
Multifunctional mesoporous silica-supported palladium nanoparticles for selective phenol hydrogenation in the aqueous phase
Fengwei
Zhang
*a and
Hengquan
Yang
*ab
aInstitute of Crystalline Materials, Shanxi University, Taiyuan 030006, PR China. E-mail: fwzhang@sxu.edu.cn; hqyang@sxu.edu.cn; Tel: +86 351 7016082
bSchool of Chemistry and Chemical Engineering, Shanxi University, Taiyuan 030006, PR China
First published on 19th September 2014
Abstract
A simple, efficient and recoverable palladium-based catalyst was successfully prepared by immobilizing palladium nanoparticles (Pd NPs) over hydrophobic core–hydrophilic shell structured mesoporous silica microspheres. The obtained catalyst was characterized by transmission electron microscopy (TEM), N2 adsorption–desorption, Fourier-transform infrared spectroscopy (FTIR), thermogravimetric analysis (TGA) and X-ray photoelectron spectroscopy (XPS). The as-prepared catalyst could rapidly convert phenol to cyclohexanone with 98.5% conversion and 97.1% selectivity under mild reaction conditions in the aqueous phase (1 atm H2, 80 °C, 3 h). The excellent catalytic performance of the Pd/MS-C3@MS-NH2 catalyst was probably attributed to the enhanced synergistic effect between highly dispersed Pd NPs and significantly decreased phenol mass transfer resistance. In addition, it could be easily recovered by centrifugation and reused 5 times without any significant loss in activity and selectivity.
Introduction
Core–shell structured multifunctional mesoporous materials have recently attracted significant attention owing to their unique characteristics, such as high specific surface area, controllable pore structure, narrow pore size distribution, and versatile surface functionalization.1–5 Thus, they are generally regarded as an ideal candidate for catalysis, adsorption, separation, energy storage and conversion, drug delivery and optics.6–12 Among them, as far as catalyst supports are concerned, great progress has been made in their functionalization, and the developed heterogeneous catalytic systems displayed extremely high catalytic activity, selectivity and stability in a variety of important organic transformations.13–20 For example, Suzuki et al. first reported the SO3H core–hydrophobic shell structured mesoporous silica microspheres through the co-condensation/expansion method, which exhibited enhanced catalytic activity for the condensation reaction of 2-methylfuran and acetone. They found that the obvious enhancement in catalytic activity due to the incorporation of hydrophobic groups was only achieved with core–shell structure, and little enhancement was observed in a randomly distributed structure.21 Yang et al. have also successfully prepared a yolk–shell multifunctional nanoreactor with basic core (−NH2) and acidic shell (−SO3H), which showed excellent catalytic performance in deacetalization–Henry cascade reaction.22As is well-known, the hydrogenation of phenol is one of the effective methods for the preparation of cyclohexanone, which serves as an important intermediate in the synthesis of caprolactam and adipic acid.23 The phenol hydrogenation route is generally undertaken through one- or two-step processes.24 The two-step process involves hydrogenation of phenol to cyclohexanol followed by dehydrogenation to cyclohexanone.25 The one-step selective phenol hydrogenation to cyclohexanone, in contrast, is more advantageous in the perspective of energy efficiency and operation cost. Recently, considerable research has been devoted to the development of highly active heterogeneous catalysts for one-step selective phenol hydrogenation.26 In particular, Han and co-workers have reported the synthesis of an unprecedented commercially available heterogeneous palladium and Lewis acid combined catalyst, which was carried out at 50 °C under 10 atm of H2 for liquid-phase phenol hydrogenation.27 However, the environmentally unfriendly co-catalysts and organic solvents were indispensable for the reaction process as the reaction pressure was relatively higher.
To overcome these problems, palladium nanoparticles supported on a variety of organic and inorganic materials, such as polymers,28 mpg-C3N4,29 N-doped ordered mesoporous carbon (NOMC),30 carbon nanotubes (CNTs),31 and carbon nanofibers (CNFs),32 have been developed and used to catalyze the hydrogenation of phenol in the aqueous phase. Nevertheless, the separation and recycling of the polymer-supported catalyst were relatively complicated and the Pd NPs were inclined to aggregate due to the high surface energy. In addition, the preparation procedures of these types of solid supports usually involve multiple steps of preparation, high pyrolysis temperature and environmentally unfriendly reactants, which consume a considerable amount of time and energy. It is also very important to have the ability to scale up the synthesis for the preparation of large quantities of materials. Therefore, the development of a more facile and greener heterogeneous catalyst with excellent phenol conversion and selectivity is still a great challenge. To our knowledge, there is no report of direct selective hydrogenation of phenol to cyclohexanone over easily recyclable multifunctional core–shell structured mesoporous silica in the aqueous phase. Herein, we report a novel hydrophobic core/hydrophilic shell structured mesoporous silica immobilized Pd NP catalyst, which acts as a highly active, water-tolerant, and easily recyclable catalyst for selective phenol hydrogenation. More importantly, it can be easily recycled and used repetitively more than 5 times without loss of catalytic efficiency.
