
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formylated chloro-bridged iridium(III) dimers as OLED materials: opening up new possibilities†
Michael Y.
Wong
a,
Guohua
Xie
b,
Clarisse
Tourbillon‡
c,
Martina
Sandroni§
c,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a,
Ifor D. W.
Samuel
b and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk; Web: http://www.zysman-colman.com Fax: +44 (0)1334 463808; Tel: +44 (0)1334 463826
bOrganic Semiconductor Centre, School of Physics and Astronomy, University of St. Andrews, North Haugh, St. Andrews, Fife KY16 9SS, UK
cDépartement de Chimie, Université de Sherbrooke, Sherbrooke, Québec, Canada J1 K 2R1
First published on 8th January 2015
Abstract
In this study, a series of four formyl-substituted chloro-bridged iridium(III) dimers were prepared. Their absorption, photophysical and electrochemical properties were studied in dichloromethane solution. It was found that as the formyl content increased on the cyclometalating ligands, emission unexpectedly became brighter. Organic light-emitting diodes (OLEDs) were fabricated using each of these iridium dimers as the emitter. The OLED fabricated using the brightest of the series, 2b, as the dopant afforded a decent external quantum efficiency (EQE) of 2.6%. This suggests that chloro-bridged iridium dimers are potential candidates as solid-state emitters.
Introduction
Neutral mononuclear cyclometalated iridium complexes have received intense attention as emitters for organic light emitting diodes (OLEDs) in lighting and visual displays.1 Their privileged use is due to the phosphorescent nature of the emission, which is mediated by the iridium metal that facilitates intersystem crossing due to spin–orbit coupling.2 Thus, unlike OLED devices based on fluorescent emitters whose internal quantum efficiencies (IQE) are capped by 25% according to spin statistics, 100% of the excitons can be harvested in phosphorescent electroluminescent devices.3 Additionally, iridium(III) complexes typically exhibit bright emission at room temperature that can be tuned across the visible spectrum as a function of substitution about and combination of ligands, possess short radiative lifetimes that mitigates undesirable triplet–triplet annihilation and are thermally and chemically stable.4The majority of syntheses of these mononuclear iridium complexes proceeds via the formation of a dichloro-bridged iridium intermediate of the form [Ir(C^N)2Cl]2 where C^N is a cyclometalating ligand such as the commonly used 2-phenylpyridinato, ppy. Despite the thousands of articles relating to the photophysical study of mononuclear iridium complexes, since the seminal work by Watts and co-workers5 almost thirty years ago, the study of these iridium dimers has often been overlooked.6 This has certainly been influenced by the fact that the parent iridium dimer, [Ir(ppy)2Cl]2, and indeed most dimers in this family, are very poorly emissive5b,7 in toluene solution while fac-Ir(ppy)3, under similar conditions, exhibits a photoluminescence quantum yield, ΦPL, ranging from 70% to near unity, depending upon the medium.8 Bryce, Monkman and co-workers6b recently reported the first examples of dichloro-bridged iridium complexes used as emissive dopants in OLEDs. Employing complexes with substituted fluorenylpyridine C^N ligands dispersed in a PVK host polymer, they could obtain OLEDs with EQEs ranging from 0.6 to 4% and power efficiencies of 0.4–3.53 lm W−1.
Monoformyl-substituted C^N ligands incorporated onto mononuclear iridium complexes have been investigated,9 particularly by Lo and co-workers10 as a tool for grafting on receptor units for bioimaging. To the best of our knowledge, no one has explored the optoelectronic properties of the dichloro-bridged dinuclear complexes bearing formyl-substituted ppy ligands. In this study, we synthesized and characterized a series of dichloro-bridged iridium dimers whose structures are shown in Chart 1. The optoelectronic properties of 1a–2b were probed as a function of the regiochemistry and number of formyl units present on the cyclometalating (C^N) ligands. Unexpectedly, when the number of formyl units increased, the ΦPL of the complex increased significantly, with 2a the brightest at 15.7% in DCM. OLED devices were fabricated using these complexes as phosphorescent dopants with performances modestly reduced in comparison to those reported by Bryce, Monkman and co-workes.6b This study corroborates their earlier report that dichloro-bridged dinuclear iridium complexes, even those simply functionalized as is the case here, can be used as the emissive layer in viable OLED devices.
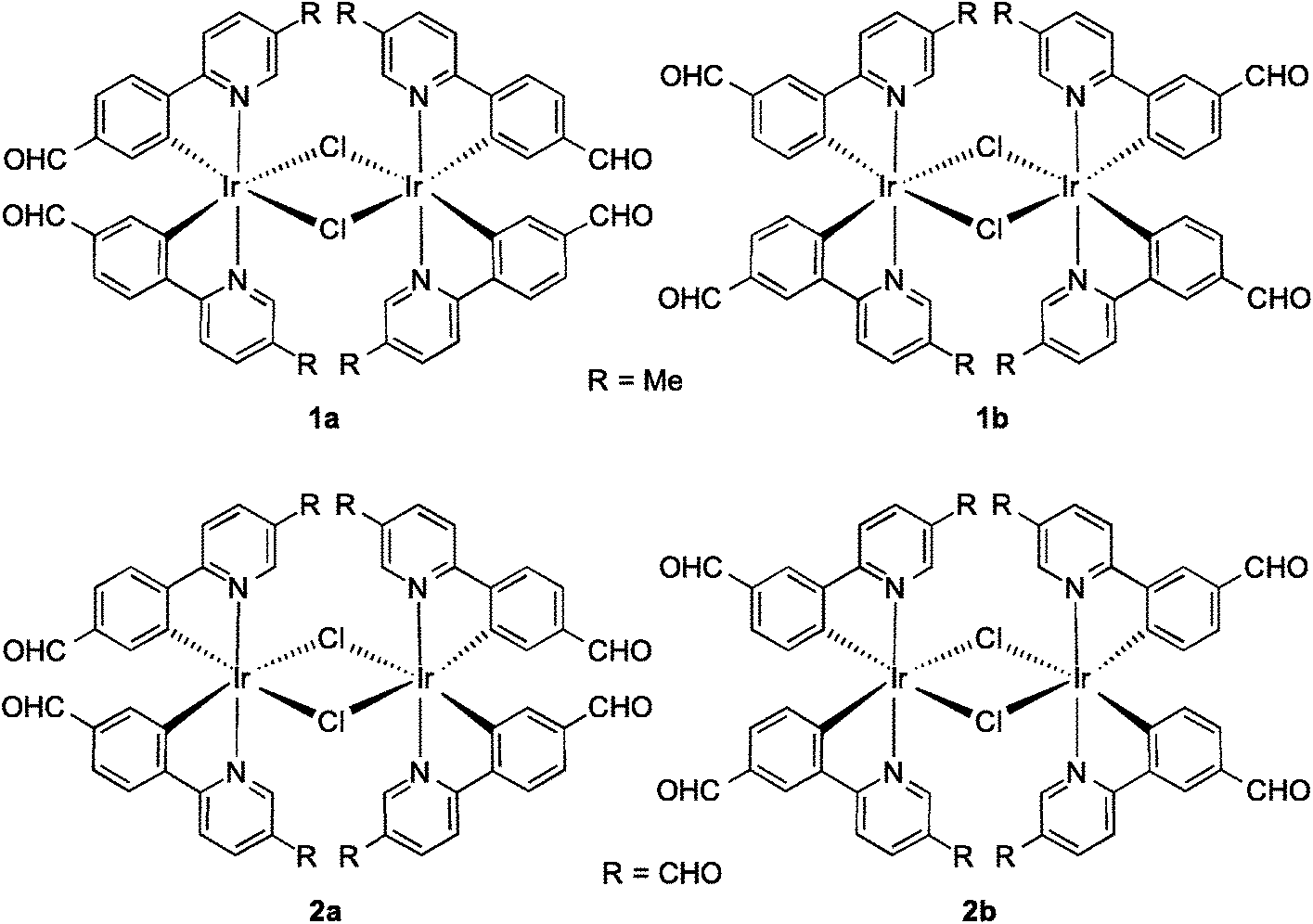 | ||
| Chart 1 Chemical structures of the complexes presented in this study. | ||
Results and discussion
Ligand and complex synthesis
The formyl-substituted cyclometalating (C^N) ligands were obtained in excellent yields (86–92%) following a Suzuki–Miyaura11 coupling between the suitably formylated arylboronic acid and either 2-bromo-5-methylpyridine or 2-bromo-5-formylpyridine (Scheme 1). The use of Pd(PPh3)4 as the catalyst resulted in much cleaner conversion to product than with Pd(PPh3)2Cl2, wherein significant homocoupling of the arylboronic acid was observed. The latter functionalized pyridine was itself obtained in 63% yield via regioselective lithium halide exchange in diethyl ether, quench with DMF and subsequent hydrolysis.12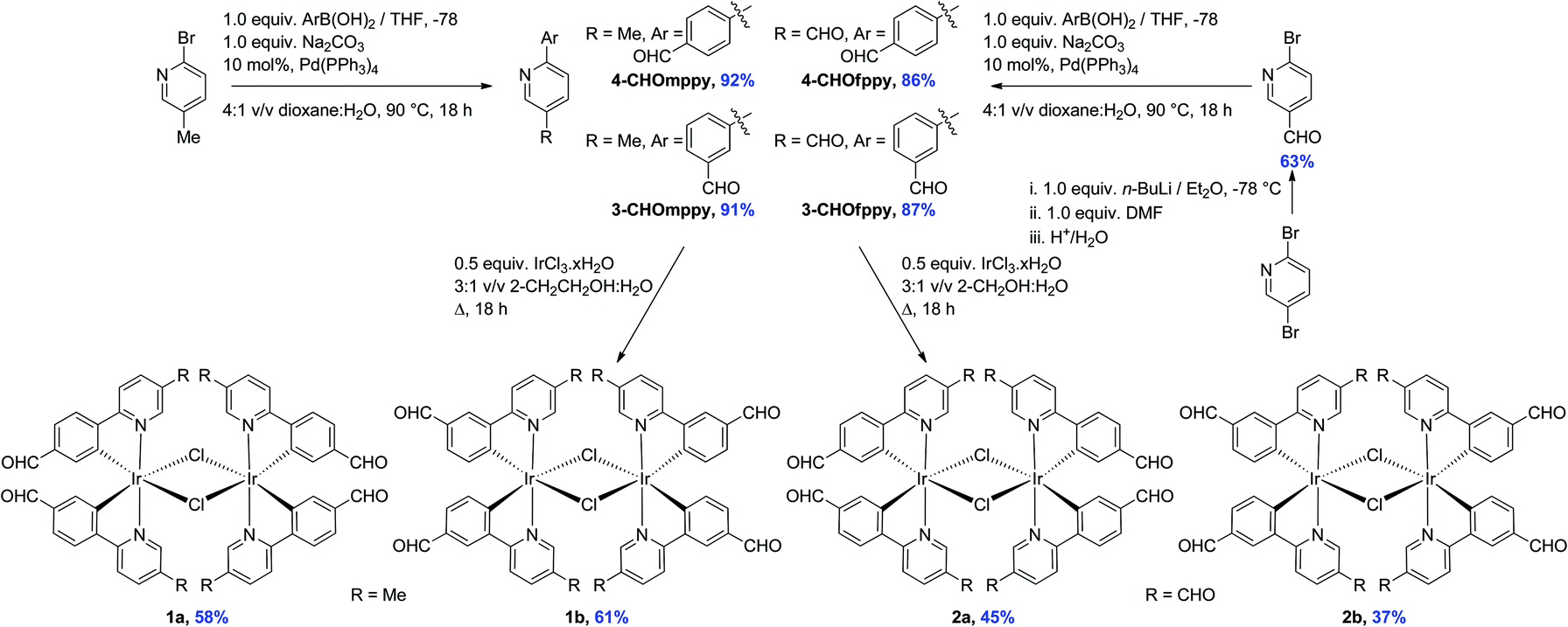 | ||
| Scheme 1 Synthesis of formyl-substituted C^N ligands and iridium dimer complexes 1a–2b. | ||
The dichloro-bridged iridium(III) dimers were prepared according to the protocol first reported by Nonoyama.13 While 1a and 1b were obtained in 58 and 61%, respectively and were analytically pure following precipitation from a refluxing 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v mixture of 2-ethoxyethanol–water, under these same conditions the synthesis of 2a and 2b resulted in very complicated 1H NMR spectra. However, when the synthesis was repeated in a refluxing 3:1 v/v mixture of 2-methoxyethanol–water, 2a and 2b precipitated from the solution and could be obtained in more modest 45 and 37% yields, respectively. Complexes 1a–2b were characterized by 1H NMR, 13C NMR, Mp, EA and HRMS. Additionally single crystals of sufficient quality for X-ray diffraction analyses for all four complexes were obtained (vide infra).
