
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis, structure, topology and catalytic application of a novel cubane-based copper(II) metal–organic framework derived from a flexible amido tripodal acid†
Anirban
Karmakar
*ab,
Clive L.
Oliver
*c,
Somnath
Roy
b and
Lars
Öhrström
a
aChemical and Biological Engineering, Physical Chemistry, Chalmers University of Technology, SE-412 96, Gothenburg, Sweden. E-mail: ohrstrom@chalmers.se
bCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: anirbanchem@gmail.com
cCentre for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, Rondebosch, Cape Town 7701, South Africa. E-mail: Clive.Oliver@uct.ac.za
First published on 26th January 2015
Abstract
A novel chiral metal–organic framework, [Cu4(HL)2(H2O)4(MeO)4]n (1), has been successfully synthesized from a tripodal flexible ligand (2S,2′S,2′′S)-2,2′,2′′-(benzenetricarbonyltris(azanediyl))tripropanoic acid (H3L). Compound 1 was characterized by IR and X-ray powder diffraction analysis. The structure was determined by X-ray single crystal diffraction analysis revealing that 1 possesses a 3D network, featuring a tetranuclear cubane-type secondary building block [Cu4(MeO)4]4+, formed via the connection of four metal ions to four methoxide ions. These secondary building blocks are linked by four different HL2− ligands to construct a porous three dimensional framework of the dia topology with one-dimensional channels. Compound 1 also acts as a heterogeneous catalyst for the diastereoselective nitroaldol (Henry) reaction, providing high yields (up to 91%) and good diastereoselectivities under ambient conditions. This catalyst can be recycled without significant loss of activity.
1. Introduction
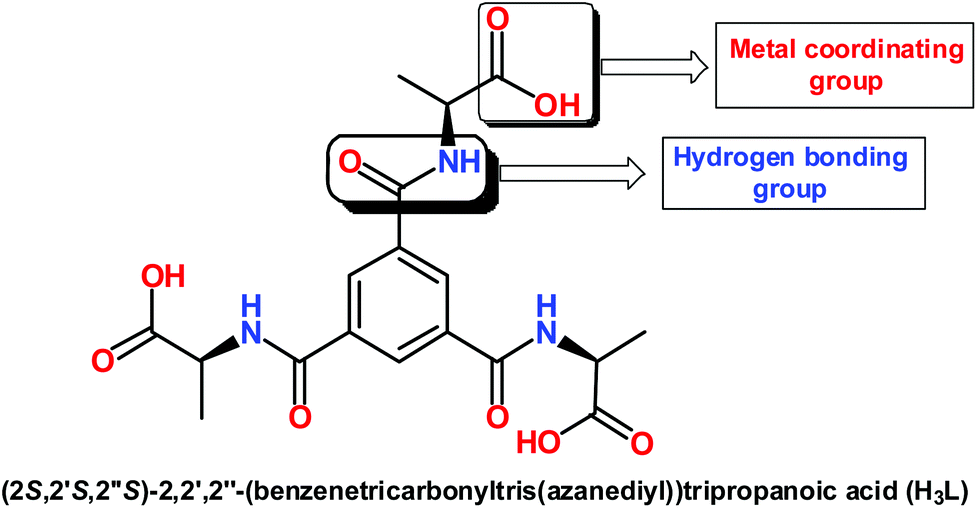
Porous metal–organic frameworks (MOFs)1 have attracted much interest in recent years due to their intriguing topologies1j–m along with potential applications in the fields of gas sorption,2 magnetism,3 nonlinear optics,4 chiral separation5 and catalysis.6 The judicious selection of appropriate linkers can give rise to the desired pore shape and size upon coordination and consequently contribute significantly to the structural design of MOFs, as well as their properties. Highly symmetrical multitopic ligands with dicarboxylate,7 tricarboxylate8 and tetracarboxylate9 functionalities may bridge mono- or multi-nuclear metal nodes, leading to stable MOFs with permanent porosity,10 while flexible ligands with additional functional groups may adopt more versatile conformations and coordination modes according to the geometric requirements of different metal ions, which may lead to unpredictable, yet interesting topologies and properties. However, only a few examples with flexible di- and tripodal ligands have been reported and the investigation of metal–organic frameworks based on them is still a great challenge.11The Henry or nitroaldol reaction is known as one of the most powerful and atom-economical reactions for C–C bond formation with various functionalized structural motifs.12 Usually this reaction is performed with homogeneous basic catalysts, such as alkali metal hydroxides, alkoxides or amines, with a rather good efficiency.13 The development of new catalysts and procedures for the Henry reaction has been constantly elaborated in view of the reduction of toxic by-products and the increase of yield and diastereoselectivity. In the last two decades many other asymmetric catalysts have been developed to convert aldehydes or α-keto esters into the corresponding nitroalkanol with good enantio- and diastereoselectivity.14 Even though high yields were achieved with reactions performed with homogeneous catalysts, the achievement of a high stereoselectivity is still challenging, and only a scant number of examples are known15 using heterogeneous catalysts. Some copper and zinc-containing metal organic frameworks16 have been reported, which actively catalyze the nitroaldol reaction. However, there is still a demand to develop new types of catalysts based on cheap and environmentally tolerable metal organic frameworks that could be easily recyclable (hence forming a heterogeneous system) and show a high efficiency under mild conditions.
Recently, a systematic investigation on a series of flexible di- and tripodal acid ligands containing various amino acid groups has been reported by us, which led to interesting hydrogen bonding networks.17 We have extended our study by synthesizing a higher dimensional motif (metal organic framework) with an easily accessible tripodal linker. Thus, two main objectives of this study were as follow: (i) to synthesize a Cu(II)-MOF using a tripodal acid linker; (ii) to apply the synthesized framework as a heterogeneous catalyst in the nitroaldol combination of nitroethane with various aldehydes. In that context, we chose (2S,2′S,2′′S)-2,2′,2′′-(benzenetricarbonyltris(azanediyl))tripropanoic acid (H3L) [Scheme 1] which is more flexible than benzene tricarboxylic acid as the carboxylic acid groups are reasonably free to rotate whilst the amide functional groups may potentially be involved in hydrogen bonding for further stabilization of the resulting structure.
 | ||
| Scheme 1 | ||
Hence, we report herein the synthesis and characterization of a new chiral copper(II) metal organic framework, prepared under hydrothermal conditions, using a flexible tripodal acid linker H3L [(2S,2′S,2′′S)-2,2′,2′′-(benzenetricarbonyltris(azanediyl))tripropanoic acid]. The structural features of the obtained Cu-MOF [Cu4(HL)2(H2O)4(MeO)4]n (1) could be established by single crystal X-ray diffraction analysis which revealed a cubane-type metal–oxygen cluster not yet observed for a metal–organic framework. The structure was also subjected to a topological study.
