
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Turning a “useless” ligand into a “useful” ligand: a magneto-structural study of an unusual family of CuII wheels derived from functionalised phenolic oximes†
Jamie M.
Frost
a,
Robert J.
Stirling
a,
Sergio
Sanz
a,
Nidhi
Vyas
b,
Gary S.
Nichol
a,
Gopalan
Rajaraman
*b and
Euan K.
Brechin
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: ebrechin@staffmail.ed.ac.uk; Tel: +44 (0)131-650-7545
bDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, 400076 Mumbai, India. E-mail: rajaraman@chem.iitb.ac.in
First published on 2nd April 2015
Abstract
While the phenolic oximes (R-saoH2) are well known for producing monometallic complexes of the type [MII(R-saoH)2] with CuII ions in near quantitative yield, their derivatisation opens the door to much more varied and interesting coordination chemistry. Here we show that combining the complimentary diethanolamine and phenolic oxime moieties into one organic framework (H4L1 and H4L2) allows for the preparation and isolation of an unusual family of [CuII]n wheels, including saddle-shaped, single-stranded [CuII8] wheels of general formula [Cu8(HL1)4(X)4]n[Y] (when n = 0, X = Cl−, NO3−, AcO−, N3−; when n = 2+ X = (OAc)2/(2,2′-bpy)2 and Y = [BF4]2) and [Cu8(HL2)4(X)4] (X = Cl−, Br−), a rectangular [Cu6(HL1)4] wheel, and a heterometallic [Cu4Na2(HL1)2(H2L1)2] hexagon. Magnetic studies show very strong antiferromagnetic exchange between neighbouring metal ions, leading to diamagnetic ground states in all cases. DFT studies reveal that the magnitude of the exchange constants are correlated to the Cu–N–O–Cu dihedral angles, which in turn are correlated to the planarity/puckering of the [CuII]n rings.
Introduction
In the early days of molecular magnetism low nuclearity complexes containing CuII ions were the focus of intense research efforts by synthetic chemists and theoreticians.1–5 The ease of synthesis of new compounds permitted the facile preparation of families of related molecules, providing simple (s = 1/2) model systems with which to test emerging theories and propose new experiments.1 In 1952 Bleaney and Bowers published a study of the magnetic properties of copper(II) acetate monohydrate,2 and some twenty three years later Hatfield and Hodgson published a magneto-structural correlation (MSC) demonstrating the linear relationship between the strength of the magnetic exchange interaction, J, and the Cu–O–Cu bridging angle in an extended family of bis(μ-hydroxido)copper(II) dimers.3,4 The first pre-designed construction of a molecule containing ferromagnetically coupled metal ions then followed in 1978 with the molecule [CuVO(fsa)2en] (where (fsa)2en4− is the bi-chelating ligand derived from the Schiff-base bis(2′-hydroxy-3′-carboxybenzylidene)-1,2-diaminoethane) in which the magnetic orbitals on the constituent metal ions were rigorously orthogonal.5 The modern era has seen researchers turning their attention to increasingly more complex systems to examine, for example, spin frustration effects in Archimedean and Platonic solids such as CuII cuboctahedra,6 and spin-electric coupling in antiferromagnetic [CuII3] triangles resulting from the interplay between exchange, spin–orbit coupling and spin chirality.7In the sixty or so years since the initial magnetic study of [Cu(OAc)2·H2O]2 numerous different ligand types have been employed to construct molecules containing multiple CuII ions.8 However, one ligand type which can be almost entirely excluded from this list are the phenolic oximes (R-saoH2), since they have been shown to have an overwhelming propensity to form mononuclear complexes of the type [Cu(R-saoH)2] in near quantitative yields.9 Indeed their remarkable selectivity for copper in the 2+ oxidation state has been exploited in industrial hydrometallurgy, with almost a quarter of the world's copper production involving salicylaldoxime-based ligands.9 This peculiar selectivity can be ascribed to the tendency of the ligands to form hydrogen bonded head-to-tail dimers mediated via intermolecular interactions between the oximic hydrogen atom on one unit and the phenolic oxygen atom of its neighbour (Fig. 1). The resulting pseudo-macrocyclic cavity facilitates metal complexation, and selectivity for CuII arises from the goodness-of-fit of this metal cation with the cavity.10
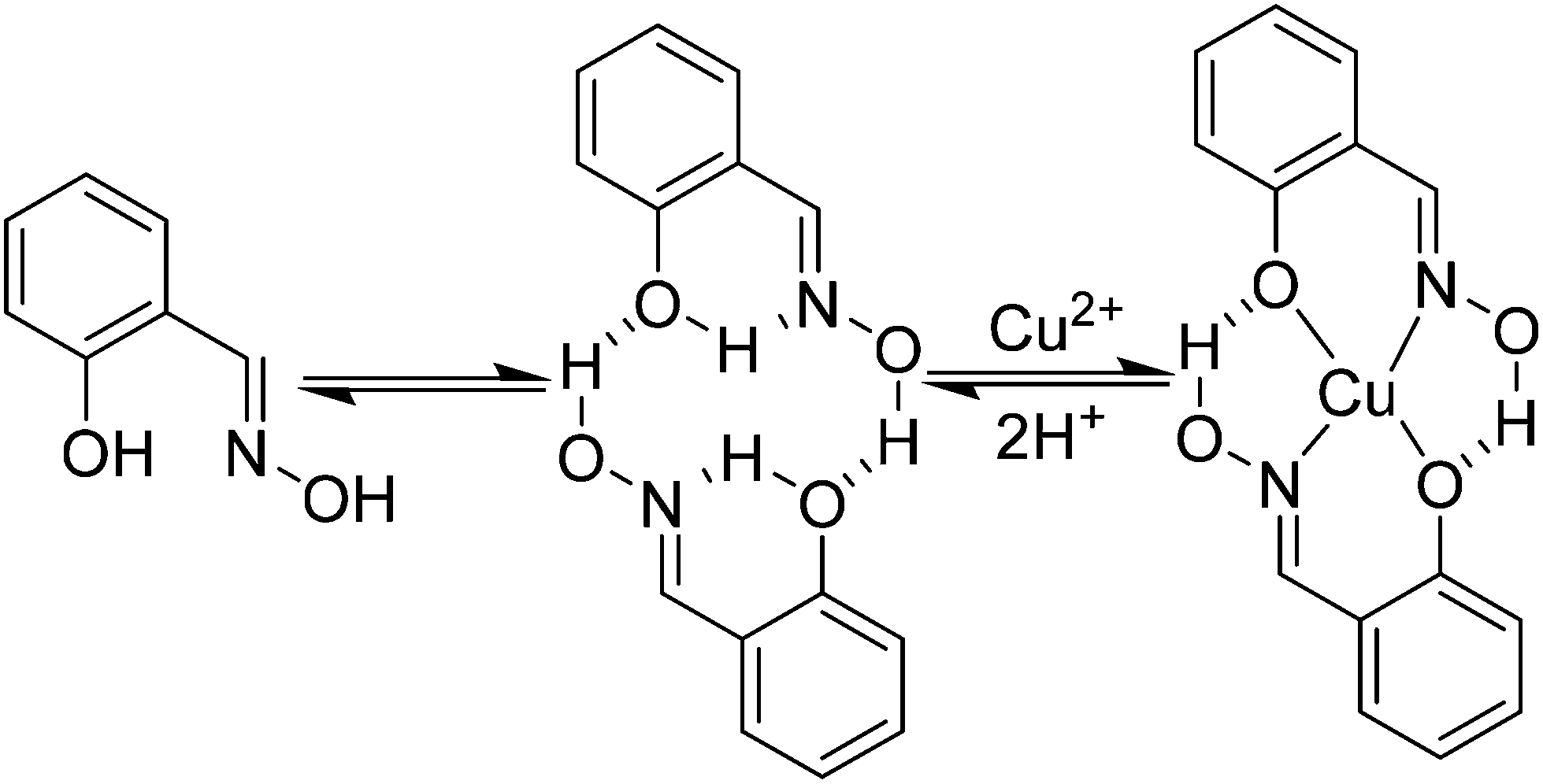 | ||
| Fig. 1 The pre-organisation of phenolic oximes for the formation of [CuII(R-saoH)2] species. | ||
Given the above, it is perhaps unsurprising that a search of the CSD reveals that there is only one example of a polymetallic CuII cage containing two or more metal ions stabilised solely by phenolic oximes.11 The complex, [Cu6(L3-2H)3(μ3-O)(μ3-OH)](PF6)3, describes two [CuII3O] triangles linked by three “double-headed” phenolic oximes, and forms when the ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal ratio employed in the reaction mixture is low; i.e. when the metal ions are present in excess.11 Perhaps the historical observation of a lack of variety in the coordination chemistry of CuII with phenolic oximes has prevented others from investigating the chemistry further, but the formation of the hexametallic CuII6 species is clear evidence that more interesting structures remain undiscovered. Indeed the [CuII3O(oxime)3] triangles of the hexametallic cage are entirely analogous to the building blocks previously observed for MnIII, FeIII and CrIII; all of which present more varied and more interesting structural and physical chemistry.12 In order to address this “misperception” and to prove that polymetallic cages of CuII can be built with this ligand type, we have adopted a synthetic approach which has already proven successful in Mn chemistry.13 While both salicylaldoxime and diethanolamine have very limited track records in CuII chemistry (a CSD search for the latter also returns only monometallic complexes) their combination into one single organic structural framework has the potential to transform the coordination abilities of both. Herein we show that this complimentary ligand approach to constructing novel polymetallic cluster compounds works rather well, by presenting the synthesis, structures and magnetic behaviour of a large and unusual family of CuII-based wheels constructed using the pro-ligands H4L1 and H4L2 (Fig. 2).
