
Epoxidation of cyclic enones with hydrogen peroxide catalysed by alkylcarboxylate-intercalated Ni–Zn mixed basic salts†
Takayoshi
Hara
,
Jun
Kurihara
,
Nobuyuki
Ichikuni
and
Shogo
Shimazu
*
Department of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi, Inage, Chiba 263-8522, Japan. E-mail: shimazu@faculty.chiba-u.jp; Fax: +81 43 290 3379; Tel: +81 43 290 3379
First published on 19th September 2014
Abstract
The interlayer spacing of layered Ni–Zn mixed basic salts (NiZn) can be precisely controlled by the intercalation of various long alkyl chain carboxylate anions into the NiZn interlayer. The butyrate-exchanged NiZn (C3H7COO−/NiZn) catalyst effectively promotes the epoxidation of various cyclic enones with aqueous hydrogen peroxide in remarkably high yields. The C3H7COO−/NiZn-catalysed epoxidation of 2-cyclohexen-1-one with an equimolar amount of H2O2 proceeds in a highly efficient manner, with 97% efficiency of H2O2 utilization. This C3H7COO−/NiZn catalyst can be reused without any loss of its catalytic activity and selectivity.
Introduction
Anion-exchange within layered inorganic materials has received much attention as a means to create various types of useful inorganic–organic nanocomposites.1 Layered double hydroxides (LDHs),2 with the formula [M2+1−xM3+x(OH)2]x+[An−x/n]x−·mH2O, where (0.51 < x < 1.00), are a representative group of this class of solids. Because of their low toxicity, LDHs have been used as drug delivery matrices by intercalating active pharmaceutical ingredients into the interlayer of the inorganic materials.3 Among the many types of LDHs, rhombohedral Mg–Al hydrotalcites (M2+: Mg2+, M3+: Al3+, and An−: CO32−) have been widely used as heterogeneous catalysts or catalyst supports.4 Because of their great potential for use in the design of high-performance heterogeneous catalysts, various strategies for the preparation of such materials have been widely reported in recent years. The acid–base properties of hydrotalcites can be controlled by changing the identity of the metal species and/or the M2+/M3+ ratio within the structure.5 Furthermore, catalytically active species, such as metal salts, organometallic complexes, or nanoparticles, can be supported on their cationic layer sheets, surfaces, or interlayer spaces.6 Recently, it has been found that environmentally benign organic transformation reactions can be achieved by using the combined catalytic systems composed of both a solid acid catalyst and a solid basic hydrotalcite.7Layered hydroxy double salts (HDSs), which consist of positively charged layers and exchangeable interlayer anions, have also received considerable interest as anion-exchangeable layered compounds.8 Compounds in the HDS family follow the general formula MA2+1−xMB2+2xAn−2x/n(OH)2·mH2O (0.15 < x < 0.25), where MA2+ and MB2+ are divalent metal cations such as Zn2+, Cu2+, Co2+, or Ni2+, and An− represents various interlayer anions. The anionic guests that can be used range from simple inorganic anions (NO3−, ClO4−, Cl−, MnO4−, SO42−, etc.) to anionic metal complexes to organic anions including drugs and dyes.9 It can be considered that HDSs are also powerful candidates for advanced nanoscaled catalysts or catalyst supports that allow for control over the location of the catalytically active metal species.
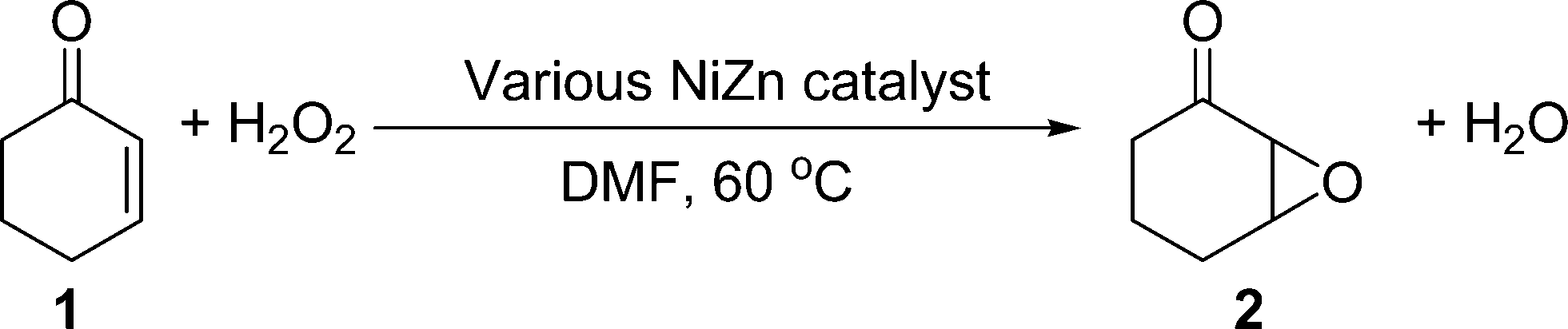
The epoxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds is one of the platform synthetic processes in basic research and industrial applications, because epoxides and their intermediates are widely employed in the synthesis of high-valued chemicals, such as cosmetics, pharmaceuticals, and polymer materials.10 The epoxidation of electron-deficient olefins in particular, such as α,β-unsaturated ketones, has been an important target for the functionalisation of ketones. It is well known that this type of epoxidation generally requires a nucleophilic oxidant, such as OOH− species.11 Many epoxidation procedures employ H2O2/Bu4NF, TBHP/DBU, mCPBA/KOH, NaOCl/cinchona alkaloid, NaBO3/THAHS, iodosyl benzene, dioxirane, or TBA2S2O8 as an oxidant,12 however, these oxidants often yield stoichiometric amounts of their deoxygenated forms as a waste by-product. In contrast, hydrogen peroxide is an ideal oxidant because water is the only by-product. Although organometallic complexes, heteropolyacids, organic molecules, and their immobilized materials can act as effective catalysts for the epoxidation of enones under neutral conditions,13 utilization of homogeneous reaction systems with H2O2 under alkaline conditions (NaOH, KOH, LiOH, Na2CO3, or K2CO3) is the most common procedure. To replace these standard homogeneous systems with more practical heterogeneous alternatives, a number of solid bases such as Mg–Al hydrotalcite, KF/alumina, hydroxyapatite, or sodalite catalysts have been developed.14
C double bonds is one of the platform synthetic processes in basic research and industrial applications, because epoxides and their intermediates are widely employed in the synthesis of high-valued chemicals, such as cosmetics, pharmaceuticals, and polymer materials.10 The epoxidation of electron-deficient olefins in particular, such as α,β-unsaturated ketones, has been an important target for the functionalisation of ketones. It is well known that this type of epoxidation generally requires a nucleophilic oxidant, such as OOH− species.11 Many epoxidation procedures employ H2O2/Bu4NF, TBHP/DBU, mCPBA/KOH, NaOCl/cinchona alkaloid, NaBO3/THAHS, iodosyl benzene, dioxirane, or TBA2S2O8 as an oxidant,12 however, these oxidants often yield stoichiometric amounts of their deoxygenated forms as a waste by-product. In contrast, hydrogen peroxide is an ideal oxidant because water is the only by-product. Although organometallic complexes, heteropolyacids, organic molecules, and their immobilized materials can act as effective catalysts for the epoxidation of enones under neutral conditions,13 utilization of homogeneous reaction systems with H2O2 under alkaline conditions (NaOH, KOH, LiOH, Na2CO3, or K2CO3) is the most common procedure. To replace these standard homogeneous systems with more practical heterogeneous alternatives, a number of solid bases such as Mg–Al hydrotalcite, KF/alumina, hydroxyapatite, or sodalite catalysts have been developed.14
We have previously developed a Ni–Zn mixed basic salt (NiZn) as a solid catalyst or catalyst support based on the design idea of “Intercalation Catalysts”.15 Ni1−xZn2x(OCOCH3)2x(OH)2·nH2O (0.15 < x < 0.25; NiZn), classified as a layered HDS, consists of positively charged layers and exchangeable interlayer anions. Our selection of NiZn as a catalyst material is motivated by its unique characteristics such as (i) simple preparation, (ii) high crystallinity, (iii) strong electrostatic interactions between guest anions and Zn2+ cations, and (iv) high anion exchange capacity, as well as (v) the ability to fine-tune the interlayer space by modifying the size of the guest anion. Recently, interlayer space-controlled NiZn with alkyl carboxylate anions intercalated into the interlayer has been found to be an effective heterogeneous Brønsted base catalyst for Knoevenagel condensation in water.15c Here, we report the successful epoxidation of cyclic enones using hydrogen peroxide in the presence of NiZn as a basic catalyst.
Results and discussion
In accordance with the previously reported literature,16 the acetate-intercalated Ni–Zn mixed basic salt, CH3COO−/NiZn, Ni0.63Zn0.37(OAc)0.37(OH)2·1.93H2O (Ni/Zn = 2.20), was synthesized.17 Anion exchange in the NiZn interlayer was performed using the same procedure in water. Treatment of CH3COO−/NiZn with various sodium alkyl carboxylates yielded anion-exchanged NiZn catalysts (Cn−1H2n−1COO−/NiZn, n = 1–10) as green powders.The powder X-ray diffraction (XRD) profiles of the synthesised NiZn catalysts are shown in Fig. 1. From the XRD results, the layered structure of NiZn was maintained in each case; however, the calculated clearance spaces of each material (C.S. = basal spacing (d001) – thickness of layer (0.46 nm)) were changed after the intercalation procedure, also shown in Fig. 1. The C.S. of each was proportional to the length of the alkyl groups intercalated (slope: 0.17 nm per CH2, Fig. 2). Taking the C.S. and the size of the pillared alkyl carboxylate anions into account, a suitable conformation of Cn−1H2n−1COO−/NiZn can be a bilayer arrangement with a tilting angle of approximately 40 degrees.