Experimental
Materials
Palladium(II) acetate (Pd(OAc)2, ≥97%), tetramethyl orthosilicate (TMOS, 98%) and sodium hydroxide (NaOH) were purchased from Sinopharm Chemical Reagent Co., Ltd. Sodium borohydride (NaBH4, 98%), (3-aminopropyl)trimethoxysilane (APTMS) and n-propyltrimethoxysilane (C3TMS) were obtained from Aladdin Chemical Reagent Co., Ltd. Cetyltrimethylammonium chloride (CTAC, 99%) was purchased from Nanjing Robiot Co., Ltd. All other chemicals were of analytical grade and were used without any further purification.Preparation of hydrophobic core–hydrophilic shell mesoporous silica
Hydrophobic core–hydrophilic shell mesoporous silica was prepared according to the method previously described by Suzuki et al. with some modification.21 Specifically, 3.52 g of CTAC and 2.5 mL of NaOH (1.0 mol L−1) were dissolved in a mixture solution of 400 mL of deionized water and 500 mL of methanol, and then 1.20 g of TMOS and 0.25 g of C3TMS were added into the above solution with vigorous stirring at room temperature. After 1 h of pre-hydrolysis, 1.20 g of TMOS and 0.20 g of APTMS were slowly added to the suspension. After 8 h of continuous stirring, the obtained white precipitate was filtered and washed thoroughly with deionized water and ethanol and dried at 100 °C for 6 h. To remove the surfactant, the as-synthesized material was dispersed in a 100 mL ethanol solution containing 1.5 mL of concentrated HCl and the mixture was stirred at 80 °C for 12 h. The resulting material was designated as MS-C3@MS-NH2. As a control, the MS@MS-NH2 and MS-NH2@MS-C3 were prepared under the same procedure except no C3TMS was added into the synthesis process and in turn the order of adding.Loading of Pd NPs on hydrophobic core–hydrophilic shell mesoporous silica
To a stirred suspension of MS-C3@MS-NH2 (1.0 g) in 30 mL of deionized water was added 65 mg of Pd(OAc)2. After stirring for 4 h (the amount of Pd(OAc)2 adsorbed onto the solid materials was calculated from the UV-Vis spectra) 20 mL of NaBH4 (120 mg) was slowly added and the suspension was stirred for another 2 h. The suspension was centrifuged and washed several times with deionized water and ethanol, resulting in Pd/MS-C3@MS-NH2 (the loading amount of Pd was 3.12 wt%).Pd/MS-C3@MS-NH2 catalysts for selective phenol hydrogenation
In a typical experiment, 80 mg of the catalyst (0.025 mmol), 0.5 mmol of phenol and 3 mL of deionized water were added into the reaction vessel. The vessel was sealed and purged three times with hydrogen to remove the air at room temperature. Then, hydrogenation was performed at 80 °C under 1 atm H2 for 3 h. After completion of the reaction, the products were extracted with ethyl acetate, followed by analysis using an Agilent 7890A gas chromatograph, and the products were identified by comparison with authentic samples.Recycling of the Pd/MS-C3@MS-NH2 catalyst
The recyclability of the Pd/MS-C3@MS-NH2 catalyst was tested for phenol-selective hydrogenation in the aqueous phase under the same reaction conditions as described above. After each run of reaction, the catalyst was separated from the reaction mixture by centrifugation, washed thoroughly with ethanol, dried at 60 °C for 2 h, and then reused directly for the next run.Characterization
Transmission electron microscopy (TEM) was performed using a FEI Tecnai G2 F20S-Twin with an accelerating voltage of 200 kV. For sample preparation, the powders were dispersed in ethanol by sonication, and one drop of the solution was dropped onto a microgrid. N2 adsorption–desorption isotherms were obtained with an ASAP2020 analyzer. Before measurement, samples were degassed under vacuum at 373 K for 6 h. The surface area of the samples was calculated by the Brunauer–Emmet–Teller (BET) method, and the pore volume and pore size distribution were calculated using the Barrett–Joyner–Elmer (BJH) model. Thermogravimetric analysis (TGA) was performed using a SETARAM Evolution 16/18 apparatus. The samples were heated on an alumina pan from 25 °C to 800 °C at a heating rate of 10 °C min−1 under nitrogen. Fourier transform infrared (FTIR) spectra were obtained using a Nicolet NEXUS 670 spectrophotometer (frequency range from 4000 to 500 cm−1) with KBr pellets. X-ray photoelectron spectra (XPS) was obtained with the PHI-5702 instrument and the C1S line at 286.6 eV was used as the binding energy reference.Results and discussion
The synthesis route of the hydrophobic core–hydrophilic shell structured Pd/MS-C3@MS-NH2 catalyst is illustrated in Scheme 1. Firstly, the hydrophobic core was prepared through the co-condensation of TMOS and C3TMS in the presence of CTAC in an alkaline methanol solution. Secondly, after a certain time of pre-hydrolysis and condensation, a mixture of TMOS and APTMS was added to the above suspension to form a layer of hydrophilic shell. The resultant materials were extracted with hot acidic alcohol solution to remove CTAC. Thirdly, palladium nanoparticles were immobilized on the MS-C3@MS-NH2 microspheres by impregnation and subsequent reduction. | ||
| Scheme 1 The synthesis procedure of the Pd/MS-C3@MS-NH2 catalyst. | ||
The respective TEM images of MS-C3@MS-NH2, Pd/MS-C3@MS-NH2, Pd/MS@MS-NH2 and Pd/MS-NH2@MS-C3 are shown in Fig. 1. From the TEM image of MS-C3@MS-NH2 (Fig. 1a), highly uniform microspheres with an average diameter of 340 nm, a shell thickness of 30 nm and perpendicularly oriented channels about 2.7 nm in diameter can be seen. For Pd/MS-C3@MS-NH2 and Pd/MS@MS-NH2 catalysts (Fig. 1b and c), the products were composed of uniform mesoporous microspheres with a particle size of 350 nm, which were similar to those of MS-C3@MS-NH2 microspheres. Additionally, it can be clearly seen that most of the palladium nanoparticles with particle sizes of about 5.8 nm for Pd/MS-C3@MS-NH2 and 5.4 nm for Pd/MS@MS-NH2 were uniformly dispersed on the surface of the multifunctional core–shell structured mesoporous silica. However, the Pd NPs were poorly distributed on the surface of the Pd/MS-NH2@MS-C3 catalyst (Fig. 1d) and they were inclined to aggregate into larger particles (about 8.1 nm).
 | ||
| Fig. 1 TEM images of (a) MS-C3@MS-NH2, (b) Pd/MS-C3@MS-NH2, (c) Pd/MS@MS-NH2 and (d) Pd/MS-NH2@MS-C3. | ||
Scanning transmission electron microscopy (STEM) analysis of the Pd/MS-C3@MS-NH2 catalyst was performed to obtain more detailed information about the structure. As shown in Fig. 2, STEM demonstrated that the as-synthesized Pd/MS-C3@MS-NH2 catalyst has a typical core–shell structure, which was in agreement with the results of TEM measurement. EDX element mapping of the sample further showed the spatial distributions of Si, O, N and Pd in the Pd/MS-C3@MS-NH2 catalyst. The strong Si and O signals across the sphere confirmed the presence of mesoporous silica, while the N and Pd signals detected in the surface region clearly suggested that they were evenly distributed on the surface of core–shell structured mesoporous silica. Moreover, our previous XPS elemental analysis results also have shown that the –NH2 groups were mainly concentrated in the shell for the Pd/MF@MN catalyst (Pd NPs supported on fluoro-functionalized core and amino-functionalized shell structured mesoporous silica).16
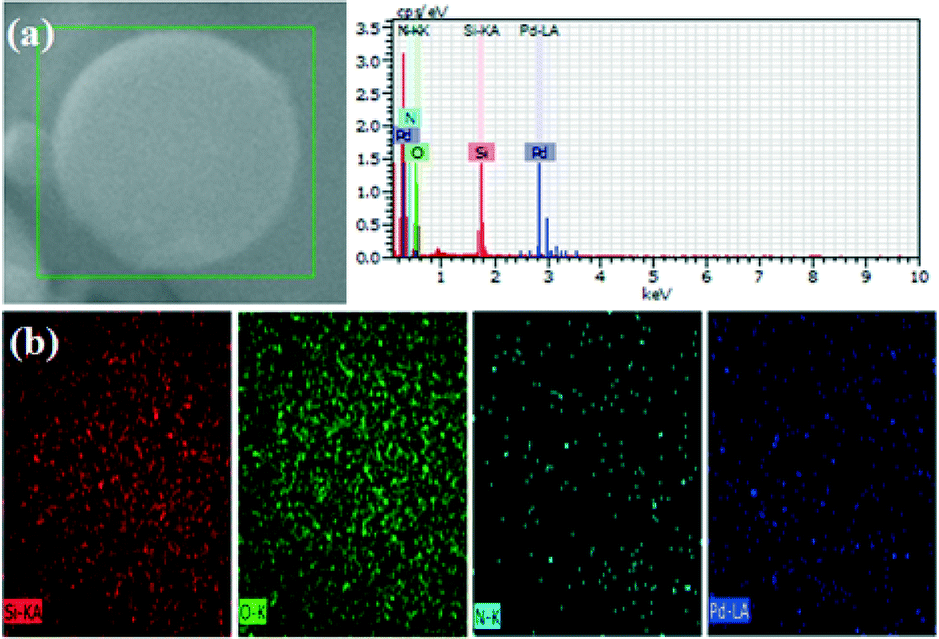 | ||
| Fig. 2 (a) Images of STEM and EDX of the Pd/MS-C3@MS-NH2 catalyst and (b) the corresponding elemental mapping of Si, O, N and Pd. | ||
In the FT-IR spectra of MS-C3@MS-NH2 and Pd/MS-C3@MS-NH2 microspheres (Fig. 3A), the peaks around 3422 cm−1, 1404 and 1072 cm−1 were assigned to the surface-absorbed water molecules, C–H bending and Si–O–Si stretching modes, respectively. The bands at 2926 and 2847 cm−1 can also be seen in these two materials, which should be attributed to the saturated C–H stretching vibrations of propyl and aminopropyl groups, respectively. Meanwhile, the presence of N–H symmetric bending vibration at 1633 cm−1 further confirmed the existence of amine groups. The results demonstrate that propyl and amino groups were successfully incorporated into the mesoporous silica microspheres and in the IR spectrum of Pd/MS-C3@MS-NH2, almost no change occurs after immobilizing Pd NPs on the MS-C3@MS-NH2 microsphere surface. The thermal stability of the Pd/MS-C3@MS-NH2 catalyst was investigated by TGA under nitrogen. As shown in Fig. 3B, the TGA curve of Pd/MS-C3@MS-NH2 exhibits 1.5 wt% weight loss at around 150 °C due to the loss of adsorbed water molecules, the second weight loss takes place above 150 °C, and about 14.1% weight loss was observed between 150 °C and 650 °C, indicating that propyl and amino groups were successfully incorporated into the mesoporous silica. The TGA curve of Pd/MS-C3@MS-NH2 further represents good thermal stability up to 150 °C.
 | ||
| Fig. 3 (A) FTIR spectra of (a) MS-C3@MS-NH2 and (b) Pd/MS-C3@MS-NH2 and (B) the TGA curve of Pd/MS-C3@MS-NH2. | ||
The N2 adsorption–desorption isotherms and the corresponding pore size distribution (inset) of MS-C3@MS-NH2 and Pd/MS-C3@MS-NH2 are shown in Fig. 4. Both the nitrogen adsorption and the desorption can be classified as type IV isotherms with a small hysteresis loop. The specific surface area, total pore volume and pore size of MS-C3@MS-NH2 were calculated to be 731 m2 g−1, 0.375 cm3 g−1 and 2.54 nm, respectively. The high specific surface area and well-developed mesoporous channels suggest that it was a good candidate as support for loading active Pd NPs. The specific surface area, total pore volume and pore size were 494 m2 g−1, 0.258 cm3 g−1 and 2.38 nm, respectively, after immobilizing Pd NPs on MS-C3@MS-NH2 microspheres, indicating that Pd loading had little effect on the specific surface area and pore size distribution of Pd/MS-C3@MS-NH2.
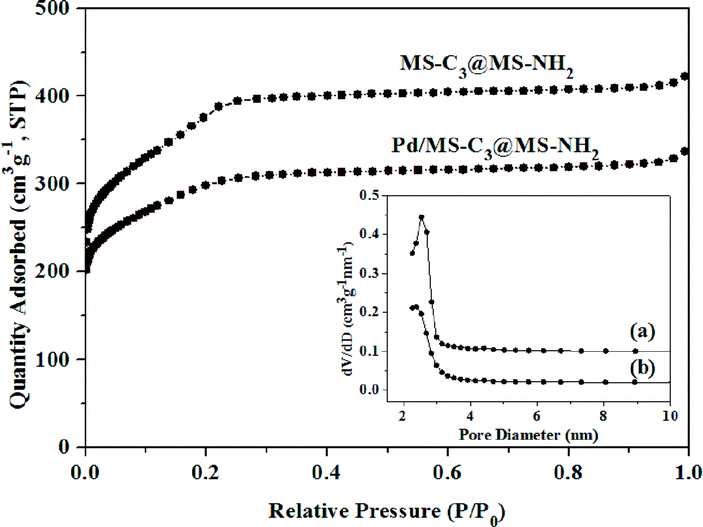 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms and pore size distributions of (a) MS-C3@MS-NH2 and (b) Pd/MS-C3@MS-NH2. | ||
The catalyst was also characterized by XPS in order to ascertain the oxidation state of Pd in the catalyst. Fig. 5 shows the Pd 3d spectrum of the catalyst. It can be seen that the Pd binding energy of Pd/MS-C3@MS-NH2 presented two sharp peaks centered at 341.6 and 336.4 eV, which were ascribed to Pd 3d3/2 and Pd 3d5/2, respectively. The results demonstrated that all of the Pd(II) species were completely converted into Pd(0) nanoparticles through chemical reduction with NaBH4.
 | ||
| Fig. 5 XPS spectrum of the Pd/MS-C3@MS-NH2 catalyst. | ||
The catalytic activity of different heterogeneous catalysts was evaluated for the selective phenol hydrogenation and the results are summarized in Table 1. As can be seen, in the absence of the catalyst and in the presence of MS-C3@MS-NH2 at 80 °C for 6 h in the aqueous phase, no product was detected (Table 1, entries 1 and 2). For Pd/MS-NH2@MS-C3 and Pd/MS@MS-NH2 catalysts, the reaction proceeds slowly with 38.2% and 78.8% phenol conversion and >99% and 98.7% cyclohexanone selectivity, respectively (Table 1, entries 3 and 4). As expected, when the reaction time increased from 3 h to 6 h, the conversion of phenol increased to 83.8% and >99%, while the selectivity for cyclohexanone decreased to 98.6% and 97.5%, respectively (Table 1, entries 5 and 6). It is worth mentioning that the reaction proceeds smoothly with 98.5% phenol conversion and 97.1% cyclohexanone selectivity in the presence of Pd/MS-C3@MS-NH2 within 3 h (Table 1, entry 7). Apparently, the hydrogenation rate of this new heterogenized catalyst was higher than that of its quasi-homogeneous counterpart (1 atm H2, 90 °C, 16 h).26a The excellent catalytic performance of Pd/MS-C3@MS-NH2 was probably due to the hydrophilic shell being not only highly dispersed and stabilized Pd NPs through amino groups but also beneficial to the uniform dispersion of the heterogeneous catalyst in water. Moreover, the hydrophobic core could rapidly concentrate phenol to the surroundings of Pd NP active sites. Along with phenol being hydrogenated to cyclohexanone, it constantly diffused into the aqueous solution from the mesoporous channel and avoided the further hydrogenation of cyclohexanone. Therefore, the catalyst can achieve the efficient, green and selective hydrogenation of phenol.
| Entry | Catalyst | T (°C) | t (h) | Conv.b (%) | Sel.b (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.5 mmol of phenol, 80 mg of catalyst (0.025 mmol) and 3.0 mL of H2O under 1 atm H2. b The conversion and selectivity were determined using an Agilent 7890A gas chromatograph. | |||||
| 1 | — | 80 | 6 | — | — |
| 2 | MS-C3@MS-NH2 | 80 | 6 | — | — |
| 3 | Pd/MS-NH2@MS-C3 | 80 | 3 | 38.2 | >99 |
| 4 | Pd/MS@MS-NH2 | 80 | 3 | 78.8 | 98.7 |
| 5 | Pd/MS-NH2@MS-C3 | 80 | 6 | 83.8 | 98.6 |
| 6 | Pd/MS@MS-NH2 | 80 | 6 | >99 | 97.5 |
| 7 | Pd/MS-C3@MS-NH2 | 80 | 3 | 98.5 | 97.1 |
Fig. 6a shows the evolution of phenol conversion and cyclohexanone selectivity with reaction temperature. As can be seen, the conversion of phenol increased from 85.2% to 99.6% as the reaction temperature increased from 50 °C to 90 °C. Although the conversion of phenol increased rapidly from 50 °C to 80 °C, the selectivity for cyclohexanone reduced gradually. Notably, a further increase in reaction temperature hardly improved the phenol conversion, but the selectivity for cyclohexanone significantly decreased from 98.2% to 86.5%. The influence of reaction time on phenol conversion and cyclohexanone selectivity is shown in Fig. 6b. The reaction was accompanied by a rapid increase in phenol conversion and a slight decrease in cyclohexanone selectivity in the first 3 h, suggesting that phenol was mainly hydrogenated to cyclohexanone in the first step. With the further increase in reaction time (from 3 h to 8 h), the phenol conversion no longer increased and cyclohexanone selectivity decreased gradually (from 97.1% to 88.2%), illustrating that the conversion of cyclohexanone into cyclohexanol plays a vital role in the second step. The influence of the S/C molar ratio on the conversion of phenol and selectivity for cyclohexanone is shown in Fig. 6c. When the molar ratio of S/C (substrate/catalyst) was 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the conversion of phenol was low due to the inadequate number of active sites. The conversion increased as the S/C molar ratio decreased from 80:1 to 20:1, and a conversion of 98.5% was achieved. However, the conversion hardly changed with a further decrease of the S/C molar ratio. Considering the cost of the catalyst and reaction rate, the suitable amount of the S/C molar ratio was 20:1. Based on the above results, the optimum conditions of selective phenol hydrogenation were obtained: reaction time 3 h, reaction temperature 80 °C and S/C molar ratio 20:1. Under the optimized conditions, the conversion of phenol could reach 98.5% and the selectivity for cyclohexanone could reach 97.1%.