:metal ratio employed in the reaction mixture is low; i.e. when the metal ions are present in excess.11 Perhaps the historical observation of a lack of variety in the coordination chemistry of CuII with phenolic oximes has prevented others from investigating the chemistry further, but the formation of the hexametallic CuII6 species is clear evidence that more interesting structures remain undiscovered. Indeed the [CuII3O(oxime)3] triangles of the hexametallic cage are entirely analogous to the building blocks previously observed for MnIII, FeIII and CrIII; all of which present more varied and more interesting structural and physical chemistry.12 In order to address this “misperception” and to prove that polymetallic cages of CuII can be built with this ligand type, we have adopted a synthetic approach which has already proven successful in Mn chemistry.13 While both salicylaldoxime and diethanolamine have very limited track records in CuII chemistry (a CSD search for the latter also returns only monometallic complexes) their combination into one single organic structural framework has the potential to transform the coordination abilities of both. Herein we show that this complimentary ligand approach to constructing novel polymetallic cluster compounds works rather well, by presenting the synthesis, structures and magnetic behaviour of a large and unusual family of CuII-based wheels constructed using the pro-ligands H4L1 and H4L2 (Fig. 2).
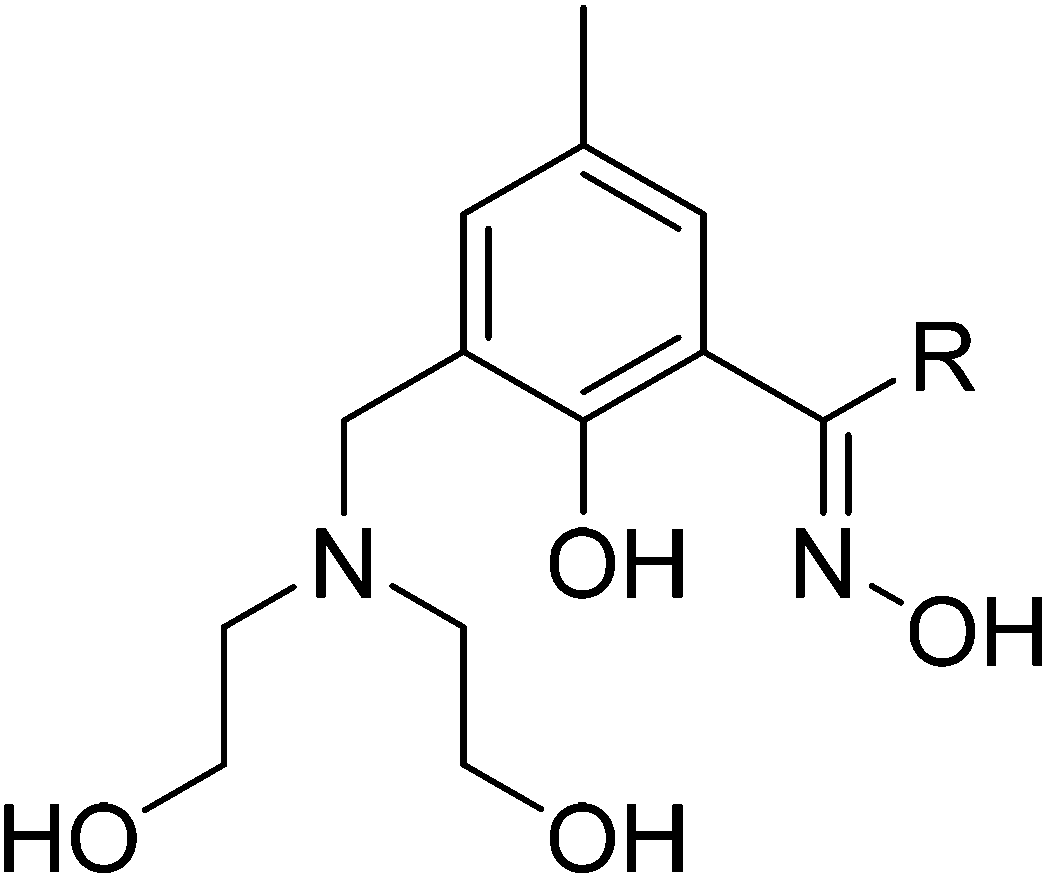 | ||
| Fig. 2 Generalised molecular structure of the pro-ligands H4L1 (R = Me) and H4L2 (R = Et). | ||
Experimental
Materials and physical measurements
All manipulations were performed under aerobic conditions using materials as received (reagent grade). The ligand H4L1 {1-(3-((bis(2-hydroxyethyl)amino)methyl)-2-hydroxy-5-methyl) phenyl)ethanone oxime} was synthesised according to published procedures.13a The synthesis of H4L2 {1-(3-((bis(2-hydroxyethyl)amino)methyl)-2-hydroxy-5-methyl)propiophenoneoxime} was achieved by adapting the literature preparation of H4L1 (see the ESI† for full details). Variable temperature, solid-state direct current (dc) magnetic susceptibility data down to 5 K were collected on a Quantum Design MPMS-XL SQUID magnetometer equipped with a 7 T dc magnet. Diamagnetic corrections were applied to the observed paramagnetic susceptibilities using Pascal's constants.Synthesis
:1, 25 mL). After 5 minutes of stirring, NEt3 (0.3 mL, 2.1 mmol) was added and the solution stirred for a further 3 h. Large green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of diethyl ether into the filtered mother liquor, over a period of 7 days. Elemental analysis (%) calculated (found) for 1: C 38.06 (37.98), H 4.57 (4.36), N 6.12 (5.91).
:1, 25 mL). After 5 minutes of stirring, NEt3 (0.3 mL, 2.1 mmol) was added and the solution stirred for a further 3 h. Green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of THF into the filtered mother liquor, over a period of 10 days. Elemental analysis (%) calculated (found) for dried 2: C 37.13 (36.99), H 4.23 (3.98), N 8.50 (8.41).
:1, 25 mL). After 5 minutes of stirring, NEt3 (0.3 mL, 2.1 mmol) was added and the solution stirred for a further 3 h. Green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of diethyl ether into the filtered mother liquor, over a period of 7 days. Elemental analysis (%) calculated (found) for dried 3: C 40.99 (40.87), H 5.38 (4.94), N 5.54 (5.11).
:1, 25 mL). After 5 minutes of stirring, NEt3 (0.3 mL, 2.1 mmol) was added and the solution stirred for a further 3 hours. Green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of diethyl ether into the filtered mother liquor, over a period of 7 days. Elemental analysis (%) calculated (found) for dried 4: C 44.21 (44.16), H 5.32 (5.24), N 6.87 (6.80).
:1, 25 mL). After 5 minutes of stirring NEt3 (0.15 mL, 1.05 mmol) was added and the solution stirred for a further 3 hours. Large green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of diethyl ether into the filtered mother liquor over a period of 7 days. Elemental analysis (%) calculated (found) for dried 5: C 37.50 (37.42), H 5.64 (5.33), N 13.05 (12.84).
:1, 25 mL). After 5 minutes of stirring, NEt3 (0.3 mL, 2.1 mmol) was added and the solution stirred for a further 3 hours. Large green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of diethyl ether into the filtered mother liquor over a period of 7 days. Elemental analysis (%) calculated (found) for dried 6: C 41.42 (41.18), H 5.32 (4.81), N 5.68 (5.17).
:1, 25 mL). After 5 minutes of stirring, NEt3 (0.3 mL, 2.1 mmol) was added and the solution stirred for a further 3 hours. Large green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of diethyl ether into the filtered mother liquor over a period of 7 days. Elemental analysis (%) calculated (found) for dried 7: C 37.52 (37.42), H 4.72 (4.55), N 5.68 (5.46).
:1, 25 mL). After 5 minutes of stirring NEt3 (0.30 ml, 2.1 mmol) was added and the solution stirred for a further 3 h. Green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of diethyl ether into the filtered mother liquor over a period of 7 days. Elemental analysis (%) calculated (found) for dried 8: C 44.89 (44.57), H 5.11 (4.88), N 7.48 (7.24).
:1, 25 mL). After 5 minutes of stirring NaOMe (216 mg, 2 mmol) was added and the solution stirred for a further 3 hours. Green, block-like X-ray quality crystals were subsequently obtained by slow diffusion of hexane into the filtered mother liquor over a period of 12 days. Elemental analysis (%) calculated (found) for dried 9: C 47.03 (46.91), H 5.71 (5.52), N 7.56 (7.22).
