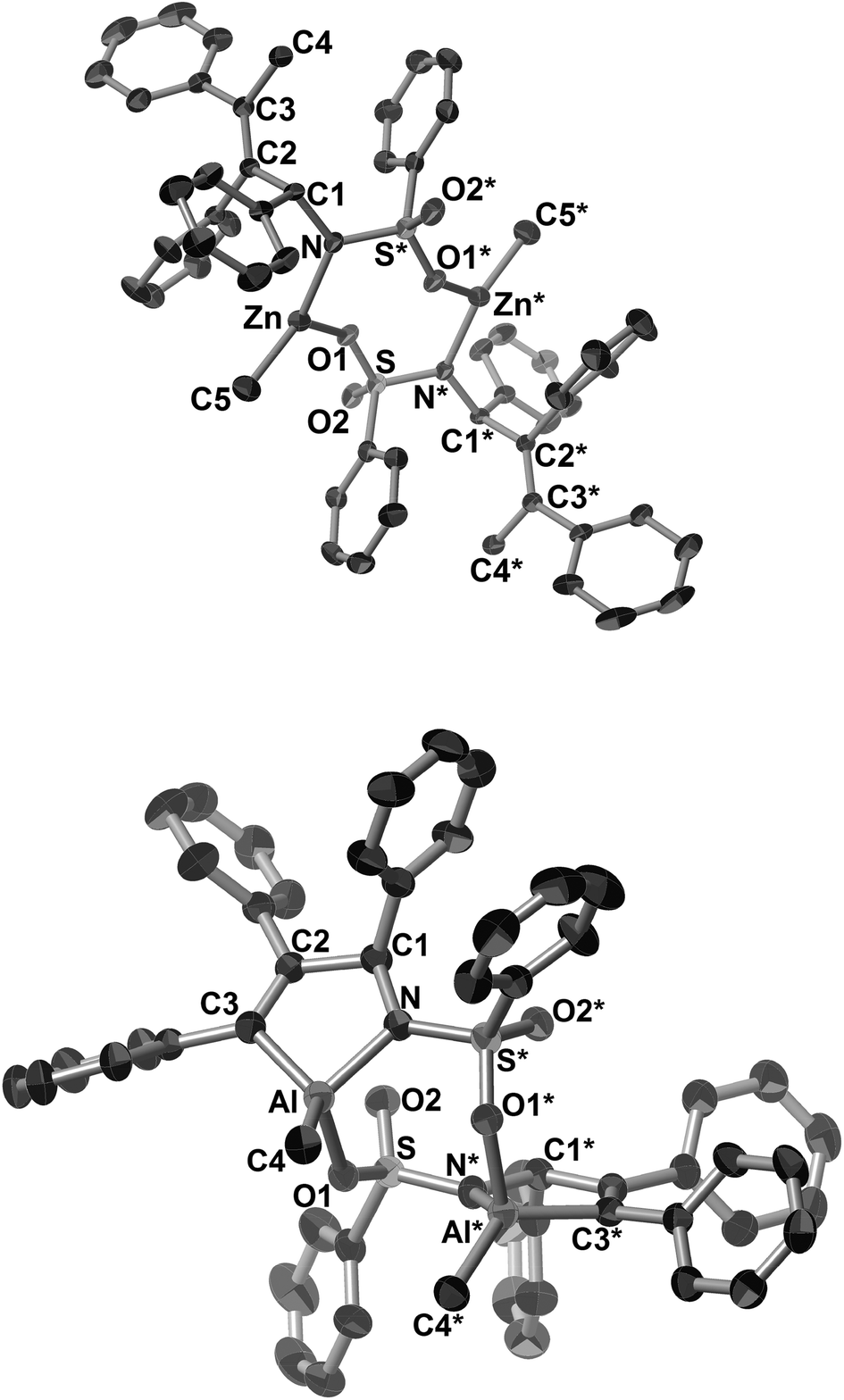 | ||
| Fig. 5 ORTEP drawings of 12aa (a) and 13aa (b) with thermal ellipsoids at the 30% probability level. H atoms and the solvated molecule (toluene) in 13aa have been omitted for clarity. Symmetry transformation used to generate equivalent atoms S* for 12aa: 2 − X, Y, 2 − Z, for 13aa: 2 − X, Y, 0.5 − Z. | ||
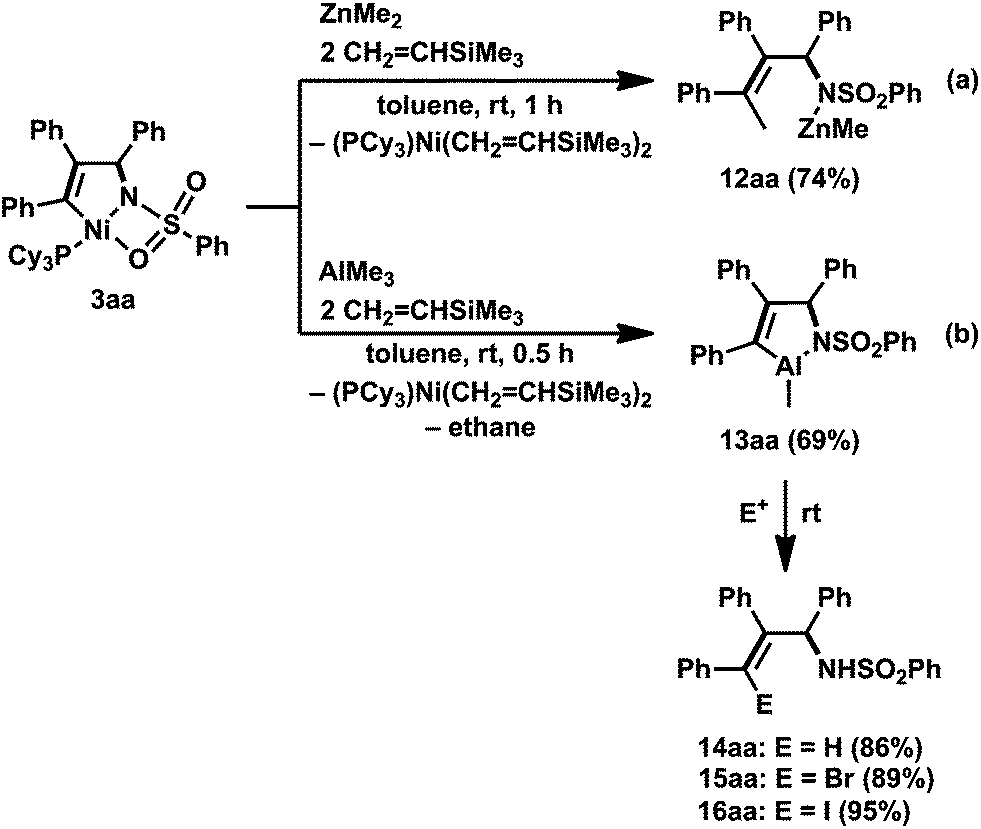 | ||
| Scheme 10 Reaction of five-membered aza-nickelacycle 3aa with methylmetal reagents. Yields of isolated products are given. Reaction conditions: for 14aa, MeOH/toluene, instance; for 15aa, NBS (2 equiv.) in CH3CN, 0.5 h; for 16aa, I2 (excess) in CH3CN, 2 h, then HCl. | ||
 | ||
| Scheme 11 Nickel(0)-catalyzed three-component coupling reaction of alkynes, imines, and organoboron reagents.13 | ||
Unexpectedly, a five-membered aza-aluminacycle (13aa) was obtained in 69% isolated yield when 3aa was treated with AlMe3 in place of ZnMe2 under identical reaction conditions (Scheme 10b).3h Monitoring of the reaction by means of 1H NMR spectroscopy indicated a concomitant generation of ethane (δH 0.80 ppm, in C6D6) and (PCy3)Ni(CH2CHTMS)2. An X-ray diffraction study of 13aa demonstrated that the aluminum atom was covalently bonded to both the carbon and the nitrogen atoms, C3 and N, respectively, to form a five-membered ring, and one methyl group, C4, also was bound to the aluminum center (Fig. 5b). As in the case of the methylzincamido 12aa, the five-membered aza-aluminacycle unit formed a dimer in the crystal lattice, and one of the oxygen atoms in the benzenesulfonyl group of 13aa was coordinated to the other aluminum atom. Unlike the three-component coupling product 12aa, the aza-aluminacycle 13aa is an organometallic reagent, in which the Al–C bond can react with electrophiles. Indeed, allylamine derivatives (14aa–16aa) could be obtained by treating 13aa with electrophiles such as proton and halogenonium (Scheme 10b).
The regeneration of the nickel(0) complex, (PCy3)Ni(CH2CHTMS)2, prompted us to develop a Ni(0)-catalyzed cyclocondensation of N-sulfonyl imine 1a, an alkyne, and AlMe3via the oxidative cyclization of 1a and the alkyne with nickel(0) as a key step. A major issue to be solved for constructing such a catalytic reaction was that the addition reaction of AlMe3 to 1a, yielding the corresponding amide (17; Table 2), also took place and was accelerated in the presence of 10 mol% of Ni(cod)2 and PCy3 (Scheme 12, right circle).16 We found that a slow addition of AlMe3 to the reaction mixture by using a syringe pump suppressed the undesired competitive reaction to give 17. Finally, the three-component cyclocondensation of 1a, 2a, and AlMe3 (slow addition, over 0.5 h) in the presence of 10 mol% of Ni(cod)2 and PCy3 afforded 13aa in 71% isolated yield (Table 2, entry 1).3h,17 Although the isolated yield of 13aa was somewhat decreased due to losses in the purification process, NMR analysis of the crude product indicated that this catalytic reaction proceeded quantitatively. In fact, protolysis of the crude product gave the corresponding allylamine 14aa in 86% isolated yield (entry 1). The same reaction conditions were applied successfully to diphenylacetylene derivatives, such as 1,2-bis(p-tolyl)acetylene (2h) and 1,2-bis(p-trifluoromethylphenyl)acetylene (2i), leading to the clean formation of 13ah and 13ai, respectively (entries 2 and 3). Furthermore, unsymmetrical alkynes were employed as coupling components in the cyclocondensation with 1a and AlMe3. Although the use of an excess (5 equiv.) amount of 1-phenyl-2-trimethylsilyl-acetylene (2j) was required for a smooth progression of the reaction, the corresponding aza-aluminacycle 13aj was formed in 85% yield as a single regioisomer (entry 4). By contrast, the reaction with 1-phenyl-1-propyne (2k) gave 13ak in 65% yield with 86:14 regioselectivity only when the slow addition of both AlMe3 and the alkyne was conducted to circumvent the insertion of the second alkyne into a five-membered aza-nickelacycle intermediate (entry 5). Dialkyl-substituted symmetrical alkynes such as 2-butyne 2b and 3-hexyne 2d did not react efficiently because of the rapid formation of the undesired 7-membered aza-nickelacycle even with the slow addition of a mixture of the alkyne and AlMe3 (entry 6). In the cases cited in runs 4, 5, and 6, the formation of 17 was observed in the 1H NMR spectra of the crude products.
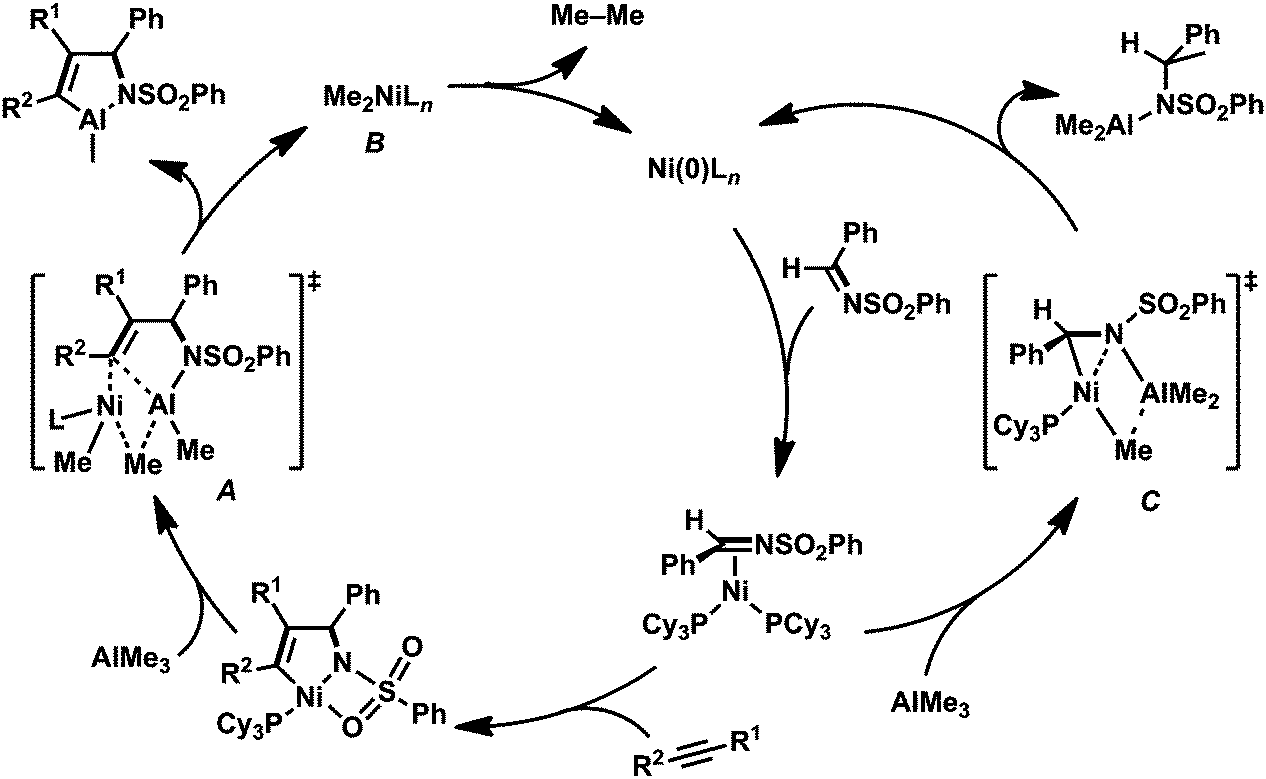 | ||
| Scheme 12 A plausible mechanism for the formation of aza-aluminacycle 13. | ||
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Alkyne 2 | Time (h) | Product | Yieldb (%) | Yield of 17c (%) |