:1, the conversion of phenol was low due to the inadequate number of active sites. The conversion increased as the S/C molar ratio decreased from 80:1 to 20:1, and a conversion of 98.5% was achieved. However, the conversion hardly changed with a further decrease of the S/C molar ratio. Considering the cost of the catalyst and reaction rate, the suitable amount of the S/C molar ratio was 20:1. Based on the above results, the optimum conditions of selective phenol hydrogenation were obtained: reaction time 3 h, reaction temperature 80 °C and S/C molar ratio 20:1. Under the optimized conditions, the conversion of phenol could reach 98.5% and the selectivity for cyclohexanone could reach 97.1%.
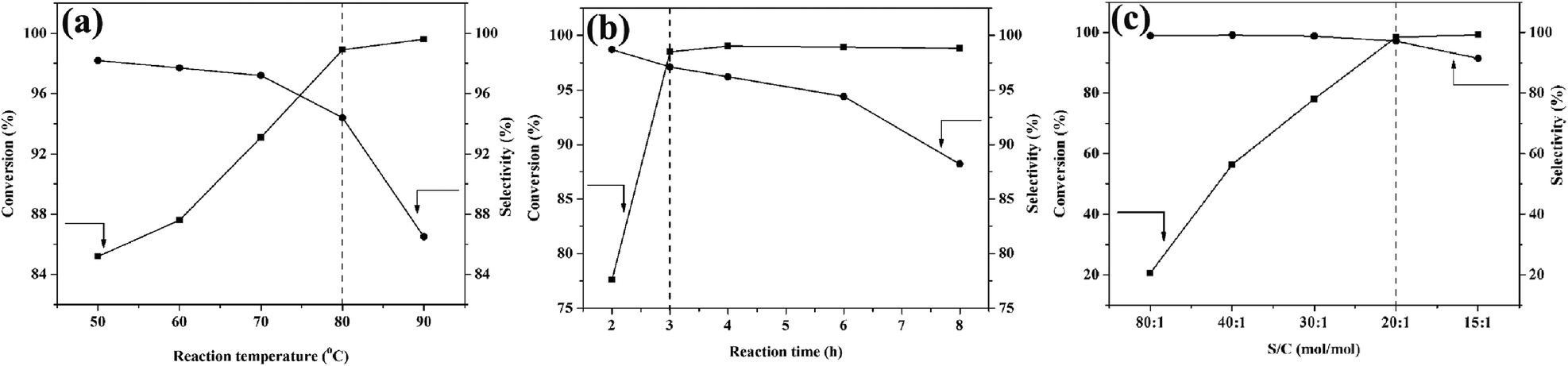 | ||
| Fig. 6 Conversion of phenol and the selectivity for cyclohexanone as functions of (a) reaction temperature, (b) reaction time and (c) S/C molar ratio. Reaction conditions: 0.5 mmol of phenol, 3.0 mL of H2O and 1 atm H2. | ||
Additional advantages of Pd/MS-C3@MS-NH2 prepared here were stability and ease of recycling because it can be facilely separated by centrifugation. Fig. 7 shows the recyclability of Pd/MS-C3@MS-NH2 for selective hydrogenation of phenol in the aqueous phase. It is worth mentioning that the catalyst still exhibited excellent catalytic activity even after running for more than five cycles. However, the slight decrease in phenol conversion with further recycle times should be due to the gradual leaching of Pd NPs from the catalyst surface (the Pd content in the catalyst was found to be 2.97 wt% based on ICP-AES analysis, indicating that ca. 4.8% of Pd was leached from the catalyst surface). The results clearly indicate that the high catalytic performance should be attributed to the small particle size and good dispersion of Pd NPs, high specific surface area and hydrophobicity of the core.