X-ray crystallography
Single crystal X-ray diffraction data of 1–9 were collected at 100 K on a Agilent Technologies SuperNova at temperatures between 200 and 120 K. See Table S1† and CIF files for full details. CCDC 991823–991828 and CCDC 1029554–1029556.Results and discussion
Complexes 1–7 (Fig. 3–5) share the same general structural framework, and so for the sake of brevity we describe 1 in detail and highlight any significant structural differences present in 2–7.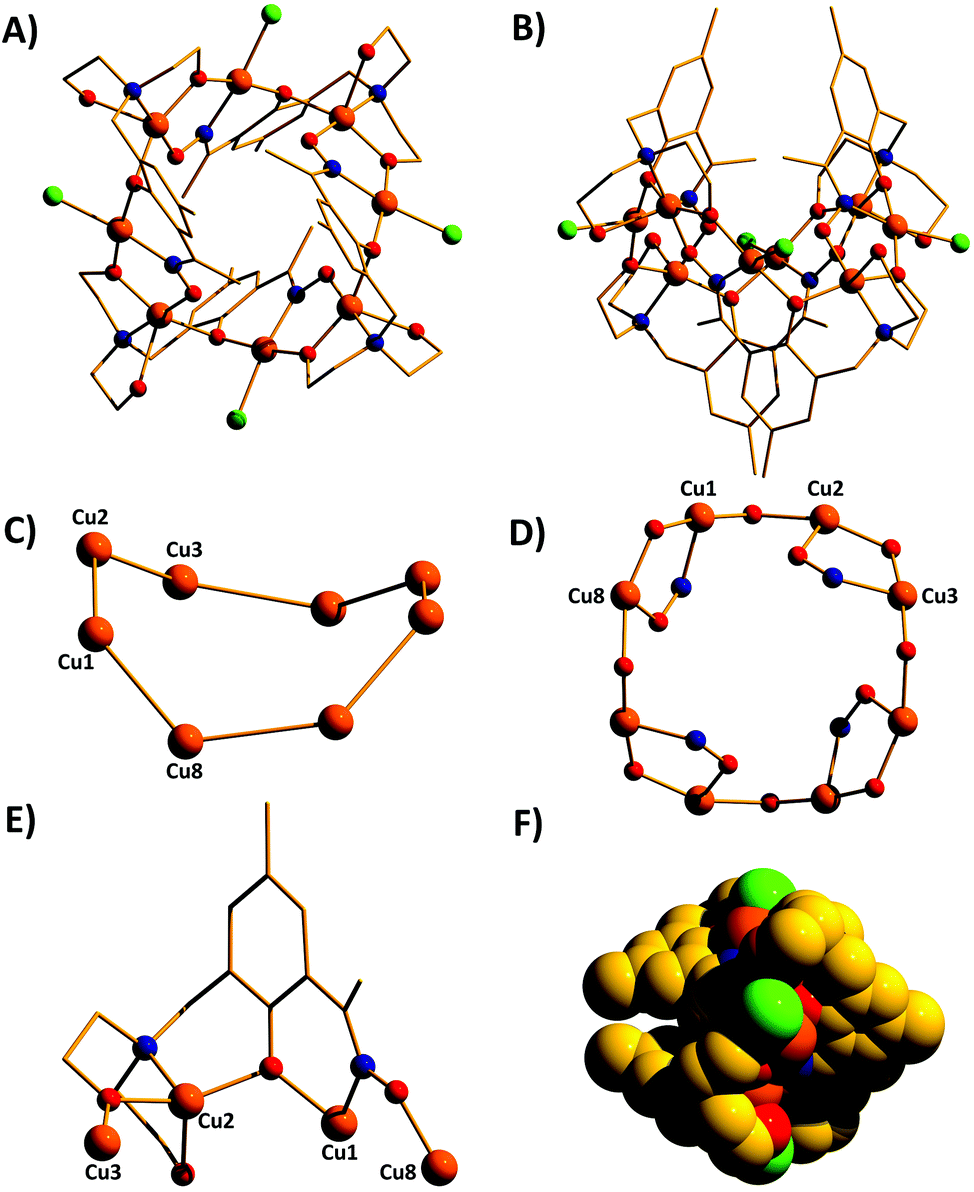 | ||
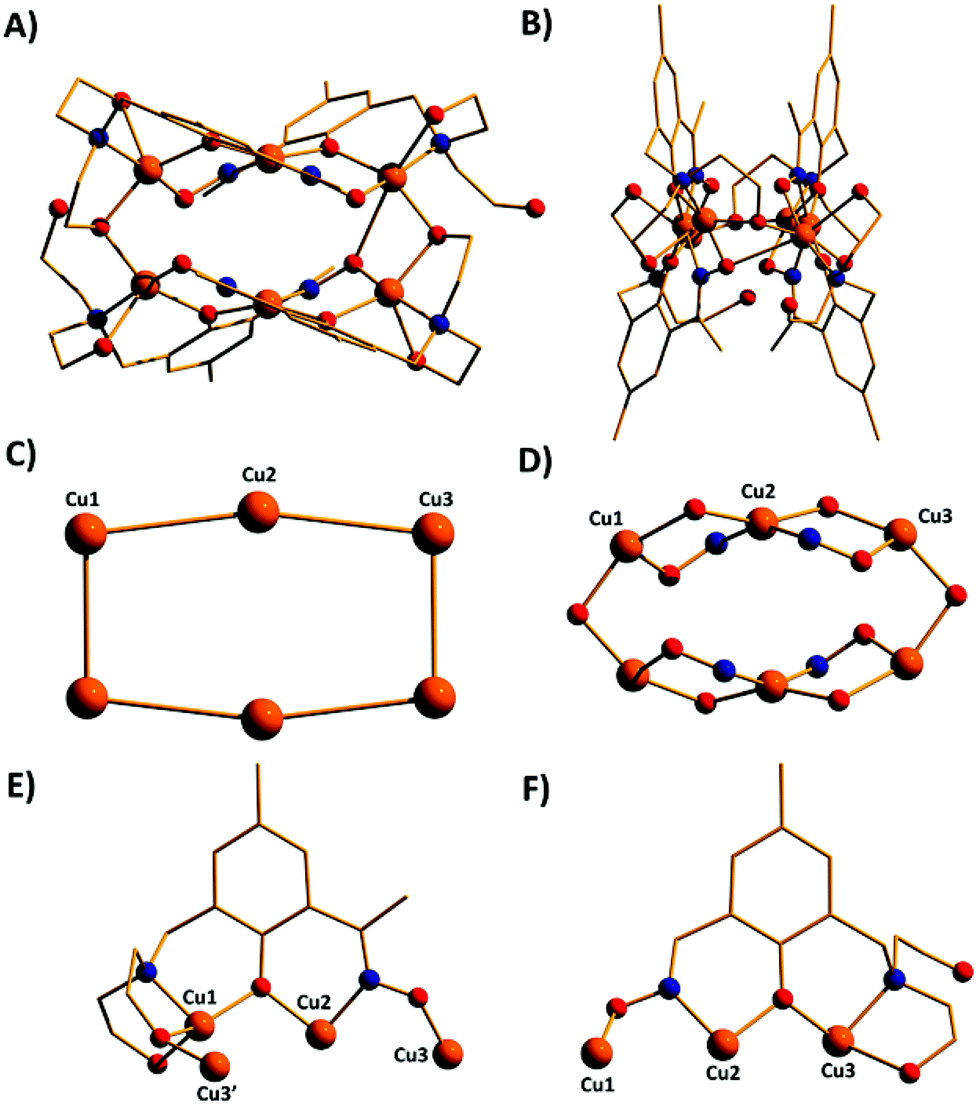
| Fig. 3 The molecular structure of complex 1 viewed perpendicular (A) and parallel (B) to the Cu8 “plane”. Colour code, Cu = orange, O = red, N = blue, C = gold, Cl = green. (C) The metallic skeleton of 1 highlighting the bowl-shape of the cluster. (D) The magnetic core of 1 showing the two different exchange interactions. (E) The bridging mode of the HL13− ligand. (F) Space-fill representation of 1. H atoms omitted for clarity. | ||
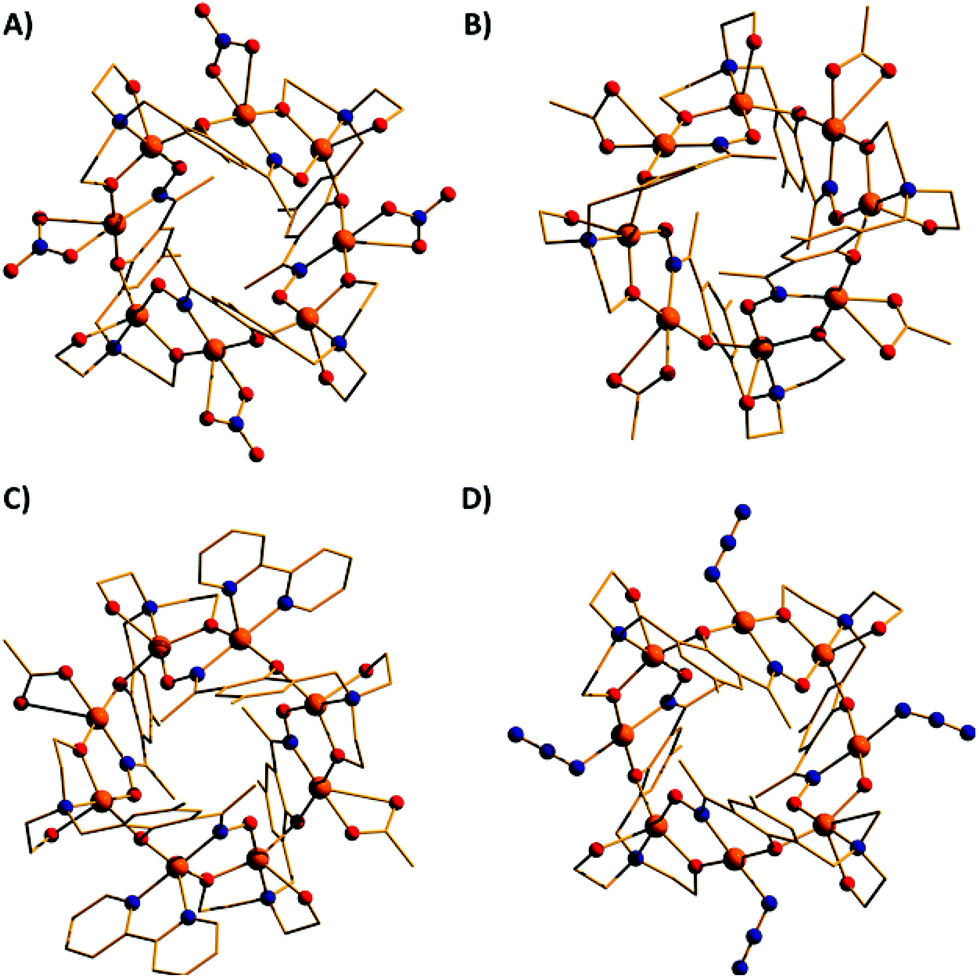 | ||
| Fig. 4 The molecular structures of complexes 2–5 (A–D). Colour code as Fig. 3. | ||
 | ||
| Fig. 5 The molecular structure of complex 8 viewed perpendicular (A) and parallel (B) to the Cu6 plane. Colour code, Cu = orange, O = red, N = blue, C = gold. (C) The metallic skeleton of 8 highlighting the bowl-shape of the cluster. (D) The magnetic core of 8 showing the two different exchange interactions. (E/F) The two bridging modes of the HL13− ligand. H atoms omitted for clarity. | ||
The metallic skeleton of complex 1 (Fig. 3) describes a saddle-like or basket shaped, single-stranded, octametallic wheel of approximate radius 7.3 Å, in which all eight CuII ions are symmetry inequivalent. The coordination geometries of the Cu ions alternate between square planar (Cu1, Cu3, Cu5, Cu7) and square-based pyramidal (Cu2, Cu4, Cu6, Cu8, τ = 0.0743–0.0131) as the wheel is circumnavigated. The HL13− ligands each bridge a total of four Cu ions: the oximic and phenolic moieties are μ-bridging (Cu–N–O–Cu, ∼19–25°; Cu–O–Cu, ∼16°), with one alkoxide arm of the diethanolamine moiety μ-bridging (Cu–O–Cu, ∼18°) and the other remaining protonated and terminally bonded (Cu–O, 2.317–2.433 Å). The remaining coordination sites on the square planar Cu ions are occupied by Cl− anions, which are H-bonded to the terminally bonded O-atoms of the diethanolamine arms (Cl⋯O, ∼3 Å). Each Cu ion is therefore linked to its neighbours by one alkoxide and one oximic –N–O– moiety on one side, and by just one phenoxide on the other. The magnetic core is thus rather simple: [Cu–OR/NO–Cu–OR]4. Two of the HL13− ligands point above and two point below the plane of the bowl, each orientated inwards toward the centre of the bowl resulting in a (C⋯C) separation of ∼3.5 Å. The closest intermolecular contacts are between the Cl− ions and the para-Me(Ph) groups of the HL13− ligands at a (Cl⋯C) distance of ∼3.7 Å. The result is that molecules of 1 pack in a serpentine-like manner when viewed down the b-axis (Fig. S2 in the ESI†).