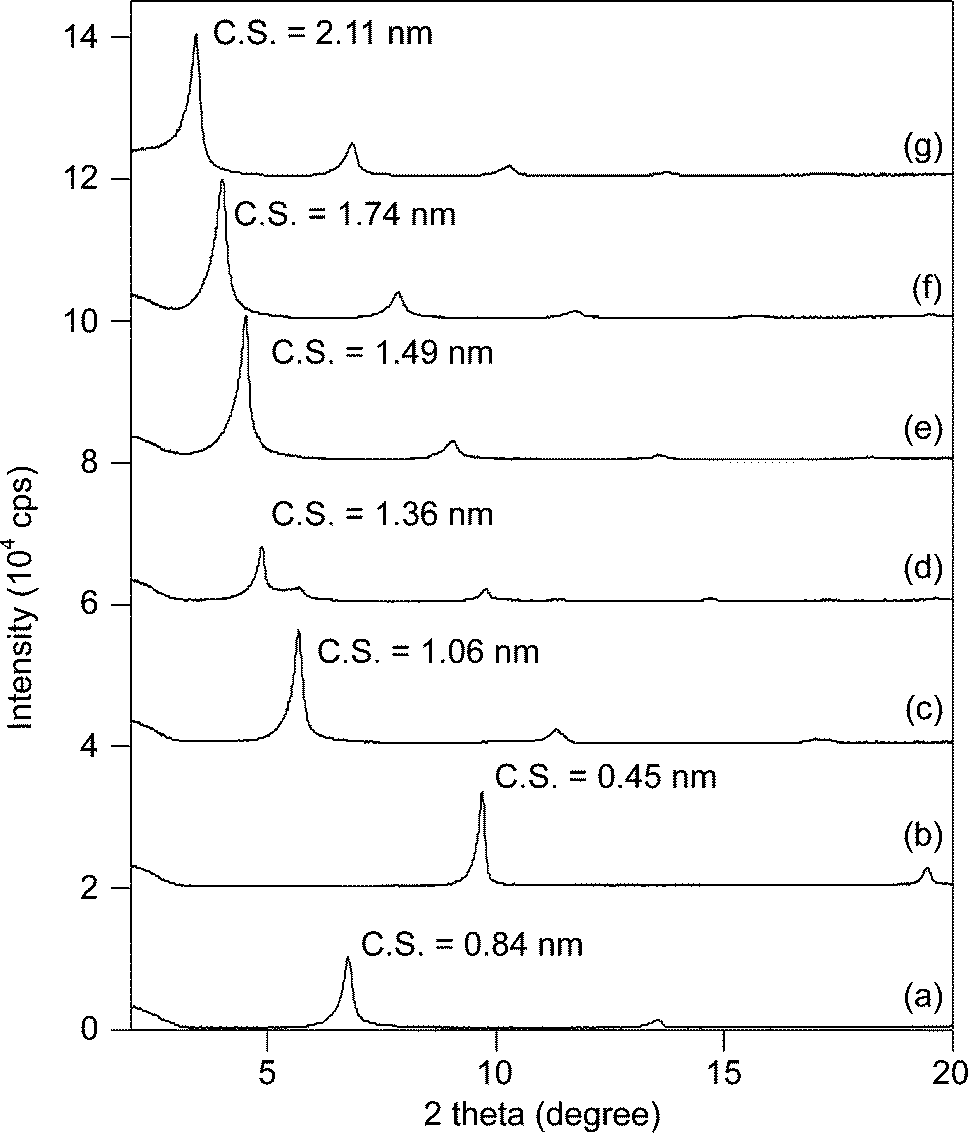 | ||
| Fig. 1 XRD profiles of (a) CH3COO−/NiZn, (b) HCOO−/NiZn, (c) C3H7COO−/NiZn, (d) recovered C3H7COO−/NiZn after epoxidation of 2-cyclohexen-1-one (1), (e) C5H11COO−/NiZn, (f) C7H15COO−/NiZn, and (g) C9H19COO−/NiZn. The C.S. values were also given. | ||
 | ||
| Fig. 2 The relation between C.S. and carbon number of alkyl groups in the carboxylate anion of Cn−1H2n−1COO−/NiZn catalysts. | ||
To explore the catalytic abilities of the anion-exchanged NiZn, the epoxidation of 2-cyclohexen-1-one (1) into 1,2-epoxycyclohexanone (2) was carried out in the presence of 4 equivalents of aqueous hydrogen peroxide (30 wt%) relative to the amount of 1. The results and the C.S. values of the Cn−1H2n−1COO−/NiZn catalysts are shown in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | C.S.b (nm) | Time (h) | Conv.c (%) | Yieldc (%) |
| a 1 (0.5 mmol), NiZn catalyst (0.05 g), DMF (2 mL), 30 wt% H2O2 aq. (4 eq. relative to the substrate), 60 °C. b Calculated from XRD data. c Determined by GC using an internal standard technique. d Ni catalyst (Ni: 0.26 mmol). e Zn catalyst (Zn: 0.13 mmol). f Hydrotalcite (0.05 g), purchased from WAKO, was used as a catalyst. g C3H7COONa (1.47 mmol) was used as a catalyst. | |||||
| 1 | HCOO−/NiZn | 0.45 | 1 | 7 | 6 |
| 2 | CH3COO−/NiZn | 0.84 | 1 | 45 | 45 |
| 3 | C3H7COO−/NiZn | 1.06 | 1 | 55 | 55 |
| 4 | C5H11COO−/NiZn | 1.49 | 1 | 28 | 28 |
| 5 | C7H15COO−/NiZn | 1.74 | 1 | 24 | 22 |
| 6 | C9H19COO−/NiZn | 2.11 | 1 | 19 | 16 |
| 7d | Ni(OCOCH3)2·4H2O | — | 3 | trace | trace |
| 8e | Zn(OCOCH3)2·2H2O | — | 3 | trace | trace |
| 9d | Ni(OH)2 | — | 3 | trace | trace |
| 10e | Zn(OH)2 | — | 3 | trace | trace |
| 11f | Mg6Al2(CO3)(OH)16·4H2O | 0.30 | 3 | 18 | 18 |
| 12g | C3H7COONa | — | 3 | 35 | 35 |
| 13 | Blank | — | 3 | no reaction | |
In the case of HCOO−/NiZn, the catalytic activity was low (entry 1), most likely due to the mass transfer limitation within the narrow interlayer space. The parent CH3COO−/NiZn catalysed this epoxidation efficiently, yielding 45% of 2 after 1 h (entry 2). It should be noted that the C3H7COO−/NiZn catalyst exhibited the highest activity, yielding only 2 in 55% yield after 1 h (entry 3).18 On the other hand, the catalysts which have more enlarged interlayer spaces such as C5H11COO−/NiZn, C7H15COO−/NiZn, and C9H19COO−/NiZn did not promote this epoxidation reaction effectively (entries 4–6). It is likely that the substrate could not diffuse smoothly into the interlayer space of these catalysts because these spaces are occupied by the alkyl chains of the guest anions. Ni(OCOCH3)2·4H2O, Zn(OCOCH3)2·2H2O, Ni(OH)2, and Zn(OH)2, the precursors of NiZn and some of the components of the brucite-like layer of NiZn, did not show any significant catalytic activity under the test reaction conditions (entries 7–10). It is well known that Mg–Al hydrotalcite can heterogeneously catalyse the epoxidation of electron-deficient olefins with H2O2.14a–c However, the reaction rate over the Mg6Al2(CO3)(OH)16·4H2O catalyst was slow under the same reaction conditions (entry 11). When C3H7COONa (1.47 mmol) was used as a catalyst under the same reaction conditions, only 35% yield of 2 was obtained after 3 h (entry 12). The epoxidation did not proceed at all without the catalyst (entry 13).
The screening of solvents to determine their effect on the epoxidation of 1 over the C3H7COO−/NiZn catalyst at 50 °C showed that N,N-dimethylformamide (DMF) was superior to the other solvents tested, giving only 2 as a product in 83% yield within 3 h (Table 2, entry 1), whereas N,N-dimethylacetamide (DMA), acetonitrile, methanol, n-hexane, 1,2-dichloroethane, toluene, and water were not effective (entries 2, 4–5, and 7–10). When a combined oxidant of benzonitrile, acetonitrile, or acetamide and hydrogen peroxide was used in the presence of the C3H7COO−/NiZn catalyst, the enone epoxidation did not proceed effectively (entries 3, 4 and 6). The fact that no benzamide or acetamide was formed in nitrile solvents indicates that the active peroxy intermediates from nitrile or amide, which were used as oxidants, were not involved in this catalytic system.19
|
|
|||
|---|---|---|---|
| Entry | Solvent | Conv.b (%) | Yieldb (%) |
| a 1 (0.5 mmol), C3H7COO−/NiZn catalyst (0.05 g), solvent (4 mL), 30 wt% H2O2 aq. (2 eq. relative to the substrate), air, 50 °C, 3 h. b Determined by GC using an internal standard technique. c With acetamide (2 mmol). | |||
| 1 | N,N-Dimethylformamide | 83 | 83 |
| 2 | N,N-Dimethylacetamide | 39 | 39 |
| 3 | Benzonitrile | 7 | 5 |
| 4 | Acetonitrile | 57 | 57 |
| 5 | Methanol | 38 | 38 |
| 6c | Methanol | 42 | 38 |
| 7 | n-Hexane | 39 | 34 |
| 8 | 1,2-Dichloroethane | 3 | 1 |
| 9 | Toluene | 9 | 8 |
| 10 | Water | 68 | 68 |

In order to further demonstrate the requirement of the heterogeneous C3H7COO−/NiZn catalyst, the catalyst was removed by filtration when epoxidation of 1 had reached almost 70% conversion (Fig. 3). After removal of the catalyst, the reaction was monitored for an additional hour, and no additional formation of 2 was observed. These results demonstrate that oxidation proceeds in the NiZn interlayer and that any dissolved anionic species are not involved in this reaction. After the epoxidation was completed, it was possible to easily separate the C3H7COO−/NiZn catalyst from the reaction mixture by simple centrifugation (Fig. 4). From the XRD profile of the recovered C3H7COO−/NiZn catalyst, it was found that the crystallinity of NiZn had decreased and its C.S. expanded from 1.06 nm to 1.36 nm (Fig. 1d) due to the intercalation of water molecules and/or DMF solvent during the reaction.20
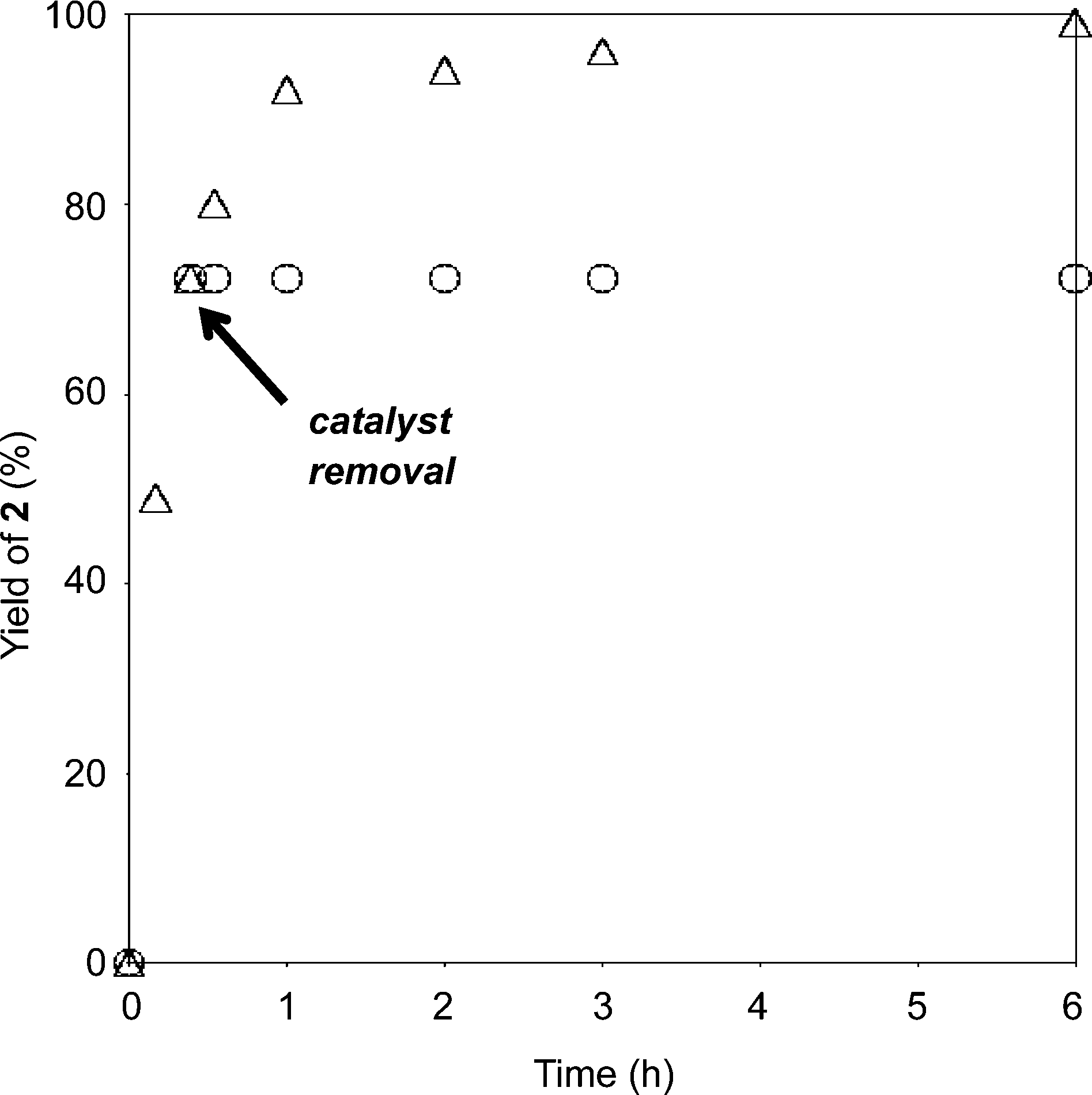 | ||
| Fig. 3 Hot filtration experiment. Reaction conditions were as follows: 1 (0.5 mmol), C3H7COO−/NiZn catalyst (0.1 g), DMF (2 mL), 30 wt% H2O2 aq. (4 eq. relative to the substrate), 60 °C. | ||
 | ||
| Fig. 4 The photograph of the C3H7COO−/NiZn catalyst (a) in the reaction mixture during catalytic epoxidation of 1 and (b) after phase separation by centrifugation. | ||
