
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
5-(Fluorodinitromethyl)-2H-tetrazole and its tetrazolates – Preparation and Characterization of New High Energy Compounds†
Ralf
Haiges
* and
Karl O.
Christe
Loker Hydrocarbon Research Institute, University of Southern California, Los Angeles, CA 90089-1661, USA. E-mail: haiges@usc.edu; Tel: +1-213-7403197
First published on 18th March 2015
Abstract
5-(Fluorodinitromethyl)-2H-tetrazole (HFDNTz) has been prepared by the cycloaddition reaction of HN3 with F(NO2)2CCN, which in turn was prepared by aqueous fluorination of sodium dinitrocyanomethanide. HFDNTz was converted into the ammonium, silver and tetraphenylphosphonium 5-(fluorodinitromethyl)tetrazolates. While the reaction of trinitroacetonitrile with HBr, followed by the treatment with NaOH, resulted in the formation of sodium dinitrocyanomethanide, the reaction of trinitroacetonitrile with aqueous ammonia produced ammonium dinitrocyanomethanide. Hydrazinium dinitromethanide was obtained from trinitroacetonitrile and hydrazine hydrate. All compounds were fully characterized by multinuclear NMR spectroscopy, IR spectroscopy and X-ray crystal structure determinations. Initial safety testing (impact and friction sensitivity) and thermal stability measurements (DTA) were also carried out.
Introduction
Tetrazoles are a fascinating class of compounds. The parent compound 1H-tetrazole has a nitrogen content of 80%, resulting in a relatively high heat of formation of about 330 kJ mol−1.1–3 At the same time, tetrazoles can exhibit astonishing high thermal stabilities which results in a wide array of application in agriculture, medicine and biology.4 For energetic materials applications, tetrazole is usually further functionalized with explosophore groups such as nitro, N-nitro, azo, or azido.5–15In recent years, much effort in energetic materials research was dedicated to the synthesis of novel energetic highly over-oxidized compounds that can be used to replace ammonium perchlorate as high-oxygen carrier in solid rocket propellant formulations. An expression that is often being used in order to indicate the degree of oxidation of a compound is the oxygen balance (OB). In its original form, the OB was defined as percentage of oxygen required for complete conversion of a molecule to carbon dioxide, water and metal oxide.16 Because if the usually high combustion temperatures in rocket engines, it is more useful to calculate the OB of a rocket propellant based on combustion to carbon monoxide instead of carbon monoxide. While energetic, tetrazoles are usually notoriously under-oxidized. 5-Nitrotetrazolate12 has an OB of 14.0% for combustion to CO but the recently investigated 5-(trinitromethyl)tetrazolate5 has an OB of 29.4%. The general disadvantage of most 5-(trinitromethyl)tetrazolates are their high impact (IS) and friction sensitivities (FS) (e.g. IS = 0.5 J and FS < 1 N for the rubidium and cesium salts).5
The instability of the trinitromethyl group toward catastrophic decomposition is well established. It has been found that the fluorodinitromethyl group –CF(NO2)2 is generally more stable than the trinitromethyl group –C(NO2)3 without incurring too much of a performance penalty. The 5-(fluorodinitromethyl)tetrazolate anion has an OB of 20.9% for combustion to N2, CO and CF4. Fluorine-containing compounds such as Teflon or Viton are common ingredients of metallized formulations for flares and pyrotechnics (e.g. MTV: Mg/Teflon/Viton). The highly exothermic formation of metal fluorides during combustion increases the flame temperature which in turn improves the performance of the formulation.
The synthesis of 5-(fluorodinitromethyl)-2H-tetrazole (HFDNTz) by reaction of fluorodinitroacetonitrile with sodium azide or trimethylsilyl azide has been reported but the obtained tetrazole was not isolated and directly converted into the sodium or ammonium salt. The tetrazole and the salts were identified only by IR and 19F NMR spectra and have not been well characterized.17,18
In this manuscript we report the synthesis and full characterization of 5-(fluorodinitromethyl)-2H-tetrazole and three salts with the energetic 5-(fluorodinitromethyl)tetrazolate anion.
Experimental part
Caution! The compounds of this work are energetic materials that might explode under certain conditions (e.g. elevated temperatures, impact, friction or electric discharge). Appropriate safety precautions,19 such as the use of shields or barricades in a fume hood and personal protection equipment (safety glasses, face shields, ear plugs, as well as gloves and suits made from leather and/or Kevlar) should be taken all the time when handling these materials. Ignoring safety precautions may lead to serious injuries!Materials and apparatus
All chemicals and solvents were obtained from Sigma-Aldrich or Alfa-Aesar and were used as supplied. NMR spectra were recorded at 298 K on Bruker AMX500 or Varian VNMRS-600s spectrometers using (CD3)2CO or D2O solutions in standard 5 mm glass tubes. Chemical shifts are given relative to neat tetramethylsilane (1H, 13C) or neat CH3NO2 (14N, 15N). Raman spectra were recorded at ambient temperatures in Pyrex glass tubes in the range of 4000–80 cm−1 on a Bruker Equinox 55 FT-RA spectrometer using a Nd-YAG laser at 1064 nm or a Cary 83 spectrometer using an Ar laser at 488 nm. Infrared spectra were recorded in the range 4000–400 cm−1 on Midac M Series or Bruker Optics Alpha ATR FT-IR spectrometers. Solid samples were recorded as KBr pellets or with an ATR attachment. The KBr pellets were prepared using an Econo mini-press (Barnes Engineering Co.). Gaseous samples were kept in a 5 cm Pyrex glass cell that was equipped with AgCl windows. Differential thermal analysis (DTA) curves were recorded using a purge of dry nitrogen gas and a heating rate of 5 °C min−1 on an OZM Research DTA552-Ex instrument with the Meavy 2.2.0 software. The sample size was 3–15 mg. The impact and friction sensitivity data were determined on an OZM Research BAM Fall Hammer BFH-10 and an OZM Research BAM Friction apparatus FSKM-10, respectively, through five individual measurements that were averaged. Both instruments were calibrated using RDX. The samples were finely powdered materials that were not sifted.X-ray crystal structure determination
The single crystal X-ray diffraction data were collected on a Bruker SMART APEX DUO diffractometer, equipped with an APEX II CCD detector, using Mo Kα radiation (TRIUMPH curved-crystal monochromator) from a fine-focus tube. The frames were integrated using the SAINT algorithm20 to give the hkl files corrected for Lp/decay. The absorption correction was performed using the SADABS program.21 The structures were solved and refined on F2 using the Bruker SHELXTL Software Package.22–25 Non-hydrogen atoms were refined anisotropically. Unless noted otherwise, the positions of hydrogen atoms attached to heteroatoms have been located from the difference electron density map. ORTEP drawings were prepared using the ORTEP-III for Windows V2.02 program.26Synthesis of trinitroacetonitrile
Under an atmosphere of nitrogen, a mixture of cyanoacetamide (16.8 g, 0.2 mol) and dichloromethane (50 mL) was cooled to 0 °C. While stirring vigorously, 100% nitric acid (32 mL) was added through an addition funnel within a time period of 2 minutes. The cyanoacetamide dissolved and a yellow two-layer mixture was obtained. While maintaining a temperature of 0 °C, oleum (20% SO3) (38 mL) was slowly added drop wise and the mixture then stirred vigorously at ambient temperature for an additional 8–10 hours until all gas-evolution stopped. The pale yellow two-phase mixture was transferred into a separatory funnel and the upper organic layer removed. The lower acid layer was extracted three times with dichloromethane (30 mL each). The combined organic layers were extracted twice with cold (−5 °C) concentrated sulphuric acid (15 mL each) and the obtained colourless solution kept over magnesium sulfate.Neat trinitroacetonitrile was obtained as a colourless solid by removing the solvent from a cold dichloromethane solution (−40 °C) on a vacuum line.
NMR (CDCl3) δ(ppm): 13C (100.54 MHz) 103.4 (s, CN), 112.1 (sept, 1J(13C14N) = 9.5 Hz, C(NO2)3); 14N (36.14 MHz) −45.4 (s, ν½ = 20 Hz, NO2), −272 (s, ν½ = 350 Hz, CN); Raman (25 °C, 50 mW): 2265 (7.8), 1628 (1.9), 1623 (2.1), 1350 (1.4), 1344 (1.4), 1276 (3.1), 1164 (1.5), 943 (4.6), 856 (6.8), 801 (0.8), 657 (0.9), 477 (2.2), 444 (2.5), 380 (6.6), 360 (10.0), 200 (3.8), 151 (6.5), 110 (7.6) cm−1.
Synthesis of sodium dinitrocyanomethanide27
The obtained solution of (NO2)3CCN in CH2Cl2 (100 mL) was added drop wise to a stirred solution of methanol (50 mL) and 48% aqueous HBr (100 mL). The mixture was stirred at ambient temperature for about 4 hours during which the solution gradually turned from colourless to reddish brown and evolved brown fumes of bromine. The pH of the mixture was then adjusted to pH 10 with 3 M NaOH in methanol. The now milky, yellow mixture was taken to dryness on a rotary evaporator. The yellow solid residue was extracted five times with ethyl acetate (70 mL each). The combined organic phases were carefully evaporated on a rotary evaporator without heating. Pumping on a vacuum line at ambient temperature for 12 hours resulted in the isolation of a yellow solid residue (yield: 18.2 g, 53.2% based on 0.2 mol cyanoacetamide). Single crystals of Na[(NO2)2CCN]·H2O were obtained by recrystallization from an ethanol–water solution.DTA: 150 °C (exotherm); NMR (acetone-d6) δ(ppm): 13C (100.54 MHz) 109.1 (s, CN), 157.0 (s, C(NO2)2); 14N (36.14 MHz) −20.6 (s, ν½ = 40 Hz, NO2), −88 (s, ν½ = 320 Hz, CN).
Raman (25 °C, 20 mW): 2244 (7.7), 2226 (4.4), 1493 (1.0), 1442 (1.5), 1433 (2.1), 1377 (8.8), 1295 (1.3), 1260 (5.7), 1250 (5.7), 1230 (10.0), 1159 (2.6), 1153 (1.8), 1070 (0.6), 1026 (0.2), 1004 (0.2), 921 (0.3), 883 (1.4), 865 (7.1), 858 (8.5), 778 (0.5), 769 (0.8), 753 (0.3), 731 (0.3), 577 (0.9), 515 (0.7), 505 (0.8), 469 (1.1), 440 (0.5), 408 (1.1), 273 (2.0), 217 (3.4), 212 (3.3), 151 (4.2), 117 (7.4), 98 (7.3) cm−1; IR (ATR): 3583 (m), 3511 (m), 2230 (ms), 1631 (m), 1611 (m), 1498 (s), 1481 (s), 1424 (m), 1376 (w), 1224 (vs), 1150 (s), 861 (w), 855 (w), 792 (vw), 773 (w), 744 (m), 569 (vw), 497 (vw), 417 (vw) cm−1.
Synthesis of ammonium dinitrocyanomethanide
A concentrated aqueous ammonia solution (5 mL) was added drop wise to a solution of (NO2)3CCN (400 mg, 2.28 mmol) in CH2Cl2 (5 mL). Immediately, a yellow reaction mixture was obtained that is weakly effervescent. After stirring for 12 hours at ambient temperature, the reaction mixture was taken to dryness on a vacuum line, resulting in a yellow solid (yield: 314 mg, 93%.) Single crystals were obtained from an aqueous solution by slow evaporation of the solvent.DTA: 240 °C (exotherm); NMR (D2O) δ(ppm): 1H (599.80 MHz) 7.2 (s br, NH4), δ(ppm): 13C (100.54 MHz) 111.8 (s, CN), 152.4 (s, C(NO2)2); 14N (36.14 MHz) −23.1 (s, ν½ = 50 Hz, NO2), −89 (s, ν½ = 300 Hz, CN), −363.4 (quint, 1J(1H14N) = 56.9 Hz, NH4); IR (ATR): 3500–2800 (br, s), 2224 (ms), 1791 (w), 1661 (m), 1610 (w sh), 1523 (w sh), 1477 (m sh), 1396 (s), 1341 (m sh), 1207 (vs), 1144 (s sh), 1110 (s), 852 (w), 827 (w), 788 (w), 772 (m), 744 (s), 566 (m), 501 (m), 433 (w) cm−1.
Synthesis of tetraphenylphosphonium dinitrocyanomethanide
A solution of PPh4Cl (0.400 g; 1.07 mmol) in water (5 mL) was added to a solution of Na[(NO2)2CCN] (0.153 g; 1.00 mmol) in water (5 mL). The precipitate was filtered off, washed with water (10 mL) and dried on a vacuum line resulting in a pale yellow solid (yield: 0.452 g; 96%). DTA: 180 °C (endotherm, melting), 240 °C (exotherm); IR (ATR): 3086 (vw), 3071 (vw), 2956 (vw), 2923 (vw), 2892 (vw), 1593 (s), 1480 (w), 1435 (s), 1395 (vw), 1358 (vw), 1314 (m), 1215 (m), 1185 (vw), 1161 (m), 1106 (s), 1073 (m), 1025 (m), 1025 (vw), 996 (m), 970 (m), 937 (vw), 835 (m), 795 (m), 754 (m), 721 (s), 688 (s), 615 (w), 523 (vs), 447 (w) cm−1.Synthesis of hydrazinium dinitromethanide
A solution of hydrazinium hydrate (1 mL) in water (1 mL) was added drop wise to a solution of trinitroacetonitrile (200 mg, 1.14 mmol) in water (5 mL). The reaction was exothermic and a colourless gas was evolved. The orange reaction mixture was allowed to evaporate under air, resulting in pale yellow [N2H5][(NO2)2CH].DTA: 134 °C (exotherm); NMR (DMSO-d6) δ(ppm): 1H (599.80 MHz) 5.6 (s br, N2H5), 8.3 (s, (NO2)2CH); 13C (150.84 MHz) 165.6 (s, CH(NO2)2); 14N (36.14 MHz) −24.1 (s, ν½ = 100 Hz, NO2), −336 (s, ν½ = 600 Hz, N2H5); IR (DTA): 3600–2800 (br, m), 1604 (w), 1585 (w), 1499 (vw), 1483 (w), 1459 (vw), 1436 (m), 1341 (w), 1298 (w), 1251 (vw sh), 1240 (vw sh), 1192 (m), 1105 (s), 1071 (m), 997 (m), 948 (w), 758 (m), 719 (s), 687 (s), 615 (vw), 568 (vw), 522 (vs), 503 (m sh), 470 (m sh), 436 (w sh), 411 (vw) cm−1.
Synthesis of fluorodinitroacetonitrile28
A solution of Na[(NO2)2CCN] (10.0 g, 65.5 mmol) in water (30 mL) was placed in a 1′′ o.d. FEP reactor equipped with a magnetic stirrer, a gas inlet and a gas outlet tube. After cooling the solution with an ice/water bath to 0 °C, a stream of 10% fluorine in nitrogen was introduced at a rate of approx. 10 l h−1. The off-gas was swept through four traps at −78 °C and then vented through a bubbler that contained perfluorinated Krytox GPL107 oil. After about 3 hours, the fluorine stream was stopped and replaced by a pure nitrogen stream. This stream was maintained for an additional two hours in order to sweep all fluorinated products into the traps. The content of all four traps was combined and fluorodinitroacetonitrile was obtained as a colourless liquid. The crude product was purified by fractional condensation using cold traps at −31 °C, −78 °C and −196 °C. The −31 °C fraction consisted of water and (NO2)3CF, and the −196 °C trap contained CO2, SiF4, and some NO2. The fluorodinitroacetonitrile (5.35 g, 55.2%) stopped in the −78 °C trap.NMR (CDCl3) δ(ppm): 13C (100.54 MHz) 104.8 (d, 2J(13C19F = 35.3 Hz, CN), 105.5 (d, quint, 1J(13C19F) = 298.1 Hz, 1J(13C14N) = 3.0 Hz, CF(NO2)2); 14N (36.14 MHz) −36.7 (d, 2J(14N19F) = 11.2 Hz, ν½ = 5 Hz, NO2), −95 (s, ν½ = 320 Hz, CN); 19F (470.55 MHz) −90.8 (quint, 2J(14N19F) = 11.3 Hz, CF(NO2)2); IR (gas-phase, 10 Torr): 2914 (vw), 2261 (m), 1632 (vs), 1297 (s), 1093 (m), 1022 (vw), 842 (w), 801 (s), 794 (s sh), 655 (w) cm−1.
Synthesis of HFDNTz
In a 250 mL round bottom flask, a mixture of NaN3 (3.00 g, 46.0 mmol) and CCl4 (50 mL) was cooled to 0 °C using an ice bath and acetic acid (15 mL) was added slowly. After 5 minutes, a solution of fluorodinitroacetonitrile (1.20 g, 8.76 mmol) in CCl4 was added slowly through an addition funnel. When the addition was complete, the mixture was stirred for 10 hours at ambient temperature. The solvent was removed using a rotary evaporator. The gel-like, colourless residue was dissolved in CH2Cl2 (20 mL) and 2 M H2SO4 (10 mL). The organic phase was removed and the aqueous phase extracted three times with CH2Cl2 (15 mL each). The combined organic phases were dried over magnesium sulphate. The solvent was carefully evaporated on a rotary evaporator. The remaining colourless, oily liquid was dried by pumping for several hours on a vacuum line at ambient temperature, resulting in a colourless solid (yield: 1.42 g, 84.3%).DTA: 110 °C (explosion); NMR (CDCl3) δ(ppm): 1H (599.80 MHz) 14.0 (s, CN4H); 13C (150.84 MHz) 115.2 (d, 1J(13C19F), = 290.1 Hz CF(NO2)2), 153.9 (d, 2J(13C19F = 25.9 Hz, CN4); 14N (36.14 MHz) −27.3 (s, ν½ = 70 Hz, NO2), −50 (s, ν½ = 350 Hz, CN4); 19F (564.33 MHz) −98.1 (s, CF(NO2)2); Raman (25 °C, 20 mW): 3100–2900 (3.9), 1701 (2.1), 1693 (2.0), 1611 (2.4), 1482 (5.7), 1424 (2.4), 1359 (4.2), 1313 (2.2), 1239 (1.6), 1210 (2.7), 1188 (2.0), 1104 (2.9), 1081 (2.0), 1057 (1.6), 1034 (1.5), 982 (6.4), 956 (3.4), 837 (10.0), 801 (4.2), 544 (2.4), 539 (2.3), 425 (3.1), 397 (3.7), 371 (8.6), 300 (2.6), 282 (2.8), 196 (3.3) cm−1; IR (KBr): 3072 (w), 3069 (vw), 3012 (w), 2911 (w), 2774 (w), 2682 (vw), 2593 (vw), 1694 (s), 1611 (vs), 1478 (w), 1422 (w), 1362 (m), 1307 (m), 1240 (s), 1211 (m), 1173 (m), 1093 (vw), 1078 (w), 1054 (m), 1031 (s), 980 (m), 883 (m), 849 (m), 836 (s), 800 (s), 745 (w), 695 (vw), 663 (vw), 594 (m), 582 (m), 549 (w), 459 (vw), 403 (vw) cm−1.
Synthesis of [NH4][FDNTz]
A solution of 20% NH3 in water (5 mL) was added to a solution of HFDNTz (400 mg, 2.08 mmol). The resulting light yellow solution was allowed to evaporate, resulting in crystalline [NH4][FDNTz] (yield: 992 mg, 93.4%).DTA: 140 °C (explosion); NMR (D2O) δ(ppm): 1H (599.80 MHz) 7.04 (s, NH4); 13C (150.84 MHz) 117.9 (d, 1J(13C19F) = 290.1 Hz CF(NO2)2), 150.8 (d, 2J(13C19F) = 23.2 Hz, CN4); 14N (36.14 MHz) −38.2 (s, ν½ = 60 Hz, NO2), −52 (s, ν½ = 350 Hz, CN4), −362.3 (quint, 1J(1H14N) = 58.7 Hz, NH4); 19F (564.33 MHz) −98.5 (s, CF(NO2)2); Raman (25 °C, 40 mW): 3200–2800 (3.4), 1685 (1.3), 1609 (2.8), 1472 (8.9), 1364 (4.1), 1326 (1.8), 1320 (1.9), 1319 (1.9), 1313 (1.9), 1309 (1.8), 1191 (5.4), 1161 (1.7), 1153 (1.7), 1102 (4.4), 1071 (2.9), 1067 (3.0), 985 (6.9), 949 (7.8), 839 (9.1), 805 (1.7), 707 (1.3), 652 (2.2), 542 (1.9), 537 (2.1), 533 (2.2), 443 (3.6), 437 (3.3), 401 (3.7), 398 (3.7), 373 (10.0), 303 (3.2), 292 (2.9), 204 (5.8), 170 (6.3), 166 (6.3) cm−1; IR (ATR): 3600–3050 (s), 2977 (m), 2931 (w), 2901 (w), 1655 (w sh), 1606 (m), 1455 (w sh), 1401 (vs), 1320 (w), 1275 (vw), 1184 (vw), 1089 (s), 1049 (vs), 980 (w), 880 (m), 836 (w), 801 (w), 643 (vw), 615 (vw), 452 (w) cm−1.
Synthesis of Ag[FDNTz]
A solution of AgNO3 (937 mg, 2.50 mmol) in water (5 mL) was added to a solution of HFDNTz (384 mg, 2.00 mmol). The resulting white precipitate was filtered off and washed with water. The white solid was dried in a vacuum in darkness (yield: 687 mg, 92.0%).DTA: 185 °C (exotherm); IR (ATR): 1644 (s), 1604 (vs), 1467 (vw), 1375 (vw), 1305 (w), 1255 (w), 1201 (w), 1114 (vw), 1042 (vw), 1004 (vw), 973 (w), 833 (w), 796 (w) cm−1.
Synthesis of Ag[FDNTz]·½NH3
Single crystals of Ag[FDNTz]·½NH3 were obtained by dissolving Ag[FDNTz] (0.243 g; 0.81 mmol) in 25% aqueous ammonia (5 mL) and letting the solution evaporate under air in darkness.DTA: 165 °C (endotherm, loss of NH3), 180 °C (exotherm); IR (ATR): 3363 (m), 3201 (m), 2339 (vw), 2261 (w), 2166 (m), 1665 (s), 1594 (vs), 1498 (vw), 1465 (w), 1356 (s), 1308 (m), 1243 (m), 1195 (w), 1112 (m), 1048 (vw), 979 (m), 834 (ms), 799 (ms), 617 (m), 538 (w) cm−1.
Synthesis of [PPh4][FDNTz]
A solution of PPh4Cl (425 mg, 2.50 mmol) in water (5 mL) was added to a solution of HFDNTz (384 mg, 2.00 mmol). The resulting white precipitate was filtered off and washed with water. The white solid was dried in a vacuum (yield: 430 mg, 98.9%).DTA: 175 °C (exotherm); NMR (CDCl3) δ(ppm): 1H (599.80 MHz) 7.5–7.9 (m, PPh4); 13C (150.84 MHz) 117.9 (d, 1J(13C19F), = 290.1 Hz CF(NO2)2), 117.5 (d, 1J(13C31P = 88.0 Hz, PPh4), 120.6 (d, 1J(13C19F = 282.0 Hz, CF(NO2)2), 130.8 (d, 2J(13C31P = 15.6 Hz, PPh4), 134.4 (d, 2J(13C31P = 10.5 Hz, PPh4), 135.9 (d, 3J(13C31P = 3.2 Hz, PPh4), 150.8 (d, 1J(13C19F = 88.0 Hz, CN4); 150.8 (d, 2J(13C19F = 23.2 Hz, CN4); 14N (36.14 MHz) −39.1 (s, ν½ = 50 Hz, NO2), −52 (s, ν½ = 380 Hz, CN4); 19F (564.33 MHz) −94.0 (s, CF(NO2)2); 31P (242.82 MHz) 23.14 (s, PPh4); IR (ATR): 3088 (vw), 3071 (vw), 2956 (vw), 2892 (vw), 1593 (s), 1480 (m), 1435 (s), 1395 (vw), 1358 (w), 1339 (vw), 1314 (m), 1215 (m), 1185 (w), 1161 (m), 1106 (s), 1073 (m), 1025 (w), 996 (m), 970 (m), 937 (vw), 835 (m), 795 (m), 754 (m), 721 (s), 688 (s), 645 (vw sh), 615 (w), 523 (vs), 447 (w), 436 (vw sh), 404 (vw) cm−1.
Results and discussion
Synthesis
The synthetic route for the preparation of 5-(fluorodinitromethyl)-2H-tetrazole (HFDNTz) is shown in Scheme 1.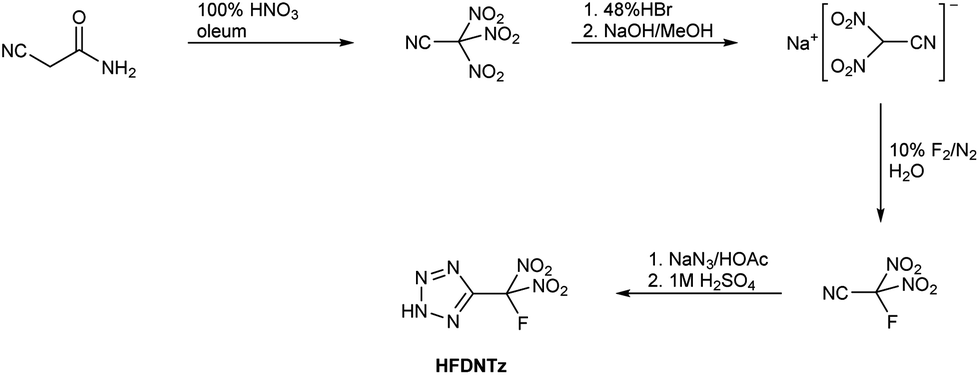 | ||
| Scheme 1 Synthesis of 5-(fluorodinitromethyl)-2H-tetrazole. | ||
Nitration of cyanoacetamide with fuming nitric acid in 20% oleum under anhydrous conditions resulted in the formation of trinitroacetonitrile.5,29 Due to the compound's reported sensitivity and high reactivity, trinitroacetonitrile was recommended to be handled only in solution.27 As a result, trinitroacetonitrile had not been fully characterized. We were able to isolate the compound by careful evaporation of the solvent from dichloromethane solutions in a vacuum at −40° C as a moisture sensitive, wax-like colourless solid. It is a very noxious lachrymator that slowly vaporizes in the air at ambient temperature.
Treatment of a trinitroacetonitrile solution in methanol with a 48% aqueous HBr solution resulted in gas evolution and the slow formation of elemental bromine. When the resulting reaction mixture was neutralized with sodium hydroxide, sodium dinitrocyanomethanide could be isolated in up to 53% yield. While this reaction had already been described in the literature, its mechanism is unknown but was assumed to involve the formation of N2O.27 Single crystals of Na[(NO2)2CCN]·H2O were obtained from an aqueous solution by slow evaporation of the solvent.
When trinitroacetonitrile was reacted with N2H4·H2O in water, a vigorous gas evolution was observed. However, no evidence for the formation of the dinitrocyanomethanide anion could be obtained. Instead, yellow crystals of [N2H5][(NO2)2CH] were formed when the resulting bright yellow solution was taken to dryness (Scheme 2). The mechanism for the formation of the dinitromethanide anion is unknown. However, it should be noted that a mixture of pure trinitroacetonitrile and neat N2H4 is hypergolic. The reaction of the two compounds is highly exothermic. On several occasions, flames and/or explosions were observed when neat hydrazine was mixed with solid trinitroacetonitrile.
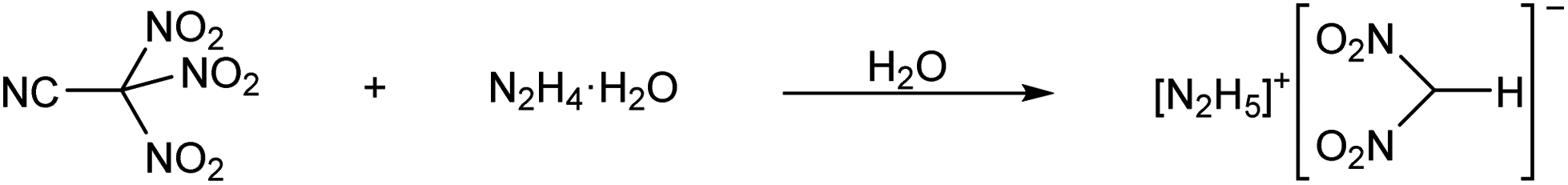 | ||
| Scheme 2 Reaction of trinitroacetonitrile with hydrazine hydrate. | ||
When a clear colourless solution of trinitroacetonitrile in dichloromethane was treated with an aqueous ammonia solution at ambient temperature, a yellow effervescing mixture was obtained. Yellow crystals of [NH4][(NO2)2CCN] were isolated in quantitative yield when the solvent was removed from the reaction mixture after 12 hours of stirring at ambient temperature. The evolved gas was identified by IR spectroscopy as N2O (Scheme 3).
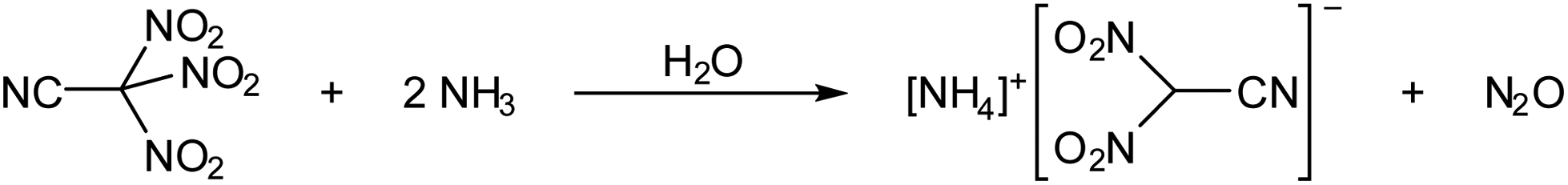 | ||
| Scheme 3 Reaction of trinitroacetonitrile with aqueous ammonia. | ||
Fluorodinitroacetonitrile, F(NO2)2CCN, was obtained by aqueous fluorination of sodium dinitrocyanomethanide with 10% F2 in N2 (Scheme 1). The off-gas of the fluorination reaction was passed trough a series of cold traps at −78 °C. Lower cold trap temperatures were not used to avoid trapping of fluorine nitrate, a possible fluorination side product. While Wiesboeck and Ruff reported only moderate yields of 29% of F(NO2)2CCN and the formation of the hydrolysis products acetonitrile and fluorodinitroacetamide when aqueous solutions with more than 3% Na[(NO2)2CCN] were fluorinated,28 we did not observe the formation of appreciable amounts on hydrolysis products even in the case of reaction mixtures containing more than 10% of Na[(NO2)2CCN]. However, we did observe a contamination of the crude F(NO2)2CCN with up to 10% fluorotrinitromethane, FC(NO2)3, and also trace amounts of CO2. Although not necessary for this work, crude F(NO2)2CCN could be purified by fractional condensation at −31 °C, −78 °C and −196 °C. The F(NO2)2CCN stopped in the −78 °C trap and was isolated as a colourless liquid with a vapour pressure of 56 Torr at 23 °C.
The 1,3-dipolar cycloaddition reaction of F(NO2)2CCN with HN3, followed by extraction with dichloromethane resulted in the isolation of HFDNTz as a colourless, hygroscopic solid in approximately 85% yield. Single crystals suitable for an X-ray crystal structure determination were obtained from a dichloromethane solution by slow evaporation of the solvent in vacuo.
HFDNTz is acidic and forms ammonium 5-(fluorodinitromethyl)tetrazolate in quantitative yield when treated with aqueous ammonia (Scheme 4).
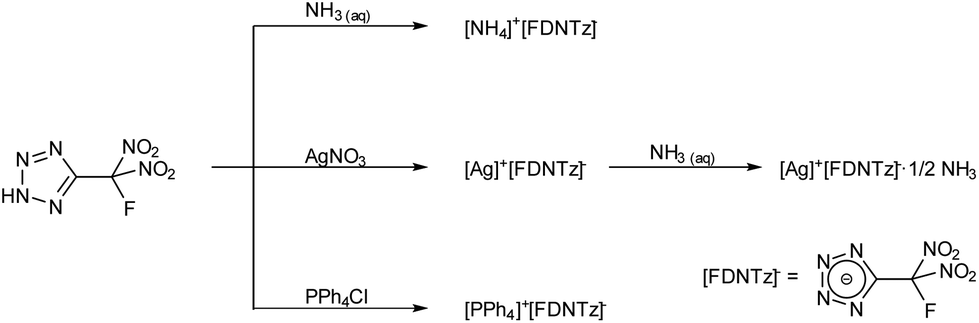 | ||
| Scheme 4 Synthesis of 5-(fluorodinitromethyl)tetrazolate (FDNTz) salts. | ||
Silver 5-(fluorodinitromethyl)tetrazolate was obtained as a white amorphous precipitate in quantitative yield when an aqueous solution of silver nitrate was added to a solution of HFDNTz in water. Colourless crystals of the ammonia adduct [Ag][FDNTz]·½NH3 were obtained by recrystallization of the amorphous precipitate from an aqueous ammonia solution. While the nitrotetrazoles of this work are energetic and must be treated with great care, both silver salts are especially treacherous and can explode upon provocation (heat or mechanical shock). The tetraphenylphosphonium (PPh4) salt of FDNTz was precipitated from an aqueous solution of HFDNTz by the addition of an aqueous PPh4Cl solution. Crystalline [PPh4][FDNTz] was obtained by recrystallization from acetone.
X-ray crystal structures
Single crystals suitable for X-ray crystal structure determinations were obtained for Na[(NO2)2CCN]·H2O, NH4[(NO2)2CCN], [PPh4][(NO2)2CCN], [N2H5][(NO2)2CH], HFDNTz, [NH4][FDNTz], [Ag][FDNTz]·½NH3, and [PPh4][FDNTz]. The relevant data and parameters for the X-ray measurements and refinements of the crystal structures are summarized in Tables 1 and 2. Further crystallographic data and representations of the unit cells for all crystal structures are given in the ESI.†| Na[(NO2)2CCN]·H2O | [NH4][(NO2)2CCN] | [PPh4][(NO2)2CCN] | [N2H5][(NO2)2CH] | |
|---|---|---|---|---|
| Formula | C2H2N3NaO5 | C2H4N4O4 | C26H20N3O4P | CH6N4O4 |
| mol wt [g mol−1] | 171.06 | 148.09 | 469.42 | 138.10 |
| Temp [K] | 100(2) | 100(2) | 104(2) | 100(2) |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | monoclinic |
| Space group | P212121 | P21/n | P21/n | P21/n |
| a [Å] | 7.5138(6) | 8.1144(12) | 11.6601(8) | 3.6434(4) |
| b [Å] | 8.1663(6) | 4.8009(8) | 14.2600(9) | 13.7827(13) |
| c [Å] | 19.9151(14) | 14.218(2) | 13.4402(9) | 10.6457(10) |
| α [°] | 90 | 90 | 90 | 90 |
| β [°] | 90 | 96.914(3) | 90.099 (1) | 98.230(2) |
| γ [°] | 90 | 90 | 90 | 90 |
| V [Å3] | 1221.99(16) | 549.85(15) | 2234.7(3) | 529.08(9) |
| Z | 8 | 4 | 4 | 4 |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| ρ calc [g cm−3] | 1.860 | 1.789 | 1.395 | 1.734 |
| μ [mm−1] | 0.237 | 0.170 | 0.163 | 0.169 |
| F(000) | 688 | 304 | 976 | 288 |
| Reflns collected | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 172 172 |
12950 |
55116 |
12436 |
| Ind reflns | 3734 | 1681 | 6814 | 1614 |
| R int | 0.0355 | 0.0265 | 0.0385 | 0.2311 |
| No. of parameters | 201 | 103 | 307 | 100 |
| R 1 [I > 2σ(I)] | 0.0253 | 0.0292 | 0.0359 | 0.0255 |
| wR2 [I > 2σ(I)] | 0.0651 | 0.795 | 0.0922 | 0.0769 |
| GOF | 1.060 | 1.053 | 1.035 | 1.087 |
| HFDNTz | [NH4][FDNTz] | Ag[FDNTz]·½NH3 | [PPh4][FDNTz] | |
|---|---|---|---|---|
| Formula | C2HFN6O4 | C2H4FN7O4 | C24H18Ag12F12N78O48 | C26H20FN6O4P |
| Mol wt [g mol−1] | 192.09 | 209.12 | 3689.60 | 530.45 |
| Temp. [K] | 100(2) | 101(2) | 100(2) | 101(2) |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | C2/c | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21 |
| a [Å] | 28.8030(7) | 13.9642(7) | 15.4270(10) | 11.8077(14) |
| b [Å] | 5.3703(1) | 8.9672(5) | 15.4526(10) | 7.1276(8) |
| c [Å] | 28.897(1) | 13.9957(7) | 20.5225(13) | 14.7721(18) |
| α [°] | 90 | 90 | 82.7270(10) | 90 |
| β [°] | 119.723(2) | 115.4716(1) | 88.1560(10) | 95.585(2) |
| γ [°] | 90 | 90 | 82.6690(10) | 90 |
| V [Å3] | 3881.71(19) | 1582.19(14) | 4812.6(5) | 1237.3(3) |
| Z | 24 | 8 | 2 | 2 |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| ρ calc [g cm−3] | 1.972 | 1.756 | 2.546 | 1.424 |
| μ [mm−1] | 0.200 | 0.174 | 2.540 | 0.165 |
| F(000) | 2304 | 848 | 3528 | 548 |
| Reflns collected | 46406 |
37464 |
22050 |
26729 |
| Ind reflns | 5932 | 4778 | 22050 |
7495 |
| R int | 0.0774 | 0.0384 | 0.0939 | 0.0451 |
| No. of parameters | 361 | 277 | 1576 | 343 |
| R 1 [I > 2σ(I)] | 0.0414 | 0.0329 | 0.0584 | 0.0435 |
| wR2 [I > 2σ(I)] | 0.0830 | 0.0786 | 0.0977 | 0.0916 |
| GOF | 1.035 | 1.026 | 0.992 | 1.023 |
From an aqueous solution, sodium dinitrocyanomethanide crystallizes as a monohydrate in the orthorhombic space group P212121 with the unit cell parameters a = 7.5138(6) Å, b = 8.1663(6) Å and c = 19.9151(14) Å. The solid-state structure of Na[(NO2)2CCN]·H2O does not consist of isolated ions but is dominated by cation–anion interactions that result in a polymeric structure. The asymmetric unit of the structure contains two formula units (Z′ = 2). One CN nitrogen atom as well as several oxygen atoms of the anion coordinate each sodium ion, which in turn is bridged to another sodium ion through a water molecule (Fig. 1).
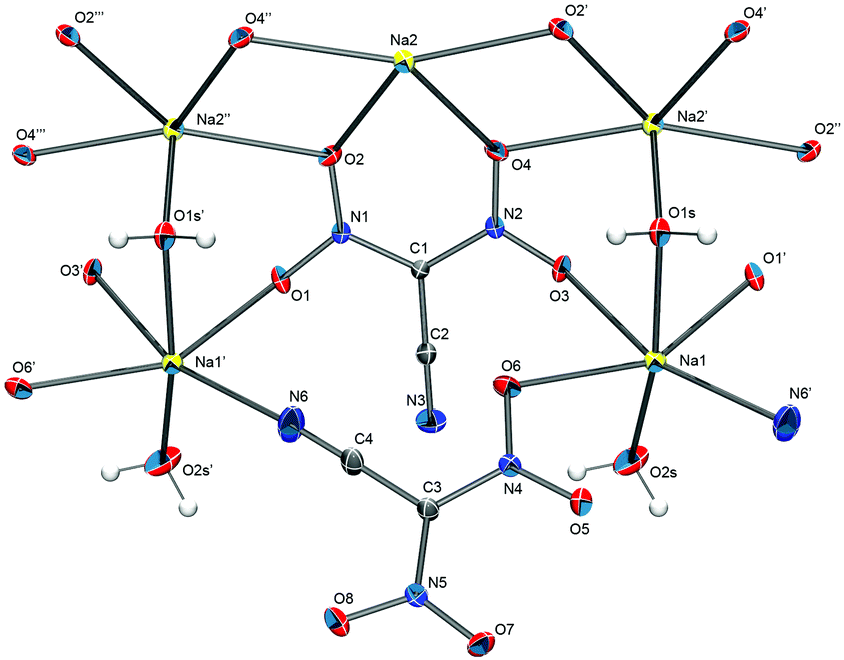 | ||
| Fig. 1 The solid-state structure of Na[(NO2)2CCN]·H2O. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atom positions were determined from the electron density map and are depicted as spheres of arbitrary radius. Selected bond distances (Å): C1–C2 1.412(2), C1–N1 1.390(2), C1–N2 1.385(2), C2–N3 1.147(2), N1–O1 1.250(2), N1–O2 1.245(2), N2–O3 1.249(2), N2–O4 1.246(2), Na1–O1′ 2.418(1), Na1–O3 2.450(1), Na1–O6 2.475(1), Na1–N6 2.421(1), Na1–O1s 2.362(1), Na1–O2s 2.292(1), Na2–O2 2.575(1), Na2–O4 2.445(1), Na2–O2′ (1), Na2′–O4 2.508(1), Na2′–O4′ 2.445(1). | ||
The ammonium salt of the dinitrocyanomethanide anion crystallizes with four formula units per unit cell (Z = 4) in the monoclinic space group P21/n. Not surprisingly, the solid-state structure of [NH4][(NO2)2CCN] contains hydrogen bonds between the ammonium ions and the oxygen atoms as well as the CN nitrogen atom of the anions (Fig. 2). The observed C–N and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) N bond distances of 1.385(2)/1.390(2) Å and 1.152(2) Å, respectively, in the anion in [NH4][(NO2)2CCN] are virtually identical to the ones observed for Na[(NO2)2CCN]·H2O (1.385(2)/1.390(2) Å and 1.147(2) Å).
N bond distances of 1.385(2)/1.390(2) Å and 1.152(2) Å, respectively, in the anion in [NH4][(NO2)2CCN] are virtually identical to the ones observed for Na[(NO2)2CCN]·H2O (1.385(2)/1.390(2) Å and 1.147(2) Å).
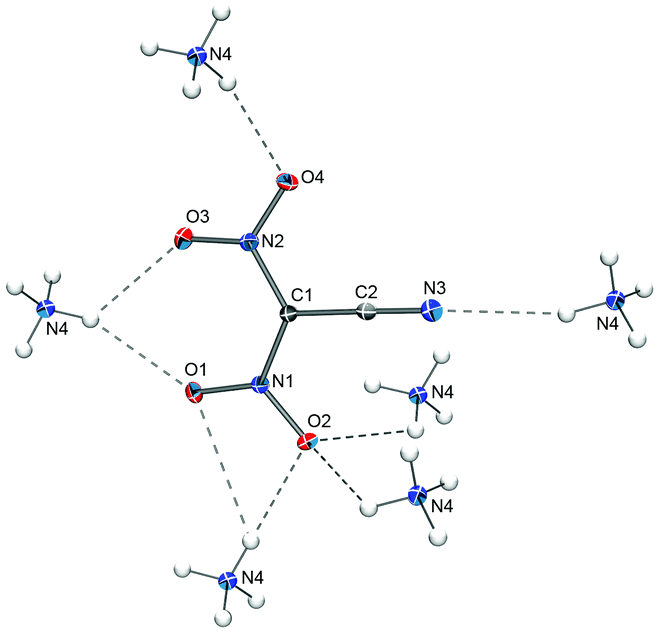 | ||
| Fig. 2 Hydrogen bonding in the solid-state structure of [NH4][(NO2)2CCN]. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atom positions were determined from the electron density map and are depicted as spheres of arbitrary radius. Selected distances (Å): C1–C2 1.412(2), C1–N1 1.385(2), C1–N2 1.402(2), C2–N3 1.152(2), N1–O1 1.240 (1), N1–O2 1.266(1), N2–O3 1.233(1), N2–O4 1.253(1), N3–N4 3.068(1) O1–N4 3.032(1), O2–N4 2.999(1)/3.130(1), O3–N4 2.841(1), O4–N4 2.920(1). | ||
Single crystals of [PPh4][(NO2)2CCN] suitable for structure determination were obtained from an acetone solution by slow evaporation of the solvent. The compound crystallizes in the monoclinic space group P21/n with four formula units in the unit cell (Z = 4). The solid state structure consists of isolated PPh4+ cations and [(NO2)2CCN]− anions (Fig. 3). The closest cation–anion interactions are 3.122(2) Å (O2⋯C19) and 3.187(2) Å (O1⋯C10). The observed C–N and CN bond distances of 1.3966(15)/1.4150(7) Å and 1.1513(17) Å, respectively, in the [(NO2)2CCN] anion are in good agreement with the ones observed for the Na+ and NH4+ salts.
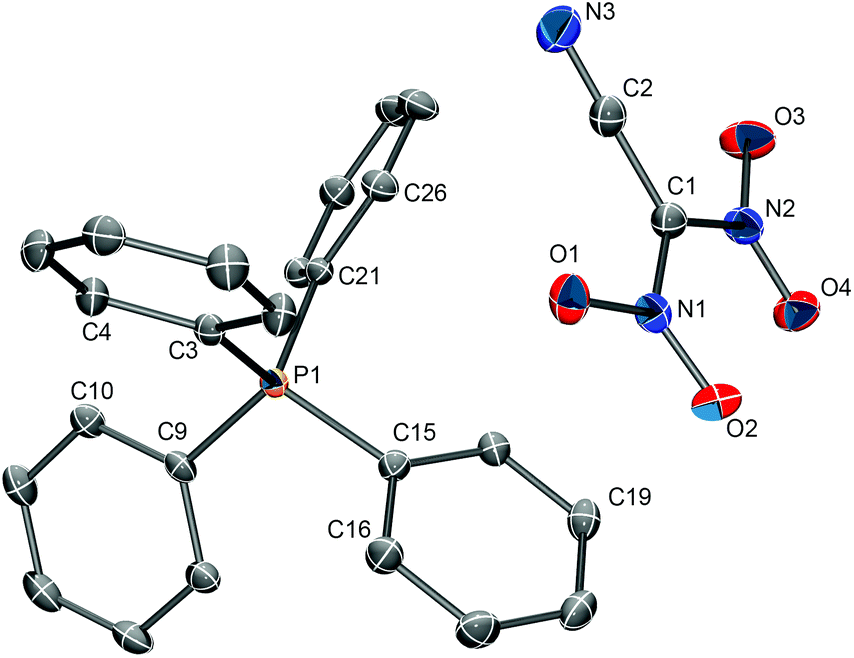 | ||
| Fig. 3 The solid-state structure of Na[(NO2)2CCN]. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms were omitted for clarity. Selected bond distances (Å): C1–C2 1.4150(17), C1–N1 1.3966(15), C1–N2 1.3994(16), C2–N3 1.1513(17), N1–O1 1.2571(14), N1–O2 1.2360(14), N2–O3 1.2518(15), N2–O4 1.2305(14). | ||
Single crystals of [N2H5][(NO2)2CH] were obtained from an aqueous solution by slow evaporation. The compound crystallizes without crystal water in the monoclinic space group P21/n (Z = 4) with the unit cell parameters a = 3.6434(4) Å, b = 13.7827(13) Å, and c = 10.6457(10) Å. The solid-state structure consists of N2H5+ cations and [(NO2)2CH]− anions (Fig. 4) that are linked through hydrogen bonds between the cation and the oxygen atoms of the anion. The observed C–N bond distances in the [(NO2)2CH]− anion of 1.368(1) Å and 1.365(1) Å are noticeable shorter than the ones observed for the [(NO2)2CCN]− anion in the PPh4+ salt (1.3966(15) and 1.4150(7) Å).
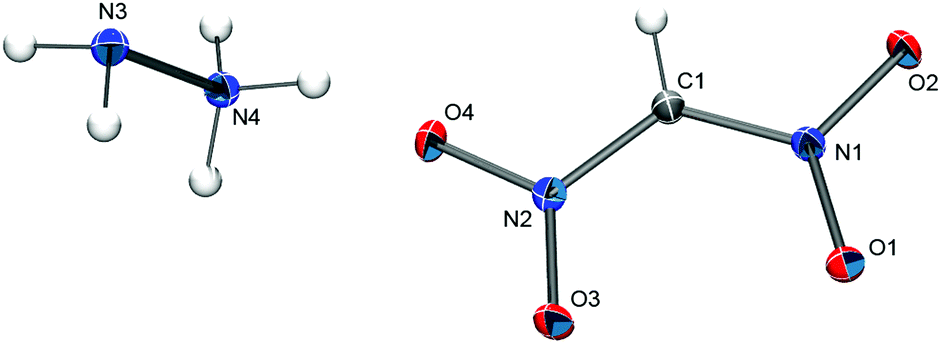 | ||
| Fig. 4 The crystal structure of [N2H5][(NO2)2CH]. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atom positions were determined from the electron density map and are depicted as spheres of arbitrary radius. Selected bond distances (Å): C1–N1 1.368(1), C1–N2 1.3647(9), N1–O1 1.2648(8), N1–O2 1.2647(8), N2–O3 1.2621(8), N2–O4 1.2648(8). | ||
Single crystals of 5-(fluorodinitromethyl)-2H-tetrazole (HFDNTz) were obtained by slow evaporation of a dichloromethane solution in vacuo at a temperature of −20 °C. The compound crystallizes in the monoclinic space group C2/c with a unit cell volume of 3881.71(19) Å3 (Z = 24). Further crystallographic details for the compound are listed in Table 2. The structure of HDNTz is depicted in Fig. 5, while the bond lengths and angles of the tetrazole ring in this compound are summarized in Table 3 together with the ones for the related tetrazolates of this work.
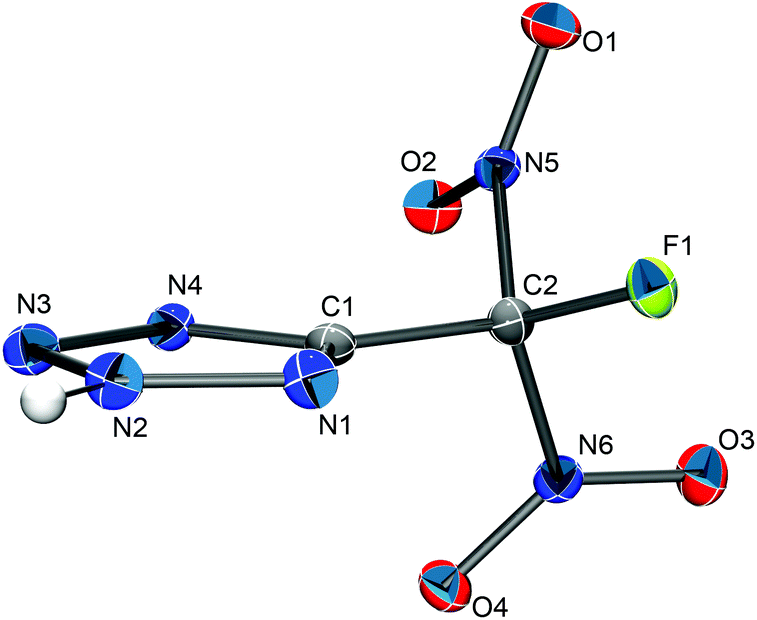 | ||
| Fig. 5 The molecular structure of 5-(fluorodinitromethyl)-2H-tetrazole. Thermal ellipsoids are shown at the 50% probability level. The position of the hydrogen atom was determined from the electron density map. It is depicted as a sphere of arbitrary radius. Selected bond distances (Å) and bond angles (°): C1–C2 1.490(2), C1–N1 1.325(2), C1–N4 1.350(2), C2–N5 1.540(2), C2–N6 1.539(2), C2–F1 1.318(2), N1–N2 1.324(2), C1–C2–F1 113.0(1), C1–C2–N5 109.7(1), C1–C2–N6 113.0(1), N1–C1–C2 122.5(2). | ||
|
|
||||
|---|---|---|---|---|
| HFDNTza | [NH4][FDNTz]a | Ag[FDNTz]·½NH3a |
[PPh4][FDNTz] | |
| a Values given for only one of the different molecules in the asymmetric unit. | ||||
| C1–C2 | 1.490(2) | 1.4850(14) | 1.463(10) | 1.476(4) |
| C1–N1 | 1.325(2) | 1.3298(13) | 1.324(9) | 1.335(4) |
| C1–N4 | 1.350(2) | 1.3357(13) | 1.325(8) | 1.319(4) |
| N1–N2 | 1.324(2) | 1.3438(12) | 1.343(8) | 1.347(3) |
| N2–N3 | 1.319(2) | 1.3224(13) | 1.327(8) | 1.311(4) |
| N3–N4 | 1.325(2) | 1.3383(12) | 1.348(7) | 1.347(3) |
| C2–F | 1.318(2) | 1.3328(11) | 1.313(8) | 1.339(4) |
| C2–N5 | 1.540(2) | 1.5466(14) | 1.540(9) | 1.543(4) |
| C2–N6 | 1.539(2) | 1.5381(14) | 1.545(9) | 1.534(4) |
| C1–N1–N2 | 100.36(14) | 103.48(8) | 104.0(6) | 102.9(2) |
| C1–N4–N3 | 105.48(14) | 103.42(8) | 104.4(5) | 103.5(2) |
| N1–N2–N3 | 115.32(14) | 109.64(8) | 109.8(6) | 110.0(2) |
| N2–N3–N4 | 105.32(13) | 109.91(8) | 108.6(6) | 109.7(2) |
| N1–C1–N4 | 113.51(15) | 113.55(9) | 113.2(6) | 113.9(2) |
| N1–C1–C2 | 122.49(15) | 124.33(9) | 125.0(7) | 119.9(2) |
| C1–C2–N5 | 109.68(13) | 113.60(8) | 113.1(6) | 114.7(2) |
| C1–C2–N6 | 113.00(13) | 112.04(8) | 108.7(6) | 110.9(2) |
| N5–C2–N6 | 104.60(12) | 103.55(8) | 103.9(5) | 102.7(2) |
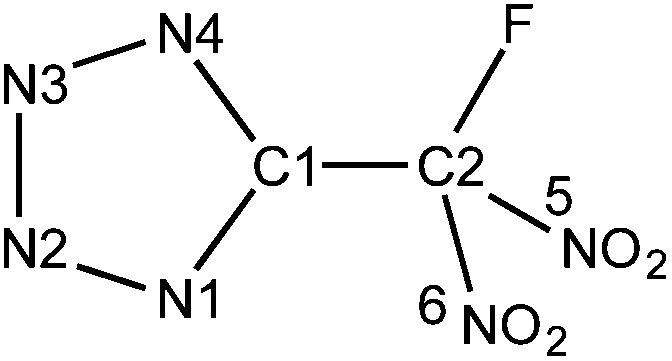
The geometry of the five-membered ring in HFDNTz is essentially identical to the one of 5-(trinitromethyl)-2H-tetrazole (HTNTz).5 Due to the strong electron withdrawing effect of the fluorodinitromethyl group, the hydrogen atom of the tetrazole moiety is exclusively located in the 2-position (N2) of the five-membered ring. Similar to HTNTz and atypical for alkyl-substituted tetrazoles, the distance between C1 and N1 (1.325(2) Å) is shorter than the one between C1 and N4 (1.350(2) Å). In addition, the three N–N distances in the five-membered ring of HFDNTz can be considered identical within their margins of error. This is in good agreement with the observed geometry of HTNTz but is unlike the distance pattern of a regular alkyl-substituted tetrazole.5 The asymmetric unit of the 5-(fluorodinitromethyl)-2H-tetrazole solid-state structure consists of three HFDNTz molecules (Z′ = 3) that are arranged in a triangular fashion with the fluorine atoms of the –CF(NO2)2 groups facing each other (Fig. 6). The three fluorine atoms form the corners of a slightly disordered regular triangle with F–F distances of 2.886(2) and 2.939(2) Å and F–F–F angles of 58.82(4) and 60.59(4)°. In addition, the HFDNTz molecules are associated through N(2)–H⋯N(4) hydrogen bonds.
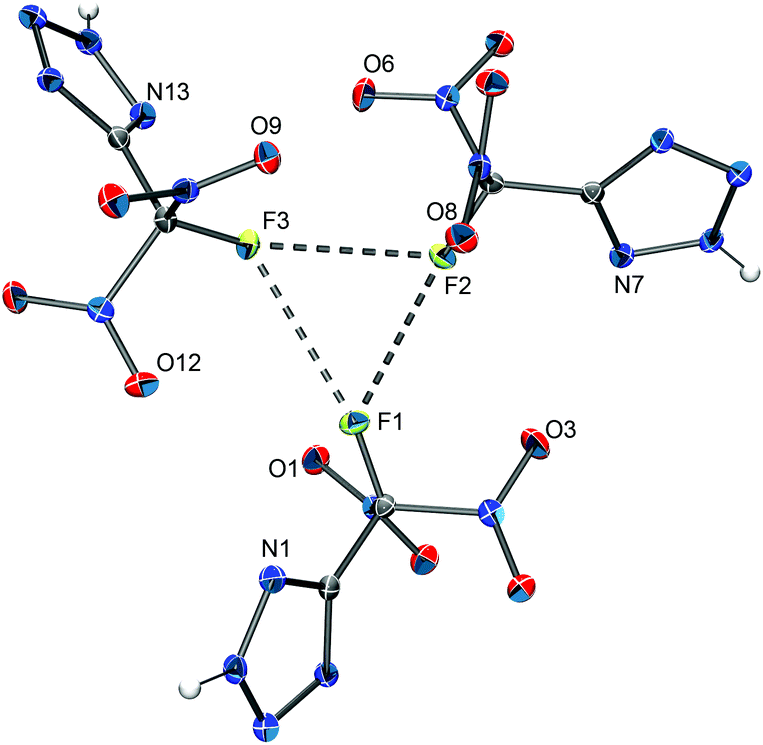 | ||
| Fig. 6 The asymmetric unit in the solid-state structure of 5-(fluorodinitromethyl)-2H-tetrazole. Thermal ellipsoids are shown at the 50% probability level. Selected distances (Å) and angles (°): F1–F2 2.886(2), F1–F3 2.939(1), F2–F3 2.939(2), F1–F2–F3 60.59(4), F2–F3–F1 58.82(4), F2–F1–F3 60.59(4). | ||
The solid-state structure of ammonium 5-(fluorodinitromethyl)tetrazolate consists of ammonium cations and FDNTz anions that are associated through hydrogen bonding (Fig. 7). Further crystallographic details of the structure are listed in Table 2, the observed bond lengths and angles for the FDNTz− anion are summarized in Table 3.
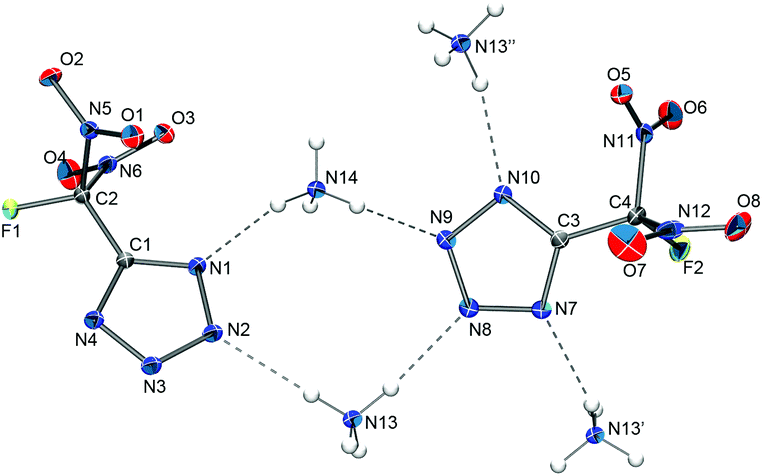 | ||
| Fig. 7 Hydrogen bonding in the solid-state structure of [NH4][FDNTz]. Hydrogen atom positions were determined from the electron density map and are depicted as spheres of arbitrary radius. Selected distances (Å): N1–N14 2.915(2), N2–N13 3.007(1), N7–N13′ 2.944(1), N8–N13, 2.982(2), N9–N14 2.965(1), N10–N13′’ 3.018(1). | ||
All attempts to grow single crystals of silver 5-(fluorodinitromethyl)tetrazolate (AgFDNTz) suitable for X-ray structure determination were unsuccessful. The crystallization of an amorphous sample of AgFDNTz from an aqueous ammonia solution resulted in crystals of the ammonia adduct Ag[FDNTz]·½NH3 instead. Selected crystallographic data of the compound are listed in Table 2. The solid-state structure of the silver salt contains [Ag(NH3)2]+ cations and polymeric anion chains. The anion chains are made up from [Ag4(FDNTz)4] units in which two silver atoms are linked in a 1,2-fashion by always two bridging FDNTz− anions. Every tetrazolate anion is coordinated to three different Ag atoms. The units are linked together by Ag(NH3) and Ag(NH3)2 units, resulting in a complex polymeric anion chain. The resulting overall structure can be described as [Ag(NH3)2]3[Ag21(NH3)6(FDNTz)24]. Part of a polymeric anion chain of the structure is depicted in Fig. 8. Further crystallographic details of the structure are listed in Table 2, the observed bond lengths and angles for the FDNTz− anion are summarized in Table 3.
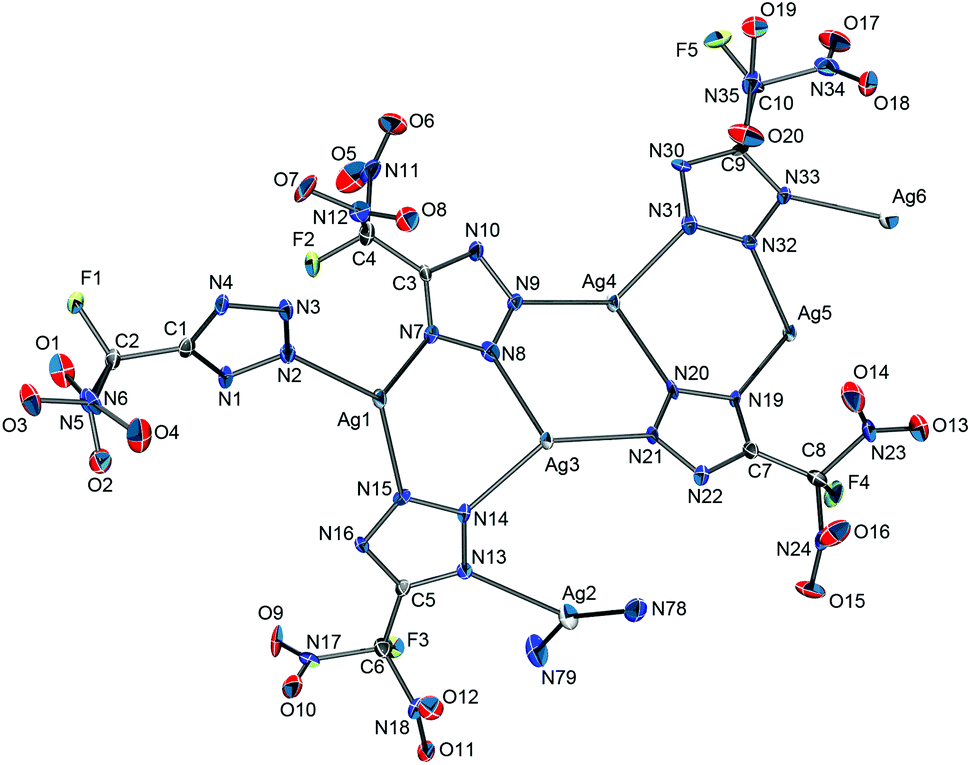 | ||
| Fig. 8 Part of the polymeric anion structure of Ag[FDNTz]·½NH3. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å): Ag1–N2, Ag1–N7, Ag1–N15, Ag2–N2, Ag2–N78, Ag2–N79, Ag3–N14, Ag3–N8, Ag3–N21. | ||
The tetraphenylphosphonium salt [PPh4][FDNTz] crystallizes in the monoclinic space group P21 with two formula units per unit cell (Z = 2). The solid-state structure consists of isolated and well-separated PPh4+ cations and FDNTz− anions (Fig. 9). The closest cation–anion distance is 2.989(3) Å (C18⋯O4). Further crystallographic details of the structure are listed in Table 2, the observed bond lengths and angles for the FDNTz− anion are summarized in Table 3. In going from the neutral tetrazole HFDNTz to the weakly coordinated tetrazolate anion in [PPh4][FDNTz], the geometry of the five-membered ring changes. While the C1–N1 distance increases only slightly (0.01 Å), the second C–N distance (C1–N4) shortens by over 0.03 Å).
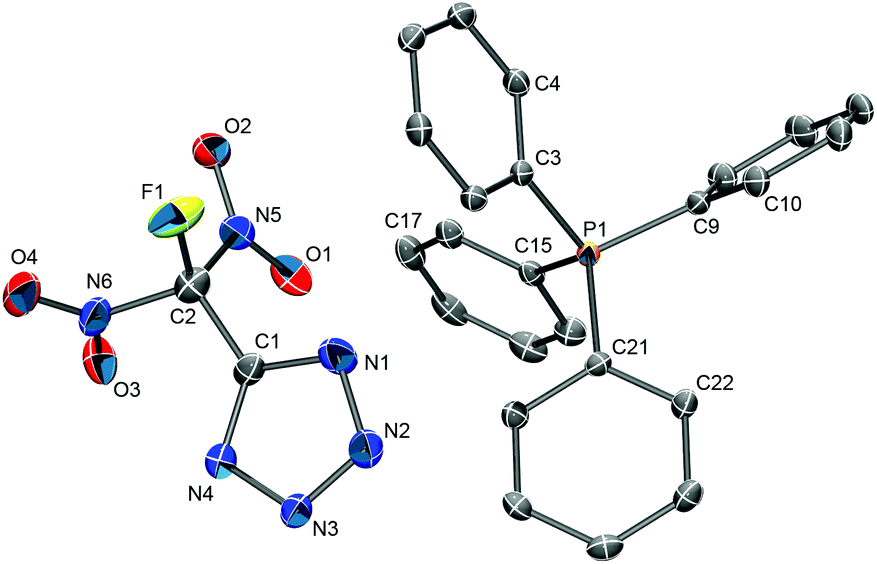 | ||
| Fig. 9 Molecular structure of [PPh4][FDNTz]. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and bond angles (°): C1–C2 1.476(4), C1–N1 1.335(4), C1–N4 1.319(4), C2–N5 1.543(4), C2–N6 1.534(4), C2–F1 1.339(4), N1–N2 1.347(4), C1–C2–F1 114.1(2), C1–C2–N5 114.7(2), C1–C2–N6 110.9(2), N1–C1–C2 119.9(3). | ||
It is interesting to note that the N1–N2 and N3–N4 distances in the anion are longer by about 0.02 Å than the ones in the parent tetrazole. The third N–N distance (N2–N3) remains essentially unchanged within the error margins.
Stability of the compounds
The impact and friction sensitivities of most of the compounds of this study were determined using a BAM Fall Hammer and BAM Friction tester. In addition, decomposition temperatures were determined through DTA scans with heating rates of 5 °C min−1. The obtained sensitivity and stability data is summarized in Table 4. The impact and friction sensitivities, and decomposition temperature of F(NO2)2CCN were not determined due to the volatility of the compound.| Compound |
T
d [°C]b |
FS [N] | IS [J] | OB [%] |
|---|---|---|---|---|
| a T d: decomposition temperature, FS: friction sensitivity, IS: impact sensitivity, OB: oxygen balance. b DTA onset. c Sample evaporates. d Endotherm at 180 °C (melting). e Explosion. f Endotherm at 165 °C (loss of NH3). | ||||
| RDX | 220 | 120 | 7.5 | −21 |
| (NO2)3CCN | —c | 112 | 12 | 17.4 |
| Na[(NO2)2CCN] | 150 | >360 | 80 | −5.2 |
| [NH4][(NO2)2CCN] | 240 | >360 | 75 | −21.6 |
| [PPh4][(NO2)2CCN] | 240d | >360 | >100 | −206.2 |
| [N2H5][(NO2)2CH] | 134 | 230 | 90 | −11.6 |
| HFDNTz | 110e | 40 | 3.5 | −2.1 |
| [NH4][FDNTz] | 140e | 50 | 4 | −13.4 |
| [Ag][FDNTz] | 185 | <2 | 2 | −1.3 |
| [Ag][FDNTz]·½NH3 | 180f | <2 | 2 | −5.2 |
| [PPh4][FDNTz] | 175 | >360 | >100 | −181.7 |
With the exception of trinitroacetonitrile, all investigated compounds are under-oxidized and have a negative oxygen balance. It is not surprising that based on the impact and friction sensitivities, the two most stable compounds, [PPh4][(NO2)2CCN] and [PPh4][FDNTz], are the ones with the lowest oxygen balances. Explosion upon heating were observed only in the case of the free tetrazole HFDNTz as well as the corresponding ammonium salt [NH4][FDNTz]. All other investigated compounds showed smooth thermal decompositions. In the case of (NO2)3CCN, it was not possible to determine a decomposition temperature because the sample evaporated completely upon heating with a nitrogen purge before its decomposition. The thermally least stable compounds are HFDNTz, [N2H5][NO2)2CH], [NH4][FDNTz], and Na[(NO2)2CCN] with decomposition temperatures of 110 °C, 134 °C, 140 °C, and 150 °C, respectively. It is interesting that [NH4][(NO2)2CCN)] shows a much higher thermal stability of 240 °C than the closely related Na[(NO2)2CCN)] (150 °C). This might be related to the different oxygen balances as well as the presence of stabilizing hydrogen bonding in the case of the ammonium salt. With the exception of the PPh4+ salt, all 5-(fluorodinitromethyl)tetrazolates and the parent 5-(fluorodinitromethyl)-2H-tetrazole are sensitive compounds. With impact sensitivities of less than 5 J and friction sensitivities of 50 Nm or less, these compounds must be considered explosion hazards that have to be handled with great care while using proper safety precautions. Both silver salts, [Ag][FDNTz] and [Ag][FDNTz]·½NH3, are especially treacherous with friction sensitivities of fewer than 2 Nm and impact sensitivities of about 2 J.
Conclusions
5-(Fluorodinitromethyl)-2H-tetrazole was prepared by a four-step synthesis starting from cyanoacetamide. Nitration of cyanoacetamide with fuming nitric acid in oleum resulted in the formation of trinitroacetonitrile, which was converted into sodium dinitrocyanomethanide by reaction with HBr, followed by treatment with NaOH. Aqueous fluorination with 10% fluorine in nitrogen resulted in the formation of fluorodinitroacetonitrile and, after reaction with HN3, in the isolation of 5-(fluorodinitromethyl)-2H-tetrazole. The tetrazole was converted into ammonium, silver and tetraphenylphosphonium 5-(fluorodinitromethyl)-2H-tetrazolate. While the treatment of trinitroacetonitrile with aqueous NH3 resulted in the isolation of [NH4][(NO2)2CCN], [N2H5][(NO2)2CH] is formed in the reaction of trinitroacetonitrile with hydrazine hydrate. Most compounds of this study have been fully characterized by their X-ray crystal structure, vibrational and multinuclear NMR spectra, their decomposition temperature, as well as friction and impact sensitivities.Note added at Proof
During the peer-review process of this manuscript, M. A. Kettner and T. M. Klapötke coincidently reported the synthesis and characterization of ammonium 5-(fluorodinitromethyl)tetrazolate.30Acknowledgements
The Office of Naval Research (ONR) and the Defence Threat Reduction Agency (DTRA) financially supported this work. We acknowledge NSF CRIF grant 1048807 for the support of an X-ray diffractometer. We thank Professors G. K. S. Prakash and G. A. Olah for their help and stimulating discussions.Notes and references
- Z. X. Chen, J. M. Xiao, H. M. Xiao and Y. N. Chiu, J. Phys. Chem. A, 1999, 103, 8062–8066 CrossRef CAS.
- A. A. Kozyro, V. V. Simirskii, A. P. Krasulin, V. M. Sevruk, G. Y. Kabo, M. L. Frenkel, P. N. Gaponik and Y. V. Grigorev, Z. Fiz. Khim., 1990, 64, 656–661 CAS.
- J. B. Pedley, R. D. Naylor and S. P. Kirby, Thermochemical data of organic compounds, Chapman and Hall, London, New York, 2nd edn, 1986 Search PubMed.
- R. N. Butler, Tetrazoles, Pergamon Press, Oxford Oxfordshire, New York, 1st edn, 1984 Search PubMed.
- R. Haiges and K. O. Christe, Inorg. Chem., 2013, 52, 7249–7260 CrossRef CAS PubMed.
- R. Haiges, C. B. Jones and K. O. Christe, Inorg. Chem., 2013, 52, 5551–5558 CrossRef CAS PubMed.
- N. Fischer, T. M. Klapötke and J. Stierstorfer, Z. Anorg. Allg. Chem., 2009, 635, 271–281 CrossRef CAS PubMed.
- A. Hammerl, T. M. Klapötke, H. Nöth, M. Warchhold and G. Holl, Propellants, Explos., Pyrotech., 2003, 28, 165–173 CrossRef CAS PubMed.
- T. M. Klapötke, Struct. Bonding, 2007, 125, 85–121 CrossRef.
- T. M. Klapötke, C. Miro Sabate and M. Rasp, J. Mater. Chem., 2009, 19, 2240–2252 RSC.
- T. M. Klapötke, C. M. Sabate and J. Stierstorfer, New J. Chem., 2009, 33, 136–147 RSC.
- T. M. Klapötke and J. Stierstorfer, J. Am. Chem. Soc., 2009, 131, 1122–1134 CrossRef PubMed.
- G. I. Koldobskii, D. S. Soldatenko, E. S. Gerasimova, N. R. Khokhryakova, M. B. Shcherbinin, V. P. Lebedev and V. A. Ostrovskii, Russ. J. Org. Chem., 1997, 33, 1771–1783 CAS.
- F. D. Marsh, J. Org. Chem., 1972, 37, 2966–2969 CrossRef CAS.
- J. Stierstorfer, T. M. Klapötke, A. Hammerl and R. D. Chapman, Z. Anorg. Allg. Chem., 2008, 634, 1051–1057 CrossRef CAS PubMed.
- W. C. Lothrop and G. R. Handrick, Chem. Rev., 1949, 44, 419–445 CrossRef CAS.
- V. Grakauskas and A. H. Albert, Nitroalkyltetrazoles UCRL 15119, Fluorochem Inc., Azusa, CA 91702, 1979 Search PubMed.
- V. Grakauskas and A. H. Albert, J. Heterocycl. Chem., 1981, 18, 1477–1479 CrossRef CAS PubMed.
- R. Haiges, J. A. Boatz, M. Yousufuddin and K. O. Christe, Angew. Chem., Int. Ed., 2007, 46, 2869–2874 CrossRef CAS PubMed.
- SAINT+ V8.34A, Bruker AXS Madison, WI Search PubMed.
- SADABS V2014/4, Bruker AXS Madison, WI Search PubMed.
- SHELXTL V2014/6, Bruker AXS Madison, WI Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- C. O. Parker, W. D. Emmons, H. A. Rolewicz and K. S. McCallum, Tetrahedron, 1962, 17, 79–87 CrossRef CAS.
- R. A. Wiesboeck and J. K. Ruff, J. Org. Chem., 1968, 33, 1257–1258 CrossRef CAS.
- A. V. Shastin, T. I. Godovikova and B. L. Korsunsky, Chem. Heterocycl. Compd., 1998, 34, 383 CrossRef CAS.
- M. A. Kettner and T. M. Klapötke, Chem. Eur. J., 2015, 21, 3755–3765 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic reports including packing diagrams. CCDC 1044179–1044181. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00291e |
| This journal is © The Royal Society of Chemistry 2015 |