The molecular structures of complexes 2–5 are shown in Fig. 4. Structural differences between 1 and 2–5, bar changes in the crystallographic/symmetry parameters (Table S1 in the ESI†) and small changes in the bond lengths and angles, are limited to the terminally bonded Cl− ions being been replaced by chelating NO3− ions in 2, chelating CH3CO2− ions in 3 and terminally bonded N3− ions in 5. In complex 4 two of the Cl− ions have been replaced by chelating CH3CO2− ions and two by chelating 2,2′-bpy molecules, with charge balance maintained through the presence of two BF4− counter anions. The chelating ligands change the geometry of the CuII ions at these sites to distorted trigonal bipyramidal, meaning that the wheels 2–4 contain eight trigonal bipyramidal metal centres, with only complex 5 maintaining the four coordinate-five coordinate pattern seen in 1.
The introduction of an Et-group (H4L2) as opposed to a Me-group (H4L1) at the oximic carbon atom of the ligand appears to exert little structural effect, with complexes 6 and 7 being largely analogous to 1, with minor changes in bond lengths and bond angles. Complexes 1–7 all contain terminally bonded or chelating anions, introduced to the reaction mixture in the copper salt. In order to examine the effect of removing this anion, we repeated the synthesis employing the methoxide salt of CuII. The reaction between anhydrous Cu(OMe)2 and H4L1 in a basic MeOH–MeCN solution afforded the homoleptic, hexametallic wheel [Cu6(HL1)4] (8; Fig. 5). The metallic skeleton of 8 describes a single-stranded hexametallic, rectangular CuII wheel. The asymmetric unit contains half the wheel, i.e. three Cu ions (Cu1–3) and two HL13− ligands. The latter are triply deprotonated as before, but coordinate differently in 8 than in 1–7. One ligand bridges a total of four and the other a total of three metal centres with the oximic and phenolic moieties μ-bridging (Cu–N–O–Cu, ∼37°, ∼48°; Cu–O–Cu; ∼108°, ∼113°) in both cases, as in 1–7. It is the alkoxide arms of the diethanolamine moiety of the two symmetry inequivalent ligands which behave differently: in one ligand, one arm is μ-bridging (Cu–O–Cu, 102.5°) with the other terminally bonded at the apical site of the Cu square-based pyramid (Cu–O, 2.35 Å); in the second, one arm is terminally bonded (Cu–O, 2.4 Å) and one non-bonded, and H-bonding to the bridging alkoxide from a neighbouring HL3− ligand (O⋯O, ∼2.6 Å). The coordination geometries of the Cu ions are thus mixed: Cu2 is four coordinate and square planar, whilst Cu1 and Cu3 are five coordinate and square-based pyramidal (τ = 0.118 and 0.0775 respectively). The metallic skeleton is near planar, as opposed to the puckered structures of 1–7, with the short and long edges of the rectangle measuring ∼3 Å and ∼6 Å, respectively. As before, one pair of HL13− ligands points above and one pair below the plane of the wheel, with the aromatic rings being separated by approximately 3.5 Å. The closest intermolecular interactions occur between the para-Me(Ph) groups of the ligand on one molecule and the terminally bonded O atom of the diethanolamine arms on its neighbour (∼3.7 Å, Fig. S3†).
A repetition of the reaction that produces complex 8, but using Cu(BF4)2·3H2O in place of anhydrous Cu(OMe)2 and employing NaOMe instead of NEt3 results in the formation of [Cu4Na2(HL1)2(H2L1)2] (9; Fig. 6). The metallic skeleton of complex 9 also describes a hexametallic wheel, but on this occasion it is heterometallic, more hexagonal than rectangular, and non-planar. The asymmetric unit includes the Cu2Na trimeric moiety, (Cu1–Na1, ∼6.4 Å; Cu1–Na1′, ∼3.2 Å) with the Na…Na diameter of the wheel being ∼6.9 Å, and the closest Cu…Cu distance ∼4.9 Å (Cu2…Cu2′). Cu1 is five coordinate and trigonal bipyramidal (τ = 0.14), whilst Cu2 is four-coordinate and square planar in geometry. The ligands are of two types: HL13− and H2L12− (Fig. 6E and 6F). The former bridges a total of four metal centres, with the oximic and phenolic moieties μ-bridging (Cu–N–O–Cu; ∼50°; Cu–O–Cu, ∼112°), one alkoxide arm of the diethylamine moiety is μ-bridging between Cu1 and Na1′, with the second arm terminally bonded to Cu1 and H-bonded to a neighbouring oximic O-atom (∼2.6 Å). The second ligand uses its oximic O-atom (O6) to bridge between Cu1 and Na1; its oximic N-atom also bonded to Cu1. The phenoxide O-atom μ-bridges between Cu2 and Na1, with both diethanolamine arms being terminally bonded to Na1. The Na+ ions are six-coordinate and in distorted octahedral geometries. There are two intramolecular H-bonding interactions per molecule; between the H-atom of the protonated diol arm of one ligand and the oximic O-atom of its symmetry equivalent (O⋯H = ∼2.6 Å°). The closest intercluster contacts are between the coordinated O-atom of one diethanolamine arm and the CH2 groups of a neighbouring arm (∼3.2 Å; Fig. S4 and S5 in the ESI†). Complex 9 is clearly rather similar in structure to complex 8, the major difference being the introduction of the larger Na+ ions in the former. This has the effect of severely twisting of the M–M–M trimer of the asymmetric unit, from ∠Cu–Cu–Cu = 166° for 8 to ∠Cu–Cu–Na = ∼146° in 9. This has two consequences for the latter complex: firstly the metallic skeleton becomes more bowl-shaped, and secondly there are no longer any coplanar ligands lying perpendicular to the M6 mean plane. Indeed the para-Me(Ph) moieties now point outward rather than inward as seen in all the previous cages (compare Fig. 5B and 6B).