:1 v/v mixture of 2-ethoxyethanol–water, under these same conditions the synthesis of 2a and 2b resulted in very complicated 1H NMR spectra. However, when the synthesis was repeated in a refluxing 3:1 v/v mixture of 2-methoxyethanol–water, 2a and 2b precipitated from the solution and could be obtained in more modest 45 and 37% yields, respectively. Complexes 1a–2b were characterized by 1H NMR, 13C NMR, Mp, EA and HRMS. Additionally single crystals of sufficient quality for X-ray diffraction analyses for all four complexes were obtained (vide infra).
Solution state characterization
Fig. 1 shows the aromatic region of the 1H NMR spectra for 1a and 4-CHOmppy (for corresponding comparison of 1H NMR for 1b, 2a and 2b with their respective C^N ligands see Fig. S19–S22, ESI†). Eight well-resolved resonances are observed in the 1H NMR of 1a in CDCl3, the result of chemical non-equivalence of the H3A/H5A protons upon cyclometalation. Structural assignments are based on 2D-COSY NMR experiments and a comparison of our 1D NMR data with those previously reported by Watts and co-workers, and more recently Schubert and co-workers.5c,14 The resonance for HA5 at δ 6.16 is significantly shifted upfield due to the formal anionic charge of the cyclometalating phenyl ring.5a,15 By contrast, HB6 is shifted downfield at δ 9.07 upon complexation.5c The regiochemistry of cyclometalation in 1b and 2b can be simply determined from the presence of HA2, which has a characteristic chemical shift at ca. δ 6 and a coupling constant of ca. 8 Hz. Thus, the formyl groups on the phenyl ring in these two complexes are located exclusively para to the iridium metal. The pattern of signals in the 1H NMR and the presence of only thirteen 13C NMR resonances also points to a single configuration coordination mode for the four complexes, with the arrangement of the nitrogen atoms of the pyridine moieties trans with respect to each other, as is typical under the reaction conditions. Though the synthesis is not stereoselective and statistical mixtures of ΔΔ, ΛΛ and ΔΛ diastereomers should exist in solution, by 1H NMR only a single set of resonances exists. Single crystal X-ray diffraction results reveal only the presence of only a racemic mixture of ΔΔ and ΛΛ isomers. (vide infra).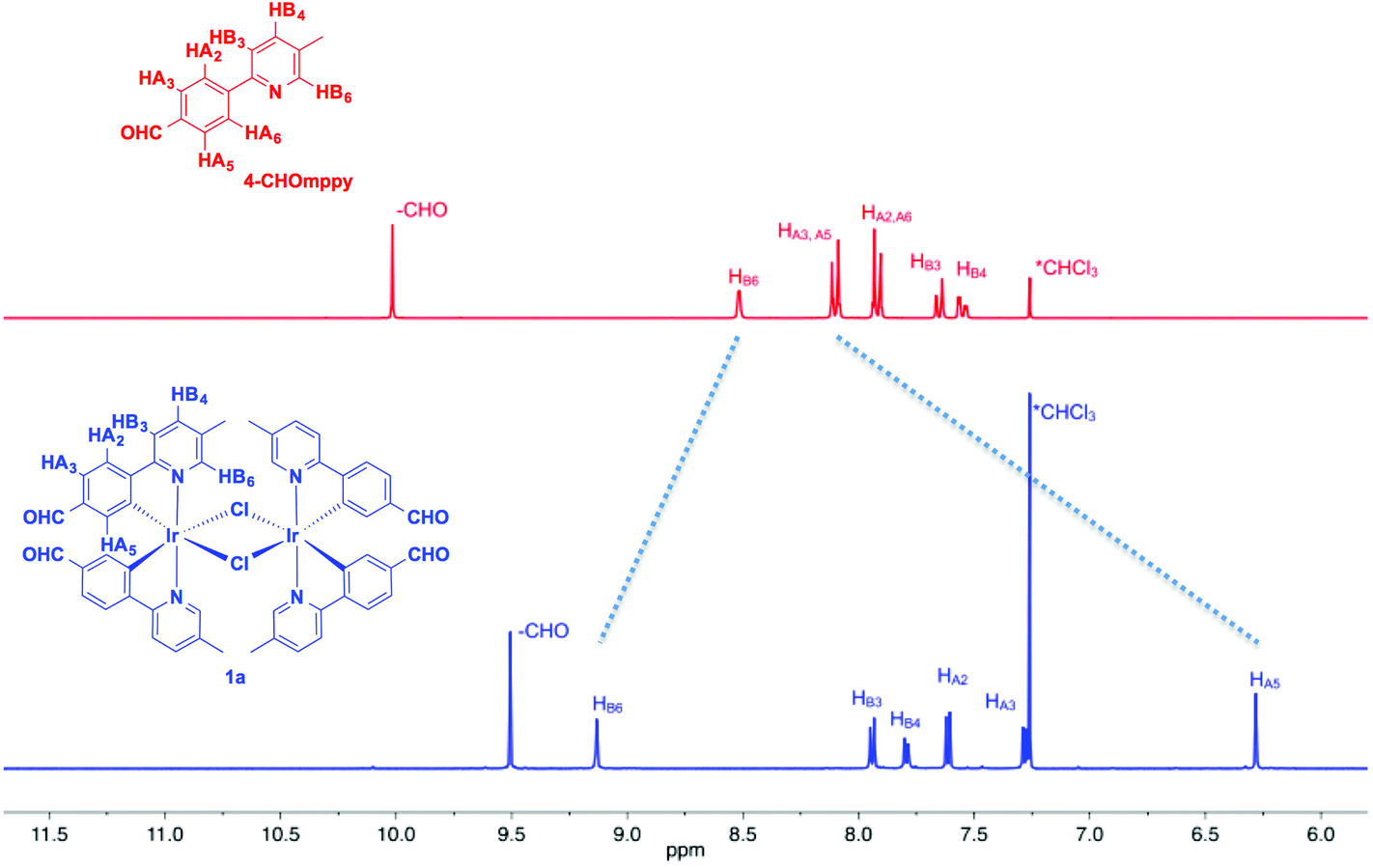 | ||
| Fig. 1 Stacked 1H NMR spectra of aromatic region 4-CHOmppy (red) and complex 1a in CDCl3 at 298 K. | ||
The MALDI HRMS show low intensity molecular ions for all four complexes. In all four mass spectra, a fragmentation peak with a mass-to-charge ratio (m/z) half that of the molecular ion is always observed, which resulted from homo-cleavage of the molecular ion under MS conditions. Similar observations have been reported in the literature.16
Solid-state characterization
All crystals were grown by vapour diffusion of diethyl ether into saturated DCM solution of the complexes. The connectivity of bonds provides an absolute proof of structure. All four complexes adopt similar dinuclear structures (Fig. 2) with compounds 1a, 1b, and 2b crystallising in the monoclinic space group P21/c, in unit cells of broadly similar sizes, and one molecule of complex in the asymmetric unit, while compound 2a crystallises in the space group P21/n, but with a unit cell of close to twice the size, and two molecules of complex in the asymmetric unit. The complexes adopt a slightly distorted octahedral coordination environment about the iridium(III) centres, with Ir–N trans to each other and Ir–C cis, in common with other examples of [Ir(C^N)2Cl]2 complexes.5c,6,15–17 While this arrangement can be readily deduced in the cases of complexes 1a, 1b, and 2b, for complex 2a, comparison of potential Ir–C/N distances, as well as the 1H NMR data, confirm the trans-N arrangement. Bond parameters about the iridium(III) are unexceptional (Fig. 2), as are the Ir⋯Ir separations [3.7867(9) to 3.8068(19) Å]. In an analogous manner to related complexes, all four complexes display racemic ΔΔ and ΛΛ diastereomers, rather than the meso ΔΛ form. All four structures showed space open to solvent within them. In 1a and 2a this could be modelled as either one (disordered) or two molecules of CH2Cl2 per complex, respectively, with no void space remaining. In 1b and 2b however, no solvent molecules could be sensibly modelled, resulting in large void spaces. In 1b, these run along the a-axis whereas in 2b, channels are narrow and do not run straight along a single axis; additional thin void spaces can be seen in the ac-plane.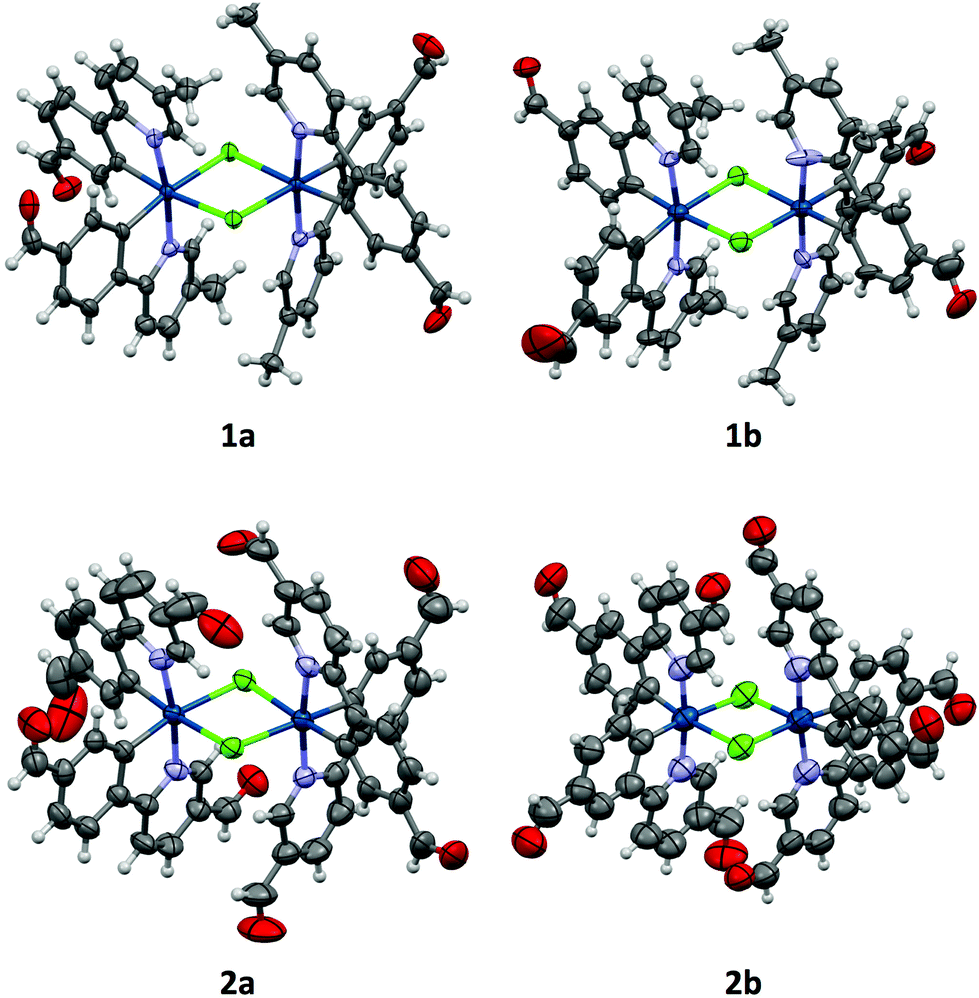 | ||
| Fig. 2 Crystal structures of complexes 1a–2b, with solvent molecules omitted. Thermal ellipsoids are drawn at the 50% probability level for 1a, 1b, and 2a, and at the 30% probability level for 2b. Bond lengths in Å: 1a Ir–C [1.989(9)–2.00(1)]; Ir–N [2.035(7)–2.059(7)]; Ir–Cl [2.494(2)–2.529(2)]; 1b Ir–C [1.99(2)–2.04(2)]; Ir–N [2.01(2)–2.06(2)]; Ir–Cl [2.498(6)–2.536(6)]; 2a Ir–C [1.988(6)–2.023(7)]; Ir–N [2.035(5)–2.058(5)]; Ir–Cl [2.510(2)–2.527(2)]; 2b Ir–C [1.87(2)–1.92(2)]; Ir–N [1.95(2)–2.00(2)]; Ir–Cl [2.491(7)–2.542(6)]. Bond angles in degrees: 1a N–Ir–C [80.2(3)–81.2(3)] Cl–Ir–Cl [81.34(7)–81.43(7)]; 1b N–Ir–C [81.5(2)–83.1(8)] Cl–Ir–Cl [81.5(2)–81.6(2)]; 2a N–Ir–C [79.9(2)–80.8(2)] Cl–Ir–Cl [81.83(5)–82.13(5)]; 2b N–Ir–C [80.8(9)–83.1(9)] Cl–Ir–Cl [81.4(2)–82.2(2)]. | ||
Various interactions are seen to be common to the four complexes, both intramolecular, for maintaining the molecular geometries seen, and intermolecular, for providing stabilising interactions between molecules. Intermolecular interactions can be seen as weak C–H⋯Cl hydrogen bonds that are present between the CH(α) adjacent to the nitrogen atom of the pyridyl rings of the C^N ligands and the bridging chlorides of the complexes [H⋯Cl distances ranging from 2.50 to 2.70 Å, with C⋯Cl separations of 3.311(8) to 3.35(2) Å]. These hydrogen bonds are complemented, in all cases except 2b, by C–H⋯π interactions between a proximal CH of a phenyl ring, and the π-system of the pyridyl of an adjacent ligand; H⋯centroid distances ranging from 2.93 to 2.94 Å, with C⋯centroid separations of 3.72(3) to 3.725(7) Å (Fig. 3).
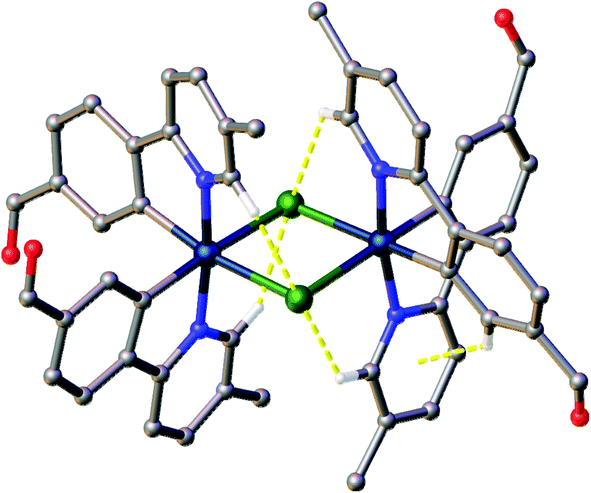 | ||
| Fig. 3 View of complex 1a showing the intramolecular interactions present in the complexes. Interactions shown as dashed yellow lines, hydrogen atoms not involved in the interactions were omitted. | ||
Common intermolecular interactions in the complexes include both interactions between the complexes and solvent, for those where the solvent could be resolved within the structure, and between adjacent complex molecules. Complex-to-solvent interactions consist of both C–H⋯π and weak C–H⋯Cl hydrogen bonds, the latter involving aryl hydrogens. Interactions between complexes follow a similar pattern. A few C–H⋯π interactions are observed, although at sufficient interatomic distances [H⋯centroid 2.73–2.93 Å, C⋯centroid 3.650(7)–3.874(10) Å] to be relatively unimportant if taken by themselves. They are reinforced, however, by sets of weak C–H⋯O hydrogen bonds, involving both aryl and (where they exist as in 1a and 1b) alkyl hydrogens, at H⋯O distances of 2.19 to 2.59 Å, and C⋯O separations of 2.999(15) to 3.542(13) Å.
As well as these sets of conserved interactions, there are some intermolecular interactions that occur in some, but not all, of these complexes. Complexes 1a, 1b, and 2b show further interaction involving their bridging chlorides. These are C–H⋯Cl hydrogen bonds involve the formyl protons of adjacent complexes. The H⋯Cl distances range from 2.51 to 2.82 Å, with corresponding C⋯O separations of 3.32(4) to 3.70(3) Å. An additional feature of the interactions in these three complexes is that none of them show π⋯π interactions (no centroid⋯centroid distances of less than 3.91 Å is present). Complex 2a, on the other hand shows quite different interactions to these (Fig. 4). The first of these is a π-stacking interaction between proximal phenyl rings of the two molecules in the asymmetric unit. The centroid⋯centroid distance is 3.767(4) Å, with an inclination between ring-planes of 0.2(4)°. This interaction is reinforced by one of the C–H⋯π interactions mentioned earlier, between a hydrogen on one of the π-stacking rings, and another ring centroid on the adjacent complex. The second set of different interactions concerns weak hydrogen bonds involving the formyl protons. Unlike in the other three complexes, in 2a these do not interact with the bridging chloride atoms, but form an extensive network of C–H⋯O interactions with other formyl groups (Fig. 4). The H⋯O distances in these interactions vary between 2.30 and 2.52 Å, with C⋯O separations of 3.102(14) to 3.346(13) Å. From the interatomic distances in these interactions it may be deduced that the molecules of complex in 2a are held together more tightly in the solid state, and interact more strongly than those in the other complexes.
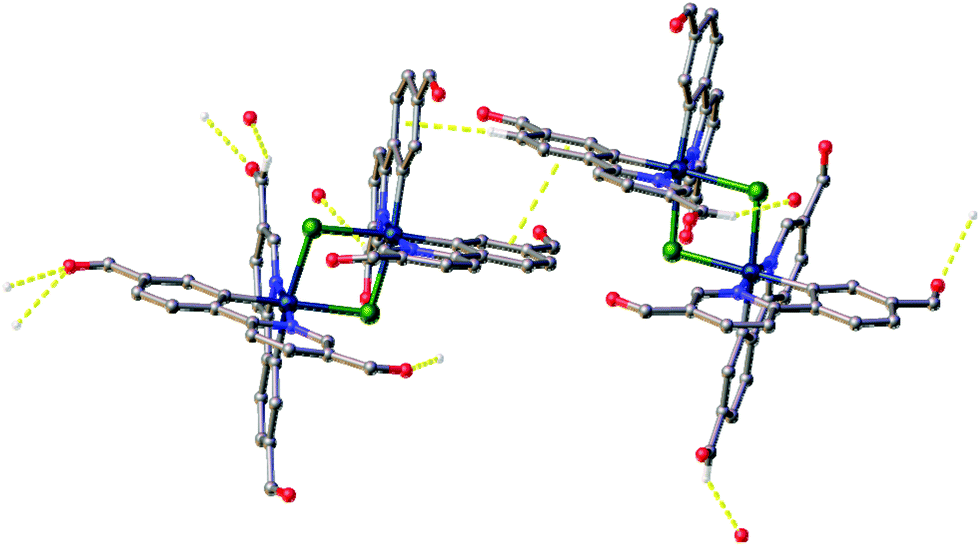 | ||
| Fig. 4 View of complex 2a showing the intermolecular interactions observed in this complex but not the others. Interactions shown as dashed yellow lines, hydrogen atoms not involved in the interactions were omitted. | ||
Electrochemical properties
The electrochemical properties of 1a–2b were studied in nitrogen-saturated DCM at room temperature using nNBu4PF6 as the supporting electrolyte and using Fc/Fc+ as an internal standard. All potentials are referenced to SCE (Fc/Fc+ = 0.46 V in DCM).18 The results are compiled in Table 1 while CV traces are shown in Fig. 5. The CV behavior was reproducible across a range of scan rates ranging from 50 to 1000 mV s−1 though at higher scan rates the redox waves broadened, rendering analysis more difficult. All complexes in this study showed essentially quasi-irreversible or irreversible electrochemistry as ipa/ipc ≪ 1.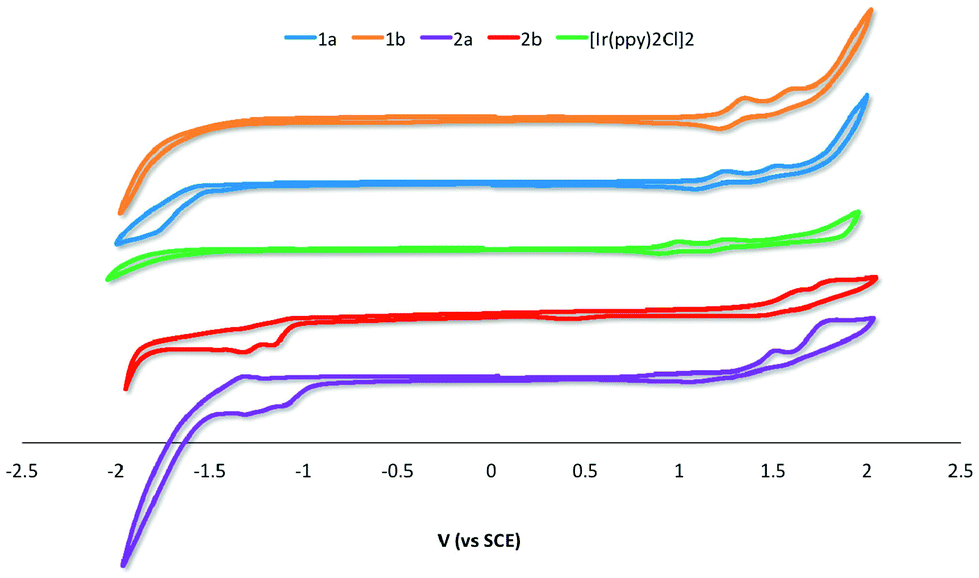 | ||
| Fig. 5 CV traces for 1a–2b and [Ir(ppy)2Cl]2 recorded at 298 K at 50 mV s−1 in deaerated DCM with 0.1 M (nBu4N)PF6. | ||
| Complex | E oxpa,1/V | E oxpa,2/V | E redpc,1/V | E redpc,2/V | ΔEredox/V | E HOMO/eV | E LUMO/eV | ΔE/eV |
|---|---|---|---|---|---|---|---|---|
| a Conditions: CV traces recorded in N2-saturated DCM solution with complex concentration at 10−4 M with 0.1 M nBu4NPF6 at 298 K; platinum wires were used as the working and counter electrodes, respectively while an Ag/AgCl reference electrode was employed; scan rate: 50 mV s−1. Values are in V vs. SCE (Fc/Fc+vs. SCE = 0.46 V).18 ΔE = −(EHOMO − ELUMO); Epa = anodic peak potential and Epc = cathodic peak potential. b The HOMO and LUMO energies were calculated using the relation EHOMO/LUMO = −(Eoxpa,1/Eredpc,1 + 4.8) eV, where Eoxpa,1/Eredpc,1 are the first oxidation or reduction peaks with respect to E1/2 of ferrocene/ferrocenium redox couple.19 c E LUMO was estimated from optical band gap, E0,0 determined from the low energy tail at 10% intensity of the lowest energy absorption band. | ||||||||
| [Ir(ppy)2Cl]2 | 1.00 | 1.26 | −5.34 | −2.85 | 2.49 | |||
| 1a | 1.25 | 1.51 | −5.59 | −3.21c | 2.38 | |||
| 1b | 1.35 | 1.61 | −5.69 | −3.11c | 2.58 | |||
| 2a | 1.46 | 1.67 | −1.10 | −1.30 | 2.56 | −5.80 | −3.24 | 2.56 |
| 2b | 1.58 | 1.72 | −1.16 | −1.31 | 2.74 | −5.92 | −3.18 | 2.74 |
The parent dimer [Ir(ppy)2Cl]2 shows two one-electron oxidation waves at 1.00 and 1.26 V indicating that there is electronic communication between the two iridium centres mediated by the chloride bridges. The oxidations are assigned to the IrIII/IrIV redox couple with significant contribution from the aryl ring. No reduction is observed within the solvent window. This profile matches that originally reported by Watts and co-workers.5b The introduction of an electron-withdrawing formyl group onto the phenyl moiety of the C^N ligand in 1a and 1b results in an expected shift to more positive potential of both oxidation waves, corresponding to a stabilization of the HOMO on each of the two iridium centers. Placement of the formyl group para to the Ir–C bonding results in a 100 mV anodic shift of both oxidation waves in 1b compared to 1a. The additional of a second formyl group at the 5-position of the pyridine fragment of the C^N ligands makes the first oxidation more difficult by 0.21 and 0.23 V for 2a and 2bversus1a and 1b, respectively.
For 1a and 1b no reduction waves are observed. However, for 2a and 2b two irreversible reduction waves are now observed pointing to a LUMO involving the 5-formyl group. The LUMO is stabilized by a further 60 mV in 2a compared to 2b; the second reduction in both of these complexes occurs at ca. −1.31 V.
The band gaps ΔE for 2a and 2b were determined from the electrochemical data and are 2.56 and 2.74 eV, respectively. The band gaps for 1a and 1b at 2.38 and 2.58 eV, respectively, were extrapolated from the tailing edge at 10% intensity of the lowest energy absorption band, corresponding to the energy of the 0,0 transition. This method certainly underestimates this parameter by at least 0.65 eV as the HOMO to LUMO transition does not account for the binding energy associated with ionization.20
UV-visible absorption spectroscopy
The UV-visible absorption spectra for 1a–2b and [Ir(ppy)2Cl]2 were recorded in aerated DCM at 298 K and are shown in Fig. 6. The results are summarized in Table S1.†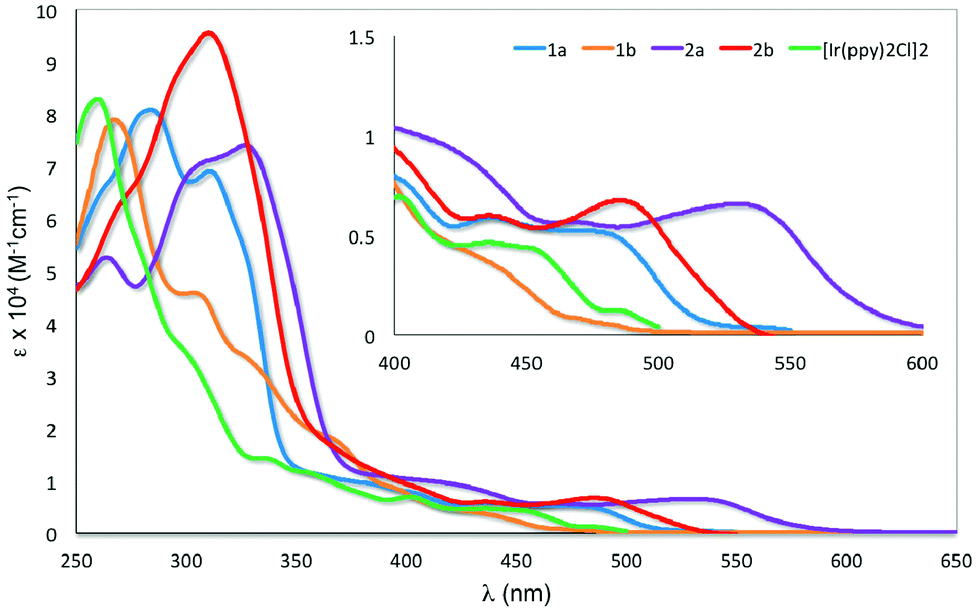 | ||
| Fig. 6 UV-visible absorption spectra of 1a–2b and [Ir(ppy)2Cl]2 in DCM at 298 K. Inset: zoomed UV-visible spectra for the region between 400–600 nm. | ||
All the iridium dimers possess intense absorption bands below ca. 350 nm corresponding to spin-allowed singlet ligand-centered (1LC) 1π–π* transitions.6a At lower energies, the absorption spectra are dominated by spin-allowed metal-to-ligand charge transfer (1MLCT) and intraligand charge transfer (1ILCT) transitions;5a,b,18 spin forbidden 3MLCT transitions are certainly also present at low energies due to the large spin–orbit coupling mediated by Ir. Similar to other homodinuclear iridium complexes,21 the molar absorptivities of these complexes are approximately double than those of neutral mononuclear iridium complexes such as [Ir(ppy)2(pic)] (pic = picolinate).22
The absorption profile for 1a and 1b are similar, with 1a being bathochromically shifted compared to 1b and [Ir(ppy)2Cl]2. This red-shift in 1a is caused by the electron-withdrawing 4-formyl group, which results in a stabilisation of both the occupied π and the unoccupied π* orbital of the C^N ligands and a smaller energy transition.23 This red-shifting of the absorption spectrum is not observed in 1b, with its HOMO–LUMO band blue-shifted compared to [Ir(ppy)2Cl]2. The change in regiochemistry of the formyl group on the phenyl fragment results in cross-conjugation with the pyridine moiety and a reduced influence on the energies of the antibonding orbitals. Additionally, the HOMO stabilising character of the 3-formyl group is more pronounced, as it is now para to the Ir–C bond. Similarly, the absorption of 2a is bathochromically shifted with respect to 2b. The magnitude of this red-shift is unexpected, particularly given the similar 1MLCT absorption maxima reported by Bryce, Monkman and co-workers for a series of fluorenyl-containing iridium dimer complexes bearing 5-fluoro- or 5-methoxy-substituted pyridine moieties on the C^N ligands.6b The greater red-shifting of the absorption spectra for 2a and 2b compared to 1a and 1b results from the presence of the second formyl group, which further stabilises the LUMO that contains significant electron density on the pyridine ring of the ligand.
Emission spectroscopy
The emission properties of the five complexes were studied in degassed DCM at 298 K with the spectra shown in Fig. 7 and the results summarized in Table 2. Their emission profiles are broad with the presence of either two well-defined emission bands or the presence of a low energy emission shoulder. The emission is assigned to a mixed 3MLCT/3LC transition.6a The very weak emission of [Ir(ppy)2Cl]2 at 520 nm reproduces that previously reported by Watts and co-workers.5c The introduction of formyl groups causes a red-shift in the emission compared to the benchmark iridium dimer. The regiochemistry and quantity of the formyl groups have a dramatic impact on the photophysical properties of these complexes. Surprisingly, in 1a the addition of the 4-formyl unit causes a red-shift of 1468 cm−1 and an accompanying order of magnitude increase in ΦPL to 2.5%. By contrast, incorporation of electron-withdrawing fluorine atoms onto the C^N ligands in both mononuclear neutral24 and charged25 complexes causes a blue-shift in the emission due to large stabilization of the HOMO. Positioning the formyl group para to the Ir–C in 1b causes a further red-shift of 1095 cm−1 compared to 1a. Emission in 1b is so weak as to preclude accurate determination of ΦPL. The introduction of a second formyl group in 2b causes a blue-shift in the emission compared to 1b. Most striking, the emission for 2a is not similarly blue-shifted compared to 1a but is red-shifted by 1395 cm−1 and is the brightest of the series of iridium dimers with a ΦPL of 15.7%. Only recently reported fluorenyl annelated phenylpyridine C^N dichloro-bridged iridium dimers have shown brighter emission than 2a for this class of complexes.6b Its emission lifetime, τe, is also an order of magnitude longer than the other complexes at nearly 2 μs. The primary reason for this enhanced emission is due to the large decrease in the non-radiative rate constant, knr caused by increased rigidity conferred by the more extensive hydrogen bonding interactions (vide supra).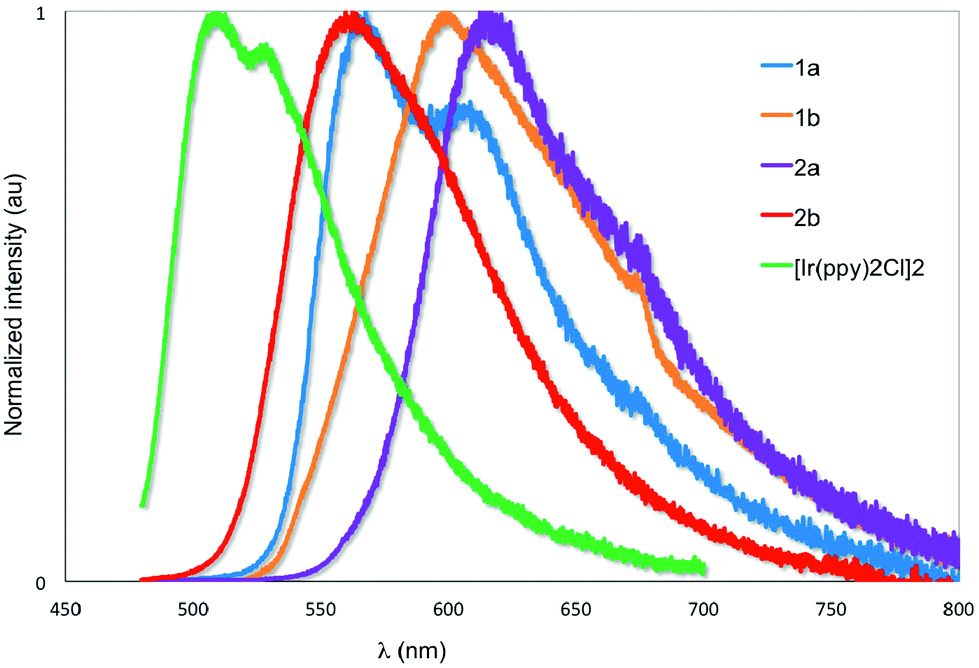 | ||
| Fig. 7 Normalized emission spectra of 1a–2b and [Ir(ppy)2Cl]2 in deaerated DCM at 298 K. λexc: 455 nm. | ||
a
| Dimer complex | λ em (nm) |
Φ
PLb (%) |
τ
ec (ns) |
k
rd (×105 s−1) |
k
nre (×105 s−1) |
|---|---|---|---|---|---|
| a Emission measured in N2-saturated DCM at room temperature. b Relative to Ru(bpy)3(PF6)2 (ΦPL = 9.5% in degassed MeCN).26 c λ exc: 470 nm. d Calculated from ΦPL = τe × kr. e Calculated from Φ = kr/(kr + knr). f Emission too weak to determine ΦPL or τe. | |||||
| [Ir(ppy)2Cl]2 | 520 | 0.1 | 125 | 0.40 | 79.60 |
| 1a | 563, 600 | 2.5 | 206 | 1.21 | 47.33 |
| 1b | 600 | Emission too weak | |||
| 2a | 611, 670(sh) | 15.7 | 1980 | 0.79 | 4.26 |
| 2b | 556 | 0.9 | 601 | 0.15 | 16.49 |
In order to better understand the photophysics of the materials and their potential for OLEDs, the photoluminescence quantum yield of the solid films was measured. Neat films were studied and compared with films in which the test complex was blended with charge transporting materials. The latter films are generally preferable for OLEDs because diluting the chromophore usually reduces concentration quenching, and because the host materials can facilitate charge transport. The films were prepared by spin-coating the materials from DCM solution onto fused silica substrates, and the results are shown in Table 3. As with solution-state measurements, 1b has minimal emission in the solid state. The other materials show much higher photoluminescence quantum yield in the blend than in the neat film. Whereas 2a was found to show the highest photoluminescence quantum yield in DCM, the brightest complex in the solid state is 2b, with a ΦPL in the doped film of 36%, more than double that of 2a. The emission maxima found in the doped films match those observed in dilute DCM solution.
| Dimer complex |
λ
PLa (nm) |
Φ
PLb (%) |
Φ
PLc (%) |
|---|---|---|---|
|
a PL measurements of blended films of CBP:PBD:complexes (60:30:10 wt%) in a 60 nm thick film.
b
Φ
PL of neat films spin-coated onto a fused silica substrate from DCM solution.
c
Φ
PL of blended films of CBP:PBD:dimer complex (60:30:10 wt%) on a fused silica substrate. CBP is 4,4′-N,N′-dicarbazole-biphenyl and PBD is 2-(tert-butylphenyl)-5-biphenyl-1,3,4-oxadiazole.
d Data not available due to weak emission.
|
|||
| 1a | 562 | 1.0 | 10.3 |
| 1b | — | — | — |
| 2a | 620 | 1.1 | 14.7 |
| 2b | 555 | 2.6 | 35.6 |
Theoretical calculations
The geometries and electronic structures of complexes 1a and 2a, as representative examples of the series, were modelled within Gaussian 0927 using density functional theory (DFT) and time-dependent DFT (TDDFT).28 These complexes were modelled using the B3LYP29 level of theory with the SBKJC-DVZ30 basis set for iridium, 6-31G* for heavy atoms directly coordinated to iridium and 3-21G* for all other atoms30a,31 in the presence of the solvent DCM.32 This methodology has been successfully used by us for other dinuclear iridium complexes.33 The calculated ground-state geometries well reproduce those found in the crystal structures with the exception of the Ir–Cl bonds, is ca. 0.1 Å longer than the experimental value. The resulting Ir⋯Ir distance of 3.95 Å is thus also overestimated.The frontier molecular orbitals for 1a and 2b are shown in Fig. 8. The calculated HOMO and LUMO energies for 1a are, respectively, −5.59 and −2.20 eV while for 2a they are −5.90 and −2.90 eV, respectively. Calculated HOMO energies agree very well with CV data in Table 1 while LUMO levels are not well reproduced. The poor prediction for 1a is most likely a function of the underestimation based on extrapolation of the LUMO level from absorption data. In 2a, the calculated LUMO is 0.34 eV higher than that obtained from the electrochemical measurement. In both complexes the HOMO is distributed across the iridium dimer with electron density on the iridium metal centers, the phenyl rings of the cyclometalating ligands and the chlorine bridge. This picture, pointing to electronic communication between the two iridium atoms, agrees well with the CV traces, which show two distinct oxidation waves, one at each iridium center. The LUMO is distributed across all of the cyclometalating ligands. In 2a electron density extends out to the formyl groups on the pyridine rings, which corroborates the assignment of the irreversible reduction in 2a as being localized on this fragment of the complex. The topologies for HOMO−1 and LUMO+1 are nearly identical to those of HOMO and LUMO, respectively.
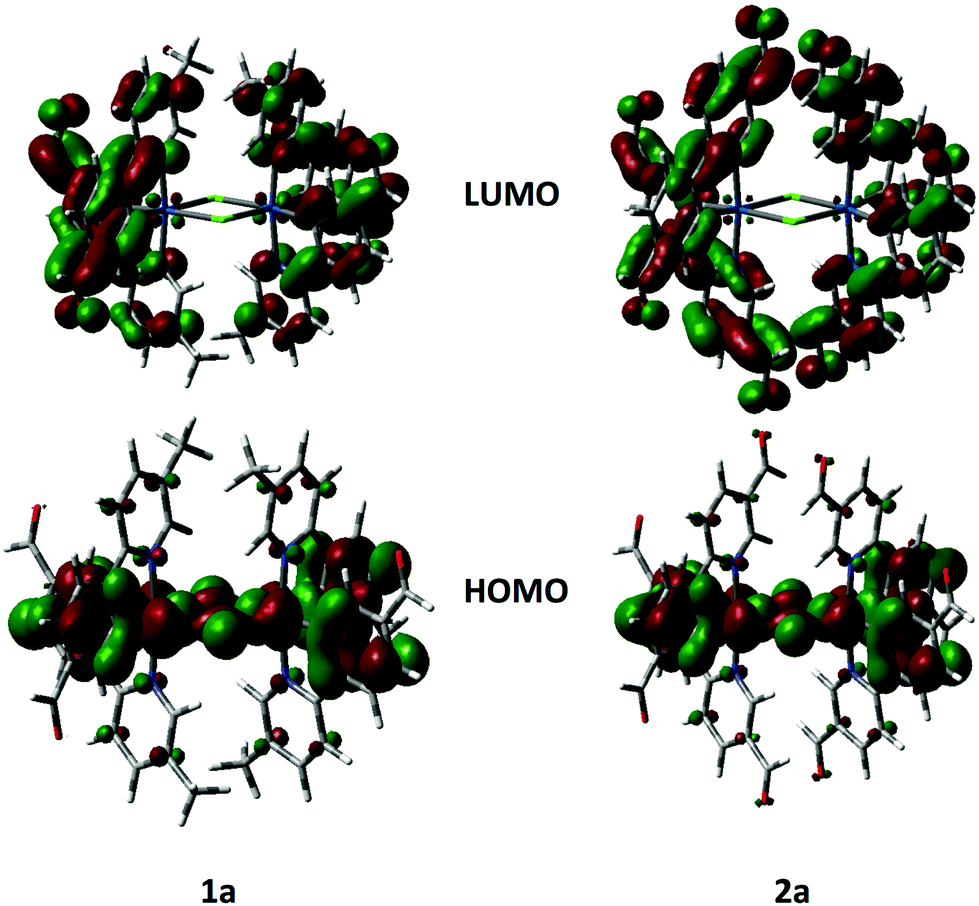 | ||
| Fig. 8 HOMO and LUMO orbitals of 1a and 2a, electron density contour plots at 0.002 e bohr−3. | ||
The predicted absorption data (Fig. S27 and S28†) obtained by TDDFT reproduce very well the gross features of the absorption spectra, including the low energy band at 540 nm observed in 2a. The calculated S0 to T1 transition obtained from TDDFT based on the S0 optimized geometries for 1a and 2a are 516 and 570 nm fit well with the very weak 3CT transitions observed in the absorption spectra.
Electroluminescence performances
In order to investigate the electrical and optical properties of the OLEDs based on these four iridium dimer complexes, we tested each of them in a device structure which was ITO/PEDOT:PSS(30 nm)/PVK(30 nm)/CBP:PBD:dimer complex (60:30:10, 60 nm)/B3PYMPM(40 nm)/Ca(20 nm)/Al(100 nm). PEDOT:PSS, PVK and B3PYMPM denote poly(3,4-ethylenedioxythiophene): poly(styrenesulfonate), poly(N-vinylcarbazole) and bis-4,6-(3,5-di-3-pyridylphenyl)-2-methylpyrimidine, respectively. PVK facilitates hole injection because its HOMO (5.6 eV)34 lies between that of ITO and the co-host materials (6.0 eV). The LUMO of PVK (2.2 eV) is slightly higher than those of CBP (2.6 eV)35 and PBD (2.3 eV) and its wide energy gap should confine excitons inside the emitting layer thus improving capture of opposite charges in the device.34,35 The electron-transporting layer B3PYMPM blocks the hole from penetration into the cathode due to its deep-lying HOMO (6.8 eV) and reduces the leakage current.36 Hence, the multi-layer helps to confine excitons inside the emitting layer as is needed for good luminous efficacy.
Fig. 9 shows the current–voltage characteristics of the devices. The devices with complexes 2a and 2b exhibited lower current density at high voltage (>7 V) than the devices made from 1a and 1b. The device based on 1b exhibited the highest current density above 7 V. However, its luminance saturated at higher driving voltage. The devices based on complex 2b possessed the lowest current density above 7 V. However the luminance of this device goes up very steadily once turned on. A high luminance, up to 5930 cd m−2 can be achieved at a driving voltage of 14.6 V. Therefore, among the four complexes, much higher luminous efficiencies can be seen from this device, i.e. 9.1 cd A−1, 2.2 lm W−1 and 2.6% of current efficiency (CE), power efficiency (PE) and external quantum efficiency (EQE), respectively (Fig. 10a–c, respectively). In contrast, the poorer efficiencies of the devices with 1a and 1b may be due to incomplete host to guest energy transfer (1a) and exciplex emission (1b), which can be inferred from the EL spectra in Fig. 11a and c. The EL spectra from the complexes 2a and 2b, were nearly independent of the current density, indicating very efficient host to guest energy transfer and nearly no exciplex contribution (Fig. 11b and d). A comparison of the electrical and optical properties is listed in Table 4.
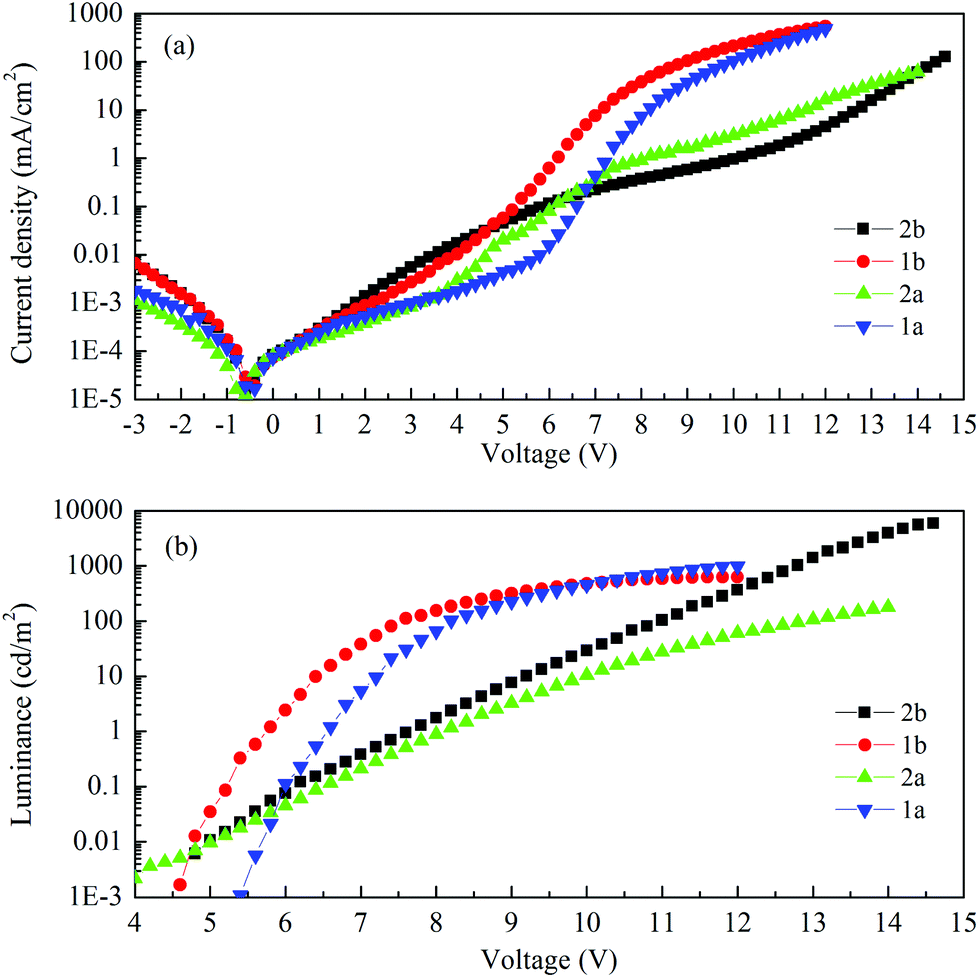 | ||
| Fig. 9 (a) Current density and (b) luminance vs. voltage characteristics of the devices. | ||
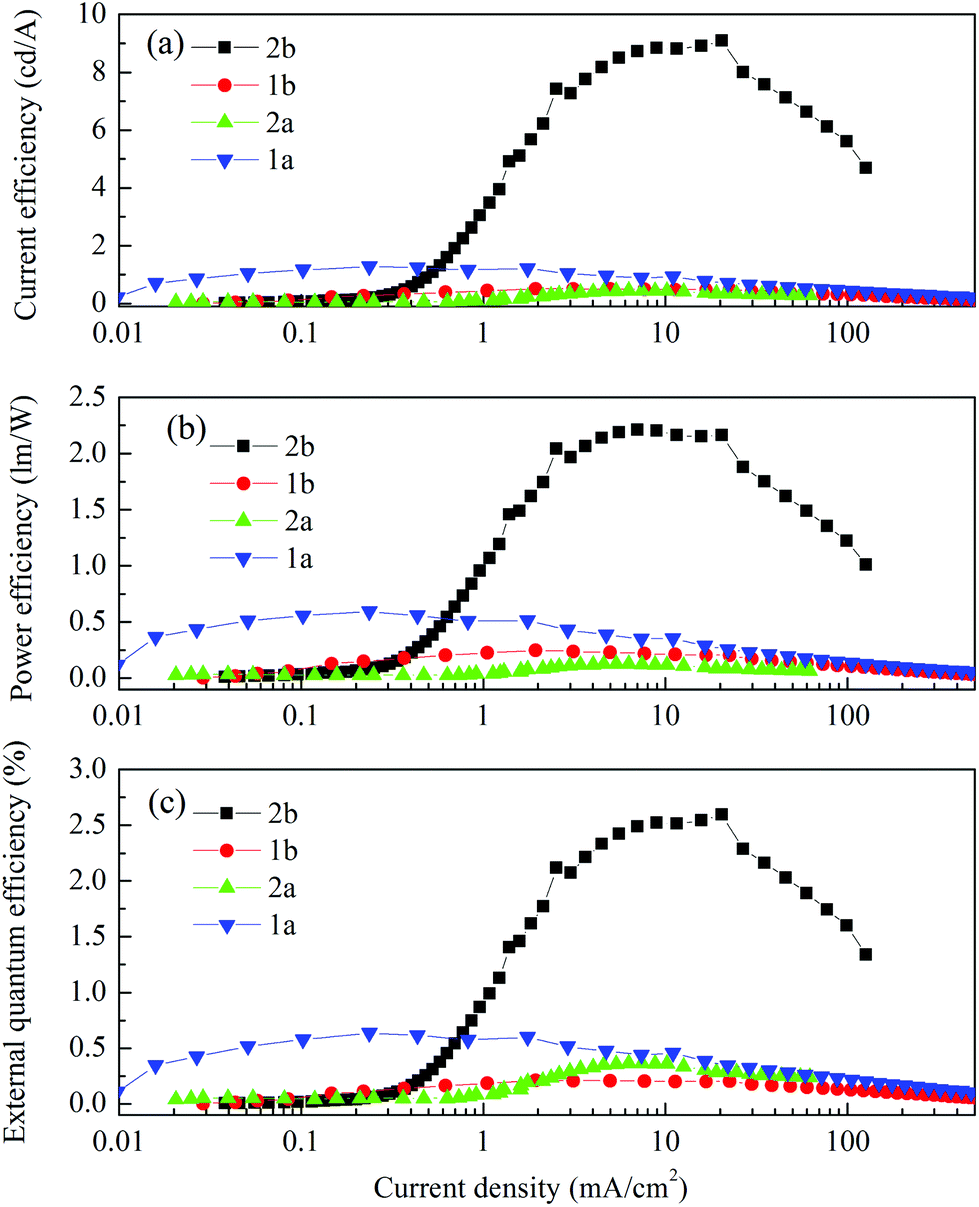 | ||
| Fig. 10 (a) Current efficiency, (b) power efficiency and (c) external quantum efficiency vs. current density characteristics of the devices. | ||
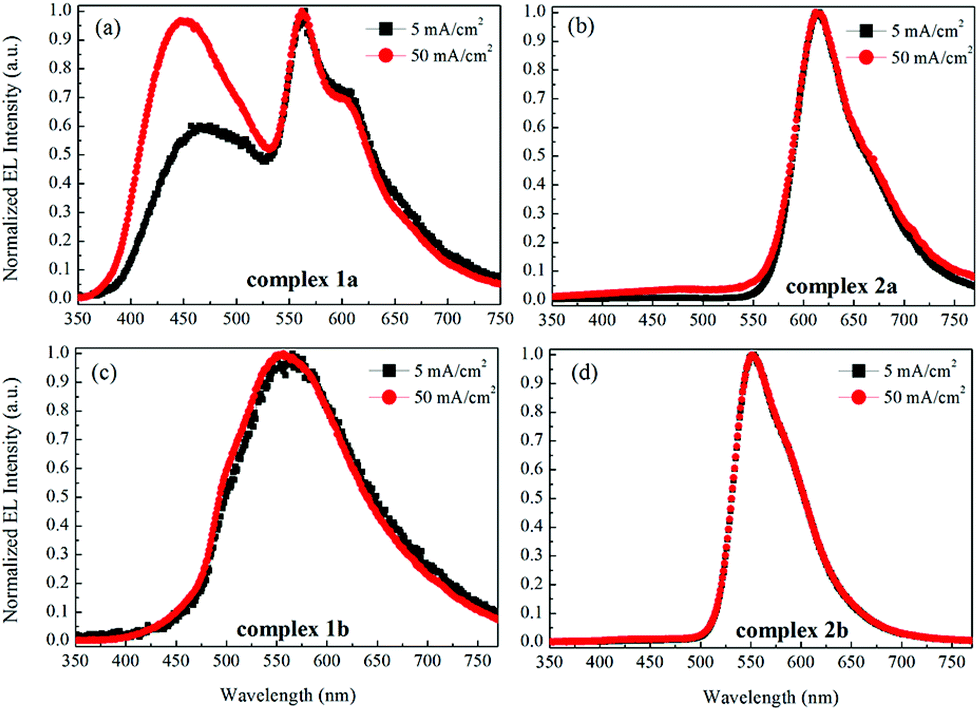 | ||
| Fig. 11 The normalized EL spectra of the devices with the emitters 1a (a), 2a (b), 1b (c) and 2b (d) at current densities of 5 and 50 mA cm−2. | ||
| Voltage at 1 cd m−2 (V) | Max. luminance (cd m−2) | Max. CE (cd A−1) | Max. PE (lm W−1) | Max. EQE (%) | λ em (nm) | CIE at 50 mA cm−2 | |
|---|---|---|---|---|---|---|---|
| a CE = current efficiency. PE = power efficiency. EQE = external quantum efficiency. CIE = The Commission Internationale de l'Eclairage coordinates. | |||||||
| 1a | 6.5 | 982 | 1.29 | 0.59 | 0.64@3 cd m−2 | 447, 562 | (0.3074, 0.3097) |
| 1b | 5.7 | 630 | 0.51 | 0.25 | 0.21@10 cd m−2 | 612 | (0.4073, 0.4966) |
| 2a | 8.1 | 178 | 0.44 | 0.13 | 0.37@28 cd m−2 | 557 | (0.5991, 0.3600) |
| 2b | 7.7 | >6000 | 9.09 | 2.16 | 2.59@1860 cd m−2 | 551 | (0.4413, 0.5420) |
There is a correlation between the EQE of our devices and solid-state photoluminescence quantum yield (Table 3). The material with the highest ΦPL (2b) also produces the OLED with the highest EQE. The complex with the lowest EQE (1b) results from its very poor emission with the lowest ΦPL among the four materials. The other two materials (1a, 2a) have intermediate photoluminescence and electroluminescence efficiencies. Of these two materials, 2a has higher ΦPL but lower external quantum efficiency in the OLED, suggesting that there is a difference in charge balance between the devices.37 We observe EL from 1b even though its PL is very weak. However, the spectrum of the EL (Fig. 11c), suggests it is due to the formation of an exciplex between CBP and B3PYMPM.
In the recent report by Bryce and Monkman on the use of dichloro-bridged iridium dimers as emitters in OLEDs, the devices [structure: ITO/PEDOT:PSS/PVK–PBD (40%)–complex (5%)/Ba/Al] achieved peak EQEs ranging from 0.6–4% with PE ranging from 0.40–3.53 lm W−1 and CE of 1.7–12 cd A−1 based on complexes that showed ΦPL of 21–41% in chlorobenzene solution.6b From Table 4, we can see that 2b exhibits the best performance of the series and shows comparable metrics to the best dichloro-bridged iridium complex reported by Bryce and Monkman.
Conclusions
In this work, the systematic synthesis, structural and optoelectronic characterization, and electroluminescent device data of a series of four dichloro-bridged iridium dimers with formyl-substituted 2-phenylpyridine C^N ligands were presented. The optoelectronic properties were found to be strongly affected by the number of formyl groups and their regiochemistry. In particular, complex 2a showed a remarkable photoluminescence quantum yield of 15.7% in DCM solution despite being the reddest emitting complex in the series. In the solid state, be it in a pristine thin film or as a dopant, complex 2b was found to be the brightest. Of the electroluminescent devices fabricated using these iridium dimers, only the OLED employing 2b as the emissive dopant gave reasonable device performance, with an EQE of 2.6%.Experimental section
General synthetic procedures
All the chemicals and reagent grade solvents were purchased and used as received. Pd(PPh3)4 was synthesized according to the literature.38 All reactions were performed under a nitrogen atmosphere using Schlenk techniques. Flash column chromatography was carried out using silica gel (Silica-P from Silicycle, 60 Å, 40–63 μm). Analytical thin-layer-chromatography (TLC) was performed with silica plates with aluminum backings (250 μm with F-254 indicator). TLC visualization was accomplished by 254/365 nm UV lamp. 1H and 13C NMR spectra were recorded on a Bruker Avance spectrometer at 500 MHz and 126 MHz, respectively. For multiplicity assignment in NMR spectra reporting, “s” stands for singlet, “d” for doublet, “t” for triplet, “m” for multiplet and “br” for broad peaks. Deuterated chloroform and methylene chloride were used for NMR spectra recording. Melting points (Mp) were measured using open-ended capillaries on an Electrothermal melting point apparatus and are uncorrected. High-resolution mass spectrometry (HRMS) was performed by EPSRC National Mass Spectrometry Service Centre (NMSSC), Swansea. Gas chromatography-mass spectrometry (GC-MS) was recorded on Hewlett Packward HP6890 series GC system tandem with 5973 Mass Selective Detector.Ligand syntheses
Light tan solid. Yield: 63%. Rf: 0.41 (EtOAc–Hexanes = 1:3 on silica). Mp: 100–101 °C (Lit. 100–101 °C).401H NMR (300 MHz, CDCl3) δ (ppm): 10.09 (s, 1 H), 8.82 (dd, J = 2.4, 0.7 Hz, 1 H), 8.02 (dd, J = 8.2, 2.4 Hz, 1 H), 7.68 (dt, J = 8.2, 0.7 Hz, 1 H). 13C NMR (76 MHz, CDCl3) δ (ppm): 189.8, 152.9, 148.7, 137.9, 131.0, 129.4. GC-MS: 13.2 min, m/z: 185 (M), 184 (M − H), 156 (M − CHO).
General procedure for Suzuki–Miyaura cross-coupling
The appropriate 2-bromopyridine (1.0 equiv.) and arylboronic acid (1.1 equiv.), sodium carbonate (3.3 equiv.) and tetrakis(triphenylphosphine)palladium(0) (5 mol%) were mixed in dioxane–water (4:1 v/v, 30 mL per 1 g bromopyridine). The mixture was degassed by freeze–pump–thaw for three cycles. It was then allowed to heat under N2 at 90 °C for 8 h. The mixture was cooled to room temperature and extracted with DCM (3×). The combined organic phase was dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica, hexanes–EtOAc–triethylamine = 80:17:3) to offer the corresponding ligand.
:3 on silica). Mp: 83–84 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 10.02 (s, 1 H), 8.51–8.52 (m, 1 H), 8.10 (dt, J = 8.3, 1.6 Hz, 2 H), 7.92 (dt, J = 8.6, 1.7 Hz, 2 H), 7.65 (d, J = 7.6 Hz, 1 H), 7.55 (ddd, J = 8.1, 2.3, 0.7 Hz, 1 H), 2.35 (s, 3 H). 13C NMR (76 MHz, CDCl3) δ (ppm): 192.3, 153.5, 150.8, 145.3, 137.9, 136.5, 133.3, 130.5, 127.6, 121.0, 18.6. HR-MS(ASAP): [M + H]+ Calculated: (C13H11NO) 198.0913; Found 198.0913.
:3 on silica). Mp: 158–159 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 10.18 (s, 1 H), 10.12 (s, 1 H), 9.18 (dd, J = 2.2, 0.8 Hz, 1 H), 8.31–8.25 (m, 3 H), 8.05–8.02 (m, 2 H), 7.99 (dt, J = 8.2, 0.7 Hz, 1 H). 13C NMR (76 MHz, CDCl3) δ (ppm): 192.1, 190.6, 160.9, 152.7, 143.7, 137.7, 137.3, 130.9, 130.6, 128.6, 121.7. HR-MS (ASAP): [M + H]+ Calculated: (C13H9NO2) 212.0706; Found 212.0706.
:3 on silica).1H NMR (300 MHz, CDCl3) δ (ppm): 10.10 (s, 1 H), 8.54–8.53 (m, 1 H), 8.47 (td, J = 1.7, 0.6 Hz, 1 H), 8.26 (ddd, J = 7.8, 1.9, 1.2 Hz, 1 H), 7.90 (dt, J = 7.7, 1.4 Hz, 1 H), 7.69 (dd, J = 8.1, 0.9 Hz, 1 H), 7.64–7.57 (m, 2 H), 2.38 (s, 3 H). 13C NMR (76 MHz, CDCl3) δ (ppm): 192.8, 153.6, 150.7, 140.6, 138.0, 137.3, 132.9, 129.9, 129.8, 128.6, 120.5, 18.6. HR-MS (ASAP): [M + H]+ Calculated: (C13H11NO) 198.0913; Found 198.0913.
:3 on silica). Mp: 123–124 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 10.17 (s, 1 H), 10.14 (s, 1 H), 9.17 (dd, J = 2.2, 0.9 Hz, 1 H), 8.60 (td, J = 1.9, 0.5, 1 H), 8.39 (ddd, J = 7.8, 2.0, 1.2 Hz, 1 H), 8.29 (dd, J = 8.3, 2.2 Hz, 1 H), 8.03–7.98 (m, 2 H), 7.70 (t, J = 8.4 Hz, 1 H). 13C NMR (76 MHz, CDCl3) δ (ppm): 192.3, 190.7, 160.9, 152.8, 139.3, 137.4, 137.3, 133.6, 131.6, 130.7, 130.2, 129.2, 121.1. HR-MS (ASAP): [M + H]+ Calculated: (C13H9NO2) 212.0706; Found 212.0706.
General procedure for the synthesis of chloro-bridged iridium dimer complexes [Ir(C^N)2Cl]2
Iridium trichloride trihydrate (1 equiv.) and the appropriate C^N ligand (2.2 equiv.) were mixed in 2-alkoxyethanol–water (v/v = 3:1, 8 mL for 100 mg IrCl3·H2O). 2-Ethoxyethanol was used for the mppy series while 2-methoxyethanol for the fppy series. The mixture was degassed via three freeze–pump–thaw cycles and was allowed to reflux for 18 h under N2. After cooling, the precipitate was filtered followed by washing with ethanol and acetone to afford the corresponding [Ir(C^N)2Cl]2 complex.
:1 on silica). Mp: not observed below 400 °C. Elemental Analysis (%); Cacld. (found): C 50.36 (50.51); H 3.25 (3.22); N 4.52 (4.58). 1H NMR (400 MHz, CD2Cl2) δ (ppm): 9.42 (s, 4 H), 9.07 (s, 4 H), 7.92 (d, J = 8.3 Hz, 4 H), 7.76 (dd, J = 8.4, 0.8 Hz, 4 H), 7.61 (d, J = 8.0 Hz, 4 H), 7.23 (dd, J = 8.0, 1.6 Hz, 4 H), 6.16 (d, J = 1.6 Hz, 4 H), 2.03 (s, 12 H). 13C NMR (100 MHz, CD2Cl2) δ (ppm): 192.2, 164.2, 151.9, 150.2, 143.7, 138.5, 135.7, 133.7, 131.6, 123.6, 122.9, 119.9, 18.5. HR-MS (ESI): M+ Calculated: (C52H40Cl2Ir2N4O4) 1240.2; Found 1240.1.
:1 on silica). Mp: 386 °C (decompose). Elemental Analysis (%); Cacld. (found): C 50.36 (50.26); H 3.25 (3.17); N 4.52 (4.57). 1H NMR (400 MHz, CD2Cl2) δ (ppm): 9.81 (s, 4 H), 9.10 (dd, J = 1.4, 0.8 Hz, 4 H), 8.08 (d, J = 8.2 Hz, 4 H), 8.05 (d, J = 1.8 Hz, 4 H), 7.87 (ddd, J = 8.3, 2.0, 0.5 Hz, 4 H), 7.08 (dd, J = 8.0, 1.8 Hz, 4 H), 6.07 (d, J = 8.0 Hz, 4 H), 2.12 (s, 12 H). 13C NMR (100 MHz, CD2Cl2) δ (ppm): 191.2, 164.3, 156.3, 151.5, 145.2, 138.8, 133.1, 131.4, 130.8, 130.1, 123.2, 119.0, 18.5. HR-MS (ESI): M+ Calculated: (C52H40Cl2Ir2N4O4) 1240.2; Found 1240.2.
:1 on silica). Mp: 374 °C (decompose). Elemental Analysis (%); Cacld. (found): C 48.19 (48.31); H 2.49 (2.29); N 4.32 (4.28). 1H NMR (400 MHz, CD2Cl2) δ (ppm): 9.74 (dd, J = 1.9, 0.8 Hz, 4 H), 9.67 (s, 4 H), 9.58 (s, 4 H), 8.37–8.31 (m, 8 H), 7.92 (d, J = 8.0 Hz, 4 H), 7.44 (dd, J = 8.1, 1.6 Hz, 4 H), 6.35 (d, J = 1.5 Hz, 4 H). 13C NMR (100 MHz, CD2Cl2) δ (ppm): 191.7, 187.6, 172.0, 153.6, 147.9, 146.3, 137.9, 137.0, 131.6, 131.2, 126.5, 123.6, 120.7. HR-MS (ESI): M+ Calculated: (C52H40Cl2Ir2N4O4) 1296.1; Found 1296.1.
:1 on silica). Mp: 337 °C (decompose). Elemental Analysis (%); Cacld. (found): C 48.19 (48.26); H 2.49 (2.60); N 4.32 (4.45). 1H NMR (300 MHz, CD2Cl2) δ (ppm): 9.84 (s, 4 H), 9.64 (s, 8 H), 8.34–8.33 (m, 8 H), 8.21 (d, J = 1.8 Hz, 4 H), 7.17 (dd, J = 8.1, 1.7 Hz, 4 H), 6.13 (d, J = 8.1 Hz, 4 H). 13C NMR (100 MHz, CD2Cl2) δ (ppm): 190.7, 187.5, 172.1, 157.5, 153.4, 143.3, 137.9, 132.1, 131.7, 131.4, 131.0, 126.4, 120.0. HR-MS (ESI): M+ Calculated: (C52H40Cl2Ir2N4O4) 1296.1; Found 1296.1.
X-Ray crystallography
Data for complexes 1a, 1b and 2a were collected at 173 K by using a Rigaku FR-X Ultrahigh brilliance Microfocus RA generator/confocal optics and Rigaku XtaLAB P200 system using Mo Kα radiation (λ = 0.71073 Å). Intensity data were collected using ω steps accumulating area detector images spanning at least a hemisphere of reciprocal space. Data for complex 2b was collected at 173 K by using a Rigaku MM-007HF High brilliance RA generator/confocal optics and Rigaku XtaLAB P100 system, with Cu Kα radiation (λ = 1.54187 Å). Intensity data were collected using ω and φ steps accumulating area detector images spanning at least a hemisphere of reciprocal space. Data for all complexes were corrected for Lorentz polarization effects. A multiscan absorption correction was applied by using CrystalClear.41 Structures were solved by Patterson methods (PATTY)42 and refined by full-matrix least-squares against F2 (SHELXL-2013).43 Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were refined using a riding model. Complex 2a showed discrete disorder in one of the formyl groups, which was modelled over two sites. Both complexes 2a and 2b showed signs of unresolvable disorder in various of the formyl groups. Restraints to some bond distances, angles and some thermal parameters were required. All calculations were performed using the CrystalStructure interface.44 Crystallographic data for the four complexes are listed in Table 5. CCDC 1026256–1026259.| 1a | 1b | 2a | 2b | |
|---|---|---|---|---|
| Formula | C53H42Cl4Ir2N4O4 | C52H40Cl2Ir2N4O4 | C54H36Cl6Ir2N4O8 | C52H32Cl2Ir2N4O8 |
| M (g mol−1) | 1410.12 | 1240.26 | 1466.06 | 1296.19 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/n | P21/c |
| a [Å] | 11.065(2) | 11.0995(19) | 22.9927(11) | 11.800(3) |
| b [Å] | 37.753(6) | 20.038(3) | 11.5838(5) | 24.431(6) |
| c [Å] | 13.197(3) | 29.679(5) | 38.7370(18) | 19.925(5) |
| β [°] | 114.701(6) | 99.998(4) | 97.2195(13) | 97.874(6) |
| V [Å3] | 5008.5(17) | 6500.7(18) | 10235.5(8) |
5690(2) |
| Z | 4 | 4 | 8 | 4 |
| ρ calcd (g cm−3) | 1.870 | 1.267 | 1.903 | 1.513 |
| μ (mm−1) | 5.695 | 4.219 | 5.583 | 9.999 |
| Measured refln. | 51348 |
97749 |
152461 |
56622 |
| Unique refln. (Rint) | 9097 (0.0756) | 11956 (0.1210) |
18712 (0.0527) |
10268 (0.1622) |
| R 1 [I > 2σ(I)] | 0.0487 | 0.1243 | 0.0367 | 0.1313 |
| wR2 (all) | 0.1378 | 0.3092 | 0.1034 | 0.3816 |
Photophysical measurements
Optically dilute solutions of concentrations on the order of 10−5 or 10−6 M were prepared in HPLC grade DCM for absorption and emission analysis, respectively. Absorption spectra were record at room temperature on a Shimadzu UV-1800 double beam spectrophotometer in a sealed quartz cuvette from Starna. Molar absorptivity values were determined from at least four solutions with concentrations varying from 3.39 × 10−6 M to 4.17 × 10−5 M followed by linear regression analysis with all results having coefficient of determination (r-squared values) being at least 0.998. Solutions were degassed via three freeze–pump–thaw cycles prior to emission analysis in a home-built quartz cuvette. Steady state emission, excitation spectra and time-resolved emission spectra were recorded at 298 K using an Edinburgh Instruments F980. Samples were excited at 455 nm for steady state measurements while at 470 nm for time-resolved measurements. Photoluminescence quantum yields were determined using the optically dilute method45 in which four sample solutions with absorbance values of 0.10, 0.080, 0.060 and 0.040 at 455 nm were used. For each sample, linearity between absorption and emission intensity was verified through linear regression analysis and additional measurements were acquired until the Pearson regression factor (R2) for the linear fit of the data set surpassed 0.9. Individual relative quantum yield values were calculated for each solution and the values reported represent the slope value. The equation Φs = Φr(Ar/As)(Is/Ir)(ns/nr)2 was used to calculate the relative quantum yield of each of the sample, where Φr is the absolute quantum yield of the reference, n is the refractive index of the solvent, A is the absorbance at the excitation wavelength, and I is the integrated area under the corrected emission curve. The subscripts s and r refer to the sample and reference, respectively. A solution of [Ru(bpy)3](PF6)2 in ACN (Φr = 0.095) was used as the external reference.26Electrochemistry measurements
Cyclic Voltammetry (CV) analysis was performed on an Electrochemical Analyzer potentiostat model CH600D from CH Instruments. Samples were prepared as DCM solutions, which were degassed by sparging with DCM-saturated nitrogen gas for 15 min prior to measurements. All measurements were performed in 0.1 M DCM solution of nBu4NPF6, which acted as the supporting electrolyte. An Ag/Ag+ electrode was used as the reference electrode while a platinum electrode and a platinum wire were used as the working electrode and counter electrode, respectively. The redox potentials are reported relative to a standard calomel electrode (SCE) with a ferrocenium/ferrocene (Fc+/Fc) redox couple as an internal reference (0.46 V vs. SCE).18Density functional theory (DFT) calculations
All calculations were performed with the Gaussian 0927 suite. The level of theory for all DFT28c,46 and TD-DFT28d–f calculations was B3LYP; excited-state triplet geometries were calculated using the unrestricted B3LYP method (UB3LYP).29b,c,47 The 6-31G* basis set48 was used for C, H and N directly linked to Iridium while the other C, H, N and F atoms where undertaken with 3-21G* basis set,30a,31a–e and the VDZ (valence double ζ) with SBKJC effective core potential basis set30 was used for Iridium. The predicted phosphorescence wavelengths were obtained by energy difference between the triplet and singlet states at their respective optimized geometries.49 The energy, oscillator strength and related MO contributions for the 100 lowest singlet–singlet and 5 lowest singlet–triplet excitations were obtained from the TD-DFT/singlets and the TD-DFT/triplets output files, respectively. The calculated absorption spectra were visualized with GaussSum 2.1 (fwhm: 1000 cm−1).50OLED fabrication and characterization
The PEDOT:PSS layer was spin-coated on pre-patterned ITO glass substrate after ultrasonic cleaning in acetone and 2-propanol consecutively and finally oxygen plasma treatment. PVK, serving as a hole transporting layer, was spin-coated directly onto PEDOT:PSS after baking at 120 °C for 10 minutes in the glove-box to remove the residual moisture. After another baking of PVK at 120 °C for 10 minutes, the emitting layer, comprising of the mixture of the iridium dimer complexes (dissolved in DCM) and hosts (dissolved in chlorobenzene), was spin-coated on PVK. An additional electron transporting layer, B3PYMPM, was thermally evaporated onto the active layer. Finally, Ca (20 nm)/Al (100 nm) as the composite cathode was thermally deposited through a shadow mask in the vacuum chamber under a pressure of 2.0 × 10−6 mbar. All the devices were encapsulated with UV epoxy resin in the glove-box. The luminance–current–voltage measurement was conducted in ambient environment by using Keithley 2400 source meter and 2000 multi-meter connected to a calibrated Si photodiode. The external quantum efficiency was calculated with the assumption of a Lambertian distribution. The electroluminescence spectrum was captured by an Andor DV420-BV CCD spectrometer. The layer thickness was measurement using a Veeco DekTak 150 surface profiler.Notes and references
- (a) H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) M. C. Gather, A. Köhnen and K. Meerholz, Adv. Mater., 2011, 23, 233 CrossRef CAS PubMed; (c) WOLEDs and Organic Photovoltaics: Recent Advances and Applications, ed. V. W. W. Yam, 2010 Search PubMed; (d) C. Fan and C. Yang, Chem. Soc. Rev., 2014, 43, 6439 RSC; (e) G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467 RSC.
- (a) E. A. Plummer, A. van Dijken, J. W. Hofstraat, L. De Cola and K. Brunner, Adv. Funct. Mater., 2005, 15, 281 CrossRef CAS; (b) C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS PubMed; (c) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed.
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS PubMed; (b) H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS PubMed.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC.
- (a) S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 106, 6647 CrossRef CAS; (b) G. A. Carlson, P. I. Djurovich and R. J. Watts, Inorg. Chem., 1993, 32, 4483 CrossRef CAS; (c) F. O. Garces, K. A. King and R. J. Watts, Inorg. Chem., 1988, 27, 3464 CrossRef CAS.
- (a) S. Bettington, M. Tavasli, M. R. Bryce, A. S. Batsanov, A. L. Thompson, H. A. A. Attar, F. B. Dias and A. P. Monkman, J. Mater. Chem., 2006, 16, 1046 RSC; (b) A. M'Hamedi, A. S. Batsanov, M. A. Fox, M. R. Bryce, K. Abdullah, H. A. Al-Attar and A. P. Monkman, J. Mater. Chem., 2012, 22, 13529 RSC.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704 CrossRef CAS PubMed.
- (a) T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290 CrossRef CAS PubMed; (b) T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813 CrossRef CAS PubMed; (c) A. Endo, K. Suzuki, T. Yoshihara, S. Tobita, M. Yahiro and C. Adachi, Chem. Phys. Lett., 2008, 460, 155 CrossRef CAS PubMed; (d) G. St-Pierre, S. Ladouceur, D. Fortin and E. Zysman-Colman, Dalton Trans., 2011, 40, 11726 RSC.
- (a) E. Holder, V. Marin, M. A. R. Meier and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1491 CrossRef CAS; (b) L. R. Alrawashdeh, A. I. Day and L. Wallace, Dalton Trans., 2013, 42, 16478 RSC; (c) Y. Ma, S. Liu, H. Yang, Y. Wu, C. Yang, X. Liu, Q. Zhao, H. Wu, J. Liang, F. Li and W. Huang, J. Mater. Chem., 2011, 21, 18974 RSC; (d) X. Cao, Y. Wu, K. Liu, X. Yu, B. Wu, H. Wu, Z. Gong and T. Yi, J. Mater. Chem., 2012, 22, 2650 RSC; (e) M. Zaarour, V. Guerchais, H. Le Bozec, C. Dragonetti, S. Righetto, D. Roberto, F. De Angelis, S. Fantacci and M. G. Lobello, Dalton Trans., 2013, 42, 155 RSC.
- (a) K. K.-W. Lo, C.-K. Chung and N. Zhu, Chem. – Eur. J., 2003, 9, 475 CrossRef CAS PubMed; (b) K. K.-W. Lo, K. Y. Zhang, S.-K. Leung and M.-C. Tang, Angew. Chem., Int. Ed., 2008, 47, 2213 CrossRef CAS PubMed; (c) S. K. Leung, K. Y. Kwok, K. Y. Zhang and K. K. Lo, Inorg. Chem., 2010, 49, 4984 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- X. Wang, P. Rabbat, P. O'Shea, R. Tillyer, E. J. J. Grabowski and P. J. Reider, Tetrahedron Lett., 2000, 41, 4335 CrossRef CAS.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767 CrossRef CAS.
- E. Holder, V. Marin, A. Alexeev and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2765 CrossRef CAS.
- D. L. Davies, M. P. Lowe, K. S. Ryder, K. Singh and S. Singh, Dalton Trans., 2011, 40, 1028 RSC.
- S. Bettington, A. L. Thompson, A. Beeby and A. E. Goeta, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m827 CAS.
- (a) L. Norel, M. Rudolph, N. Vanthuyne, J. A. G. Williams, C. Lescop, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Angew. Chem., Int. Ed., 2010, 49, 99 CrossRef CAS PubMed; (b) F. O. Garces, K. Dedeian, N. L. Keder and R. J. Watts, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 1117 CrossRef; (c) L.-Q. Chen, C.-L. Yang and J.-G. Qin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m513 Search PubMed; (d) E. Mesto, F. Scordari, M. Lacalamita, L. De Cola, R. Ragni and G. M. Farinola, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 480 CAS; (e) L. Chen, C. Yang, M. Li, J. Qin, J. Gao, H. You and D. Ma, Cryst. Growth Des., 2006, 7, 39 CrossRef; (f) E. Baranoff, H. J. Bolink, E. C. Constable, M. Delgado, D. Haussinger, C. E. Housecroft, M. K. Nazeeruddin, M. Neuburger, E. Orti, G. E. Schneider, D. Tordera, R. M. Walliser and J. A. Zampese, Dalton Trans., 2013, 42, 1073 RSC; (g) E. C. Constable, C. D. Ertl, C. E. Housecroft and J. A. Zampese, Dalton Trans., 2014, 43, 5343 RSC; (h) C. Xu, H. M. Li, Z. Q. Xiao, Z. Q. Wang, S. F. Tang, B. M. Ji, X. Q. Hao and M. P. Song, Dalton Trans., 2014, 43, 10235 RSC; (i) Z.-Q. Wang, C. Xu, X.-M. Dong, Y.-P. Zhang, X.-Q. Hao, J.-F. Gong, M.-P. Song and B.-M. Ji, Inorg. Chem. Commun., 2011, 14, 316 CrossRef CAS PubMed; (j) K. A. McGee and K. R. Mann, Inorg. Chem., 2007, 46, 7800 CrossRef CAS PubMed; (k) C. Xu, Z.-Q. Wang, X.-M. Dong, X.-Q. Hao, X.-M. Zhao, B.-M. Ji and M.-P. Song, Inorg. Chim. Acta, 2011, 373, 306 CrossRef CAS PubMed; (l) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed; (m) V. Krisyuk, A. Turgambaeva, J. Lee and S.-W. Rhee, Transition Met. Chem., 2005, 30, 786 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17 RSC.
- E. S. Andreiadis, D. Imbert, J. Pécaut, A. Calborean, I. Ciofini, C. Adamo, R. Demadrille and M. Mazzanti, Inorg. Chem., 2011, 50, 8197 CrossRef CAS PubMed.
- B. Beyer, C. Ulbricht, D. Escudero, C. Friebe, A. Winter, L. González and U. S. Schubert, Organometallics, 2009, 28, 5478 CrossRef CAS.
- C. E. Welby, L. Gilmartin, R. R. Marriott, A. Zahid, C. R. Rice, E. A. Gibson and P. I. Elliott, Dalton Trans., 2013, 42, 13527 RSC.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS PubMed.
- S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514 CrossRef CAS PubMed.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, 7.0 ed., Wallingford, CT, 2009.
- (a) P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef; (b) W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef; (c) In The Challenge of d and f Electrons, ed. D. R. Salahub and M. C. Zerner, ACS, Washington, DC, 1989 Search PubMed; (d) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS PubMed; (e) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS; (f) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- (a) J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939 CrossRef CAS; (b) W. J. Stevens, W. J. Basch and M. Krauss, J. Chem. Phys., 1984, 81, 6026 CrossRef PubMed; (c) W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Can. J. Chem., 1992, 70, 612 CrossRef CAS; (d) T. R. Cundari and W. J. Stevens, J. Chem. Phys., 1993, 98, 5555 CrossRef CAS PubMed.
- (a) M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 2797 CrossRef CAS; (b) W. J. Pietro, M. M. Francl, W. J. Hehre, D. J. Defrees, J. A. Pople and J. S. Binkley, J. Am. Chem. Soc., 1982, 104, 5039 CrossRef CAS; (c) K. D. Dobbs and W. J. Hehre, J. Comput. Chem., 1986, 7, 359 CrossRef CAS; (d) K. D. Dobbs and W. J. Hehre, J. Comput. Chem., 1987, 8, 861 CrossRef CAS; (e) K. D. Dobbs and W. J. Hehre, J. Comput. Chem., 1987, 8, 880 CrossRef CAS; (f) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS PubMed; (g) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (h) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS; (i) P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209 CrossRef CAS; (j) M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- (a) L. Donato, C. E. McCusker, F. N. Castellano and E. Zysman-Colman, Inorg. Chem., 2013, 52, 8495 CrossRef CAS PubMed; (b) A. M. Soliman, D. Fortin, P. D. Harvey and E. Zysman-Colman, Dalton Trans., 2012, 41, 9382 RSC.
- X. H. Yang, F. Jaiser, B. Stiller, D. Neher, F. Galbrecht and U. Scherf, Adv. Funct. Mater., 2006, 16, 2156 CrossRef CAS.
- J. Kalinowski, M. Cocchi, D. Virgili, V. Fattori and J. A. G. Williams, Adv. Mater., 2007, 19, 4000 CrossRef CAS.
- S. Lee, K.-H. Kim, D. Limbach, Y.-S. Park and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 4105 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS PubMed.
- D. R. Coulson, L. C. Satek and S. O. Grim, in Inorg. Synth, ed. F. A. Cotton, 2007, p. 121 Search PubMed.
- A. Bouillon, J.-C. Lancelot, V. Collot, P. R. Bovy and S. Rault, Tetrahedron, 2002, 58, 2885 CrossRef CAS.
- P. H. Bernardo, T. Sivaraman, K. F. Wan, J. Xu, J. Krishnamoorthy, C. M. Song, L. Tian, J. S. Chin, D. S. Lim, H. Y. Mok, V. C. Yu, J. C. Tong and C. L. Chai, J. Med. Chem., 2010, 53, 2314 CrossRef CAS PubMed.
- CrystalClear-SM Expert v. 2.1, The Woodlands, Texas, USA, 2010–2014 Search PubMed.
- P. T. Beurskens, G. Beurskens, R. de Gelder, S. Garcia-Granda, R. O. Gould, R. Israel and J. M. M. Smits, DIRDIF-99, University of Nijmegen, The Netherlands, 1999 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- CrystalStructure v4.1, The Woodlands, Texas, USA, 2014 Search PubMed.
- (a) G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991 CrossRef CAS; (b) S. Fery-Forgues and D. Lavabre, J. Chem. Educ., 1999, 76, 1260 CrossRef CAS.
- (a) P. Hohenberg and W. Kohn, Phys. Rev., 1964, B136, 864 CrossRef; (b) W. Kohn and L. J. Sham, Phys. Rev., 1965, A140, 1133 CrossRef; (c) R. G. Parr and W. Yang, Density-functional theory of atoms and molecules, Oxford Univ. Press, Oxford, 1989 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223 CrossRef CAS PubMed.
- (a) S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2010, 49, 5625 CrossRef CAS PubMed; (b) M. S. Lowry, W. R. Hudson, R. A. Pascal Jr. and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129 CrossRef CAS PubMed.
- N. M. O'Boyle, GaussSum 2.0, Dublin City University, Dubin Ireland, 2006, Available at http://gausssum.sf.net Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1026256–1026259. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03127j |
| ‡ Current address: Laboratoire PPSM - CNRS UMR 8531, ENS Cachan, 61 Av du Président Wilson, 94235 Cachan Cedex France. |
| § Current address: CEMCA UMR CNRS 6521, Université de Bretagne Occidentale, 6 av. Victor Le Gorgeu, Brest, France, 29200. |
| This journal is © The Royal Society of Chemistry 2015 |