| a General conditions: 1a, 2 (0.30 mmol each), and Ni(cod)2/PCy3 (0.03 mmol) were reacted in toluene (10.0 mL) at rt. After dropwise addition of AlMe3, the reaction mixture was stirred until the color derived from aza-nickelacycle 3 (typically purple) disappeared. b Isolated yield as allylamines 14 after protolysis. The values in parentheses are of isolated 13. c Cited yields, determined by 1H NMR, were of the corresponding protonated products. d The reaction was conducted using 1.5 mmol of 2j. e The reaction was conducted with concomitant addition of AlMe3 and 2k. f The minor regioisomer (11%) was also obtained. g Formation of a 1,2-dihydropyridine derivative was observed. | |||||
| 1 | 2a (R1, R2 = Ph) | 1.0 | 13aa | 86 (71) | — |
| 2 | 2h (R1, R2 = p-MeC6H4) | 6.0 | 13ah | 85 (82) | — |
| 3 | 2i (R1, R2 = p-CF3C6H4) | 3.0 | 13ai | 90 (99) | — |
| 4d | 2j (R1 = SiMe3, R2 = Ph) | 3.0 | 13aj | 85 (73) | 7 |
| 5e | 2k (R1 = Me, R2 = Ph) | 3.0 | 13ak | 65 (44) | 13 |
| 6 g | 2d (R1, R2 = Et) | 2.5 | 13ad | 27 | 59 |
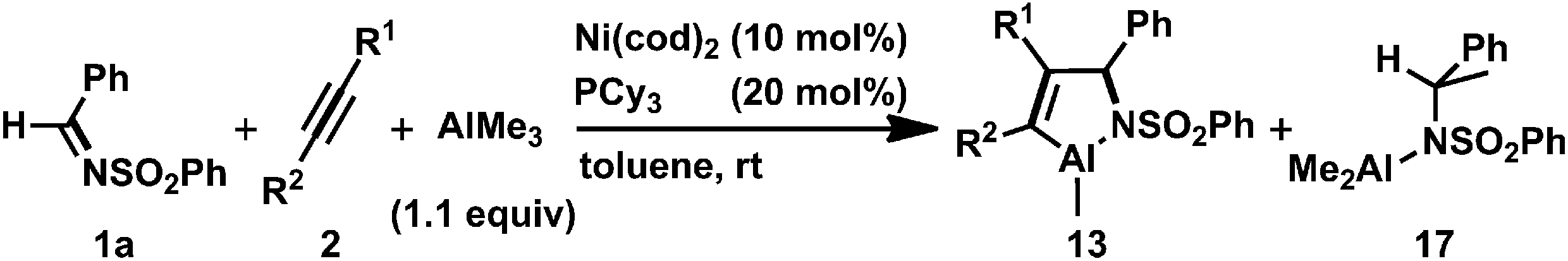
Based on the results of the stoichiometric reactions, the cyclocondensation reaction might proceed via the mechanism depicted in Scheme 12.3h As previously mentioned, the oxidative cyclization of an imine and an aldehyde with nickel gave a five-membered aza-nickelacycle. The transmetalation between the aza-nickelacycle and AlMe3 afforded a transient intermediate (A). Then, the nucleophilicity of the methyl group in A was high enough to allow the sequential transmetalation between nickel and aluminum, yielding the desired aza-aluminacycle 13 and a dimethyl nickel(II) intermediate (B). Reductive elimination from the dimethyl nickel(II) intermediate B might release ethane for the regeneration of a Ni(0) species.
However, if the nucleophilicity of the methyl group was insufficient, a reductive elimination from A might proceed to give a three-component coupling product, such as 12.13 We also confirmed that in the THF solution, trimethylaluminum can serve as an alkylmetal reagent in a three-component coupling reaction to give the corresponding amide. The key to the success of this catalytic reaction was the slow addition of AlMe3. Without this slow addition, the yield was significantly decreased as a result of the direct reaction of N-sulfonyl imine 1a with AlMe3 to give the side-reaction product 17. To the best of our knowledge, this is the first example of the catalytic formation of aza-aluminacyclopentenes, although cycloalumination of either olefins or acetylenes mediated by Cp2Zr derivatives has been used in the preparation of organoaluminum compounds.18 It should be mentioned that Montgomery and co-workers developed a related nickel-catalyzed cyclocondensation reaction of aldehydes, alkynes, and dialkylsilanes, leading to oxasilacyclopentens (Scheme 13a).19 In addition, Zhou and co-workers demonstrated the nickel-catalyzed reductive coupling of imines and alkynes with ZnEt2 as a reductant, providing allylic amines with a trisubstituted olefin moiety (Scheme 13b).20 Rhodium- or iridium-catalyzed reductive coupling reactions of imines and alkynes in the presence of H2 have been developed by Krische's group.1g,21
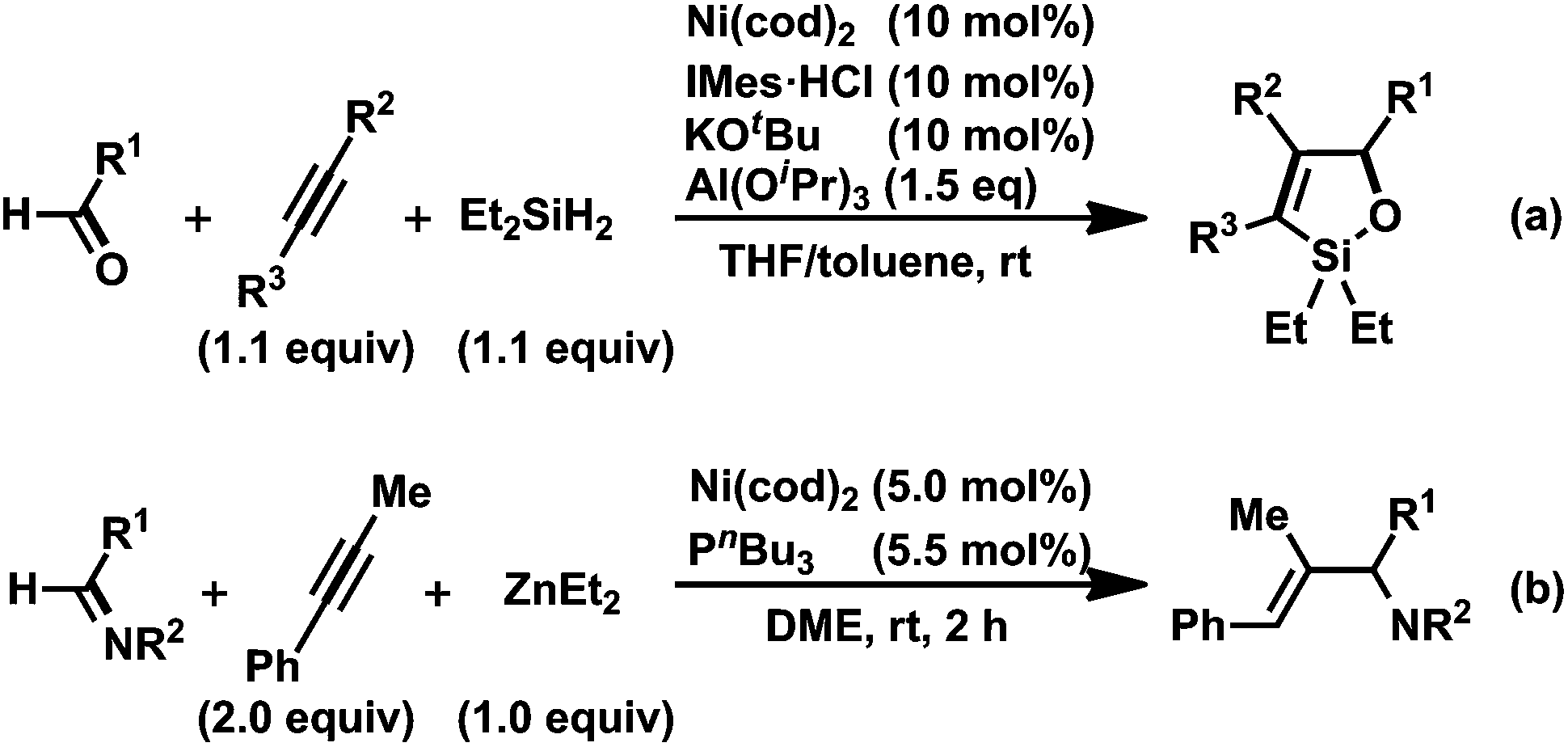 | ||
| Scheme 13 (a) Nickel(0)-catalyzed cyclocondensation reaction of aldehydes, alkynes, and Et2SiH2 and (b) nickel(0)-catalyzed reductive coupling of imines, alkynes, and ZnEt2.15,16 | ||
5. Nickel(0)-catalyzed formation of γ-lactams via [2 + 2 + 2] carbonylative cycloaddition reaction of an imine and an alkyne
Although a hetero-Pauson–Khand (or aza-Pauson–Khand) reaction, the transition-metal-catalyzed or mediated carbonylative cycloaddition of an imine, either an alkyne or an alkene, and CO, is known as a straightforward method for constructing a γ-lactam skeleton, a transition-metal-catalyzed carbonylative cycloaddition leading to α,β-unsaturated γ-lactams has historically been somewhat limited in the well-established Pauson–Khand reaction.8,22 In particular, despite the fact that treating the five-membered aza-nickelacycle 3 with CO indisputably took place to give α,β-unsaturated γ-lactams (Schemes 2 and 4), such nickel-catalyzed transformation reactions are totally hampered under a CO atmosphere due to the formation of catalytically unreactive nickel carbonyl complexes such as Ni(CO)3L. In order to establish a nickel(0)-catalyzed carbonylative cycloaddition, strict control of the CO concentration is assumed to be required. The CO concentration should be high enough to react with the aza-nickelacycle intermediate, yet simultaneously low enough not to generate a catalytically unreactive nickel tricarbonyl complex.Against this backdrop, the reaction of 3aa with phenyl formate and NEt3 was first conducted in various solvents at 60 °C (Scheme 14a).3o Phenyl formate has an interesting reactivity that allows it to generate CO in situ in the presence of organic/inorganic bases, and its application to transformation reactions involving carbonylation using CO generated in situ has been independently reported by Tsuji et al.23 and Manabe et al.24 As a result, 3aa was smoothly transferred into α,β-unsaturated γ-lactams 4aa in DMF-d7 and CD3CN, both of which were reported as suitable solvents to generate CO from phenyl formate.23,24a The formation of PhOH and Ni(CO)3(PCy3) was also observed by 1H and 31P NMR analyses. However, the reaction in C6D6, with a moderate efficiency of CO generation,23,24a afforded a rather complicated mixture that contained 4aa (37%), PhOH, and a trace amount of Ni(CO)3(PCy3).25 The carbonylation of 3aa generated in situ via the oxidative cyclization of 1a and 2a with Ni(cod)2/PCy3 was then investigated, and 4aa was again formed in excellent yield in DMF-d7, whereas CD3CN did not afford a positive result due to the poor solubility of Ni(cod)2 (Scheme 14b).
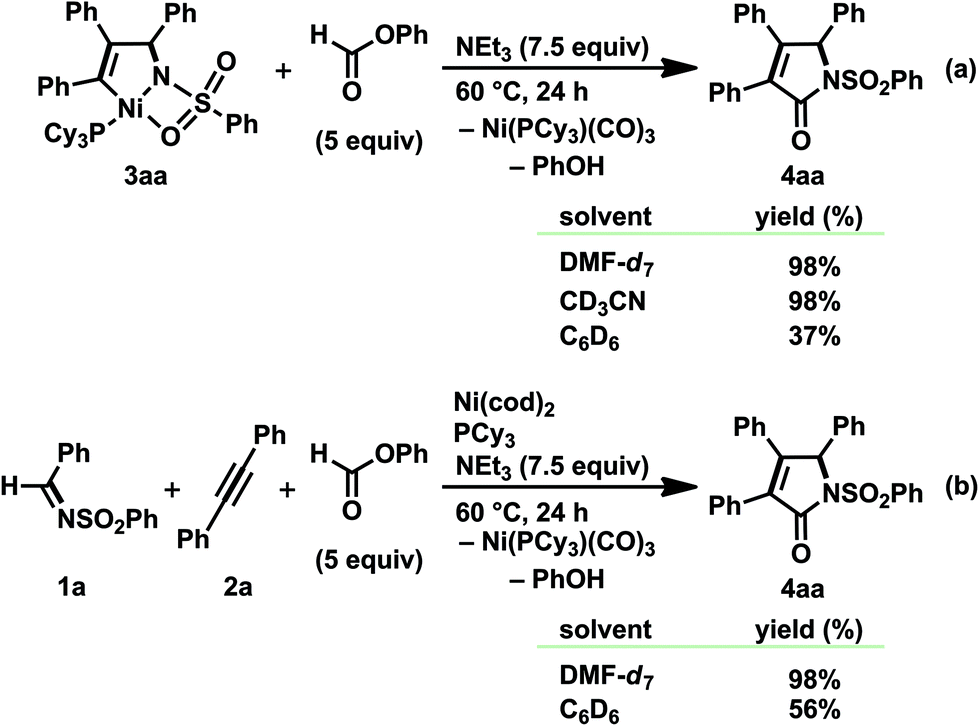 | ||
| Scheme 14 Reactions of (a) isolated or (b) in situ prepared five-membered aza-nickelacycle 3aa with phenyl formate in the presence of NEt3. Yields, determined by 1H NMR spectroscopy, are given. | ||
In contrast to the aforementioned stoichiometric reactions, an attempt at a nickel-catalyzed reaction in DMF was sluggish; the desired γ-lactam 4ac was not obtained at all from the reaction of 1a, 4-octyne 2c (1.0 equiv.), phenyl formate (1.5 equiv.), and NEt3 (2.0 equiv.) in the presence of Ni(cod)2 and PCy3 (10 mol% and 20 mol%, respectively).3o This was probably due to the rapid and quantitative formation of Ni(CO)3(PCy3) based on the amount of Ni(0). Therefore, the reaction was conducted in C6D6 in order to lower the rate of the in situ generation of CO. As a result, 4ac was formed in 44% yield at 60 °C over a period of 48 h.25 Elevating the reaction temperature to 70 °C and employing 2 equiv. of 2c promoted the reaction efficiency, and the yield of 4ac was improved to 74% (48 h), which was determined to be our optimum conditions. It should be mentioned that the choice of both the ligand and base was crucial for the smooth formation of 4ac; employing other tertiary phosphines or IPr26 dramatically diminished the yield of 4ac, and DBU, DMAP, and quinuclidine were not suitable under the presented conditions.
A variety of α,β-unsaturated γ-lactams 4 were prepared by the nickel(0)-catalyzed [2 + 2 + 1] carbonylative cycloaddition of imines 1 and alkynes 2 with phenyl formate (Scheme 15).3o Both alkyl-substituted symmetrical alkynes 2a and 2c as well as aryl-substituted ones, such as 1,2-bis(p-anisyl)acetylene (2l) and 1,2-bis(p-fluorophenyl)acetylene (2m), gave the corresponding γ-lactams (4aa, 4ac, 4al, and 4am) in moderate to good isolated yields. Neither bis(trimethylsilyl)acetylene (2n) nor dimethyl acetylenedicarboxylate (2o) gave the corresponding products probably due to difficulties with the simultaneous coordination of the alkyne 2n or 2o and 1a to nickel(0). In the former reaction, the coordination of 2n to nickel was hampered under these conditions, whereas the facile cyclotrimerization of 2o took place in the latter reaction.3e As anticipated from the regioselective formation of 11 (Scheme 8), 4ag was formed as a single regioisomer from 2-methyl-1-hexen-3-yne, 2g. In addition, 1-phenyl-2-trimethylsilyl-acetylene, 2j, also gave 4aj as a sole regioisomer in 69% yield. On the other hand, the reaction employing 1-phenyl-1-propyne, 2k, as an unsymmetric alkyne proceeded to afford 4ak in 83% yield as a mixture of regioisomers with a ratio of 89/11, and this ratio was comparable to that observed in the cyclocondensation reaction using 2k (Table 2, entry 5). The regioselectivity observed in these reactions with 2j and 2k might be due to the contribution of an η3-benzyl structure in a possible intermediate.27
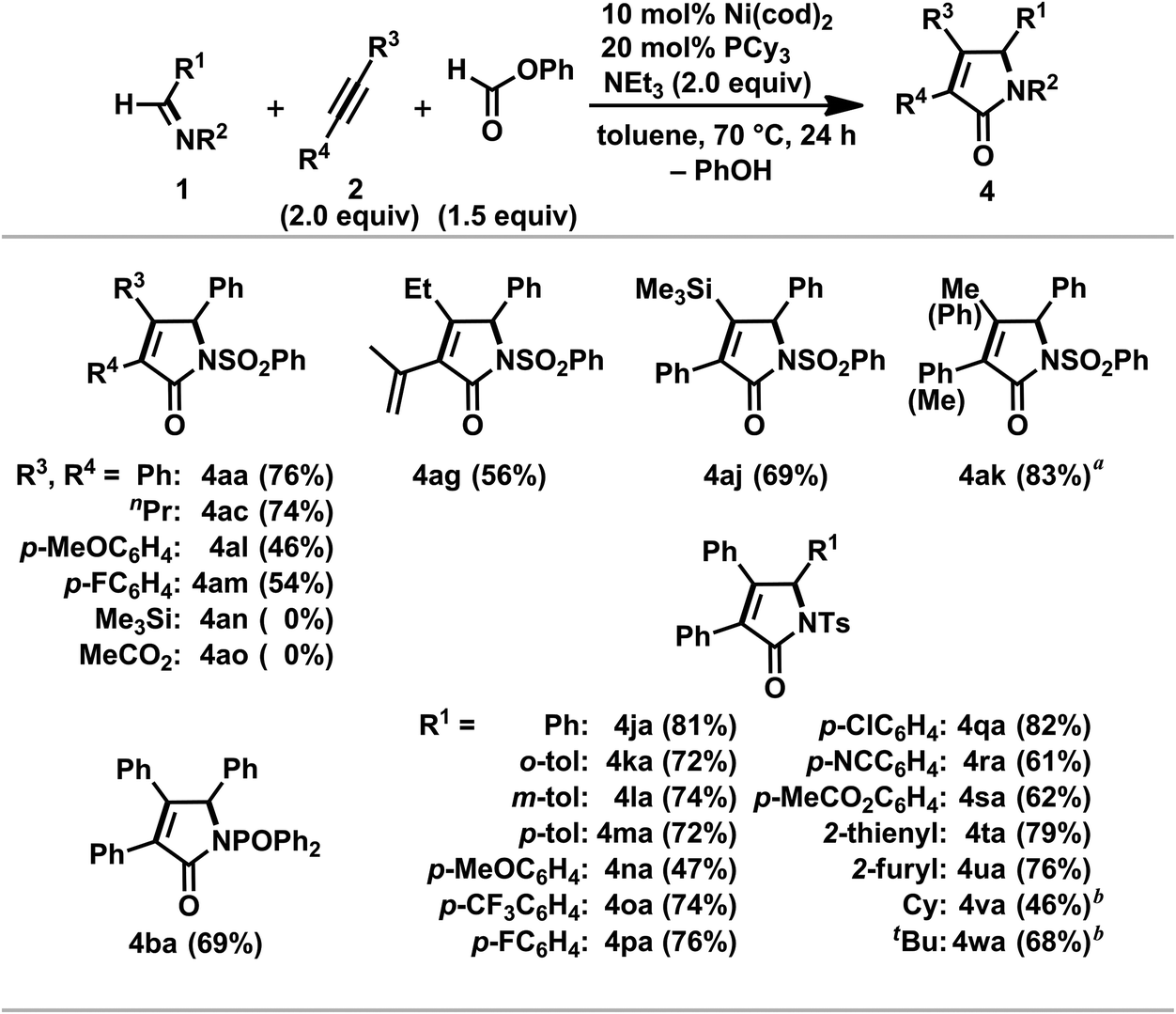 | ||
| Scheme 15 Nickel(0)/PCy3-catalyzed [2 + 2 + 1] carbonylative cycloaddition reaction of imines 1 and alkynes 2 with phenyl formate. General conditions: 1 (0.40 mmol), 2 (0.80 mmol), phenyl formate (0.60 mmol), NEt3 (0.80 mmol) and Ni(cod)2/PCy3 (0.04 and 0.08 mmol, respectively) were reacted in toluene (1.0 mL) at 70 °C for 24 h. Yields of isolated products are given. (a) The structure of the minor regioisomer was depicted in parentheses (major/minor = 89/11). (b) Ni(cod)2/PCy3 (0.08 and 0.16 mmol, respectively) was employed. | ||
The substrate scope with respect to imines was investigated with diphenylacetylene 2a under the optimal conditions.3o The catalytic reaction with N-phosphinyl imine 1b proceeded to give 4ba in 69% yield. A variety of N-benzylidene-toluenesulfonamide derivatives (1j–s) were applicable to the present catalytic system to yield the corresponding γ-lactams (4ja–sa) in good to high yields whereas a significant decrease in the yield of 4na was found in the case of 1n bearing an electron-rich arene ring. The thienyl- and furyl-substituted imines (1t and 1u) also gave 4ta and 4ua in 79% and 76% yields, respectively. Although an increase in the catalyst loading was required, alkyl-substituted N-tosylimines, such as CyCHNTs (1v) and tBuCHNTs (1w), participated in the carbonylative [2 + 2 + 1] cycloaddition reaction to give N-Cy- and N-tBu-γ-lactams (3na and 3oa) in 46% and 68% yields, respectively.
The reaction of carbonylative [2 + 2 + 1] cycloaddition reaction products 4aa or 4ja with a phosphide anion28 resulted in the removal of the N-arylsulfonyl groups, yielding a synthetically valuable N-protonated γ-lactam 18 in excellent yield (Scheme 16). Boc-protection of 18 was successfully achieved by treating with Boc2O and DMAP to give N-Boc-γ-lactam 19, which is regarded as an important synthetic intermediate.29,30 Combined with these derivatizations, the present catalytic system would afford a wide range of γ-lactams without the use of toxic CO gas and expensive transition metals under harsh reaction conditions, which were often found in the reports of related work.8d,22
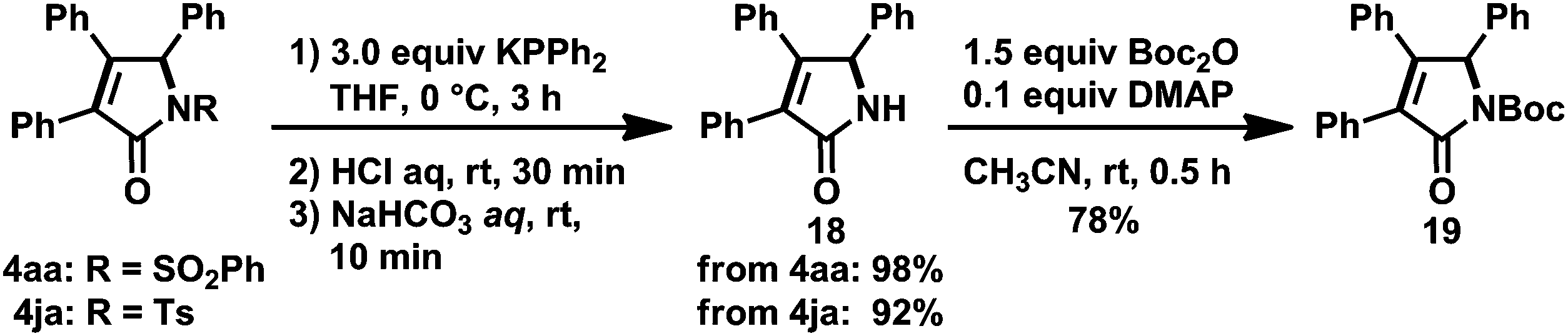 | ||
| Scheme 16 Synthesis of N-protonated and N-Boc-substituted γ-lactams 18 and 19. Yields of isolated products are given. | ||
6. Conclusion and outlook
Continuous efforts on the development of transition-metal-catalyzed cycloaddition and multicomponent coupling reactions have been made to allow the rapid preparation of highly complicated organic molecules from a variety of unsaturated compounds. As highlighted in this Perspective, catalytic transformation reactions that involve five-membered aza-nickelacycle intermediates generated via the oxidative cyclization of an imine and an alkyne with nickel(0) have been developed over the past few decades. The ingenious design of either N-substituents of imines or the ligand that coordinates to nickel, indeed, expands remarkably the scope of imine derivatives in practical synthetic applications. We are hopeful that the presented reactions will help provide further opportunities to develop novel catalytic transformations of imines, and will contribute to further the progress in this field of chemistry.Acknowledgements
We thank the following for support: MEXT, JST (ACT-C), Asahi Glass Foundation, Nippon Oil Corporation (ENEOS Hydrogen Trust Fund), and the Frontier Research Base for Global Young Researchers, Osaka University.Notes and references
- For selected recent reviews, see: (a) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901 CrossRef CAS PubMed; (b) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787 CrossRef CAS PubMed; (c) S. Kotha, E. Brahmachary and K. Lahiri, Eur. J. Org. Chem., 2005, 4741 CrossRef CAS PubMed; (d) P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307 CrossRef CAS PubMed; (e) K. Tanaka, Synlett, 2007, 1977 CrossRef CAS PubMed; (f) B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085 RSC; (g) E. Skucas, M.-Y. Ngai, V. Komanduri and M. J. Krische, Acc. Chem. Res., 2007, 40, 1394 CrossRef CAS PubMed; (h) T. Shibata and K. Tsuchikama, Org. Biomol. Chem., 2008, 6, 1317 RSC; (i) B. R. Galan and T. Rovis, Angew. Chem., Int. Ed., 2009, 48, 2830 CrossRef CAS PubMed; (j) H. A. Reichard, M. McLaughlin, M. Z. Chen and G. C. Micalizio, Eur. J. Org. Chem., 2010, 391 CrossRef CAS PubMed.