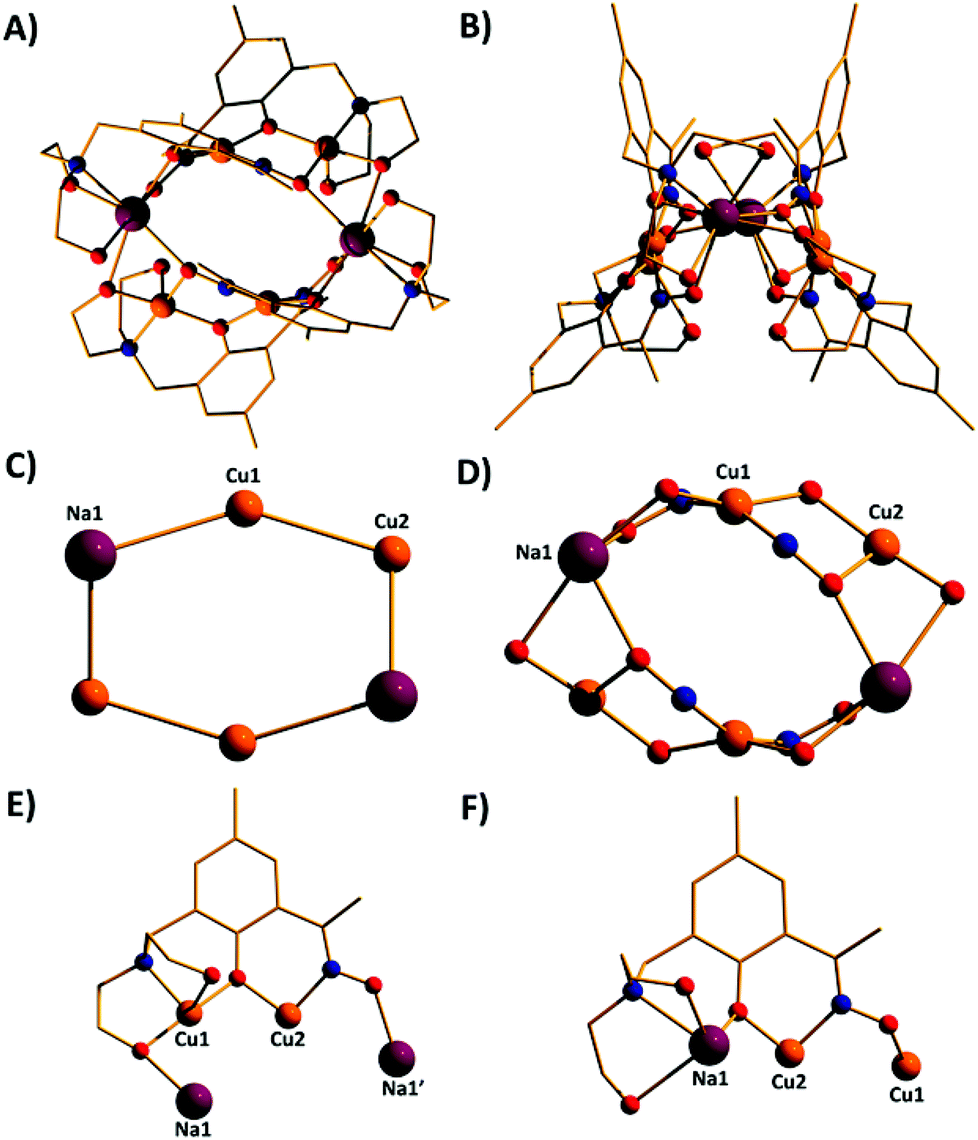 | ||
| Fig. 6 The molecular structure of complex 9 viewed perpendicular (A) and parallel (B) to the Cu4Na2 “plane”. Colour code, Cu = orange, Na = purple, O = red, N = blue, C = gold. (C) The metallic skeleton of 9 highlighting the bowl-shape of the cluster. (D) The magnetic core of 9 showing the two different exchange interactions. (E) The bridging mode of the H1L3− ligand (F) The bridging mode of the H1L3− ligand. H atoms omitted for clarity. | ||
It is difficult at this stage to definitively correlate the changes in syntheses, as summarised in Fig. 7, to the observed structural changes. The simplest reaction is that between Cu(OMe)2 and H4L1, which produces [Cu6(HL)4] (8) and MeOH. The same product results from reactions employing different alkoxide precursors. Thus it seems the alcohol produced does not bond to the CuII ions. When the anion of the metal salt is changed to one that is commonly found to chelate, bond terminally or even bridge between metal centres, or if that anion is added separately to the reaction mixture (halide, pseudohalide, carboxylate), the nuclearity of the cage increases to [Cu8] despite the fact that this anion is non-bridging. Much of this seems rather counter-intuitive at first: one might expect the removal of terminally bonded anions to promote increased bridging of HL13− and/or to encourage further deprotonation to L14−. However this does not occur, and even the addition of an excess of base does not achieve this transformation. We can only surmise that the addition of a bonding anion forces a change in the metal geometry which in turn forces a change in the bonding mode of the ligand and thus to different cluster nuclearities and topologies. The introduction of the alkali metal ion Na+, in the form of NaOMe, suggests other heterometallic cages can be made by simply employing alternative group 1 or group 2 alkoxides, although we have not investigated this further.
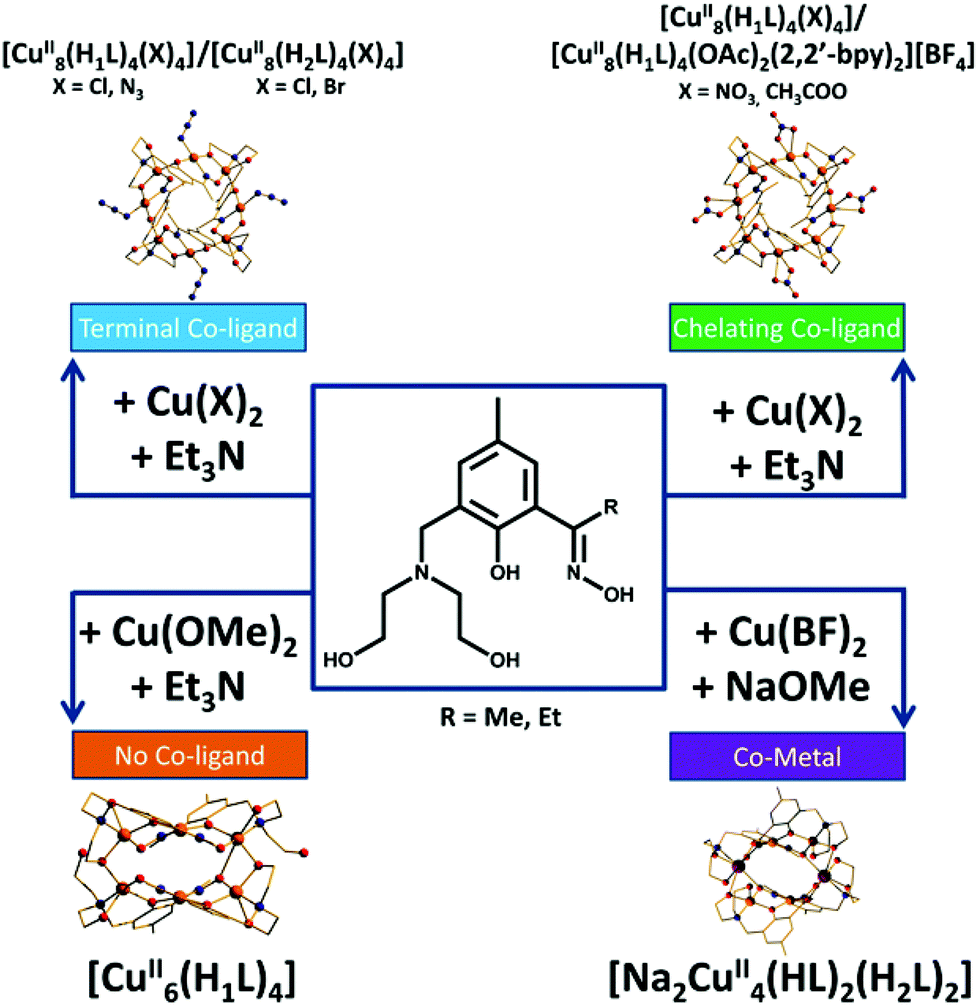 | ||
| Fig. 7 Summarising the effect changes in CuX2 starting material has on product formation with H4L1/2. | ||
A search of the CSD reveals that there are thirteen examples of O-bridged, single-stranded homo-metallic CuII wheels of nuclearity ≤ 8 in the literature. Of these, seven are octametallic14–20 and six are hexametallic.21–26 There are many more examples of both octametallic and hexametallic single-stranded Cu wheels which contain bridging motifs that do not feature oxygen atoms.27 To the best of our knowledge there are no reported examples of single-stranded heterometallic wheels of nuclearity six with a Cu:M (where M is an alkali metal cation) ratio of 4:2. Complexes 1–7 therefore become only the 12th examples of O-bridged single-stranded octametallic CuII wheels, complex 8 only the 7th example of a single-stranded hexametallic CuII wheel, and complex 9 the first example of a single-stranded heterometallic Na/CuII wheel. Apart from their rather beautiful structural aesthetics, molecular wheels have long held fascination for both chemists and physicists, since they can often act as model compounds for the study of quantum effects and spin frustration.28 and have recently been identified as potential candidates for quantum information processing.29
Magnetochemistry
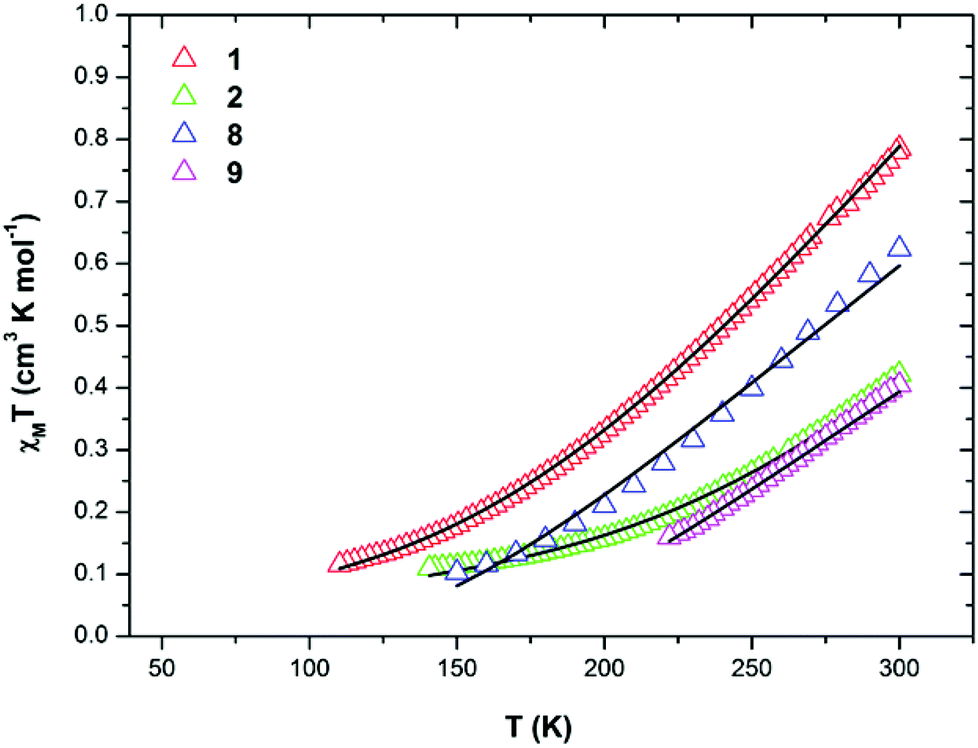 | ||
| Fig. 8 The magnetic susceptibility data, as χMT, versus T at B = 0.1 T for complexes 1 (red), 2 (green), 8 (blue) and 9 (magenta). The solid black lines are fits of the experimental data. The best fit 3J model is shown for 8. See text for details. | ||
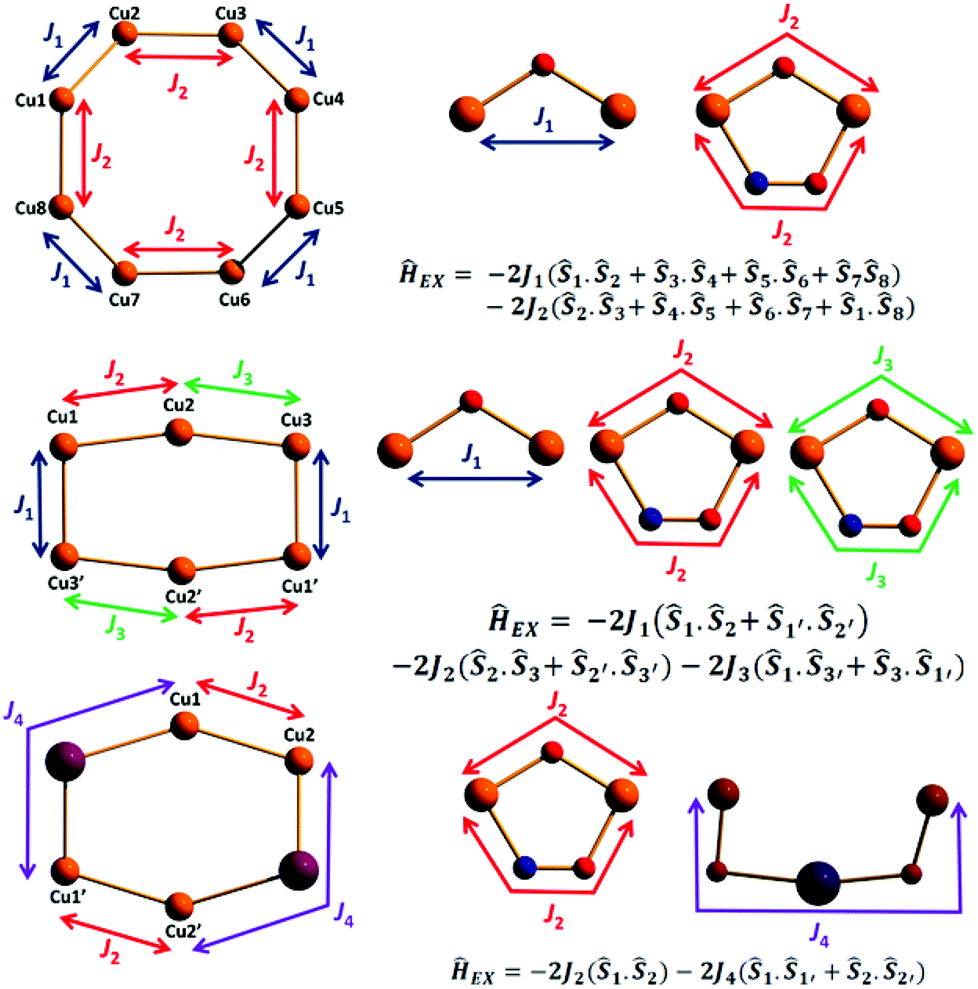 | ||
| Fig. 9 The magnetic cores of the representative Cu8, Cu6 and Cu4Na2 metal cores (left) and the models representing the exchange component of the spin-Hamiltonians which their experimental susceptibility data were fitted to (right). For the experimental fit of 9J4 = 0. | ||
| Complex/method | J 1 (cm−1) | J 2 (cm−1) | J 3 (cm−1) | J 4 (cm−1) |
|---|---|---|---|---|
| 1/Fit | −20.1 | −457 | — | — |
| 1/DFT | −45.0 | −320 | — | — |
| 2/Fit | −38.9 | −302 | — | — |
| 2/DFT | −46.6 | −278 | — | — |
| 8/Fit (2J) | −200 | −247 | −247 | — |
| 8/Fit (3J) | −107 | −239 | −260 | — |
| 8/DFT | +5.50 | −25.9 | −120 | — |
| 9/Fit | — | −271 | — | — |
| 9/DFT | — | −95.8 | — | +0.08 |
Theoretical studies
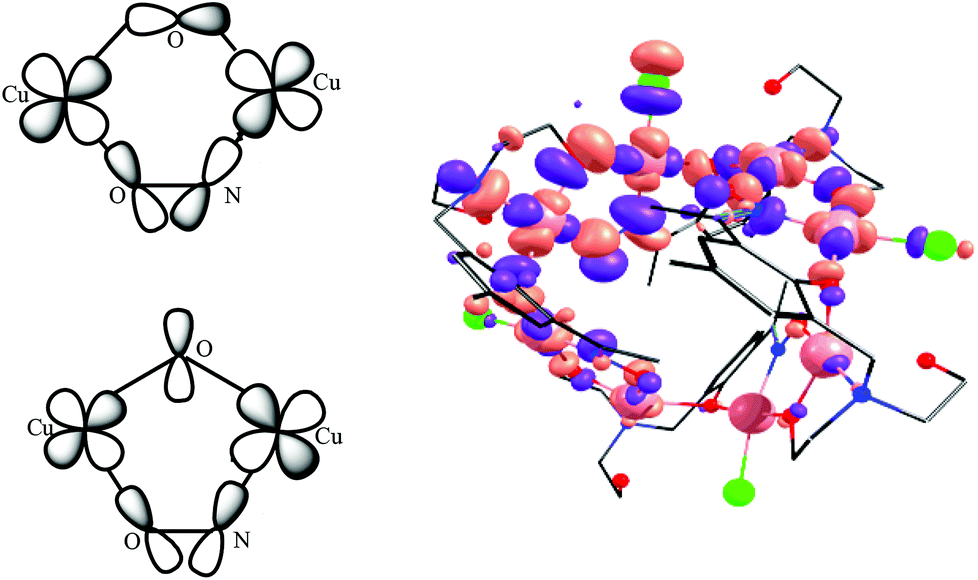 | ||
| Fig. 10 Schematic representation of the complementarity effect exhibited by the oxime bridge with symmetric and antisymmetric combinations of dx2−y2 orbitals (left) and a representative MO diagram depicting the antisymmetric combination of the dx2−y2 orbitals (right). | ||
The overlap integrals, computed as 0.08 and 0.20 for J1 and J2, respectively, confirm this. Computed spin density plots for complexes 1, 8, and 9 are shown in Fig. 11. The Cu(II) centres in complex 1 possess spin densities of 0.63 [Cu(1)] and 0.59 [Cu(2)], values substantially lower than the expected value of 1.0, revealing significant delocalisation (and hence strong exchange) of the unpaired spin onto the bridging ligands. The spin density on the bridging groups are also significant, 0.12 on the O-atom of the μ-alkoxide (J1), and 0.16 on the O-atom of the μ-alkoxide, and 0.13 and 0.10 for O and N atoms of the oxime bridge in the J2 interaction.
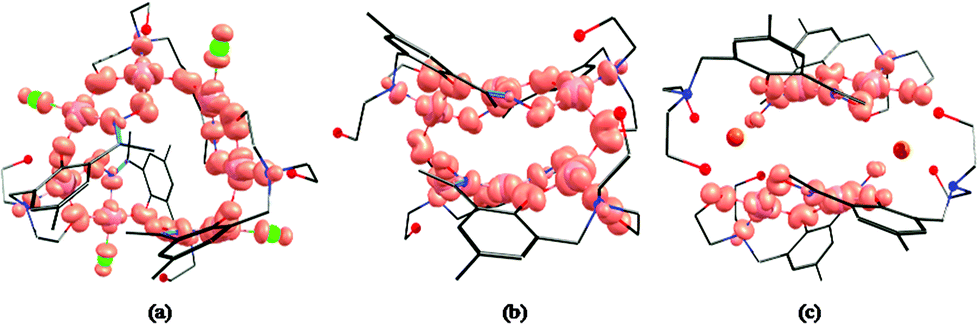 | ||
| Fig. 11 B3LYP computed spin density plots of complex 1 (a), 8 (b) and 9 (c). | ||
For complex 2 calculations yield a similar set of exchange constants, although the J2 interaction is computed to be slightly smaller (Table 1). The decrease in the magnitude of the J2 interaction compared to complex 1 is found to correlate to the Cu–O–N–Cu dihedral angle. In complex 2 the bulkier, chelating nitrate groups cause a twist in the Cu–N–O–Cu dihedral leading to weaker dx2−y2 − dx2−y2 overlap and a reduction in the J values. The spin density on the Cu(II) centres in complex 2 are plotted in Fig. S7.†
For complex 8 calculations yield J1 = +5.53 cm−1, J2 = −25.9 cm−1 and J3 = −120 cm−1. The J1 interaction is computed to be weakly ferromagnetic in nature, in contrast to that fitted experimentally. Here the Cu–O–Cu angle is relatively small (102.2°) and along with the large out-of-plane shift of the O–C(alkyl) group from the Cu–O–Cu–O plane (∼40.0°) the geometry is approaching that theoretically predicted to mediate ferromagnetic exchange;44 the crossover area likely being the source of error in the theoretical calculation. J2 and J3 are antiferromagnetic in nature with J3 being approximately five times larger. This is again correlated to the difference in the Cu–N–O–Cu dihedral angle, with smaller angles yielding stronger antiferromagnetic coupling. The magnitude of the J2 and J3 interactions are much smaller than that computed for complexes 1 and 2 due to the smaller ring size – the Cu–N–O–Cu moieties being far more twisted than that found in complexes 1 and 2. For complex 9 the computed exchange constants are J2 = −95.8 cm−1 and J4 = +0.08 cm−1. The former is consistent with that observed in previous complexes, and the latter as expected for an interaction mediated through a diamagnetic Na+ ion. Computed overlap integrals (0.26 and 0.005 for J2 and J4, respectively) are consistent with this picture (Fig. S8†). In all four complexes the strongest antiferromagnetic coupling constant is underestimated by DFT, the differences being particularly obvious when the other J values are small in magnitude. The underestimation of the exchange interactions in Cu(II) complexes by DFT has been highlighted previously,45 and is thus a limiting factor in the accurate estimation of absolute values.
Since the Cu–N–O–Cu dihedral has been found to play an important role in determining the J values in this family of complexes, we have developed a magneto-structural correlation based on a dimeric model of complex 9 shown in the inset of Fig. 12. By varying the Cu–N–O–Cu dihedral angle (x) [with the Cu–O–Cu angle fixed] from 0–80° an exponential correlation was found, with the smallest dihedral angles leading to the strongest AF exchange, and the largest dihedral angles leading to the weakest AF exchange. Fitting the data to the expression
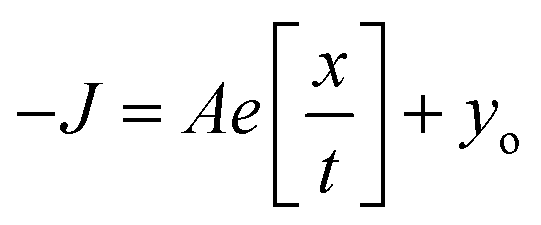
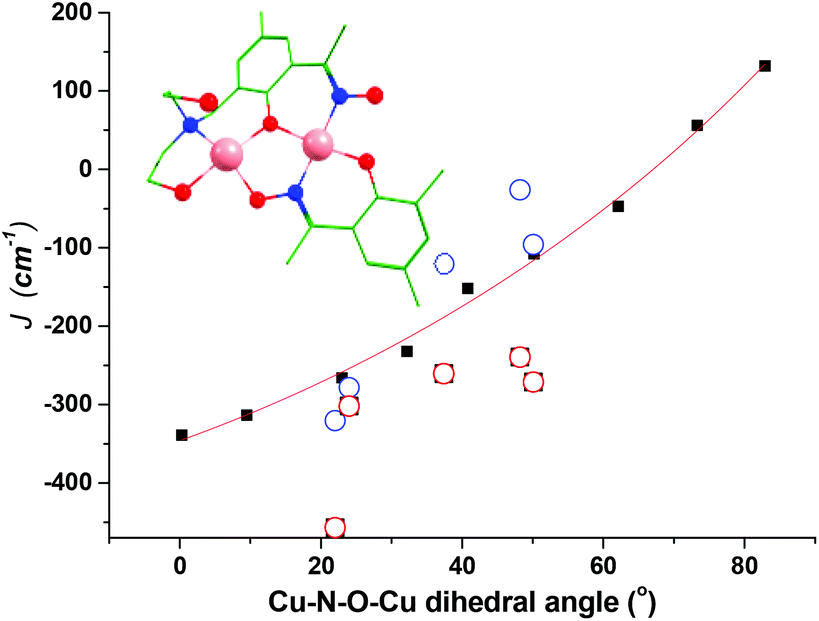 | ||
| Fig. 12 Magnetostructural correlation developed by DFT calculations by varying the Cu–N–O–Cu dihedral angle in a model complex based on complex 9. The solid black squares are computed points and the red line the best fit to these points. The hollow blue circles are the DFT calculated values for the full structure, whilst the red circles are the experimentally fitted exchange constants. | ||
Conclusions
The simple reaction of Cu(II) salts with diethanolamine-functionalised phenolic oximes has resulted in seven new examples of single-stranded octametallic Cu(II) wheels, one new example of a single-stranded hexametallic Cu(II) wheel, and the first reported example of a single-stranded hexametallic mixed-metal Na2Cu4 wheel. The construction of polymetallic cages from ligand moieties normally associated with producing monometallic Cu(II) compounds is clear evidence that combining complimentary ligand types into one organic framework is a sensible methodology for building polymetallic cluster compounds. In each case the interaction between neighbouring metal ions was shown to be strongly antiferromagnetic. DFT calculations suggest the origin to be a complementarity effect due to the presence of the oxime bridge, in which smaller Cu–N–O–Cu dihedral angles yield larger exchange constants, which are inherently linked to the planarity of the wheel. A magnetostructural correlation developed even suggests that such Cu(II) wheels can be made to display intramolecular ferromagnetic exchange if the Cu–N–O–Cu torsion angles can be of the order of 60° or larger. Current efforts are aimed at synthetic strategies to achieve just that.Acknowledgements
The authors acknowledge the EPSRC (UK). GR would like to thank the AISRF and DST Nanomission (SR/NM/NS-1119/2011) for funding. NV would like to thank the DST for a fast track fellowship.Notes and references
- D. Gatteschi, O. Kahn and R. D. Willett, Magnetostructural Correlations in Exchange Coupled Systems, D. Reidel, Dordrecht, 1985 Search PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
- V. H. Crawford, H. W. Richardson, J. R. Wasson, D. J. Hodgson and W. E. Hatfield, Inorg. Chem., 1976, 15, 2107 CrossRef CAS.
- W. E. Hatfield, Comments Inorg. Chem., 1981, 1, 105 CrossRef CAS.
- O. Kahn, J. Galy, P. Tola and H. Coudanne, J. Am. Chem. Soc., 1978, 100, 3931 CrossRef CAS.
- A. Honecker and M. E. Zhitomirsky, JCPS, 2009, 145, 012082 Search PubMed.
- M. Triff, F. Troiani and D. Loss, Phys. Rev. Lett., 2008, 101, 217201 CrossRef.
- See for example: (a) C. P. Landee and R. E. Greenery, Inorg. Chem., 1986, 25, 3771 CrossRef CAS; (b) G. D. Fallon and K. S. Murray, Inorg. Chim. Acta, 1985, 96, L53 CrossRef CAS; (c) H. Uekusa, S. Ohba, T. Tokii, Y. Muto, M. Kato, S. Husbye, O. W. Steward, S. Chang, J. P. Rose, J. F. Pletcher and I. Suzuki, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 650 CrossRef; (d) R. Costa, P. R. Moreira, S. Youngme, K. Siriwong, N. Wannarit and F. Illlas, Inorg. Chem., 2010, 49, 285 CrossRef CAS PubMed; (e) T. Yamase, H. Ishikawa, H. Abe, K. Fukaya, H. Nojiri and H. Takeuchi, Inorg. Chem., 2012, 51, 4606 CrossRef CAS PubMed.
- P. A. Tasker, P. G. Plieger and L. C. West, Compr. Coord. Chem. II, 2004, 9, 759 CAS.
- A. G. Smith, P. A. Tasker and D. J. White, Coord. Chem. Rev., 2003, 241, 61 CrossRef CAS.
- M. Wenzel, R. S. Forgan, A. Faure, K. Mason, P. A. Tasker, S. Piligkos, E. K. Brechin and P. G. Plieger, Eur. J. Inorg. Chem., 2009, 4613 CrossRef CAS.
- See for example: (a) R. Inglis, C. J. Milios, L. F. Jones, S. Piligkos and E. K. Brechin, Chem. Commun., 2012, 48, 181 RSC; (b) K. Mason, I. A. Gass, F. J. White, G. S. Papaefstathiou, E. K. Brechin and P. A. Tasker, Dalton Trans., 2011, 40, 2875 RSC; (c) P. Chaudhuri, M. Hess, E. Reutschler, T. Weyhermuller and U. Florke, New J. Chem., 1998, 553 RSC.
- See for example: (a) S. Sanz, J. M. Frost, T. Rajeshkumar, S. J. Dalgarno, G. Rajaraman, W. Wernsdorfer, J. Schnack, P. J. Lusby and E. K. Brechin, Chem. – Eur. J., 2014, 20, 3010 CrossRef CAS PubMed; (b) S. Sanz, J. M. Frost, M. B. Pitak, S. J. Coles, S. Piligkos, P. J. Lusby and E. K. Brechin, Chem. Commun., 2014, 50, 3310 RSC; (c) J. M. Frost, S. Sanz, T. Rajeshkumar, M. B. Pitak, S. J. Coles, G. Rajaraman, W. Wernsdorfer, J. Schnack, P. J. Lusby and E. K. Brechin, Dalton Trans., 2014, 43, 10690 RSC.
- G. Mezei, P. Baran and R. G. Raptis, Angew. Chem., Int. Ed., 2004, 43, 574 CrossRef CAS PubMed.
- R. Acevedo-Chavez, M. E. Costas and R. Escudero, J. Solid State Chem., 1997, 132, 24 CrossRef CAS.
- (a) D. A. Fowler, A. V. Mossine, C. M. Beavers, S. J. Teat, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2011, 133, 11069 CrossRef CAS PubMed; (b) H. Kumari, A. V. Mossine, S. R. Kline, C. L. Dennis, D. A. Fowler, S. J. Teat, C. L. Barnes, C. A. Deakyne and J. L. Atwood, Angew. Chem., Int. Ed., 2012, 51, 1452 CrossRef CAS PubMed.
- G. A. Ardizzoia, M. A. Angaroni, G. La Monica, F. Cariati, M. Moret and N. Masciocch, J. Chem. Soc., Chem. Commun., 1991, 1021 Search PubMed.
- A. Mukherjee, I. Rudra, M. Nethaji, S. Ramasesha and A. R. Chakravarty, Inorg. Chem., 2004, 42, 463 CrossRef.
- L. Zherlitsyna, N. Auner and M. Bolte, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, 199 Search PubMed.
- H. Kumari, A. V. Mossine, S. R. Kline, C. L. Dennis, D. A. Fowler, S. J. Teat, C. L. Barnes, C. A. Deakyne and J. L. Atwood, Angew. Chem., Int. Ed., 2012, 51, 1452 CrossRef CAS PubMed.
- M. A. Castro, M. Rusjan, D. Vega, O. Pena, T. Weyhermuller, F. D. Cukiernik and L. D. Slep, Inorg. Chim. Acta, 2011, 379, 499 CrossRef PubMed.
- S. D. Bunge, J. A. Ocana, T. L. Cleland and J. L. Steele, Inorg. Chem., 2009, 48, 4619 CrossRef CAS PubMed.
- A. A. Mohamed, S. Ricci, A. Burini, R. Galassi, C. Santini, G. M. Chiarella, D. Y. Melgarejo and J. P. Fackler Jr., Inorg. Chem., 2011, 50, 1014 CrossRef CAS PubMed.
- L. F. Jones, C. A. Kilner, M. P. de Miranda, J. Wolowska and M. A. Halcrow, Angew. Chem., Int. Ed., 2007, 46, 4073 CrossRef CAS PubMed.
- J. R. Carruthers, K. Prout and F. J. C. Rossotti, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2044 CrossRef.
- B. F. Hoskins, R. Robson and P. Smith, J. Chem. Soc., Chem. Commun., 1990, 488 RSC.
- See for example: (a) S. T. Onions, S. L. Heath, D. J. Price, R. W. Harrington, W. Clegg and C. J. Matthews, Angew. Chem., Int. Ed., 2004, 43, 1814 CrossRef CAS PubMed; (b) D. Dragancea, V. B. Arion, S. Shova, E. Rentschler and N. V. Gerbeleu, Angew. Chem., Int. Ed., 2005, 44, 7938 CrossRef CAS PubMed; (c) J. Xaio, B. Y. Liu, G. Wei and X. C. Huang, Inorg. Chem., 2011, 50, 11032 CrossRef PubMed; (d) X. C. Huang, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2004, 126, 31218 Search PubMed.
- See for example: (a) J. Schnack, Dalton Trans., 2010, 39, 4677 RSC; (b) J. Schnack, H. J. Schmidt, J. Richter and J. Schulenburg, Eur. Phys. J. B, 2001, 24, 475 CrossRef CAS PubMed; (c) I. Rousochatzakis, A. M. Lauchli and F. Mila, Phys. Rev. B: Condens. Matter, 2008, 77, 094420 CrossRef; (d) J. Schnack and R. Schnalle, Polyhedron, 2009, 28, 1620 CrossRef CAS PubMed.
- See for example: (a) J. Jaklič and P. Prelovsěk, Phys. Rev. B: Condens. Matter, 1994, 49, 5065 CrossRef; (b) J. Schnack and O. Wendland, Eur. Phys. J. B, 2010, 78, 535 CrossRef CAS; (c) J. Schnack, P. Hage and H.-J. Schmidt, J. Comput. Phys., 2008, 227, 4512 CrossRef PubMed.
- S. Piligkos, ITO-MAGFIT, Department of Chemistry, University of Copenhagen, 2010 Search PubMed.
- A. Bencini and D. Gatteschi, Electron Paramagnetic Resonance of Exchange Coupled Systems, Springer, Heidelberg, 1990 Search PubMed.
- See for example: (a) D. Gaynor, Z. A. Starivoka, S. Ostrovsky, W. Haase and K. B. Nolan, Chem. Commun., 2002, 506 RSC; (b) H. Saarinen, M. Orama and J. Korvenranta, Acta Chem., Scand., 1989, 43, 834 CrossRef CAS PubMed.
- See for example: (a) A. Escuer, G. Vlahopoulou, S. P. Perlepes and F. A. Mautner, Inorg. Chem., 2011, 50, 2468 CrossRef CAS PubMed; (b) P. Chaudhuri, Coord. Chem. Rev., 2003, 43, 143 CrossRef; (c) C. J. Milios, T. C. Stamatatos and S. P. Perlepes, Polyhedron, 2006, 25, 134 CrossRef CAS PubMed; (d) P. A. Angardis, P. Baran, R. Boca, F. Cervantes-Lee, W. Haase, G. Mezei, R. G. Raptis and R. Werner, Inorg. Chem., 2002, 41, 2219 CrossRef PubMed.
- See for example: (a) H. L. Zhu, C. X. Ren and X. M. Chen, J. Coord. Chem., 2002, 55, 667 CrossRef CAS; (b) G. A. V. Albada, I. Mutikainen, U. Turpeinen and J. Reedijk, Polyhedron, 2004, 23, 993 CrossRef PubMed; (c) M. Gonzalez-Alvarez, G. Alzuet, J. Borras, S. Garcia-Granda and J. M. Montejo-Bernardo, J. Inorg. Biochem., 2003, 96, 443 CrossRef CAS; (d) V. K. Bhardwai, N. Aliaga-Alcalde, M. Corbella and G. Hundal, Inorg. Chim. Acta, 2010, 97, 363 Search PubMed; (e) S. Munoz, J. Pons, J. Ros, M. Font-Bardia, C. A. Kilner and M. A. Halcrow, Inorg. Chim. Acta, 2011, 373, 211 CrossRef CAS PubMed; (f) M. Drillon, A. Grand and P. Rey, Inorg. Chem., 1990, 29, 771 CrossRef CAS; (g) H. E. LeMay Jr., D. J. Hodgson, P. Preuttiangkura and L. J. Theriot, J. Chem. Soc., Dalton Trans., 1979, 781 RSC.
- See for example: (a) M. Sutradhar, T. R. Barman, J. Klanke, M. G. B. Drew and E. Rentschler, Polyhedron, 2013, 53, 48 CrossRef CAS PubMed; (b) E. S. Koumousi, C. P. Raptopoulou, S. P. Perlepes, A. Escuer and T. C. Stamatatos, Polyhedron, 2010, 29, 204 CrossRef CAS PubMed; (c) M. Maekawa, S. Kitigawa, Y. Nakao, S. Sakamoto, A. Yatani, W. Mori, S. Kashino and M. Munakata, Inorg. Chim. Acta, 1999, 20, 293 Search PubMed; (d) A. Yatani, M. Fujii, Y. Nakao, S. Kashino, M. Kinoshita, W. Mori and S. Suzuki, Inorg. Chim. Acta, 2001, 316, 127 CrossRef CAS; (e) Y. Song, X. T. Chen, C. G. Zheng, D. R. Zhu, X. Z. You and L. H. Weng, Transition Met. Chem., 2001, 26, 247 CrossRef CAS; (f) A. Escuer, G. Vlahopoulo, S. P. Perlepes, M. Font-Bardia and T. Clavet, Dalton Trans., 2011, 40, 225 RSC; (g) J. P. Naskar, C. Biswas, B. Guhathakurta, N. Aliaga-Alcade, L. Lu and M. Zhu, Polyhedron, 2011, 30, 2310 CrossRef CAS PubMed; (h) V. Mathrubootham, A. W. Addison, K. T. Holman, E. Sinn and L. K. Thompson, Dalton Trans., 2009, 8111 RSC; (i) P. Dhal, M. Nandy, D. Sadhukhan, E. Zangrando, G. Pilet, C. J. Gomez-Garica and S. Mitra, Dalton Trans., 2013, 42, 14545 RSC; (j) L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Dalton Trans., 2014, 43, 7589 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- (a) A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef PubMed; (b) A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS PubMed.
- E. Ruiz, S. Alvarez, A. Rodriguez-Fortea, P. Alemany, Y. Pouillon and C. Massobrio, in Magnetism: Molecules to Materials, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2001, vol. II, p. 227 Search PubMed.
- T. Cauchy, E. Ruiz and S. Alvarez, J. Am. Chem. Soc., 2006, 128, 15722 CrossRef CAS PubMed.
- N. Berg, T. Rajeshkumar, S. M. Taylor, E. K. Brechin, G. Rajaraman and L. F. Jones, Chem. – Eur. J., 2012, 18, 5906 CrossRef CAS PubMed.
- T. Rajeshkumar, H. V. Annadata, M. Evangelisti, S. K. Langley, N. F. Chilton, K. S. Murray and G. Rajaraman, Inorg. Chem., 2015, 54, 1661 CrossRef CAS PubMed.
- (a) E. Ruiz, P. Alemany, S. Alvarez and J. Cano, Inorg. Chem., 1997, 36, 3683 CrossRef CAS PubMed; (b) T. Rajeshkumar, H. V. Annadata, M. Evangelisti, S. K. Langley, N. F. Chilton, K. S. Murray and G. Rajaraman, Inorg. Chem., 2015, 54, 1661 CrossRef CAS PubMed.
- (a) E. Ruiz, P. Alemany, S. Alvarez and J. Cano, Inorg. Chem., 1997, 36, 3683 CrossRef CAS PubMed; (b) E. Ruiz, S. Alvarez, A. Rodriguez-Fortea, P. Alemany, Y. Pouillon and C. Massobrio, in Magnetism: Molecules to Materials II, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2001, p. 227 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, crystallographic details, packing diagrams and further details of the DFT calculations. CCDC 991823–991828 and 1029554–1029556. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00884k |
| This journal is © The Royal Society of Chemistry 2015 |