
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biologically active [Pd2L4]4+ quadruply-stranded helicates: stability and cytotoxicity†
Samantha M.
McNeill
a,
Dan
Preston
b,
James E. M.
Lewis
b,
Anja
Robert
a,
Katrin
Knerr-Rupp
a,
Danyon O.
Graham
a,
James R.
Wright
b,
Gregory I.
Giles
*a and
James D.
Crowley
*b
aDepartment of Pharmacology and Toxicology, University of Otago, P.O. Box 913, Dunedin, New Zealand. E-mail: gregory.giles@otago.ac.nz
bDepartment of Chemistry, University of Otago, PO Box 56, Dunedin, New Zealand. E-mail: jcrowley@chemistry.otago.ac.nz; Fax: +64 3 479 7906; Tel: +64 3 479 7731
First published on 14th May 2015
Abstract
There is emerging interest in the anti-proliferative effects of metallosupramolecular systems due to the different size and shape of these metallo-architectures compared to traditional small molecule drugs. Palladium(II)-containing systems are the most abundant class of metallosupramolecular complexes, yet their biological activity has hardly been examined. Here a small series of [Pd2(L)4](BF4)4 quadruply-stranded, dipalladium(II) architectures were screened for their cytotoxic effects against three cancer cell lines and one non-malignant line. The helicates exhibited a range of cytotoxic properties, with the most cytotoxic complex [Pd2(hextrz)4](BF4)4 possessing low micromolar IC50 values against all of the cell lines tested, while the other helicates displayed moderate or no cytotoxicity. Against the MDA-MB-231 cell line, which is resistant to platinum-based drugs, [Pd2(hextrz)4](BF4)4 was 7-fold more active than cisplatin. Preliminary mechanistic studies indicate that the [Pd2(hextrz)4](BF4)4 helicate does not induce cell death in the same way as clinically used metal complexes such as cisplatin. Rather than interacting with DNA, the helicate appears to disrupt the cell membrane. These studies represent the first biological characterisation of quadruply-stranded helicate architectures, and provide insight into the design requirements for the development of biologically active and stable palladium(II)-containing metallosupramolecular architectures.
Introduction
Spurred on by the success of small molecular inorganic drugs1 such as cisplatin2 (cis-[Pt(NH3)2Cl2], CDDP)3 there has been an upsurge of interest in the biological properties4 of metallosupramolecular architectures.5 This is driven by the hope that the different size and shape of these metallosupramolecular systems, compared to small molecule drugs, will lead to cytotoxic agents with novel biological mechanisms of action. The biological properties of a range of self-assembled architectures including metallo-macrocycles,6 cages/prisms7 and helicates have been examined. It is the metallo-helicates,8 however, that have received particular attention. Building on early work from Lehn and co-workers,9 which had shown that cationic double-helical copper(I) complexes interact strongly with DNA, Hannon and co-workers carried out a series of pioneering studies on the biological properties of the triply-stranded [Fe2L3]4+ (L = N,N′-(methanediyldibenzene-4,1-diyl)bis[1-(pyridin-2-yl)methanimine]) helicate.10 This tetracationic cylinder interacts strongly with duplex DNA, binding in the major groove,11 but interestingly it can display alternative binding modes including non-covalent complexation at the center of three-way (Y-shaped) DNA12 and RNA13 junctions. The binding of [Fe2L3]4+ to DNA has also been shown to induce coiling of the nucleic acid structure.14 Additionally, Hannon and co-workers found that these tetracationic iron(II) cylinders and related ruthenium(II) systems display both anti-cancer15 and anti-bacterial16 properties. The biological properties of other related triply-stranded helicate systems have also been examined. These molecules have been shown to bind G-quadruplex DNA,17 inhibit Aβ amyloid aggregation18 and also display cytotoxic (anti-cancer19 and anti-bacterial19b) properties. While it is clear from these studies that cationic helicate architectures can strongly interact with DNA, a recent report from Scott and co-workers20 has suggested that DNA binding is not always responsible for the observed biological activity.Palladium(II) containing systems21 represent the most common class of metallosupramolecular architectures. However, despite considerable interest in small palladium(II) complexes,22 the biological properties of palladium-containing metallosupramolecular architectures have hardly been examined.6i,23
Herein, as part of our interests in metallosupramolecular architectures24 and the biological applications of metal complexes,25 we screened four quadruply-stranded, dipalladium(II) architectures for their antiproliferative effects against three different cancer cell lines, and compared this activity against non-malignant cells. It is shown that the most stable dipalladium(II) helicate displays low micromolar IC50 values against all the tested cell lines. Furthermore, mechanistic studies indicate that the helicate does not cause cell death by interacting with DNA, rather it seems to disrupt the cell membrane. To the best of our knowledge this is the first reported cytotoxic study of quadruply-stranded helicate architectures.
Results and discussion
Synthesis and stability
The ligands (tripy,27bntrz24g and hextrz24e) and complexes ([Pd2(tripy)4](BF4)4,26 [Pd2(bntrz)4](BF4)4,24h [Pd2(hextrz)4](BF4)4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24e) were generated using previously reported procedures (Fig. 1 and ESI†). The new bistriazole ligand (pegtrz) and its corresponding dipalladium helicate [Pd2(pegtrz)4](BF4)4 were synthesised using the conditions24e previously exploited to generate the hextrz ligand and helicate (Fig. 1 and ESI†).
24e) were generated using previously reported procedures (Fig. 1 and ESI†). The new bistriazole ligand (pegtrz) and its corresponding dipalladium helicate [Pd2(pegtrz)4](BF4)4 were synthesised using the conditions24e previously exploited to generate the hextrz ligand and helicate (Fig. 1 and ESI†).
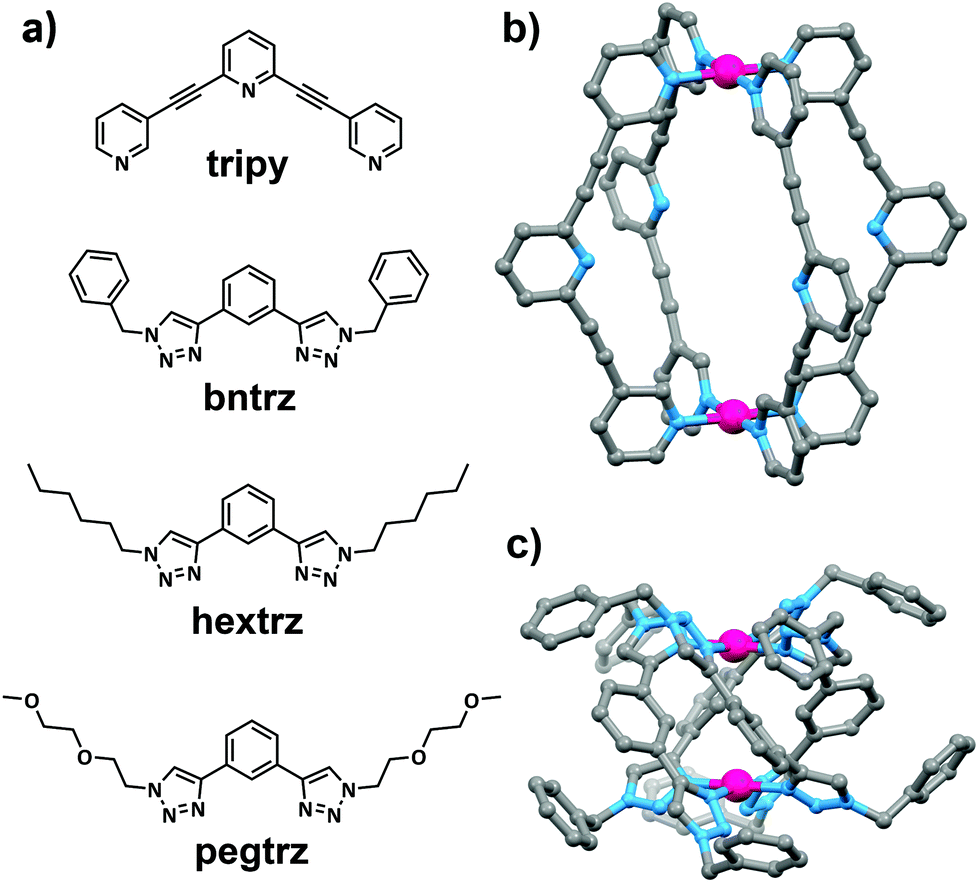 | ||
| Fig. 1 (a) Chemical structures of the pyridine- (tripy) and triazole-(bntrz, hextrz and pegtrz) based ligands; the molecular structures of (b) [Pd2(tripy)4](BF4)426 and (c) [Pd2(bntrz)4](BF4)424h architectures. Anions, solvent molecules and hydrogen atoms have been omitted for clarity. | ||
Before carrying out cytotoxicity measurements the stability of the [Pd2(L)4](BF4)4 architectures in the presence of common biological nucleophiles (chloride (Cl−), histidine and cysteine) was determined using 1H NMR competition experiments. Solutions (d6-DMSO/D2O, 3:2, 2.5 mM) of the [Pd2(L)4](BF4)4 architectures were treated with 8 equivalents of one of the nucleophiles and the course of the reaction monitored, using time-course 1H NMR spectroscopy, for a period of 20 hours (Table 1 and ESI†). Under these conditions the [Pd2(tripy)4](BF4)4 cage rapidly decomposed. The half-life for the decomposition of the palladium(II)-tripyridyl complex with histidine was 5 minutes. The half-lives for the corresponding reactions with the chloride (Cl−) and cysteine nucleophiles were less than one minute. This rapid decomposition behaviour is consistent with previous experiments using chloride and 4-dimethylaminopyridine in acetonitrile (CH3CN) solutions.26
:2 d6-DMSO/D2O, 298 K, 500 MHz)
| [Pd2(L)4](BF4)4 architecture | Nucleophile | 13C δa | ||
|---|---|---|---|---|
| Chloride | Histidine | Cysteine | ||
| a Chemical shift of Pd(II) probe complex carbene carbon (ESI). | ||||
| [Pd2(tripy)4](BF4)4 | <1 min | 5 min | <1 min | 158.7 |
| [Pd2(bntrz)4](BF4)4 | >20 h | 12 h | 30 min | 161.3 |
| [Pd2(hextrz)4](BF4)4 | >20 h | >20 h | >20 h | 161.4 |
| [Pd2(pegtrz)4](BF4)4 | >20 h | 12 h | 20 min | 161.4 |
Compared to the [Pd2(tripy)4](BF4)4 system the triazole-based helicates [Pd2(bntrz)4](BF4)4, [Pd2(hextrz)4](BF4)4 and [Pd2(pegtrz)4](BF4)4 were all considerably more stable. All of the triazole-based helicates displayed good stability against chloride (Cl−) nucleophiles, with the half-lives for the decomposition reactions all over 20 hours. The [Pd2(bntrz)4](BF4)4 and [Pd2(pegtrz)4](BF4)4 helicates were less stable in the presence of histidine and cysteine. The half-lives for the helicate decomposition reactions with histidine were both approximately 12 hours, while a much more rapid disassembly of the [Pd2(bntrz)4](BF4)4 and [Pd2(pegtrz)4](BF4)4 helicates occurred with the sulphur-containing cysteine nucleophile (t1/2 = 30 and 20 minutes, respectively). Interestingly, the [Pd2(hextrz)4](BF4)4 helicate was long-lived in the presence of all the nucleophiles tested (t1/2 = >20 hours for all the nucleophiles). No cage decomposition was observed in the presence of chloride (Cl−), and only 13% of the [Pd2(hextrz)4](BF4)4 cage was broken down when exposed either to histidine or cysteine.
It appears that both electronic and steric factors are responsible for the observed differences in the stability of the [Pd2(L)4](BF4)4 architectures. The electron donor strength of the pyridyl and triazolyl ligands was determined using the palladium(II) probe complexes reported by Huynh and co-workers (ESI†).28 The 13C carbene carbon chemical shift of these probe complexes suggested that the triazolyl ligands are all more electron-rich and therefore “better” ligands than the pyridyl system (Table 1 and ESI†). The observed higher ligand donor strength of the triazole ligands relative to the pyridyl system undoubtedly contributes to the greater stability of the triazole based helicates relative to the pyridyl cage. Inspection of the molecular structures (Fig. 1) of the [Pd2(L)4](BF4)4 architectures indicates that steric factors also potentially help stabilize the triazole-based helicates. The [Pd2(tripy)4](BF4)4 architecture adopts a lantern shape that contains an open/accessible internal cavity. This allows the nucleophiles access to the square planar palladium(II) ions of the cage either from the “outside” face of the cage or through the central cavity of the architecture. Conversely, the helical twist of the triazole-based architectures means that there is no open central cavity in these systems. As such the nucleophiles can only attack the palladium(II) ions of the helicates from the “outside” face of the architectures. This steric effect in combination with the greater ligand donor strength of the triazole ligands leads to the higher stability of the helicates relative to the tripyridyl cage system.
The difference in the stabilities of the three triazole-based helicates is more subtle. Each system has the same helicate structure and the 13C carbene chemical shift of the probe complexes suggested that the donor strength of all the triazolyl ligands was very similar (Table 1 and ESI†). Despite these structural and electronic similarities, the stability testing showed that the [Pd2(hextrz)4](BF4)4 complex was considerably more stable than the [Pd2(bntrz)4](BF4)4 and [Pd2(pegtrz)4](BF4)4 helicates against the amino acid nucleophiles histidine and cysteine. We postulate that this enhanced stability is connected to the presence of the hydrophobic hexyl substituents in the [Pd2(hextrz)4](BF4)4 complex. Presumably these hydrophobic chains aggregate together in the polar d6-DMSO/D2O solvent mixture used for stability studies and reduce the access of the nucleophiles to the palladium(II) ions of the helicate, thereby increasing the kinetic stability of this system relative to the other helicates.
Helicate cytotoxicity
To assess biological activity the cytotoxicities (as half-maximal inhibitory concentrations (IC50)) of the individual ligands tripy, bntrz, hextrz and pegtrz, along with their respective [Pd(L)4](BF4)4 architectures, were determined against four different cell lines: A549 (lung cancer), CDDP resistant MDA-MB-231 (breast cancer),29 DU-145 (prostate cancer), and MCF10A (immortalized non-malignant breast tissue) (Table 2).| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| A549 | MDA MB231 | DU145 | MCF10A | |
| “—” signifies not determined. | ||||
| [Pd2(tripy)4](BF4)4 | 41.4 ± 3.9 | 56.7 ± 2.2 | 70.1 ± 13.8 | 71.4 ± 3.9 |
| [Pd2(bntrz)4](BF4)4 | 55.5 ± 1.2 | 18.1 ± 2.7 | 34.1 ± 0.9 | 51.9 ± 0.7 |
| [Pd2(hextrz)4](BF4)4 | 6.9 ± 0.9 | 6.0 ± 0.6 | 3.4 ± 0.4 | 8.1 ± 1.2 |
| [Pd2(pegtrz)4](BF4)4 | >100 | — | — | — |
| tripy | 95.3 ± 9.7 | >100 | >100 | >100 |
| bntrz | >100 | >100 | >100 | 88.6 ± 5.4 |
| hextrz | 28.5 ± 2.6 | 89.8 ± 10.7 | 28.5 ± 1.3 | 18.1 ± 3.1 |
| pegtrz | >100 | — | — | — |
The ligands tripy, bntrz and pegtrz and the [Pd2(pegtrz)4](BF4)4 helicate exhibited minimal cytotoxicity under the conditions studied (IC50 > 80 μM). The ligand hextrz and both [Pd2(tripy)4](BF4)4 and [Pd2(bntrz)4](BF4)4 helicates displayed increased cytotoxicity, with IC50 values ranging from 70 to 18 μM against all of the cell lines, while the [Pd2(hextrz)4](BF4)4 helicate displayed high cytotoxicity (IC50 = 3–8 μM against all cell lines studied). The [Pd(CH3CN)4](BF4)4 complex alone did not display any cytotoxicity (IC50 > 100 μM), indicating that the observed cytotoxicities were due to the helicate, rather than palladium(II) or BF4− administration.
There was no correlation between helicate cytotoxicity and cell line; for example [Pd2(tripy)4](BF4)4 was almost twice as potent against the A549 cell line compared to DU-145, while [Pd2(bntrz)4](BF4)4 was more potent against the DU-145 line than A549. None of the compounds displayed any selectivity towards cancerous phenotypes, with all of the helicates exhibiting approximately similar IC50 values against non-malignant MCF 10A cells as towards the tumour-derived cell lines. When compared, under identical conditions, to the clinically used platinum complex cisplatin (CDDP, experimentally derived IC50 of 41.2 ± 3.9 μM against the MDA-MB-231 cell line) [Pd2(tripy)4](BF4)4 was less potent, while [Pd2(bntrz)4](BF4)4 and [Pd2(hextrz)4](BF4)4 were 2.3 and 6.8 fold more potent, respectively.
As [Pd2(hextrz)4](BF4)4 displayed the lowest IC50 value (Table 2) and also the highest stability (Table 1), it was possible that an intact [Pd2(L)4](BF4)4 helicate structure was required for high cytotoxicity. A comparison of the ligand hextrz and [Pd2(hextrz)4](BF4)4 cytotoxicity data supported this hypothesis. As the palladium(II) metallocenters are coordinated by four ligands, if ligand dissociation was required for cytotoxicity then the helicate IC50 would be four-fold less than the IC50 of the individual ligands (Fig. 2). While [Pd2(hextrz)4](BF4)4 was consistently more toxic than hextrz, a four-fold ratio was only observed against A549 cells (Fig. 2). Additionally, the ratio of ligand to helicate toxicity varied substantially between cell lines, from approximately 2 for the MCF10A cells to 15 for the MDA-MB-231's (Fig. 2), demonstrating that there was no correlation between ligand and helicate cytotoxicity.
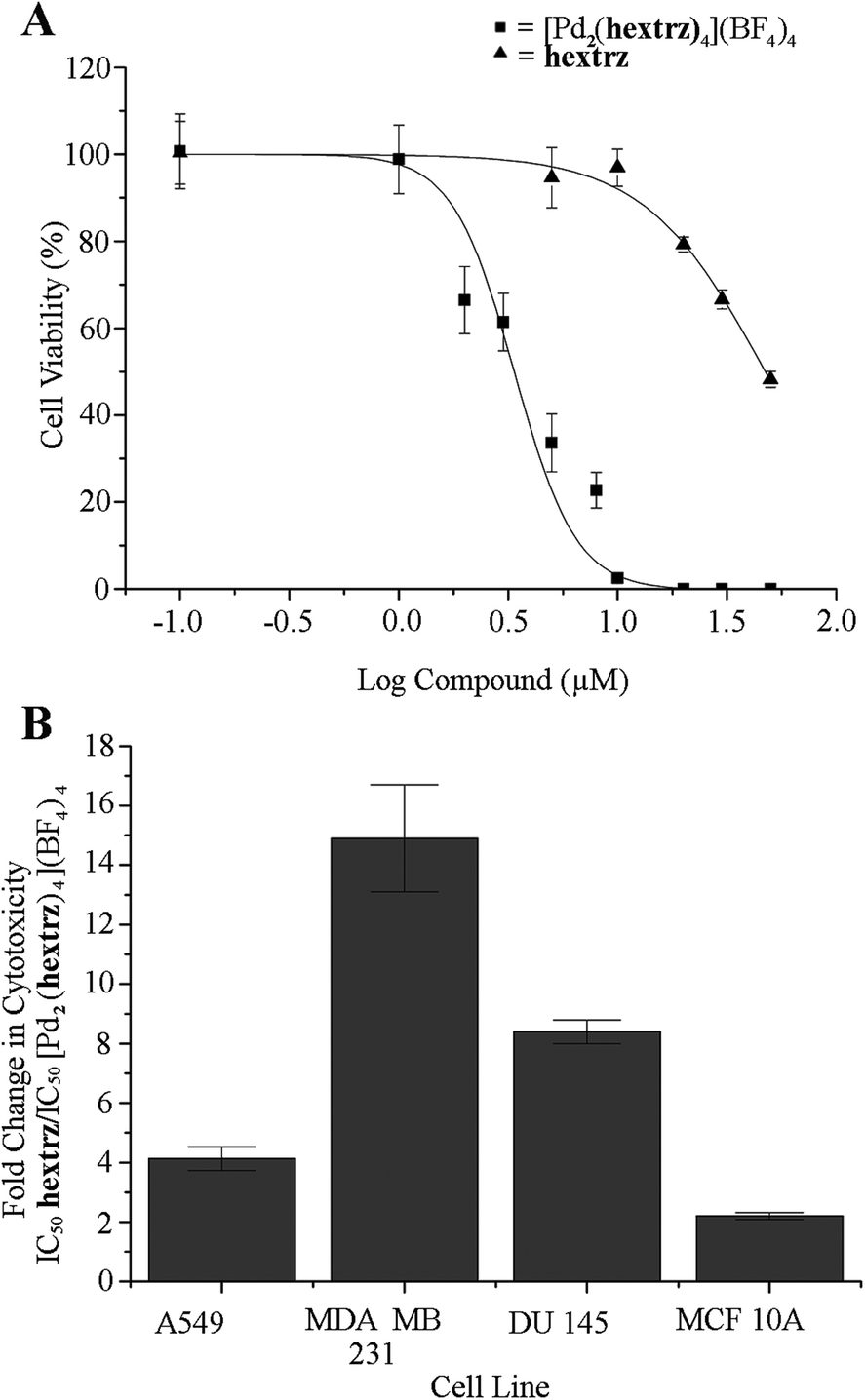 | ||
| Fig. 2 [Pd2(hextrz)4](BF4)4 cytotoxicity is not due to helicate decomposition. Cells were treated with [Pd2(hextrz)4](BF4)4 or hextrz for 24 hours before cell viability analysis. (A) Against DU-145 cells the IC50 for the ligand (28.5 μM) was 8 fold higher than for the helicate (3.4 μM); if helicate toxicity was due to rapid decomposition to release the ligand, then the stoichiometry of the complex would predict a 4-fold ratio in IC50. (B) A comparison of the IC50 ratio between ligand and helicate across the four cell lines demonstrated no correlation between helicate and ligand toxicity. Data points are expressed as means ± SEM where n = 6. | ||
Time course of helicate action – a comparison with CDDP
To further identify the mechanistic basis of the high cytotoxicity displayed by [Pd2(hextrz)4](BF4)4, a time course analysis was performed to determine the time required for the onset of cytotoxicity (Fig. 3). No further difference in cytotoxicity was observed between 2 and 48 hours, indicating that by 2 hours the administration of the helicate had resulted in the rapid onset of cell death; however, after 2 hours the helicate induced no further toxicity to the surviving cells. This cytotoxic mode of cell death is distinct from classical metal complexes such as CDDP, which display increasing cytotoxicity over time.30 CDDP toxicity is a result of DNA binding, causing errors in the cell replication process which results in the initiation of programmed cell death via apoptosis. As CDDP toxicity is dependent upon the replication status of individual cells, it selectively kills cells as they undergo cell division, resulting in a staggered cell death over several days.31 In contrast, the rapid onset of cell death with [Pd2(hextrz)4](BF4)4 provides evidence that the helicate is killing cells by a mechanism unrelated to DNA damage.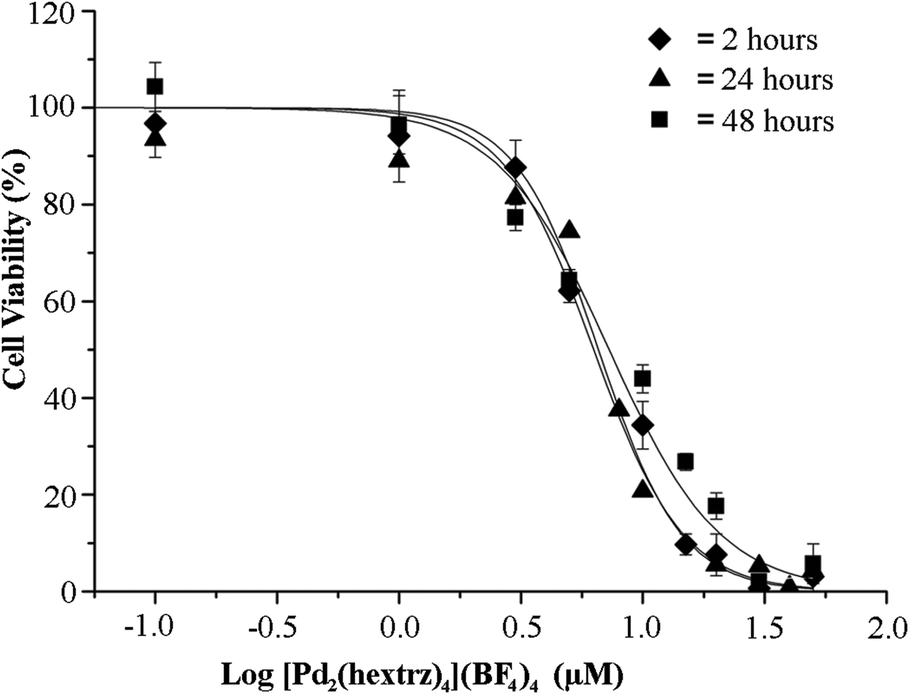 | ||
| Fig. 3 Cell death caused by [Pd2(hextrz)4](BF4)4 occurs within 2 hours post administration. A549 cells were treated with 0–50 μM [Pd2(hextrz)4](BF4)4 for 2, 24 or 48 hours, and cell viability assessed. There was no significant difference in IC50 between time points. Data are expressed as means ± SEM (n = 6). | ||
[Pd2(hextrz)4](BF4)4 treatment causes a rapid loss of cell membrane integrity
We next aimed to identify the potential biological targets of the [Pd2(hextrz)4](BF4)4 helicate. As rapid cell death is often associated with a compromised membrane integrity, we evaluated whether helicate induced cell death was attributable to membrane damage. For these studies we measured the release of the intracellular enzyme lactate dehydrogenase (LDH) into the extracellular medium. Extracellular levels of LDH are usually present at low concentrations, attributable to a small population of cells undergoing spontaneous death in culture systems.32 However, these extracellular LDH levels rapidly increase if the cell membrane is disrupted, due to the release of intracellular enzyme into the extracellular space. The assay therefore provides a convenient method for rapidly assessing membrane integrity.32Consistent with a mechanism involving membrane damage, a time and concentration dependent increase in extracellular LDH levels was observed immediately following [Pd2(hextrz)4](BF4)4 administration (Fig. 4). At a relatively high concentration of 50 μM (7 fold above the IC50), a rapid elevation in LDH activity was observed, which peaked at 10 minutes. However, at lower concentrations of 5 and 10 μM, continuous increases in LDH release were observable at 10, 30 and 60 minutes, indicating that around the IC50 concentration membrane damage was a continual process that occurred over a sustained time period (Fig. 4).
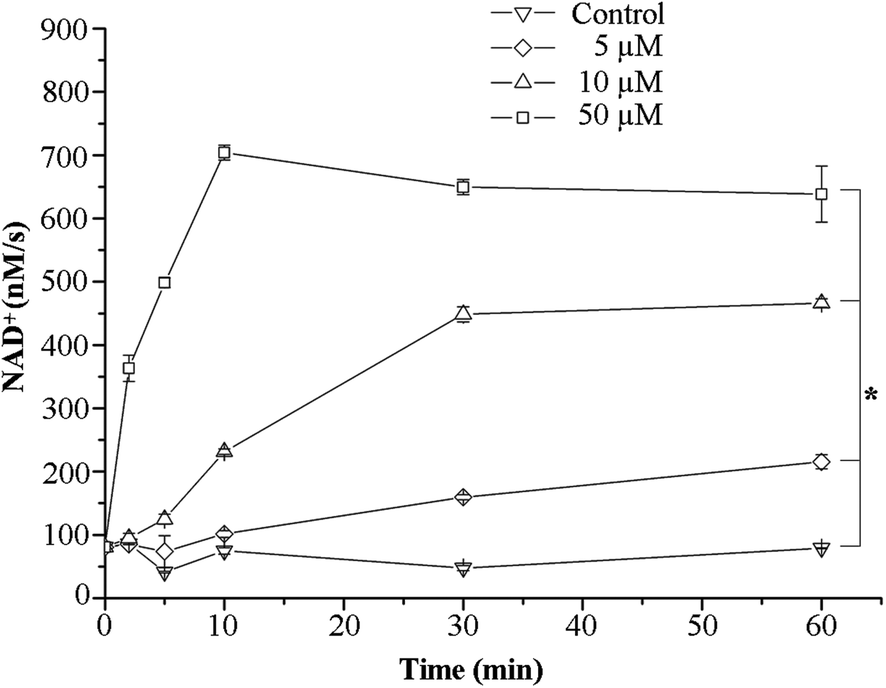 | ||
| Fig. 4 [Pd2(hextrz)4](BF4)4 causes rapid disruption of the cell membrane. A549 cells were treated with [Pd2(hextrz)4](BF4)4 and the change in extracellular LDH levels calculated by measuring enzyme activity. Data represents extracellular LDH activity at each time point and are expressed as means ± SEM (n = 3). * = p < 0.05. | ||
To investigate the extent to which LDH release was a diagnostic indicator of the viability of the entire cell population, we next conducted double labelling experiments with the dyes Hoechst 33342 and propidium iodide. Hoechst 33342 is a membrane permeable dye used for cell counting as it effectively labels all cells. In contrast, propidium iodide is a cell membrane impermeable dye, which is only taken up by non-viable cells with compromised cell membrane integrity. In combination with Hoechst 33342 it can therefore be used to distinguish between dead and viable cells within a heterogenous population.33 In A549 cells this double label analysis confirmed that, by 10 minutes, a 10 μM concentration of [Pd2(hextrz)4](BF4)4 (slightly above the 24 hour IC50 of 7 μM) was sufficient to have killed >85% of the cell population, while a 50 μM concentration induced 100% cell death (Fig. 5). The phase contrast image demonstrated that [Pd2(hextrz)4](BF4)4 induced cell death was accompanied by a rounding of the cells, a characteristic feature of cell damage that can accompany necrosis34 (Fig. 5). In contrast 5 μM [Pd2(hextrz)4](BF4)4 (slightly below the 24 hour IC50) only induced 1.5% cell death at 10 min, although rounding of the cell population was observable in the phase contrast image, indicating that cell death processes had potentially been initiated in a greater number of cells. Therefore a small switch in helicate concentration, from slightly below to slightly above the IC50 value, could result in the sudden onset of cell death. The mechanism underlying this remarkably rapid switch in activity is currently unknown. In comparison, administration of the hextrz ligand alone, even at 100 μM (4-fold higher than the 24 hour IC50), resulted in no cell death at 10 min, confirming that the helicate and its cognate ligand induced cytotoxicity via distinct molecular mechanisms (Fig. 5).
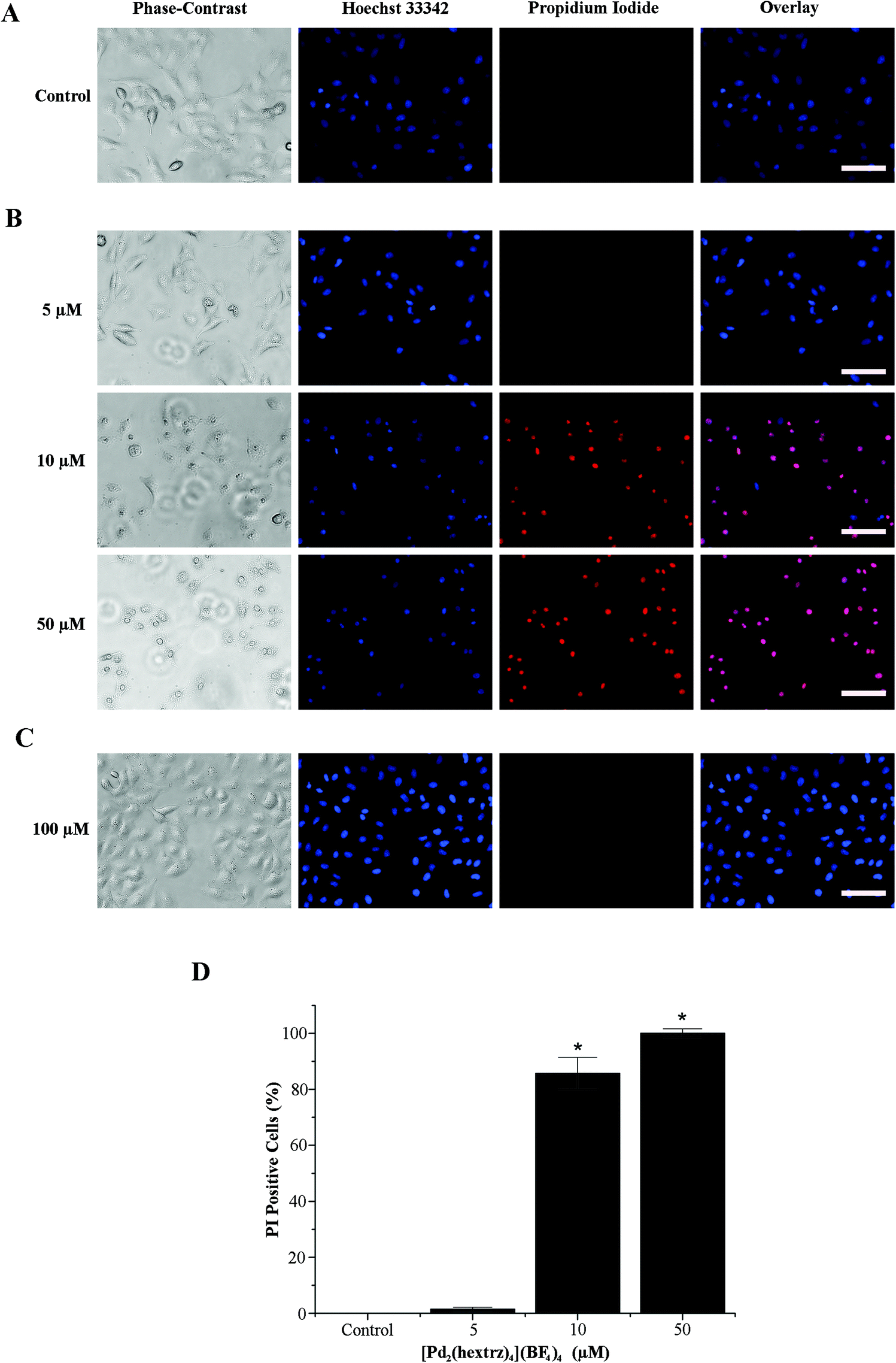 | ||
| Fig. 5 [Pd2(hextrz)4](BF4)4 induces rapid membrane damage. A549 cells were treated with helicate or ligand for 10 min and then double stained with Hoechst 33342 and propidium iodide. (A) Control cells (0.15% DMSO). (B) Cells treated with 5–50 μM of [Pd2(hextrz)4](BF4)4. (C) Cells treated with hextrz. The representative images were acquired with a 20× objective, scale bars (white) indicate 50 μm. (D) The percentage of propidium iodide (PI) positive cells were calculated from fluorescence images obtained for each treatment condition (n = 3), data is expressed as means ± SEM. * = p < 0.05 as compared to control. | ||
Conclusions
Herein the biological activities of four quadruply-stranded dipalladium architectures were studied. The palladium(II)-based helicates exhibited a range of cytotoxic properties, with the most cytotoxic complex [Pd2(hextrz)4](BF4)4 possessing low micromolar IC50 values against all of the cell lines tested, while the other helicates displayed moderate or no cytotoxicity. Stability studies indicated that the cytotoxic effect of the dipalladium architectures correlated well with the stability of these systems in the presence of biological nucleophiles, suggesting that it is intact helicate that is responsible for the biological activity.Disappointingly, the [Pd2(hextrz)4](BF4)4 helicate displayed no selectivity towards cancerous phenotypes; the compound exhibited approximately similar IC50 values against non-malignant MCF 10A cells as towards the tumour-derived cell lines. Against the MDA-MB-231 cell line, which is resistant to platinum-based drugs, [Pd2(hextrz)4](BF4)4 was 7-fold more active than cisplatin and, when compared to the IC50 values against the other cell lines, there was no evidence of resistance. This suggests that the helicate does not induce cell death in the same way as clinically used metal complexes such as cisplatin. Preliminary mechanistic investigations with the [Pd2(hextrz)4](BF4)4 helicate revealed that it induced cell death within minutes, accompanied by a loss of cellular membrane integrity. While these observations are consistent with the helicate acting as a metallo-detergent,35 there are several potential alternative mechanisms of membrane damage, including pore formation36 and changes to the fluidity of the lipid bilayer.37 With the current data the exact cause of the membrane damage is not clear. Because the cytotoxic mechanism of the helicate was loss of cellular membrane integrity, the compound exhibited no selectivity towards cancer cells compared to non-malignant cells. This lack of specificity towards cancer cells prevents the immediate application of this complex as an anti-cancer agent.
However, the current work suggests that suitably designed stable palladium(II)-containing metallosupramolecular architectures (especially quadruply-stranded helicates) can display biological activity. As such we are now examining other palladium(II)-containing architectures and quadruply-stranded helicates containing other metals ions in search of more selective anti-cancer agents.
Acknowledgements
This work was supported by an Otago Medical Research Foundation Laurenson Award (LA307). The authors thank the Department of Pharmacology and Toxicology and the Department of Chemistry, University of Otago for additional funding. DP and JEML thank the University of Otago for PhD scholarships. DP thanks Otago Medical Research Foundation for a McQueen Summer Studentship.Notes and references
- (a) K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed; (b) M. Patra, G. Gasser and N. Metzler-Nolte, Dalton Trans., 2012, 41, 6350–6358 RSC; (c) M. Patra and G. Gasser, ChemBioChem, 2012, 13, 1232–1252 CrossRef CAS PubMed; (d) C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS; (e) C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC; (f) N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- (a) E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed; (b) N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC; (c) J. Reedijk, Eur. J. Inorg. Chem., 2009, 1303–1312 CrossRef CAS.
- (a) B. Rosenberg, Platinum Met. Rev., 1971, 15, 42–51 CAS; (b) B. Rosenberg and L. VanCamp, Cancer Res., 1970, 30, 1799–1802 CAS; (c) H. C. Harder and B. Rosenberg, Int. J. Cancer, 1970, 6, 207–216 CrossRef CAS PubMed; (d) B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed; (e) B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- (a) B. Therrien, CrystEngComm, 2015, 17, 484–491 RSC; (b) B. Therrien, Chem. – Eur. J., 2013, 19, 8378–8386 CrossRef CAS PubMed; (c) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464–2474 CrossRef CAS PubMed; (d) B. Therrien, Top. Curr. Chem., 2012, 319, 35–56 CrossRef CAS PubMed; (e) M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243–2261 CrossRef CAS.
- (a) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed; (b) M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC; (c) T. Nakamura, H. Ube and M. Shionoya, Chem. Lett., 2013, 42, 328–334 CrossRef CAS; (d) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC; (e) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (f) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (g) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- (a) I. V. Grishagin, J. B. Pollock, S. Kushal, T. R. Cook, P. J. Stang and B. Z. Olenyuk, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18448–18453 CrossRef CAS PubMed; (b) D. Schilter, T. Urathamakul, J. L. Beck, M. J. Hannon and L. M. Rendina, Dalton Trans., 2010, 39, 11263–11271 RSC; (c) H. Ahmad, D. Ghosh and J. A. Thomas, Chem. Commun., 2014, 50, 3859–3861 RSC; (d) A. Dubey, J. W. Min, H. J. Koo, H. Kim, T. R. Cook, S. C. Kang, P. J. Stang and K.-W. Chi, Chem. – Eur. J., 2013, 19, 11622–11628 CrossRef CAS PubMed; (e) X.-H. Zheng, H.-Y. Chen, M.-L. Tong, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2012, 48, 7607–7609 RSC; (f) X.-H. Zheng, Y.-F. Zhong, C.-P. Tan, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2012, 41, 11807–11812 RSC; (g) A. Terenzi, C. Ducani, V. Blanco, L. Zerzankova, A. F. Westendorf, C. Peinador, J. M. Quintela, P. J. Bednarski, G. Barone and M. J. Hannon, Chem. – Eur. J., 2012, 18, 10983–10990 CrossRef CAS PubMed; (h) A. Mishra, H. Jung, J. W. Park, H. K. Kim, H. Kim, P. J. Stang and K.-W. Chi, Organometallics, 2012, 31, 3519–3526 CrossRef CAS PubMed; (i) A. Mishra, S. Ravikumar, S. H. Hong, H. Kim, V. Vajpayee, H. W. Lee, B. C. Ahn, M. Wang, P. J. Stang and K.-W. Chi, Organometallics, 2011, 30, 6343–6346 CrossRef CAS PubMed; (j) N. P. E. Barry, F. Edafe and B. Therrien, Dalton Trans., 2011, 40, 7172–7180 RSC; (k) V. Vajpayee, Y. H. Song, Y. J. Yang, S. C. Kang, T. R. Cook, D. W. Kim, M. S. Lah, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Organometallics, 2011, 30, 6482–6489 CrossRef CAS PubMed; (l) F. Linares, M. A. Galindo, S. Galli, M. A. Romero, J. A. R. Navarro and E. Barea, Inorg. Chem., 2009, 48, 7413–7420 CrossRef CAS PubMed; (m) M. A. Galindo, M. Angustias Romero and J. A. R. Navarro, Inorg. Chim. Acta, 2009, 362, 1027–1030 CrossRef CAS; (n) R. Kieltyka, P. Englebienne, J. Fakhoury, C. Autexier, N. Moitessier and H. F. Sleiman, J. Am. Chem. Soc., 2008, 130, 10040–10041 CrossRef CAS PubMed; (o) M. Mounir, J. Lorenzo, M. Ferrer, M. J. Prieto, O. Rossell, F. X. Avilès and V. Moreno, J. Inorg. Biochem., 2007, 101, 660–666 CrossRef CAS PubMed; (p) D. K. Orsa, G. K. Haynes, S. K. Pramanik, M. O. Iwunze, G. E. Greco, J. A. Krause, D. M. Ho, A. L. Williams, D. A. Hill and S. K. Mandal, Inorg. Chem. Commun., 2007, 10, 821–824 CrossRef CAS; (q) P. J. Barnard, L. E. Wedlock, M. V. Baker, S. J. Berners-Price, D. A. Joyce, B. W. Skelton and J. H. Steer, Angew. Chem., Int. Ed., 2006, 45, 5966–5970 CrossRef CAS PubMed.
- (a) L. E. H. Paul, B. Therrien and J. Furrer, J. Biol. Inorg. Chem., 2015, 20, 49–59 CrossRef CAS PubMed; (b) L. E. H. Paul, B. Therrien and J. Furrer, Org. Biomol. Chem., 2015, 13, 946–953 RSC; (c) W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625–631 RSC; (d) P. R. Symmers, M. J. Burke, D. P. August, P. I. T. Thomson, G. S. Nichol, M. R. Warren, C. J. Campbell and P. J. Lusby, Chem. Sci., 2015, 6, 756–760 RSC; (e) Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189–1193 RSC; (f) L. E. H. Paul, J. Furrer and B. Therrien, J. Organomet. Chem., 2013, 734, 45–52 CrossRef CAS; (g) J. Freudenreich, C. Dalvit, G. Süss-Fink and B. Therrien, Organometallics, 2013, 32, 3018–3033 CrossRef CAS; (h) J. W. Yi, N. P. E. Barry, M. A. Furrer, O. Zava, P. J. Dyson, B. Therrien and B. H. Kim, Bioconjugate Chem., 2012, 23, 461–471 CrossRef CAS PubMed; (i) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed; (j) F. Schmitt, N. P. E. Barry, L. Juillerat-Jeanneret and B. Therrien, Bioorg. Med. Chem. Lett., 2012, 22, 178–180 CrossRef CAS PubMed; (k) M. A. Furrer, F. Schmitt, M. Wiederkehr, L. Juillerat-Jeanneret and B. Therrien, Dalton Trans., 2012, 41, 7201–7211 RSC; (l) N. P. E. Barry, O. Zava, W. Wu, J. Zhao and B. Therrien, Inorg. Chem. Commun., 2012, 18, 25–28 CrossRef CAS; (m) N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Chem. – Eur. J., 2011, 17, 9669–9677 CrossRef CAS PubMed; (n) O. Zava, J. Mattsson, B. Therrien and P. J. Dyson, Chem. – Eur. J., 2010, 16, 1428–1431 CrossRef CAS PubMed; (o) B. Therrien, G. Suess-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed; (p) V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Chem. Commun., 2011, 47, 5184–5186 RSC; (q) V. Vajpayee, S. m. Lee, J. W. Park, A. Dubey, H. Kim, T. R. Cook, P. J. Stang and K.-W. Chi, Organometallics, 2013, 32, 1563–1566 CrossRef CAS PubMed.
- R. A. Kaner and P. Scott, Future Med. Chem., 2015, 7, 1–4 CrossRef CAS PubMed.
- B. Schoentjes and J.-M. Lehn, Helv. Chim. Acta, 1995, 78, 1–12 CrossRef CAS.
- M. J. Hannon, C. L. Painting, A. Jackson, J. Hamblin and W. Errington, Chem. Commun., 1997, 1807–1808 RSC.
- E. Moldrheim, M. Hannon, I. Meistermann, A. Rodger and E. Sletten, J. Biol. Inorg. Chem., 2002, 7, 770–780 CrossRef CAS PubMed.
- (a) D. R. Boer, J. M. C. A. Kerckhoffs, Y. Parajo, M. Pascu, I. Uson, P. Lincoln, M. J. Hannon and M. Coll, Angew. Chem., Int. Ed., 2010, 49, 2336–2339 CrossRef CAS PubMed; (b) J. Malina, M. J. Hannon and V. Brabec, Chem. – Eur. J., 2007, 13, 3871–3877 CrossRef CAS PubMed; (c) L. Cerasino, M. J. Hannon and E. Sletten, Inorg. Chem., 2007, 46, 6245–6251 CrossRef PubMed; (d) A. Oleksi, A. G. Blanco, R. Boer, I. Usón, J. Aymamí, A. Rodger, M. J. Hannon and M. Coll, Angew. Chem., Int. Ed., 2006, 45, 1227–1231 CrossRef CAS PubMed.
- S. Phongtongpasuk, S. Paulus, J. Schnabl, R. K. O. Sigel, B. Spingler, M. J. Hannon and E. Freisinger, Angew. Chem., Int. Ed., 2013, 52, 11513–11516 CrossRef CAS PubMed.
- (a) I. Meistermann, V. Moreno, M. J. Prieto, E. Moldrheim, E. Sletten, S. Khalid, P. M. Rodger, J. C. Peberdy, C. J. Isaac, A. Rodger and M. J. Hannon, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5069–5074 CrossRef CAS PubMed; (b) M. J. Hannon, V. Moreno, M. J. Prieto, E. Moldrheim, E. Sletten, I. Meistermann, C. J. Isaac, K. J. Sanders and A. Rodger, Angew. Chem., Int. Ed., 2001, 40, 880–884 CAS.
- (a) A. C. G. Hotze, N. J. Hodges, R. E. Hayden, C. Sanchez-Cano, C. Paines, N. Male, M.-K. Tse, C. M. Bunce, J. K. Chipman and M. J. Hannon, Chem. Biol., 2008, 15, 1258–1267 CrossRef CAS PubMed; (b) G. I. Pascu, A. C. G. Hotze, C. Sanchez-Cano, B. M. Kariuki and M. J. Hannon, Angew. Chem., Int. Ed., 2007, 46, 4374–4378 CrossRef CAS PubMed; (c) A. C. G. Holtze, B. M. Kariuki and M. J. Hannon, Angew. Chem., Int. Ed., 2006, 45, 4839–4842 CrossRef CAS PubMed.
- A. D. Richards, A. Rodger, M. J. Hannon and A. Bolhuis, Int. J. Antimicrob. Agents, 2009, 33, 469–472 CrossRef CAS PubMed.
- (a) J. Wang, Y. Chen, J. Ren, C. Zhao and X. Qu, Nucleic Acids Res., 2014, 42, 3792–3802 CrossRef CAS PubMed; (b) C. Zhao, J. Geng, L. Feng, J. Ren and X. Qu, Chem. – Eur. J., 2011, 17, 8209–8215 CrossRef CAS PubMed; (c) H. Yu, X. Wang, M. Fu, J. Ren and X. Qu, Nucleic Acids Res., 2008, 36, 5695–5703 CrossRef CAS PubMed.
- (a) M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS PubMed; (b) H. Yu, M. Li, G. Liu, J. Geng, J. Wang, J. Ren, C. Zhao and X. Qu, Chem. Sci., 2012, 3, 3145–3153 RSC.
- (a) A. D. Faulkner, R. A. Kaner, Q. M. A. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797–803 CrossRef CAS PubMed; (b) S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2012, 4, 31–36 CrossRef CAS PubMed.
- V. Brabec, S. E. Howson, R. A. Kaner, R. M. Lord, J. Malina, R. M. Phillips, Q. M. A. Abdallah, P. C. McGowan, A. Rodger and P. Scott, Chem. Sci., 2013, 4, 4407–4416 RSC.
- (a) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (b) N. B. Debata, D. Tripathy and D. K. Chand, Coord. Chem. Rev., 2012, 256, 1831–1945 CrossRef CAS; (c) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 371–380 CrossRef PubMed.
- (a) A. R. Kapdi and I. J. S. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751–4777 RSC; (b) A. Garoufis, S. K. Hadjikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 1384–1397 CrossRef CAS.
- A. Mishra, S. Chang Lee, N. Kaushik, T. R. Cook, E. H. Choi, N. Kumar Kaushik, P. J. Stang and K.-W. Chi, Chem. – Eur. J., 2014, 20, 14410–14420 CrossRef CAS PubMed.
- (a) J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833–1843 RSC; (b) J. E. M. Lewis and J. D. Crowley, Supramol. Chem., 2014, 26, 173–181 CrossRef CAS; (c) J. E. M. Lewis, C. J. McAdam, M. G. Gardiner and J. D. Crowley, Chem. Commun., 2013, 49, 3398–3400 RSC; (d) J. E. M. Lewis and J. D. Crowley, Aust. J. Chem., 2013, 66, 1447–1454 CrossRef CAS; (e) S. Ø. Scott, E. L. Gavey, S. J. Lind, K. C. Gordon and J. D. Crowley, Dalton Trans., 2011, 40, 12117–12124 RSC; (f) K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328–330 RSC; (g) M. L. Gower and J. D. Crowley, Dalton Trans., 2010, 39, 2371–2378 RSC; (h) J. D. Crowley and E. L. Gavey, Dalton Trans., 2010, 39, 4035–4037 RSC; (i) J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612–623 RSC; (j) J. D. Crowley, I. M. Steele and B. Bosnich, Eur. J. Inorg. Chem., 2005, 3907–3917 CrossRef CAS; (k) J. D. Crowley and B. Bosnich, Eur. J. Inorg. Chem., 2005, 2015–2025 CrossRef CAS; (l) J. D. Crowley, A. J. Goshe and B. Bosnich, Chem. Commun., 2003, 2824–2825 RSC.
- (a) S. V. Kumar, W. K. C. Lo, H. J. L. Brooks and J. D. Crowley, Inorg. Chim. Acta, 2015, 425, 1–6 CrossRef CAS; (b) A. Noor, G. S. Huff, S. V. Kumar, J. E. M. Lewis, B. M. Paterson, C. Schieber, P. S. Donnelly, H. J. L. Brooks, K. C. Gordon, S. C. Moratti and J. D. Crowley, Organometallics, 2014, 33, 7031–7043 CrossRef CAS; (c) S. K. Vellas, J. E. M. Lewis, M. Shankar, A. Sagatova, J. D. A. Tyndall, B. C. Monk, C. M. Fitchett, L. R. Hanton and J. D. Crowley, Molecules, 2013, 18, 6383–6407 CrossRef CAS PubMed.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- K. J. Kilpin, M. L. Gower, S. G. Telfer, G. B. Jameson and J. D. Crowley, Inorg. Chem., 2011, 50, 1123–1134 CrossRef CAS PubMed.
- (a) H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS; (b) D. Yuan and H. V. Huynh, Organometallics, 2012, 31, 405–412 CrossRef CAS; (c) Q. Teng and H. V. Huynh, Inorg. Chem., 2014, 53, 10964–10973 CrossRef CAS PubMed; (d) J. R. Wright, P. C. Young, N. T. Lucas, A.-L. Lee and J. D. Crowley, Organometallics, 2013, 32, 7065–7076 CrossRef CAS PubMed.
- B. D. Lehmann, J. A. Bauer, X. Chen, M. E. Sanders, A. B. Chakravarthy, Y. Shyr and J. A. Pietenpol, J. Clin. Invest., 2011, 121, 2750–2767 CAS.
- H. Alborzinia, S. Can, P. Holenya, C. Scholl, E. Lederer, I. Kitanovic and S. Wölfl, PLoS One, 2011, 6, e19714 CAS.
- L. M. Levasseur, H. K. Slocum, Y. M. Rustum and W. R. Greco, Cancer Res., 1998, 58, 5749–5761 CAS.
- H. T. Wolterbeek and A. J. G. M. van der Meer, Assay Drug Dev. Technol., 2005, 3, 675–682 CrossRef CAS PubMed.
- N. Atale, S. Gupta, U. C. S. Yadav and V. Rani, J. Microsc., 2014, 255, 7–19 CrossRef CAS PubMed.
- T. V. Berghe, N. Vanlangenakker, E. Parthoens, W. Deckers, M. Devos, N. Festjens, C. J. Guerin, U. T. Brunk, W. Declercq and P. Vandenabeele, Cell Death Differ., 2010, 17, 922–930 CrossRef PubMed.
- D. Lichtenberg, H. Ahyayauch and F. M. Goñi, Biophys. J., 2013, 105, 289–299 CrossRef CAS PubMed.
- M. R. Popoff, FEBS J., 2011, 278, 4602–4615 CrossRef CAS PubMed.
- T. Váradi, J. Roszik, D. Lisboa, G. Vereb, J. M. Molina-Guijarro, C. M. Galmarini, J. Szöllősi and P. Nagy, Eur. J. Pharmacol., 2011, 667, 91–99 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI contains the experimental procedures, 1H, 13C and DOSY NMR, ESMS, and crystallographic data. CCDC 1045884–1045889. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01259g |
| This journal is © The Royal Society of Chemistry 2015 |