The catalytic performance of this framework as a heterogeneous catalyst, in terms of activity, heterogeneity, and recyclability, was successfully tested for the nitroaldol (Henry) reaction of nitroethane with various aldehydes in a methanolic medium.
2. Results and discussion
2a. Syntheses and characterization
The hydrothermal reaction of H3L with copper(II) nitrate trihydrate in the presence of dimethyl formamide and methanol leads to the formation of [Cu4(HL)2(H2O)4(MeO)4]n (1) [L = (2S,2′S,2′′S)-2,2′,2′′-(benzenetricarbonyltris(azanediyl))tripropanoate].In the IR spectra, the characteristic strong bands of the coordinated carboxylate groups of 1 appear at 1530 cm−1 and 1360 cm−1 for the asymmetric and the symmetric stretching, respectively. C–O stretching of the coordinated carboxylate group is observed at 1270 cm−1. A strong band at 1637 cm−1 is due to ν(CO) of the uncoordinated carboxylic acid,18 whereas ν(OH) of water molecules is in the 3303 cm−1 region. The band at 1580 cm−1 is attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the amide group. Metal organic framework 1 was also characterized by single crystal X-ray diffraction and powder X-ray diffraction analysis.
O stretching vibration of the amide group. Metal organic framework 1 was also characterized by single crystal X-ray diffraction and powder X-ray diffraction analysis.
Thermogravimetric analysis was also performed on framework 1 (Fig. S5, ESI†). Framework 1 shows a weight loss of 5.30% between 71 and 141 °C, corresponding to the loss of two molecules of water (calcd: 5.60%). Upon further heating, framework 1 exhibits a weight loss of 9.40% in the 180–239 °C temperature range, which accounts for the total removal of two coordinated methoxy molecules (calcd: 9.60%). The remaining material then starts to decompose from 241 °C until 492 °C.
2b. Crystal structure analysis
Single-crystal X-ray diffraction studies reveal that [Cu4(HL)2(H2O)4(MeO)4]n (1) crystallizes in a chiral space group (the tetragonal space group P43212), which is expected due to the chirality of the ligand. The asymmetric unit contains two copper(II) ions, one HL2− ligand, two water molecules and two methoxide ions (Fig. 1). The formulation of this framework is based on the typical Jahn–Teller distortion of Cu(II) (vide infra) and the Cu–O distances [1.945(4)–1.951(4) Å], which correspond closer to those observed for Cu–OMe (1.94 Å) than for Cu–O(H)Me (1.99 Å).19a In addition, the EPR results below confirm that the copper ions are in the 2+ oxidation state. | ||
| Fig. 1 Asymmetric unit of 1 with the partial atom labeling scheme. | ||