- For reviews on the nickel-catalyzed reactions via (hetero)nickelacycle intermediate, see: (a) Modern Organo Nickel Chemistry, ed. Y. Tamaru, Willey-VCH, Weinheim, Germany, 2005 Search PubMed; (b) S.-I. Ikeda, Angew. Chem., Int. Ed., 2003, 42, 5120 CrossRef CAS PubMed; (c) J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890 CrossRef CAS PubMed; (d) R. M. Moslin, K. Miller-Moslin and T. F. Jamison, Chem. Commun., 2007, 4441 RSC; (e) S.-S. Ng, C.-Y. Ho, K. D. Schleicher and T. F. Jamison, Pure Appl. Chem., 2008, 80, 929 CrossRef CAS PubMed; (f) K. Tanaka and Y. Tajima, Eur. J. Org. Chem., 2012, 3715 CrossRef CAS PubMed; (g) J. Montgomery, Organonickel Chemistry, in Organometallics in Synthesis: Fourth Manual, ed. B. H. Lipshutz, Wiley, Hoboken, N.J., 2013, pp. 319–428 Search PubMed; (h) S. Ogoshi, Yuki Gosei Kagaku Kyokaishi, 2013, 71, 14 CrossRef CAS; (i) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- For examples of our related work, see: (a) S. Ogoshi, M. Oka and H. Kurosawa, J. Am. Chem. Soc., 2004, 126, 11802 CrossRef CAS PubMed; (b) S. Ogoshi, M. Ueta, T. Arai and H. Kurosawa, J. Am. Chem. Soc., 2005, 127, 12810 CrossRef CAS PubMed; (c) S. Ogoshi, M. Nagata and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 5350 CrossRef CAS PubMed; (d) S. Ogoshi, K.-i. Tonomori, M.-a. Oka and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 7077 CrossRef CAS PubMed; (e) S. Ogoshi, H. Ikeda and H. Kurosawa, Angew. Chem., Int. Ed., 2007, 46, 4930 CrossRef PubMed; (f) S. Ogoshi, H. Ikeda and H. Kurosawa, Pure Appl. Chem., 2008, 80, 1115 CrossRef CAS; (g) T. Tamaki, M. Nagata, M. Ohashi and S. Ogoshi, Chem. – Eur. J., 2009, 15, 10083 CrossRef CAS PubMed; (h) M. Ohashi, O. Kishizaki, H. Ikeda and S. Ogoshi, J. Am. Chem. Soc., 2009, 131, 9160 CrossRef CAS PubMed; (i) M. Ohashi, H. Saijo, T. Arai and S. Ogoshi, Organometallics, 2010, 29, 6534 CrossRef CAS; (j) T. Tamaki, M. Ohashi and S. Ogoshi, Chem. Lett., 2011, 40, 248 CrossRef CAS; (k) Y. Hoshimoto, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 4668 CrossRef CAS PubMed; (l) A. Nishimura, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2012, 134, 15692 CrossRef CAS PubMed; (m) Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2012, 51, 10812 CrossRef CAS PubMed; (n) Y. Hoshimoto, T. Ohata, M. Ohashi and S. Ogoshi, Chem. – Eur. J., 2014, 20, 4105 CrossRef CAS PubMed; (o) Y. Hoshimoto, T. Ohata, Y. Sasaoka, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2014, 136, 15877 CrossRef CAS PubMed.
- A very recent review on oxa-nickalacycles, see: Y. Hoshimoto, M. Ohashi and S. Ogoshi, Acc. Chem. Res. DOI:10.1021/acs.accounts.5b00061.
- For seminal studies on isolation of nickelalactams generated from an alkyne, an isocyanate, and nickel(0), see: (a) H. Hoberg and B. W. Oster, J. Organomet. Chem., 1982, 234, C35 CrossRef CAS; (b) H. Hoberg and B. W. Oster, J. Organomet. Chem., 1983, 252, 359 CrossRef CAS; A seminal study on nickel-catalyzed reaction of two alkynes and a carbodiimide to give a 2-imino-1,2-dihydropyridine, see: (c) H. Hoberg and G. Burkhart, Synthesis, 1979, 525 CrossRef CAS.
- For reviews on the preparation of dihydropyridines, see: (a) M. P. Silva, P. A. M. M. Varandas and A. M. S. Silva, Synthesis, 2013, 3053 CrossRef PubMed; (b) R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141 RSC; (c) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223 CrossRef CAS; (d) U. Eisnerand and J. Kuthan, Chem. Rev., 1972, 72, 1 CrossRef.
- For reviews on the preparation of allylic amines, see: (a) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931 RSC; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (c) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed; (d) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed. See also ref. 1g.
- For reviews on the preparation of γ-lactams via hetero-Pauson–Khand (or aza-Pauson–Khand) reactions, see: (a) R. R. Torres, The Pauson–Khand Reaction: Scope, Variations and Applications, John Wiley & Sons, UK, 2012 Search PubMed; (b) I. Omae, Coord. Chem. Rev., 2011, 255, 139 CrossRef CAS PubMed; (c) J. H. Park, K.-M. Chang and Y. K. Chung, Coord. Chem. Rev., 2009, 253, 2461 CrossRef CAS PubMed; (d) N. Chatani, Chem. Rec., 2008, 8, 201 CrossRef CAS PubMed.
- K.-i. Fujita, M. Yamashita, F. Puschmann, M. M. Alvarez-Falcon, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9044 CrossRef CAS PubMed.
- (a) J. Kovach, M. Peralta, W. W. Brennessel and W. D. Jones, J. Mol. Struct., 2011, 992, 33 CrossRef CAS PubMed; (b) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- The precipitates were not fully identified; however, the product might be an imine-IPr adduct or its enamine-type isomer. See: (a) D. A. DiRocco, K. M. Oberg and T. Rovis, J. Am. Chem. Soc., 2012, 134, 6143 CrossRef CAS PubMed; (b) M. He and J. W. Bode, Org. Lett., 2005, 7, 3131 CrossRef CAS PubMed.
- L. Adak, W. C. Chan and N. Yoshikai, Chem. – Asian J., 2011, 6, 359 CrossRef CAS PubMed.
- (a) S. J. Patel and T. F. Jamison, Angew. Chem., Int. Ed., 2003, 42, 1364 CrossRef CAS PubMed; (b) S. J. Patel and T. F. Jamison, Angew. Chem., Int. Ed., 2004, 43, 3941 CrossRef CAS PubMed.
- (a) P. Liu, P. McCarren, P. H.-Y. Cheong, T. F. Jamison and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2050 CrossRef CAS PubMed; (b) K. M. Miller, T. Luanphaisarnnont, C. Molinaro and T. F. Jamison, J. Am. Chem. Soc., 2004, 126, 4130 CrossRef CAS PubMed.
- (a) P. A. Wender, T. M. Pederson and M. J. C. Scanio, J. Am. Chem. Soc., 2002, 124, 15154 CrossRef CAS PubMed; (b) M. Amatore, D. Leboeuf, M. Malacria, V. Gandon and C. Aubert, J. Am. Chem. Soc., 2013, 135, 4576 CrossRef CAS PubMed.
- K. Biswas, O. Prieto, P. J. Goldsmith and S. Woodward, Angew. Chem., Int. Ed., 2005, 44, 2232 CrossRef CAS PubMed.
- By contrast, slow addition of ZnMe2 was unnecessary due to no occurrence of the corresponding direct addition in the nickel-catalyzed three-component coupling reaction.
- Selected reviews, see: (a) U. M. Dzhemilev and A. G. Ibragimov, Russ. Chem. Rev., 2000, 69, 121 CrossRef CAS PubMed; (b) U. M. Dzhemilev and A. G. Ibragimov, J. Organomet. Chem., 1994, 466, 1 CrossRef CAS.
- R. D. Baxter and J. Montgomery, J. Am. Chem. Soc., 2008, 130, 9662 CrossRef CAS PubMed.
- C.-Y. Zhou, S.-F. Zhu, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 10955 CrossRef CAS PubMed.
- (a) J.-R. Kong, C.-W. Cho and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 11269 CrossRef CAS PubMed; (b) E. Skucas, J. R. Kong and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 7242 CrossRef CAS PubMed; (c) A. Barchuk, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 8432 CrossRef CAS PubMed; (d) M.-Y. Ngai, A. Barchuk and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 12644 CrossRef CAS PubMed.
- For selected recent reports on the synthesis of lactams via carbonylative cycloaddition reaction, see: (a) X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447 CrossRef CAS PubMed; (b) T. Iwata, F. Inagaki and C. Mukai, Angew. Chem., Int. Ed., 2013, 52, 11138 CrossRef CAS PubMed and references therein; (c) T. Fukuyama, N. Nakashima, T. Okada and I. Ryu, J. Am. Chem. Soc., 2013, 135, 1006 CrossRef CAS PubMed and references therein.
- T. Fujihara, T. Hosoki, Y. Katafuchi, T. Iwai, J. Terao and Y. Tsuji, Chem. Commun., 2012, 48, 8012 RSC.
- (a) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2012, 14, 3100 CrossRef CAS PubMed; (b) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2013, 15, 5370 CrossRef CAS PubMed; (c) T. Ueda, H. Konishi and K. Manabe, Angew. Chem., Int. Ed., 2013, 52, 8611 CrossRef CAS PubMed.
- The formation of an unidentified compound, whose 31P resonance appeared at δ 41.8, was observed under these stoichiometric conditions conducted in C6D6 (Scheme 14a). However, the formation of this compound was not observed under the catalytic conditions.
- IPr was found to react with phenyl formate as a base. Thus, rapid formation of Ni(CO)3(IPr) was observed in the presence of IPr as a ligand.
- A similar regioselectivity was observed in Ni(0)-catalyzed reactions. See: (a) A. Herath, W. Li and J. Montgomery, J. Am. Chem. Soc., 2008, 130, 469–471 CrossRef CAS PubMed; (b) A. Herath, W. Li and J. Montgomery, J. Am. Chem. Soc., 2009, 131, 17024–17029 CrossRef PubMed; (c) S. Ogoshi, A. Nishimura and M. Ohashi, Org. Lett., 2010, 12, 3450–3452 CrossRef CAS PubMed.
- S. Yoshida, K. Igawa and K. Tomooka, J. Am. Chem. Soc., 2012, 134, 19358 CrossRef CAS PubMed.
- For examples of N-Boc-substituted α,β-unsaturated γ-lactams as synthetic intermediates, see: (a) K. P. Jayasundera, H. Kinoshita and K. Inomata, Chem. Lett., 1998, 27, 1227 CrossRef; (b) D. Farran, I. Parrot, J. Martinez and G. Dewynter, Angew. Chem., Int. Ed., 2007, 46, 7488 CrossRef CAS PubMed; (c) M. Storgaard, F. Z. Dörwald, B. Peschke and D. Tanner, J. Org. Chem., 2009, 74, 5032 CrossRef CAS PubMed; (d) G. Chaubet, T. Coursindel, X. Morelli, S. Betzi, P. Roche, Y. Guari, A. Lebrun, L. Toupet, Y. Collette, I. Parrot and J. Martinez, Org. Biomol. Chem., 2013, 11, 4719 RSC.
- For examples, see: (a) G. Casiraghi, G. Rassu, P. Spanu and L. Pinna, J. Org. Chem., 1992, 57, 3760 CrossRef CAS; (b) G. Rassu, F. Zanardi, L. Battistini, E. Gaetani and G. Casiraghi, J. Med. Chem., 1997, 40, 168 CrossRef CAS PubMed; (c) F. Zanardi, L. Battistini, M. Nespi, G. Rassu, P. Spanu, M. Cornia and G. Casiraghi, Tetrahedron: Asymmetry, 1996, 7, 1167 CrossRef CAS; (d) G. Rassu, L. Pinna, P. Spanu, F. Zanardi, L. Battistini and G. Casiraghi, J. Org. Chem., 1997, 62, 4513 CrossRef CAS; (e) L. Planas, J. Pérard-Viret and J. Royer, J. Org. Chem., 2004, 69, 3087 CrossRef CAS PubMed; (f) C. Curti, A. Sartori, L. Battistini, G. Rassu, F. Zanardi and G. Casiraghi, Tetrahedron Lett., 2009, 50, 3428 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |