
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A shortcut to high-affinity Ga-68 and Cu-64 radiopharmaceuticals: one-pot click chemistry trimerisation on the TRAP platform†
Zsolt
Baranyai
a,
Dominik
Reich
b,
Adrienn
Vágner
a,
Martina
Weineisen
b,
Imre
Tóth
a,
Hans-Jürgen
Wester
b and
Johannes
Notni
*b
aDepartment of Inorganic and Analytical Chemistry, University of Debrecen, Egyetem tér 1, Debrecen, H-4032, Hungary
bLehrstuhl für Pharmazeutische Radiochemie, Technische Universität München, Walther-Meissner-Strasse 3, D-85748 Garching, Germany. E-mail: johannes.notni@tum.de; Web: http://www.prc.ch.tum.de
First published on 5th May 2015
Abstract
Due to its 3 carbonic acid groups being available for bioconjugation, the TRAP chelator (1,4,7-triazacyclononane-1,4,7-tris(methylene(2-carboxyethylphosphinic acid))) is chosen for the synthesis of trimeric bioconjugates for radiolabelling. We optimized a protocol for bio-orthogonal TRAP conjugation via Cu(I)-catalyzed Huisgen-cycloaddition of terminal azides and alkynes (CuAAC), including a detailed investigation of kinetic properties of Cu(II)–TRAP complexes. TRAP building blocks for CuAAC, TRAP(alkyne)3 and TRAP(azide)3 were obtained by amide coupling of propargylamine/3-azidopropyl-1-amine, respectively. For Cu(II) complexes of neat and triply amide-functionalized TRAP, the equilibrium properties as well as pseudo-first-order Cu(II)-transchelation, using 10 to 30 eq. of NOTA and EDTA, were studied by UV-spectrophotometry. Dissociation of any Cu(II)–TRAP species was found to be independent on the nature or excess of a competing chelator, confirming a proton-driven two-step mechanism. The respective thermodynamic stability constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KML: 19.1 and 17.6) and dissociation rates (k: 38 × 10−6 and 7 × 10−6 s−1, 298 K, pH 4) show that the Cu(II) complex of the TRAP-conjugate possesses lower thermodynamic stability but higher kinetic inertness. At pH 2–3, its demetallation with NOTA was complete within several hours/days at room temperature, respectively, enabling facile Cu(II) removal after click coupling by direct addition of NOTA trihydrochloride to the CuAAC reaction mixture. Notwithstanding this, an extrapolated dissociation half life of >100 h at 37 °C and pH 7 confirms the suitability of TRAP-bioconjugates for application in Cu-64 PET (cf. t1/2(Cu-64) = 12.7 h). To showcase advantages of the method, TRAP(DUPA-Pep)3, a trimer of the PSMA inhibitor DUPA-Pep, was synthesized using 1 eq. TRAP(alkyne)3, 3.3 eq. DUPA-Pep-azide, 10 eq. Na ascorbate, and 1.2 eq. Cu(II)-acetate. Its PSMA affinity (IC50), determined by the competition assay on LNCaP cells, was 18-times higher than that of the corresponding DOTAGA monomer (IC50: 2 ± 0.1 vs. 36 ± 4 nM), resulting in markedly improved contrast in Ga-68-PET imaging. In conclusion, the kinetic inertness profile of Cu(II)–TRAP conjugates allows for simple Cu(II) removal after click functionalisation by means of transchelation, but also confirms their suitability for Cu-64-PET as demonstrated previously (Dalton Trans., 2012, 41, 13803).
KML: 19.1 and 17.6) and dissociation rates (k: 38 × 10−6 and 7 × 10−6 s−1, 298 K, pH 4) show that the Cu(II) complex of the TRAP-conjugate possesses lower thermodynamic stability but higher kinetic inertness. At pH 2–3, its demetallation with NOTA was complete within several hours/days at room temperature, respectively, enabling facile Cu(II) removal after click coupling by direct addition of NOTA trihydrochloride to the CuAAC reaction mixture. Notwithstanding this, an extrapolated dissociation half life of >100 h at 37 °C and pH 7 confirms the suitability of TRAP-bioconjugates for application in Cu-64 PET (cf. t1/2(Cu-64) = 12.7 h). To showcase advantages of the method, TRAP(DUPA-Pep)3, a trimer of the PSMA inhibitor DUPA-Pep, was synthesized using 1 eq. TRAP(alkyne)3, 3.3 eq. DUPA-Pep-azide, 10 eq. Na ascorbate, and 1.2 eq. Cu(II)-acetate. Its PSMA affinity (IC50), determined by the competition assay on LNCaP cells, was 18-times higher than that of the corresponding DOTAGA monomer (IC50: 2 ± 0.1 vs. 36 ± 4 nM), resulting in markedly improved contrast in Ga-68-PET imaging. In conclusion, the kinetic inertness profile of Cu(II)–TRAP conjugates allows for simple Cu(II) removal after click functionalisation by means of transchelation, but also confirms their suitability for Cu-64-PET as demonstrated previously (Dalton Trans., 2012, 41, 13803).
Introduction
Today, functional molecular imaging is an indispensable part of personalised healthcare. Integrated scanners for simultaneous spatial mapping of morphology/anatomy as well as functional aspects, such as metabolism or receptor expression, have experienced a particularly dynamic evolution in recent years. Their high value for medical diagnostics is rooted in an outstanding superposition (registration) of functional and anatomical images, allowing for precise localisation and functional analysis of lesions and, subsequently, individual planning of tailored therapies. While integration of functional nuclear imaging, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT), with computed tomography (CT) has already become a clinical standard,1 the new possibilities associated with the recent combination of PET with magnetic resonance imaging (MRI) into a full-body scanner have thrilled both researchers and medical practitioners,2–4 not least because MRI can also provide certain types of functional information. Since a comparable progress is currently observed for small-animal imaging devices, multimodal 3D-imaging becomes more and more attractive for preclinical research and life sciences in general. Here, optical methods,5 such as fluorescence/luminescence,6 Cerenkov7,8 or photoacoustic9 imaging, play an important role as well, because the smaller size of investigated subjects is compatible with the limited penetration depth of light. With multi-purpose small animal scanners, meaningful and compelling in vivo data can be obtained quickly, thus saving time and money in medical and pharmaceutical development.As the capabilities of functional imaging are primarily determined by the performance and diversity of the corresponding probes, efforts directed at suitable tracers have been increased in recent time. In terms of PET tracers, this development is further promoted by the fact that some medically useful positron-emitting metal radionuclides have recently become widely available, such as 89Zr,1064Cu,11 and 68Ga.12,13 Of these, gallium-68 is particularly attractive for preclinical and translational research, because it is obtained from 68Ge/68Ga-radionuclide generators with a very long shelf life (up to 1 year).14–18 Such a generator can be eluted several times a day, resulting in low radionuclide costs per experiment, particularly for high workload. Structures to be labelled are usually conjugates of biomolecules with a chelator, into which ionic 68GaIII is introduced simply by complex formation. Compared to most traditional syntheses of 11C- or 18F-labelled compounds, such one-step 68Ga-radiolabelling procedures are generally fast (10–15 min) and quite similar for different compounds. In almost all cases, slightly adjusted standard protocols can be applied, for which purpose a wide range of automated 68Ga-labelling modules (including technical support) are commercially available.19 In other words, the key challenge in making a 68Ga radiopharmaceutical consists in synthesising the desired chelator conjugate as a precursor—for the rest, out-of-the-box solutions are at hand.
In view of these prospects, recent years have seen a variety of novel 68Ga chelators, optimised for efficient labelling and facile bioconjugation.20–30 We proposed 1,4,7-triazacyclononane-1,4,7-tris(2-carboxyethyl-methylenephosphinic acid) (TRAP, Scheme 1) as a suitable platform for elaboration of 68Ga radiopharmaceuticals.31–34 Apart from the fact that this compound shows high efficiency in GaIII complexation35,36 and possesses unique selectivity for the GaIII ion,37 its symmetrical structure with three carboxylic acid moieties not involved in complex formation allows for decoration with up to three functional molecules, such as peptide receptor ligands, fluorophors, other metal binding structures, etc.35,38–41 Hence, TRAP is chosen for a straightforward design of radiolabelled homo- or heteromultimeric conjugates, obviating the need for branching linkers or additional multimeric scaffolds.
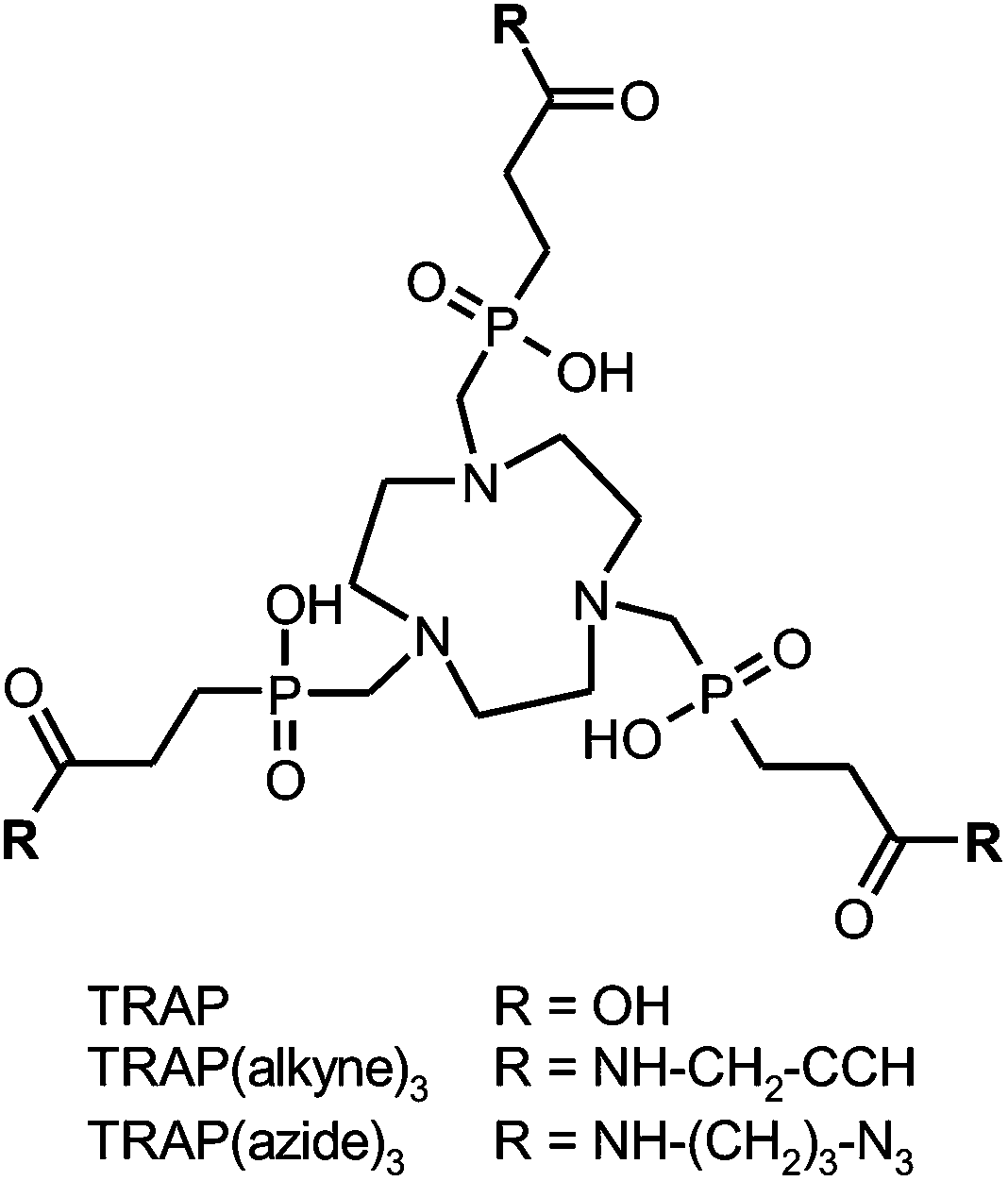 | ||
| Scheme 1 Structural formulae of TRAP and derivatives for functionalisation by means of CuI-catalyzed Huisgen 1,3-dipolar cycloaddition (“Click chemistry”). | ||
During the first few attempts to synthesise TRAP-based multimeric conjugates by amide formation, it was observed that common peptide coupling protocols, consisting of transformation of carboxylates into active esters and subsequent reaction with an amine, could not be applied without modification. This is because active esters of TRAP are not stable in the long term, which is probably related to the close proximity of the phosphinate moieties. We found that the problem can be solved by using excess amine and adding a uronium-type coupling reagent as the last component.32 Clearly, this approach prohibits the presence of certain functional groups in substrates, particularly carboxylic acids and aliphatic alcohols. Consequently, such substrates must be appropriately protected, which, at best, might be just inconvenient, but can also turn out to be a rather complicated task. An obvious workaround consists of using a different coupling chemistry. In a proof-of-principle experiment employing the propargylamine conjugate TRAP(alkyne)3 (Scheme 1), we have already shown that the CuII-catalyzed 1,3-bipolar Huisgen-cycloaddition of terminal alkynes and azides (commonly dubbed “click chemistry”)42 can be applied,35 with the major drawback being that the reaction yields the copper complexes of the conjugates. Although the copper ion could be removed from the chelate cage with sulfide, this method spoiled the product with colloidal copper sulfide precipitates severely perturbing the workup. Furthermore, sulfide is a reducing agent and cleaves disulfide bridges, further limiting the method's scope of application. Another common method, the demetallation with cyanide, is not attractive as well. The use of this highly toxic reagent is disfavoured in the last synthetic step towards pharmaceutical grade compounds, for it might complicate release procedures in a GMP production setting because the absence of traces of free cyanide had to be proven and certified for each batch.
Since a solution for all these problems was expected to substantially expand the field of application for TRAP, and thus pave the way towards a great variety of useful PET tracers, we set out to establish a simple and robust protocol for click chemistry functionalisation. As we focused on CuII removal by transchelation and, therefore, kinetic inertness of CuII complexes of TRAP, the results are also of immediate relevance for 64Cu-labelled TRAP radiopharmaceuticals and their applicability. Moreover, our study might serve as a blueprint for conjugation reactions involving other chelator scaffolds intended to be functionalised via click reactions.43
Results and discussion
The click reaction
Employing the model reaction of TRAP(alkyne)3 (Scheme 1) with benzyl azide, a variety of reaction conditions were tested in order to determine the optimum and limitations. CuII-acetate was used in combination with a 10-fold excess of sodium ascorbate for in situ formation of catalytically active CuI. Other sources of CuII, such as CuSO4, were found suitable as well, on the condition of complete solubility in the chosen solvent system. Due to chelate formation with TRAP, more than one equivalent of CuII is required in general; however, an excess of more than 1.2 eq. provided no further advantage. Complete interconversion of all alkyne sites was achieved with a 10% excess of the azide component per alkyne (3.3 eq. in total). This compares favourably to the amide functionalisation, which usually requires about 5 eq. of the respective amine in order to obtain the trimeric conjugate only,32,44,45 and renders the click synthesis particularly attractive for valuable biomolecules. Furthermore, we found that the reaction was completed after 30 min in many solvents, namely methanol, ethanol, 2-propanol, DMSO, 1,4-dioxane, and aqueous mixtures thereof. We noted that in pure acetonitrile, formation of an emulsion was observed, resulting in an incomplete reaction. This indicates the fact that solubility of all components, that is, reactants and intermediates of the stepwise conjugation, is recommended to ensure a smooth reaction, rendering the choice of a solvent of key importance.Altogether, the “click trimerisation” based on TRAP(alkyne)3 is best conducted using a small excess of azide (3.3 eq.) and CuII (1.2 eq.), as well as a generous allowance of sodium ascorbate (>10 eq.), and can be performed in a wide variety of polar solvents or (aqueous) solvent mixtures. We found that under optimal conditions (pure starting compounds, no precipitation of any component during the reaction), turnover is virtually quantitative in less than one hour, and only the trimer and traces of residual azide substrates are observed in the reaction control (HPLC). Finally, we also prepared the corresponding TRAP derivative with opposite Huisgen functionality, TRAP(azide)3 (Scheme 1), to broaden the scope of the method by enabling trimerisation of biological vectors with alkyne groups as well.
Solution thermodynamics
Although the structure of any bifunctional chelator is changed in the course of conjugation, the associated effects on thermodynamic stability and kinetic inertness of metal complexes often receive little attention. Apart from the obvious impact of modifications directly on the chelating unit, such as the conversion of DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) to DOTA-monoamide46 upon synthesis of DOTA-conjugates,47 conjugation on distal functional groups is widely regarded to have no significant consequences.However, since kinetic inertness of complexes is essentially determined by competitive interaction of metal ions and protons with the ligand's donor atoms, we considered it to be of relevance that TRAP possesses three carboxylates located near the chelating moiety, acting as proton acceptors, which are not present in the respective conjugates. Hence, thermodynamic and kinetic studies were performed for TRAP as well as for TRAP(CHX)332 (Scheme 2), serving as a model for trimeric TRAP conjugates.
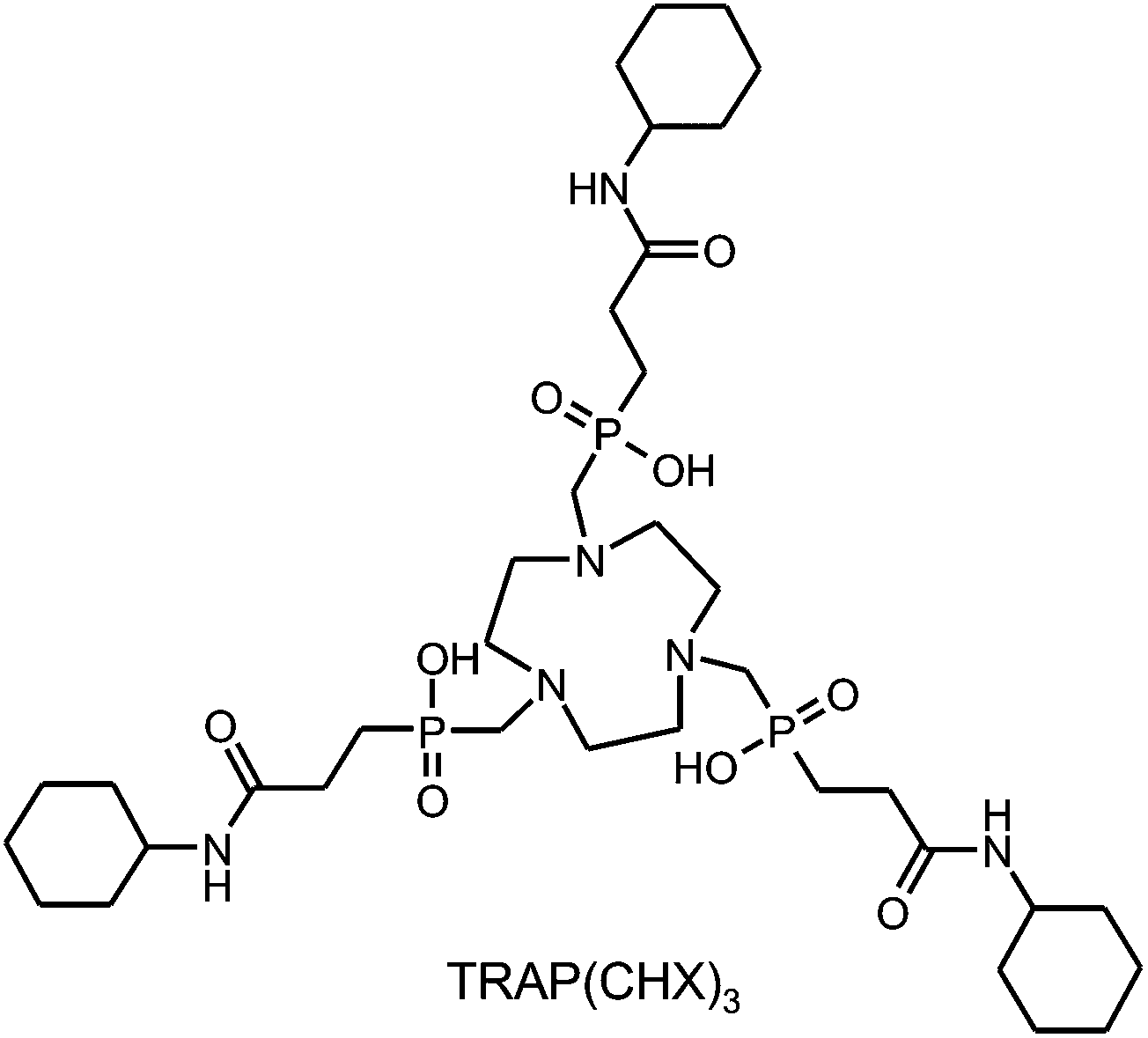 | ||
| Scheme 2 Structural formula of TRAP(CHX)3. | ||
The influence of conjugation becomes apparent upon comparison of the first protonation constants of the compounds that characterise the protonation of the ring nitrogen (logK1H, see Table 1). While the value for TRAP (11.74) is smaller than that for NOTA (12.16), TRAP(CHX)3 (10.92) is even less basic but still ranges above EDTA (9.34). Since the protonation constants characterising the most basic donor sites of ligands (K1H) generally correlate with thermodynamic stability constants (KML) of their metal complexes, it comes as no surprise that the respective values for the CuII complexes decrease in the same order from Cu(NOTA) over Cu(TRAP) to Cu(TRAP(CHX)3), possessing logKML values of 22.44, 19.09 and 17.63, respectively (see Table 2). The same trend is observed for other divalent metal ions, further substantiating the notion of a different behaviour of TRAP and its conjugates.
K5H/logK6H and logK2H/logK3H, respectively. NOTA = 1,4,7-triazacyclononane-1,4,7-triacetic acid, EDTA = ethylenediamine-tetraacetic acid
| TRAP(CHX)3 | TRAP | NOTA | EDTA | ||||
|---|---|---|---|---|---|---|---|
| NaCl | NaCl | Me4NCl34 | NaCl | Me4NCl48 | NaCl | Me4NCl49 | |
| logK1H |
10.92(1) | 11.74(3) | 11.48 | 12.16(2) | 13.17 | 9.34(1) | 10.11 |
| logK2H |
3.66(2) | 5.46(2) | 5.44 | 5.75(3) | 5.74 | 6.12(1) | 6.19 |
| logK3H |
1.55(8) | 4.80(2) | 4.84 | 3.18(2) | 3.22 | 2.85(2) | 2.87 |
| logK4H |
— | 4.16(2) | 4.23 | 1.90(2) | 1.96 | 2.18(2) | 2.26 |
| logK5H |
— | 3.49(2) | 3.45 | — | — | 1.77(2) | — |
| logK6H |
— | 1.50(2) | 1.66 | — | — | — | — |
KML for non-protonated complexes with divalent metals, determined at 25 °C in NaCl, I = 0.15 M. Data have been obtained using UV/Vis spectroscopy (a), potentiometry (b), or NMR (c). Literature data for potentiometry in Me4NCl, I = 0.1 M, are shown for comparison. Stepwise protonation constants of the complexes, as well as details on experimental procedures and UV/VIS or NMR signals used for calculation, are given in the ESI
The three macrocyclic chelators readily form CuII-complexes in acidic solution (pH 1.5 and lower). Upon raising the pH, red shifts of absorption maxima in VIS spectra were observed for these compounds (for details see the ESI†), indicating similar structural changes associated with certain protonation equilibria. Red and blue absorption shifts in CuII-complexes are generally associated with ligand coordination and decoordination, respectively, in the axial position.50 Accordingly, known solid state structures of CuII–NOTA complexes give rise to the assumption that protonation of one axial carboxylate in N3O3-hexacoordinate [Cu(NOTA)]−51 results in de-coordination of carboxylic acid, followed by formation of a [Cu(HNOTA)] structure comprising a pentacoordinate CuII centre with a tetragonal-pyramidal geometry.52 In view of the comparable changes in VIS spectra of NOTA and the phosphinate complexes, we propose a similar structural change for the latter, that is, a more or less complete de-coordination of one phosphonic acid upon protonation.
The involved species [Cu(TRAP(CHX)3)]− and [Cu(H3TRAP))]− exhibit protonation constants (logKH) of 1.97 and 1.58, respectively, revealing a similar basicity of the coordinating phosphinates among each other as well as in comparison with those of the free chelators (1.55 and 1.50, see Table 1; a complete list of experimentally determined protonation constants of the complexes is given in the ESI†). Apparently, the key process of side arm phosphinate protonation occurs at comparable pH values for both complexes, because there is only a relatively weak interaction between the phosphinate oxygen donor atoms and CuII.
Kinetic inertness & transchelation
In order to establish a procedure for removal of CuII from TRAP conjugates, we investigated transchelation of CuII towards two chelators with known suitability for CuII coordination, namely EDTA and NOTA.52 Reactions were monitored by means of UV spectrophotometry at the absorption bands of Cu(EDTA) and Cu(NOTA) complexes over a pH range of 1.7–4, using 10–30 eq. of EDTA or NOTA, and kinetic parameters were calculated from these data (see the ESI†).Fig. 1 shows that the obtained pseudo-first-order rate constants at 298 K are independent from the identity or excess of the competing chelator, and increase with decreasing pH. Consequently, the transchelation is recognised as a three-step process, initiated by protonation of the complex and followed by its spontaneous dissociation as the rate-determining step, thereafter free Cu2+ is captured quickly by the scavenger ligand (Scheme 3). Such rationale is well in line with established concepts, because, while transchelation from complexes of open-chain ligands can occur via concerted mechanisms with intermediate formation of ternary complexes,53 dissociation of metals from pendant-arm macrocycles is mostly governed by stepwise mechanisms via formation of protonated intermediates.54,55
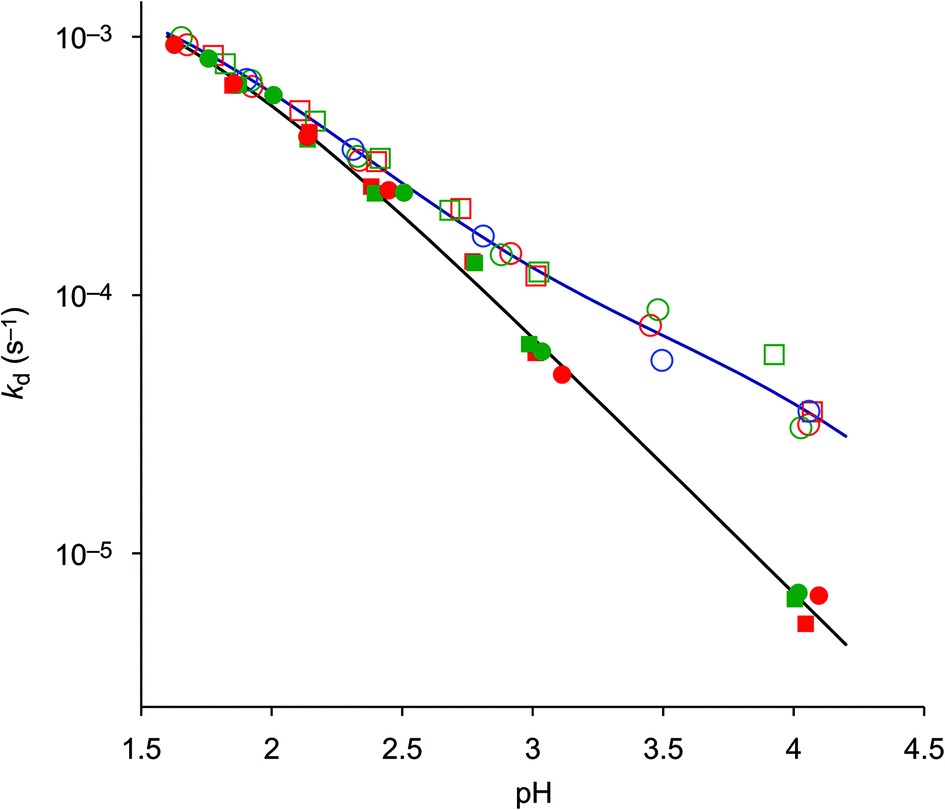 | ||
| Fig. 1 Pseudo-first-order rate constants at 25 °C (298 K) for the reaction of CuII complexes of TRAP (empty symbols) and TRAP(CHX)3 (filled symbols), with 10 (red), 20 (green) and 30 (blue) equivalents of EDTA (squares) and NOTA (circles). Respective kd functions were calculated from kinetic and equilibrium data. | ||
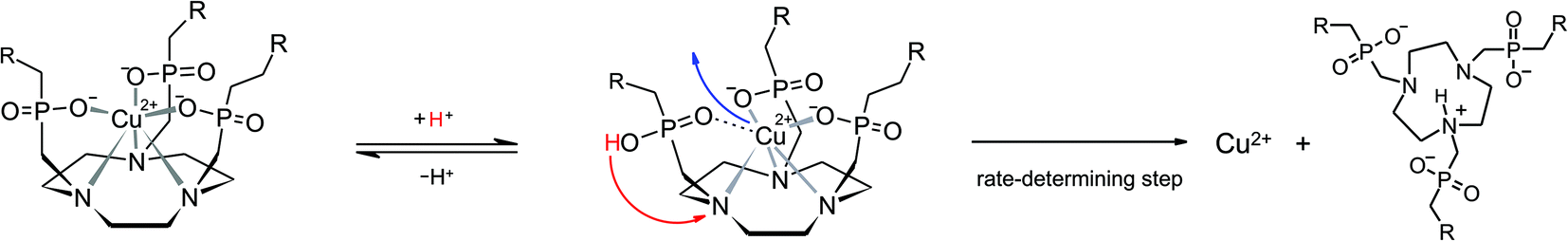 | ||
| Scheme 3 Proposed mechanism for the proton-assisted dissociation of CuII–TRAP complexes. The colored arrows indicate the structural rearrangements occurring during the rate-determining step. The driving force of the entire reaction is a practically irreversible complexation of CuII by an excess scavenger ligand (not shown). | ||
In order to gain further insight into the underlying reaction mechanisms and the species involved, pseudo-first-order rate constants were measured at various temperatures over the range of 288–323 K, and the Eyring activation parameters were calculated for complex species with different protonation states (Table 3, for details see the ESI†). As expected, a comparison of the rate constants at 298 K confirms that the formally uncharged species [Cu(HTRAP(CHX)3)] and [Cu(H4TRAP))], which most likely comprise pentacoordinate CuII and one non-coordinating phosphinic acid donor group (Scheme 3), contribute most to dissociation in acidic media. Since these species are, according to their aforementioned protonation constants in the range of 1.6 to 1.9, highly abundant at pH 1.5, and their activation parameters are identical with respect to the error margins (Table 3), the observed levelling of the overall dissociation rate constants below pH 2 (Fig. 1) is well explained.
| [Cu(H3TRAP)]− | [Cu(H4TRAP)] | [Cu(HTRAP(CHX)3)] | [Cu(TRAP(CHX)3)]− | |
|---|---|---|---|---|
| k 298 K (s−1) | (6.2 ± 0.3) × 10 −5 | (2.1 ± 0.1) × 10 −3 | (2.2 ± 0.1) × 10 −3 | (7 ± 2) × 10 −7 |
| ΔH‡ (kJ mol−1) | 77 ± 8 | 67 ± 3 | 69 ± 5 | 88 ± 10 |
| ΔS‡ (J mol−1 K−1) | −68 ± 10 | −71 ± 8 | −65 ± 10 | −65 ± 30 |
| ΔG‡298 K (kJ mol−1) | 97.2 | 88.2 | 88.3 | 108 |
The more the abundances of the deprotonated species [Cu(H3TRAP)]− and [Cu(TRAP(CHX)3)]− increase with increasing pH, the more their individual dissociation rate constants (Table 3) become relevant for the overall rates. Despite the rate constant of [Cu(H3TRAP)]− (6.2 × 10−5 s−1) being more than one order of magnitude lower than that of the uncharged [Cu(H4TRAP)] (2.1 × 10−3 s−1), its contribution is substantial and dominates dissociation of Cu(TRAP) above pH 3.5 (Fig. 2). In contrast, the rate constant for the corresponding [Cu(TRAP(CHX)3)]− (7 × 10−7 s−1) is so low that its contribution is negligible up to pH 4, which explains the observed divergence of overall dissociation rates for Cu(TRAP) and Cu(TRAP(CHX)3) above pH > 2.5 (Fig. 1) and, therefore, the increased inertness of the conjugate complexes.
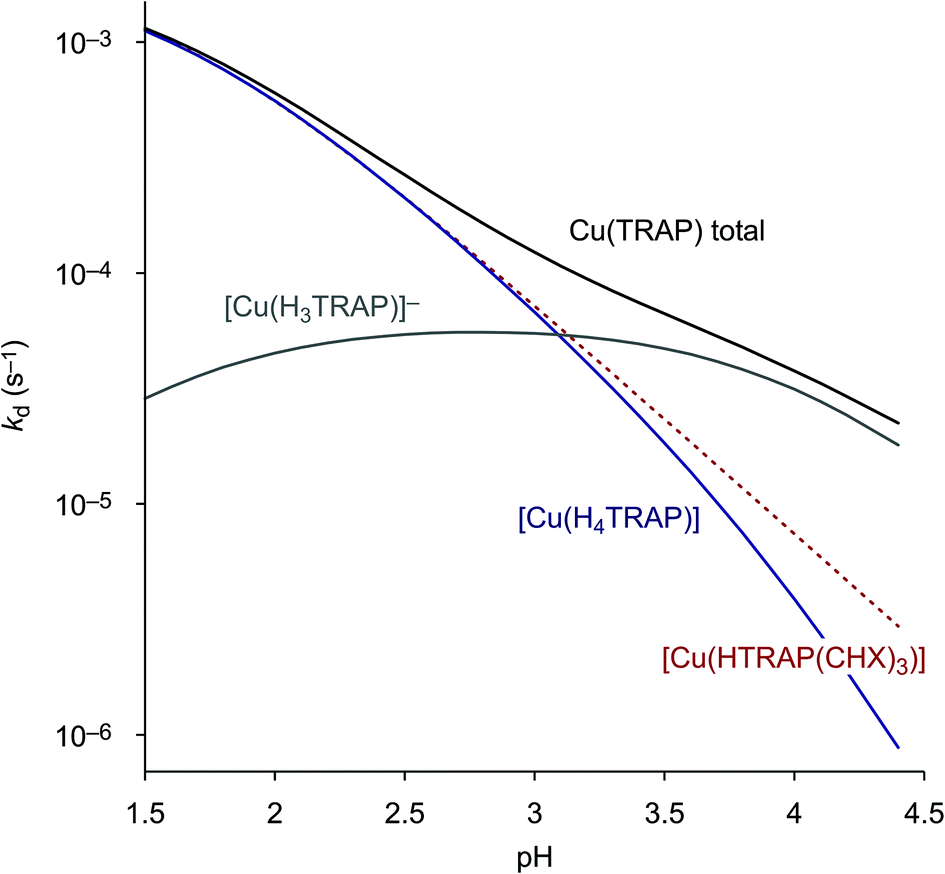 | ||
| Fig. 2 Contributions of the spontaneous dissociation of the different Cu(TRAP) complexes to the overall, pH-dependent reaction rate. In this pH range, the curve for [Cu(HTRAP(CHX)3)] (dotted line) is equivalent to the overall rate for Cu(TRAP(CHX)3) (see Fig. 1), as it is the only contributing species. | ||
The pH-dependent half lives of dissociation, calculated and extrapolated on the basis of equilibrium and kinetic data, finally allow us to draw some important conclusions. Fig. 3 illustrates that functionalisation of TRAP results in the significantly increased kinetic inertness of the corresponding CuII complexes above pH 2. Furthermore, removal of CuII from TRAP conjugates is best performed at pH 2–3, where τ1/2 is sufficiently short to achieve complete demetallation overnight at r.t., or within a few hours at elevated temperatures. On the other hand, an extrapolated τ1/2 of >100 h for Cu(TRAP(CHX)3) at pH 7 and at body temperature strongly corroborates the general suitability of TRAP conjugates for 64Cu-PET imaging.56
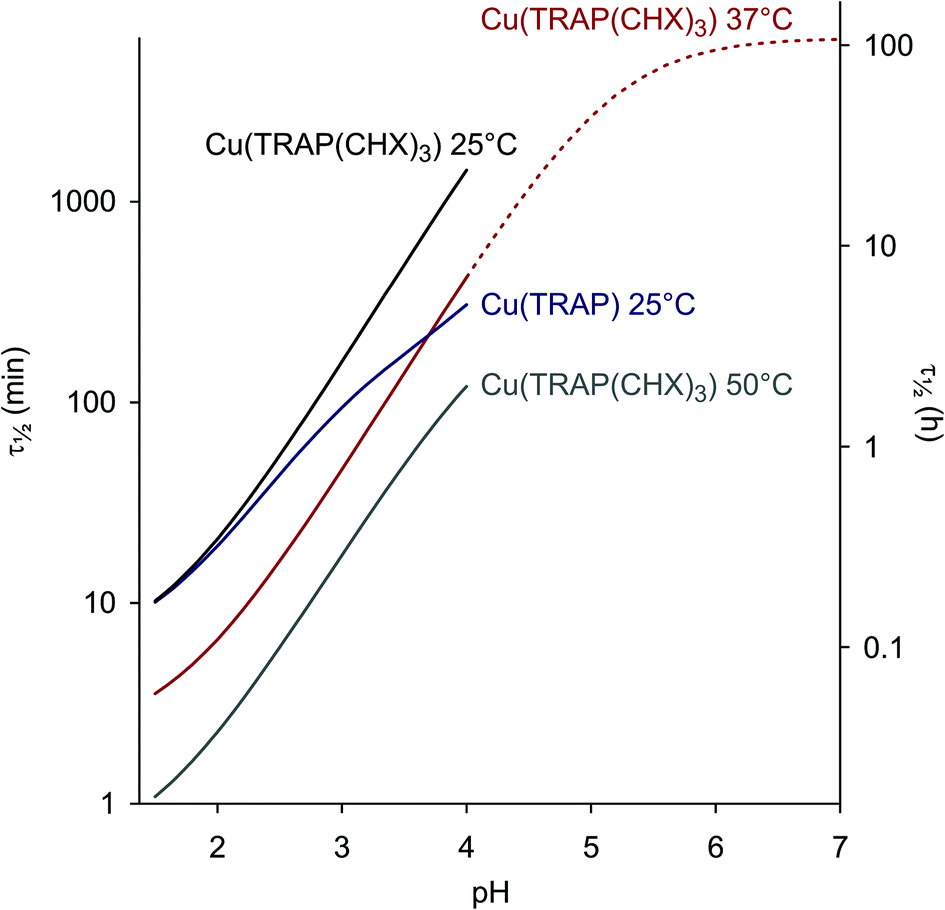 | ||
| Fig. 3 Dissociation half lives as functions of pH (τ1/2 = ln2/kd, see also ESI, eqn (18) and (23)†). An extrapolation is shown for Cu(TRAP(CHX)3) at 37 °C and above pH 4. | ||
Application
During kinetic measurements, CuII transchelation to EDTA was found incomplete (ca. 90%). This is not surprising because the thermodynamic and conditional stability of the CuII–EDTA complexes is comparably low (logKML = 19.02(3), 3.15(1), and 2.04(1) were measured for [Cu(EDTA)]2−, [Cu(HEDTA)]− and [Cu(H2EDTA)]), resulting in partial dissociation of Cu(EDTA) at pH 2–3, similar to Cu(TRAP)). Hence, EDTA is only suitable for demetallation when applied in a very high stoichiometric excess. Notwithstanding this, CuII could be completely removed from Cu(TRAP(CHX)3) by means of diafiltration (continuous ultrafiltration),32 using a 1 mM solution of H4EDTA (pH 2.8) as the running buffer, because this approach allowed for successive removal of Cu(EDTA) from the equilibrium.
Despite being considerably more expensive, NOTA was identified as the reagent of choice for removal of CuII from TRAP conjugates, owing to two important characteristics which efficiently suppress the back reaction of the transchelation equilibrium. First, its CuII complex is considerably more stable than Cu(EDTA) or any CuII–TRAP complex (Table 2), providing a strong driving force for the entire reaction. Second, the protonated complex [Cu(HNOTA)] is kinetically inert,52 meaning that it does not show a tendency towards spontaneous dissociation like Cu(EDTA) or the phosphinate complexes. We found that demetallation of CuII-containing TRAP conjugates could be achieved by adding NOTA directly to the reaction mixture and adjusting the pH to 2–3 with dilute HCl, once the click coupling is finished. In practice, addition of NOTA trihydrochloride has been found to be most convenient, since the NOTA-bound HCl readily adjusts the pH of the solution to the required acidic range, obviating the need for any further reagent. Altogether, the favorable properties of NOTA justify its application despite the high price, as this is of secondary importance in view of the micromolar amounts of substrates commonly handled during synthesis of precursors for radiolabelling.
In order to demonstrate the utility and convenience of the devised one-pot procedure, it was applied for the synthesis of a trimeric conjugate of DUPA-Pep which otherwise would have been much more difficult to obtain (Scheme 4). DUPA-Pep57 is a urea-type enzyme inhibitor which has earlier been labelled with 99mTc and used for in vivo SPECT imaging of prostate-specific membrane antigen (PSMA, EC 3.4.17.21, synonyms: glutamate carboxypeptidase II, NAALADase),58 a hydrolytic zinc enzyme that is overexpressed by human prostate cancers.59 For trimerisation, a terminal azide was introduced by standard peptide coupling in solution, using pre-activated 5-azidopentanoic acid as the building block. Now, the trimer was synthesised by applying our two-step, one-pot procedure (Scheme 4). Conjugation and demetallation were virtually quantitative according to HPLC/MS; unfortunately, final HPLC purification considerably decreased the overall yield.
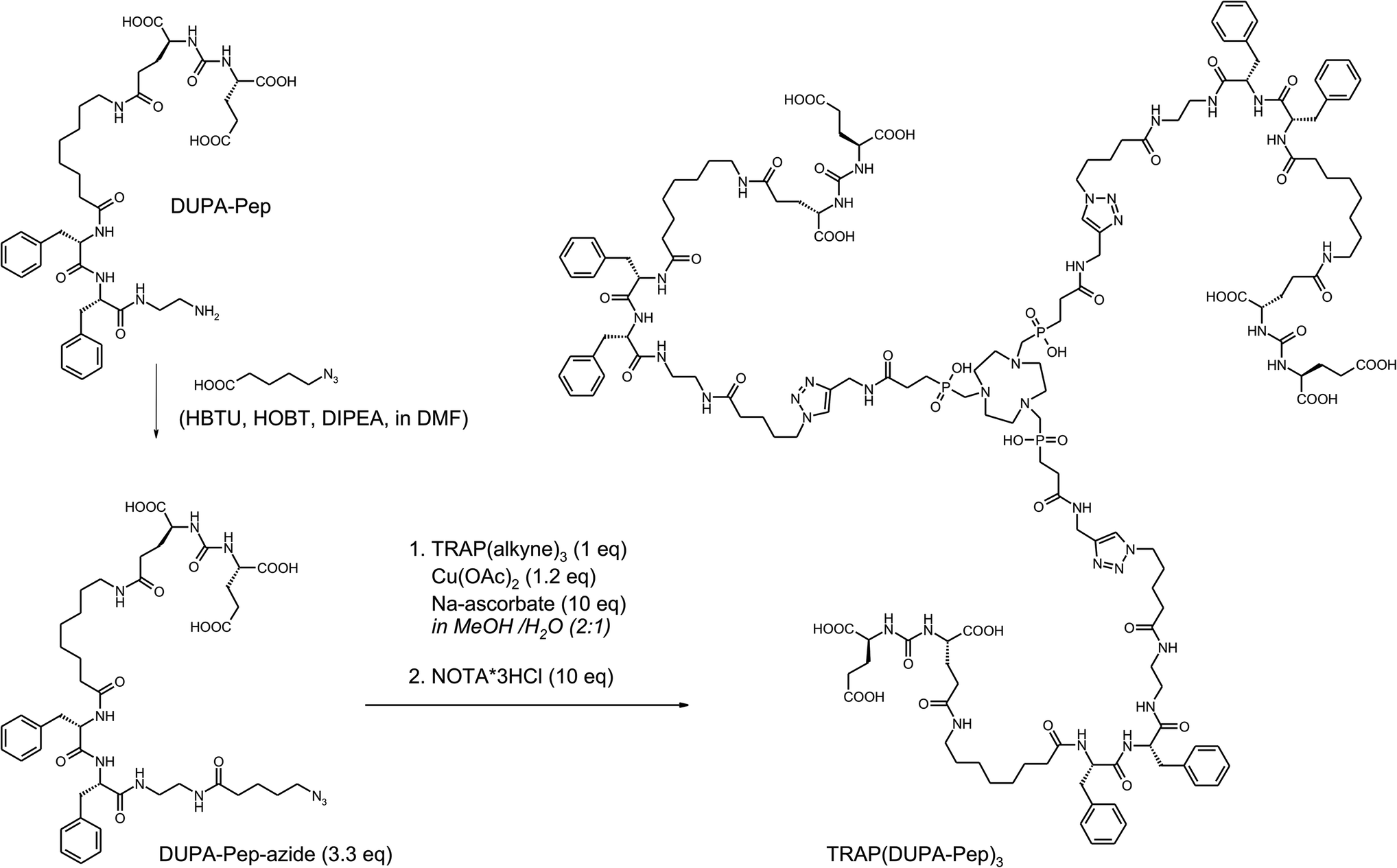 | ||
| Scheme 4 Synthesis of TRAP(DUPA-Pep)3 for 68Ga-labelling and subsequent in vivo mapping of PSMA expression using PET. | ||
Notwithstanding this, a comparison of in vitro and in vivo data for GaIII-complexes of TRAP(DUPA-Pep)3 and DOTAGA-DUPA-pep (Scheme 5), a structurally related monomeric chelator conjugate synthesised as a reference, demonstrated the possible advantages of trimeric conjugates over the corresponding monomers. Affinities to PSMA were determined by cellular displacement assays on LNCaP cells (human prostate carcinoma). IC50 values of 36 ± 4 nM and 2 ± 0.1 nM for the monomer and the trimer, respectively, translate to an impressive 18-fold increase in affinity.
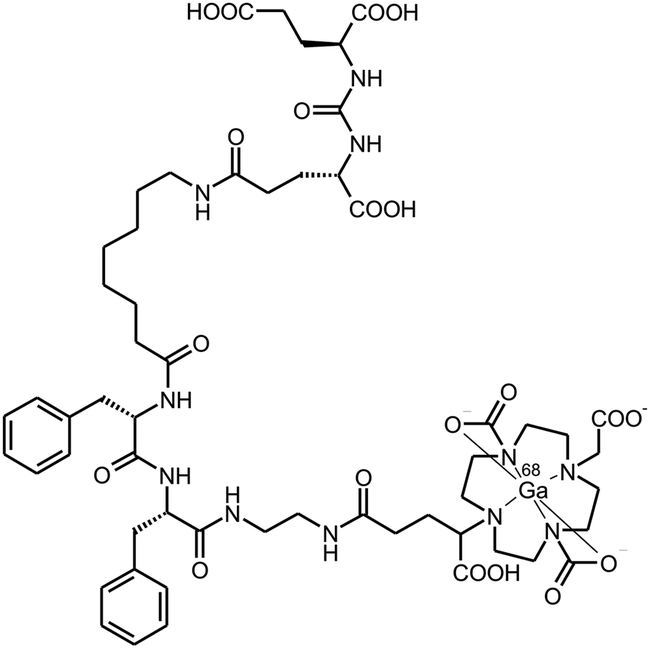 | ||
| Scheme 5 68Ga-DOTAGA-DUPA-Pep, a PSMA-targeting 68Ga-radiopharmaceutical used as the reference monomer. | ||
PET images were acquired for both 68Ga-labelled compounds, with CD-1 athymic nude mice bearing LNCaP xenografts on the right shoulder as subjects. Fig. 4 shows that trimerisation resulted in substantially increased tumor uptake, accompanied by a lower background. However, in this particular case, the increased tumor-to-background ratio most probably does not only rely on improved target affinity. According to the recent literature, the poor performance of the monomer is owing to degradation of DUPA-Pep in vivo, which, in turn, is rooted in metabolic instability of the L-Phe–L-Phe structural motif.60 Interconnection of several of these unstable moieties in 68Ga-TRAP(DUPA-Pep)3 most likely results in a prolonged time for complete loss of targeting properties of the entire construct, as more than one metabolic cleavage per radiolabel is required. This might be the reason for the superior retention of activity at the target, observed for 68Ga-TRAP(DUPA-Pep)3. This finding is of particular interest, because it suggests that it might be generally possible to obtain useful (radio)pharmaceuticals from metabolically unstable targeting molecules by means of multimerisation. However, confirmation of this hypothesis certainly requires more such multimers as examples, the synthesis of which could be greatly facilitated using the TRAP platform and applying our one-pot click chemistry protocol.
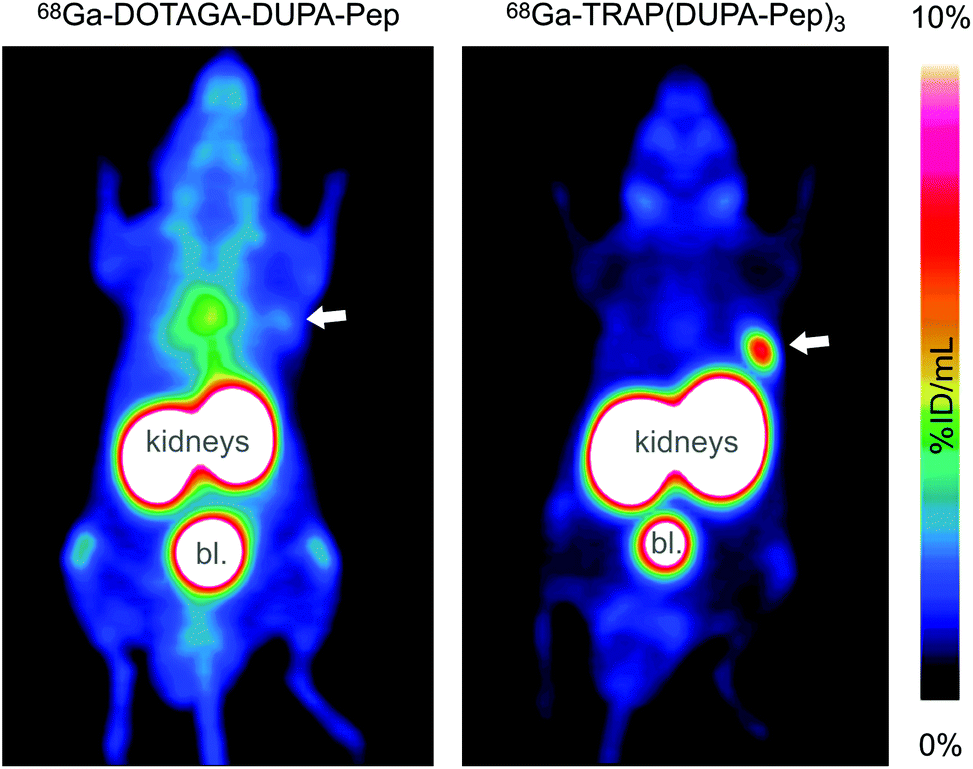 | ||
| Fig. 4 PET images (maximum intensity projections) of CD-1 athymic nude mice bearing LNCaP tumor xenografts on the right shoulder, acquired 90 min after injection of 68Ga-DOTAGA-DUPA-Pep (left) and 68Ga-TRAP(DUPA-Pep)3 (right). Tumor positions are indicated by arrows. High activity uptake in kidneys and bladder (denoted bl.) is owing to physiological PSMA expression and/or excretion. The high background level in the left image is caused by unspecific distribution of activity due to metabolic instability of 68Ga-DOTAGA-DUPA-Pep.60 | ||
Conclusion
The opposing trends of thermodynamic stability and kinetic inertness, observed for the CuII complexes of TRAP and TRAP-conjugates, are another example for the generally known but frequently unheeded fact that equilibrium constants are of little predictive value concerning kinetic inertness, and thus, in vivo stability, of metal complexes. For example, similar observations have been described recently for CuII complexes of cyclam-based diphosphinates and diphosphonates.61 We would thus like to emphasise again that for a valid interpretation or prognosis of in vivo behaviour, investigation of fundamental kinetics is of key importance. In addition, our results illustrate that modifications on remote functional groups (i.e., conjugation sites) of a given bifunctional chelator can significantly affect the metal-binding properties of its primary coordination site.In this respect, our data corroborate and explain a previous study on the stability of some 64Cu-labelled TRAP-type chelators, which showed that the 64CuII complex of a TRAP-peptide trimer is much more stable upon challenging with aq. Na2EDTA (pH 4.5) than that of neat TRAP (85% vs. 50% of intact chelate, respectively, after 12 h).56 However, it should not be disregarded that particularly for 64CuII complexes, mechanisms other than proton-assisted ones can contribute to demetallation in vivo, for example, interaction with other metal cations (transmetallation)62 or enzymatic cleavage,63 which further complicates the matter.
In summary, we devised a simple and convenient one-pot procedure for synthesis of TRAP-based trimeric chelator conjugates by means of click chemistry. Using the frameworks TRAP(alkyne)3 and TRAP(azide)3, the CuAAC reaction with 3.3 eq. of a biomolecule with the respective opposite Huisgen functionality, 1.2 eq. of CuII, and excess sodium ascorbate quickly affords the trimer. This can easily be demetallated by addition of excess NOTA trihydrochloride, owing to the pronounced thermodynamic stability and kinetic inertness of the CuII–NOTA complex. At this stage, it is worth noting that introduction of azide functions to peptides on solid supports is not only feasible by using commercially available building blocks, such as 5-azidopentanoic acid, as shown above. The recently described direct conversion of the N-terminus of a resin-bound peptide into an azide64 ideally complements our method, paving the way towards rapid access to trimeric peptidic radiopharmaceuticals. The prediction that such structures will frequently exhibit enhanced target affinity and improved imaging properties, as exemplified in this study, appears justified in view of the wealth of pertinent experience with multimerised targeting vectors.35,44,57,65–71
Experimental
A complete and detailed description of Materials and methods is provided in the ESI,† while a brief account is given here. TRAP32 was prepared as described. TRAP(CHX)3,32 TRAP(alkyne)335 and TRAP(azide)3 were prepared according to a published procedure,32 by employing a modified peptide coupling protocol (reagent: HATU, base: DIPEA, solvent: DMSO) and the respective amines (cyclohexylamine, propargylamine, 3-azidopropylamine). DOTAGA-DUPA-Pep was prepared from commercial DUPA-Pep and decorated with DOTAGA using DOTAGA-anhydride.72
Equilibrium studies (protonation and stability constants) were performed by pH-potentiometry, UV-spectrophotometry, and 1H- and 31P-NMR spectroscopy in 0.15 M NaCl. Rates of ligand exchange reactions were determined for 0.2 mM solutions of Cu(TRAP) and Cu(TRAP(CHX)3) by monitoring the formation of Cu(NOTA) and Cu(EDTA) with UV-spectrophotometry at 263 and 243 nm, at temperatures of 15, 25, 37, and 50 °C, and pH values in the range of 1.5–4, which were kept constant using dichloroacetic acid (DCA) (pH range 1.5–2.5), chloroacetic acid (MCA) (pH range 2.5–3.5) and 1,4-dimethylpiperazine (DMP) (pH = 3.1–4.1) buffers (0.01 M). Pseudo-first-order conditions were achieved by employing 10–30 eq. of NOTA and 10–20 eq. of EDTA.
Non-radioactive GaIII complexes of DOTAGA-DUPA-Pep and TRAP(DUPA-Pep)3 for determination of affinity were prepared by adding 2 mM gallium nitrate (0.5 mL) to the same volume of 2 mM solutions of the conjugates. Complete complex formation occurred immediately and was confirmed by ESI-MS. IC50 values were determined by a cellular displacement assay on LNCaP (human prostate carcinoma) cells, using the radioiodinated urea-type PSMA inhibitor ([125I]I-BA)KuE for competition.60
68Ga labelling was performed using the non-processed generator eluate (1 M HCl, pH adjusted to 2–3 with HEPES) of a SnO2-based 68Ge/68Ga-generator (iThemba LABS, SA) with subsequent purification of the radiopharmaceuticals by solid-phase extraction, employing a fully automated module.44 This procedure is readily transferable to clinical settings.73 PET imaging was performed on a Siemens Inveon small animal PET system as described.35 Approx. 12 MBq of 68Ga-DOTAGA-DUPA-Pep (≈0.3 nmol) or 68Ga-TRAP(DUPA-Pep)3 (≈0.15 nmol) respectively were administered to CD-1 athymic nude mice bearing LNCaP xenografts on the right shoulder. PET data were acquired 60 min p.i. for 15 min, and images were reconstructed by using OSEM3D algorithm.
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant #NO822/4-1 and SFB 824, project Z1), by the European Union and the State of Hungary (co-financed by the European Social Fund in the framework of TÁMOP 4.2.4. A/2-11-1-2012-0001 ‘National Excellence Program’, grant #A2-MZPD-12-0038, and by the Hungarian Scientific Research Found (OTKA K109029 and K84291) is gratefully acknowledged. The authors furthermore thank Prof. Markus Schwaiger (Department of Nuclear Medicine, TUM) for providing laboratory space and granting access to imaging devices; Sybille Reder, Markus Mittelhäuser and Marco Lehmann for assistance with animal PET; Prof. Ernő Brücher and Dávid Horváth (University of Debrecen) for their advice on equilibrium and kinetic measurements; Simon Moosmang, Simon Schmid, Veronika Stiegler and Anja Wacker for laboratory assistance.References
- T. Beyer, D. W. Townsend, T. Brun, P. E. Kinahan, M. Charron, R. Roddy, J. Jerin, J. Young, L. Byars and R. Nutt, J. Nucl. Med., 2000, 41, 1369–1379 CAS.
- H. Herzog, Z. Med. Phys., 2012, 22, 281–298 CrossRef PubMed.
- C. Rischpler, S. G. Nekolla, I. Dregely and M. Schwaiger, J. Nucl. Med., 2013, 54, 402–415 CrossRef CAS PubMed.
- A. Drzezga, M. Souvatzoglou, M. Eiber, A. J. Beer, S. Fürst, A. Martinez-Möller, S. G. Nekolla, S. Ziegler, C. Ganter, E. J. Rummeny and M. Schwaiger, J. Nucl. Med., 2012, 53, 845–855 CrossRef PubMed.
- D. Razansky, N. C. Deliolanis, C. Vinegoni and V. Ntziachristos, Curr. Pharm. Biotechnol., 2012, 13, 504–522 CAS.
- C. Darne, Y. J. Lu and E. M. Sevick-Muraca, Phys. Med. Biol., 2014, 59, R1–R64 CrossRef PubMed.
- Y. D. Xu, H. G. Liu and Z. Cheng, J. Nucl. Med., 2011, 52, 2009–2018 CrossRef PubMed.
- P. T. K. Chin, M. M. Welling, S. C. J. Meskers, R. A. V. Olmos, H. Tanke and F. W. B. van Leeuwen, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, 1283–1291 CrossRef CAS PubMed.
- L. M. Nie and X. Y. Chen, Chem. Soc. Rev., 2014, 43, 7132–7170 RSC.
- M. A. Deri, B. M. Zeglis, L. C. Francesconi and J. S. Lewis, Nucl. Med. Biol., 2013, 40, 3–40 CrossRef CAS PubMed.
- T. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- C. Decristoforo, R. D. Pickett and A. Verbruggen, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39, S31–S40 CrossRef PubMed.
- J. Notni, Nachr. Chem., 2012, 60, 645–649 CrossRef CAS PubMed.
- M. W. Greene and W. D. Tucker, Int. J. Appl. Radiat. Isot., 1961, 12, 62–63 CrossRef CAS.
- C. Loc'h, B. Maziere and D. Comar, J. Nucl. Med., 1980, 21, 171–173 Search PubMed.
- J. Schuhmacher and W. Maier-Borst, Int. J. Appl. Radiat. Isot., 1981, 32, 31–36 CrossRef CAS.
- K. Zhernosekov, M. Harfensteller, J. Moreno, O. Leib, O. Buck, A. Tuerler, R. Henkelmann and T. Nikula, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, S251 Search PubMed.
- E. de Blois, H. S. Chan, C. Naidoo, D. Prince, E. P. Krenning and W. A. P. Breeman, Appl. Radiat. Isot., 2011, 69, 308–315 CrossRef CAS PubMed.
- S. Boschi, F. Lodi, C. Malizia, G. Cicoria and M. Marengo, Appl. Radiat. Isot., 2013, 76, 38–45 CrossRef CAS PubMed.
- Z. Baranyai, F. Uggeri, A. Maiocchi, G. B. Giovenzana, C. Cavallotti, A. Takács, I. Tóth, I. Bányai, A. Bényei, E. Brücher and S. Aime, Eur. J. Inorg. Chem., 2013, 147–162 CrossRef CAS PubMed.
- J. Schuhmacher, G. Klívenyi, W. E. Hull, R. Matys, H. Hauser, H. Kalthoff, W. H. Schmiegel, W. Maier-Borst and S. Matzku, Nucl. Med. Biol., 1992, 19, 809–824 CAS.
- E. Boros, C. L. Ferreira, J. F. Cawthray, E. W. Price, B. O. Patrick, D. W. Wester, M. J. Adam and C. Orvig, J. Am. Chem. Soc., 2010, 132, 15726–15733 CrossRef CAS PubMed.
- G. Tircsó, E. T. Benyó, E. H. Suh, P. Jurek, G. E. Kiefer, A. D. Sherry and Z. Kovács, Bioconjugate Chem., 2009, 20, 565–575 CrossRef PubMed.
- C. L. Ferreira, D. T. T. Yapp, D. Mandel, R. K. Gill, E. Boros, M. Q. Wong, P. Jurek and G. E. Kiefer, Bioconjugate Chem., 2012, 23, 2239–2246 CrossRef CAS PubMed.
- J. Notni, K. Pohle, J. A. Peters, H. Görls and C. Platas-Iglesias, Inorg. Chem., 2009, 48, 3257–3267 CrossRef CAS PubMed.
- K. P. Eisenwiener, M. I. M. Prata, I. Buschmann, H. W. Zhang, A. C. Santos, S. Wenger, J. C. Reubi and H. R. Mäcke, Bioconjugate Chem., 2002, 13, 530–541 CrossRef CAS PubMed.
- A. N. Singh, W. Liu, G. Hao, A. Kumar, A. Gupta, O. K. Oz, J.-T. Hsieh and X. Sun, Bioconjugate Chem., 2011, 22, 1650–1662 CrossRef CAS PubMed.
- F. L. Guerra Gomez, T. Uehara, T. Rokugawa, Y. Higaki, H. Suzuki, H. Hanaoka, H. Akizawa and Y. Arano, Bioconjugate Chem., 2012, 23, 2229–2238 CrossRef PubMed.
- J. Šimeček, J. Notni, T. G. Kapp, H. Kessler and H. J. Wester, Mol. Pharm., 2014, 11, 1687–1695 CrossRef PubMed.
- J. Šimeček, O. Zemek, P. Hermann, J. Notni and H. J. Wester, Mol. Pharm., 2014, 11, 3893–3903 CrossRef PubMed.
- J. Notni, J. Šimeček and H. J. Wester, ChemMedChem, 2014, 9, 1107–1115 CrossRef CAS PubMed.
- J. Notni, P. Hermann, J. Havlíčková, J. Kotek, V. Kubíček, J. Plutnar, N. Loktionova, P. J. Riss, F. Rösch and I. Lukeš, Chem. – Eur. J., 2010, 16, 7174–7185 CrossRef CAS PubMed.
- (a) J. Šimeček, J. Notni, V. Kubíček and P. Hermann, Nucl. Med. Biol., 2010, 37, 679 CrossRef PubMed; (b) J. Notni, J. Šimeček, P. Hermann and H. J. Wester, J. Labelled Compd. Radiopharm., 2011, 54, S407 Search PubMed.
- J. Šimeček, M. Schulz, J. Notni, J. Plutnar, V. Kubíček, J. Havlíčková and P. Hermann, Inorg. Chem., 2012, 51, 577–590 CrossRef PubMed.
- J. Notni, J. Šimeček, P. Hermann and H. J. Wester, Chem. – Eur. J., 2011, 17, 14718–14722 CrossRef CAS PubMed.
- J. Notni, K. Pohle and H. J. Wester, EJNMMI Res., 2012, 2, 28 CrossRef CAS PubMed.
- J. Šimeček, P. Hermann, H. J. Wester and J. Notni, ChemMedChem, 2013, 8, 95–103 CrossRef PubMed.
- J. Notni, P. Hermann, I. Dregely and H. J. Wester, Chem. – Eur. J., 2013, 19, 12602–12606 CrossRef CAS PubMed.
- J. Notni, P. Hermann, I. Dregely and H. J. Wester, J. Labelled Compd. Radiopharm., 2013, 56, S62 Search PubMed.
- J. Notni, P. Hermann, I. Dregely, M. Schwaiger and H. J. Wester, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, S229 Search PubMed.
- J. Notni, J. Plutnar and H. J. Wester, EJNMMI Res., 2012, 2, 13 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- E. Kriemen, E. Ruf, U. Behrens and W. Maison, Chem. – Asian J., 2014, 9, 2197–2204 CrossRef CAS PubMed.
- J. Notni, K. Pohle and H. J. Wester, Nucl. Med. Biol., 2012, 39, 777–784 CrossRef PubMed.
- J. Notni, R. Braren, A. J. Beer, K. Steiger, A. Schlitter, I. Esposito, J. Siveke, M. Erkan, M. Schwaiger and H. J. Wester, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, S131 Search PubMed.
- V. Kubíček, J. Havlíčková, J. Kotek, G. Tircsó, P. Hermann, É. Tóth and I. Lukeš, Inorg. Chem., 2010, 49, 10960–10969 CrossRef PubMed.
- (a) G. J. Stasiuk and N. J. Long, Chem. Commun., 2013, 49, 2732–2746 RSC; (b) N. Viola-Villegas and R. P. Doyle, Coord. Chem. Rev., 2009, 253, 1906–1925 CrossRef CAS PubMed.
- B. Drahoš, V. Kubíček, C. S. Bonnet, P. Hermann, I. Lukeš and É. Tóth, Dalton Trans., 2011, 40, 1945–1951 RSC.
- K. Kumar, C. A. Chang, L. C. Francesconi, D. D. Dischino, M. F. Malley, J. Z. Gougoutas and M. F. Tweedle, Inorg. Chem., 1994, 33, 3567–3575 CrossRef CAS.
- E. Prenesti, P. G. Daniele, S. Berto and S. Toso, Polyhedron, 2006, 25, 2815–2823 CrossRef CAS PubMed.
- S. Pant, M. T. W. Hearn and K. Saito, Aust. J. Chem., 2010, 63, 502–506 CrossRef CAS.
- A. C. Bényei , Cambridge Crystallographic Data Centre (CCDC) database entry #1025548, 2014.
- J. F. Morfin and É. Tóth, Inorg. Chem., 2011, 50, 10371–10378 CrossRef CAS PubMed.
- P. Lubal, M. Kývala, P. Hermann, J. Holubová, J. Rohovec, J. Havel and I. Lukeš, Polyhedron, 2001, 20, 47–55 CrossRef CAS.
- I. Voráčová, J. Vaněk, J. Pasulka, Z. Střelcová, P. Lubal and P. Hermann, Polyhedron, 2013, 61, 99–104 CrossRef PubMed.
- J. Šimeček, H. J. Wester and J. Notni, Dalton Trans., 2012, 41, 13803–13806 RSC.
- S. A. Kularatne, K. Wang, H.-K. R. Santhapuram and P. S. Low, Mol. Pharm., 2009, 6, 780–789 CrossRef CAS PubMed.
- J. R. Mesters, C. Barinka, W. Li, T. Tsukamoto, P. Majer, B. S. Slusher, J. Konvalinka and R. Hilgenfeld, EMBO J., 2006, 25, 1375–1384 CrossRef CAS PubMed.
- (a) A. Ghosh and W. D. Heston, J. Cell. Biochem., 2004, 91, 528–539 CrossRef CAS PubMed; (b) D. A. Silver, I. Pellicer, W. R. Fair, W. D. Heston and C. Cordon-Cardo, Clin. Cancer Res., 1997, 3, 81–85 CAS.
- M. Weineisen, J. Šimeček, M. Schottelius, M. Schwaiger and H. J. Wester, EJNMMI Res., 2014, 4, 36 CrossRef PubMed.
- M. Paúrová, J. Havlíčková, A. Pospíšilová, M. Vetrík, I. Císařová, H. Stephan, H.-J. Pietzsch, M. Hruby, P. Hermann and J. Kotek, Chem. – Eur. J., 2015, 21, 4671–4687 CrossRef PubMed.
- V. Maheshwari, J. L. J. Dearling, S. T. Treves and A. B. Packard, Inorg. Chim. Acta, 2012, 393, 318–323 CrossRef CAS PubMed.
- K. Zarschler, M. Kubeil and H. Stephan, RSC Adv., 2014, 4, 10157–10164 RSC.
- M. B. Hansen, T. H. M. van Gurp, J. C. M. van Hest and D. W. P. M. Löwik, Org. Lett., 2012, 14, 2330–2333 CrossRef CAS PubMed.
- H. Kessler, M. Schudok and A. Haupt, in Peptides 1988 (Proc. 20th Europ. Pept. Symp., Sept. 4–9, 1988, Tübingen), ed. G. Jung and E. Bayer, Walter de Gruyter, Berlin, New York, 1989, 664–666 Search PubMed.
- H. J. Wester and H. Kessler, J. Nucl. Med., 2005, 46, 1940–1945 CAS.
- G. Thumshirn, U. Hersel, S. L. Goodman and H. Kessler, Chem. – Eur. J., 2003, 9, 2717–2725 CrossRef CAS PubMed.
- C. Wängler, S. Maschauer, O. Prante, M. Schäfer, R. Schirrmacher, P. Bartenstein, M. Eisenhut and B. Wängler, ChemBioChem, 2010, 11, 1–15 CrossRef PubMed.
- B. C. Lee, B. S. Moon, J. S. Kim, J. H. J. H. S. Park, J. A. Katzenellenbogen and S. E. Kim, RSC Adv., 2013, 3, 782–792 RSC.
- I. Dijkgraaf, C. B. Yim, G. M. Franssen, R. C. Schuit, G. Luurtsem, S. Liu, W. J. G. Oyen and O. C. Boerman, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 128–137 CrossRef CAS PubMed.
- T. Poethko, M. Schottelius, G. Thumshirn, U. Hersel, M. Herz, G. Henriksen, H. Kessler, M. Schwaiger and H. J. Wester, J. Nucl. Med., 2004, 45, 892–902 CAS.
- C. Bernhard, M. Moreau, D. Lhenry, C. Goze, F. Boschetti, Y. Rousselin, F. Brunotte and F. Denat, Chem. – Eur. J., 2012, 18, 7834–7841 CrossRef CAS PubMed.
- A. Al-Ibraheem, R. A. Bundschuh, J. Notni, A. Buck, A. Winter, H. J. Wester, M. Schwaiger and K. Scheidhauer, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 2005–2013 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt00576k |
| This journal is © The Royal Society of Chemistry 2015 |