The epoxidation of various cyclic enones using the C3H7COO−/NiZn catalyst was carried out in DMF at 60 °C (Table 3). 2-Cyclopenten-1-one and 1 were successfully oxidised into their corresponding epoxyketones within 3 and 6 h, respectively (entries 1 and 3). The products expected from an aldol-type condensation were scarcely observed under the reaction conditions when performed over the C3H7COO−/NiZn catalyst. The recovered C3H7COO−/NiZn catalyst was found to be reusable without any loss of its activity and selectivity at least four times (Table 1, entry 3 and Table 3, entries 4–7). After the 4th recycling experiment (Table 3, entry 7), any nickel and zinc species and butylate anion leached into the reaction mixture were in amounts small enough to be undetectable by atomic absorption spectroscopy and ion chromatography. The reactivity is affected by various factors such as the orbital energy in LUMO of substrates, contact probability, the formation rate of HOO−, inhibition of side reaction, and so on. Especially, the reactivity of enones can be explained by using the orbital energies in LUMO of the substrates.21 Because the nucleophilic attack of HOO− to the substrate is known as the rate-determining step,22 LUMO energies of substrates are critical to explain the difficulty of the epoxidation of enones. In the case of the epoxidation of β-substituted cyclic enones,23 such as 3-methyl-2-cyclopenten-1-one (LUMO energy: −0.128 eV) and 3-methyl-2-cyclohexen-1-one (−0.121 eV), the reaction rates were decreased (entries 2 and 8). The lower reactivity of these compounds compared with the non-substituted ones (1: −0.116 eV, 2-cyclopenten-1-one: −0.126 eV) might be explained by the steric hindrance about the CC double bond of substrates. The epoxidation of α-isophorone hardly occurs (entry 11) due to the steric hindrance of the methyl group at the β-position and the gem-dimethyl group.24
| Entry | Substrate | Product | Time (h) | Conv.b (%) | Yieldb (%) |
|---|---|---|---|---|---|
| a Substrate (0.5 mmol), C3H7COO−/NiZn catalyst (0.05 g), DMF (2 mL), 30 wt% H2O2 aq. (4 eq. relative to the substrate), 60 °C, air. b Determined by GC using an internal standard technique. c Value in parenthesis is isolated yield. d 1st reuse. e 2nd reuse. f 3rd reuse. g 4th reuse. h Methanol (2 mL) was used as a solvent. | |||||
| 1 |

|

|
3 | 97 | 97 |
| 2 |

|
|
12 | 62 | 62 |
| 3 |

|
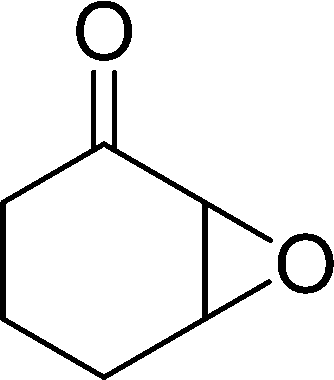
|
6 | 98 | 98 (91)c |
| 4d | 1 | 55 | 55 | ||
| 5e | 1 | 56 | 55 | ||
| 6f | 1 | 54 | 54 | ||
| 7g | 1 | 53 | 52 | ||
| 8 |

|

|
12 | 70 | 70 |
| 9h |
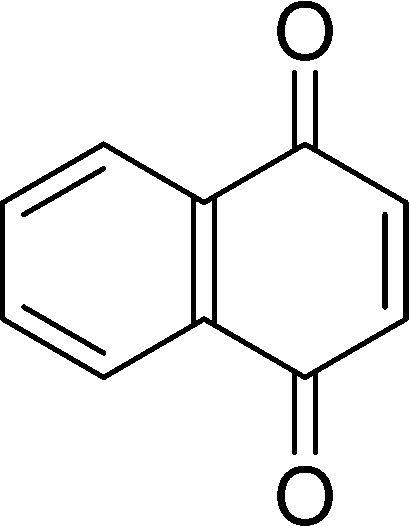
|

|
5 | 97 | 96 |
| 10 |

|
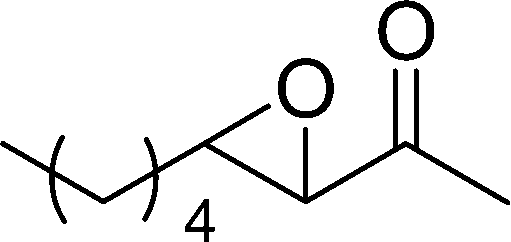
|
9 | 16 | 11 |
| 11 |

|
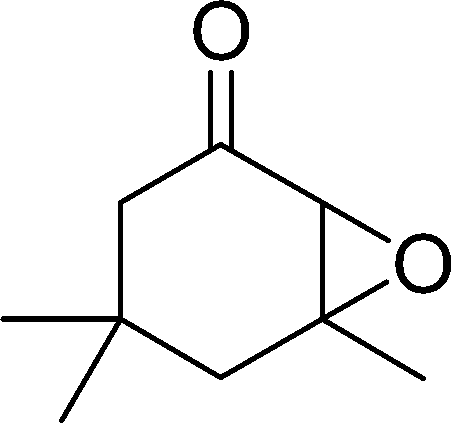
|
24 | 5 | n.d |
| 12 |
|

|
24 | 13 | 10 |
| 13 |

|

|
24 | 20 | 20 |
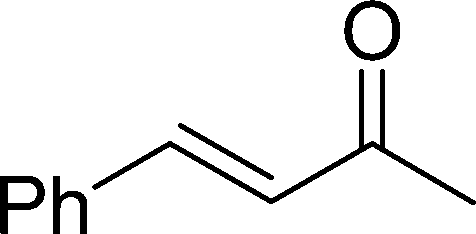
The β,β-disubstituted acyclic enone, (R)-(+)-pulegone, with a high LUMO energy of 0.273 eV, was not transformed into an epoxyketone, even after 24 h. In contrast, the C3H7COO−/NiZn-catalysed epoxidation of 1,4-naphthoquinone, which has a low LUMO orbital energy (−1.529 eV), totally proceeded, and the corresponding product was formed almost quantitatively (entry 9). The high reactivity of the substrates possessing low LUMO orbital energies supports the hypothesis that the catalytic epoxidation process occurs via the nucleophilic attack of a perhydroxyl species. Conversely, 3-nonen-2-one with a high LUMO orbital energy25 gave an extremely low yield of the corresponding epoxyketone (entry 10). In addition, the epoxidation of bulky substrates such as benzalacetone and chalcone resulted in only 10% and 20% yields of 2,3-epoxy-1,3-diphenylpropanone and 2,3-epoxy-1,3-diphenyl propanone, respectively (entries 12 and 13).
Moreover, the C3H7COO−/NiZn-catalyzed epoxidation of 1 with an equimolar amount of H2O2 proceeded in a highly effective manner,26 giving 97% of 2 at 60 °C in 8 h, with 97% efficiency of H2O2 utilization, as highlighted in Scheme 1.27
 | ||
| Scheme 1 C3H7COO−/NiZn-catalysed epoxidation of 1 with an equimolar amount of H2O2. Reaction conditions: 1 (0.5 mmol), C3H7COO−/NiZn catalyst (0.05 g), 30 wt% H2O2 aq. (0.5 mmol), DMF (2 mL), 60 °C, 8 h. | ||
Based on these results, the most plausible mechanism for C3H7COO−/NiZn-catalysed epoxidation is as follows. A perhydroxyl anion, which is formed by the reaction between the base site of NiZn and H2O2, attacks the β-olefinic carbon atom of an α,β-unsaturated ketone. Following the ring enclosure, the corresponding epoxyketone is formed and anion exchange between OH− and C3H7COO− takes place.
Conclusions
We conclude that the C3H7COO−/NiZn catalytic system presented here allows simple and practical epoxidation of cyclic enones, forming fine chemicals and pharmaceuticals in a manner that meets the increasing demand for environmentally friendly chemical processes.Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (25420824).Notes and references
- (a) J. Liang, R. Ma and T. Sasaki, Dalton Trans., 2014, 43, 10355 RSC; (b) N. Takahashi and K. Kuroda, J. Mater. Chem., 2011, 21, 14336 RSC; (c) C. Taviot-Gueho, Y. Feng, A. Faour and F. Leroux, Dalton Trans., 2010, 39, 5994 RSC; (d) A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191 RSC.
- (a) F. Bergaya, G. Lagaly and M. Vayer, in Developments in Clay Science – Volume 5B Handbook of Clay Science: Techniques and Applications, ed. F. Bergaya and G. Lagaly, 2013, p. 333 Search PubMed; (b) D. G. Evans and R. C. T. Slade, in Structure and Bonding, ed. X. Duan and D. G. Evans, Springer-Verlag, Berlin, 2006, vol. 119, p. 1 Search PubMed; (c) In Handbook of layered Materials, ed. P. S. Braterman, Z. P. Xu and F. Yarberry, Marcel Dekker, New York, 2004, p. 373 Search PubMed; (d) In Layered Double Hydroxides: Present and Future, ed. V. Rives, Nova Science Publishers, New York, 2001 Search PubMed.
- (a) E. Conterosito, G. Croce, L. Palin, C. Pagano, L. Perioli, D. Viterbo, E. Boccaleri, G. Paul and M. Milanesio, Phys. Chem. Chem. Phys., 2013, 15, 13418 RSC; (b) G. Carja, G. Lehutu, L. Dartu, M. Mertens and P. Cool, Appl. Clay Sci., 2012, 65–66, 37 CrossRef CAS PubMed; (c) M. D. Urena-Amate, N. D. Boutarbouch, M. M. Socias-Viciana and E. Gonzalez-Pradas, Appl. Clay Sci., 2011, 52, 368 CrossRef CAS PubMed; (d) H. Nakayama, K. Kuwano and M. Tsuhako, J. Phys. Chem. Solids, 2007, 69, 1552 CrossRef PubMed; (e) H. Tamura, J. Chiba, M. Ito, T. Takeda and S. Kikkawa, Solid State Ionics, 2004, 172, 607 CrossRef CAS PubMed.
- For reviews and accounts, see (a) S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2013, 15, 2026 RSC; (b) K. Takehira and T. Shishido, Catal. Surv. Asia, 2007, 11, 1 CrossRef CAS; (c) F. Figueras, M. L. Kantam and B. M. Choudary, Curr. Org. Chem., 2006, 10, 1627 CrossRef CAS; (d) D. Tichit and B. Coq, CATTECH, 2003, 7, 206 CrossRef CAS; (e) K. Kaneda, K. Yamaguchi, K. Mori, T. Mizugaki and K. Ebitani, Catal. Surv. Jpn., 2000, 4, 31 CrossRef CAS.
- D. P. Debecker, E. M. Gaigneaux and G. Busca, Chem. – Eur. J., 2009, 15, 3920 CrossRef CAS PubMed.
- (a) A. C. Gomes, S. M. Bruno, A. A. Valente, I. S. Gonçalves and M. Pillinger, J. Organomet. Chem., 2013, 744, 53 CrossRef CAS PubMed; (b) Y. Kuroda, Y. Miyamoto, M. Hibino, K. Yamaguchi and N. Mizuno, Chem. Mater., 2013, 25, 2291 CrossRef CAS; (c) K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008 CrossRef CAS PubMed; (d) K. Motokura, N. Hashimoto, T. Hara, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2012, 13, 2416 RSC; (e) A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2011, 50, 2986 CAS; (f) S. K. Jana, Y. Kubota and T. Tatsumi, J. Catal., 2008, 255, 40 CrossRef CAS PubMed; (g) J. Palomeque, F. Figueras and G. Gelbard, Appl. Catal., A, 2006, 300, 100 CrossRef CAS PubMed; (h) M. L. Kantam, B. V. Prakash, C. R. V. Reddy and B. Sreedhar, Synlett, 2005, 2329 CrossRef CAS PubMed.
- (a) M. Shirotori, S. Nishimura and K. Ebitani, Catal. Sci. Technol., 2014, 4, 971 RSC; (b) K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2005, 127, 9674 CrossRef CAS PubMed.
- (a) R. M. R. Bull, C. Markland, G. R. Williams and D. O'Hare, J. Mater. Chem., 2011, 21, 1822 RSC; (b) J. T. Rajamathi, A. Arulraj, N. Ravishankar, J. Arulraj and M. Rajamathi, Langmuir, 2008, 24, 11164 CrossRef CAS PubMed; (c) G. G. C. Arizaga, K. G. Satyanarayana and F. Wypych, Solid State Ionics, 2007, 178, 1143 CrossRef CAS PubMed; (d) J.-H. Yang, Y.-S. Han, M. Park, T. Park, S.-J. Hwang and Y.-S. Choy, Chem. Mater., 2007, 19, 2679 CrossRef CAS; (e) H. Morioka, H. Tagaya, M. Karasu, J. Kadokawa and K. Chiba, Inorg. Chem., 1999, 38, 4211 CrossRef CAS; (f) S. Yamanaka, T. Sako, K. Seki and M. Hattori, Solid State Ionics, 1992, 53–56, 527 CrossRef CAS.
- (a) T. V. Sathisha, B. E. K. Swamy, B. N. Chandrashekar, N. Thomas and B. Eswarappa, J. Electroanal. Chem., 2012, 674, 57 CrossRef CAS PubMed; (b) E. Kandare and J. M. Hossenlopp, Inorg. Chem., 2006, 45, 3766 CrossRef CAS PubMed; (c) R. Rojas, C. Barriga, M. A. Ulibarri, P. Malet and V. Rives, J. Mater. Chem., 2002, 12, 1071 RSC; (d) H. Nishizawa and K. Yuasa, J. Solid State Chem., 1998, 141, 229 CrossRef CAS; (e) M. Meyn, K. Beneke and C. Lagaly, Inorg. Chem., 1993, 32, 1209 CrossRef CAS; (f) R. Rojas, M. A. Ulibarri, C. Barriga and V. Rives, Microporous Mesoporous Mater., 2008, 112, 262 CrossRef CAS PubMed; (g) E. Kandare and J. M. Hossenlopp, J. Phys. Chem. B, 2005, 109, 8469 CrossRef CAS PubMed; (h) J. Arulraj, J. T. Rajamathi, K. R. Prabhu and M. Rajamathi, Solid State Sci., 2007, 9, 812 CrossRef CAS PubMed; (i) N. Nityashree and M. Rajamathi, J. Phys. Chem. Solids, 2013, 74, 1164 CrossRef CAS PubMed.
- (a) B. S. Lanes and K. Burgess, Chem. Rev., 2003, 103, 2457 CrossRef PubMed; (b) S. T. Oyama, in Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, ed. S. T. Oyama, Elsevier, Amsterdam, 2008, p. 1 Search PubMed.
- (a) G. B. Payne and P. H. Williams, J. Org. Chem., 1961, 26, 651 CrossRef CAS; (b) A. Russo and A. Lattanzi, Synthesis, 2009, 1551 CAS; (c) M. J. Porter and J. Skidmore, Chem. Commun., 2000, 1215 RSC.
- (a) M. Miyashita, T. Suzuki and A. Yoshikoshi, Chem. Lett., 1987, 285 CrossRef CAS; (b) V. K. Yadav and K. K. Kapoor, Tetrahedron, 1995, 51, 8573 CrossRef CAS; (c) R. J. L. Garcia, C. Fajardo, A. Fraile and M. R. Martin, J. Org. Chem., 2005, 70, 4300 CrossRef PubMed; (d) B. Lygo and P. G. Wainwright, Tetrahedron Lett., 1998, 39, 1599 CrossRef CAS; (e) T. S. Straub, Tetrahedron Lett., 1995, 36, 663 CrossRef CAS; (f) K. M. McQuaid and T. R. R. Pettus, Synlett, 2004, 2403 CAS; (g) D. Yang, M.-K. Wong and Y.-C. Yip, J. Org. Chem., 1995, 60, 3887 CrossRef CAS; (h) S.-G. Yang, J.-P. Hwang, M.-Y. Park, K. Lee and Y.-H. Kim, Tetrahedron, 2007, 63, 5184 CrossRef CAS PubMed.
- (a) W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638 CrossRef; (b) O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196 CrossRef CAS; (c) K. Kamata, K. Sugahara, K. Yonehara, R. Ishimoto and N. Mizuno, Chem. – Eur. J., 2011, 17, 7549 CrossRef CAS PubMed; (d) O. Lifchits, M. Mahlau, C. M. Reisinger, A. Lee, C. Farès, I. Polyak, G. Gopakumar, W. Thiel and B. List, J. Am. Chem. Soc., 2013, 135, 6677 CrossRef CAS PubMed; (e) X. Wang, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2008, 130, 6070 CrossRef CAS PubMed; (f) N. J. Schoenfeldt, A. W. Korinda and J. M. Notestein, Chem. Commun., 2010, 46, 1640 RSC; (g) K. Yamaguchi, C. Yoshida, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 530 CrossRef CAS PubMed.
- (a) K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Org. Chem., 2000, 65, 6897 CrossRef CAS PubMed; (b) B. M. Choudary, M. L. Kantam, B. Bharathi and C. V. Reddy, Synlett, 1998, 1203 CrossRef CAS PubMed; (c) C. Cativiela, F. Figueras, J. M. Fraile, J. I. Garcia and J. A. Mayoral, Tetrahedron Lett., 1995, 36, 4125 CrossRef CAS; (d) J. Palomeque, J. Lopez and F. Figueras, J. Catal., 2002, 211, 150 CrossRef CAS; (e) U. R. Pillai and E. Sahle-Demessie, Appl. Catal., A, 2004, 261, 69 CrossRef CAS PubMed; (f) N. Hiyoshi, Appl. Catal., A, 2012, 419–420, 164 CrossRef CAS PubMed.
- (a) T. Hara, J. Sawada, Y. Nakamura, N. Ichikuni and S. Shimazu, Catal. Sci. Technol., 2011, 1, 1376 RSC; (b) T. Hara, M. Ishikawa, J. Sawada, N. Ichikuni and S. Shimazu, Green Chem., 2009, 11, 2034 RSC; (c) T. Hara, J. Kurihara, N. Ichikuni and S. Shimazu, Chem. Lett., 2010, 39, 304 CrossRef CAS.
- S. Yamanaka, K. Ando and M. Ohashi, Mater. Res. Soc. Symp. Proc., 1995, 371, 131 CrossRef CAS PubMed.
- The chemical formula of the synthesized CH3COO−/NiZn was confirmed by elemental analysis, XRF, and TG-DTA. The anion exchange capacity was found to be 2.44 mmol g−1..
- In the case of simple alkenes such as cyclooctene, 1-decene, and styrene, the corresponding epoxides were not obtained at all..
- (a) K. Yamaguchi, K. Ebitani and K. Kaneda, J. Org. Chem., 1999, 64, 2966 CrossRef CAS; (b) K. Yamaguchi, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 1999, 23, 799 RSC.
- The molecular size of DMF and water are ca. 0.43 nm and 0.15 nm, respectively. The results of molecular modeling are shown in the ESI.†.
- The electrophilicity of enones can be described with LUMO orbital energies, see R. J. Loncharich, F. K. Brown and K. N. Houk, J. Org. Chem., 1989, 54, 1129 CrossRef CAS.
- H. O. House and R. S. So, J. Am. Chem. Soc., 1958, 80, 2428 CrossRef CAS.
- Kaneda and co-workers calculated the LUMO orbital energy of enones, shown in ref. 14a..
- (R)-(−)-Carvone also did not react under these reaction conditions..
- In ref. 14a, the LUMO energies of 4-hexen-3-one were calculated to be −0.008 eV (cis) and −0.047 eV (trans)..
- K. Kamata, K. Yonehara, Y. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964 CrossRef CAS PubMed.
- The decomposition of H2O2 might be inhibited by the buffering effect of the solid NiZn catalyst..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy01063a |
| This journal is © The Royal Society of Chemistry 2015 |