Both copper(II) centres have square pyramidal geometry (τ5 = 0.06 for Cu1 and τ5 = 0.04 for Cu2) and the coordination sphere of each copper(II) center is occupied by one carboxylate oxygen atom of a HL2− unit [Cu1–O2, 1.969(4) Å; Cu2–O5, 1.977(4) Å], three methoxide O-atoms and a water molecule. The carboxylate oxygen, two methoxide O-atoms and the water molecule occupy the equatorial sites and the axial site coordinated by the remaining methoxide ion. As expected and due to Jahn–Teller distortion19b–e the axial Cu–O bond distance is slightly longer [Cu1–O1M1, 2.424(4) Å; Cu2–O1M2, 2.405(4) Å] than the equatorial ones. The equatorial Cu–O bond lengths vary from 1.945(4) to 2.004(5) Å (Table S2†). Selected bond distances and angles of 1 are listed in Table S2 (ESI†).
In this framework tetranuclear cubane-type clusters of [Cu4(MeO)4]4+ (Fig. 2B) serve as secondary building block units (SBUs) and are located about 2-fold rotation axes. In the literature very few copper(II) complexes having methoxo-bridged20a–f and oxygen-bridged20g–j cubane-type tetranuclear clusters are reported, but to our knowledge, no metal–organic frameworks involving any type of oxygen bridge with the same cubane motif as observed for 1 have been reported, albeit that it has been observed for discrete clusters.20d
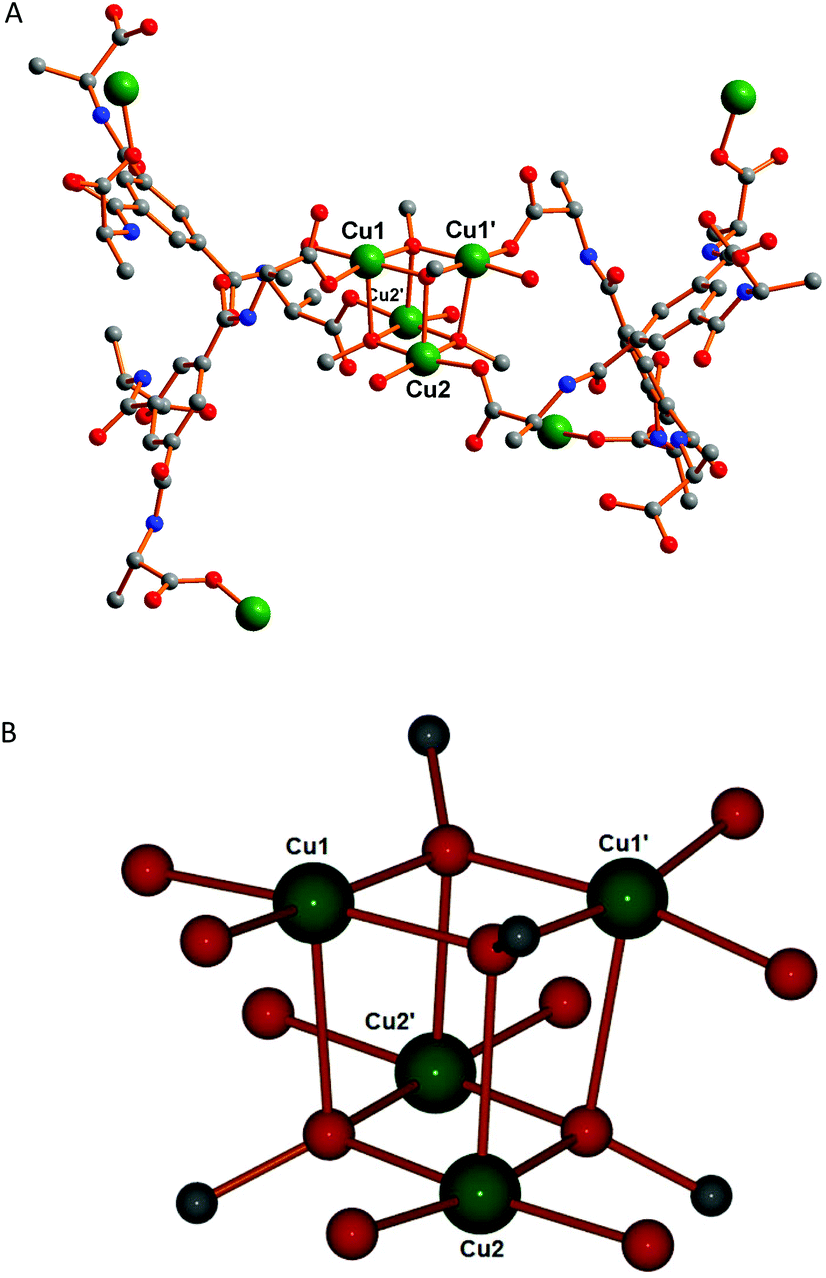 | ||
| Fig. 2 (A) Coordination scheme around the tetranuclear copper(II) core in 1. (B) Structure of the tetranuclear cubane-type clusters of the [Cu4(MeO)4]4+ unit showing the metal coordination spheres. | ||
Every tetranuclear cluster is associated with four HL2− ligands, four molecules of water and four μ3-methoxy ions. The carboxylate groups of ligand HL2− are associated with the two neighbouring tetranuclear clusters, while the carboxylic acid group remains uncoordinated and orients itself towards the open channels.
The packed view of 1 is characterized by open channels along the crystallographic c axis (Fig. 3) with approximate dimensions of 6 × 7 Å2 and a void space of 16.2% per unit cell according to PLATON.34 The structure of 1 is also stabilized by hydrogen bonding interactions, three amide hydrogen atoms of ligand HL2− are hydrogen bonded with three amide oxygen atoms of neighboring HL2− ligands via N1–H1⋯O1#1 2.9581(2) Å, N2–H2⋯O7#2 2.9302(2) Å and N3A–H3A⋯O4#3 3.0067(2) Å, #1 1/2 − y, 1/2 + x, −1/4 + z; #2 y, x, 1 − z; #3 −1/2 + y, 1/2 − x, 1/4 + z interactions. Hydrogen bonding interactions between carboxylate oxygen atoms O2 and O0AA (as acceptors) and the OW2 hydrogen atoms of water molecules (as bifurcated donors) are also observed [OW2–H1W2⋯O2#4 3.1165(2) Å, OW2–H2W2⋯O3#5 2.9566(2) Å, #4 1/2 + x, 1/2 − y, 1/4 − z; #5 1 − y, −x, 1/2 − z]. In addition, intermolecular C–H⋯O contacts are relevant and help in stabilizing the structure (Table S3, ESI†).
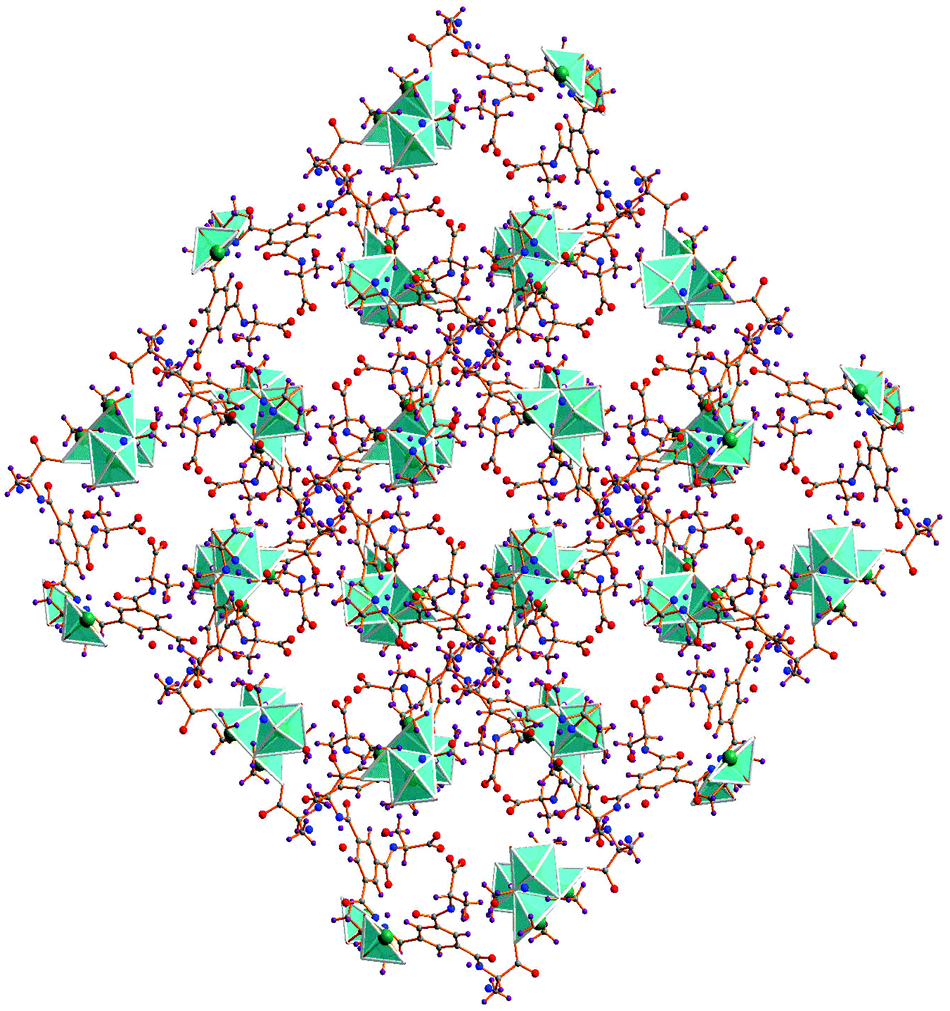 | ||
| Fig. 3 A representation of the 3D network of 1. | ||
The topology of 1 was determined by considering the Cu4O4 units as vertices and the ligand as a linear connector in analyzing the network with SYSTRE.21a,b This operation reveals the ubiquitous diamond or dia topology, the most common of all four connected nets.21b The structure with an overlaid network is shown in Fig. 4. It is noteworthy that although 1 crystallizes in an enantiomorphous space group the dia net is not one of the inherently chiral network topologies (qtz is the most common of these that are four-connected). As observed in the literature the topology of the net is not necessarily chiral even if the space group is chiral.22
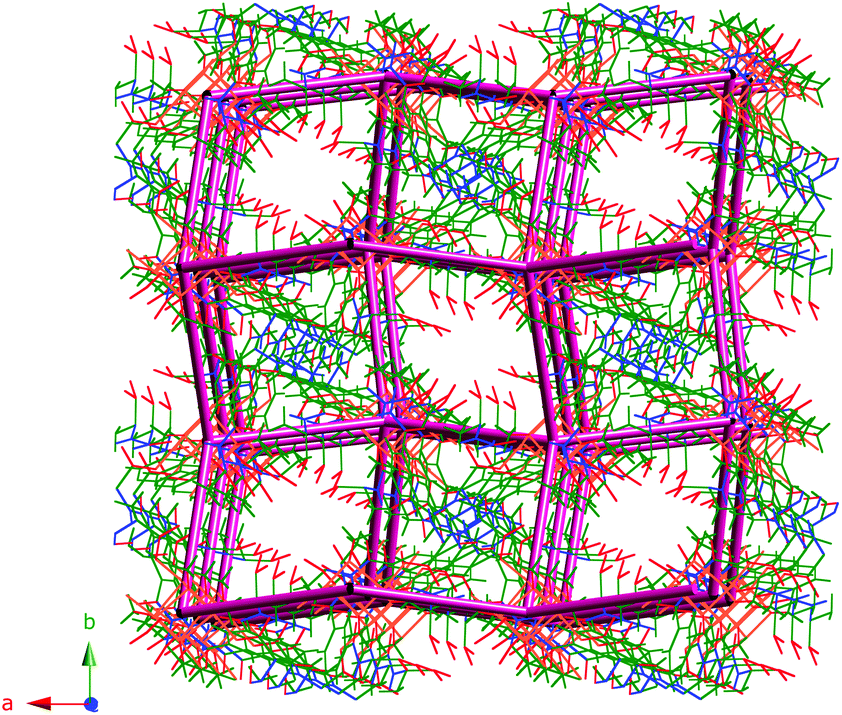 | ||
| Fig. 4 Structure of compound 1 with the underlying dia topology shown in purple. Solvent water molecules are not shown. | ||
3. Catalytic activity of 1 in the Henry reaction
We have tested the catalytic activity of the copper MOF 1 as a solid heterogeneous catalyst in the nitroaldol (or Henry) reaction of nitroethane with various aldehydes. In a typical reaction, a mixture of aldehyde (0.50 mmol), nitroethane (2.50 mmol), 3 mol% of Cu-MOF catalyst and methanol contained in a capped glass vessel was stirred at 70 °C for 24 h. The mixture was then centrifuged to remove the solid catalyst. The evaporation of the solvent gave the crude product which was a mixture of the β-nitroalkanol diastereoisomers (syn and anti forms, with the predominance of the former) which were analyzed by 1H NMR.The optimization of the reaction conditions (temperature, reaction time, the amount of catalyst, solvent) was carried out in a model nitroethane–benzaldehyde system with 1 as the catalyst (Scheme 2 with typical reaction conditions; Table 1).
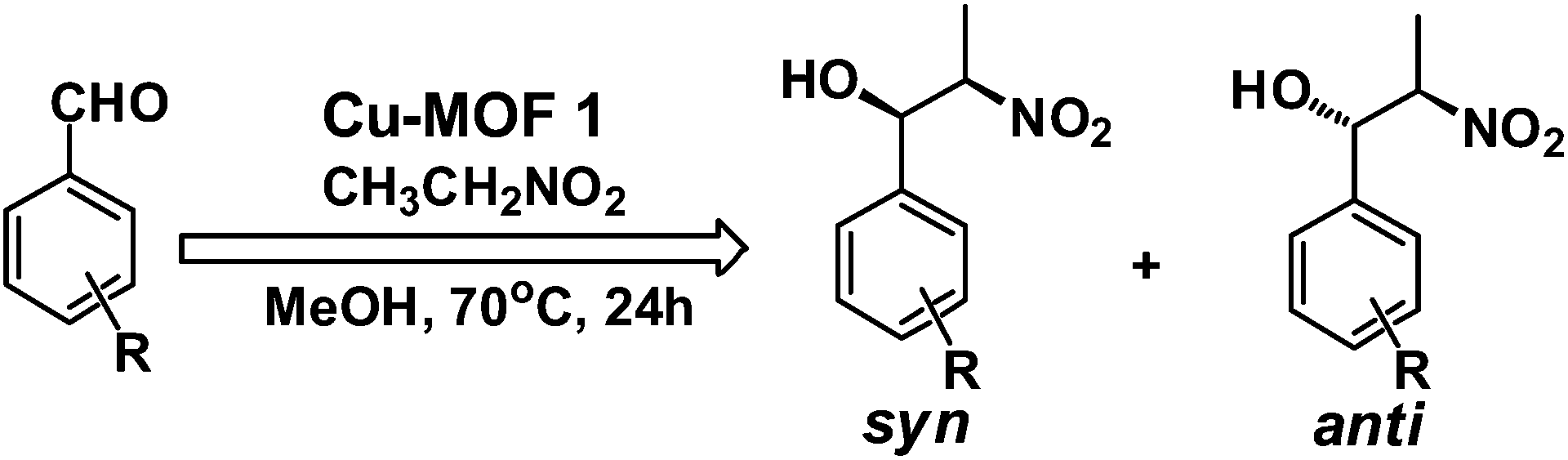 | ||
| Scheme 2 Nitroaldol (Henry) reaction. | ||
| Entry | Catalyst | Time (h) | Amount of the catalyst (mol%) | T (°C) | Solvent | Yieldb (%) | Selectivityc (syn/anti) | TONd |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 3.0 mol% of catalyst 1, benzaldehyde (0.50 mmol), nitroethane (2.50 mmol) and methanol (2 mL). b The number of moles of β-nitroalkanol per mole of the aldehyde × 100. c Calculated by 1H NMR. d The number of moles of β-nitroalkanol per mole of the catalyst (2 Cu per mole). e Nitromethane was used as the substrate; f Nitropropane was used as the substrate. | ||||||||
| 1 | 1 | 2 | 3.0 | 70 | MeOH | 40 | 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23 :23 |
13.3 |
| 2 | 1 | 4 | 3.0 | 70 | MeOH | 58 | 76:24 |
19.3 |
| 3 | 1 | 6 | 3.0 | 70 | MeOH | 65 | 75:25 |
21.6 |
| 4 | 1 | 8 | 3.0 | 70 | MeOH | 72 | 75:25 |
24.0 |
| 5 | 1 | 10 | 3.0 | 70 | MeOH | 78 | 75:25 |
26.0 |
| 6 | 1 | 24 | 3.0 | 70 | MeOH | 89 | 75:25 |
29.6 |
| 7 | 1 | 48 | 3.0 | 70 | MeOH | 91 | 76:24 |
30.3 |
| 8 | 1 | 24 | 1.0 | 70 | MeOH | 58 | 77:23 |
58.0 |
| 9 | 1 | 24 | 5.0 | 70 | MeOH | 79 | 79:21 |
15.8 |
| 10 | 1 | 24 | 7.0 | 70 | MeOH | 81 | 78:22 |
11.5 |
| 11 | 1 | 24 | 3.0 | 70 | CH3CN | 12 | 79:21 |
4.0 |
| 12 | 1 | 24 | 3.0 | 70 | THF | 45 | 80:20 |
15.0 |
| 13 | 1 | 24 | 3.0 | 70 | Water | 84 | 80:20 |
28.0 |
| 14 | 1 | 24 | 3.0 | 20 | MeOH | 5 | 79:21 |
1.6 |
| 15 | 1 | 24 | 3.0 | 30 | MeOH | 35 | 77:23 |
11.7 |
| 16 | 1 | 24 | 3.0 | 50 | MeOH | 71 | 78:22 |
23.7 |
| 17 | 1 | 24 | 3.0 | 110 | MeOH | 63 | 69:31 |
21.0 |
| 18 | Blank | 24 | — | 70 | MeOH | — | — | — |
| 19 | Cu(NO3)2·3H2O | 24 | 3.0 | 70 | MeOH | 24 | 82:18 |
8.0 |
| 20 | H3L | 24 | — | 70 | MeOH | — | — | — |
| 21e | 1 | 24 | 3.0 | 70 | MeOH | 87 | — | 29.0 |
| 22f | 1 | 24 | 3.0 | 70 | MeOH | 23 | 91:9 |
7.7 |
When 3 mol% of solid complex 1 is used as the catalyst at 70 °C, a conversion of 89% (syn/anti: 75:25) of benzaldehyde into β-nitroalkanol is reached (entry 6, Table 1) after 24 h. Upon extending the reaction time to 48 h, the reaction yield increased slightly (91%, syn/anti: 76:24). The plot of yield versus time for the Henry reaction of benzaldehyde and nitroethane with complex 1 is presented in Fig. 5A.
 | ||
| Fig. 5 (A) Plots of β-nitroalkanol yield vs. time for the Henry reaction of benzaldehyde and nitroethane in the presence of compound 1 as the catalyst. (B) Kinetic profiles in four consecutive reaction cycles employing 1 as the catalyst. | ||
A blank reaction was performed (the absence of any metal source; Table 1, entry 18) using benzaldehyde as the substrate, at 70 °C and in methanol. No β-nitroalkanol was detected after a reaction time of 24 h. The nitroaldol reaction also did not take place when using compound H3L instead of catalyst 1 (Table 1, entry 20). We have also checked the reactivity of Cu(NO3)2·3H2O in methanol medium and the obtained reaction yield was much lower, i.e. ca. 24% (entry 19, Table 1).
We have also tested the effects of temperature, catalyst amount and solvents. The increase in the amount of catalyst 1 from 1.0 to 3.0 mol% enhances the product yield from 58 to 89%, but a further increase in the amount of the catalyst leads to a slight decrease of the reaction yield (entries 8–10, Table 1).
To select the most suitable solvent, experiments with various solvents (CH3CN, THF, MeOH and water) have been carried out with 1 and the results (Table 1, entries 6 and 11–13) indicate that methanol (conversion of 89%) is the best polar solvent, whereas the worst one is CH3CN (12% conversion). In water and THF, yields of 84% or 45%, respectively, were obtained (Table 1, entries 12 and 13).
Varying the temperature from 20 to 70 °C improved the yield of β-nitroalkanol from 5 to 89% but a further temperature increase had a negative effect (entries 14–17, Table 1). The systems exhibit diastereoselectivity towards the syn isomer, typically leading to syn/anti molar ratios in the 82:18 to 78:22 range using nitroethane as the substrate. The selectivity in syn and anti products is an important issue in the Henry reaction and efforts have been focused on the development of catalytic diastereo- or enantio-selective processes.23 The size of the nitroalkane chain also affects the yield, e.g. with nitropropane the conversion was only 23% (entry 22, Table 1), whilst with nitroethane or nitromethane conversion values of 89% or 87%, respectively, were obtained (entries 6 and 21, Table 1).
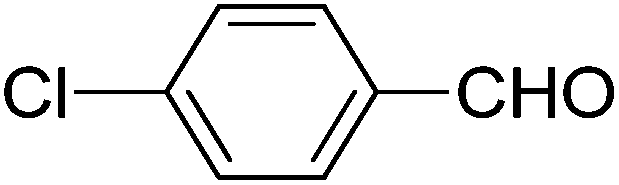
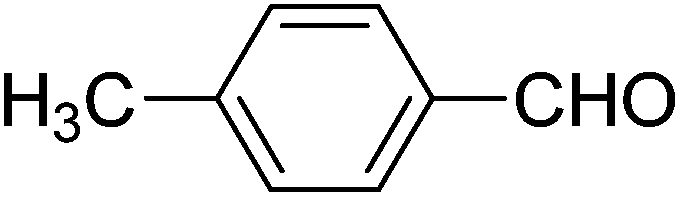
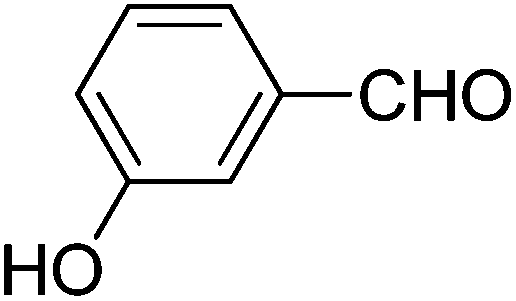
We also examined the catalytic activity of 1 with different types of substituted aromatic and aliphatic aldehydes in the reaction with nitroethane. The results are summarized in Table 2. p-Nitrobenzaldehyde produced the maximum yield (95%), while the lowest yield (27%) was obtained for p-methoxybenzaldehyde. The yields for two aliphatic aldehydes were good (84–85%) but lower than that for benzaldehyde.
| Aldehyde | Yieldb (%) | Selectivityc (syn/anti) | TONd |
|---|---|---|---|
| a Reaction conditions: 3.0 mol% of catalyst 1, aldehyde (0.50 mmol), nitroethane (2.5 mmol) and methanol (2 mL). b The number of moles of β-nitroalkanol per mole of the aldehyde × 100. c Calculated by 1H NMR. d The number of moles of β-nitroalkanol per mole of the catalyst. | |||

|
95 | 78:22 |
31.7 |

|
27 | 76:24 |
9.0 |
|
81 | 71:29 |
27.0 |
|
34 | 79:21 |
11.3 |
|
47 | 77:23 |
15.7 |
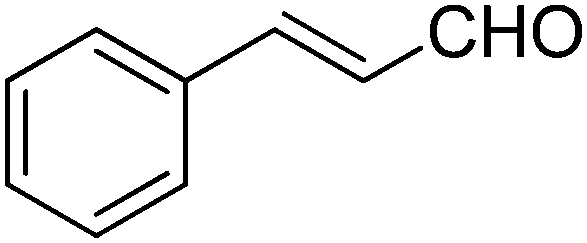
|
52 | 87:13 |
17.3 |
| CH3CHO | 85 | 78:12 |
28.3 |
| CH3CH2CHO | 84 | 75:24 |
28.0 |
Competition reactions involving equal molar amounts of p-nitrobenzaldehyde and acetaldehyde were conducted to probe whether catalysis occurs primarily on the surface of the MOF or within the pores. If catalysis were occurring to a significant extent within pores, we might well expect the 3-nitrobutan-2-ol product to be formed preferentially over the 2-nitro-1-(4-nitrophenyl)propan-1-ol, since the internal cavities of framework 1 would have selectively adsorbed small molecules (acetaldehyde) over large molecules (p-nitrobenzaldehyde) due to its smaller pore size (6 Å × 7 Å channels). We have observed that p-nitro benzaldehyde produces more yield than acetaldehyde. Also we have used various sizes of different aldehydes but we did not observe any selectivity for a particular aldehyde, the aryl aldehydes bearing electron-withdrawing groups exhibit higher reactivities than those having electron-donating moieties, which may be related to an increase in the electrophilicity of the substrate in the former case. Thus, it may be possible that catalysis occurs mainly on the surface of the MOF and not on the pores of framework 1.
In order to examine the stability of 1 in the Henry reaction, it was recycled in four consecutive experiments, and it was observed that its activity remained almost the same (Fig. 5B). FT-IR and powder X-ray diffraction of catalyst 1 performed before and after the reaction indicated that the structure of the solid was retained (Fig. S1†). In addition, the filtered solution, after the separation of the catalyst, was evaporated and the amount of copper was determined, being only 0.012% of the amount used in the reaction, thus ruling out any significant leaching of the catalyst. This test also indicates the significant stability of framework 1 during the catalytic reaction.
Although there are some reports on coordination polymers16 which are catalytically active for this kind of reaction, the yields and selectivity are usually higher for our compound as compared to other metal organic frameworks. Recently, Chuan-De Wu et al. reported that a 3D porous metal–organic framework constructed from two kinds of pyridine carboxylates and copper(II) ions, in the reaction of 4-nitrobenzaldehyde and nitroethane, leads to an overall yield of only 78% after 36 h reaction time (in our case 95% in 24 h).16c Moreover, our catalyst 1 exhibits a marked selectivity towards the syn diastereoisomer (Table 2, syn:anti of 78:22) which was not reported in other cases. In comparison with other reported heterogeneous catalysts for the Henry reaction, our catalyst has the advantages of being relatively cheap and easy-to-prepare.
4. EPR analysis
The EPR spectrum of 1 was recorded in the solid state, Fig. 6(a), at room temperature and (b) at 77 K. The X-band EPR spectrum of 1 depicts a broad band centred at g = 2.15 (3171 G for ν = 9.526 GHz) which corresponds to the transition ΔMS = ±1. No clearly detectable zero field splitting or half-field signals were observed. The frozen solution spectrum, Fig. 6(b), shows a band centred at g = 2.14 which also corresponds to the transition ΔMS = ±1. By simulation of the spectra, the following g values were obtained: g1 = 2.05, g2 = 2.17 and g3 = 2.23. | ||
| Fig. 6 First derivative EPR spectra of 1 at room temperature (a) and at 77 K (b). | ||
The geometric parameter G, which is a measure of the exchange interaction between the copper centres in the polycrystalline compound, is calculated using the equation: for rhombic spectra G = (g3 − 2.003)/(g⊥ − 2.003), where g⊥ = (g1 + g2)/2. If G < 4.0, a considerable exchange interaction is indicated in the solid complexes.24 The G value for compound 1 in the polycrystalline state at 77 K is 2.12. For complexes of this type, the parameter R can be indicative of the predominance of the dz2 or dx2–y2 orbital in the ground state, where R = (gz − gy)/(gy − gx). If R > 1, the greater contribution to the ground state arises from the dz2 orbital and the structure is closer to a trigonal bipyramid than to a square pyramid. Instead, if R < 1, a greater contribution to the ground state arises from the dx2–y2 orbital and the structure is closer to a square pyramid than to a trigonal bipyramid. In the present case, R = 0.5, being compatible with the X-ray diffraction data. The solution spectrum could not be obtained due to the insolubility of the framework 1 in common organic solvents. This result unequivocally confirms the oxidation state as Cu(II) as inferred in section 2b.
5. Conclusion
We successfully isolated a copper(II) metal organic framework derived from a tripodal flexible ligand (2S,2′S,2′′S)-2,2′,2′′-(benzenetricarbonyltris(azanediyl))tripropanoic acid (H3L) under hydrothermal conditions. The single-crystal X-ray diffraction experiment revealed that this framework consists of tetranuclear cubane-type clusters of [Cu4(MeO)4]4+ acting as secondary building blocks. The tetranuclear cubane of copper(II) is formed via the connection of four metal ions by four μ3-bridged methoxide ions. This metal–oxygen motif, although found for discrete metal-clusters, has not been observed as part of a metal–organic framework hitherto. The framework has one dimensional channels along the c axis with approximate dimensions of 6 × 7 Å2 and a void space of 16.2% per unit cell volume.Framework 1 effectively catalyzes the Henry reaction of nitroethane with various aldehydes producing the corresponding β-nitroalkanols in high yields with a significant stereoselectivity towards the syn diastereomer. We have also proved the stability and recyclability of the catalysts. The syn/anti ratio of the nitroaldol products depends on various factors such as the amount of the catalyst, the electrophilicity of the substrates and the reaction conditions.
The above observations provide further evidence that simple Cu(II) complexes can be utilized as effective heterogeneous catalysts in the important types of reactions in this study. Further explorations of the use of this catalyst family in other organic transformations, as well as mechanistic investigations, are ongoing.
6. Experimental
All the chemicals were obtained from commercial sources and used as received. (2S,2′S,2′′S)-2,2′,2′′-(Benzenetricarbonyltris(azanediyl))tripropanoic acid (H3L) was prepared according to ref. 17. The infrared spectra (4000–400 cm−1) were recorded on a Bruker IFS-125 spectrometer using KBr pellets; abbreviations: s = strong, m = medium, w = weak, bs = broad and strong, mb = medium and broad. Powder X-ray diffraction (PXRD) was conducted on a D8 Advance Bruker AXS (Bragg Brentano geometry) theta–2theta diffractometer, with copper radiation (Cu Kα, λ = 1.5406 Å) and a secondary monochromator, operated at 40 kV and 40 mA. The flat plate configuration was used and the typical data collection range was between 5° and 35° 2θ. Thermogravimetric analysis was carried out with a Perkin-Elmer Instrument system (STA6000) at a heating rate of 10 °C min−1 under a dinitrogen atmosphere, in the range of room temperature to ca. 600 °C. The X-band EPR spectra were recorded either at room temperature (RT) or at liquid nitrogen (LN) temperature (77 K) on a Bruker ESP 300E spectrometer. The ESP 300E spectrometer was operated at ∼9.51 GHz with a frequency modulation of 100 kHz. While keeping the resolution either at 2048 points or at 4096 points, the microwave power was adjusted to 20 dB attenuations and the receiver gain was set to either 3.0 × 105 or 4.0 × 105. To improve the signal to noise ratio, 10 scans of each sample were accumulated. The spectral acquisition parameters were constant for each experiment. All measurements were performed using 3 mm quartz tubes (Wilmad 707-SQ-250M). The EPR spectra were simulated using an EPR simulation program.256a. Synthesis of 1
A mixture of H3L (42 mg, 0.1 mmol) and Cu(NO3)2·3H2O (24 mg, 0.1 mmol) was dissolved in 4 mL of MeOH–DMF (1:0.1 v/v) and placed in a capped glass vessel. Then 0.1 ml of 1 (M) KOH is added into it and mixed thoroughly, a blue precipitate appeared immediately. The precipitate was dissolved by adding 0.5 ml of NH4OH drop-wise until the solution turned deep blue. The resulting mixture was sealed in an 8 mL glass vessel and heated at 80 °C for 48 h. After cooling to room temperature the reaction mixture was kept for slow evaporation. After 7 days deep blue block crystals appeared. The yield was 34% (based on Cu), which was calculated based on the crystalline material obtained. A lower reaction yield of Cu(II) frameworks was also observed in some previously reported studies.26 FT-IR (KBr, cm−1): 3303 (bs), 1637 (s), 1530 (s), 1450 (w), 1407 (m), 1360 (s), 1270 (w), 1090 (w), 724 (s).
6b. Procedure for the nitroaldol (Henry) reaction catalyzed by 1
In a typical reaction, a mixture of aldehyde (0.50 mmol), nitroethane (2.50 mmol) and catalyst 1 (9.70 mg, 3 mol%) was taken in a capped glass vessel, and then 2.0 mL of methanol were added into it. The mixture was heated at 70 °C for 24 h, and subsequently quenched by centrifugation and filtration at room temperature. The filtrate was extracted with dichloromethane. The organic extracts were collected over anhydrous sodium sulfate; subsequent evaporation of the solvent gave the crude product. The product was dissolved in DMSO-d6 and analyzed by 1H NMR. The yield of the β-nitroalkanol product (relative to the aldehyde) was established typically by taking into consideration the relative amounts of these compounds, as given by 1H NMR and previously reported.16d The ratio between the syn and anti isomers was also determined by 1H NMR spectroscopy. In the 1H NMR spectra, the values of vicinal coupling constants (for the β-nitroalkanol products) between the α-N–C–H and the α-O–C–H protons help identifying the isomers, being J = 7–9 or 3.2–4 Hz for the syn or anti isomers, respectively.27 The 1H-NMR spectra and the calculation of the yield and selectivity for framework 1 in the Henry reaction are presented in ESI (Fig. S4†).In order to perform the recycling experiment, first we washed the used catalyst (separated by centrifugation and filtration of the supernatant solution) with methanol and dried it in air. It was then reused for the nitroaldol reaction as described above.
6c. X-ray crystal structure determination
A suitable crystal of 1 was immersed in cryo-oil, mounted on a Nylon loop, and measured at a temperature of 100(2) K using an Oxford Cryostream 700. The X-ray diffraction data were collected on a Bruker DUO APEX II CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The data reduction and cell refinement were performed using SAINT-Plus28 and scaled for absorption effects by multi-scan using SADABS.29 The space group was determined from systematic absences by XPREP30 and confirmed using the program Layer.31 The structures were solved by direct methods using SHELXS-9732 and subsequently refined by full-matrix least-squares methods, based on F2 values against all reflections.33 Non-hydrogen atoms were refined with anisotropic displacement parameters, whilst hydrogen atoms were placed geometrically, except for water hydrogen atoms which were placed from difference Fourier maps. Thermal parameters of hydrogen atoms were constrained to Uiso = 1.2Ueq (parent atom). Excess electron density in the channels that could not be modelled was ‘cleaned’ using the SQUEEZE routine in PLATON.34 CCDC 1024691 for compound 1 contains the supplementary crystallographic data for this paper.Acknowledgements
AK expresses his gratitude to the Fundação para a Ciência e a Tecnologia for post-doctoral fellowships (Ref. No. SFRH/BPD/76192/2011), LÖ and CLO thank the Swedish Research Links program for Southern Africa, LÖ is also grateful for funding from the Swedish Science Council and Chalmers Tekniska Högskola and CLO thanks the National Research Foundation of South Africa for funding.References
-
(a) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
; (b) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef
-
(a) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS PubMed
-
(a) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC
-
(a) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS PubMed
-
(a) M. Padmanaban, P. Müller, C. Lieder, K. Gedrich, R. Grünker, V. Bon, I. Senkovska, S. Baumgärtner, S. Opelt, S. Paasch, E. Brunner, F. Glorius, E. Klemm and S. Kaskel, Chem. Commun., 2011, 47, 12089–12091 RSC
-
(a) A. U. Czaja, N. Trukhan and U. Muller, Chem. Soc. Rev., 2009, 38, 1284 RSC
-
(a) A. Schaate, S. Klingelhoefer, P. Behrens and M. Wiebcke, Cryst. Growth Des., 2008, 8, 3200–3205 CrossRef CAS
-
(a) D. Sun, S. Ma, Y. Ke, D. J. Collins and H. Zhou, J. Am. Chem. Soc., 2006, 128, 3896–3897 CrossRef CAS PubMed
-
(a) Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS PubMed
-
(a) B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS PubMed
-
(a) H.-T. Zhang, Y.-Z. Li, T.-W. Wang, E. N. Nfor, H.-Q. Wang and X.-Z. You, Eur. J. Inorg. Chem., 2006, 3532–3536 CrossRef CAS
-
(a) F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS
-
(a) R. Ballini, G. Bosica and P. Forconi, Tetrahedron, 1996, 52, 1677–1684 CrossRef CAS
-
(a) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561–2574 CrossRef CAS
-
(a) A. Bulut, A. Aslan and O. Dogan, J. Org. Chem., 2008, 73, 7373–7375 CrossRef CAS PubMed
-
(a) X.-M. Lin, T.-T. Li, Y.-W. Wang, L. Zhang and C.-Y. Su, Chem. – Asian J., 2012, 7, 2796–2804 CrossRef CAS PubMed
- A. Karmakar, C. L. Oliver, A. E. Platero-Prats, E. Laurila and L. Öhrström, CrystEngComm, 2014, 16, 8243–8251 RSC
-
(a)
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley & Sons, New York, 5th edn, 1997 Search PubMed
-
(a) A. Nimmermark, L. Öhrström and J. Reedijk, Z. Kristallogr., 2013, 228, 311–317 CrossRef CAS
-
(a) R. W. Saalfrank, C. Spitzlei, A. Scheurer, H. Maid, F. W. Heinemann and F. Hampel, Chem. – Eur. J., 2008, 14, 1472–1481 CrossRef CAS PubMed
-
(a) O. Delgado Friedrichs and M. O'Keeffe, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2003, 60, 351–360 CrossRef PubMed
-
(a) M. T. Johnson, Z. Džolić, M. Cetina, M. Lahtinen, M. S. G. Ahlquist, K. Rissanen, L. Öhrström and O. F. Wendt, Dalton Trans., 2013, 42, 8484–8491 RSC
-
(a) V. J. Bulbule, G. K. Jnaneshwara, R. R. Deshmukh, H. B. Borate and V. H. Deshpande, Synth. Commun., 2001, 31, 3623–3626 CrossRef CAS PubMed
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS
- S. Roy, R. J. Butcher, M. S. El Fallah, J. Tercero and J. C. Pessoa, Polyhedron, 2013, 53, 269–277 CrossRef CAS PubMed
- Z. Lu, H. Xing, R. Sun, J. Bai, B. Zheng and Y. Li, Cryst. Growth Des., 2012, 12, 1081–1084 CAS
- V. J. Bulbule, V. H. Deshpande, S. Velu, A. Sudalai, S. Sivasankar and V. T. Sathe, Tetrahedron, 1999, 55, 9325–9332 CrossRef CAS
-
Bruker AXS Inc., Madison, Wisconsin, USA, 2004
-
G. M. Sheldrick, University of Göttingen, Göttingen, Germany, 1996
-
Bruker AXS Inc., Madison, Wisconsin, USA, 2003
- L. J. Barbour, J. Appl. Crystallogr., 1999, 351–352 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1024691. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03087g |
| This journal is © The Royal Society of Chemistry 2015 |