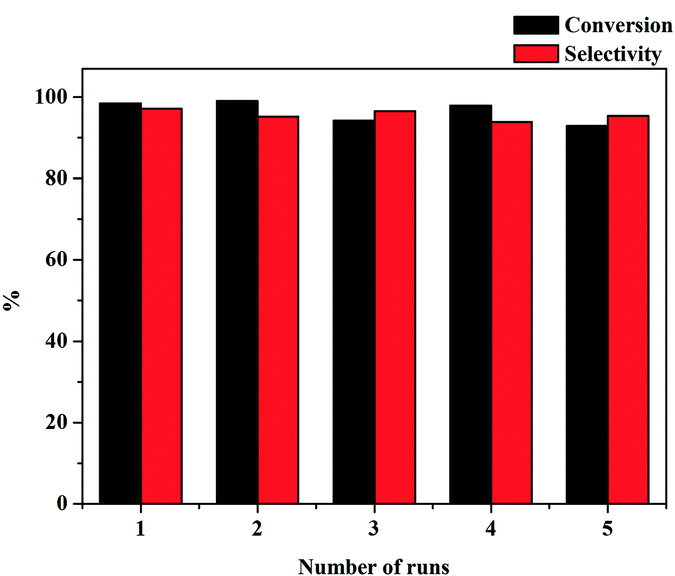 | ||
| Fig. 7 Recycling tests of the Pd/MS-C3@MS-NH2 catalyst in selective phenol hydrogenation. Reaction conditions: 0.5 mmol of phenol, 3.0 mL of H2O, 80 mg of catalyst, 1 atm H2, 80 °C, 3 h. | ||
Conclusion
In conclusion, a simple and environmentally friendly approach was reported for the synthesis of hydrophobic core–hydrophilic shell structured Pd/MS-C3@MS-NH2 nanocomposite catalysts, which exhibited superior catalytic activity and stability toward the selective hydrogenation of phenol in water. The reasons could be attributed to the synergistic effect derived from binary organic functional groups since the amino groups promoted the uniform dispersion of Pd NPs inside the pore channels while the propyl groups decreased the diffusion limitation of phenol in water. Thus, it can be seen that this synthesis strategy provided a useful idea for the fabrication of other nanoparticle-supported catalysts with high specific surface area, ordered mesoporous channel and hydrophobic/hydrophilic functionality, which should be highly promising in diverse catalytic reactions.Acknowledgements
This work was supported by the Scientific Research Start-up Funds of Shanxi University (023151801002) and the National Natural Science Foundation of China (20903064, 221173137).Notes and references
- D. Niu, Z. Ma, Y. Li and J. Shi, J. Am. Chem. Soc., 2010, 132, 15144–15147 CrossRef CAS PubMed.
- X. Liang, J. Li, J.-B. Joo, A. Gutiérrez, A. Tillekaratne, I. Lee, Y. Yin and F. Zaera, Angew. Chem., Int. Ed., 2012, 51, 8034–8036 CrossRef CAS PubMed.
- L. Xu, H.-G. Peng, K. Zhang, H. Wu, L. Chen, Y. Liu and P. Wu, ACS Catal., 2013, 3, 103–110 CrossRef CAS.
- Z. Li, L. Mo, Y. Kathiraser and S. Kawi, ACS Catal., 2014, 4, 1526–1536 CrossRef CAS.
- F. Zhang, X. Wu, C. Liang, X. Li, Z. Wang and H. Li, Green Chem., 2014, 16, 3768–3777 RSC.
- (a) S. Ikurumi, S. Okada, K. Nakatsuka, T. Kamegawa, K. Mori and H. Yamashita, J. Phys. Chem. C, 2014, 118, 575–581 CrossRef CAS; (b) S. Okada, K. Mori, T. Kamegawa, M. Che and H. Yamashita, Chem. – Eur. J., 2011, 17, 9047–9051 CrossRef CAS PubMed; (c) S. Okada, S. Ikurumi, T. Kamegawa, K. Mori and H. Yamashita, J. Phys. Chem. C, 2012, 116, 14360–14367 CrossRef CAS.
- L. Zhang, S. Z. Qiao, Y. G. Jin, Z. G. Chen, H. C. Gu and G. Q. Lu, Adv. Mater., 2008, 20, 805–809 CrossRef CAS.
- J. P. Ge, Y. X. Hu and Y. D. Yin, Angew. Chem., Int. Ed., 2007, 46, 7428–7431 CrossRef CAS PubMed.
- F. Zhang, R. Che, X. M. Li, C. Yao, J. P. Yang, D. K. Shen, P. Hu, W. Li and D. Y. Zhao, Nano Lett., 2012, 12, 2852–2858 CrossRef CAS PubMed.
- X. Q. Huang, S. H. Tang, B. J. Liu, B. Ren and N. F. Zheng, Adv. Mater., 2011, 23, 3420–3425 CrossRef CAS PubMed.
- M. Feyen, C. Weidenthaler, R. Güttel, K. Schlichte, U. Holle, A.-H. Lu and F. Schüth, Chem. – Eur. J., 2011, 17, 598–605 CrossRef CAS PubMed.
- Z. J. Zhang, L. M. Wang, J. Wang, X. M. Jiang, X. H. Li, Z. J. Hu, Y. L. Ji, X. C. Wu and C. Y. Chen, Adv. Mater., 2012, 24, 1418–1423 CrossRef CAS PubMed.
- J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Yang, E. Strounina, G. Q. Lu and S. Z. Qiao, Adv. Funct. Mater., 2012, 22, 591–599 CrossRef CAS.
- W. Li and D. Zhao, Adv. Funct. Mater., 2013, 25, 142–149 CrossRef CAS.
- (a) P. Li, C.-Y. Cao, Z. Chen, H. Liu, Y. Yu and W.-G. Song, Chem. Commun., 2012, 48, 10541–10543 RSC; (b) P. Li, C.-Y. Cao, H. Liu, Y. Yu and W.-G. Song, J. Mater. Chem. A, 2013, 1, 12804–12810 RSC; (c) H. G. Peng, L. Xu, H. Wu, K. Zhang and P. Wu, Chem. Commun., 2013, 49, 2709–2711 RSC.
- H. Yang, X. Jiao and S. Li, Chem. Commun., 2012, 48, 11217–11219 RSC.
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- Q. Zhang, I. Lee, J.-B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef CAS PubMed.
- Z.-A. Qiao, P. Zhang, S.-H. Chai, M. Chi, G. M. Veith, N. C. Gallego, M. Kidder and S. Dai, J. Am. Chem. Soc., 2014, 136, 11260–11263 CrossRef CAS PubMed.
- P. Zhang, Y. Gong, H. Li, Z. Chen and Y. Wang, Nat. Commun., 2013, 4, 1593 CrossRef PubMed.
- T. M. Suzuki, T. Nakamura, E. Sudo, Y. Akimoto and K. Yano, J. Catal., 2008, 258, 265–272 CrossRef CAS PubMed.
- Y. Yang, X. Liu, X. Li, J. Zhao, S. Bai, J. Liu and Q. Yang, Angew. Chem., Int. Ed., 2012, 51, 9164–9168 CrossRef CAS PubMed.
- (a) I. Dodgson, K. Griffin, G. Barberis, F. Pignataro and G. Tauszik, Chem. Ind., 1989, 830–833 CAS; (b) Kirk-Othmer Encyclopedia of Chemical Technology, ed. M. Howe-Grant, Wiley, New York, 1991 Search PubMed.
- M. Chatterjee, H. Kawanami, M. Sato, A. Chatterjee, T. Yokoyama and T. Suzuki, Adv. Synth. Catal., 2009, 351, 1912–1924 CrossRef CAS.
- V. Z. Fridman and A. A. Davydov, J. Catal., 2000, 195, 20–30 CrossRef CAS.
- (a) H. Liu, Y. Li, R. Luque and H. Jiang, Adv. Synth. Catal., 2011, 353, 3107–3113 CrossRef CAS; (b) D. Zhang, Y. Guan, E. J. M. Hensen, L. Chen and Y. Wang, Catal. Commun., 2013, 41, 47–51 CrossRef CAS PubMed.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- (a) J.-F. Zhu, G.-H. Tao, H.-Y. Liu, L. He, Q.-H. Sun and H.-C. Liu, Green Chem., 2014, 16, 2664–2669 RSC; (b) A. Chen, G. Zhao, J. Chen, L. Chen and Y. Yu, RSC Adv., 2013, 3, 4171–4175 RSC.
- (a) Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed; (b) Y. Li, X. Xu, P. Zhang, Y. Gong, H. Li and Y. Wang, RSC Adv., 2013, 3, 10973–10982 RSC.
- Z. Li, J. Liu, C. Xia and F. Li, ACS Catal., 2013, 3, 2440–2448 CrossRef CAS.
- J. Chen, W. Zhang, L. Chen, L. Ma, H. Gao and T. Wang, ChemPlusChem, 2013, 78, 142–148 CrossRef CAS.
- A. Chen, Y. Li, J. Chen, G. Zhao, L. Ma and Y. Yu, ChemPlusChem, 2013, 78, 1370–1378 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |