
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in alkene hydro-functionalisation promoted by homogeneous catalysts based on earth abundant elements: formation of C–N, C–O and C–P bond
Violeta
Rodriguez-Ruiz
a,
Romain
Carlino
a,
Sophie
Bezzenine-Lafollée
a,
Richard
Gil
a,
Damien
Prim
b,
Emmanuelle
Schulz
ac and
Jérôme
Hannedouche
*ac
aUniv Paris-Sud, ICMMO, UMR 8182, Orsay, F-91405, France. E-mail: jerome.hannedouche@u-psud.fr
bUniversité Versailles Saint-Quentin, ILV, UMR8180, 45, avenue des Etats-Unis, Versailles, F-78035, France
cCNRS, Orsay, F-91405, France
First published on 16th March 2015
Abstract
This Perspective article provides an overview of the recent advancements in the field of intra- and inter-molecular C–N, C–O and C–P bond formation by hydroamination, hydroalkoxylation, hydrophosphination, hydrophosphonylation or hydrophosphinylation of unactivated alkenes, including allenes, 1,3-dienes and strained alkenes, promoted by (chiral) homogeneous catalysts based on earth abundant elements of the s and p blocks, the first row transition metals and the rare-earth metals. The relevant literature from 2009 until late 2014 has been covered.
 Violeta Rodriguez-Ruiz | Violeta Rodriguez-Ruiz received her B.Sc. in Pharmacy at the University of Valencia, Spain. She got her M.Sc. in Chemistry at the Technical Institute of Chemistry (ITQ, UPV-CSIC), where she carried out her Ph.D. at Prof. Corma's group. In 2011, she obtained a Marie Curie-ITN Experienced Researcher fellowship to work at the “Institut Galien Paris-Sud” (Faculty of Pharmacy, CNRS-Université Paris-Sud). In 2012, she joined the “Laboratory of Molecular Catalysis” (ICMMO, Faculty of Sciences, CNRS-Université Paris-Sud) as a Post-Doctoral Fellow. Since 2014, she works at the “Laboratory for Vascular Translational Science” (Institut Galilée, Université Paris13-INSERM) as Temporary Lecturer and Research Assistant. |
 Romain Carlino | Romain Carlino was born in 1988 in Monaco. He obtained his diploma from Ecole Nationale Supérieure de Chimie de Clermont-Ferrand and his master degree at Université Blaise Pascal in 2013. He is currently a PhD student at ICMMO (Université Paris-Sud) working on rare-earth complexes for catalytic applications in hydroalkoxylation reaction under the supervision of Dr Sophie Bezzenine-Lafollée and Dr Richard Gil. |
 Sophie Bezzenine-Lafollée | Sophie Bezzenine-Lafollée received her Ph.D. degree from Université Paris VI in 1998. After a postdoctoral stay on asymmetric synthesis with Pr. P.Müller at the University of Geneva, Switzerland, she spent two years in the laboratory of Pr. J. Ardisson at the University of Cergy-Pontoise working on total synthesis. She became Maître de Conférence in Orsay University in 2001, she firstly worked with Drs F. Guibé and J. Collin on lanthanides chemistry. Recently, she collaborated with Pr. V. Gandon on silver-free two-component approach in gold catalysis and since 2010, she works with Dr R. Gil on enantioselective catalysis with rare earths complexes. |
 Richard Gil | Richard Gil studied chemistry at Université Paris-Sud (Orsay, France) where he received his PhD in 1993. His thesis work was under the supervision of Professor Jean-Claude Fiaud and dealt with enantioselective palladium-catalysed reactions. He then spent one post-doctoral year (1993–1994) at Imperial College London under the group of Pr. Susan E. Gibson. In 1995, he went to Université de Cergy-Pontoise and became Maître de Conférences in 1996. In 2002, he returned to ICMMO where he joined the Dr Jacqueline Collin group in which he has been interested in lanthanide-catalysed reactions. His research interests include enantioselective reactions catalysed by rare earths. |
 Damien Prim | Damien Prim received his PhD at the University Paul Verlaine of Metz in 1994 under the guidance of Pr. G. Kirsch. He joined Pr. L. Ghosez group at the University Catholique de Louvain, Belgium, for a one year postdoctoral training. After four years (1995–1999) as Assistant Professor at the University Paul Verlaine, he moved to the University Pierre et Marie Curie, Paris, as CNRS research associate (1999–2001) and joined the University of Versailles in 2001. His current research interests concern homogeneous catalysis, multitask ligands and their application to the synthesis of helical, twisted and planar molecular architectures. |
 Emmanuelle Schulz | Emmanuelle Schulz graduated from ESCIL in Lyon and received her Ph.D. degree in 1992 for studies concerning the total synthesis of Strigol (Pr. P. Welzel, Ruhr-Universität Bochum/Université de Lyon). After a postdoc at RP Agro, she joined the group of Pr. M. Lemaire and obtained a position at the CNRS. Since 2000, she is at ICMMO (Université Paris Sud). Her research interests are mainly directed towards asymmetric catalysis. She particularly explores the enantioselective hydroamination reaction promoted by chiral rare-earth based catalysts. New procedures for the easy recovery and reuse of chiral organometallic catalysts are also developed in her group. |
 Jérôme Hannedouche | After completing a PhD degree under the supervision of Pr. Martin Wills (University of Warwick, UK) in 2003, Jérôme Hannedouche undertook a 2-year postdoctoral training with Pr. Olivier Riant (UCL, Belgium). In October 2006, he was appointed at the Institut de Chimie Moléculaire et des Matériaux d'Orsay (ICMMO) (Université Paris-Sud, France) as a CNRS fellow researcher. His research interests lie in the development of novel synthetic methodologies in asymmetric catalysis, mainly for C–N bond formation via hydroamination reaction. |
1. Introduction
The wide applications of nitrogen-, oxygen- and phosphorus-based organic compounds in various domains such as pharmaceuticals, fine and bulk chemicals, agrochemicals, biological and coordination chemistry or catalysis amongst others have driven the interest of the scientific community with a strong demand for the development of sustainable, more selective and efficient processes for their syntheses. Among the plethora of synthesis routes, the addition of E–H (E = N, O, P) across an unactivated carbon–carbon double bond, the more generally called hydrofunctionalisation (or hydroelementation)1 reaction of unactivated alkene, promoted by a metal or metal-free catalyst is a very promising field of research towards the development of an economic and environmentally friendly synthetic methodology. Indeed, the hydrofunctionalisation reaction provides a 100% atom efficiency and waste-free process from relatively low-cost and ubiquitous starting materials. Furthermore, catalysis brings the opportunity to fine-tune the regio-, chemo- and stereoselectivity of the overall transformation by a perfect synergy between the metal and the ligand(s) or by the direct design of a metal-free system. However, despite the inherent interesting features of the transformation, it is very challenging to control the regioselectivity of this reaction providing exclusively the Markovnikov or the anti-Markovnikov adduct. As a general trend, Markovnikov selectivity is the most common selectivity noticed in the addition reactions of N–H and O–H onto unactivated alkenes while the addition of P–H provides usually the anti-Markovnikov product. For the corresponding addition reaction, few contributions exhibit the opposite regioselectivity.2–4 These features highlight the challenges to access both regioisomeric products which are all of synthetic value. Additionally to the regioselectivity, the control of stereoselectivity of the newly generated stereogenic carbon of the branched product is also an important issue of this process. For example, the intermolecular and highly enantio- and regioselective addition of amines to simple aliphatic alkenes still remains one of the most unresolved problems in alkene hydroamination5 as is the control of the enantioselectivity in alkene hydroalkoxylation.3d–e,6 The last decades have witnessed an intensive research activity focusing on the development of metal-based and metal-free catalysts to promote the addition of E–H across an unactivated carbon–carbon double bond intra- and intermolecularly. Although tremendous progresses have been gained from the study of precious metal catalysts,1 the growing request for novel, more economical and eco-friendly catalytic systems has stimulated the exploration of earth abundant elements as alternatives to address some of the issues.This Perspective article will outline some of the recent advancements in the field of intra- and inter-molecular C–N, C–O and C–P bond formation by hydroamination, hydroalkoxylation, hydrophosphination, hydro-phosphonylation or hydrophosphinylation of unactivated alkenes, including allenes, 1,3-dienes and strained alkenes, promoted by (chiral) catalytic systems based on earth abundant elements. The hydration of alkenes is not included and the reader should refer to reported reviews on the topic.7 This article will focus on systems based on Group 1–3 elements, the most relevant first-row transition metal elements of the field (titanium, iron, cobalt, nickel, copper and zinc) and also the block p element aluminum. It is beyond the scope of this article to comprehensively review all the research activity of this broad field and literature coverage has been mainly limited from 2009 until 2014, even if a few representative reports appeared earlier are included. This article has been divided according to the Group, the element within the Group and the type of C–E (E = N, O, P) bond formed in the transformation. For a broader overview, the latest developments in metal free-catalysed C–E (E = N, O, P) bond formation by alkene hydrofunctionalisation have also been included and will start this Perspective.
2. Metal-free catalysts
2.1 C–N bond formation
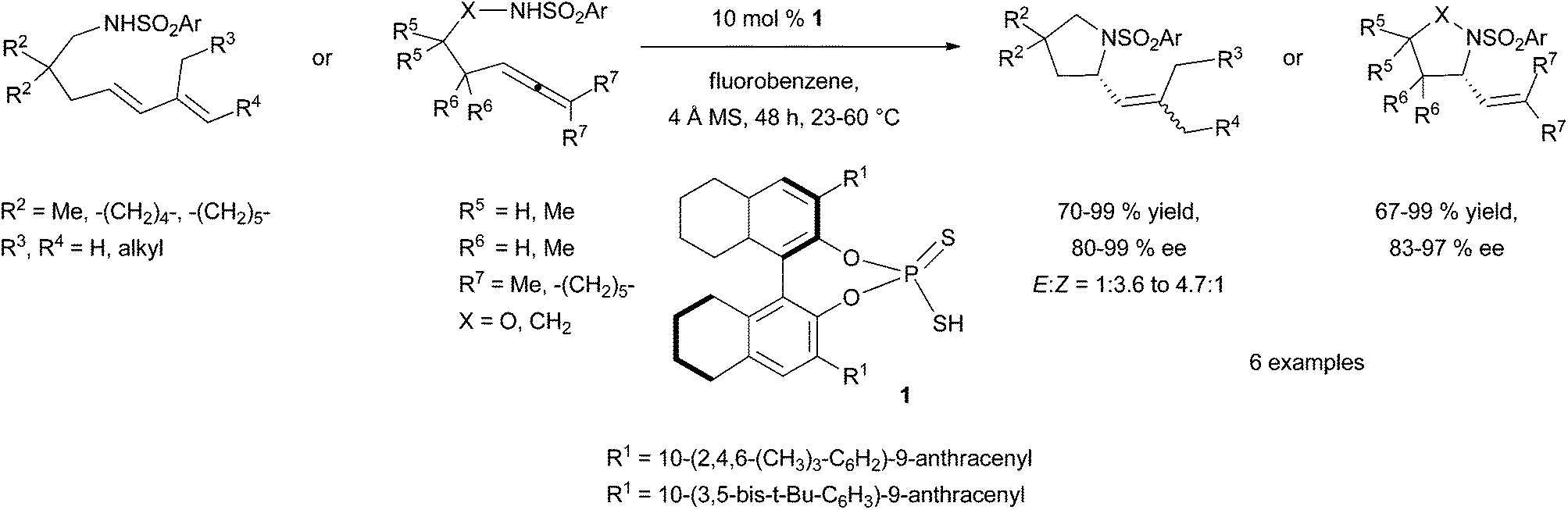 | ||
| Scheme 1 Enantioselective intramolecular hydroamination of amines tethered to 1,3- and 1,2-dienes with chiral dithiophosphoric acids. | ||
This strategy was successfully developed by the perfect match between the chiral tethering catalyst and both alkene and amine partners. Extensive screening of aldehydes and ketones have identified benzyloxyacetaldehyde 2 as the appropriate metal-free tethering catalyst for intermolecular Cope-type hydroamination of allylic amines and α-benzylhydroxylamine (20 mol%) at room temperature (Scheme 2). Mechanistic studies suggest that the hydroamination step is the rate-limiting step of the catalytic cycle and lead to the identification that paraformaldehyde (5–10 mol%) is a more efficient metal-free catalyst providing higher yields of hydroamination products.14 The use of 20 mol% of (R)-glyceraldehyde 3 allowed the development of an enantioselective variant of this process to furnish enantioenriched vicinal diamine derivatives in high yields and with ee values of up to 87% after 24 h at room temperature (Scheme 2). The reaction is proposed to proceed through the formation of a mixed aminal intermediate resulting from nucleophilic attack of the nitrone (resulting from aldehyde-hydroxylamine condensation) by the allylic amine. This methodology is so far restricted to simple allylic amines as homoallylic amines and disubstituted allylic amines led to catalyst deactivation.
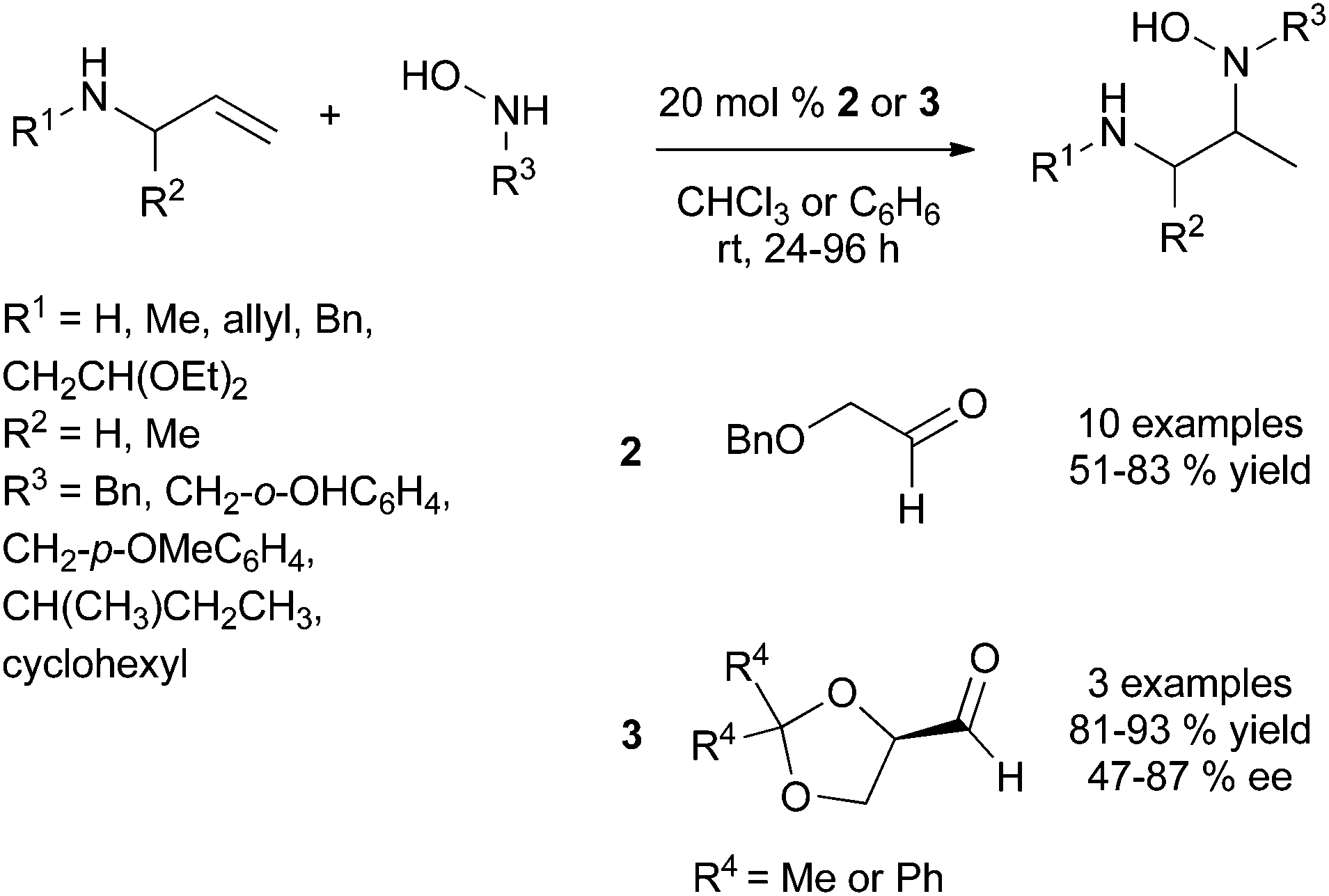 | ||
| Scheme 2 Intermolecular Cope-type hydroamination catalysed by achiral and chiral aldehydes. | ||
More recently, the group of Jacobsen has investigated the use of chiral thioureas to stabilise the polar transition state of the Cope-type intramolecular hydroamination reaction through hydrogen-bonding interactions (Scheme 3).15 In this context, extensive screening of a variety of chiral thiourea catalysts bearing polarisable and conformationally constrained aromatic components was conducted and led to the identification of thiourea 4 as the optimal catalyst structure. Various 4-alkenyl-1-hydroxylamines bearing in some cases electronically different (E)-styryl moieties, cyclise in good to high yields and enantioselectivities under the optimal mild conditions displayed in Scheme 3. It is suggested that additional cation–π interactions between the cationic transition state and the arene of the catalyst occur in the process.
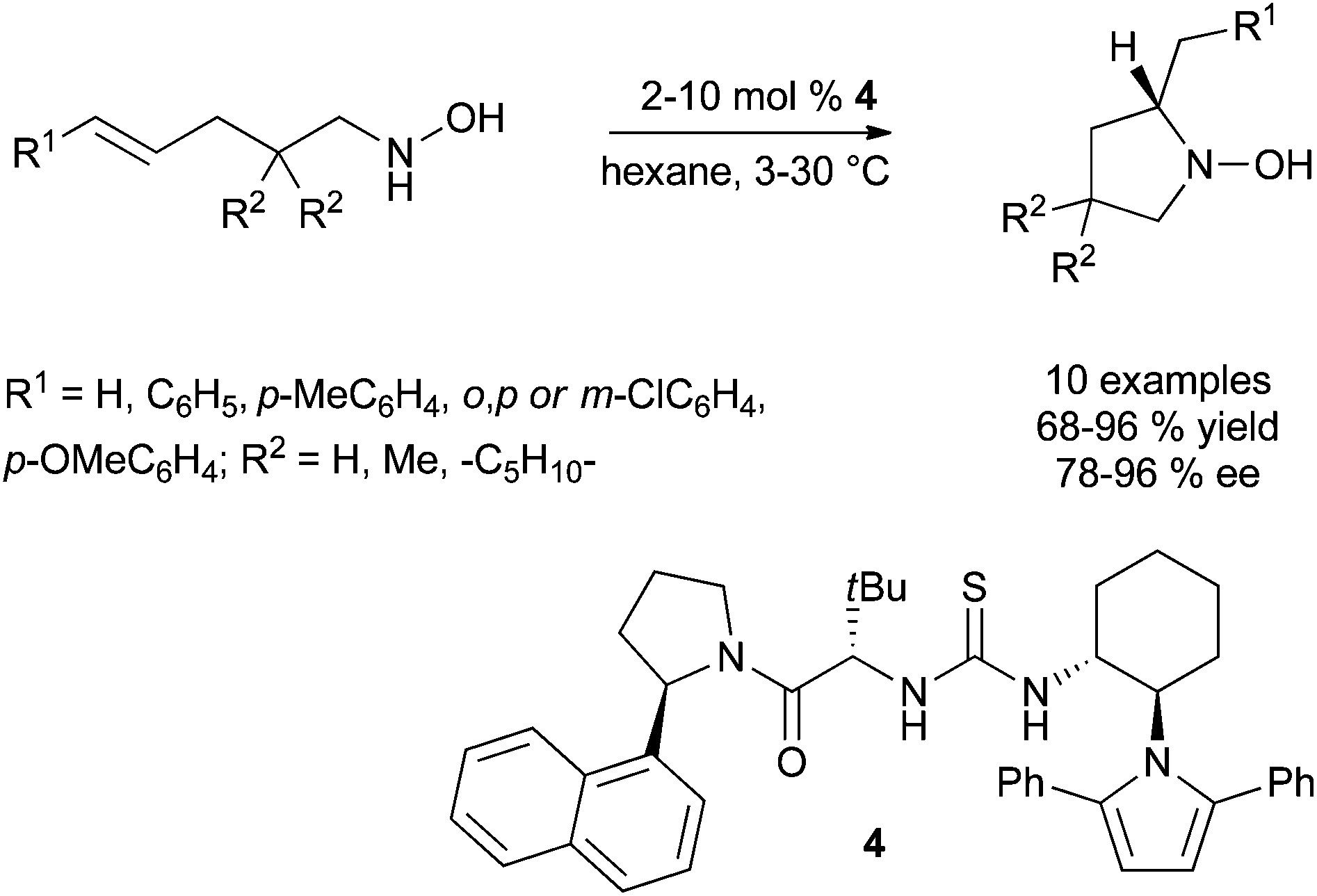 | ||
| Scheme 3 Enantioselective thiourea-catalysed intramolecular Cope-type hydroamination. | ||
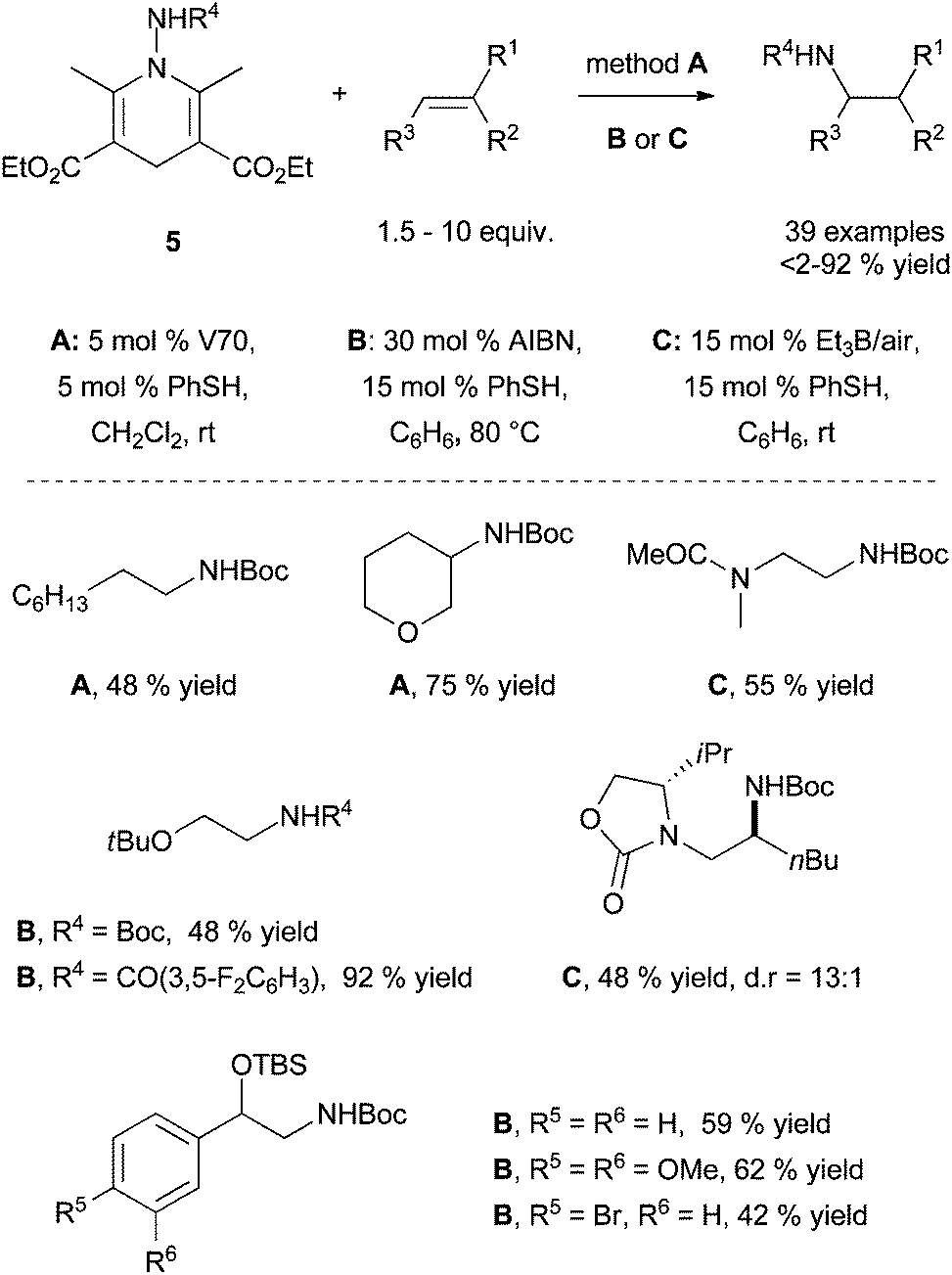 | ||
| Scheme 4 Intermolecular radical transfer hydroamination with N-aminated Hantzsch ester 5 using polarity-reversal catalysis. | ||
The process involves a regioselective N-radical addition onto the alkene to deliver a C-radical (Scheme 5). In the presence of a catalytic amount of a thiol as polarity reversal catalyst, efficient H-transfer occurs giving the hydroamination product and forming a thiyl radical. Subsequently, the latter undergoes hydrogen abstraction from the N-aminated dihydropyridine to regenerate the thiol catalyst and form 6. Homolytic cleavage of the weak N–N bond of 6 should regenerate the N-centered radical and liberate pyridine derivative 7. The polarity reversal agent allows an efficient hydrogen-transfer reaction to the C-centered radicals.
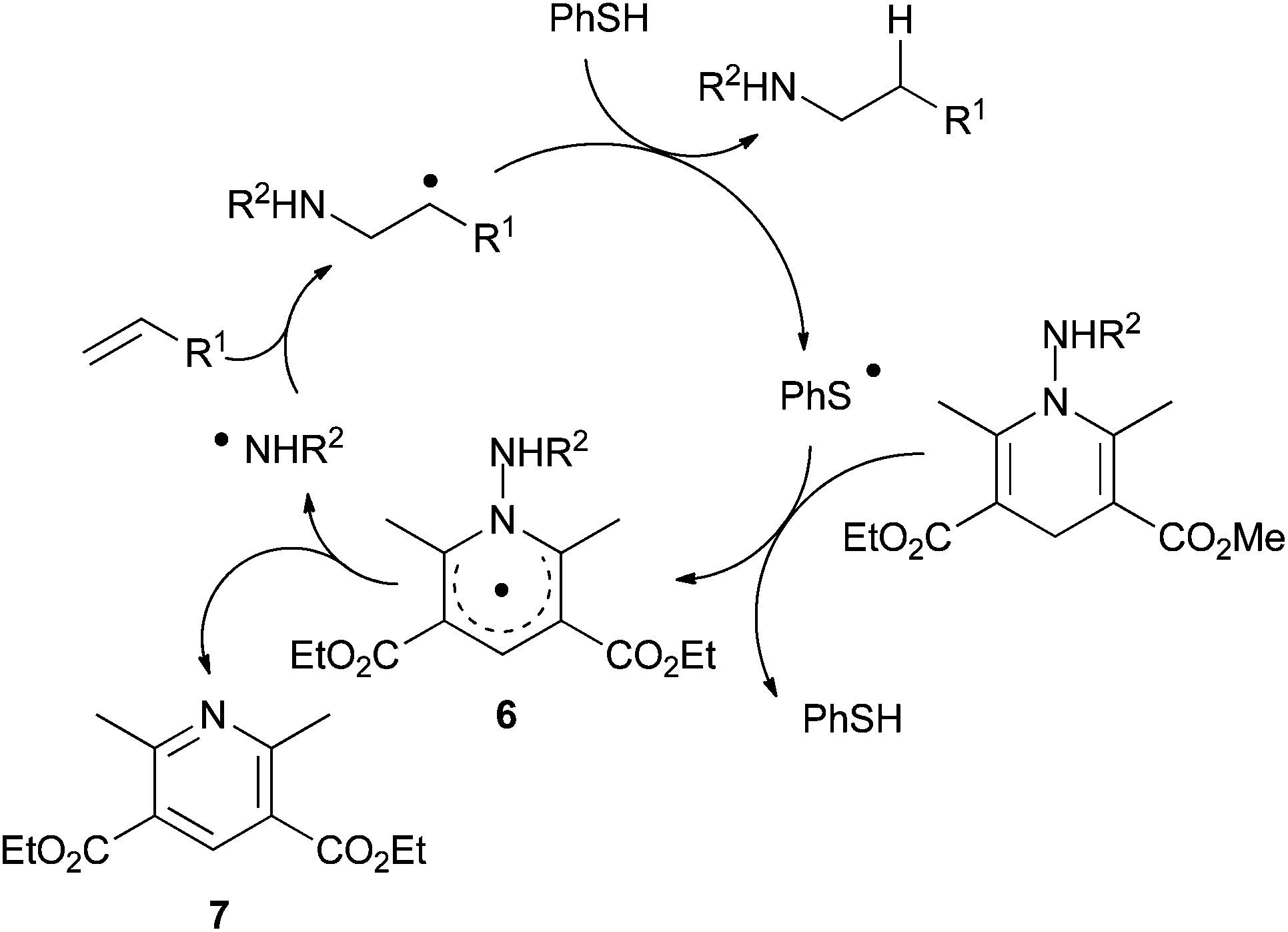 | ||
| Scheme 5 Proposed mechanism for intermolecular radical transfer hydroamination with N-aminated Hantzsch ester 5 using polarity-reversal catalysis. | ||
The group of Nicewicz has also reported an alternative radical anti-Markovnikov intramolecular hydroamination process based on single electron oxidation of olefins using a mild two-component organic photoredox catalyst system.19 Inspired by their previous work on direct intramolecular anti-Markovnikov addition of alcohols to alkenes (vide infra)3a and after optimisation studies, the group provides a very efficient metal-free method for the cyclohydroamination of electron-poor amines tethered to alkenes under mild conditions using Fukuzumi acridinium photoredox catalyst 8·BF4− and phenylthiol as hydrogen-donor under irradiation (Scheme 6).
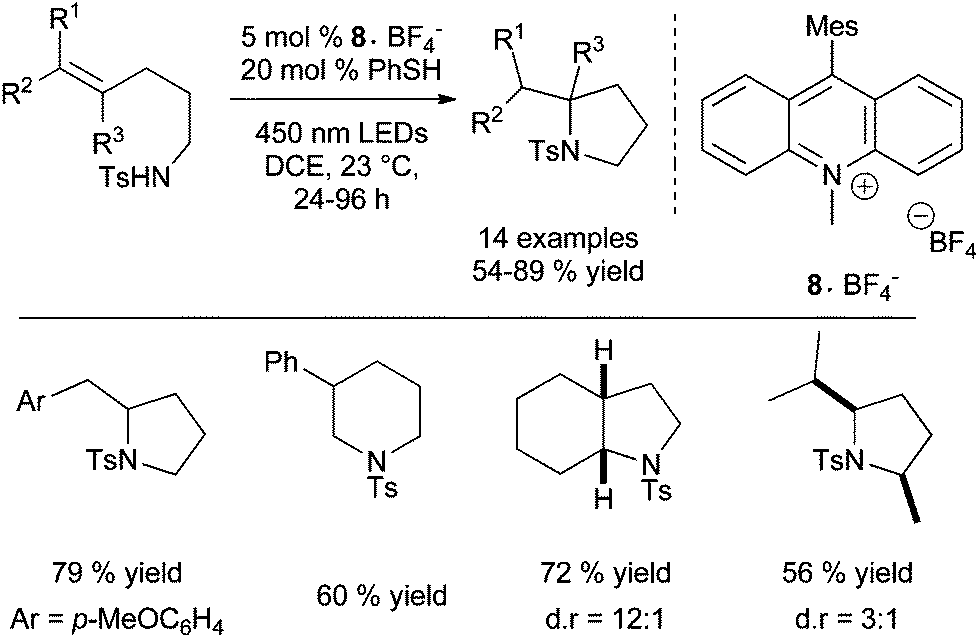 | ||
| Scheme 6 Intramolecular anti-Markovnikov hydroamination of tosylamines catalysed by Fukuzumi acridinium photoredox catalyst. | ||
Under these conditions, electron-rich amines are prone to oxidation and are poor substrates. This method which provides exclusively the anti-Markovnikov product does not require geminal substituents on the tether to favour cyclisation. Control experiments reveal that the reaction does not proceed by N-centered radical as in Studer's work (Scheme 4) or via a thiol–ene pathway. Furthermore, the methodology was expanded to the intermolecular anti-Markovnikov hydroamination of unactivated alkenes and trifluoromethanesulfonamide using 8·BF4−, diphenyl disulfide instead of phenylthiol and 2,6-lutidine as catalytic additive (Scheme 7).20 Under these modified conditions, hydroamination products are obtained in moderate to good yields with exclusive anti-Markovnikov selectivity. In contrast to the intramolecular version, no reactivity is observed with sulphonamides or tert-butyl carbamate. The use of odourless solid diphenyldisulfide offers a practical advantage over the highly toxic liquid thiophenol. The method is applicable to α- and β-substituted styrenes and di- and tri-substituted aliphatic alkenes and tolerates a variety of functional groups as illustrated in Scheme 7. Additionally, pyrazole, indazole and 1,2,3-triazole are also efficient partners for the anti-Markovnikov hydroamination of styrenic substrates. This photoredox process is proposed to proceed via an analogous mechanism as described for the hydroalkoxylation reaction (vide infra).
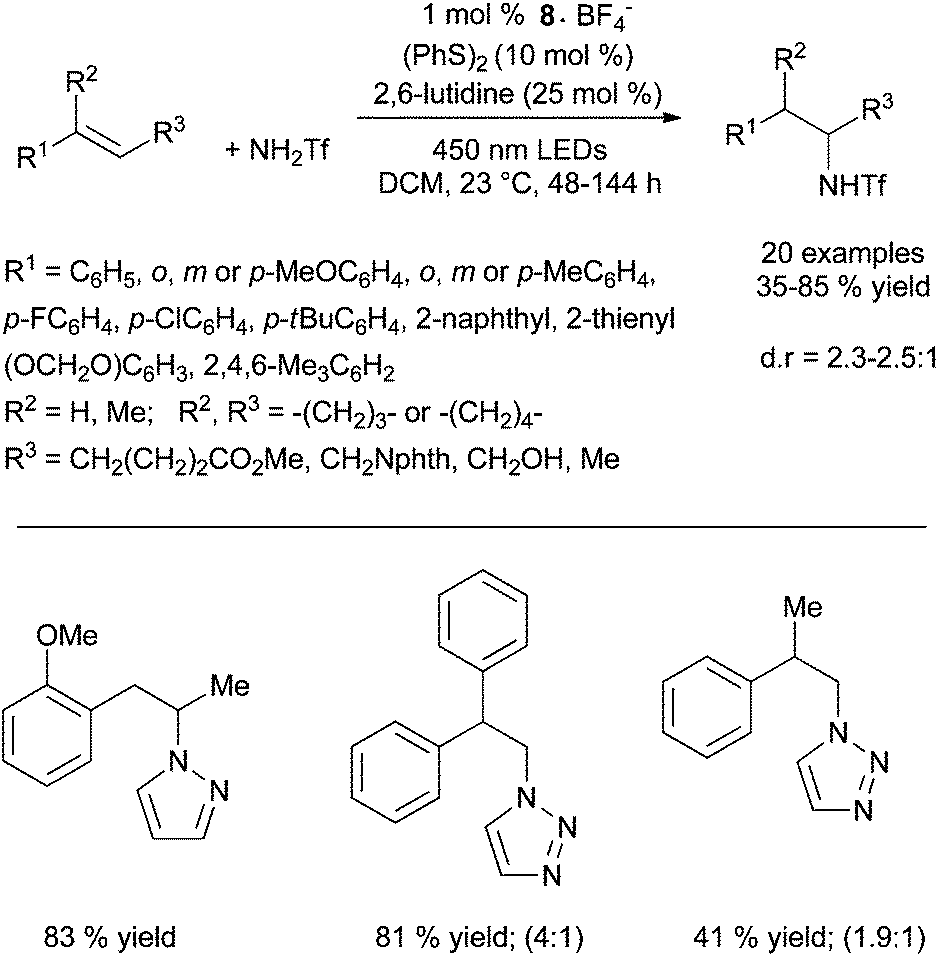 | ||
| Scheme 7 Intramolecular anti-Markovnikov hydroamination of trifluoromethanesulfonamide, pyrazole and 1,2,3-triazole catalysed by Fukuzumi acridinium photoredox catalyst. | ||
2.2 C–O bond formation
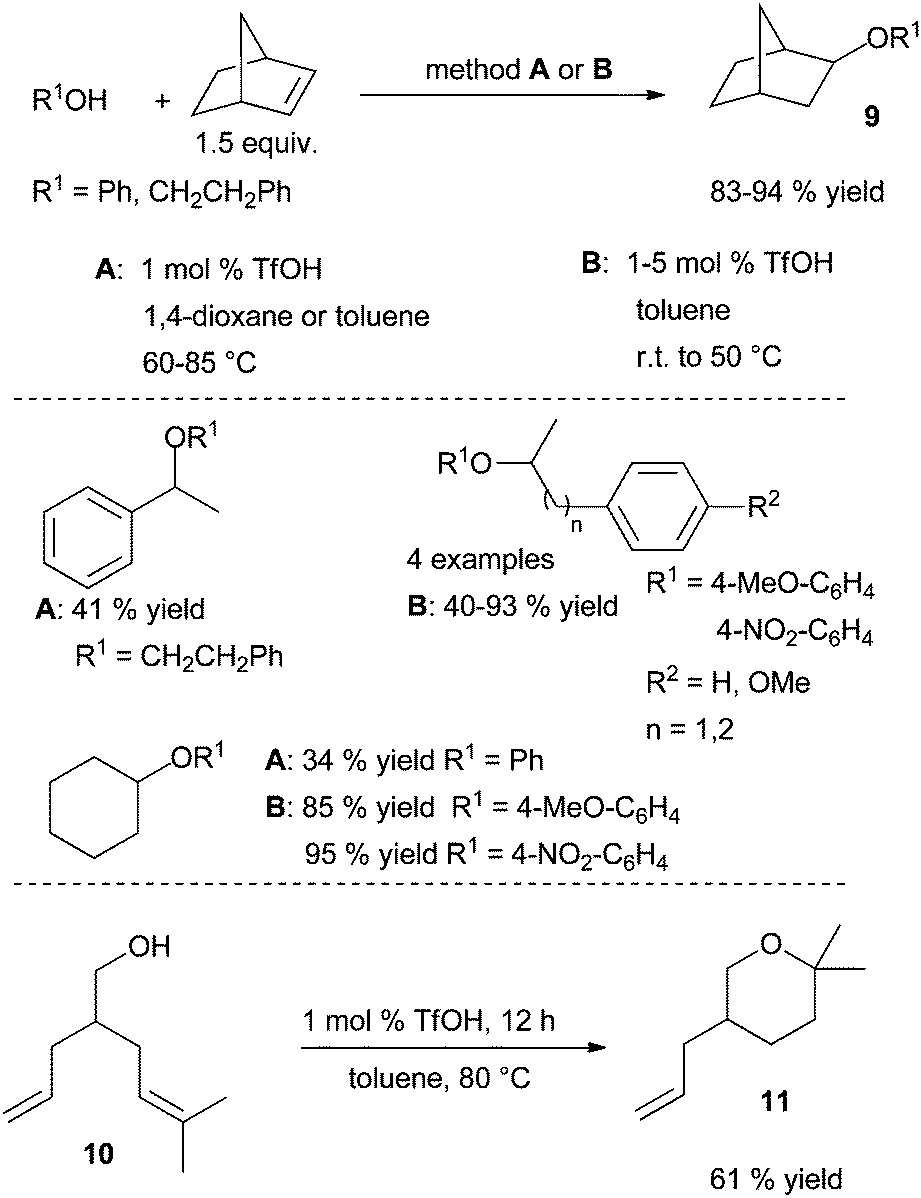 | ||
| Scheme 8 Inter and intramolecular hydroalkoxylation of alkenes catalysed by TfOH. | ||
In 2011, the group of Hintermann highlighted that in alkene hydroalkoxylation promoted by metal triflate catalysts under the experimental conditions studied, triflic acid is generated as the true catalyst species.23 The same year, Ryu et al. showed that imidazolium and triazolium ionic liquids (IL) 12 are good promotors in intramolecular hydroalkoxylation of alcohols tethered to alkenes (Scheme 9).24 Although the four ILs 12 tested (R5 = H, R6 = Bu, X = CH; R5 = Me, R6 = (CH2)4SO3H, X = CH, R5 = Bu, R6 = (CH2)4SO3H, X = CH, R5 = Bu, R6 = (CH2)4SO3H, X = N), have shown similar efficiency in the initial NMR studies, only the two last tethered-SO3H ILs 12 (R5 = Bu) were more effective in large-scale reactions and led to similar results. The recyclability of these functionalised ionic liquids was also demonstrated as one of them was reused in two cycles without significant loss of efficiency. Moreover, the authors described an interesting application of the methodology in the synthesis of (±)-centrolobine catalysed by 12 (R5 = Bu, R6 = (CH2)4SO3H, X = CH) and by its analogue (R5 = Bu, R6 = (CH2)4SO3H, X = N) giving the product of the intramolecular hydroalkylation step with 77% and 82% yields respectively. (−)-Centrolobine is an antiprotozoal natural product containing a 2,6-cis-disubstituted tetrahydropyran pattern and the intramolecular hydroalkoxylation catalysed by 12 gives exactly a total selectivity in favour to the cis stereoisomer (Scheme 9).
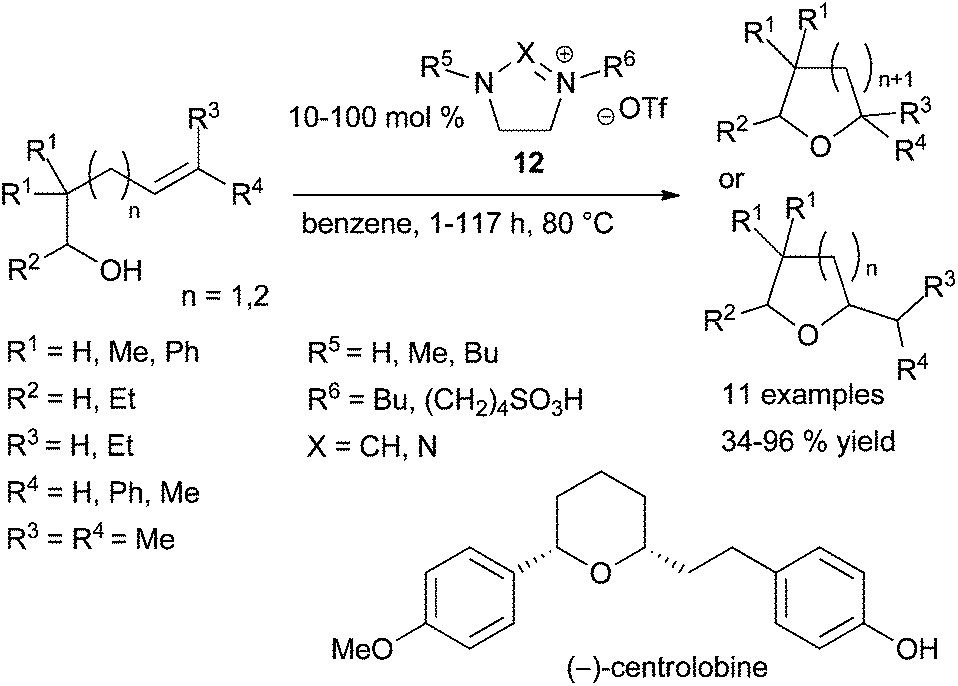 | ||
| Scheme 9 Intramolecular hydroalkoxylation of alkenes catalysed by imidazolium and triazolium salts. | ||
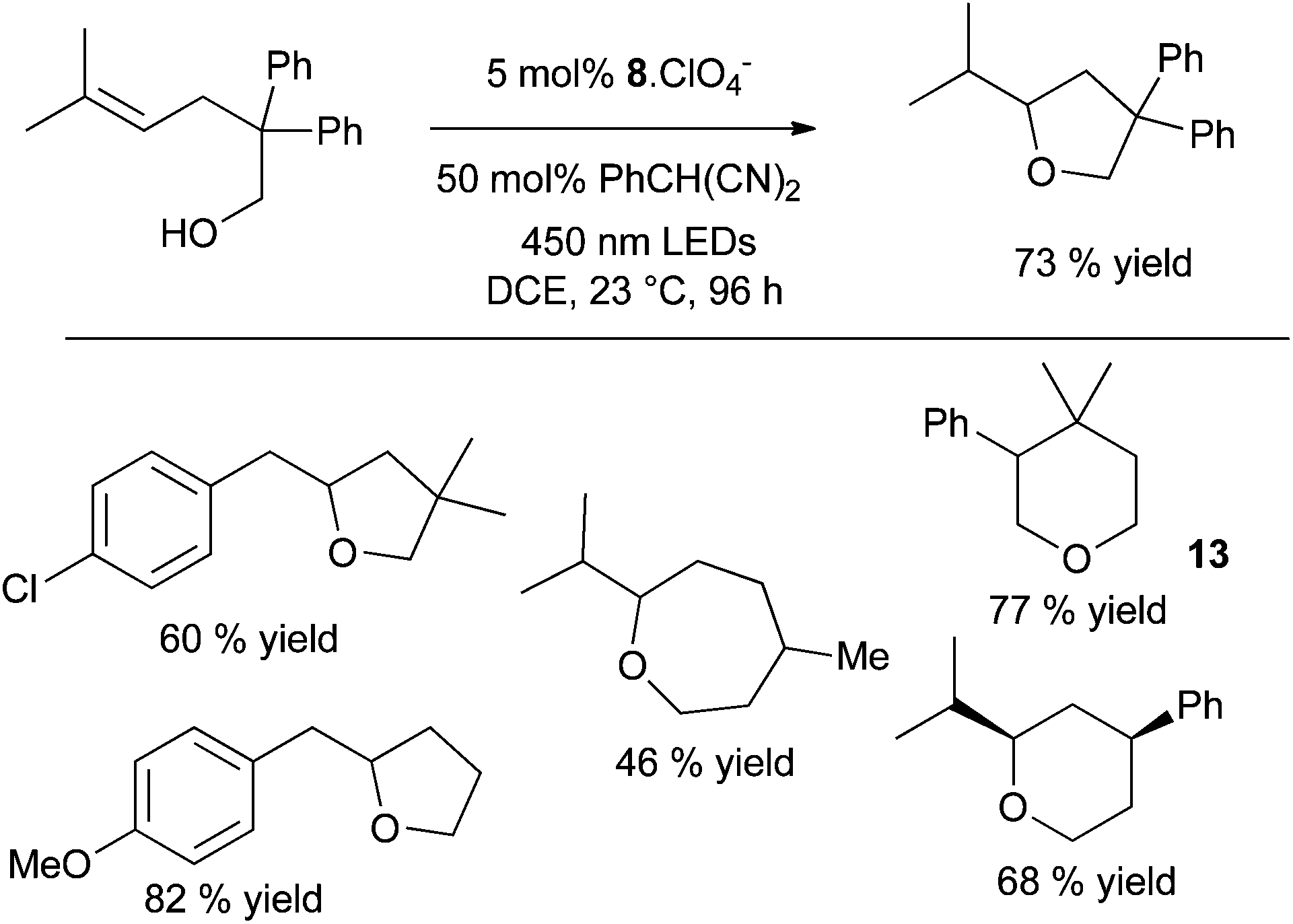 | ||
| Scheme 10 Anti-Markovnikov organic photoredox hydroalkoxylation of alkenes. | ||
The authors are still studying the mechanism25 but they assume that after a step of photo-activation, the mesityl acridinium catalyst oxidises the alkene in a radical cation intermediate which is cyclised to form a radical oxonium. After hydrogen atom transfer and deprotonation the desired product is obtained. The authors confirmed the link between both catalytic cycles, the photoredox one and the H atom transfer (HAT), the 2-phenylmalononitrile radical (Ered = +0.19 V vs. SCE, MeCN) oxidizing the acridinyl radical (Eox1 = −0.57 V vs. SCE, MeCN) in a fast process. (Scheme 11).
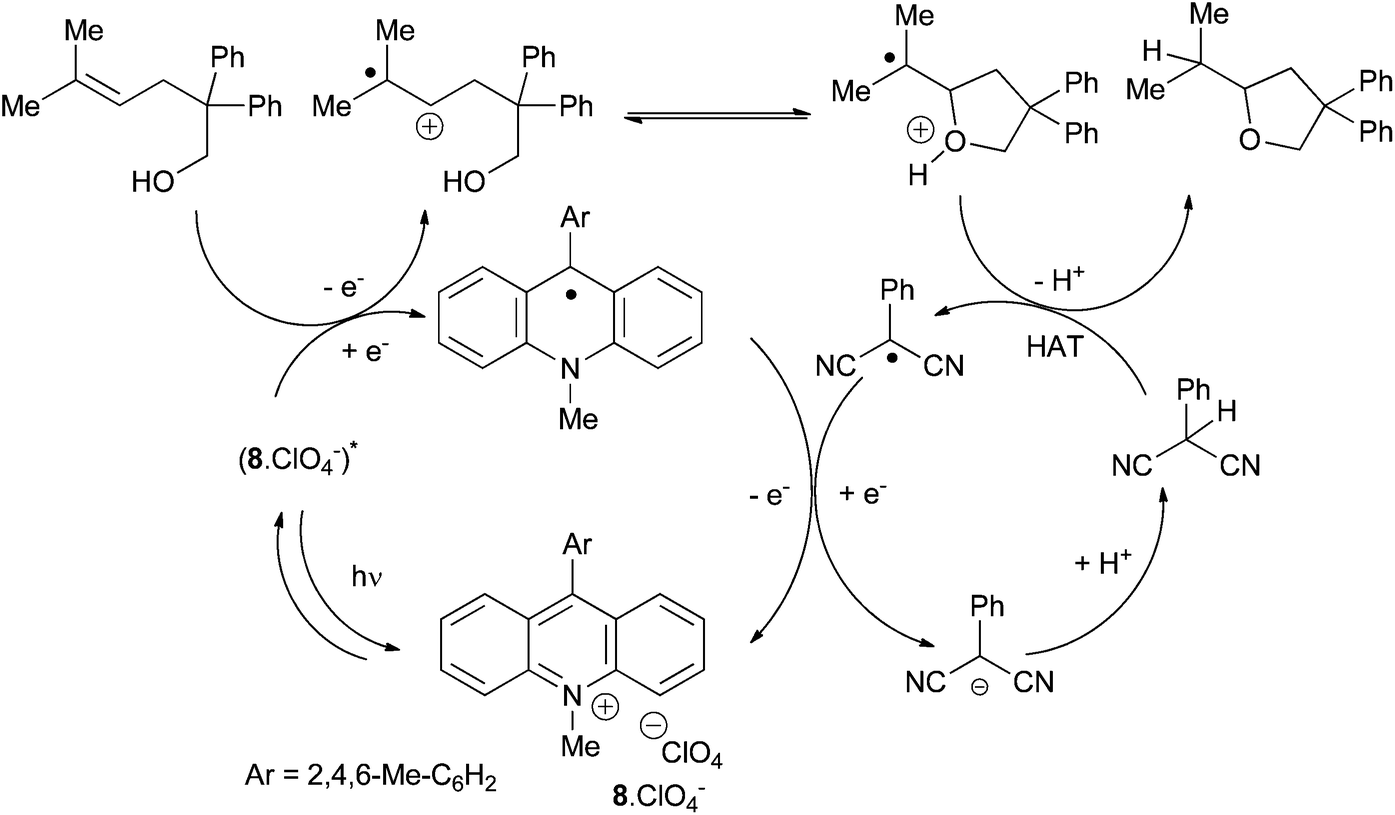 | ||
| Scheme 11 Proposed mechanism of organic photoredox hydroalkoxylation of alkenes. | ||
2.3 C–P bond formation
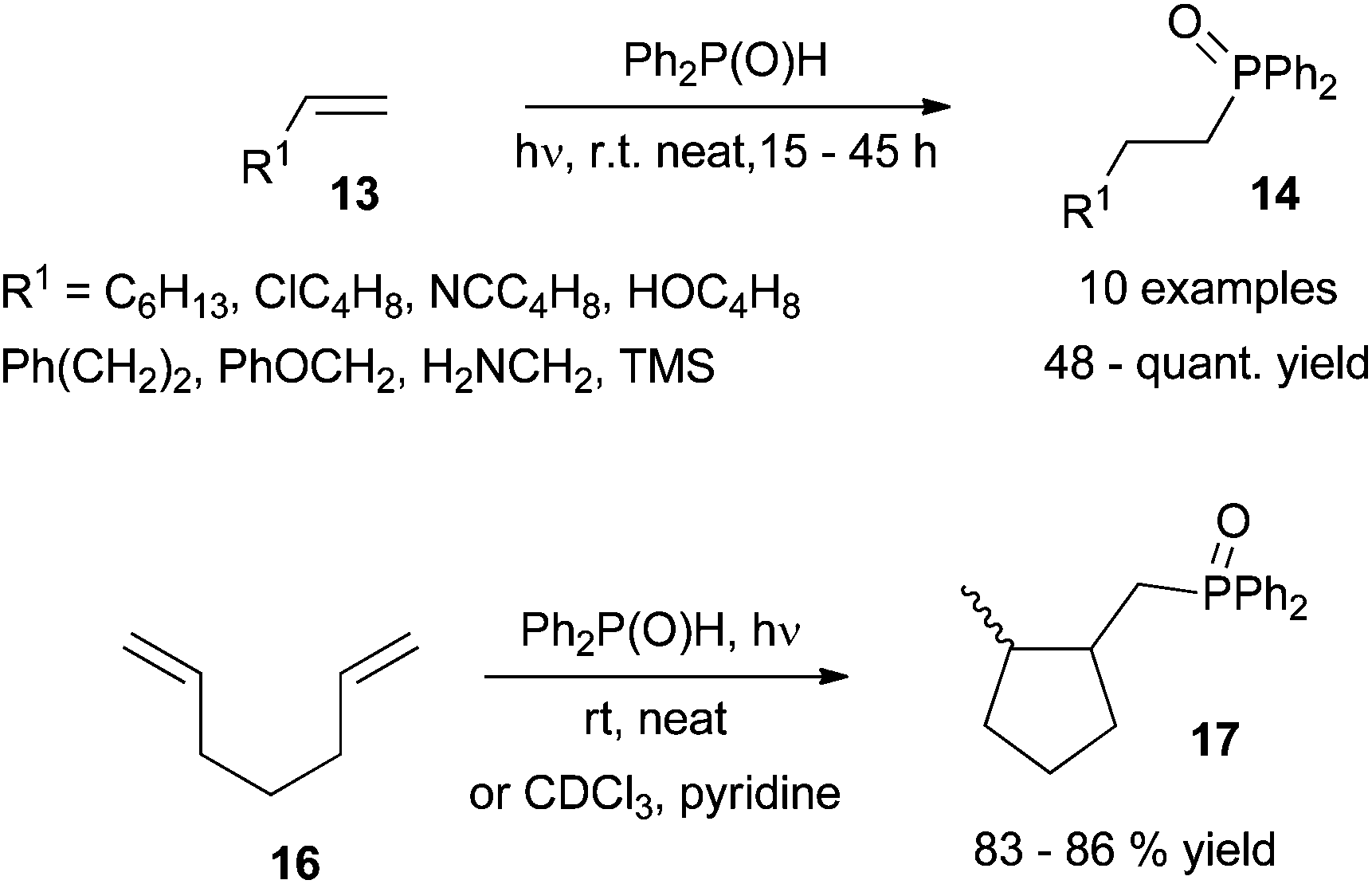
In the context of C–P bond formation, appealing photoinduced hydrophosphinylation of alkenes has recently been described by Ogawa et al.26 The former “Pudovik reaction”27 has been successfully adapted to the addition of R2P(O)–H to carbon–carbon double bonds and proceeds regioselectively under photoirradiation to cleanly afford bisarylalkylphosphine oxides. Irradiation with a xenon lamp in a 5 to 1 ratio of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/P reactant is required to ensure optimal results at room temperature. The transformation appears completely regioselective and affords fair to quantitative yields with an attractive functional group tolerance as shown in Scheme 12 (top, 13→14). The transformation is also successful for internal alkenes or in the presence of bulky substituents such as cyclohexane and dihydropyran leading to the corresponding phosphine oxides in 71% to quantitative yields respectively.
C/P reactant is required to ensure optimal results at room temperature. The transformation appears completely regioselective and affords fair to quantitative yields with an attractive functional group tolerance as shown in Scheme 12 (top, 13→14). The transformation is also successful for internal alkenes or in the presence of bulky substituents such as cyclohexane and dihydropyran leading to the corresponding phosphine oxides in 71% to quantitative yields respectively.
 | ||
| Scheme 12 Photoinduced hydrophosphinylation of alkenes. | ||
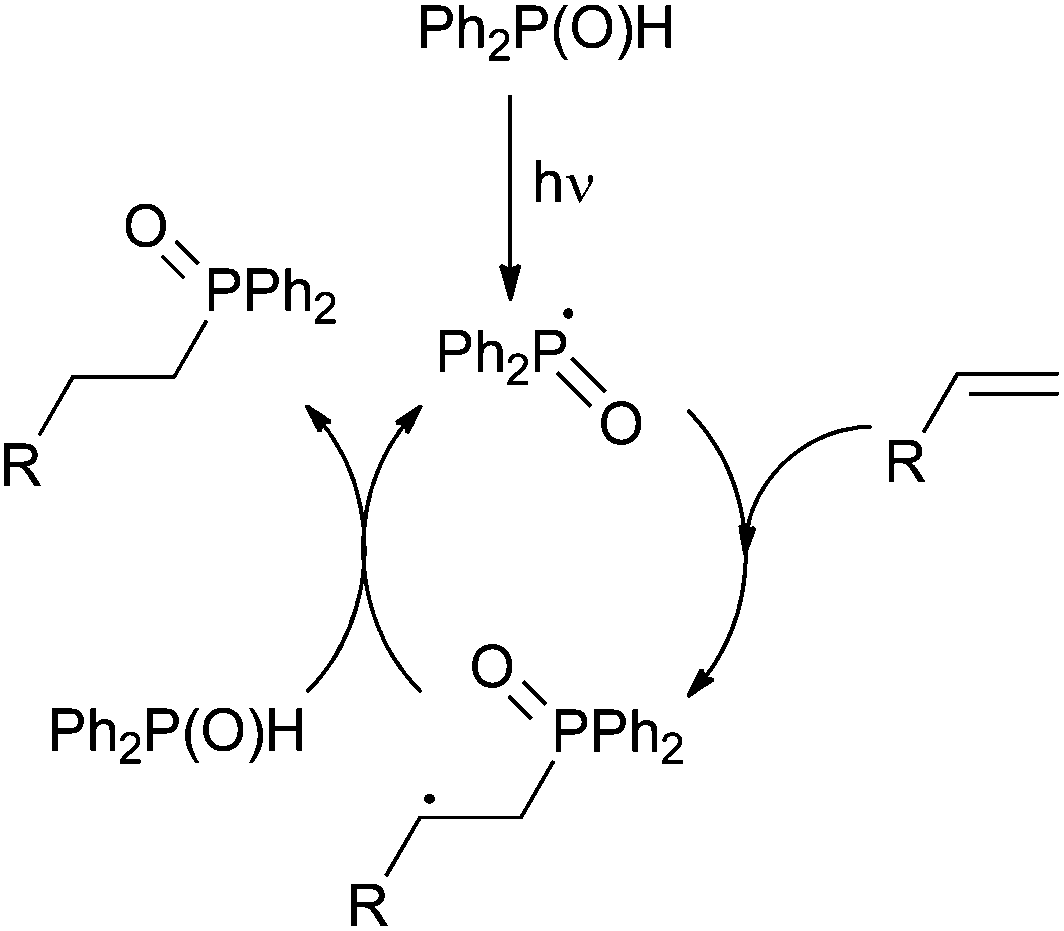
The methodology allows access to cyclopentane derivatives via photoinduced formation of a C–P bond and subsequent exo-trig cyclisation from 1,6-cycloheptadiene precursor 16 (Scheme 12, bottom). Mixtures of cis/trans disubstituted cyclopentanes 17 are obtained, the former cis isomer being usually produced as the major one regardless of the conditions used: neat or in solution using pyridine as an additive. A possible mechanism involves the formation of key phosphinoyl radical followed by selective formation of an exo-C–P bond (Scheme 13). Hydrogen abstraction from a second diphenylphosphine oxide molecule affords the expected hydrophosphinylation adduct and a further phosphinoyl radical.
 | ||
| Scheme 13 Proposed mechanism for photoinduced hydrophosphinylation of alkenes. | ||
3. Group 1-based catalysts
3.1 C–N bond formation
The hydroamination promoted by alkali metals is an old transformation28 and numerous efforts have been drawn for developing new catalytic systems allowing mild reaction conditions.29 Recent developments include the readily cyclohydroamination transformation of simple aminoalkene derivatives by catalytic quantities of nBuLi solutions, delivering the corresponding nitrogen-containing heterocycles in high yield (Scheme 14). The formation of the new C–N bond albeit requires heating the reaction mixture to 110 °C, but the scope of the reaction is wide as demonstrated by the preparation of 25 different products by this procedure.30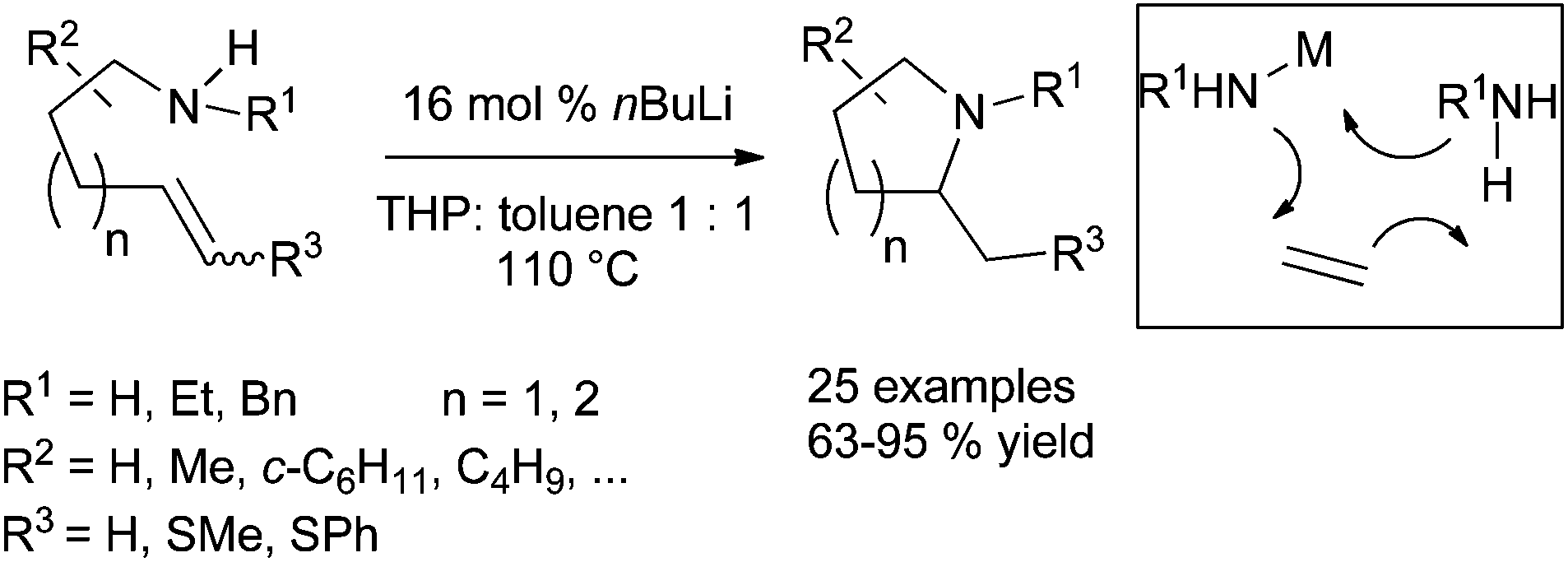 | ||
| Scheme 14 Base-catalysed cyclohydroamination reaction. | ||
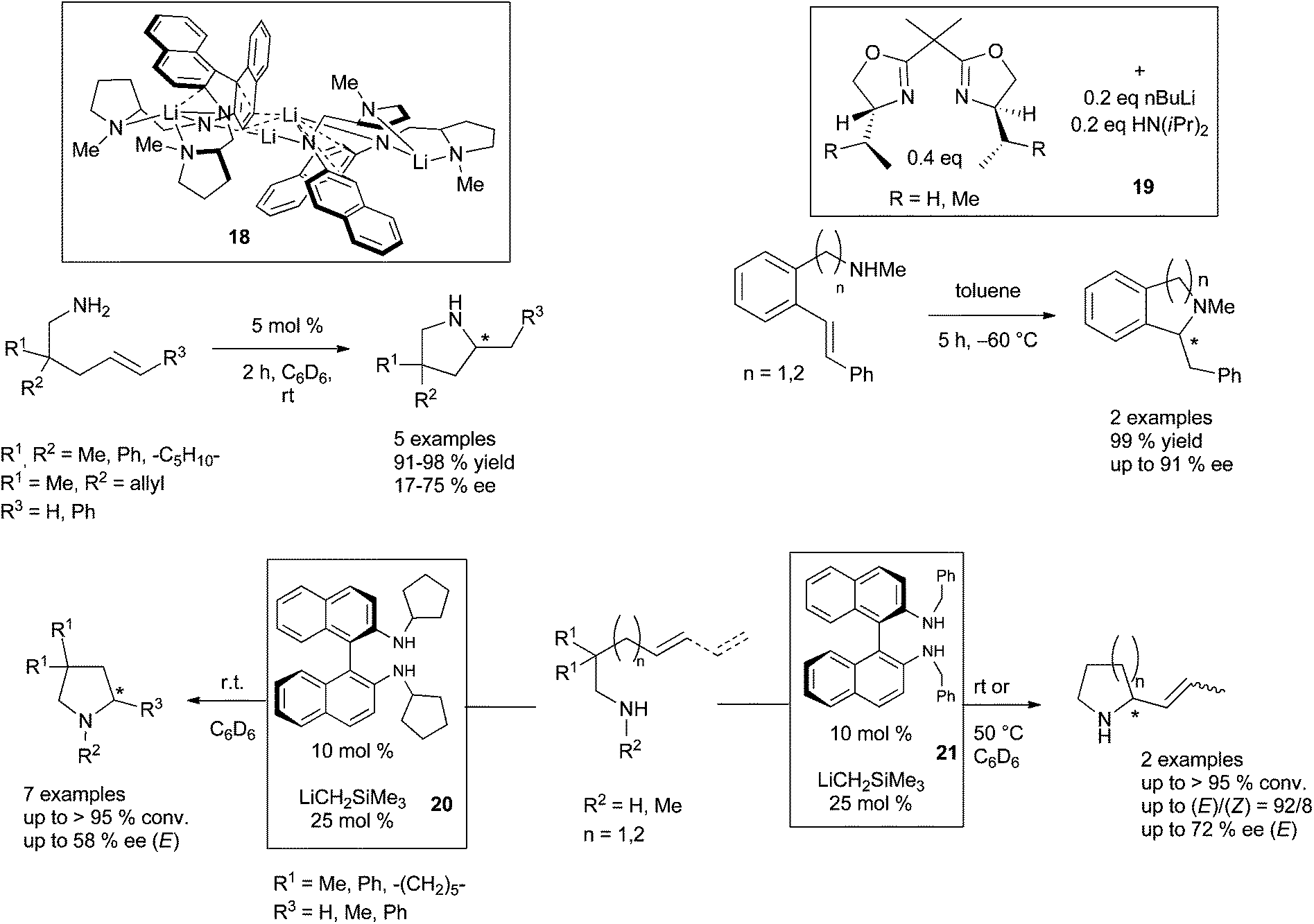
The more difficult intermolecular version was studied by the group of Hultzsch and concerned the hydroamination of vinylarenes.31 The use of 2 mol% of LiN(SiMe3)2–TMEDA as catalytic system promoted the hydroamination of vinylarene derivatives with primary and secondary amines (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) at 120 °C and led to the anti-Markovnikov product with high selectivity. Attempts to perform enantioselective intermolecular hydroamination, by replacing TMEDA by (−)-sparteine and reacting α- and β-methylstyrene with benzylamines remained unfortunately unsatisfactory (≤14% ee). Jaspers and Doye have further interestingly demonstrated that KOH is also a suitable strong base to catalyse the addition of various arylamines to styrene derivatives in dry DMSO, a methodology that they could illustrate with 17 examples.32 The challenge of introducing enantioselectivity by chiral alkali bases has until now only been successfully overcome for intramolecular transformations. Only three examples are known, with one of them involving the use of an isolated lithium-based complex. A particularly noteworthy result indeed issued from the group of Hultzsch which prepared a dilithium diamide derivative, as a dimer, from a symmetrically N-methylpyrrolidine-substituted binaphthylamine ligand (see 18, Scheme 15).33 Due to its dimeric structure, according to the authors, this catalyst promoted the cyclisation of aminopentene derivatives into the corresponding pyrrolidines with up to 75% ee. Tomioka and his group described the formation of scalemic N-methylisoquinoline and isoindoline by combination of the precursor aminomethylolefins with catalytic amounts of chiral bis(oxazolines) and nBuLi.34 Additional diisopropylamine was necessary for the in situ prepared catalyst to afford a complete conversion, and the targeted products were isolated in up to 91% ee (see 19, Scheme 15). Some of us have reported that chiral lithium salts, easily prepared by in situ combination of N-substituted (R)-(+)-1,1′-binaphthyl-2,2′-diamine ligands and [(trimethylsilyl)methyl]lithium delivered efficient pre-catalytic species for the asymmetric hydroamination of amino-1,3-dienes and aminoalkenes.35 The most efficient catalyst for the cyclohydroamination of aminoalkenes (see 20, Scheme 15) delivered expected pyrrolidines at room temperature, in up to 58% ee. A similar catalytic system (see 21, Scheme 15) produced 2-propenyl-pyrrolidine and -piperidine with the highest stereo- and enantioselectivities described to date.
:1 ratio) at 120 °C and led to the anti-Markovnikov product with high selectivity. Attempts to perform enantioselective intermolecular hydroamination, by replacing TMEDA by (−)-sparteine and reacting α- and β-methylstyrene with benzylamines remained unfortunately unsatisfactory (≤14% ee). Jaspers and Doye have further interestingly demonstrated that KOH is also a suitable strong base to catalyse the addition of various arylamines to styrene derivatives in dry DMSO, a methodology that they could illustrate with 17 examples.32 The challenge of introducing enantioselectivity by chiral alkali bases has until now only been successfully overcome for intramolecular transformations. Only three examples are known, with one of them involving the use of an isolated lithium-based complex. A particularly noteworthy result indeed issued from the group of Hultzsch which prepared a dilithium diamide derivative, as a dimer, from a symmetrically N-methylpyrrolidine-substituted binaphthylamine ligand (see 18, Scheme 15).33 Due to its dimeric structure, according to the authors, this catalyst promoted the cyclisation of aminopentene derivatives into the corresponding pyrrolidines with up to 75% ee. Tomioka and his group described the formation of scalemic N-methylisoquinoline and isoindoline by combination of the precursor aminomethylolefins with catalytic amounts of chiral bis(oxazolines) and nBuLi.34 Additional diisopropylamine was necessary for the in situ prepared catalyst to afford a complete conversion, and the targeted products were isolated in up to 91% ee (see 19, Scheme 15). Some of us have reported that chiral lithium salts, easily prepared by in situ combination of N-substituted (R)-(+)-1,1′-binaphthyl-2,2′-diamine ligands and [(trimethylsilyl)methyl]lithium delivered efficient pre-catalytic species for the asymmetric hydroamination of amino-1,3-dienes and aminoalkenes.35 The most efficient catalyst for the cyclohydroamination of aminoalkenes (see 20, Scheme 15) delivered expected pyrrolidines at room temperature, in up to 58% ee. A similar catalytic system (see 21, Scheme 15) produced 2-propenyl-pyrrolidine and -piperidine with the highest stereo- and enantioselectivities described to date.
 | ||
| Scheme 15 Asymmetric cyclohydroamination of aminoalkenes and aminodienes promoted by chiral lithiated bases. | ||
Significant progress has therefore taken place in recent years, motivated by the use of cheap and easily available alkali bases. Their use necessary implies a reduced functional tolerance; nevertheless great advances have been reported in the area of enantioselective cyclohydroamination. The main hurdle to succeed in catalysing the corresponding asymmetric intermolecular version remains still to be crossed.
4. Group 2-based catalysts
4.1 C–N bond formation
Hydroamination reaction promoted by alkaline earth metals has been for a long time underexploited, albeit these species represent a benign series of elements and remain inexpensive. Alkaline earth metals possess indeed a great number of features common to the elements of the 4f series, long known to promote this reaction. They indeed share large radii and an important electropositive character of the cation. The group of Hill reported thus in 2005 the efficient calcium-mediated room temperature intramolecular hydroamination catalysis promoted by a synthetically easily accessible β-diketiminato calcium bis(trimethylsilyl)amide complex.36 Since this pioneering work, various ligands have been further developed towards the synthesis of heteroleptic compounds, including aminotroponiminate complexes.37 Comparative activity studies have been published between the different alkaline earth metals, with preliminary results indicating a decrease in the rate with increasing ion radius of the metal center, for the cyclohydroamination reaction. The main challenge that has been tackled concerns the isolation and characterisation of well-defined species for controlling the Schlenk equilibrium which often occurs, due to the labile nature of the ligands.Focusing on the chemistry of β-diketiminate derivatives, Barrett, Hill et al. prepared and compared the activity of different group 2 pre-catalysts:38 the magnesium methyl and the calcium and strontium silylamide β-diketiminate derivatives, [{ArNC(Me)CHC(Me)NAr}MX-(THF)n] (M = Mg, X = CH3, n = 0; M = Ca, X = N(SiMe3)2, n = 0 or 1; M = Sr, X = N(SiMe3)2, n = 1, Ar = 2,6-diisopropylphenyl), the bistrimethylsilylamides (THF solvated or not) and the bistrimethylsilylalkyl species (solvated) for Mg, Ca, Sr and Ba. They revealed that Ba pre-catalysts were not efficient and alkyl derivatives decomposed at high temperature and thus conclude amido species to be more valuable promoters. Although a general reactivity in the order Ca > Sr > Mg was proven, the authors noticed the important influence of the presence (or not) of THF, depending on the nature of the ligand and the substrate, for the same metal. Mechanistic studies demonstrated the tricky influence of the M2+ cation on both the thermodynamic and the kinetic course of the reaction, according to the ligand and the reactant structure. Trying to tame this reactivity, further development of new structurally different pre-catalysts has been more recently published.
For instance, Carpentier, Sarazin et al. have been able to prepare and characterise amido and alkyl iminoanilide alkaline earth complexes 22 and 23 that proved to be kinetically stable in solution (Scheme 16).39 The hydrocarbyl complexes were very active pre-catalysts for both the cyclohydroamination of 2,2-dimethyl-pent-4-enylamine (I) (with a reactivity trend Ca > Sr > Ba, Table 1, entries 1–6) and intermolecular hydroamination (with an interesting inverse tendency Ca < Sr < Ba, see Table 1, entries 7–12) showing no ligand redistribution. These complexes were particularly efficient for the intermolecular anti-Markovnikov hydroamination of alkenes, with an unprecedented huge activity for Ba derivatives, proposed to occur through a six-centered concerted mechanistic pathway. 2,5-Bis{N-(2,6-diisopropylphenyl)iminomethyl} pyrrolyl amido complexes of Ca and Sr 24 were prepared from the corresponding heteroleptic iodo complexes, possessing the symmetrically κ3-coordinated ligand in solution (Scheme 16).40 These pre-catalysts were nevertheless less active in the cyclohydroamination reaction (Table 1, entries 13–14). Hill et al. described the use of sterically-hindered bis(imino)-acenaphthene ligands.41
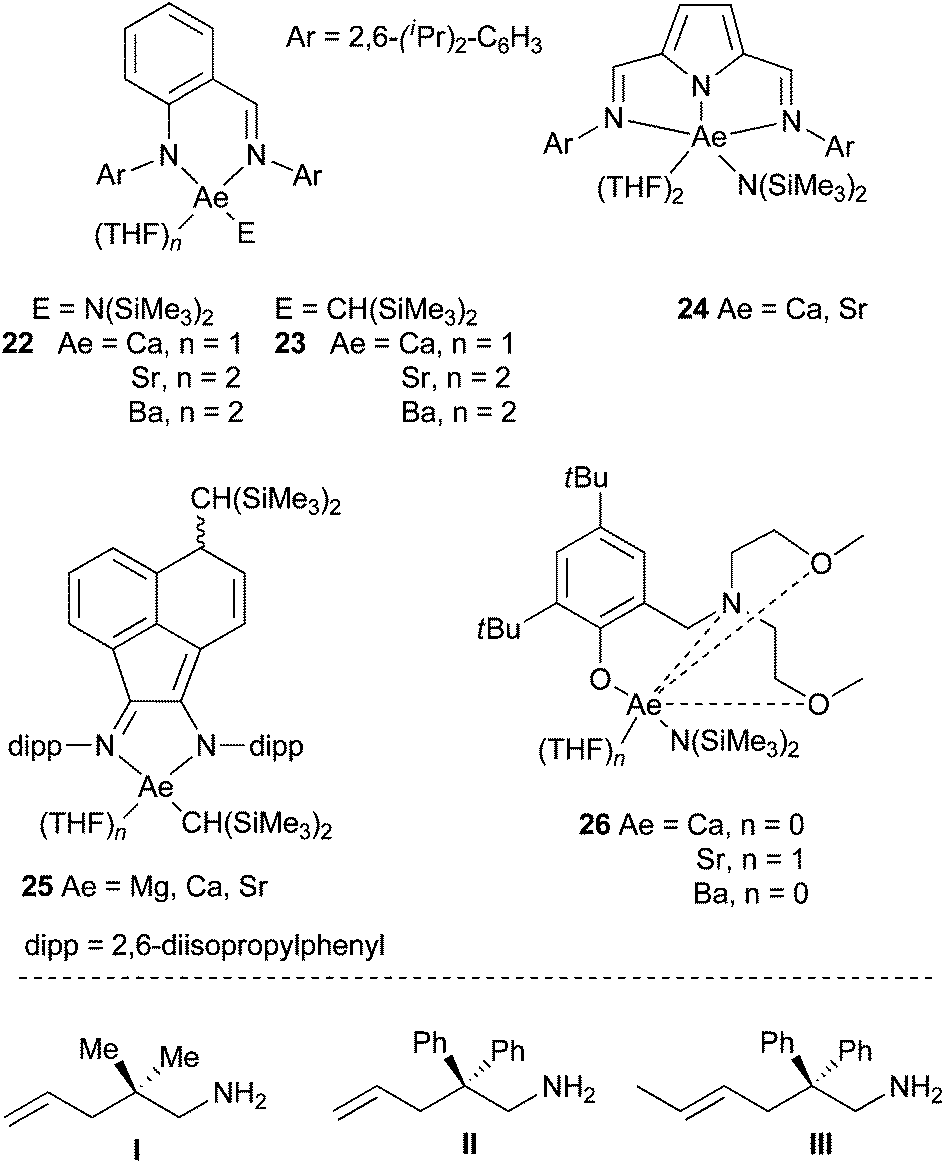 | ||
| Scheme 16 Heteroleptic alkaline earth complexes active in the cyclo- and intermolecular hydroamination. | ||
| Entry | Substrate | Catalyst (Ae) (mol%) | Time (h) | T (°C) | Conv (%) |
|---|---|---|---|---|---|
| a All cyclohydroamination reactions are performed in C6D6, the intermolecular version are run in neat conditions, without additional solvents. | |||||
| 1 | I | 22 (Ca) (1) | 4 min | 25 | 90 |
| 2 | I | 22 (Sr) (2) | 7 min | 60 | 97 |
| 3 | I | 22 (Ba) (2) | 1 | 60 | 96 |
| 4 | I | 23 (Ca) (1) | <2 min | 25 | >99 |
| 5 | I | 23 (Sr) (1) | <3 min | 25 | 98 |
| 6 | I | 23 (Ba) (1) | 0.3 | 25 | 96 |
| 7 | Styrene/pyrrolidine | 22 (Ca) (2) | 0.5 | 60 | 65 |
| 8 | Styrene/pyrrolidine | 22 (Sr) (2) | 0.5 | 60 | 75 |
| 9 | Styrene/pyrrolidine | 22 (Ba) (2) | 0.5 | 60 | 95 |
| 10 | Styrene/pyrrolidine | 23 (Ca) (2) | 0.5 | 60 | 90 |
| 11 | Styrene/pyrrolidine | 23 (Sr) (2) | 5 min | 60 | 85 |
| 12 | Styrene/pyrrolidine | 23 (Ba) (2) | 5 min | 60 | 97 |
| 13 | I | 24 (Ca) (5) | 0.5 | 25 | 92 |
| 14 | I | 24 (Sr) (5) | 2 | 60 | 92 |
| 15 | II | 25 (Mg) (0.5) | 18 | 60 | <5 |
| 16 | II | 25 (Ca) (0.5) | 0.25 | 25 | >99 |
| 17 | II | 25 (Sr) (0.5) | 1 | 25 | 76 |
| 18 | III | 25 (Ca) (5) | 5 | 25 | 95 |
| 19 | II | 26 (Ca) (1.6) | 0.5 | 80 | >99 |
| 20 | II | 26 (Sr) (1.6) | 1 | 80 | 99 |
| 21 | II | 26 (Ba) (1.6) | 1 | 80 | 10 |
They succeeded in the synthesis of alkyl derivatives, that showed enhanced stability towards Schlenk-type ligand redistribution, because of the high steric demand of the bis(trimethylsilyl)methyl substituent. These complexes proved to be highly reactive in the cyclohydroamination of aminoalkenes, as exemplified for the hydroamination of the benchmark substrate, 2,2-diphenyl-pent-4-enylamine (II) (Table 1, entries 15–17). The calcium complex 25 (Ae = Ca) was especially active even to promote the cyclisation of challenging substrates such as III, displaying a disubstituted olefin (Table 1, entry 18). Another type of ligand, based on aminophenolate backbone has also been prepared (26, Scheme 16),42 and their activity in the cyclohydroamination of aminoalkenes evaluated and compared to β-diketiminato complexes. Here again the activity for Ca-, Sr- and Ba-derivatives followed the same trend but these new complexes were by far less active (Table 1, entries 19–21).
Since the Schlenk equilibrium could be controlled, efforts to perform this reaction in an enantioselective manner by using chiral ligands have been attempted. First studies in 2008 have been published by Harder et al.43 who synthesised chiral heteroleptic calcium complexes from enantiopure bis(oxazolines) or chiral β-diketimine derivatives. The corresponding amide complexes have been isolated and characterised by X-ray analysis, but they underwent to some extent Schlenk equilibrium to form catalytically inactive [Ca(L)2] complexes, and an ee lower than 10% was obtained for cyclisation of substrate II. Ward and his group could improve the selectivity to reach 26% ee for the same reaction, preparing calcium complexes supported by chiral diamines44 and developing also enantiopure bisimidazoline amido derivatives.45 A significant change in the enantioselectivity values was provided by the use of amido calcium complexes 27–29 bearing bis(oxazolinylphenyl)amine ligands, exhibiting very low ligand redistribution and affording the targeted pyrrolidine compound in up to 48% ee (Scheme 17).46
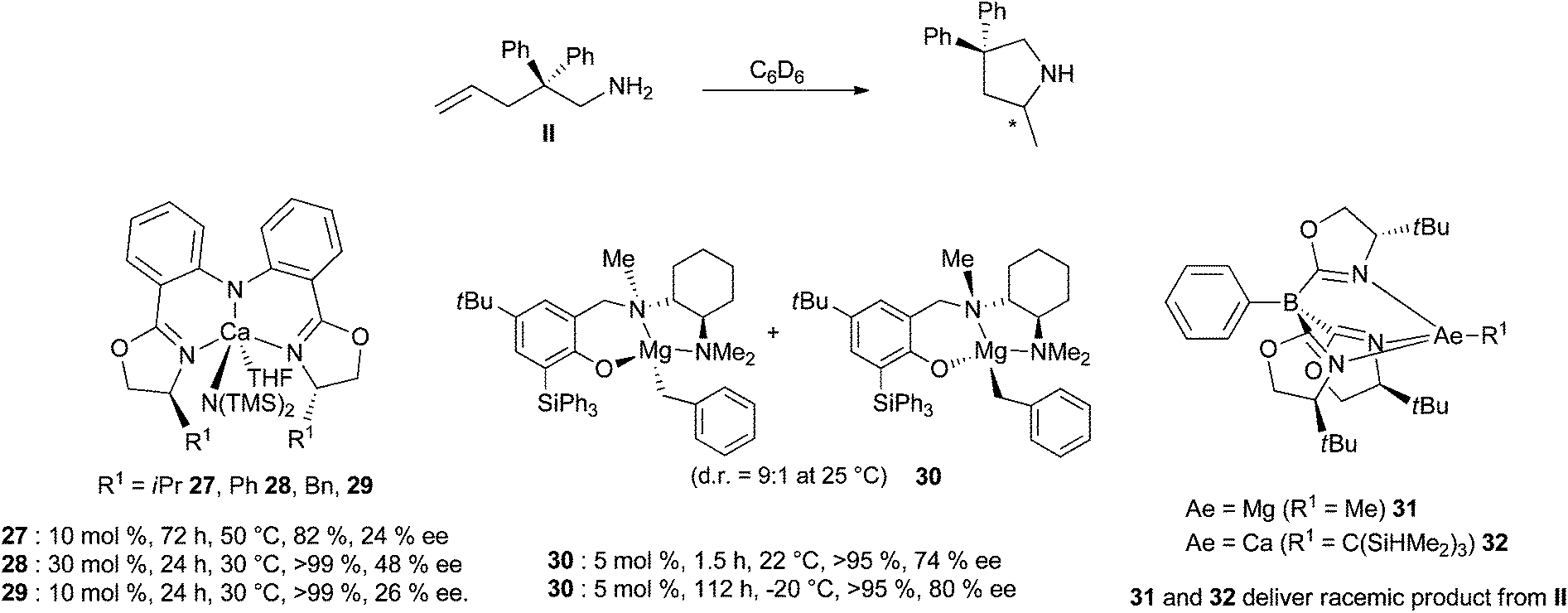 | ||
| Scheme 17 Enantiopure chiral heteroleptic alkaline earth complexes for promoting enantioselective cyclohydroamination. | ||
Accordingly, the group of Hultzsch has focused on the preparation of magnesium derivatives. They were obtained as chiral bimetallic complexes from reaction of nBu2Mg using L-proline-derived diamidobinaphthyl ligands. The magnesium complexes showed moderate to high catalytic activity, but the enantioselectivity of the reaction was limited to 14% ee due to facile ligand exchange reaction and/or protolytic ligand cleavage upon addition of the substrate.47 Furthermore, alkyl magnesium complexes containing multidentate phenoxyamine ligands delivered efficient catalysts for the cyclohydroamination reaction, showing no ligand exchange, and allowing complete conversion at room temperature for substrate II (2 mol% cat, 3 h).48 They were thus structurally good candidates towards the development of chiral catalysts; chiral phenoxyamine ligands incorporating a chelating cyclohexyldiamine arm such as in complex 30 (Scheme 17) have therefore been prepared and in the presence of the bulky triphenylsilyl substituent ligand, exchange processes were eliminated.49 This magnesium catalyst delivered the highest enantioselectivity reported to date for the alkaline earth-based hydroamination catalysis, delivering 2-methyl-4,4-diphenyl-pyrrolidine with up to 92% ee. Sadow et al. have discovered that ToMMgMe complexes (ToM = tris(4,4-dimethyl-2-oxazolinyl)phenylborate) are also active precatalysts for the cyclohydroamination reaction (substrate II, 50 °C, 10 mol%, 12 h).50 Accordingly, chiral bulky tris(oxazolinyl)borato magnesium and calcium compounds 31–32 have been prepared (Scheme 17) but these catalysts proved however poorly enantioselective (with the analogous substrate, bearing a cyclohexyl group, the Mg complex yielded the pyrrolidine with 93% conversion and 36% ee, cat 10 mol%, 26 h, 60 °C).51 The challenging preparation of scalemic products is still ongoing by the use of alkaline earth-based complexes, with a great demand for selective complexes, highly stable towards the Schlenk equilibrium.
4.2 C–O bond formation
In order to develop an inexpensive and mild protocol, catalytic systems were also studied for hydroalkoxylation. Recently, two research groups have reported the efficiency of these elements for the intramolecular hydroalkoxylation of allenol and alkenol-type substrates respectively. In 2012, Barrett et al. described the hydroalkoxylation of allenols catalysed by alkaline earth bis(trimethylsilyl)amides (Scheme 18).52 According to the metal and the experimental conditions, the isomerisation of the exo-cyclic to the endo-cyclic double bond occurred after the first step of hydroalkoxylation. The reaction is efficient with calcium complex on monosubstituted allenols, however with 3,5-disubstituted pent-3,4-dien-1-ol, reactions are very slow and need higher temperature (110 °C for 24–72 h with 8% conversion). No exo-cyclic double bond product is formed with starting allenols bearing substituents in the 2-position.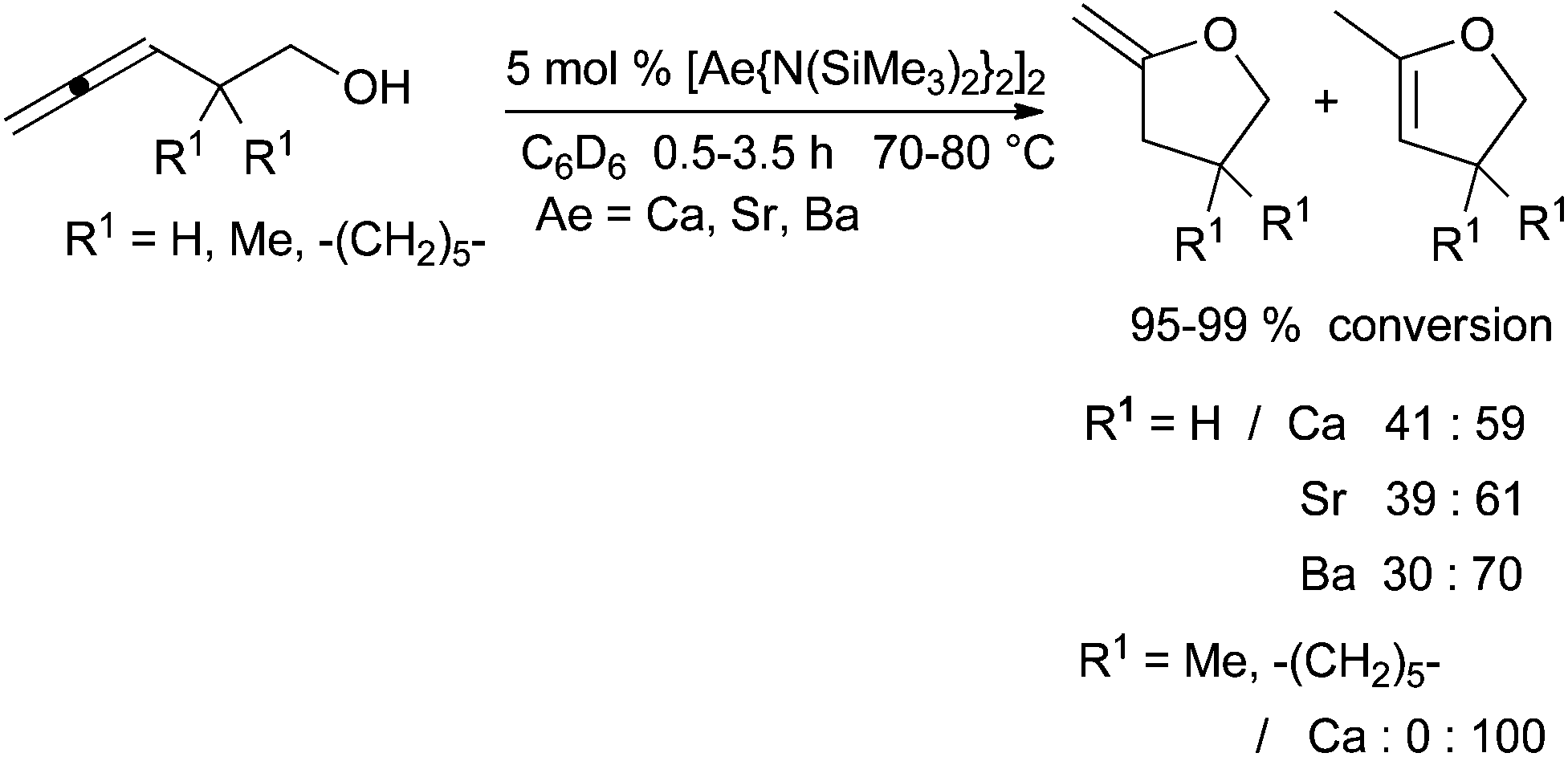 | ||
| Scheme 18 Intramolecular hydroalkoxylation of allenols catalysed by alkaline earth amides. | ||
The same year, Niggemann et al. described the intramolecular hydroalkoxylation of alkenols catalysed by Ca(NTf2)2 (Scheme 19).53a The additive Bu4NPF6 promotes the solubility of the calcium complex CaNTf2PF6 produced after anion exchange; this complex gives higher reaction rates.53b
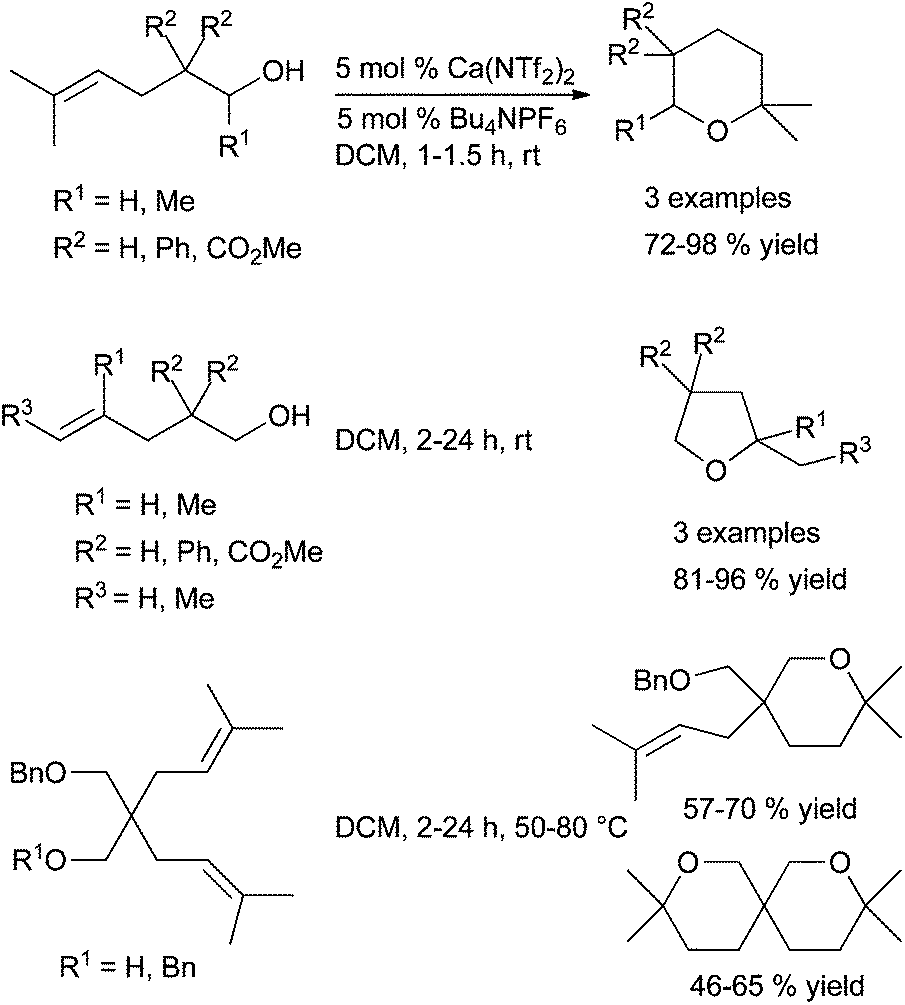 | ||
| Scheme 19 Intramolecular hydroalkoxylation of allenols catalysed by alkaline earth amides. | ||
After prolonged reaction time, this catalytic system allows the deprotection of benzylether groups and in a second step the hydroalkoxylation reaction leading to the spirobicyclic ether. Except for this deprotection step, the association of Ca(NTf2)2-Bu4NPF6 allows very mild reaction conditions since the hydroalkoxylation is carried out at room temperature in most cases with a high yield of cycloisomerisation. An exclusive Markovnikov regioselectivity is observed with mono, 1,1-di and trisubstituted double bonds. The attack of hydroxyl group on the 1,2-disubstituted double bond occurs with lower rates and various regioselectivities. As a general trend, the reactivity of the unsaturated alcohol depends strongly on both the double bond substitution pattern and the degree of angle compression brought by the substituents.
4.3 C–P bond formation
Alkyl and amide alkaline earth complexes (Ca, Sr, Ba) based on iminoanilide or bisiminoanilide ligands have been revealed recently to be efficient (pre)catalysts for intermolecular hydrophosphinations of styrene derivatives. Independently developed by Cui et al. and Carpentier et al.,54,39b both families 22–23 proved stable in solution and produced regioselectively anti-Markovnikov adducts in Scheme 20. Yields are impressive for both the introduction of PPh2 and PCy2 fragments at low catalyst loadings (1–5 mol%). Conversions are depending on the ability of styrene to bind to the metal centre and thus on the electronic contribution of substituents installed at the styrene substrate. As expected, electron-withdrawing groups are preferred, leading to the following reactivity trend: CF3 > Cl > H > Me > tBu > OMe. A second key point is the metal size. Indeed, the ionic radius deeply influences hydrophosphination within a same ligand series with the following order of reactivity Ba > Sr > Ca. A similar trend was noticed in alkene hydroamination promoted by the same series of complexes (Scheme 16). Hydrophosphination of conjugated dienes (isoprene and myrcene) were also examined (Scheme 20). In this case, the regioselectivity seems largely depending on two main factors: (i) the steric congestion imposed by the ligand, (ii) the identity of the metal centre. 3,4-Addition products are favoured using iminoanilide-Ca or -Sr complexes 22–23 (Ae = Ca, Sr). In contrast, 1,4-addition adducts are obtained using more bulky iminoanilide-Ba 22–23 (Ae = Ba) or bisiminoanilide-Ca complexes 33. Moreover, alkylcatalysts 23 are more active than the amide analogues 22 leading to faster conversions and higher yields.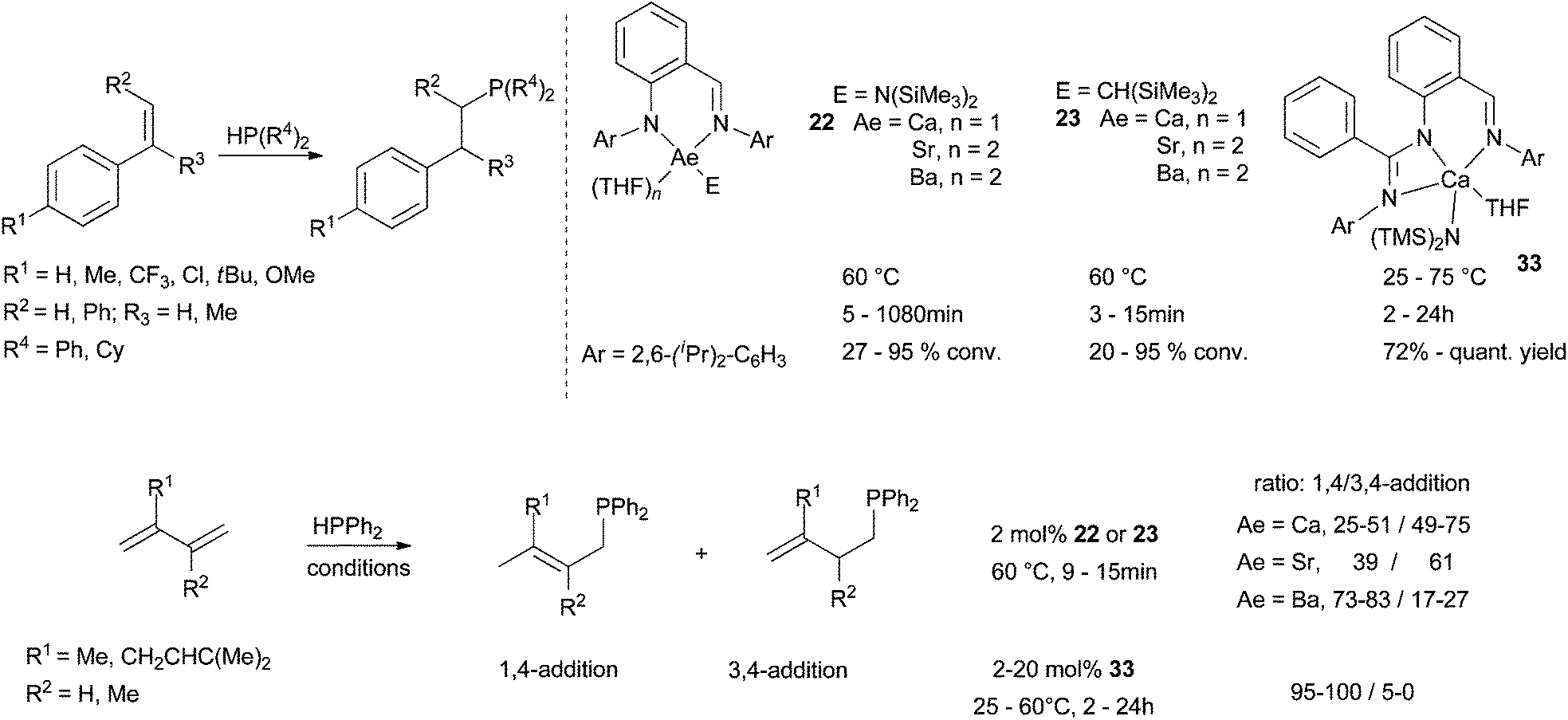 | ||
| Scheme 20 Group 2-catalysed intermolecular hydrophosphination of styrene derivatives and conjugated dienes. | ||
5. Group 3-based catalysts
5.1 C–N bond formation
Seminal work arose from the group of Marks in the late eighties,55 who discovered the extraordinary ability of rare-earth based complexes to promote the hydroamination of unactivated olefin with various amines. Due to their non-toxicity, their relative abundance in nature, the stereoelectronic tunability of their coordination sphere and the variation of the metal ionic radius, numerous complexes have been prepared from diverse ancillary ligands allowing large optimisation variables.56 Metallocene and then non-metallocene complexes have been shown efficient promoters for both intra- and intermolecular hydroamination. Some challenges still exist, however, that justify the search for new catalysts; demanding substrates, possessing for instance internal non-activated olefins or lacking substituents for Thorpe-Ingold effect, are tough to cyclise; large rings are not easy to obtain; the ratio alkene/amine = 1/1 is hardly maintainable for intermolecular hydroamination; asymmetric intermolecular hydroamination is barely described. The main developments in the last past year have been devoted to the preparation of mono-cyclopentadienyl complexes, abandoning metallocenes towards an electronically less saturated and sterically less crowded metallic site. Marks et al. for instance prepared phenylene-bridged half-lanthanocenes as binuclear complexes for the hydroamination of various substrates. Steric contribution of the adjacent metal-containing fragment enhanced greatly the stability towards ligand redistribution, but diminished unfortunately the catalytic activity vs. the mononuclear analogues.57 Chen and his group published further the synthesis and characterisation of half-sandwich indenyl rare-earth dialkyl complexes of Y, Lu and Dy 34 (Scheme 21). X-ray diffraction demonstrated η5-hapticity of indenyl ligands in all complexes that were all active, albeit at 60 °C, for the cyclohydroamination of substrate I (Scheme 21, and Table 2, entries 1–3).58 Other mono-cyclopentadienyl pre-catalysts have been targeted possessing additional coordinating elements for the rare-earth metal. Hence, Sundermeyer et al. synthesised cyclopentadienylphosphazene dialkyl complexes of rare-earth 35 (Scheme 21) demonstrating that the ligand could stabilise dialkyls over the full range of group 3 and lanthanide cation radii.59 Due to the strongly chelating constrained geometry of the cyclopentadienyl-phosphazene ligands, only η1,η5-coordination was observed for all rare-earth elements used, without any additional solvent chelation. Intramolecular hydroamination of substrate I occurred successfully with a reactivity trend ranging in the order Lu < Y < Sm < Nd < Pr (Scheme 21, and Table 2, entries 4–8).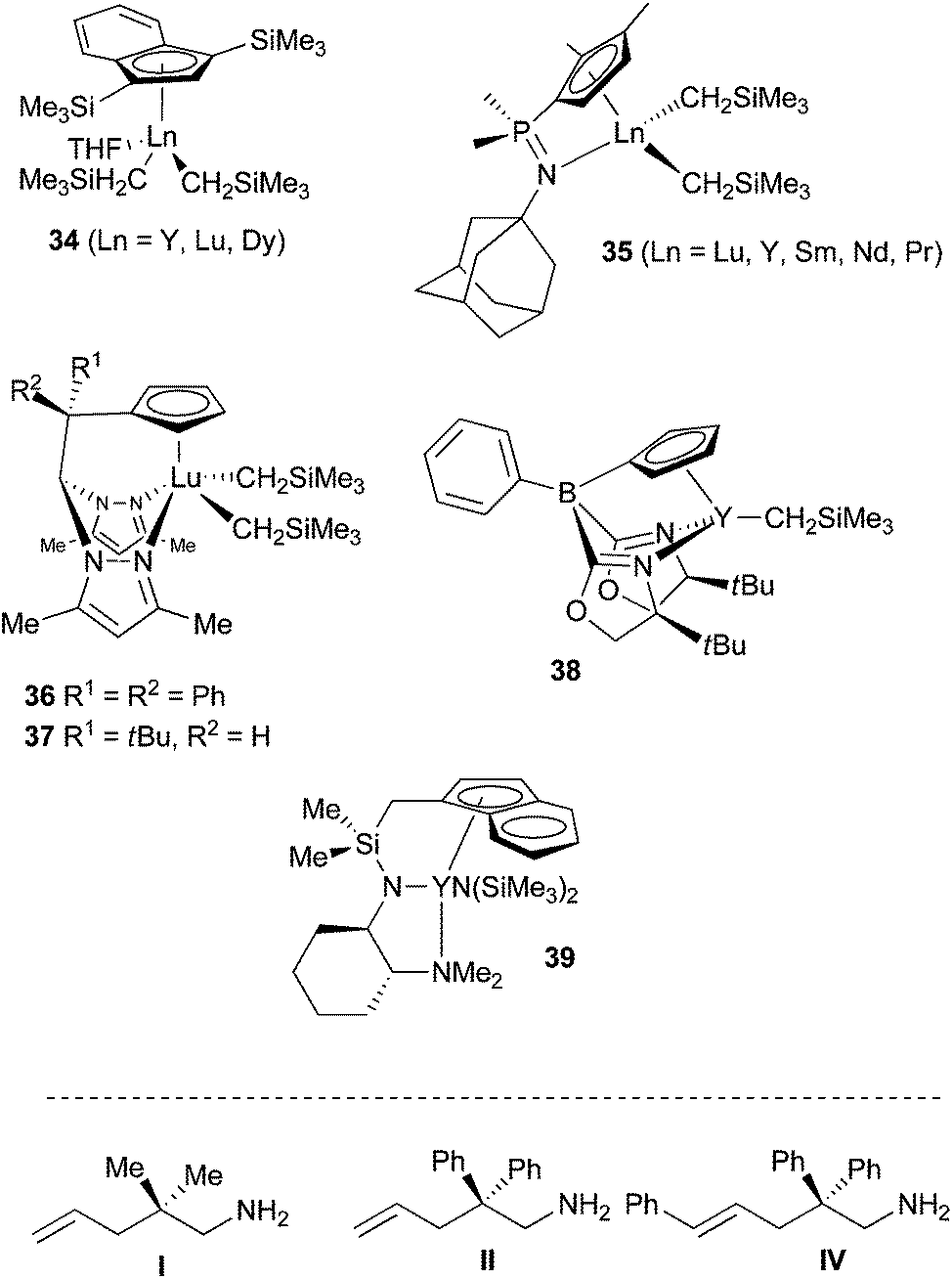 | ||
| Scheme 21 Cyclopentadienyl- or indenyl-based rare earth complexes for hydroamination of aminoalkenes. | ||
| Entry | Substrate | Catalyst (Ln) (mol%) | Time (h) | T (°C) | Conv (%) | eea (%) |
|---|---|---|---|---|---|---|
| a n.a. = non applicable. | ||||||
| 1 | I | 34 (Y) (1) | 0.5 | 60 | 98 | n.a. |
| 2 | I | 34 (Lu) (1) | 0.8 | 60 | 98 | n.a. |
| 3 | I | 34 (Dy) (1) | 0.3 | 60 | 98 | n.a. |
| 4 | I | 35 (Lu) (6) | 14 | 60 | >92 | n.a. |
| 5 | I | 35 (Y) (5) | 4.5 | 27 | >93 | n.a. |
| 6 | I | 35 (Sm) (5) | 1 | 27 | >93 | n.a. |
| 7 | I | 35 (Nd) (5.3) | 0.5 | 27 | >93 | n.a. |
| 8 | I | 35 (Pr) (5.3) | 0.4 | 27 | >93 | n.a. |
| 9 | II | 36 (1) | 1.5 | 20 | 98 | n.a. |
| 10 | II | 37 (1) | 2 | 20 | 98 | n.a. |
| 11 | II | 38 (5) | 0.2 | 20 | >99 | 94 |
| 12 | II | 39 (2) | 0.8 | 20 | 98 | 85 |
| 13 | IV | 39 (2) | 9.6 | 20 | 99 | 97 |
Neutral heteroscorpionate dialkyl lutetium complexes such as 36 have been developed by the group of Otero and Lara-Sánchez.60 Chiral complex 37 could also be interestingly prepared as an enantioenriched species, by spontaneous resolution and preferential crystallisation of one enantiomer, due to the formation of a conglomerate by supramolecular CH–π interactions (Scheme 21). This precatalyst promoted the intramolecular hydroamination of substrate II at room temperature with up to 64% ee. Considering enantioselective catalysis, important results were described by Sadow et al. who performed the intramolecular hydroamination of substrate II in the presence of an oxazolinylborato diamido yttrium complex 38, to yield the product in 94% ee (S).61 Remarkably, the analogous Zr-complex possessing the same ancillary ligand also delivered this high enantioselectivity but furnished the opposite enantiomer (R). Although a similar mechanistic route was postulated for both metals involving related transition states, the stereoinduction must be issued from dissimilar mechanisms. Very recently, tridentate amido-indenyl-based yttrium complex 39 has been reported containing enantiopure 1,2-cyclohexanediamine as chiral backbone (Scheme 21).62 This perfectly characterised chiral catalyst afforded very high selectivities for the cyclization of both II (85% ee) and the di-substituted substrate IV (97% ee).
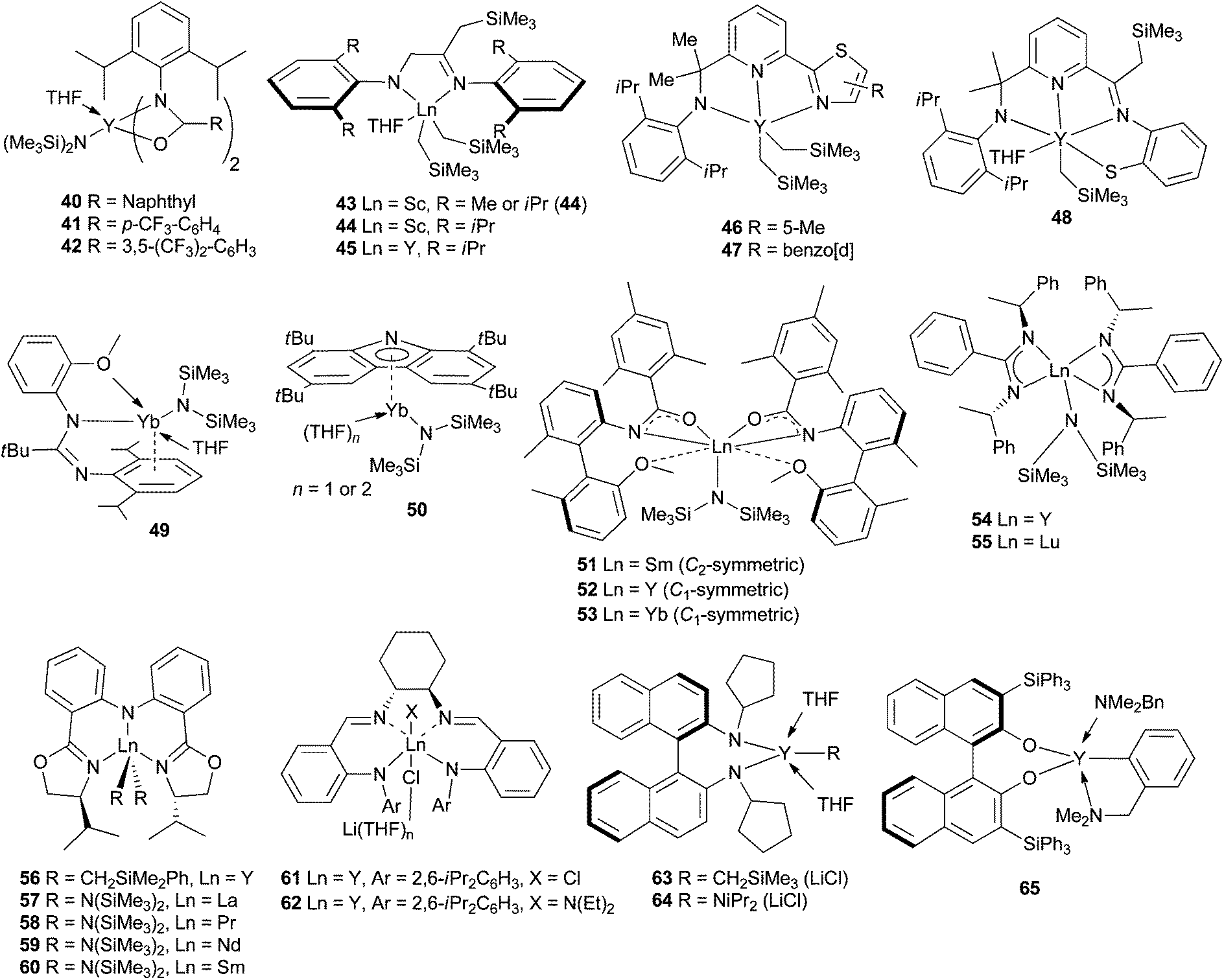
Non-metallocene complexes have also been the subject of much effort, in the search of activity and selectivity. Bis- and mono(amidate) complexes of yttrium 40–42 were reached by reaction of the amide proligand and Y(N(SiMe3)2)3 (Scheme 22).63 Specifically, in the presence of electron-withdrawing groups on the ligand, enhanced catalytic activity was observed (compare entries 1–3 in Table 3). The monoamidate derivative 40 synthesised from the naphthyl-substituted ligand, delivered the targeted pyrrolidine from I with analogous activity (2.5 h, 83%). Amido-imino and diamido scandium and yttrium complexes 43–45, prepared by intramolecular alkylation of α-diimine ligands by treatment with alkyl complexes M(CH2SiMe3)3-(THF)2 could be obtained, according to the steric bulk of the aryl-diimine ligand. The best activity in the benchmark reaction was obtained with the more sterically hindered complex (Table 3, entries 4–6).64N−,N,N monoanionic dialkyl yttrium complexes 46–47 supported by thiazole-containing amidopyridinate ligands were prepared and fully characterised. Cyclohydroamination of substrate II was performed with these neutral and the corresponding cationic complexes, with the cationic forms showing more important activities (Table 3, entries 7–9). Generation of a monoalkyl complex interestingly occurred by metal to ligand alkyl migration and benzothiazole ring opening, resulting in a monoalkyl aryl thiolate yttrium complex 48 supported by a tetradentate N−,N,N,S− dianionic ligand. Unfortunately this new species revealed to be a poor catalyst.65 Of less conventional way, divalent heteroleptic ytterbium amido complexes 49–50 supported by amidinate and 1,3,6,8-tetra-tBu-carbazol-9-yl ligands respectively have been prepared by the groups of Carpentier and Trifonov.66 The presence of an OMe group on one of the aryl substituents of the amidinate ligand led to an unprecedented κ1-N,κ2-O,η6-arene coordination, instead of the classic κ2-N,N′ mode in complex 49. Similarly, replacing a simple carbazol-9-yl by the tetra tBu analogue resulted in the switch of coordination mode from σ to π (50). These complexes were interestingly tested in the intermolecular hydroamination between styrene and pyrrolidine and were active catalysts at 60 °C, albeit with a necessary long reaction time (Table 3, entries 10–11).
 | ||
| Scheme 22 Non-cyclopentadienyl based rare earth complexes for hydroamination of aminoalkenes. | ||
| Entry | Substrate | Catalyst (mol%) | Time (h) | T (°C) | Conv (%) | eea (%) |
|---|---|---|---|---|---|---|
| a n.a. = non applicable. | ||||||
| 1 | I | 40 (10) | 4.5 | 25 | 88 | n.a. |
| 2 | I | 41 (10) | 2.5 | 25 | 88 | n.a. |
| 3 | I | 42 (10) | 2.5 | 25 | 93 | n.a. |
| 4 | I | 43 (5) | 5 | 60 | 53 | n.a. |
| 5 | I | 44 (5) | 5 | 60 | >95 | n.a. |
| 6 | I | 45 (5) | 0.2 | 20 | >95 | n.a. |
| 7 | II | 46 (5) | 1.5 | 40 | 11 | n.a. |
| 8 | II | 46 Cationic (5) | 1.5 | 40 | 97 | n.a. |
| 9 | II | 47 Cationic (5) | 0.75 | 80 | >99 | n.a. |
| 10 | Styrene/pyrrolidine | 49 (5) | 31 | 60 | 85 | n.a. |
| 11 | Styrene/pyrrolidine | 50 (5) | 31 | 60 | 96 | n.a. |
| 12 | I | 51 (5) | 16 | 60 | 95 | 66 |
| 13 | I | 52 (5) | 16 | 60 | 97 | 54 |
| 14 | I | 53 (5) | 16 | 60 | 90 | 50 |
| 15 | I | 54 (5) | 6.5 | 60 | >95 | 62 |
| 16 | I | 55 (5) | 35 | 60 | 92 | 75 |
| 17 | II | 56 (5) | 0.25 | 22 | >99 | 43 |
| 18 | II | 57 (5) | 0.25 | 22 | >99 | 5 |
| 19 | II | 58 (5) | 1 | 22 | >99 | 16 |
| 20 | II | 59 (5) | 0.2 | 22 | >99 | 12 |
| 21 | II | 60 (5) | 1 | 22 | >99 | 20 |
| 22 | I | 61/nBuLi (5) | 27 | 25 | 95 | 90 |
| 23 | I | 62 (5) | 29 | 25 | 95 | 90 |
| 24 | II | 63 (6) | 2.3 | 25 | >99 | 66 |
| 25 | II | 63/LiCl (6) | 1.5 | 25 | >99 | 77 |
| 26 | II | 64 (6) | 2 | 25 | >99 | 65 |
| 27 | II | 64/LiCl (6) | 1 | 25 | 92 | 72 |
Chiral biphenyl-based tridentate amidate ligands allowed the synthesis of various complexes according to the size of the lanthanide ion.67 For Sm, the formation of a C2-symmetric bis-ligated amidate complex 51 occurred whereas for Y and Yb, C1-symmetric bis-ligated amidate complexes 52–53, were privileged as demonstrated by X-ray diffraction analyses. These catalysts promoted the cyclization of I with up to 66% ee, at 60 °C (Table 3, entries 12–14). Chiral bis(benzamidinate)-amido complexes of yttrium 54 and lutetium 55 have been synthesised in one step from the corresponding Ln-amides in the group of Roesky. These complexes showed additional axial chirality and they crystallised as diastereomerically pure compounds.68 The yttrium complex was more active but less selective than the Lu one and the pyrrolidine issued from I could be obtained in up to 75% ee (Table 3, entries 15 and 16). By far more active but less selective complexes have been very recently described by Ward et al., through coordination chemistry of the bis(oxazolinylphenyl)amide (BOPA) ligand with lanthanide alkyl and amides (56–60, Ln = Y, La, Pr, Nd, Sm).69 The yttrium alkyl derivative led to the synthesis of the pyrrolidine in 43% ee. The corresponding amido complex was not reactive, but other Ln-derived amido complexes could promote the reaction, albeit with low selectivities (Table 3, entries 18–21). Contrarily, excellent results in terms of enantioselectivities have been reported by the group of Mu, who discovered the utility of chiral tetra-azane ligands to prepare efficient scandium and yttrium complexes from salt metathesis of MCl3. X-Ray diffraction studies showed a distorted-octahedral coordination environment, the metal centre sharing the Cl with a LiCl(THF)n moiety. From the chloride complexes, the authors could generate active hydroamination catalysts with in situ addition of nBuLi or Me3SiCH2Li on complex 61 (Scheme 22), and the corresponding amido complexes 62 were also catalysts for this reaction delivering up to 90% ee.70 Some of us have discovered and studied the activity in asymmetric intramolecular hydroamination reaction, of a new family of structurally defined heterobimetallic rare earth lithium ate complexes and corresponding neutral species, based on N-substituted binaphthylamido ligands.71 In the specific case of yttrium complexes, cooperative effects between Y and Li were highlighted for both alkyl and amido neutral complexes 63 and 64 delivering enhanced enantioselectivities in the presence of LiCl, for cyclohydroamination reactions (Table 3, entries 24–27).72 The same ligand family was used for the in situ generation of binaphthylamido alkyl yttrium ate complexes that were interestingly demonstrated to be very active catalysts for the anti-Markovnikov addition between styrenyl or vinylpyridine derivatives and secondary amines.73 The challenge for performing this reaction in an enantioselective manner has been surpassed by Hultzsch and coworkers by preparing a triphenylsilyl-substituted binaphtholate alkyl yttrium complex 65. Up to now, this is the only rare earth catalyst able to perform asymmetric intermolecular hydroamination of unactivated alkenes with simple amines following Markovnikov regioselectivity; up to 61% ee was reached for the addition of benzylamine on 1-pentene.
5.2 C–O bond formation
Lanthanide halides and triflates have been used as Lewis acids-type catalysts for the C–O bond formation by hydroalkoxylation as well as lanthanide amides as Brønsted base. In 2002, Rosini et al. described the use of cerium(III) chloride in the intramolecular alkene hydroalkoxylation. This methodology allows transformations of substrates bearing ester functional groups. These reactions are carried out in boiling acetonitrile giving the corresponding products in fair to good yields. It seems that CeI3 would be the real promotor but the yields are better when 10 mol% CeCl3·7H2O–NaI were used. Moreover, lesser yields and longer reaction times were observed when 1.5 equivalents of the promotor is used (Scheme 23). The role of water albeit crucial is not well understood.74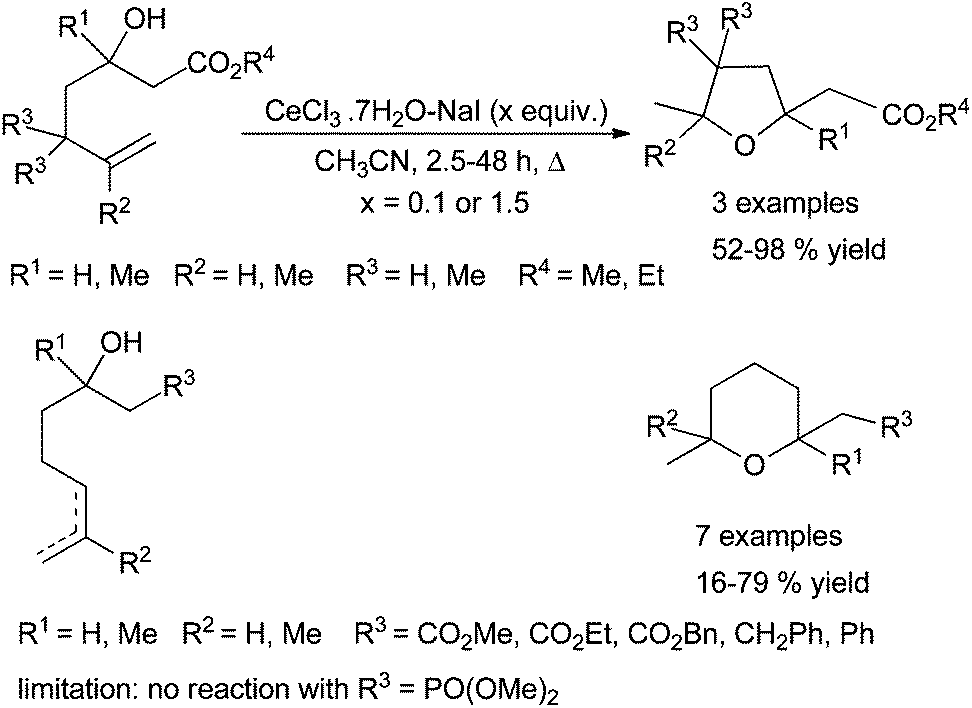 | ||
| Scheme 23 Intramolecular hydroalkoxylation of alkenols catalysed by cerium(III) chloride heptahydrate in presence of sodium iodide. | ||
In 2007, building on its strong expertise on the application of lanthanide amides in hydroamination, the group of Marks developed the use of La[N(TMS)2]3 and Yb(OTf)3 in the cyclisation of allenols and alkenols respectively with Markovnikov-type selectivity (Scheme 24).75 Lanthanide triflates are more efficient than lanthanide amides for hydroalkoxylation. Application of La[N(TMS)2]3 have been tested by Alcaide et al. on chiral β,γ- and γ,δ-allendiols leading to hydroxymethylfurane unlike Pt, Pd and Au salts.76
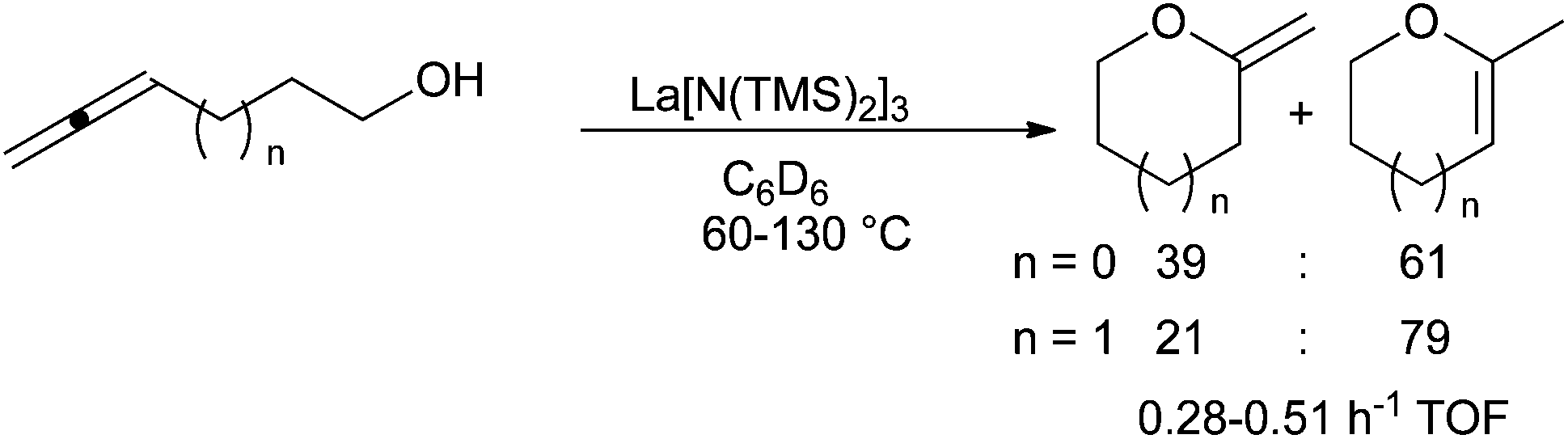 | ||
| Scheme 24 Intramolecular hydroalkoxylation of allenols catalysed by La[N(TMS)2]3. | ||
In the course of studies, Dzudza and Marks et al. have also reported that lanthanide triflates could be associated with ionic liquid (IL) to improve the catalytic efficiency of the functionalisation of alkenes (Scheme 25).77 ILs have been chosen for their low volatility, their high polarity and their potential environmental benefit. Various room temperature ionic liquids (RTILs) have been tested and the best results were obtained with [1-methyl-1,3-diazabicyclo-[5.4.0]undec-7-enium] [OTf] noted [C2mim] [OTf]. The use of these conditions allows a low catalytic charge with only 1 mol% of Ln(OTf)3. The catalytic activities, expressed by the turnover frequencies (h−1), depend on the ionic radius of the lanthanide (Ln = Yb, Sm, La). The smaller the radius is, the higher the activity is and ytterbium triflate affords the highest activities.
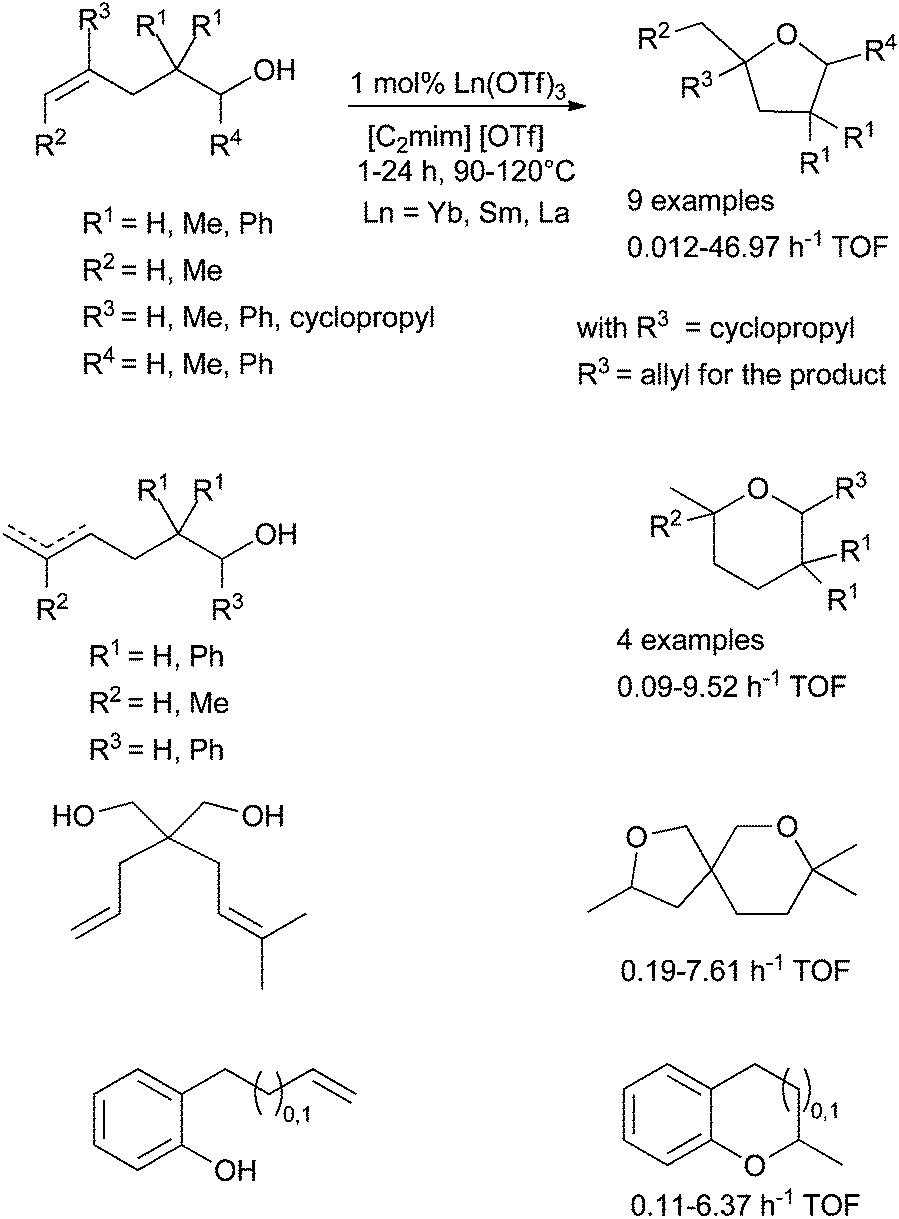 | ||
| Scheme 25 Intramolecular hydroalkoxylation of alkenols catalysed by lanthanide triflate in ionic liquid. | ||
Finally, efficient methods have been developed in intermolecular hydroalkylation between isoprene and methanol using a strong association between lanthane triflate and phosphoric acid (Scheme 26, method A).78
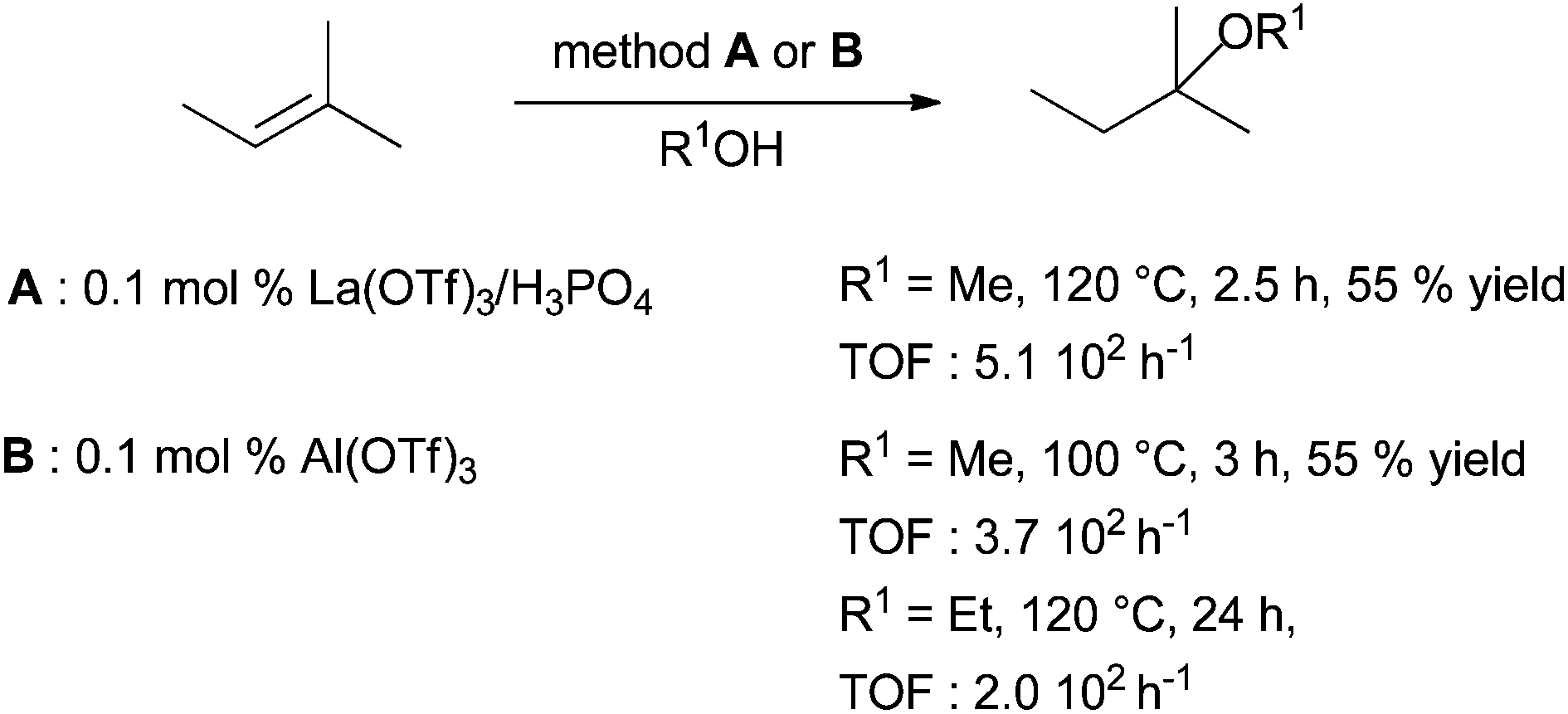 | ||
| Scheme 26 Intermolecular hydroalkoxylation of isoprene with methanol catalysed by lanthanide triflate associated to phosphoric acid or aluminium triflate. | ||
5.3 C–P bond formation
Intermolecular hydrophosphination of styrene and phenyl- and diphenyl-phosphine was achieved using well defined Yb and Eu complexes 49, 50, 66 and 67 as shown in Schemes 22 and 27.54,79 All reactions were found to be regioselective affording the anti-Markovnikov adducts. Noteworthy, unusual π-coordination in Yb complexes showed performance in hydrophosphination of styrene as high as more classical complexes.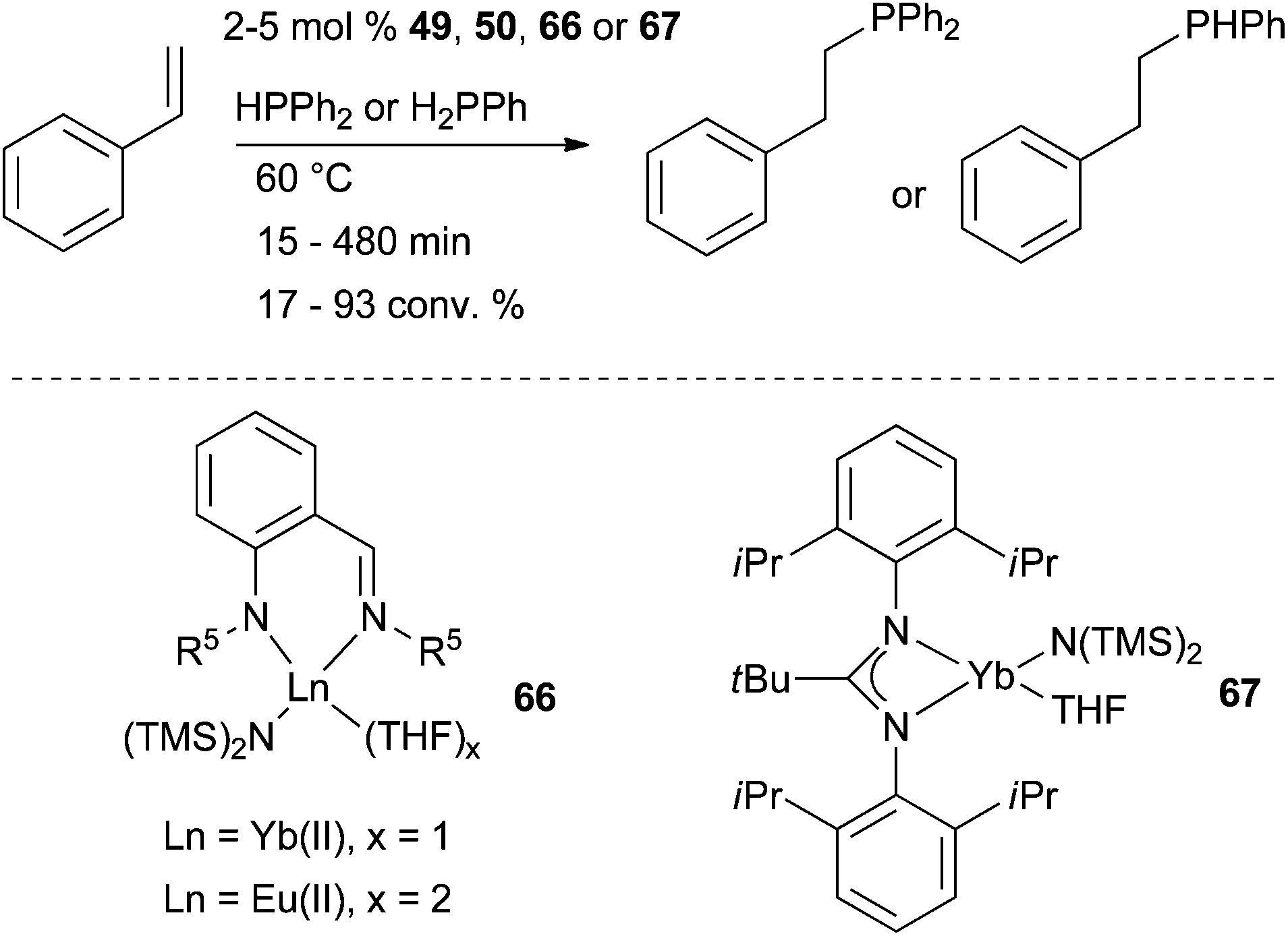 | ||
| Scheme 27 Intermolecular hydrophosphination of styrene and phenyl- and diphenyl-phosphine catalysed by ytterbium(II) and europium(II) complexes. | ||
6. First-row transition metal catalysts
6.1 Titanium
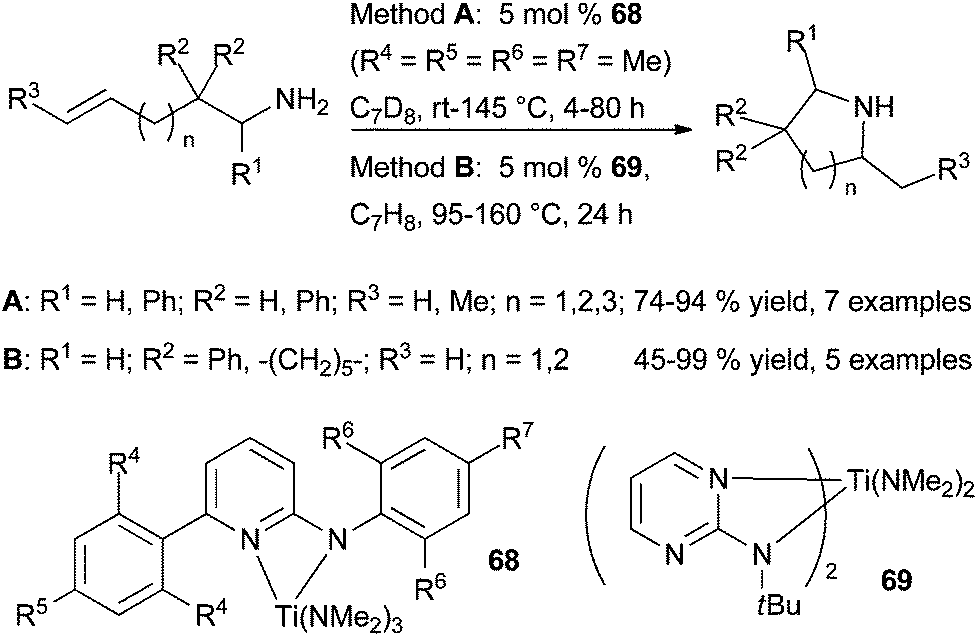 | ||
| Scheme 28 Titanium-catalysed cyclohydroamination of aminoalkenes. | ||
The same year, Doye et al. disclosed the preparation of well-defined bis[2-(tert-butylamino)pyrimidine]bis(dimethylamino) titanium complex (69) and its application as catalyst for the hydroamination of alkynes and cyclohydroamination of primary amines tethered to simple alkenes.83 This bis-ligated (aminopyrimidinato)titanium catalyst allows the formation of pyrrolidines and piperidines in moderate to high yields from gem-disubstituted aminoalkenes under however harsher conditions than 68.
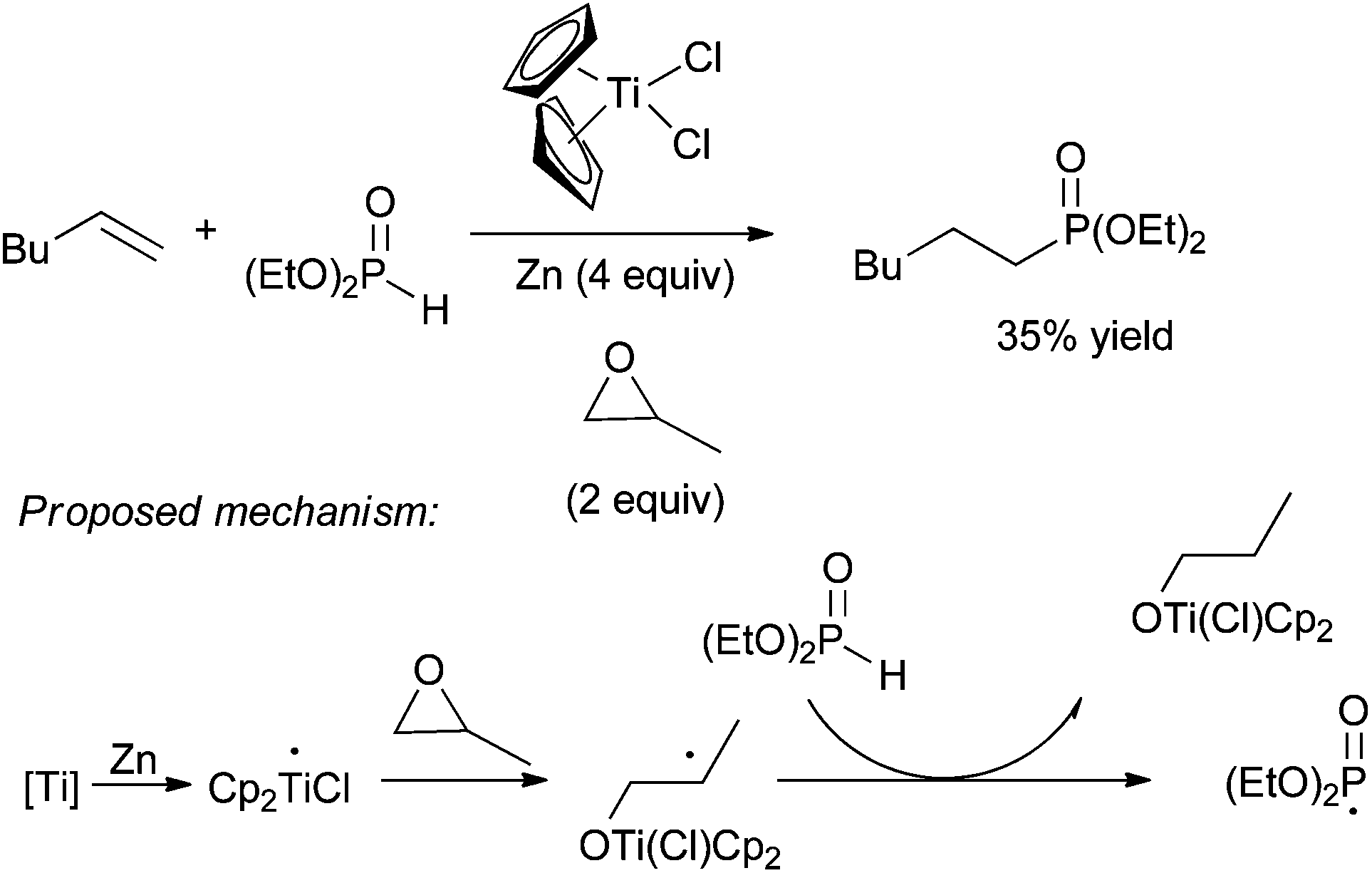 | ||
| Scheme 29 Hydrophosphonylation of hex-1-ene catalysed by Cp2TiCl2. | ||
6.2 Iron
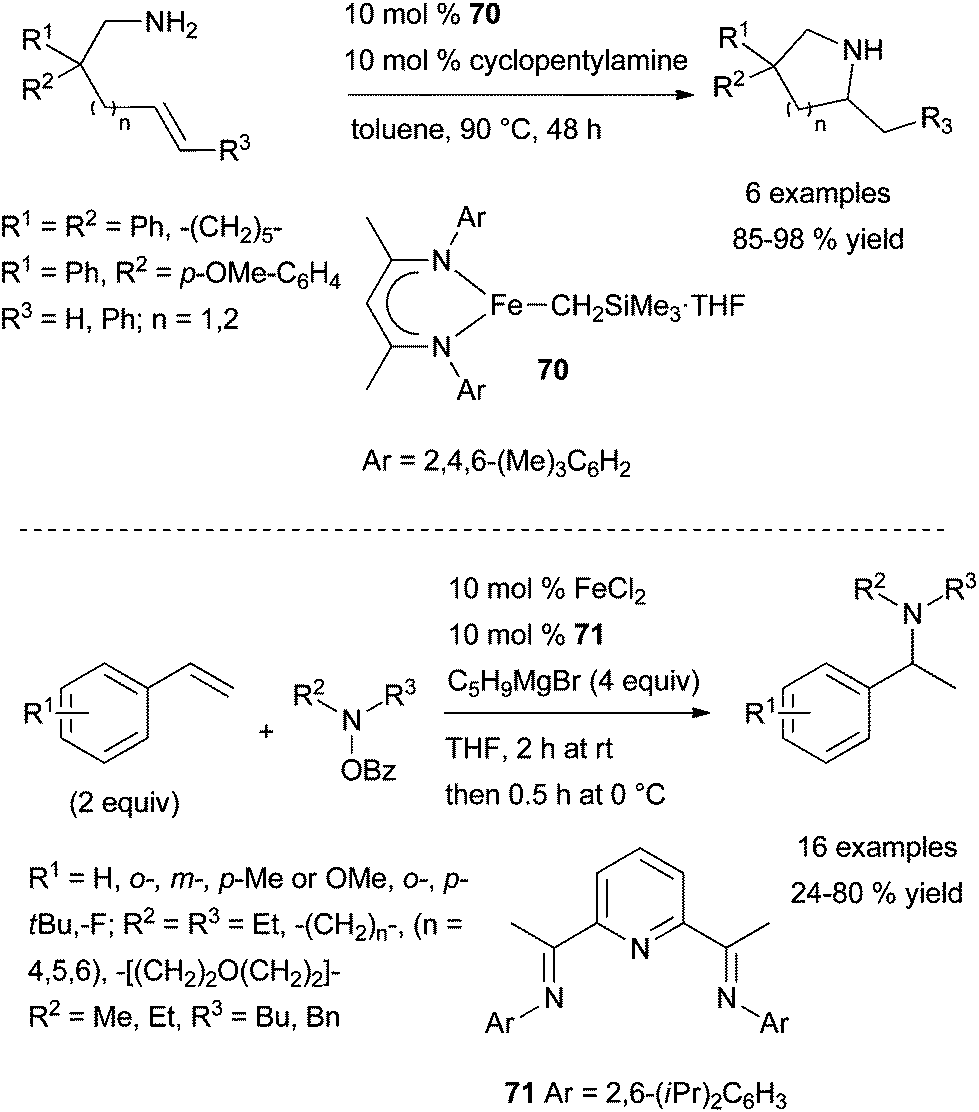 | ||
| Scheme 30 Iron-catalysed intra- and intermolecular Markovnikov hydroamination of primary amines tethered to alkenes and styrenes. | ||
Last year, Yang et al. disclosed the first report of iron-catalysed electrophilic amination of unactivated alkenes by an umpolung approach.91 Following optimisation studies, a catalytic mixture of FeCl2·71 (10 mol%), in the presence of cyclopentylmagnesium bromide as the reducing agent were identified as optimal for the regioselective Markovnikov hydroamination of styrene with O-benzoyl-N-hydroxypiperidine as the electrophilic nitrogen source (Scheme 30, bottom). This protocol which requires the slow addition of the electrophilic amine, is applicable to a variety of electron-poor and -rich substituted styrenes and delivers the branched amines in moderate to excellent yields. However, it is inefficient for the transformation of styrenes bearing a chloro or a trifluoromethyl group on the aromatic ring, α- and β-methylstyrenes, and aliphatic terminal alkenes.
 | ||
| Scheme 31 Catalytic inter- and intramolecular hydroalkoxylation of alkenes by iron chloride in the presence of TsOH. | ||
Through many transformations, the high activity and selectivity of iron have been demonstrated in the literature. In 2011, B. Alcaide and P. Almendros have illustrated an efficient chemodivergent metal-controlled methodology during the oxycyclisation study of β-lactam enallenols. The cyclisation of enallenols catalysed by precious metal salts – [PtCl2(CH2CH2)]2 and AuCl3 – affords the allene cycloisomerisation adduct whereas the chemoselectivity with FeCl3 is in favour of the alkenol activation. It was the first example of a total selectivity in favour of an olefin moiety vs. allene functionality. Moreover with FeCl3, regiospecificity for five membered rings was observed affording exclusively the β-lactam-tetrahydrofuran hydride (Scheme 32). It is worth noting that complete conversions can be obtained with BiCl3, InCl3 or HfCl4 but in these cases stoichiometric amounts of activators are needed.94 A limitation of the system concerns β-lactam enallenols containing tetrasubstituted olefins. For these substrates, the carbonyl group becomes more electrophilic than the tetrasubstituted alkene leading to lactones instead of the hydroalkoxylation products.95
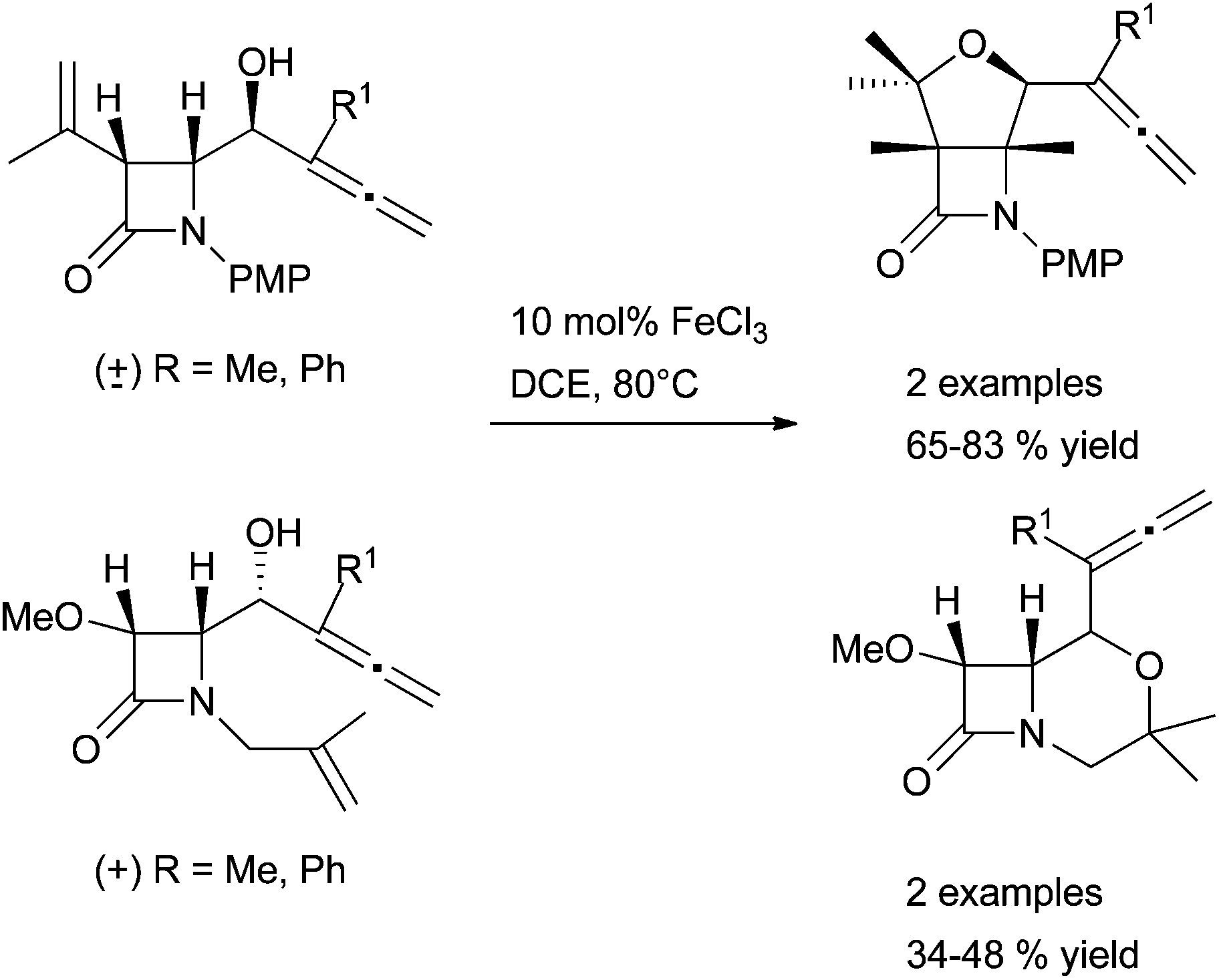 | ||
| Scheme 32 FeCl3-catalysed hydroalkoxylation of β-lactam enallenols. | ||
The chemodivergent metal-controlled methodology developed by B. Alcaide and P. Almendros was also applied to non-β-lactam enallenols. During this work, full chirality transfer from syn and anti enallenols was observed (Scheme 33). However, with an enallenol moiety tethered to an aromatic ring, a carbocylisation/dehydration sequence occurs.
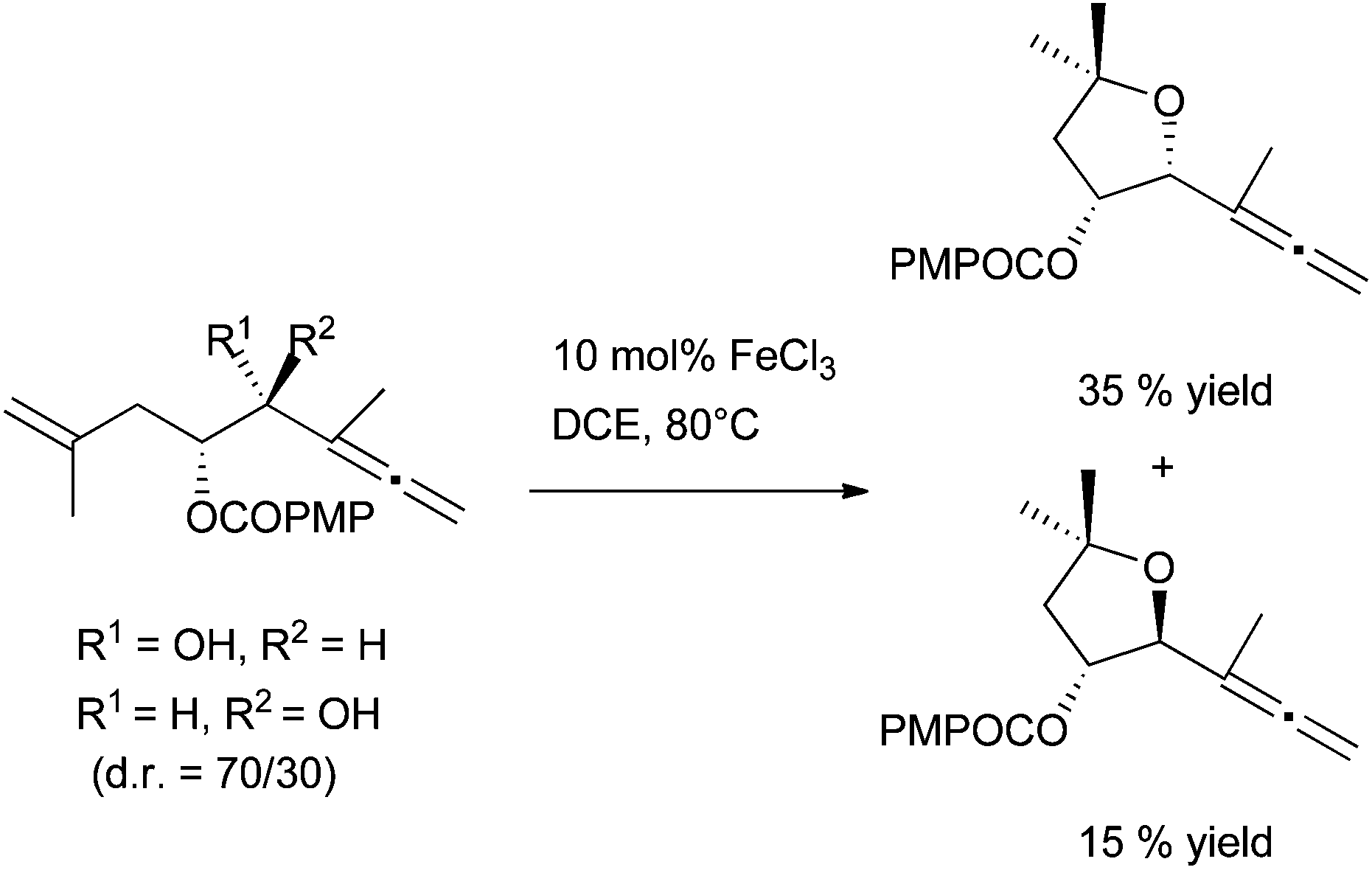 | ||
| Scheme 33 FeCl3-catalysed hydroalkoxylation of non-β-lactam enallenols. | ||
However, the hydroalkoxylation of allenol catalysed by an iron catalytic system is also possible. In fact, a highly efficient catalytic system for hydrofunctionalisation of trialkylated δ-allenyl alcohols and amines was reported by Kang et al. in 2012.88 The activities of the iron catalytic systems depend on their counterion and reaction conditions, and hydrofunctionalisation may be followed or not by the isomerisation of the cyclised product (Scheme 34). As indicated in section 6.2.1, this catalytic system was also applied in hydroamination of N-tosyl aminoallenes.88
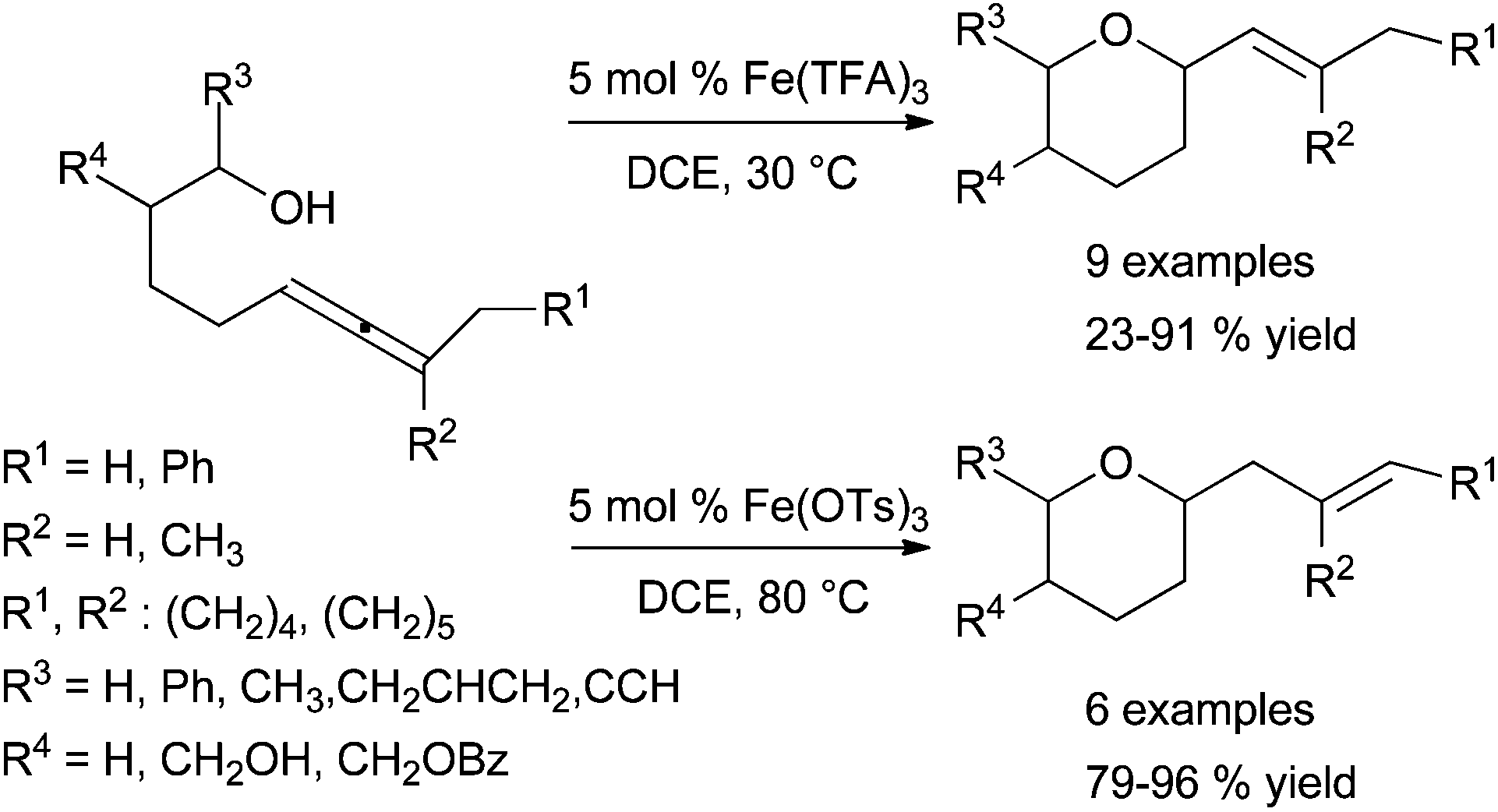 | ||
| Scheme 34 Hydroalkoxylation reaction catalysed by Fe(TFA)3 and Fe(OTs)3 for the synthesis of tetrahydropyran. | ||
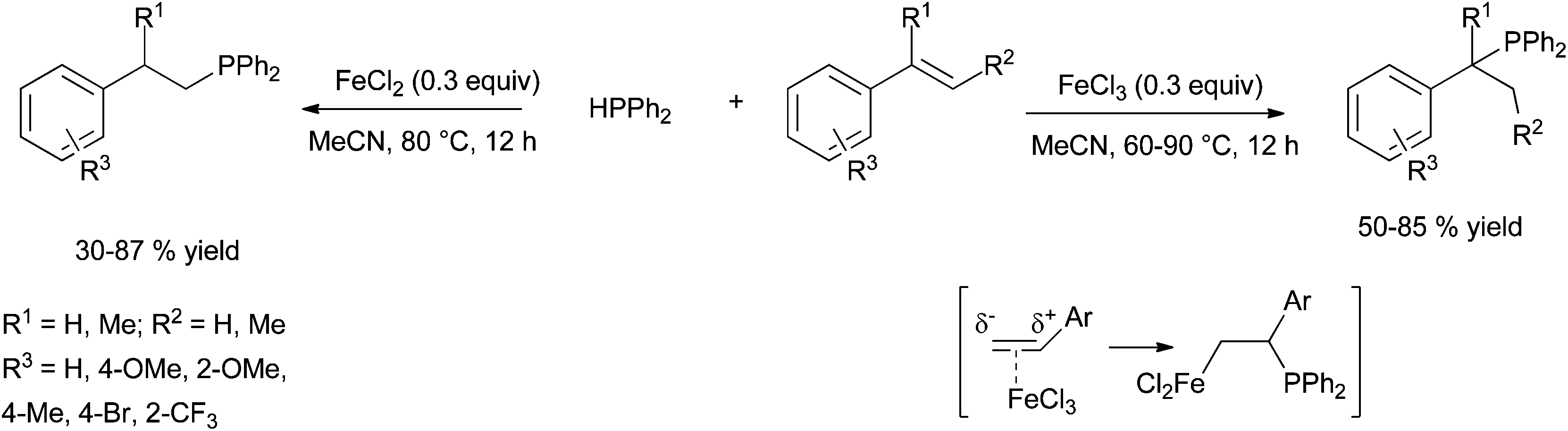 | ||
| Scheme 35 FeCl2- versus FeCl3-catalysed hydrophosphination of styrene derivatives: a switchable regioselectivity. | ||
Air stable salen-capped iron catalyst 72 has recently been developed by Webster et al.96 providing a new entry towards hydrophosphination reactions (Scheme 36). Indeed, hydrophosphination proceeds with low catalyst loading (0.5%) at room temperature to afford the anti-Markovnikov adduct. Isolated yields (from 43% to quantitative) show that the transformation appears poorly sensitive to the electronic effects of substituents installed at the styrene substrate. Preliminary mechanistic studies suggest that the reaction is unlikely to be radical mediated and rule out disproportionation of [salen-Fe]-O species into monomeric Fe(II)-based catalysts prior to substrate activation.
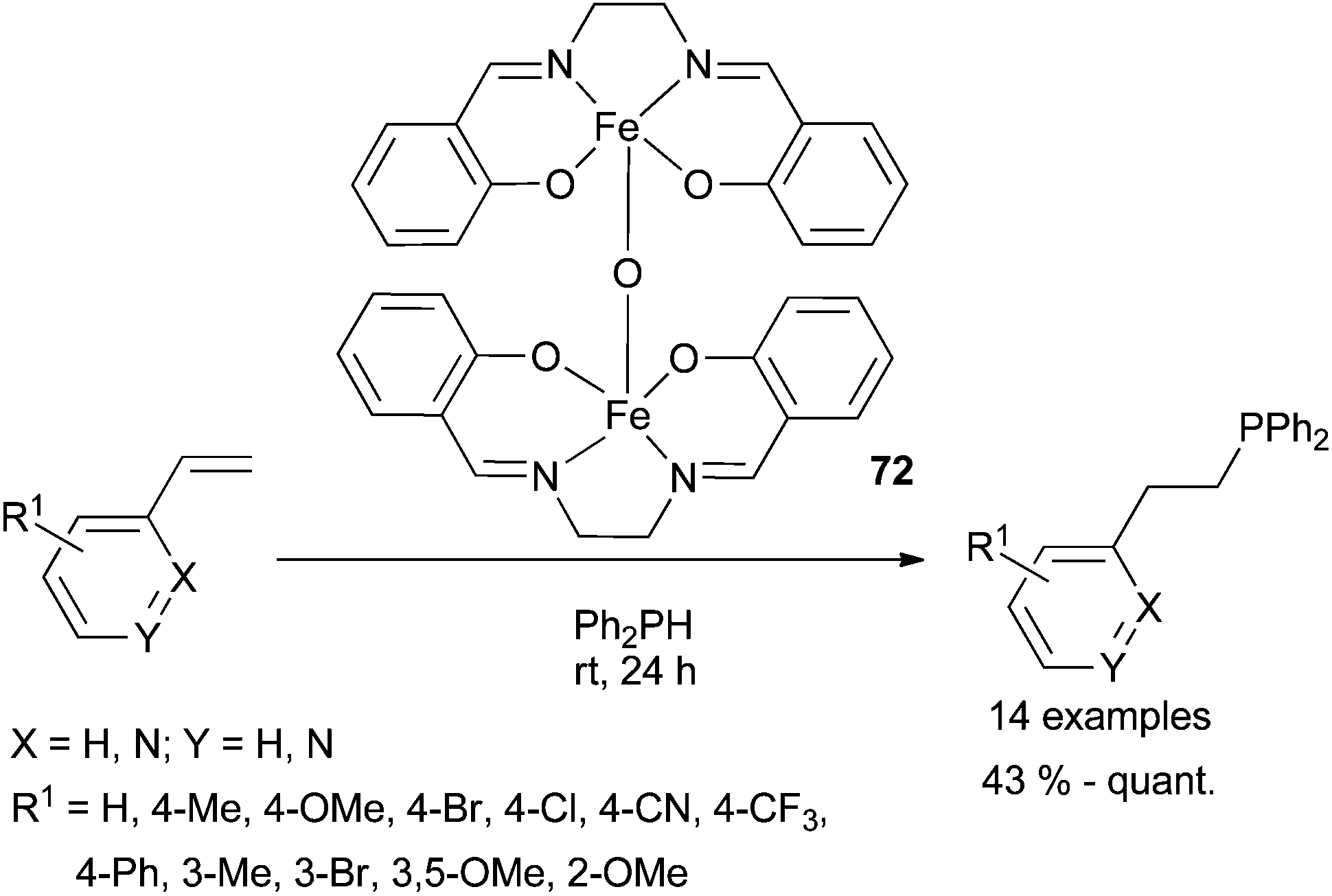 | ||
| Scheme 36 Anti-Markovnikov hydrophosphination of styrene and vinyl pyridine derivatives catalysed by an air stable salen-capped iron catalyst. | ||
6.3 Cobalt
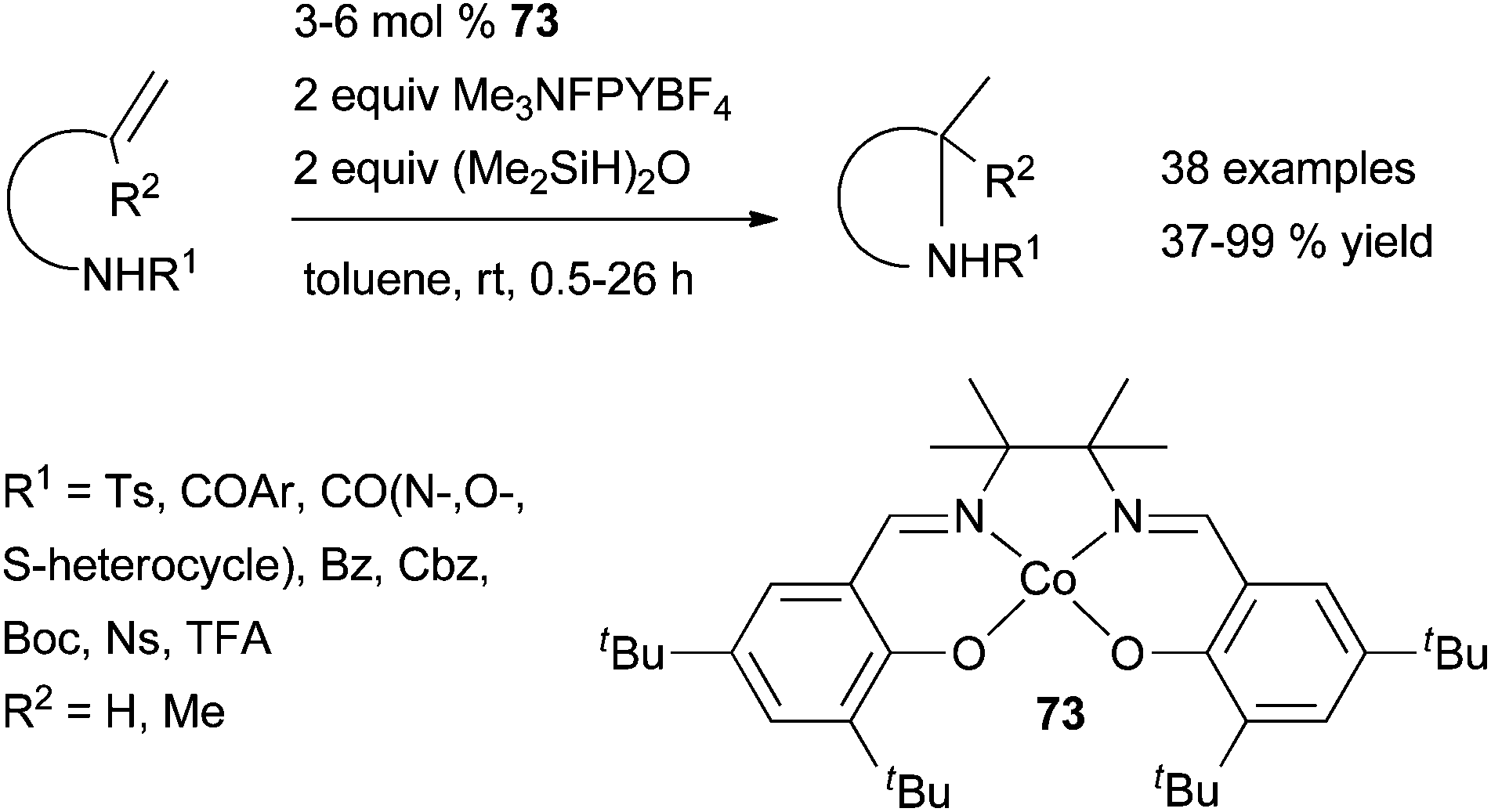 | ||
| Scheme 37 Cobalt-catalysed intramolecular hydroamination of secondary protected amines tethered to terminal alkenes. | ||
This protocol has a high functional group tolerance and impressively broad scope allowing the formation of 3-, 5-, 6- and even 7-membered heterocycles from various N-protected aminoalkenes, biased or unbiased toward cyclisation. Nevertheless, the methodology is unsuitable for the reaction of N-benzyl or free amines as well as internal olefins. From the work on cobalt-catalysed hydroalkoxylation, it might be speculated that the reaction proceeds through the formation of a carbocation intermediate, arising from oxidation of a carbon radical intermediate by a cobalt(III) species. The carbon radical results from a homolytic cleavage of a cobalt(III) alkyl species. The latter is generated by regioselective migratory insertion of the alkene moiety into the Co–H bond of a Co(III) hydride.
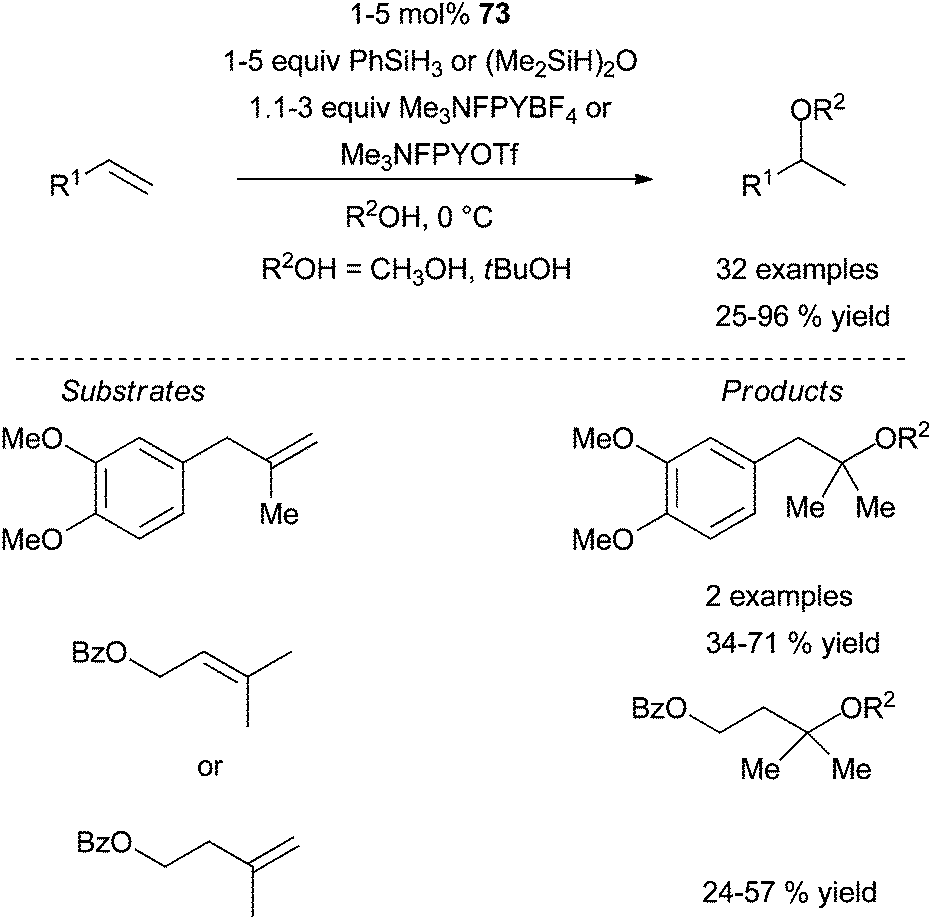 | ||
| Scheme 38 Cobalt-catalysed intermolecular hydroalkoxylation of mono- di- and tri-substituted olefins. | ||
It is interesting to note that under these catalytic conditions, the selectivity is in favour of the hydroalkoxylation versus hydroamination as represented in Scheme 39.98 However, a switch of selectivity can be achieved by the appropriate choice of the alcohol protecting group.
 | ||
| Scheme 39 O- versus N-selectivity in the cobalt-catalysed intramolecular hydroamination of protected aminoalkenes. | ||
6.4 Nickel
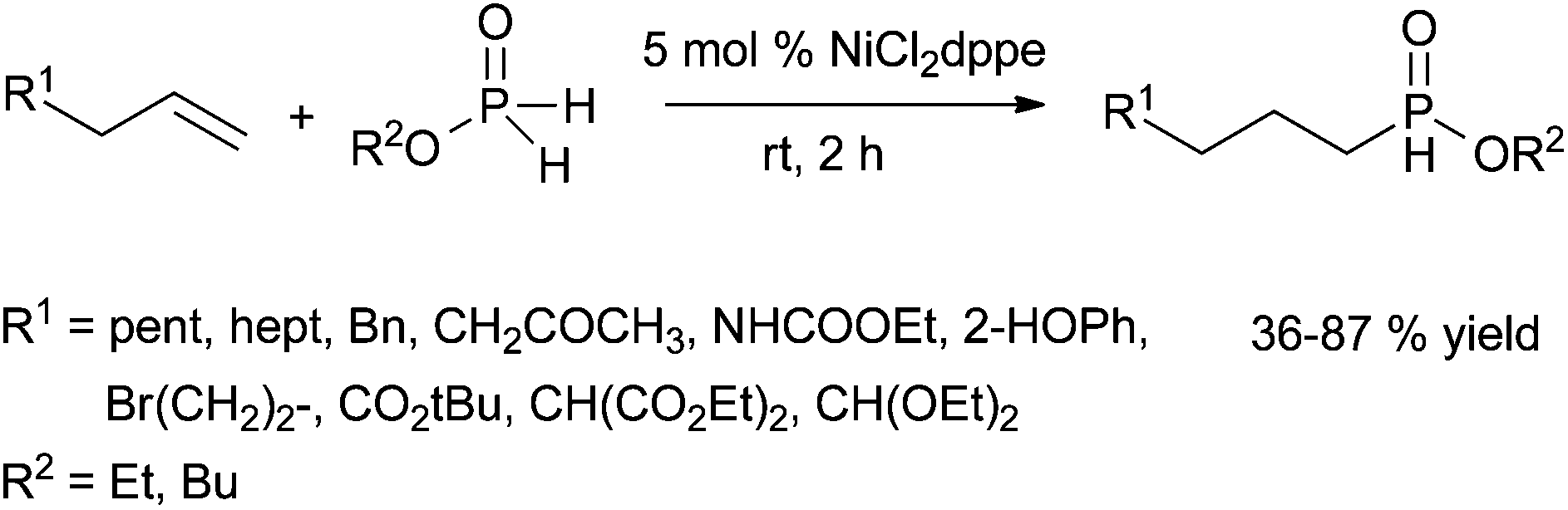 | ||
| Scheme 40 Nickel-catalysed intermolecular hydrophosphinylation of linear unactivated alkenes (dppe = 1,2-bis(diphenylphosphino)ethane). | ||
6.5 Copper
 | ||
| Scheme 41 Copper(I)-catalysed intramolecular hydroamination of aminoalkenes. | ||
More recently, a convenient copper(II)-catalysed synthesis of chiral 2-methylindoles using a Cu(OTf)2·(R,R)-Ph-Box catalyst was disclosed as the first report of enantioselective copper-catalysed alkene hydroamination.107 Despite the moderate to high level of enantioinduction (72–90% ee), this methodology affords the corresponding N-sulfonyl-2-methylindolines in low to moderate yields (37–70%) and requires the stoichiometric use of co-reagents (H-atom donor, oxidant and base). Further mechanistic investigations of alkene hydroamination catalysed by the combination of a copper halide, a diphosphine ligand and a silver salt underlines that a Brønsted acid might be the prominent catalytic species under these reaction conditions.108 This study brought to light that the development of truly copper-catalysed hydroamination processes are challenging, mainly for asymmetric applications.
Recently, the groups of Hirano, Miura109a and Buchwald109b have independently reported a highly regio- and enantioselective copper-catalysed intermolecular hydroamination of styrenes under very mild reaction conditions and with exclusive Markovnikov regioselectivity. These reports exploit an alternative umpolung electrophilic amination approach110,111 to classical alkene hydroamination using easily accessible O-benzoylhydroxylamines as electrophilic amination reagents and led to a remarkable breakthrough in the field. Building on their previous work on regio- and stereo-specific copper-catalysed aminoboration of styrenes,112 Hirano, Miura et al. have screened several copper salts/mono-and bidentate ligand combinations in the presence of various hydride sources and base for the hydroamination of styrene with O-benzoyl-N,N-diethylhydroxylamine.109a
It was identified that the combination Cu(OAc)2·75 (Scheme 42) (10 mol%) in the presence of polymethylhydrosiloxane (PMHS) and LiOtBu was optimal and could be applied to the regioselective Markovnikov hydroamination of functionalised styrenes and trans-β-substituted styrenes with O-benzoylhydroxylamines, affording the corresponding racemic amines in good to excellent yields at room temperature. Impressively, a slightly modified catalytic system (CuCl·(S,S)-Me-Duphos (76) or CuCl·(R,R)-Ph-BPE (77)) furnishes chiral amines with enantiomeric excesses ranging from 66 to 94% ee (Scheme 42, left). Unfortunately, these copper-based systems reported by Hirano, Miura and co-workers are not effective for the electrophilic amination of cis-β-substituted styrenes and, more importantly of terminal aliphatic alkenes.
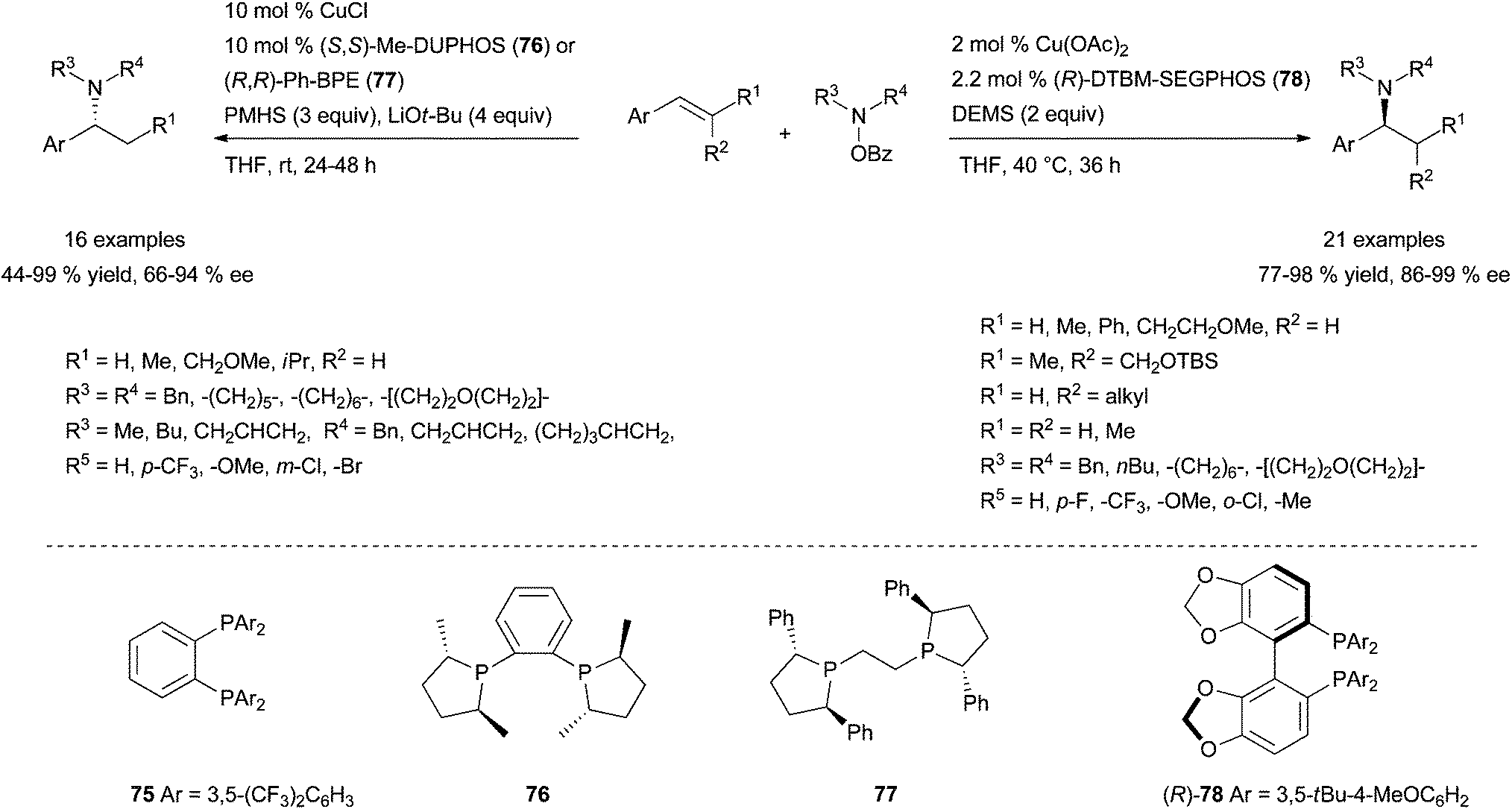 | ||
| Scheme 42 Regio- and enantioselective intermolecular copper-catalysed electrophilic amination of styrenes with O-benzoyl hydroxylamines. | ||
Exploiting an analogue umpolung strategy, the group of Buchwald has brilliantly published a highly efficient and related chiral system for the regio- and enantioselective copper-catalysed electrophilic amination of various aromatic alkenes.109b The reported Cu(OAc)2·(R)-DTBM-Segphos (78) system, in the presence of diethoxymethylsilane (DEMS) as hydride source, has a broader scope than Hirano and Miura's catalytic system with similar Markovnikov selectivity and level of enantioinduction (Scheme 42, right). In addition to styrenes, β,β-disubstituted styrenes or β-substituted styrenes featuring a trans or a cis configuration are elegantly converted into the branched amines with high enantioinduction, under the described reaction conditions.
Furthermore, the reported Cu(OAc)2·(±)-DTBM-Segphos (78) system has the exclusive ability to provide linear tertiary amines in high yields from the anti-Markovnikov electrophilic amination of terminal aliphatic alkenes with O-benzoyl hydroxylamines, (Scheme 43).109b Only a few metal systems have demonstrated such a regioselectivity with the scope of alkenes often limited to vinylarene derivatives, highlighting the achievement made by Buchwald's group. More recently, the group has expanded the methodology to the synthesis of a range of functionalised β-chiral amines in excellent yields with high enantioselectivities as the first report of enantioselective anti-Markovnikov hydroamination.113 These reactions are proposed to occur by a regio- and stereo-selective syn-migratory alkene insertion into the Cu–H bond of a Cu–H intermediate. The subsequent C–N bond formation might occur with retention of stereochemistry.
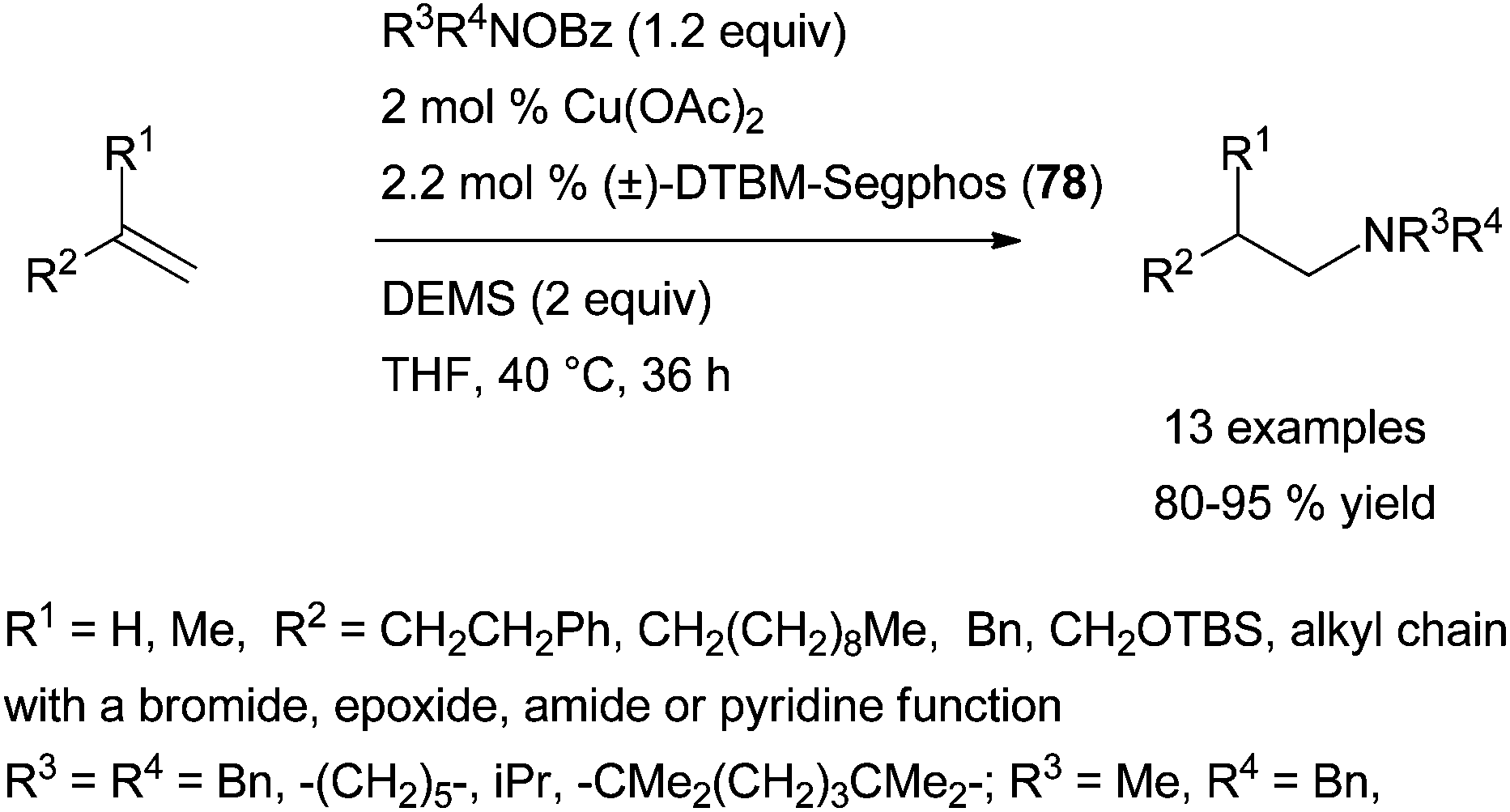 | ||
| Scheme 43 Copper(II)-catalysed anti-Markovnikov electrophilic amination of terminal aliphatic alkenes with O-benzoyl hydroxylamines. | ||
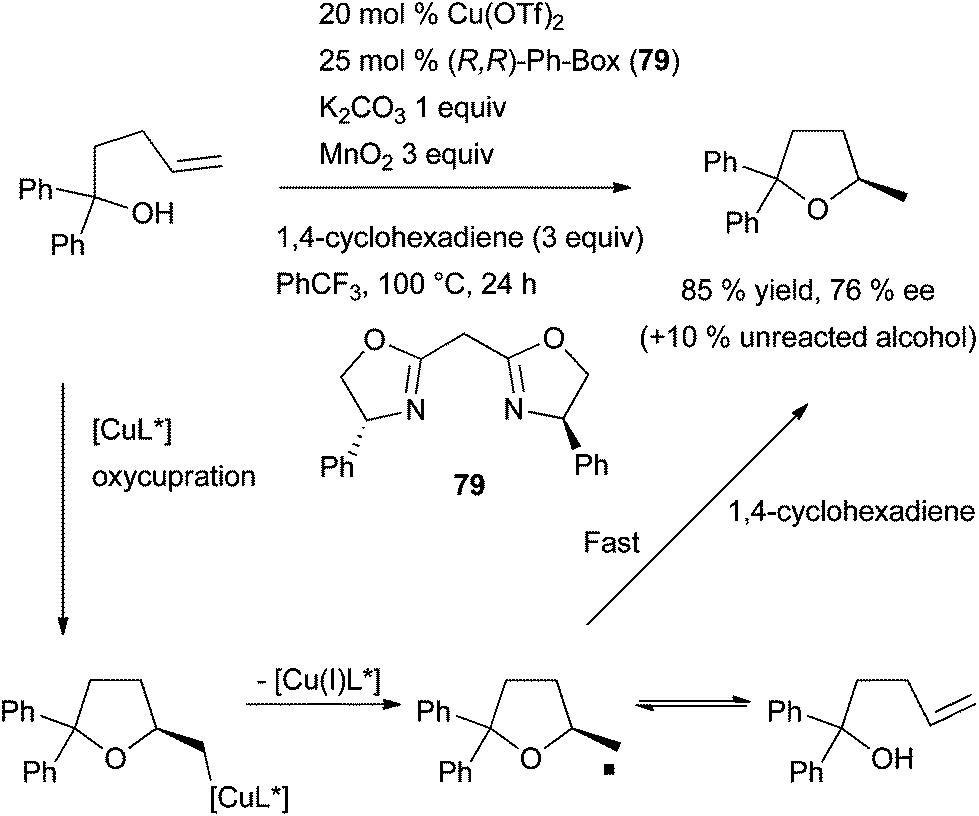 | ||
| Scheme 44 Enantioselective intramolecular hydroalkylation of alkenes catalysed by a copper complex. | ||
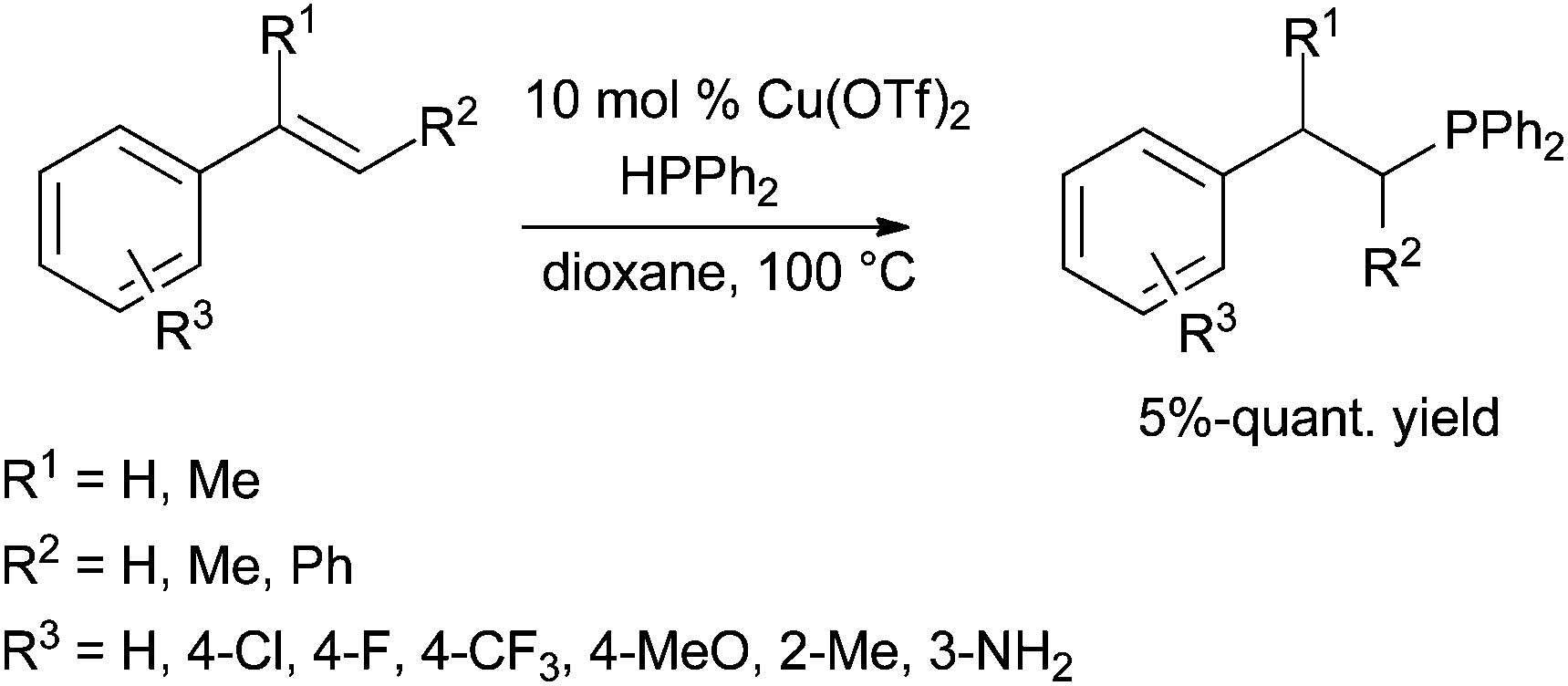 | ||
| Scheme 45 Copper-catalysed intermolecular hydrophosphination of styrene derivatives. | ||
6.6 Zinc
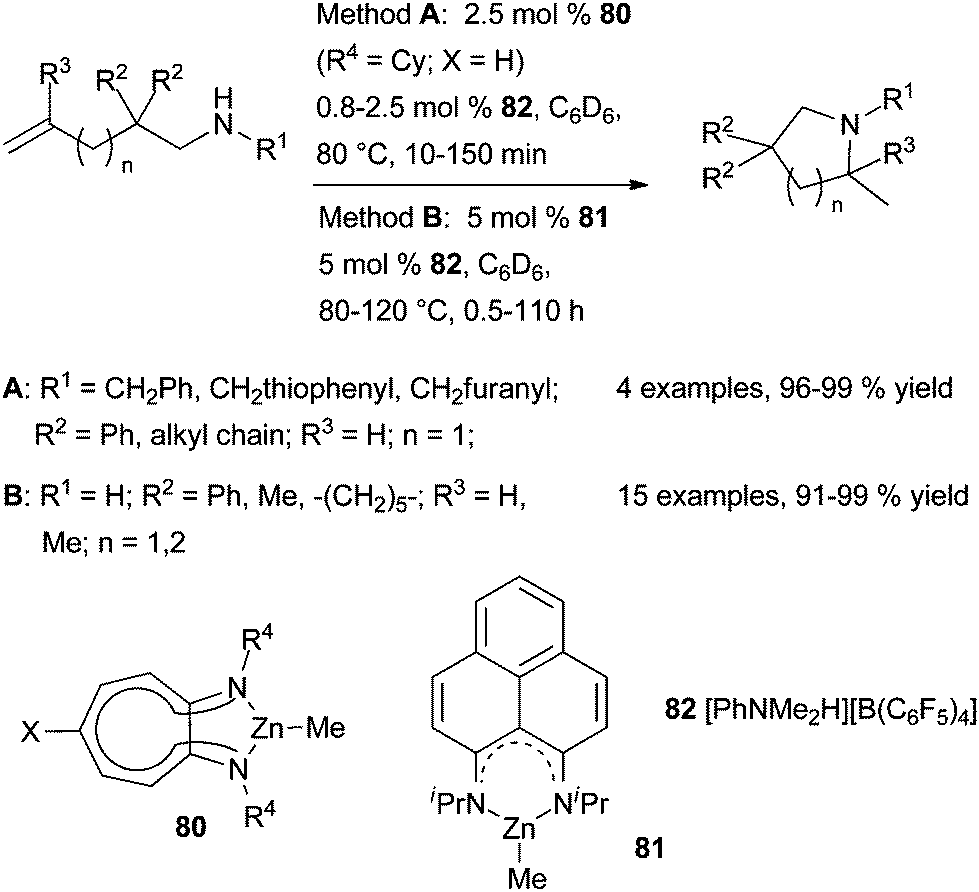 | ||
| Scheme 46 Zinc(II)-catalysed cyclohydroamination of amines tethered to alkenes. | ||
More recently, Mandal et al. have reported the synthesis, characterisation and catalytic application of two organozinc complexes that contain symmetrical phenalenyl-based N,N-ligands such as 81 (Scheme 46).121 Both three-coordinate complexes have a well-defined structure with a zinc atom having a trigonal-planar coordination sphere. Nevertheless, catalyst 81 displays similar catalytic activity to that of 80 for the cyclohydroamination of functionalised primary and secondary aminoalkenes.
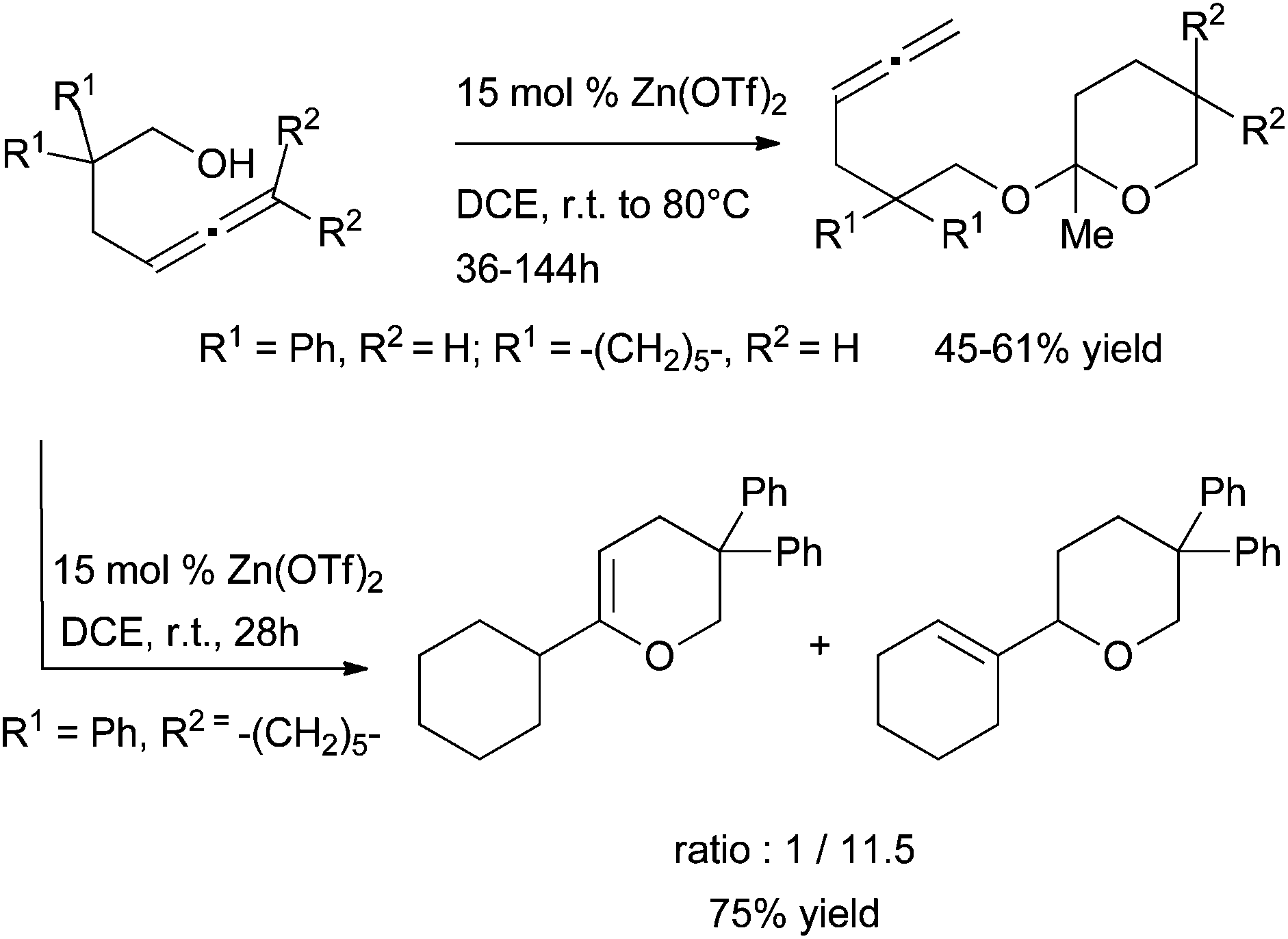 | ||
| Scheme 47 6-exo-dig cyclisation of γ-allenols in the presence of Zn(OTf)2. | ||
7. Aluminium catalysts
Despite being the most abundant metal and the third most abundant element, after oxygen and silicon in the Earth's crust, aluminium has been poorly explored as a catalyst for alkene hydrofunctionalisation. Only a few examples have been reported for the formation of C–N and C–O bonds for specific applications.7.1 C–N bond formation
In 2010, the group of Bergman disclosed the first report of aluminium-catalysed hydroamination of unactivated alkenes using the structurally well-defined four-coordinate aluminium complex 83 bearing a dianionic phenyl-diamine based ligand (Scheme 48).123 The latter is synthesised from commercially available [Al(NMe2)3]2 as a monomer thanks to the bulky aryl groups on the amine functions. The structurally-defined complex 83 (10 mol%) has the ability to promote the cyclohydroamination of primary aminoalkenes bearing geminal substituents on the backbone to form 5- and 6-membered rings in moderate to good yield under harsh conditions. Control experiments reveal that an acid catalysis was not operating in the process. From a stoichiometric reaction between 83 and 2,2-diphenylpent-4-ene-1-amine, the resting state of the catalyst is proposed to bear an aluminium-amide species resulting from loss of two molecules of HNMe2. More recently, Wehmschulte et al. have evaluated the catalytic activity of cationic four-coordinate dialkylaluminum and m-terphenylalkylaluminum compounds in the cyclohydroamination of primary amines tethered to alkenes.124 It was found that the most sterically crowded compound 84 was the most active of the series for the cyclisation of primary aminoalkenes, leading to the formation of pyrrolidines in moderate to high yields at 130 °C (Scheme 48). The cationic nature of these compounds is primordial for an efficient reactivity. A secondary amine tethered to alkene could also be cyclised under analogous conditions.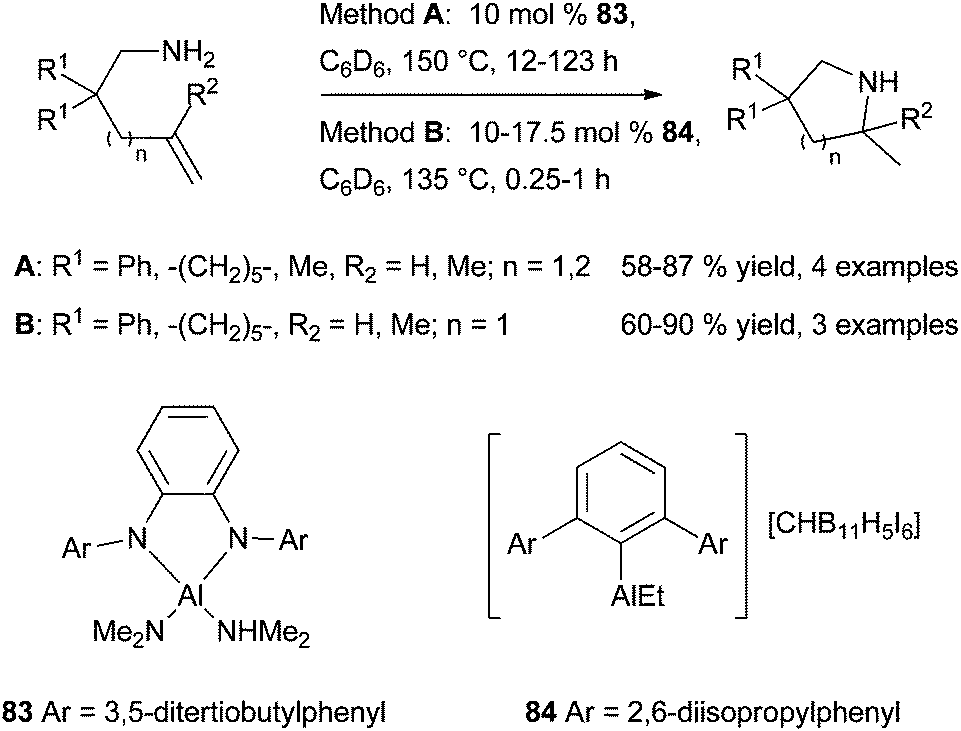 | ||
| Scheme 48 Aluminium-catalysed cyclohydroamination of aminoalkenes. | ||
7.2 C–O bond formation
Aluminium(III) derivatives are also able to catalyse the cyclisation of unsaturated alcohols. The pioneering work of Pons and Duñach et al. in this field have shown that Al(OTf)3 is the more efficient of all of them.22a,125,126 A large number of cyclic ethers were obtained with this metal triflate in high yields with Markovnikov regioselectivity (one example is depicted in Scheme 49). Theoretical calculations support the hypothesis of Lewis superacid type catalysis. The key step for the activation of the unactivated double bond should be the increased acidity of the hydroxyl proton. The intermolecular hydroalkoxylation of alkenes and catalysed by Al(OTf)3 was also reported by the group of Williams in 2012 (Scheme 26, method B).78 More recently, Hintermann et al. have disclosed that Al(OiPr)3 is also a powerful catalyst for the cyclisation of 2-allylphenols.127 However, these fast transformations require higher temperatures than the conditions described early on by Pons and Duñach et al. With Al(OiPr)3, hidden Brønsted acid catalysis can be definitely eliminated. A mechanism similar to metal-catalysed hydroamination reactions can be considered. In their study, the authors clearly reported the limitations of the Al(OiPr)3 catalyst in this type of cyclisation. The substrates form stable chelates with Al(III) and benzylic alcohol could not be cyclised (Scheme 49).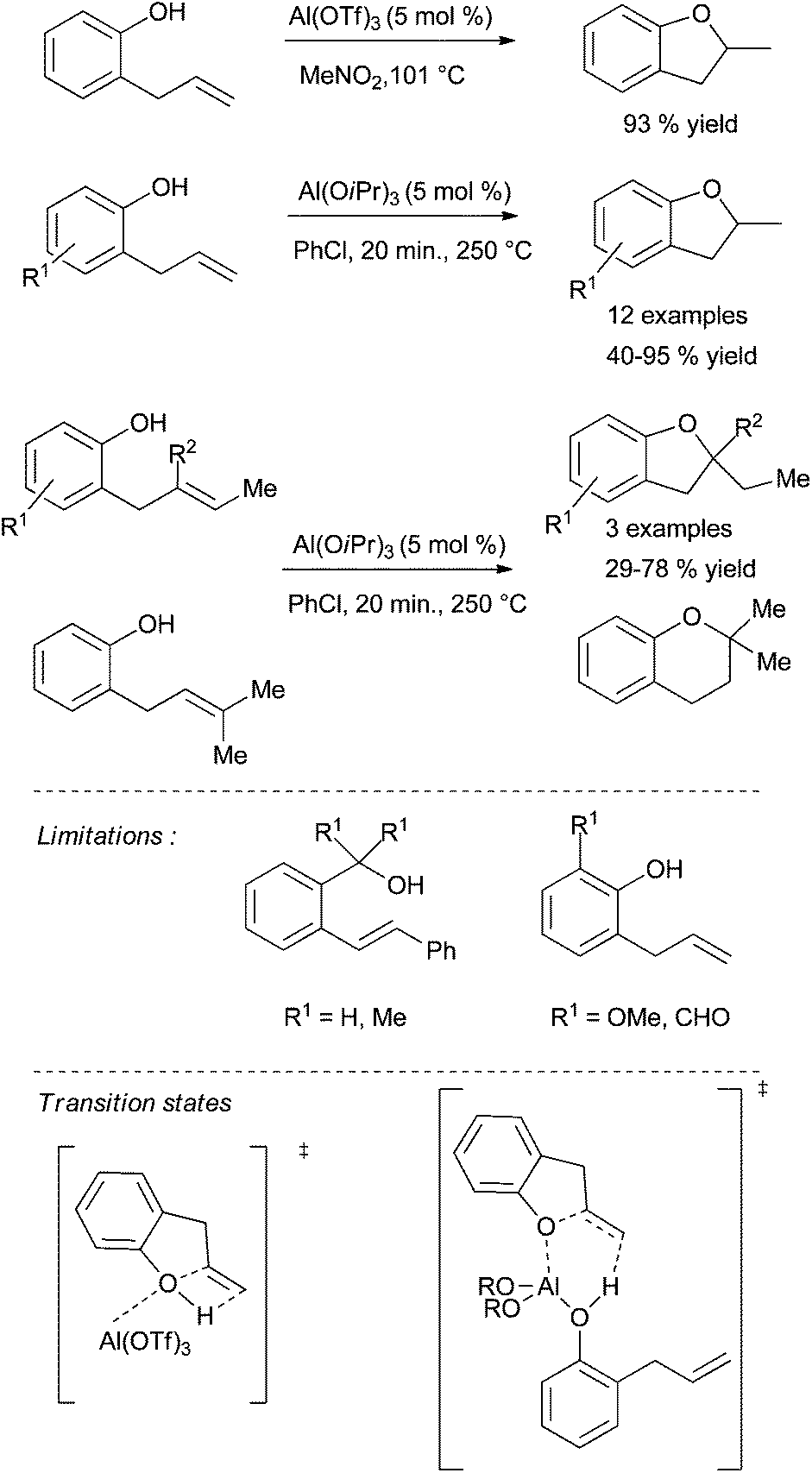 | ||
| Scheme 49 Hydroalkoxylation of 2-allylphenol catalysed by Al(OTf)3 and Al(OiPr)3. | ||
8. Conclusion and outlook
This article has reviewed some of the most recent results for performing the hydrofunctionalisation of alkenes with cheap, non-toxic, abundant and readily available catalytic species. The number of cited articles, and their diversity, is particularly revealing of the awareness of the need for elegant, inexpensive and direct methods to create these new carbon–heteroatom connections. New C–N, C–O or C–P bonds can be formed starting from various precursors able to add to carbon–carbon double bonds. By far, many more examples illustrate the C–N bond formation which is probably due to several reasons; nitrogen-containing compounds are valuable scaffolds towards the preparation of biologically active products, which substantiate an active search to find effective and green synthetic procedures. Furthermore, the nitrogen-starting compounds capable of reacting with olefins are actually diverse in their structure, and may be easily modulated, for instance, to tame their basicity. This is much less obvious, of course, if one refers to alcohols, and to a lesser extent, to phosphorus-containing derivatives. The hydrofunctionalisation reaction is concerned with notions of chemoselectivity, regioselectivity and enantioselectivity. Variable responses have been made, according to both the bond to be formed and the catalyst used, but comparison of the successes and shortcomings can be drawn.The construction of a new carbon–heteroatom link tolerates a wide range of functional groups as long as the promoters belong to the “organocatalysts” series or to the first-row transition metals. This is not accurate anymore if metals of Groups 1, 2 or 3 are concerned. In these cases, indeed, the associated metal-acidic site is too much sensitive to stand the presence of diverse functionalities and the substrates are almost exclusively carbon-containing compounds. Some exceptions can be found for the hydroalkoxylation reaction which can be performed on substrates bearing for instance ester groups, if the catalyst is a non-sensitive lanthanide halide or Ca(NTf2)2.
Some interesting chemoselective reactions have been reported, for the catalysts issued from the first-row. For instance, the copper-catalysed intramolecular hydroamination of alkenes has been performed on substrates containing hydroxyl groups, and the desired hydroxylated pyrrolidine and piperidine derivatives were obtained in high yield, without it being necessary to protect the OH groups. In a less straightforward way, but following nevertheless a simple procedure, the cobalt-catalysis (associated with a silane derivative and N-fluoropyridinium salt), of an olefin bearing a free hydroxyl group and a deactivated amine, delivers exclusively the hydroalkoxylation product. If an acetyl moiety is introduced as hydroxyl protecting group, the reactivity is interestingly reversed towards the sole formation of the hydroamination product. Iron catalysis led also to exciting results in terms of chemoselectivity. The oxycyclisation of β-lactam enallenols has been indeed demonstrated to occur entirely on the alkene group. This reactivity completes remarkably the catalysis described in the presence of noble metals (platinum or gold salts) which promote the allene cycloisomerisation.
An important point that governs the hydrofunctionalisation reaction is its selectivity towards a regioselective Markovnikov or an anti-Markovnikov addition. The control of this selectivity is tricky and is mainly governed by the structure of the catalyst but also the substrate backbone. Overwhelmingly, the formation of new C–P bonds has been demonstrated to follow an anti-Markovnikov pathway. Indeed photoinduced catalysis, and alkaline earth-, rare earth-, first row (Ti, Ni, Cu) metal-based complexes delivered the linear products from terminal olefins (mainly styrene derivatives). One example challenges this list, since iron-catalysis was demonstrated to allow providing a choice of one or the other regioisomer, according to the oxidation state of the iron centre, as chloride salts, devoid of any ligand. In contrast, the hydroalkoxylation delivers mainly products resulting from Markovnikov addition. Unique examples of anti-Markovnikov addition of an alcohol onto an olefin arose from photoredox catalysis, with the reverse selectivity being explained by the stability of intermediary radical species. A much more mixed state of the art issued from the results described for the hydroamination reaction. The well-studied intramolecular version of this reaction delivers, in the presence of the presented catalytic systems, exclusively products with Markovnikov selectivity. In the case of the intermolecular transformation, the selectivity of the reaction is highly related to both the structure of the involved olefinic substrate and the catalytic species. For instance, the rare-earth catalysed intermolecular hydroamination of styrene delivers the linear anti-Markovnikov product, whereas the same reaction performed with simple alkenyl derivatives lead to the branched Markovnikov compound. This is due to an aryl directing interaction of the weakly coordinating arene π-system and the electrophilic lanthanide centre, in the first case. Another example may be found in the copper-catalysed hydroamination reaction, in the presence of electrophilic amines, which occurs through Markovnikov or anti-Markovnikov selectivity, according to the structure of the engaged substrate.
Introducing enantioselectivity through formation of new carbon–heteroatom bonds is highly desirable and it has been the subject of numerous efforts with varying success. Research dedicated to performing asymmetric hydroamination reactions are already longstanding, and one may state that the control of enantioselectivity in cyclohydroamination transformations has already found numerous solutions. Indeed, in the case of alkenes tethered to simple primary or secondary amines, a large variety of examples are summarised in this article, implying catalysts with a great diversity in their structure and in the involved mechanistic pathway, which lead to nitrogen-containing heterocycles with very high enantiopurity. The use of chiral organic Brønsted acids is much more in its infancy for this reaction, but according to the huge development of such species for other reactions, one may expect future interesting advances in this reactivity. For the related intermolecular version, control of the enantioselectivity remains challenging, at least for alkali, alkaline earth and rare earth-based catalysts. In this context, nice recent examples involving the use of cheap and abundant copper catalysts have brought interesting answers, even if it must be specified that the reaction is in this case based on the use of electrophilic amines, in an umpolung-type transformation. For both of the other reactions, the formation of new C–O or C–P bonds, the state of the art really differs from what is known in the related C–N bond formation. As far as we know, only one recent example of intramolecular enantioselective hydroalkoxylation has been reported with a copper-bisoxazoline catalyst. The formation of scalemic phosphorus-containing products by such hydrofunctionalisation reaction is not known, probably in close connection to the preferred anti-Markovnikov regioselectivity. Such transformations could thus be studied with specific styrenyl-derived substrates, but a jump in reactivity must be found. The search for enantioselective formation of C–O or C–P bonds is still highly challenging.
Some catalytic systems have already been studied for promoting the three different hydrofunctionalisation reactions; this is the case for some group 2 and group 3 elements. Iron- and copper-promoted catalysis has also found successful solutions in each case. Some examples of nickel-catalysis show that this metal can promote the three reactions, albeit it has been hardly investigated. On the other hand, some catalysts used for one transformation may be promising to promote the other bond formations. This is probably the case for the less sensitive species, which can accommodate the presence of all the reactive functional groups present in the different substrates. There is still place for the search for the universal catalyst, but this review article already demonstrated that earth abundant elements are highly efficient for these valuable transformations, as direct addition reactions, allowing for the direct introduction of heteroelements into carbon compounds for various applications.
Acknowledgements
We thank ANR JCJC DHAMFER (ANR-11-JS07-012-01), project P2MOC (ANR-11-BS07-030-01), CHARMMMAT labex (ANR-11-LABEX-0039), MENSR, Univ Paris Sud, University of Versailles St Quentin and CNRS for financial support.Notes and references
- For general and more specialised reviews on hydrofunctionalisation reaction: (a) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed; (b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS PubMed; (c) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed; (d) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed; (e) S. Greenberg and D. W. Stephan, Chem. Soc. Rev., 2008, 37, 1482 RSC; (f) C. J. Weiss and T. J. Marks, Dalton Trans., 2010, 39, 6576 RSC; (g) A. S. K. Hashmi and M. Bührle, Aldrichimica Acta, 2010, 43, 27 CAS; (h) K. D. Hesp and M. Stradiotto, ChemCatChem, 2010, 2, 1192 CrossRef CAS PubMed; (i) J. G. Taylor, L. A. Adrio and K. K. Hii, Dalton Trans., 2010, 39, 1171 RSC; (j) J. S. Yadav, A. Antony, T. S. Rao and B. V. S. Reddy, J. Organomet. Chem., 2011, 696, 16 CrossRef CAS PubMed; (k) A. Hamasaki, E. Yamamoto, H. Itoh and M. Tokunaga, J. Organomet. Chem., 2011, 696, 202 CrossRef CAS PubMed; (l) N. T. Patil, R. D. Kavthe and V. S. Shinde, Tetrahedron, 2012, 68, 8079 CrossRef CAS PubMed; (m) J. Hannedouche and E. Schulz, Chem. – Eur. J., 2013, 19, 4972 CrossRef CAS PubMed; (n) S. A. Pullarkat and P.-H. Leung, Top. Organomet. Chem., 2013, 43, 145 CrossRef CAS; (o) Hydrofunctionalization, ed. V. P. Ananikov and M. Tanaka, Top. Organomet. Chem., Springer, Berlin, Heidelberg, 2013, vol. 43, pp. 1–325 Search PubMed; (p) J. Le Bras and J. Muzart, Chem. Soc. Rev., 2014, 43, 3003 RSC; (q) M. P. Munoz, Chem. Soc. Rev., 2014, 43, 3164 RSC.
- For rare examples of late-transition metal catalysed hydroamination with anti-Markovnikov selectivity: (a) M. Utsunomiya, R. Kuwano, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 5608 CrossRef CAS PubMed; (b) A. Takemiya and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 6042 CrossRef CAS PubMed; (c) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702 CrossRef CAS PubMed; (d) C. Munro-Leighton, S. A. Delp, N. M. Alsop, E. D. Blue and T. B. Gunnoe, Chem. Commun., 2008, 111 RSC; (e) M. Beller, H. Trauthwein, M. Eichberger, C. Breindl, J. Herwig, T. E. Müller and O. R. Thiel, Chem. – Eur. J., 1999, 5, 1306 CrossRef CAS.
- For rare examples of anti-Markovnikov hydroalkoxylation selectivity: (a) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577 CrossRef CAS PubMed; (b) K. Mizuno, I. Nakanishi, N. Ichinose and Y. Otsuji, Chem. Lett., 1989, 1095 CrossRef CAS; (c) K. Mizuno, T. Tamai, T. Nishiyama, K. Tani, M. Sawasaki and Y. Otsuji, Angew. Chem., Int. Ed. Engl., 1994, 33, 2113 CrossRef PubMed; (d) Y. Inoue, T. Okano, N. Yamasaki and A. Tai, J. Chem. Soc., Chem. Commun., 1993, 718 RSC; (e) S. Asaoka, T. Kitazawa, T. Wada and Y. Inoue, J. Am. Chem. Soc., 1999, 121, 8486 CrossRef CAS.
- For rare examples of P–H addition reaction with Markovnikov selectivity: (a) M. C. Hoff and P. Hill, J. Org. Chem., 1959, 24, 356 CrossRef CAS; (b) M. R. Douglass and T. J. Marks, J. Am. Chem. Soc., 2000, 122, 1824 CrossRef CAS; (c) L. Routaboul, F. Toulgoat, J. Gatignol, J.-F. Lohier, B. Norah, O. Delacroix, C. Alayrac, M. Taillefer and A.-C. Gaumont, Chem. – Eur. J., 2013, 19, 8760 CrossRef CAS PubMed.
- For seminal reports on metal-catalysed enantio- and regioselective intermolecular hydroamination of simple aliphatic alkenes, see: (a) Z. Zhang, S. Du Lee and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 5372 CrossRef CAS PubMed; (b) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed.
- Y. Miller, L. Miao, A. S. Hosseini and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 12149 CrossRef CAS PubMed.
- For recent reviews on the topic: (a) J. Guoa and P. Teo, Dalton Trans., 2014, 43, 6952 RSC; (b) L. Hintermann, ChemCatChem, 2012, 4, 321 CrossRef CAS PubMed; (c) Ref. 1k .
- (a) C. M. Haskins and D. W. Knight, Chem. Commun., 2002, 2724 RSC; (b) B. Schlummer and J. F. Hartwig, Org. Lett., 2002, 4, 1471 CrossRef CAS PubMed; (c) L. L. Anderson, J. Arnold and R. G. Bergman, J. Am. Chem. Soc., 2005, 127, 14542 CrossRef CAS PubMed; (d) Z. Li, J. Zhang, C. Brouwer, C.-G. Yang, N. W. Reich and C. He, Org. Lett., 2006, 8, 4175 CrossRef CAS PubMed; (e) D. C. Rosenfeld, S. Shekhar, A. Tazkeyima, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179 CrossRef CAS PubMed; (f) L. Ackermann, L. T. Kasparb and A. Althammer, Org. Biomol. Chem., 2007, 5, 1975 RSC; (g) Z. S. Qureshi, K. M. Deshmukh, P. J. Tambade, K. P. Dhake and B. M. Bhanage, Eur. J. Org. Chem., 2010, 6233 CrossRef CAS PubMed; (h) Y.-M. Wang, T.-T. Li, G.-Q. Liu, L. Zhang, L. Duan, L. Li and Y.-M. Li, RSC Adv., 2014, 4, 9517 RSC; (i) G.-Q. Liu, B. Cui, H. Sun and Y.-M. Li, Tetrahedron, 2014, 70, 5696 CrossRef CAS PubMed.
- L. Ackermann and A. Althammer, Synlett, 2008, 995 CrossRef CAS PubMed.
- N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. Dean Toste, Nature, 2011, 470, 245 CrossRef CAS PubMed.
- For highlight of this work, see: I. Dion and A. M. Beauchemin, Angew. Chem., Int. Ed., 2011, 50, 8233 CrossRef CAS PubMed.
- A. M. Beauchemin, Org. Biomol. Chem., 2013, 11, 7039 CAS.
- M. J. MacDonald, D. J. Schipper, P. J. Ng, J. Moran and A. M. Beauchemin, J. Am. Chem. Soc., 2011, 133, 20100 CrossRef CAS PubMed.
- N. Guimond, M. J. MacDonald, V. Lemieux and A. M. Beauchemin, J. Am. Chem. Soc., 2012, 134, 16571 CrossRef CAS PubMed.
- A. R. Brown, C. Uyeda, C. A. Brotherton and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 6747 CrossRef CAS PubMed.
- (a) J. Kemper and A. Studer, Angew. Chem., Int. Ed., 2005, 44, 4914 CrossRef CAS PubMed; (b) J. Guin, C. Mück-Lichtenfeld, S. Grimme and A. Studer, J. Am. Chem. Soc., 2007, 129, 4498 CrossRef CAS PubMed.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC.
- (a) J. Guin, R. Frölich and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 779 CrossRef CAS PubMed; (b) C.-M. Chou, J. Guin, C. Mück-Lichtenfeld, S. Grimme and A. Studer, Chem. – Asian J., 2011, 6, 1197 CrossRef CAS PubMed.
- T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588 CrossRef CAS PubMed.
- T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198 CrossRef CAS PubMed.
- (a) X. Franck, B. Figadère and A. Cavé, Tetrahedron Lett., 1997, 38, 1413 CrossRef CAS; (b) K. Miura, S. Okajima, T. Hondo, T. Nakagawa, T. Takahashi and A. Hosomi, J. Am. Chem. Soc., 2000, 122, 11348 CrossRef CAS.
- (a) S. Antoniotti, S. Poulain-Martini and E. Duñach, Synlett, 2010, 2973 CAS; (b) L. Coulombel and E. Duñach, Green Chem., 2004, 6, 499 RSC.
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353 CrossRef CAS PubMed.
- Y. Jeong, D.-Y. Kim, Y. Choi and J.-S. Ryu, Org. Biomol. Chem., 2011, 9, 374 CAS.
- (a) D. A. Nicewicz and D. S. Hamilton, Synlett, 2014, 1191 CrossRef PubMed; (b) N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024 CrossRef CAS PubMed.
- S.-I. Kawaguchi, A. Nomoto, M. Sonoda and A. Ogawa, Tetrahedron Lett., 2009, 50, 624 CrossRef CAS PubMed.
- A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 81 CrossRef CAS.
- B. W. Howk, E. L. Little, S. L. Scott and G. M. Whitman, J. Am. Chem. Soc., 1954, 76, 1899 CrossRef CAS.
- J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795 CrossRef CAS.
- (a) C. Quinet, P. Jourdain, C. Hermans, A. Ates, I. Lucas and I. E. Markó, Tetrahedron, 2008, 64, 1077 CrossRef CAS PubMed; (b) C. Quinet, L. Sampoux and I. E. Markó, Eur. J. Org. Chem., 2009, 1806 CrossRef CAS PubMed.
- P. Horrillo-Martínez, K. C. Hultzsch, A. Gil and V. Branchadell, Eur. J. Org. Chem., 2007, 3311 CrossRef PubMed.
- D. Jaspers and S. Doye, Synlett, 2011, 1444 CAS.
- P. Horrillo Martínez, K. C. Hultzsch and F. Hampel, Chem. Commun., 2006, 2221 RSC.
- T. Ogata, A. Ujihara, S. Tsuchida, T. Shimizu, A. Kaneshige and K. Tomioka, Tetrahedron Lett., 2007, 48, 6648 CrossRef CAS PubMed.
- (a) J. Deschamp, C. Olier, E. Schulz, R. Guillot, J. Hannedouche and J. Collin, Adv. Synth. Catal., 2010, 352, 2171 CrossRef CAS PubMed; (b) J. Deschamp, J. Collin, J. Hannedouche and E. Schulz, Eur. J. Org. Chem., 2011, 3329 CrossRef CAS PubMed.
- M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042 CrossRef CAS PubMed.
- S. Datta, M. T. Gamer and P. W. Roesky, Organometallics, 2008, 27, 1207 CrossRef CAS.
- M. Arrowsmith, M. R. Crimmin, A. G. M. Barrett, M. S. Hill, G. Kociok-Köhn and P. A. Procopiou, Organometallics, 2011, 30, 1493 CrossRef CAS.
- (a) B. Liu, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2012, 51, 4943 CrossRef CAS PubMed; (b) B. Liu, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2013, 19, 13445 CrossRef CAS PubMed.
- J. Jenter, R. Köppe and P. W. Roesky, Organometallics, 2011, 30, 1404 CrossRef CAS.
- (a) M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2011, 30, 1291 CrossRef CAS; (b) M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2014, 33, 206 CrossRef CAS.
- B. Liu, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2013, 19, 2784 CrossRef CAS PubMed.
- F. Buch and S. Harder, Z. Naturforsch., B: Chem. Sci., 2008, 63, 169 CAS.
- J. S. Wixey and B. D. Ward, Chem. Commun., 2011, 47, 5449 RSC.
- J. S. Wixey and B. D. Ward, Dalton Trans., 2011, 40, 7693 RSC.
- T. D. Nixon and B. D. Ward, Chem. Commun., 2012, 48, 11790 RSC.
- P. Horrillo-Martínez and K. C. Hultzsch, Tetrahedron Lett., 2009, 50, 2054 CrossRef PubMed.
- X. Zhang, T. J. Emge and K. C. Hultzsch, Organometallics, 2010, 29, 5871 CrossRef CAS.
- X. Zhang, T. J. Emge and K. C. Hultzsch, Angew. Chem., Int. Ed., 2012, 51, 394 CrossRef CAS PubMed.
- J. F. Dunne, D. B. Fulton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2010, 132, 17680 CrossRef CAS PubMed.
- S. R. Neal, A. Ellern and A. D. Sadow, J. Organomet. Chem., 2011, 696, 228 CrossRef CAS PubMed.
- C. Brinkmann, A. G. M. Barrett, M. S. Hill, P. A. Procopiou and S. Reid, Organometallics, 2012, 31, 7287 CrossRef CAS.
- (a) A. K. Diba, J.-M. Begouin and M. Niggemann, Tetrahedron Lett., 2012, 53, 6029 CrossRef PubMed; (b) S. Haubenreisser and M. Niggemann, Adv. Synth. Catal., 2011, 353, 469 CrossRef CAS PubMed.
- H. Fu and C. Cui, Organometallics, 2012, 31, 1208 CrossRef.
- M. R. Gagne and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 4108 CrossRef CAS.
- For a review, see: S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed.
- H. F. Yuen and T. J. Marks, Organometallics, 2009, 28, 2423 CrossRef CAS.
- X. Xu, Y. Chen, J. Feng, G. Zou and J. Sun, Organometallics, 2010, 29, 549 CrossRef CAS.
- N. K. Hangaly, A. R. Petrov, K. A. Rufanov, K. Harms, M. Elfferding and J. Sundermeyer, Organometallics, 2011, 30, 4544 CrossRef CAS.
- A. Otero, A. Lara-Sánchez, C. Nájera, J. Fernández-Baeza, I. Márquez-Segovia, J. A. Castro-Osma, J. Martínez, L. F. Sánchez-Barba and A. M. Rodríguez, Organometallics, 2012, 31, 2244 CrossRef CAS.
- K. Manna, M. L. Kruse and A. D. Sadow, ACS Catal., 2011, 1, 1637 CrossRef CAS.
- Z. Chai, D. Hua, K. Li, J. Chu and G. Yang, Chem. Commun., 2014, 50, 177 RSC.
- L. J. E. Stanlake and L. L. Schafer, Organometallics, 2009, 28, 3990 CrossRef CAS.
- H. Kaneko, H. Tsurugi, T. K. Panda and K. Mashima, Organometallics, 2010, 29, 3463 CrossRef CAS.
- D. M. Lyubov, L. Luconi, A. Rossin, G. Tuci, A. V. Cherkasov, G. K. Fukin, G. Giambastiani and A. A. Trifonov, Chem. – Eur. J., 2014, 20, 3487 CrossRef CAS PubMed.
- I. V. Basalov, S. C. Roşca, D. M. Lyubov, A. N. Selikhov, G. K. Fukin, Y. Sarazin, J.-F. Carpentier and A. A. Trifonov, Inorg. Chem., 2014, 53, 1654 CrossRef CAS PubMed.
- Q. Wang, F. Zhang, H. Song and G. Zi, J. Organomet. Chem., 2011, 696, 2186 CrossRef CAS PubMed.
- P. Benndorf, J. Kratsch, L. Hartenstein, C. M. Preuss and P. W. Roesky, Chem. – Eur. J., 2012, 18, 14454 CrossRef CAS PubMed.
- S. D. Bennett, B. A. Core, M. P. Blake, S. J. A. Pope, P. Mountford and B. D. Ward, Dalton Trans., 2014, 43, 5871 RSC.
- Y. Zhang, W. Yao, H. Li and Y. Mu, Organometallics, 2012, 31, 4670 CrossRef CAS.
- J. Hannedouche, J. Collin, A. Trifonov and E. Schulz, J. Organomet. Chem., 2011, 696, 255 CrossRef CAS PubMed.
- Y. Chapurina, R. Guillot, D. Lyubov, A. Trifonov, J. Hannedouche and E. Schulz, Dalton Trans., 2013, 42, 507 RSC.
- S. Germain, E. Schulz and J. Hannedouche, ChemCatChem, 2014, 6, 2065 CrossRef CAS PubMed.
- E. Marotta, E. Foresti, T. Marcelli, F. Peri, P. Righi, N. Scardovi and G. Rosini, Org. Lett., 2002, 4, 44 CrossRef PubMed.
- (a) X. Yu, S. Y. Seo and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 7244 CrossRef CAS PubMed; (b) C. J. Weiss and T. J. Marks, Dalton Trans., 2010, 39, 6576 RSC.
- B. Alcaide, P. Almendros, R. Carrascosa and T. Martinez del Campo, Chem. – Eur. J., 2010, 16, 13243 CrossRef CAS PubMed.
- (a) A. Dzudza and T. J. Marks, Chem. – Eur. J., 2010, 16, 3403 CrossRef CAS PubMed; (b) A. Dzudza and T. J. Marks, Org. Lett., 2009, 11, 1523 CrossRef CAS PubMed.
- D. B. G. Williams, M. S. Sibiya and P. S. van Heerden, Fuel Process. Technol., 2012, 94, 75 CrossRef CAS PubMed.
- I. V. Basalov, S. C. Rosca, D. M. Lyubov, A. N. Slikhov, G. K. Fukin, Y. Sarazin, J.-F. Carpentier and A. A. Trifonov, Inorg. Chem., 2014, 53, 1654 CrossRef CAS PubMed.
- (a) D. V. Gribkov and K. C. Hultzsch, Angew. Chem., Int. Ed., 2004, 43, 5542 CrossRef CAS PubMed; (b) P. D. Knight, I. Munslow, P. N. O. Shaughnessy and P. Scott, Chem. Commun., 2004, 894 RSC; (c) H. Kim, P. H. Lee and T. Livinghouse, Chem. Commun., 2005, 5205 RSC; (d) D. A. Watson, M. Chiu and R. G. Bergman, Organometallics, 2006, 25, 4731 CrossRef CAS PubMed; (e) A. L. Gott, A. J. Clarke, G. J. Clarkson and P. Scott, Organometallics, 2007, 26, 1729 CrossRef CAS; (f) M. C. Wood, D. C. Leitch, C. S. Yeung, J. A. Kozak and L. L. Schafer, Angew. Chem., Int. Ed., 2006, 46, 354 CrossRef PubMed; (g) G. Zi, F. Zhang, L. Xiang, Y. Chen, W. Fang and H. Song, Dalton Trans., 2010, 39, 4048 RSC; (h) A. L. Reznichenko and K. C. Hultzsch, Organometallics, 2010, 29, 24 CrossRef CAS; (i) K. Manna, S. Xu and A. D. Sadow, Angew. Chem., Int. Ed., 2011, 50, 1865 CrossRef CAS PubMed; (j) K. Manna, W. C. Everett, G. Schoendorff, A. Ellern, T. L. Windus and A. D. Sadow, J. Am. Chem. Soc., 2013, 135, 7235 CrossRef CAS PubMed.
- (a) J. A. Bexrud, J. D. Beard, D. C. Leitch and L. L. Schafer, Org. Lett., 2005, 7, 1959 CrossRef CAS PubMed; (b) C. Müller, C. Loos, N. Schulenberg and S. Doye, Eur. J. Org. Chem., 2006, 2499 CrossRef PubMed; (c) A. V. Lee and L. L. Schafer, Organometallics, 2006, 25, 5249 CrossRef CAS; (d) K. Marcsekova, C. Loos, F. Rominger and S. Doye, Synlett, 2007, 2564 CAS; (e) C. Müller, W. Saak and S. Doye, Eur. J. Org. Chem., 2008, 2731 CrossRef PubMed; (f) K. Graebe, F. Pohlki and S. Doye, Eur. J. Org. Chem., 2008, 4815 CrossRef CAS PubMed; (g) G. Zi, X. Liu, L. Xiang and H. Song, Organometallics, 2009, 28, 1127 CrossRef CAS; (h) B. Lian, T. P. Spaniol, P. Horrillo-Martínez, K. C. Hultzsch and J. Okuda, Eur. J. Inorg. Chem., 2009, 429 CrossRef CAS PubMed; (i) G. Zi, F. Zhang, L. Xiang, Y. Chen, W. Fang and H. Song, Dalton Trans., 2010, 39, 4048 RSC; (j) R. O. Ayinla and L. L. Schafer, Dalton Trans., 2011, 40, 7769 RSC; (k) D. Jaspers, W. Saak and S. Doye, Synlett, 2012, 2098 CAS; (l) T. R. Helgert, T. Keith Hollis and E. J. Valente, Organometallics, 2012, 31, 3002 CrossRef CAS.
- E. Chong, S. Qayyum, L. L. Schafer and R. Kempe, Organometallics, 2013, 32, 1858 CrossRef CAS.
- C. Brahms, P. Tholen, W. Saak and S. Doye, Eur. J. Org. Chem., 2013, 7583 CrossRef CAS PubMed.
- P. W. Roesky, Angew. Chem., Int. Ed., 2009, 48, 4892 CrossRef CAS PubMed.
- J. Dörfler and S. Doye, Angew. Chem., Int. Ed., 2013, 52, 1806 CrossRef PubMed.
- C. Midrier, M. Lantsoght, J.-N. Volle, J.-L. Pirat and D. Virieux, Tetrahedron Lett., 2011, 52, 6693 CrossRef CAS PubMed.
- (a) K. Komeyama, T. Morimoto and K. Takaki, Angew. Chem., Int. Ed., 2006, 45, 2938 CrossRef CAS PubMed; (b) J. Michaux, V. Terrasson, S. Marque, J. Wehbe, D. Prim and J.-M. Campagne, Eur. J. Org. Chem., 2007, 2601 CrossRef PubMed; (c) X. Cheng, Y. Xia, H. Xei, B. Xu, C. Zhang, Y. Li, G. Qian, X. Zhang, K. Li and W. Li, Eur. J. Org. Chem., 2008, 1929 CrossRef CAS PubMed.
- M. S. Jung, W. S. Kim, Y. H. Shin, H. J. Jin, Y. S. Kim and E. J. Kang, Org. Lett., 2012, 14, 6262 CrossRef CAS PubMed.
- E. Bernoud, P. Oulié, R. Guillot, M. Mellah and J. Hannedouche, Angew. Chem., Int. Ed., 2014, 53, 4930 CrossRef CAS PubMed.
- (a) T. J. J. Sciarone, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2002, 1580 RSC; (b) J. Vela, S. Vaddadi, T. R. Cundari, J. M. Smith, E. A. Gregory, R. J. Lachicotte, C. J. Flaschenriem and P. L. Holland, Organometallics, 2004, 23, 5226 CrossRef CAS.
- C. B. Huehls, A. Lin and J. Yang, Org. Lett., 2014, 16, 3620 CrossRef CAS PubMed.
- K. Komeyama, T. Morimoto, Y. Nakayama and K. Takaki, Tetrahedron Lett., 2007, 48, 3259 CrossRef CAS PubMed.
- F. Ke, Z. Li, H. Xiang and X. Zhou, Tetrahedron Lett., 2011, 52, 318 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, T. Martínez del Campo, M. C. Redondo and I. Fernández, Chem. – Eur. J., 2011, 17, 15005 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros and M. T. Quirós, Adv. Synth. Catal., 2011, 353, 585 CrossRef CAS PubMed.
- K. J. Gallagher and R. L. Webster, Chem. Commun., 2014, 50, 12109 RSC.
- H. Shigehisa, T. Aoki, S. Yamaguchi, N. Shimizu and K. Hiroya, J. Am. Chem. Soc., 2013, 135, 10306 CrossRef CAS PubMed.
- H. Shigehisa, N. Koseki, N. Shimizu, M. Fujisaw, M. Niitsu and K. Hiroya, J. Am. Chem. Soc., 2014, 136, 13534 CrossRef CAS PubMed.
- J. Pawlas, Y. Nakao, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 3369 CrossRef PubMed.
- S. Bigot, M. S. Ibn El Alami, A. Mifleur, Y. Castanet, I. Suisse, A. Mortreux and M. Sauthier, Chem. – Eur. J., 2013, 19, 9785 CrossRef CAS PubMed.
- S. Ortial, H. C. Fisher and J.-L. Montchamp, J. Org. Chem., 2013, 78, 6599 CrossRef CAS PubMed.
- D. Prim, J.-M. Campagne, D. Joseph and B. Andrioletti, Tetrahedron, 2002, 58, 2041 CrossRef CAS.
- J. G. Taylor, N. Whittall and K. K. Hii, Org. Lett., 2006, 8, 3561 CrossRef CAS PubMed.
- C. Munro-Leighton, S. A. Delp, N. M. Alsop, E. D. Blue and T. B. Gunnoe, Chem. Commun., 2008, 111 RSC.
- H. Ohmiya, T. Moriya and M. Sawamura, Org. Lett., 2009, 11, 2145 CrossRef CAS PubMed.
- H. Ohmiya, M. Yoshida and M. Sawamura, Synlett, 2010, 2136 CAS.
- B. W. Turnpenny, K. L. Hyman and S. R. Chemler, Organometallics, 2012, 31, 7819 CrossRef CAS PubMed.
- C. Michon, F. Medina, F. Capet, P. Roussel and F. Agbossou-Niedercorn, Adv. Synth. Catal., 2010, 35, 3293 CrossRef PubMed.
- (a) Y. Miki, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830 CrossRef CAS PubMed; (b) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746 CrossRef CAS PubMed.
- X. Yan, X. Yang and C. Xi, Catal. Sci. Technol., 2014, 4, 4169 CAS.
- For lead references in copper-catalyzed electrophilic amination; see: (a) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680 CrossRef CAS PubMed; (b) R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, J. Am. Chem. Soc., 2012, 134, 6571 CrossRef CAS PubMed.
- N. Matsuda, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2013, 135, 4934 CrossRef CAS PubMed.
- S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 15913 CrossRef CAS PubMed.
- J. G. Taylor, N. Whittall and K. K. (Mimi) Hii, Chem. Commun., 2005, 5103 RSC.
- A. Leyva-Perez, J. A. Vidal-Moya, J. R. Cabrero-Antonino, S. S. Al-Deyab, S. I. Al-Resayes and A. Corma, J. Organomet. Chem., 2011, 696, 362 CrossRef CAS PubMed.
- A. Zulys, M. Dochnahl, D. Hollmann, K. Löhnwitz, J.-S. Herrmann, P. W. Roesky and S. Blechert, Angew. Chem., Int. Ed., 2005, 44, 7794 CrossRef CAS PubMed.
- (a) M. Dochnahl, J.-W. Pissarek, S. Blechert, K. Löhnwitz and P. W. Roesky, Chem. Commun., 2006, 3405 RSC; (b) M. Dochnahl, K. Löhnwitz, J.-W. Pissarek, M. Biyikal, S. R. Schulz, S. Schön, N. Meyer, P. W. Roesky and S. Blechert, Chem. – Eur. J., 2007, 13, 6654 CrossRef CAS PubMed; (c) M. Dochnahl, K. Löhnwitz, J.-W. Pissarek, P. W. Roesky and S. Blechert, Dalton Trans., 2008, 2844 RSC; (d) K. Löhnwitz, M. J. Molski, A. Lühl, P. W. Roesky, M. Dochnahl and S. Blechert, Eur. J. Inorg. Chem., 2009, 1369 CrossRef PubMed.
- M. Dochnahl, K. Löhnwitz, A. Lühl, J.-W. Pissarek, M. Biyikal, P. W. Roesky and S. Blechert, Organometallics, 2010, 29, 2637 CrossRef CAS.
- J. Jenter, A. Lühl, P. W. Roesky and S. Blechert, J. Organomet. Chem., 2011, 696, 406 CrossRef CAS PubMed.
- J.-W. Pissarek, D. Schlesiger, P. W. Roesky and S. Blechert, Adv. Synth. Catal., 2009, 351, 2081 CrossRef CAS PubMed.
- (a) A. Mukherjee, T. K. Sen, P. K. Ghorai, P. P. Samuel, C. Schulzke and S. K. Mandal, Chem. – Eur. J., 2012, 18, 10530 CrossRef CAS PubMed; (b) A. Mukherjee, T. K. Sen, P. K. Ghorai and S. K. Mandal, Organometallics, 2013, 32, 7213 CrossRef CAS.
- J. L. Arbour, H. S. Rzepa, A. J. P. White and K. K. (Mimi) Hii, Chem. Commun., 2009, 7125 RSC.
- J. Koller and R. G. Bergman, Chem. Commun., 2010, 46, 4577 RSC.
- M. Khandelwal and R. J. Wehmschulte, J. Organomet. Chem., 2012, 696, 4179 CrossRef CAS PubMed.
- L. Coulombel, M. Weïwer and E. Duñach, Eur. J. Org. Chem., 2009, 5788 CrossRef CAS PubMed.
- L. Coulombel, M. Rajzmann, J.-M. Pons, S. Olivero and E. Duñach, Chem. – Eur. J., 2006, 12, 6356 CrossRef CAS PubMed.
- J. Schlûter, M. Blazejak and L. Hintermann, ChemCatChem, 2013, 5, 3309 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |