
High three-way catalytic activity of rhodium particles on a Y-stabilized La-containing ZrO2 support: the effect of Y on the enhanced reducibility of rhodium and self-regeneration†
Hisaya
Kawabata
*ab,
Yuki
Koda
a,
Hirosuke
Sumida
a,
Masahiko
Shigetsu
a,
Akihide
Takami
a and
Kei
Inumaru
b
aAdvanced Materials Research Field, Technical Research Center, Mazda Motor Corporation, 3-1 Shinchi, Fuchu-cho, Aki-gun, Hiroshima 730-8670, Japan. E-mail: kawabata.h@mazda.co.jp; Fax: +81 82 252 5342; Tel: +81 82 252 5068
bDepartment of Applied Chemistry, Graduate School of Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashi-Hiroshima 739-8527, Japan. E-mail: inumaru@hiroshima-u.ac.jp; Fax: +81 82 424 5494; Tel: +81 82 424 7741
First published on 22nd September 2014
Abstract
A novel, highly active three-way catalyst, rhodium supported on Y- and La-added zirconia (Rh/Zr–Y–La–O), was found in this study. Rh/Zr–Y–La–O showed superior performance to a previously reported Rh on La-added ZrO2 (Rh/Zr–La–O) catalyst (Kawabata et al., Chem. Commun., 2013, 49(38), 4015; Catal. Sci. Technol. 2014, 4(3), 697).21,22 The effects of Y addition to ZrO2-based supports were investigated in detail. CO temperature-programmed reduction and in situ Fourier transform infrared spectra of adsorbed NO species indicated that Y addition to La-containing ZrO2 enhanced the reducibility of rhodium supported on the catalyst and that more metallic Rh was exposed on the surface after the oxidation of Rh/Zr–Y–La–O than of Rh/Zr–La–O. Before and after an aging treatment at 1273 K that simulated 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 km travelled by vehicles, Rh/Zr–Y–La–O showed high steam reforming activity. After the aging treatment, Rh/Zr–Y–La–O was deactivated using an oxidation treatment, but its three-way catalysis activity was completely regenerated after a short (5 min) exposure to steam reforming reaction conditions, demonstrating self-regeneration capability. After the aging treatment, Rh/Zr–Y–La–O showed higher rhodium dispersion than other catalysts. This was attributed to the high surface area of the support after aging and the stabilization of ZrO2 by the addition of Y. Our findings highlight the role of catalyst supports in designing effective three-way catalysts with high tolerance to the oxidative conditions in new vehicles and engines.
000 km travelled by vehicles, Rh/Zr–Y–La–O showed high steam reforming activity. After the aging treatment, Rh/Zr–Y–La–O was deactivated using an oxidation treatment, but its three-way catalysis activity was completely regenerated after a short (5 min) exposure to steam reforming reaction conditions, demonstrating self-regeneration capability. After the aging treatment, Rh/Zr–Y–La–O showed higher rhodium dispersion than other catalysts. This was attributed to the high surface area of the support after aging and the stabilization of ZrO2 by the addition of Y. Our findings highlight the role of catalyst supports in designing effective three-way catalysts with high tolerance to the oxidative conditions in new vehicles and engines.
1. Introduction
Three-way catalysis for automotive exhaust purification is an important process that has been extensively studied for several decades.1–11 Since emission regulations are constantly becoming more stringent worldwide,12,13 there has been consistent focus on improving three-way catalytic activity and durability. The conditions to which a catalyst is subjected in automotive exhaust are severe: the temperatures and gas phase compositions quickly change over wide ranges. The most influential condition impeding catalytic activity is the atmospheric changes in oxidant and reductant concentrations. Recently, a new type of engine operation has been commercialized to minimize fuel consumption; the engine automatically switches off when the car stops for a short time. This new operation causes highly transient and fluctuating conditions, including high oxygen concentrations that deactivate three-way catalysts. Tolerance to oxidative conditions is crucial for designing a highly efficient three-way catalyst for practical applications.2Rhodium is a key catalytic component in three-way catalysts for the effective conversion of CO, hydrocarbons and NOx to H2O, CO2 and N2.10,14 However, rhodium has a number of drawbacks under oxidative conditions.3,4 In the case of alumina supports, rhodium readily reacts with the support to form different phases at high temperatures. Rhodium is known to form oxides with various oxidation states, structures and morphologies (rafts or plate-like structures) on alumina supports.15,16 Changes in size, shape and phase induced by rhodium interactions with alumina supports significantly affect the activity of the catalysts.17,18 Bond formation between Rh and the support is a possible way to suppress the sintering behaviour of rhodium.19 Zirconia has weaker interactions with rhodium than alumina, and zirconia-modified alumina has been reported to be useful for controlling the activity of rhodium.20
Previously, we have demonstrated the high catalytic activity of rhodium supported on La-containing ZrO2 (Rh/Zr–La–O) for the elimination of NOx, CO and hydrocarbons through a three-way catalytic process from a synthetic automotive exhaust gas.21,22 The catalyst showed better performance than other catalysts, especially after oxidative pre-treatment, although the catalyst did not contain oxygen-storage components such as CeO2. The Rh/Zr–La–O catalyst showed high activity in the steam reforming reaction, even after an aging treatment that mimicked a mileage of 80000 km for real vehicles. Exposure to steam reforming reaction conditions reduced the oxidized Rh on the Zr–La–O support, refreshing the deactivated catalyst. The state of rhodium was not only affected by interactions between the metal particles and the support, but also by the steam reforming reaction occurring over the catalyst. After the aging treatment, the Rh/Zr–La–O catalyst exhibited rhodium sintering and phase separation of the support, indicating that further work is required to develop more efficient catalysts.
In this study, we prepared a novel, highly active three-way catalyst, Rh supported on yttrium- and lanthanum-added zirconia (Rh/Zr–Y–La–O). We also investigated the effect of yttrium addition to ZrO2-based supports in detail. Yttrium is a typical stabilizing element for zirconia.23 The redox properties and dispersion of rhodium and the crystal structure of the support were significantly affected by the addition of Y. The importance of the steam reforming reaction was also demonstrated for the yttrium-added catalyst Rh/Zr–Y–La–O. These investigations aimed to identify the reason for the excellent three-way catalytic activity of Rh/Zr–Y–La–O.
2. Experimental
2.1. Catalyst preparation
The yttrium-stabilized ZrO2 was prepared by an ammonia-assisted co-precipitation method where zirconium nitrate ZrO(NO3)2·6H2O (60.8 g, >98%, Nacalai Tesque) and yttrium nitrate Y(NO3)3·6H2O (8.7 g) were dissolved in 200 g of water and an aqueous NH4OH solution (1%, Wako Pure Chemical Industries, Ltd., Osaka, Japan) was added to raise the pH (≥12). The precipitate was filtered, dried at 423 K for 2 h in air, and then calcined at 773 K for 2 h in air to produce the ZrO2–Y2O3 mixed oxide. The ZrO2–La2O3 and Y-stabilized ZrO2–La2O3 mixed oxides were also prepared by the ammonia-assisted co-precipitation method from Y(NO3)3·6H2O and La(NO3)3·6H2O (Rare Metallic Co., Ltd., Tokyo, Japan). For the preparation of Y-stabilized ZrO2–La2O3, lanthanum nitrate, yttrium nitrate and zirconium nitrate were dissolved in water before adding an aqueous NH4OH solution. The concentrations of Y and La were 10% and 5 wt% as oxides, respectively. The Y, La and Y–La mixed oxides were designated as Zr–Y–O, Zr–La–O and Zr–Y–La–O, respectively. The ZrO2 support (Brunauer–Emmett–Teller (BET) surface area: 52.3 m2 g−1) was purchased from Kishida Chemical Co., Ltd. (Osaka, Japan). Rhodium (0.33 wt% as rhodium metal) was loaded onto the oxide support by impregnation using an aqueous solution of Rh(NO3)3·H2O (Daiichi Kigenso Kogyo, Osaka, Japan). The obtained Rh-loaded oxide supports were dried at 393 K for 12 h in air and subsequently calcined in air at 773 K. For the evaluation of the three-way catalytic reaction, the Rh-loaded ZrO2 mixed oxide catalysts were deposited on a cordierite honeycomb. To do this, the Rh-loaded powder catalysts were mixed with distilled water and colloidal zirconia (ZSL-10D, Daiichi Kigenso Kogyo, Osaka, Japan) was used to bind the catalyst powder to the cordierite honeycomb. The slurry-coated cordierite honeycomb was dried at 473 K for 2 h in air and then calcined at 773 K for 2 h in air. The amount of catalyst coated on the cordierite honeycomb was 100 g dm−3. The resulting products were termed “fresh” catalysts.2.2. Three-way catalytic reactions
The activity of the Rh-based catalysts was evaluated using a fixed-bed continuous flow reactor. The reaction gas was a mixture of 500 ppm C3H6, 1000 ppm NO, 0.7% CO, 0.2% H2, 0.6% O2, 13.6% CO2 and 10% H2O. Nitrogen was used as a diluent. The gas composition mimicked the actual exhaust gas emitted at ≈3000 rpm engine operation. The flow rate of the reaction gas was 26 dm3 min−1, corresponding to a gas hourly space velocity of 60000 h−1. Prior to catalytic performance measurements, the catalysts were pre-treated under a flow of the gas mixture at 773 K for 10 min and then cooled to 373 K under a flow of nitrogen. The three-way catalytic activity was evaluated over a range of temperatures from 373–773 K (at a heating rate of 30 K min−1). The products were monitored using a flame ionization detector for hydrocarbons, infrared absorption for CO, and chemical luminescence for NO using a Horiba MEXA-9100 system (Kyoto, Japan).
2.3. Aging procedure
Accelerated durability tests, or aging treatments, were conducted by treating the fresh catalysts at 1273 K for 24 h under a 2% O2 and 10% H2O atmosphere (diluent: N2). This aging treatment simulated empirically a mileage of 80000 km for real vehicles.
2.4. Steam reforming reaction
The reaction gas was a mixture of 660 ppm C3H6 and 2% H2O (molar ratio of H2O/C = 10). Nitrogen was used as a diluent. The flow rate of the steam reforming reaction gas was 26 dm3 min−1, corresponding to a gas hourly space velocity of 60000 h−1. The activity of the steam reforming reaction was measured using a fixed-bed continuous flow reactor, the same as that for three-way catalytic activity measurements. Prior to the activity evaluations, the catalyst was pre-treated under a three-way catalytic reaction gas at 773 K for 10 min and then cooled to 373 K under a flow of nitrogen.
2.5. Characterization
For each of the characterization techniques described in sections 2.5.1–2.5.5, the catalysts were used in powder form without the cordierite honeycomb.| 3CO + Rh2O3 → 3CO2 + 2Rh | (1) |
:1 CO:surface Rh stoichiometry.30 The rhodium particle size was estimated using the dispersion value from eqn (2),31| L = f × M/(ρ × NA × π × r2 × D) | (2) |
3. Results
3.1. Crystal phases of supports
Structural alterations caused by the introduction of Y and La to ZrO2 were studied by XRD, as shown in Fig. 1. For fresh Rh/ZrO2, only the monoclinic ZrO2 phase was detected (Fig. 1a). The addition of either Y or La to ZrO2 stabilized the high-temperature ZrO2 phases; the cubic ZrO2 phase was formed for Rh/Zr–Y–O (Fig. 1c), and the tetragonal ZrO2 phase was formed for Rh/Zr–La–O (Fig. 1e). When both Y and La were added to ZrO2, the structure became cubic ZrO2. Regardless of the presence of La, addition of Y to zirconia caused the formation of the cubic phase (Fig. 1c and g). No diffraction peak of Rh was detectable for the fresh catalysts.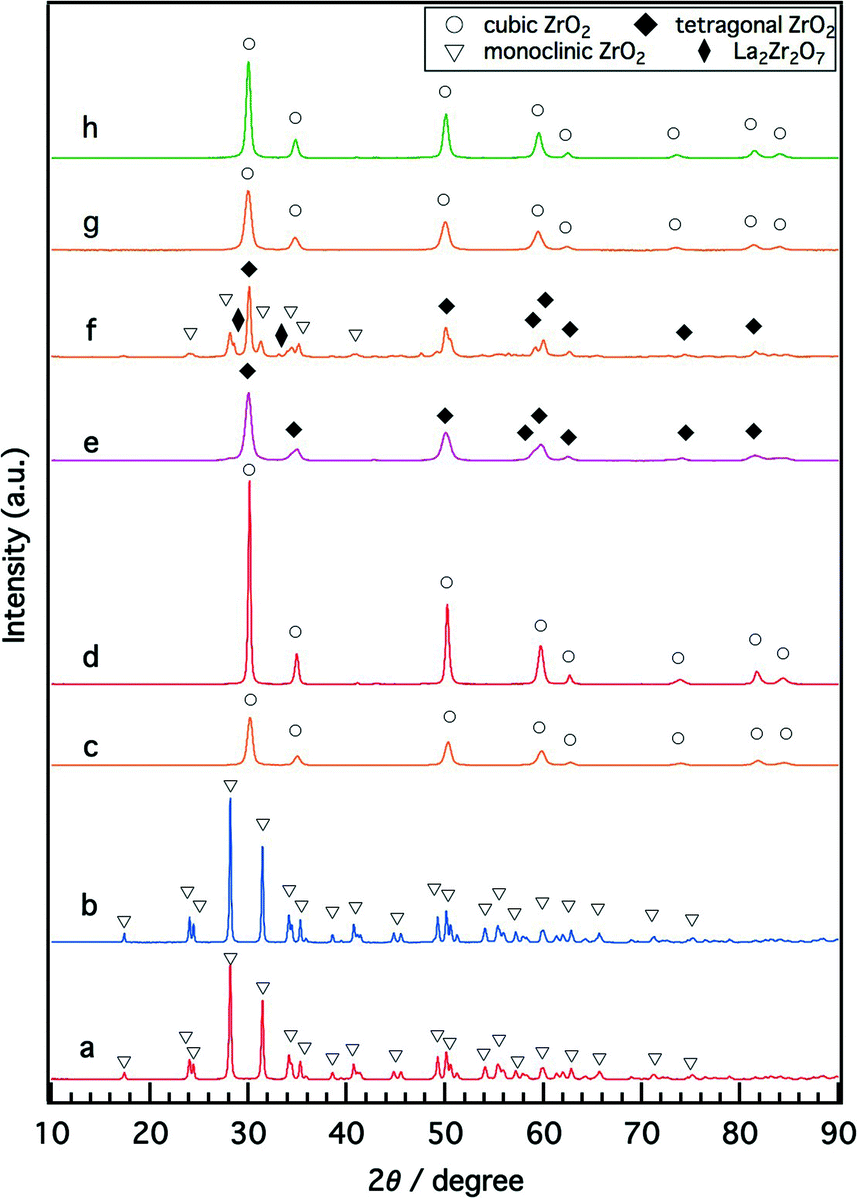 | ||
| Fig. 1 XRD patterns of (a) fresh Rh/ZrO2, (b) aged Rh/ZrO2, (c) fresh Rh/Zr–Y–O, (d) aged Rh/Zr–Y–O, (e) fresh Rh/Zr–La–O, (f) aged Rh/Zr–La–O, (g) fresh Rh/Zr–Y–La–O and (h) aged Rh/Zr–Y–La–O. Aging was conducted at 1273 K under a 2% O2 and 10% H2O/N2 atmosphere for 24 h. | ||
After the aging treatment at 1273 K, the structure of Rh/ZrO2 did not change from monoclinic ZrO2 (Fig. 1b). For Rh/Zr–La–O after the aging treatment, the tetragonal ZrO2 transformed into three phases. One was tetragonal ZrO2, which was the major phase, and the others were monoclinic ZrO2 and La2Zr2O7, which were the minor phases (Fig. 1f). La2Zr2O7 had a pyrochlore structure with a 1:1 molar ratio of La and Zr. In contrast, as shown in Fig. 1d and h, the cubic ZrO2 phase was maintained, even after the aging treatment, for Rh/Zr–Y–O and Rh/Zr–Y–La–O. The introduction of Y to ZrO2 resulted in the formation of the cubic ZrO2 phase for both the fresh and the aged catalysts. Rhodium diffraction peaks of the aged catalysts were very weak, but the Rh 111 peak was detectable for some aged catalysts (Fig. S1 in the ESI†).
3.2. Effects of oxidation treatment on the performance of the three-way rhodium-based catalysts
Because of recently developed engine operations, catalytic activity after exposure to oxidative conditions is an important criterion for applications in real vehicles. We have previously reported that Rh/Zr–La–O showed higher three-way catalytic activity compared with Rh/ZrO2 and rhodium supported on other lanthanide (Ce, Pr or Nd)-added ZrO2.21,22 In this study, the catalytic activities were evaluated for Y-stabilized ZrO2 and for Y-stabilized Zr–La–O using two different pre-treatments. The first was a treatment under the reaction gas (simulated automotive exhaust) and the second was under oxidative conditions. For the fresh catalysts, their activities after treatment under the reaction gas and after the oxidation pre-treatment are shown in Fig. 2 and 3, respectively. Fig. 4 and 5 present the activities of the aged catalysts after the two different pre-treatments. In each figure, the top panel shows hydrocarbon conversion and the bottom panel shows NOx conversion. Corresponding CO conversions are also provided in the ESI† Fig. S2 to S5.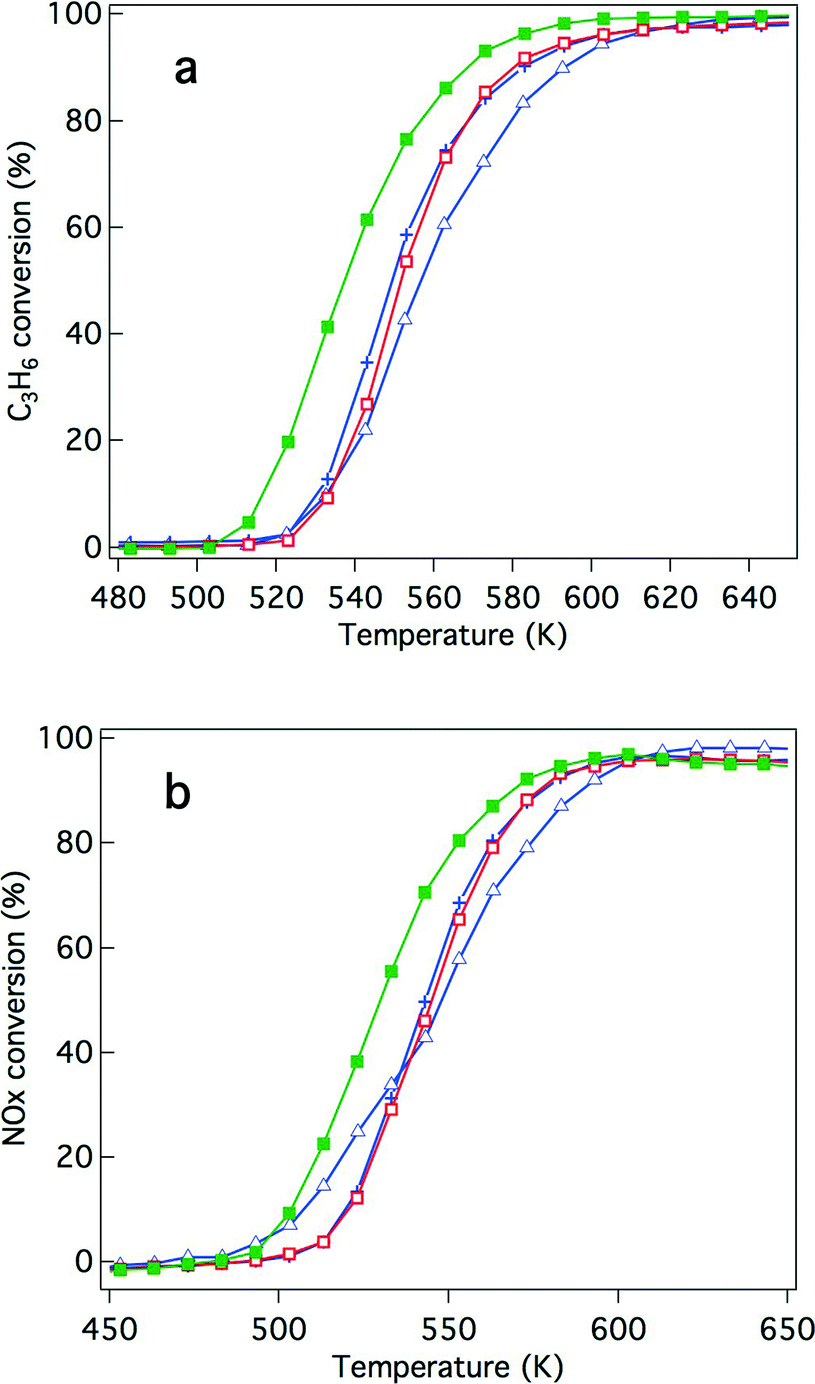 | ||
| Fig. 2 Conversion of the fresh three-way catalysts that underwent the reaction gas pre-treatment for (a) C3H6 conversion and (b) NO conversion. The catalysts studied were (+) Rh/ZrO2, (△) Rh/Zr–Y–O, (□) Rh/Zr–La–O, and (■) Rh/Zr–Y–La–O. | ||
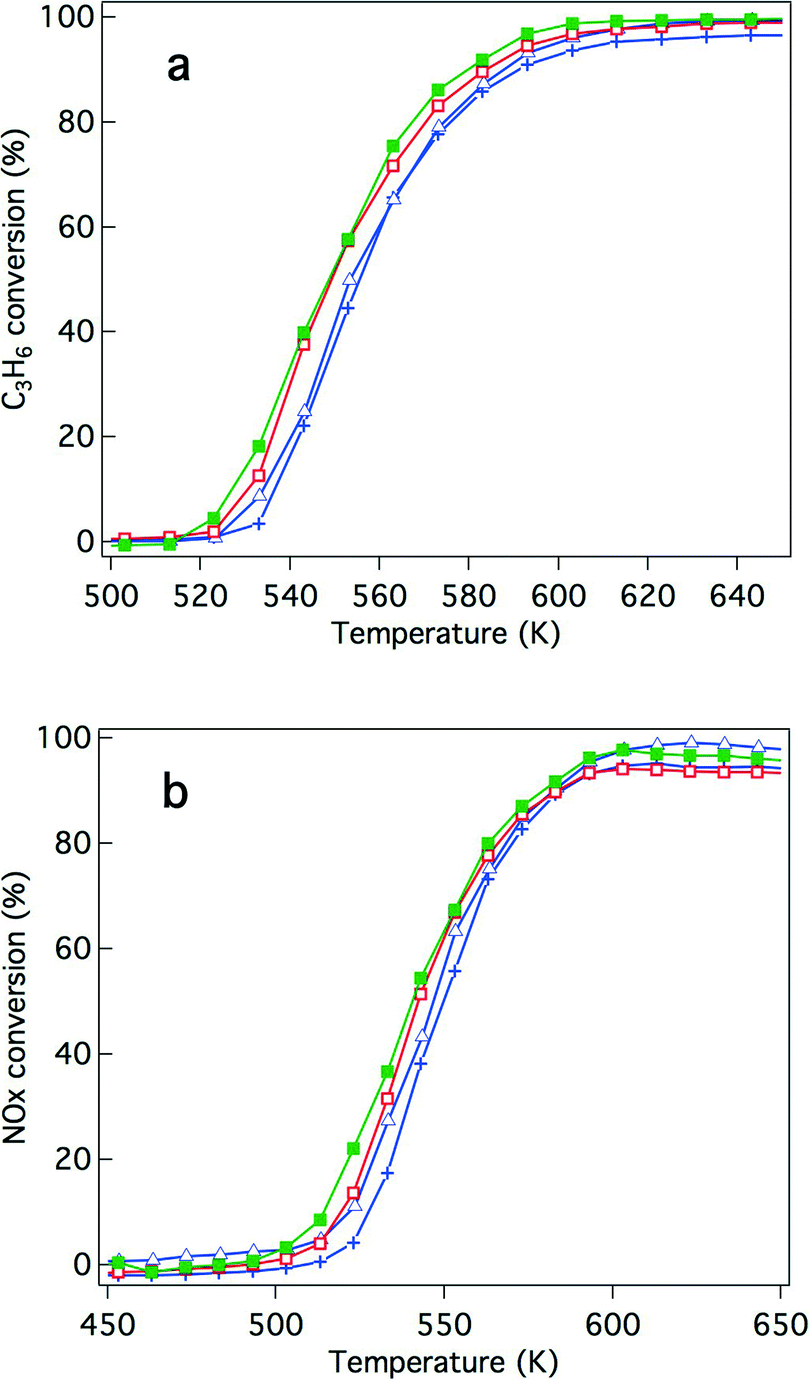 | ||
| Fig. 3 Conversion of the fresh three-way catalysts that underwent the oxidation treatment for (a) C3H6 conversion and (b) NO conversion. The catalysts studied were (+) Rh/ZrO2, (△) Rh/Zr–Y–O, (□) Rh/Zr–La–O, and (■) Rh/Zr–Y–La–O. | ||
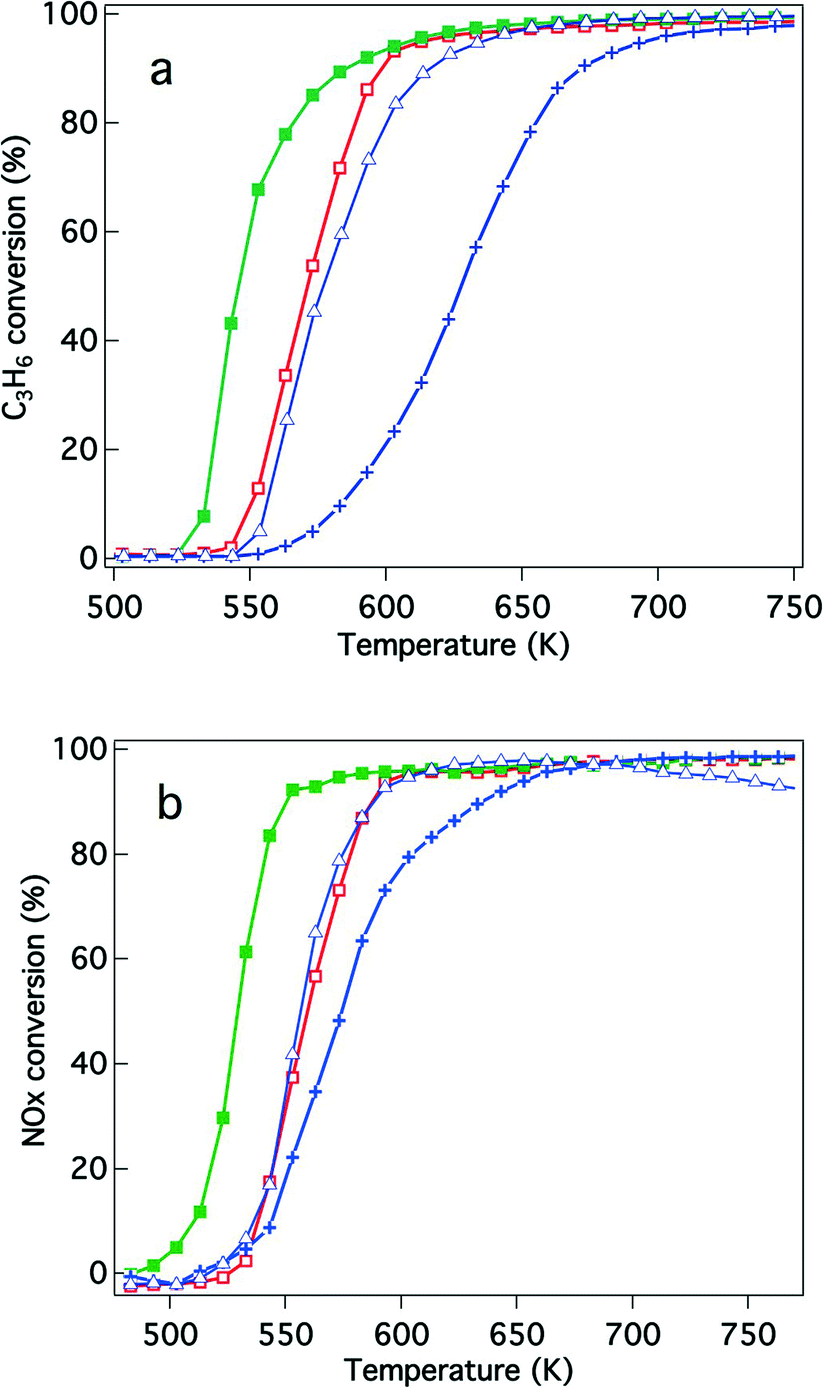 | ||
| Fig. 4 Conversion of the aged three-way catalysts that underwent the reaction gas pre-treatment for (a) C3H6 conversion and (b) NO conversion. The catalysts studied were (+) Rh/ZrO2, (△) Rh/Zr–Y–O, (□) Rh/Zr–La–O, and (■) Rh/Zr–Y–La–O. | ||
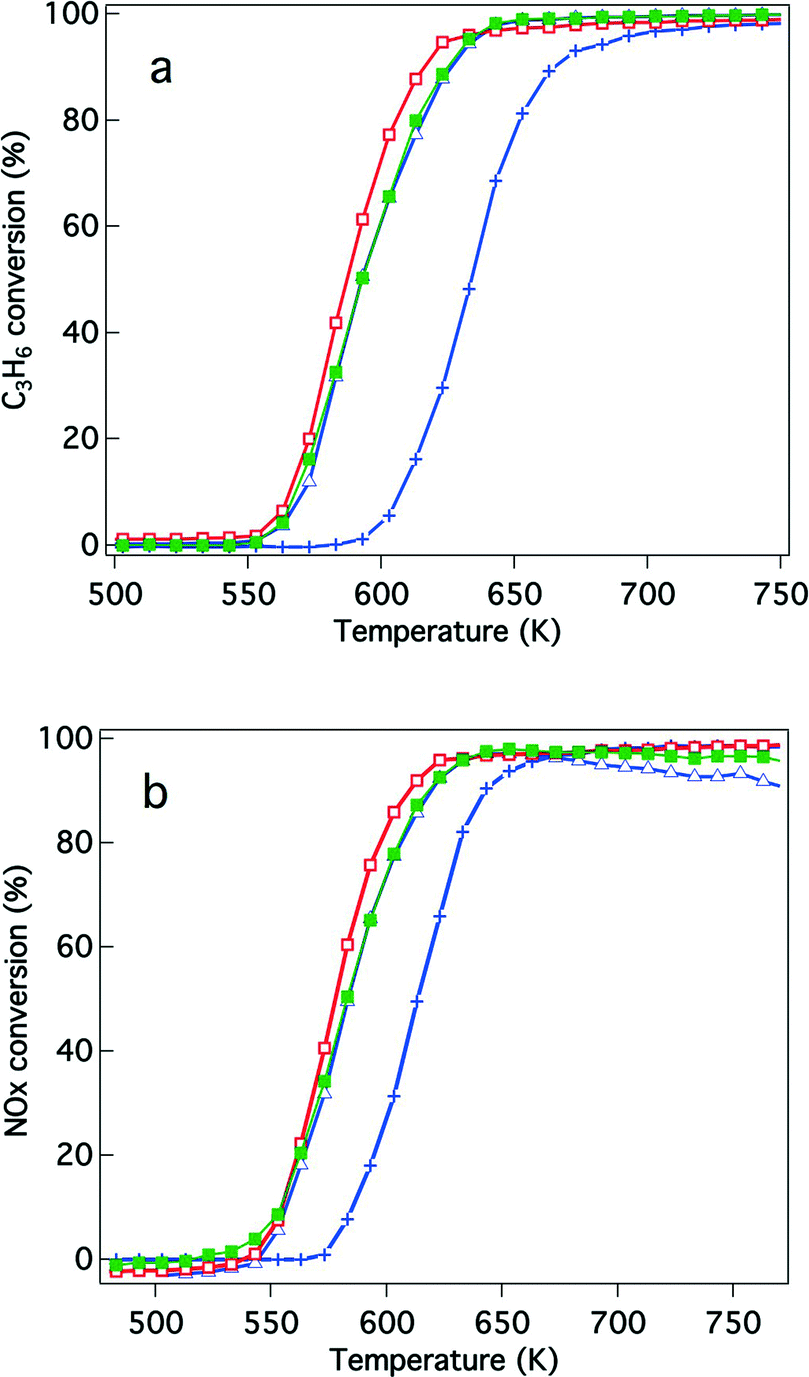 | ||
| Fig. 5 Conversion of the aged three-way catalysts that underwent the oxidation treatment for (a) C3H6 conversion and (b) NO conversion. The catalysts studied were (+) Rh/ZrO2, (△) Rh/Zr–Y–O, (□) Rh/Zr–La–O, and (■) Rh/Zr–Y–La–O. | ||
First, the effects of Y addition on the activity of the fresh catalysts were studied. After the reaction gas pre-treatment, the activity of Rh/Zr–Y–O decreased compared with that of Rh/ZrO2 for both hydrocarbon conversion (Fig. 2a) and NOx conversion (Fig. 2b). For Y addition to Rh/Zr–La–O, hydrocarbon and NOx conversions were both enhanced, as shown in Fig. 2a and b, respectively. This indicates that Rh/Zr–Y–La–O had higher catalytic activity than the previously reported Rh/Zr–La–O catalyst.
The effects of Y addition on the activities of the fresh catalysts that underwent the oxidative pre-treatment were also investigated. Fig. 3 presents the results of the activity tests. After the oxidative pre-treatment, Rh/Zr–Y–O showed almost the same hydrocarbon conversion activity as Rh/ZrO2 (Fig. 3a) and the activity of Rh/Zr–Y–La–O was comparable to that of Rh/Zr–La–O. For NOx conversion, Y addition slightly enhanced the activity of Rh/Zr–Y–La–O and Rh/Zr–Y–O compared with that of the catalyst without Y (Fig. 3b). Rh/Zr–Y–La–O had the best catalytic activity after the oxidative pre-treatment.
NO conversion started at 500 K, which was a lower temperature compared to that for the C3H6 conversion (520 K). The conversion of CO started at almost the same temperature as NO conversion (Fig. S2 to S5 in the ESI†). NO must have reacted with CO and/or H2 in the reaction gas before C3H6 started to convert.
The effects of Y addition on the activities of the aged catalysts were also studied, as shown in Fig. 4. After the reaction gas pre-treatment, Rh/Zr–Y–O had higher hydrocarbon and NOx conversions than Rh/ZrO2. Also, Rh/Zr–Y–La–O had higher hydrocarbon and NOx conversions than Rh/Zr–La–O (Fig. 4a and b). Among the tested catalysts, Rh/Zr–Y–La–O had the highest activity for hydrocarbon and NOx conversions. After the oxidative pre-treatment of the aged catalysts (Fig. 5), the activity of Rh/Zr–Y–La–O was comparable to, or slightly lower than, that of Rh/Zr–La–O. Rh/Zr–Y–O also had activities similar to Rh/Zr–Y–La–O. Rh/ZrO2 had much lower activity than the other catalysts under these conditions.
We measured the inlet and outlet temperatures of the catalyst bed at the working state of three-way catalysis by using thermocouples. When the conversion of C3H6 and NO started in a three-way catalytic reaction at around 520 K, the outlet temperature showed higher values than the inlet temperature (see Fig. S6 in the ESI†), indicating the occurrence of an exothermic reaction (e.g. hydrocarbon oxidation). When the temperature increased up to 673 K, however, the outlet temperature became lower than the inlet temperature. This phenomenon demonstrated that the endothermic reaction such as the steam reforming reaction was taking place during the three-way catalysis.
3.3. Rhodium particle sizes and dispersion on different supports
Fig. 6 presents the TEM images of the catalysts before and after the aging treatment. Fig. 6a–d are images of fresh catalysts Rh/ZrO2, Rh/Zr–Y–O, Rh/Zr–La–O and Rh/Zr–Y–La–O after the three-way catalytic run at 773 K, respectively. Fig. 6e–h are images of the aged catalysts presented in the same order as in Fig. 6a–d. The particle size range of the support was 10–20 nm for Zr–Y–O (Fig. 6b), while for ZrO2 the particle size range was 50–70 nm (Fig. 6a). Particle sizes of the Zr–La–O and Zr–Y–La–O supports both ranged from 10–20 nm (Fig. 6c and d). For the fresh catalysts, the oxide supports formed smaller particles when La or Y was added. After the aging treatment, the particle size ranges for ZrO2 and Zr–Y–O were 50–100 nm, indicating that the support particles of the fresh catalysts sintered to form larger particles during the aging treatment (Fig. 6e and f). Conversely, the particle size ranges for Zr–La–O and Zr–Y–La–O were 10–20 nm, even after the aging treatment, implying that the La-containing supports do not readily sinter. The surface areas of these catalysts were in agreement with their particle sizes (Table 1). The BET surface areas of Rh/ZrO2 and Rh/Zr–Y–O, in which support particle growth was observed, were 16.4 and 21.4 m2 g−1, respectively. Rh/Zr–La–O and Rh/Zr–Y–La–O, which had smaller particle sizes, had higher surface areas of 29.6 and 51.2 m2 g−1, respectively. The largest surface area was measured for Rh/Zr–Y–La–O, and the decrease in the surface area of this catalyst due to the aging treatment was small (from 75.3 m2 g−1 to 51.2 m2 g−1) compared with those in the other catalysts.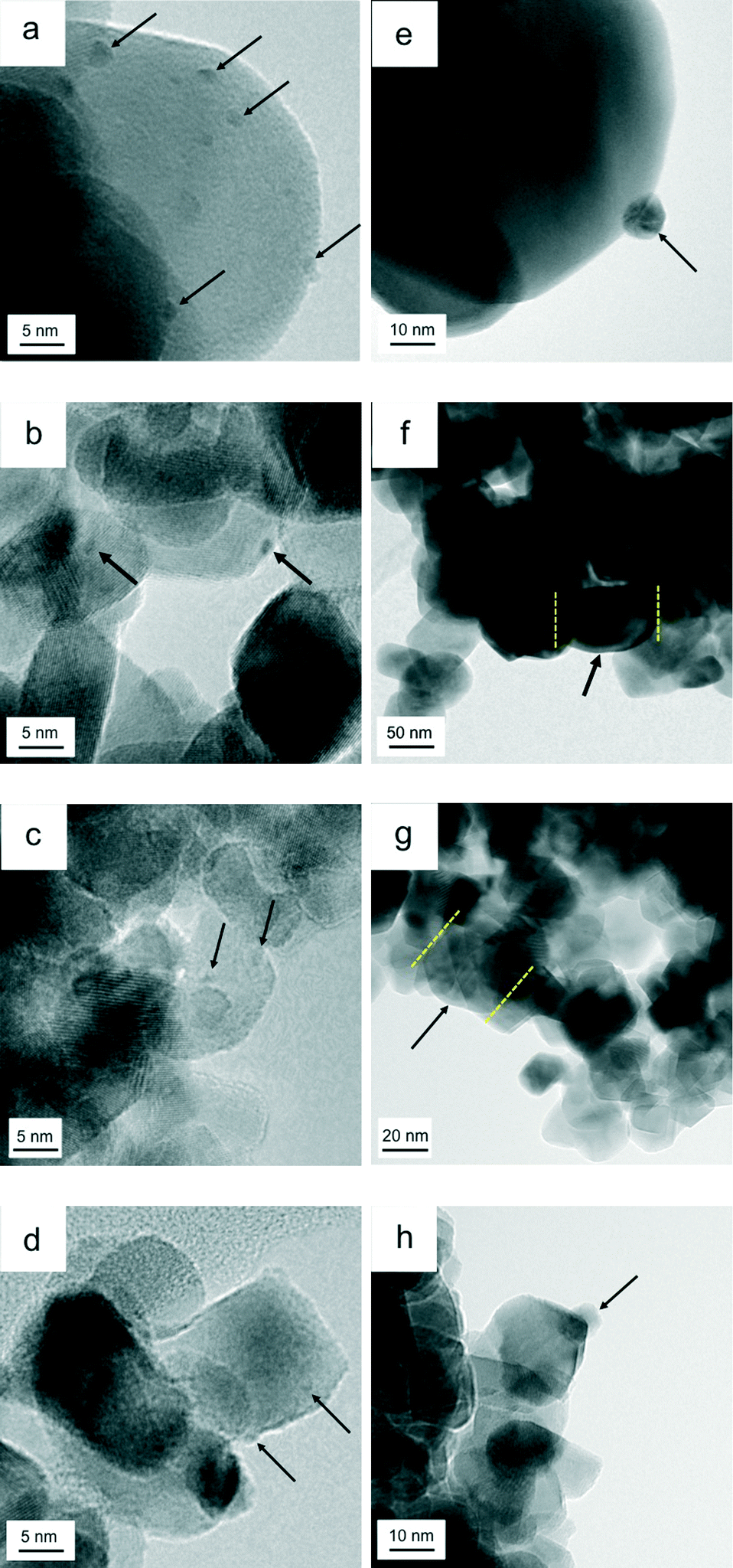 | ||
| Fig. 6 TEM images of (a) fresh Rh/ZrO2, (b) fresh Rh/Zr–Y–O, (c) fresh Rh/Zr–La–O, (d) fresh Rh/Zr–Y–La–O, (e) aged Rh/ZrO2, (f) aged Rh/Zr–Y–O, (g) aged Rh/Zr–La–O, and (h) aged Rh/Zr–Y–La–O. Aging was conducted at 1273 K under 2% O2 and 10% H2O/N2 for 24 h. | ||
The effect of Y addition on the rhodium particle size is even more important. The rhodium particle size was approximately 1–2 nm for the fresh catalysts prepared in this study, as shown in Fig. 6a–d (the arrows indicate Rh particles). Table 1 lists the dispersion of Rh estimated by CO adsorption at 300 K for both the fresh and the aged catalysts. Rhodium dispersion in the fresh catalysts was greater than 50% and Rh particle sizes estimated from the dispersion were approximately 2 nm, which was consistent with the TEM observations. Rhodium particle sizes did not appear to be affected by the addition of Y. The presence of rhodium was confirmed by energy-dispersive X-ray (EDX) analysis of all of the fresh and aged catalysts. For aged Rh/ZrO2, spherical rhodium particles with diameters of approximately 10 nm were observed (Fig. 6e). Aged Rh/Zr–Y–O contained larger rhodium particles (diameter = 70 nm), as shown in Fig. 6f (EDX data are given in Fig. S7 in the ESI†). For aged Rh/Zr–La–O, large rhodium particles (30–100 nm, most likely thin, plate-like particles based on their pale contrasts) were observed (Fig. 6g, EDX data are given in Fig. S8 in the ESI†). No nanosized rhodium particles were detected in Rh/Zr–Y–O and Rh/Zr–La–O. In the TEM image of Rh/Zr–Y–La–O (Fig. 6h), a number of small rhodium particles (approximately 5 nm in size) were observed, while the particle size estimated by Scherrer's equation using the Rh 111 diffraction peak (Fig. S1 in the ESI†) was approximately 80 nm, suggesting that larger particles were present in areas outside that covered in the TEM analysis. Although Rh dispersion for all of the aged catalysts was quite low (below 10%), aged Rh/Zr–Y–La–O exhibited the highest dispersion among the aged catalysts tested in this study. Rhodium particle sizes were calculated from the dispersion, assuming that the particles were spherical. The values were 22.6, 27.7, 32.6 and 15.8 nm for Rh/ZrO2, Rh/Zr–Y–O, Rh/Zr–La–O and Rh/Zr–Y–La–O, respectively. The smaller mean rhodium size of Rh/Zr–Y–La–O was consistent with the TEM observations.
3.4. Temperature-programmed reduction by CO (CO-TPR)
Temperature-programmed reduction profiles of the fresh catalysts are presented in Fig. 7. The O/Rh atomic ratios calculated from the TPR data are shown in Table 2. A higher O/Rh ratio (1.48) was measured for fresh Rh/Zr–Y–O than for Rh/ZrO2 (0.14). In our previous study, the oxidation state of rhodium on ZrO2 analyzed by X-ray photoelectron spectroscopy (XPS) was trivalent after being oxidized by 5% O2. The difference between the low O/Rh ratio for Rh/ZrO2 and the presence of trivalent rhodium from XPS results indicated that only the surface of rhodium particles was oxidized to Rh3+.22 Conversely, the O/Rh ratio was 1.48 for Rh/Zr–Y–O, indicating that all of the rhodium particles were almost fully oxidized. Similarly, the amount of CO2 generated during the TPR process increased by the addition of Y to Rh/Zr–La–O (Fig. 7c and d). A larger O/Rh ratio of 1.20 was measured for Rh/Zr–Y–La–O, while Rh/Zr–La–O had a lower O/Rh ratio of 0.70. The TPR profile also changed upon Y addition to Rh/Zr–La–O. It should be noted that Rh/Zr–Y–La–O was more readily reduced at low temperature than Rh/Zr–La–O (Fig. 7c and d). These results indicate, for the fresh catalysts, that Y addition caused Rh to be more readily oxidized and the reducibility of Rh was also enhanced by the addition of Y to the La-containing catalyst. Fig. 8 presents the CO-TPR profiles of the aged catalysts. A sharp peak was observed at 520 K for Rh/ZrO2 and Rh/Zr–Y–O, as shown in Fig. 8a and b, respectively. Conversely, the aged Rh/Zr–La–O and Rh/Zr–Y–La–O catalysts exhibited a broad reduction profile starting at around 500 K (Fig. 8c and d). No significant effect of Y addition on the reduction profiles was detected for the aged catalysts.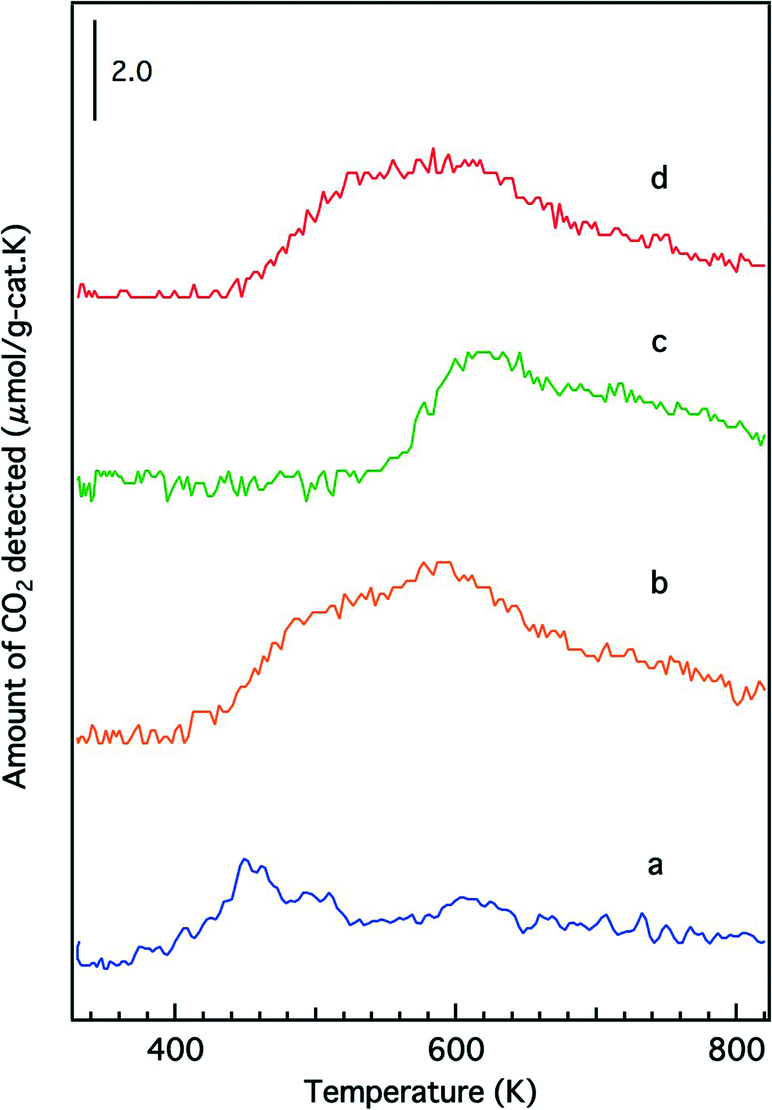 | ||
| Fig. 7 CO-TPR profiles of the fresh catalysts: (a) Rh/ZrO2, (b) Rh/Zr–Y–O, (c) Rh/Zr–La–O and (d) Rh/Zr–Y–La–O. | ||
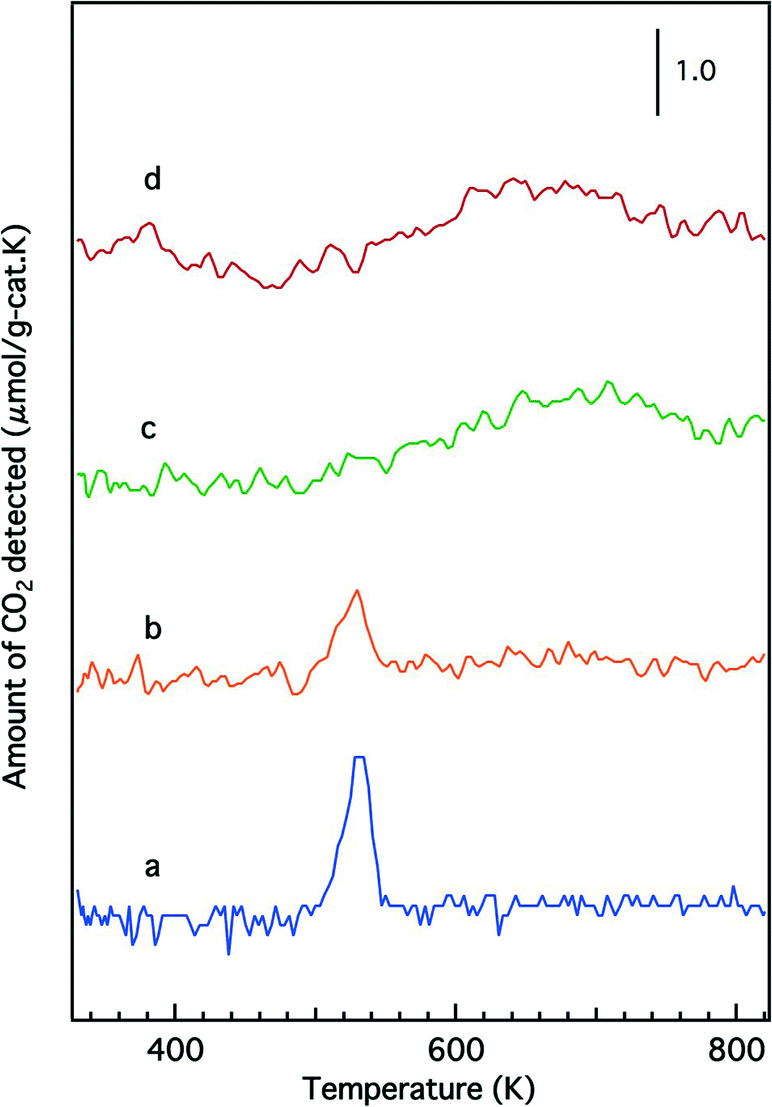 | ||
| Fig. 8 CO-TPR profiles of the aged catalysts: (a) Rh/ZrO2, (b) Rh/Zr–Y–O, (c) Rh/Zr–La–O and (d) Rh/Zr–Y–La–O. | ||
3.5. Infrared spectra
Although XPS is a powerful tool for investigating the oxidation state of rhodium, the Y 3p3/2 signal at 311 eV overlaps with the Rh 3d5/2 signal at 307 eV. Therefore, in the case of Y-containing materials, information about rhodium was not accessible, especially when the loading of rhodium was as low as 0.33 wt%, as was the case in this study. For this reason, we employed NO molecules as probes for the Rh metallic state because it is widely accepted that NO readily forms Rh0–NO adsorbates on rhodium particles.24,25Fig. 9 presents the IR absorption spectra of the Rh/Zr–La–O and Rh/Zr–Y–La–O catalysts after exposure to NO at various temperatures. Several bands associated with NOx adsorbates were clearly detected for the La-containing catalysts. Bands at 1894, 1857, 1533, 1242 and 1185 cm−1 were observed for Rh/Zr–La–O (Fig. 9a). The band at 1857 cm−1 was assigned to Rh0–NOδ+,24 and the bands from 1533 to 1185 cm−1 were attributed to nitrate species.26 The band at 1894 cm−1 was assigned to RhI–NO+.27 In the case of Rh/Zr–Y–La–O, the intensity of the Rh0–NO band at 1848 cm−1 was higher than that for Rh/Zr–La–O at temperatures from 473 to 573 K (magnified bands in Fig. 9). These results indicate that the amount of Rh0 on the surface was higher for Rh/Zr–Y–La–O than for Rh/Zr–La–O.
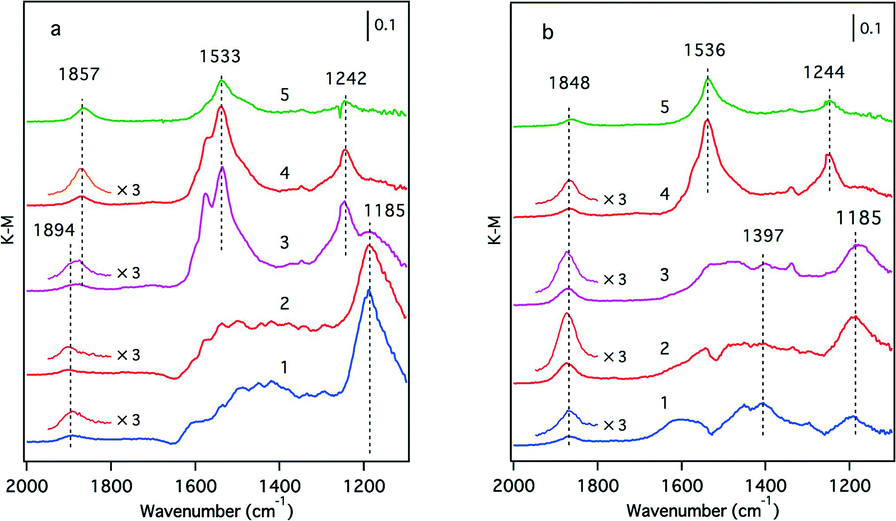 | ||
| Fig. 9 IR spectra of NO absorbed on fresh (a) Rh/Zr–La–O and (b) Rh/Zr–Y–La–O. Spectra were taken at (1) 373 K, (2) 473 K, (3) 573 K, (4) 673 K and (5) 773 K. | ||
3.6. Steam reforming reaction activity and its effect on three-way catalysis
The steam reforming reaction is influential in three-way catalysis for enhancing NOx conversion by producing hydrogen as a reductant. This reaction is known to proceed over Rh catalysts.28,29 Previously, we reported that exposure to the steam reforming reaction reduced the oxidized rhodium and recovered the activity of the catalyst.22 In this study, the effect of Y addition on this reaction was examined. Fig. 10 presents C3H6 conversion in the steam reforming reaction over the fresh catalysts. The addition of Y to ZrO2 resulted in a dramatic decrease in the activity of this reaction. Yttrium addition to La-containing ZrO2 decreased the activity by a small amount, but the catalyst still maintained high activity. Fig. 11 presents C3H6 conversion in the steam reforming reaction over the aged catalysts. Both Rh/Zr–La–O and Rh/Zr–Y–La–O exhibited superior activity to Rh/ZrO2 and Rh/Zr–Y–O. It is evident that when Y was added to the support, the presence of La was highly effective in maintaining the catalyst's high steam reforming reaction activity for both fresh and aged catalysts.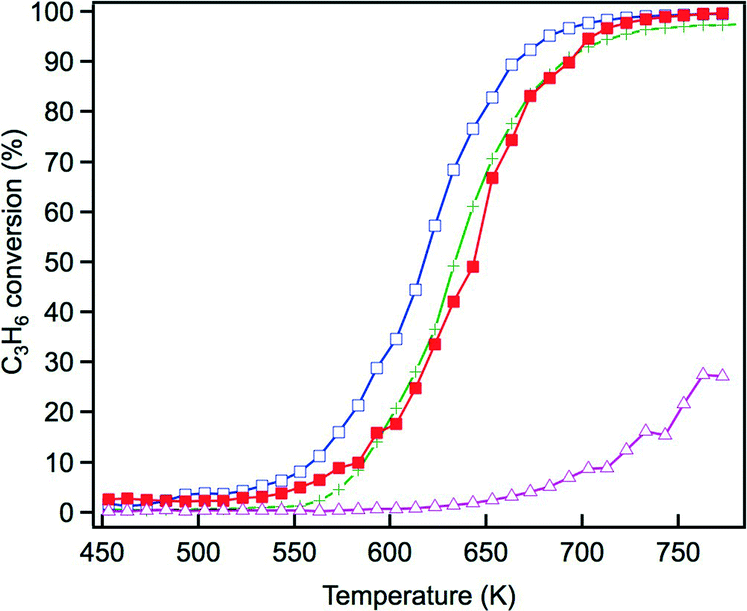 | ||
| Fig. 10 C3H6 conversion in the steam reforming reaction over the fresh Rh catalysts: (+) Rh/ZrO2, (△) Rh/Zr–Y–O, (□) Rh/Zr–La–O and (■) Rh/Zr–La–Y–O. | ||
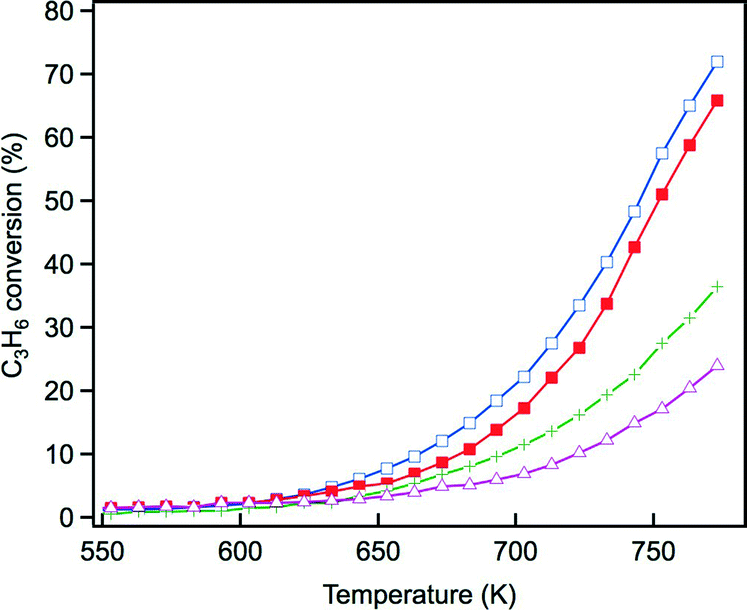 | ||
| Fig. 11 C3H6 conversion in the steam reforming reaction over the aged Rh catalysts: (+) Rh/ZrO2, (△) Rh/Zr–Y–O, (□) Rh/Zr–La–O and (■) Rh/Zr–La–Y–O. | ||
The influence of the steam reforming reaction on the activity of the three-way catalysts Rh/Zr–Y–O and Rh/Zr–Y–La–O was investigated to identify the impact of Y addition on the reaction. Fig. 12a and b present the results for the aged Rh/Zr–Y–O and Rh/Zr–Y–La–O, respectively. First, the three-way catalytic activity of the aged catalysts was examined without any pre-treatment (curve 1 in each figure). The next experiments examined the three-way catalytic activity of the aged catalysts after pre-treatment under 5% O2 flow at 773 K for 5 min (curves 2 in each figure). The final experiments investigated the three-way catalytic activity of the aged catalysts that were oxidized under 5% O2 flow at 773 K for 5 min followed by steam reforming reaction at 520 K for 5 min (curves 3 in each figure). The oxidative pre-treatment caused dramatic decreases in the three-way catalytic activities of the two tested catalysts. In the case of Rh/Zr–Y–O, which had low steam reforming activity, recovery of the three-way catalytic activity after the steam reforming reaction was limited (Fig. 12a, curve 3). Conversely, Rh/Zr–Y–La–O showed a complete recovery (Fig. 12b, curve 3) after the steam reforming reaction from the low activity after the oxidation treatment (Fig. 12b, curve 2). Besides hydrogen and CO generated by the steam reforming reaction, C3H6 also recovered the three-way catalytic activity of Rh/Zr–La–O (results not shown). Recovery of the catalyst by the steam reforming reaction included the contribution of C3H6 as a reductant. These experiments demonstrated that the regenerative effect of the steam reforming reaction worked well for Rh/Zr–Y–La–O, as was also observed for Rh/Zr–La–O in our previous study.21
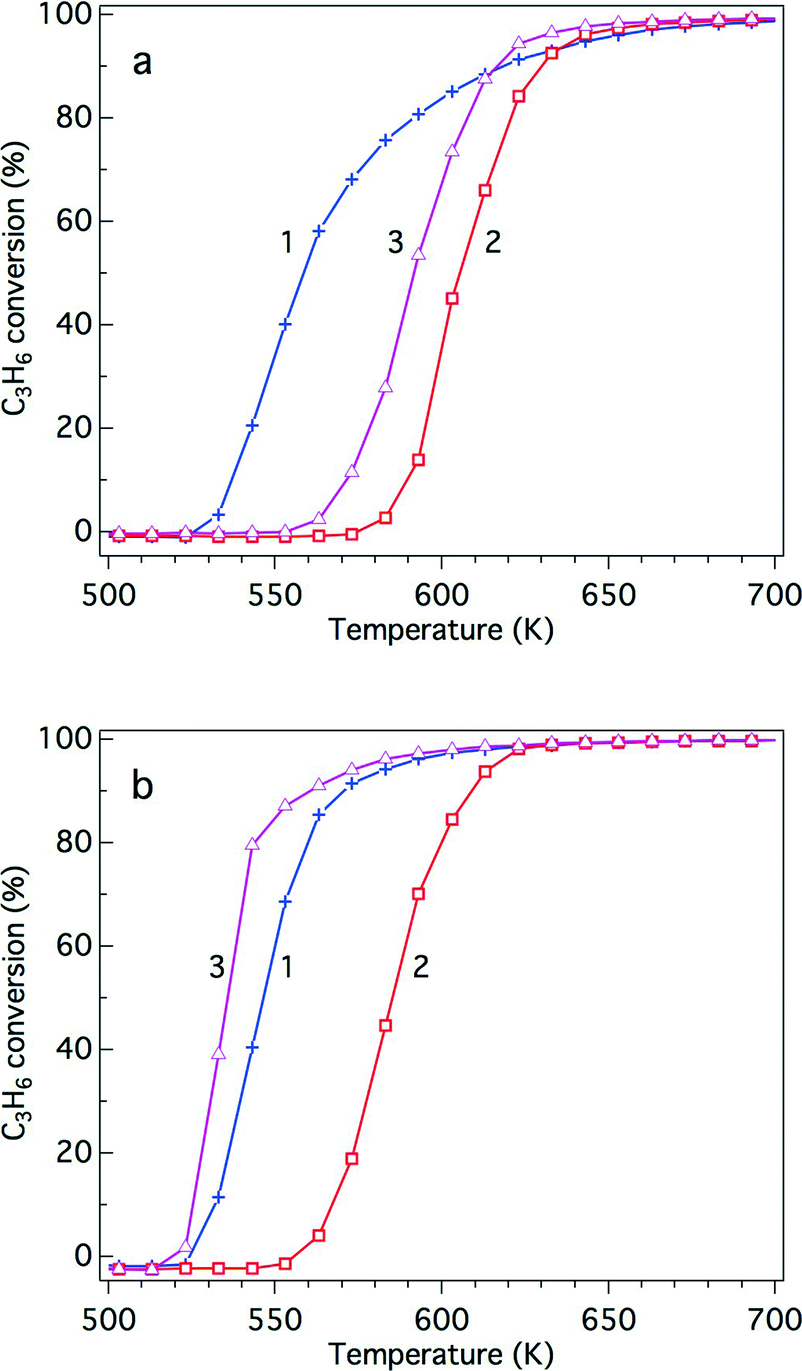 | ||
| Fig. 12 Effect of the steam reforming reaction on the three-way catalytic C3H6 conversion over the (a) aged Rh/Zr–Y–O and (b) aged Rh/Zr–Y–La–O catalysts. Three types of pre-treatment were conducted: (1) no pre-treatment, (2) pre-treatment with 5% oxygen at 773 K for 5 min, and (3) pre-treatment with 5% oxygen at 773 K for 5 min followed by the steam reforming reaction at 520 K for 5 min. | ||
4. Discussion
We have previously reported that rhodium supported on Zr–La–O was highly active under fresh and the aged conditions.22 Rhodium supported on Zr–La–O maintained its low oxidation state during the three-way catalytic reaction and after the oxidative treatment. This performance was not achieved when other lanthanides (Ce, Pr, or Nd) were added to the zirconia supports. After the aging treatment, the Rh/Zr–La–O catalyst exhibited higher activity in the steam reforming reaction. It was confirmed that the catalytic activity was recovered because oxidized rhodium was reduced by the steam reforming reaction. The aging treatment transformed the structure of the Zr–La–O support from tetragonal to three phases: monoclinic ZrO2, tetragonal ZrO2 and pyrochlore (La2Zr2O7). The treatment also altered the rhodium particle size from a few nanometers to 30–100 nm. In this study, Y was added to the ZrO2-based supports to stabilize the structure and its effect was investigated based on the oxidation state of Rh, activity for the steam reforming reaction and the three-way catalytic performance. As a result, it was found that Rh/Zr–Y–La–O showed excellent three-way catalytic performance that was better than that of the previously reported Rh/Zr–La–O catalyst.21,22For the fresh Rh/ZrO2 and Rh/Zr–Y–O catalysts, Y addition to ZrO2 enhanced the oxidation of Rh, as demonstrated by the results of the O/Rh ratio derived from CO-TPR. The O/Rh ratio increased from 0.14 for Rh/ZrO2 to 1.48 for Rh/Zr–Y–O (Table 2). The O/Rh ratio of 1.48 for Rh/Zr–Y–O was almost the stoichiometric value corresponding to the formation of fully oxidized Rh2O3 particles. Also, the introduction of Y induced a dramatic decrease in the steam reforming activity of the fresh catalysts (Fig. 10). As a result, the three-way catalytic activity of Rh/Zr–Y–O was low. The oxidation states of Rh for the two aged catalysts were similar to each other; the O/Rh ratios for Rh/ZrO2 and Rh/Zr–Y–O were 0.20 and 0.12, respectively. The sharp TPR peaks (Fig. 8a and b) suggest that only the surfaces of rhodium particles were oxidized to Rh2O3 in each aged sample. The steam reforming activities were very low for the aged catalysts. In particular, Rh/Zr–Y–O had the lowest activity (Fig. 11). The limited regeneration was demonstrated by the aged Rh/Zr–Y–O catalyst, as shown in Fig. 12a. The lower three-way catalytic activities of these two catalysts can be readily explained by their low regeneration. The reason why the aged Rh/Zr–Y–O showed three-way catalytic activity higher than the aged Rh/ZrO2 is unclear.
The enhancement of rhodium oxidation by Y addition was also observed for fresh Rh/Zr–Y–La–O. Also, the introduction of Y to Zr–La–O improved the reducibility of rhodium oxides (lower reduction temperature at CO-TPR). Enhancement of rhodium oxidation was indicated by the increased O/Rh ratio from 0.70 for Rh/Zr–La–O to 1.20 for Rh/Zr–Y–La–O (Table 2). In our previous report,22 the oxidation state of rhodium on Zr–La–O after oxidative pre-treatment averaged between Rh0 and Rh3+ (Fig. S9 in the ESI†). These results indicate that rhodium particles became more readily oxidized after Y addition. The CO-TPR profile of the fresh Rh/Zr–Y–La–O showed that rhodium on Zr–Y–La–O was more readily reduced than that on Zr–La–O. Specifically, the reduction profile of Rh/Zr–La–O began at approximately 540 K, but in the case of Rh/Zr–Y–La–O, the profile started from a much lower temperature (440 K, Fig. 7c and d). IR spectra of adsorbed NO also revealed that the band assigned to Rh0–NO+ was enhanced by Y addition, especially at around 473 K, the temperature at which the three-way catalytic reaction started (Fig. 9b). This is also an indication of the presence of rhodium(0) on the surface, highlighting that the rhodium on Zr–Y–La–O was reduced more readily than that on Rh/Zr–La–O. Also, Rh/Zr–Y–La–O demonstrated high steam reforming activity that was comparable to that of Rh/Zr–La–O (Fig. 10). Therefore, the regeneration of Rh was likely pronounced during the three-way catalysis over Rh/Zr–Y–La–O.
Y addition did not affect rhodium oxidation for the aged catalysts, as indicated by the results of the CO-TPR profiles (Fig. 8). Aged catalysts contain larger Rh particles as was indicated by their low Rh dispersion. Thus, it is unlikely that the oxide support influences the oxidation of rhodium through interactions between the Rh particles and the support. The XRD results for Rh/Zr–Y–La–O after the aging treatment showed that Y addition prevented phase separation (Fig. 1h). Stabilization of the support was also reflected in the changes in BET surface areas before and after the aging treatment. The surface area of Rh/Zr–La–O decreased to 29.6 m2 g−1, while Rh/Zr–Y–La–O had a surface area as high as 51.2 m2 g−1. The relatively high rhodium dispersion of the aged Rh/Zr–Y–La–O (Table 1) was likely due to the high surface area of the support after the aging treatment. The small rhodium particles in the TEM images correlated well with the dispersion of aged Rh/Zr–Y–La–O (Fig. 6). The high steam reforming activity was maintained, even after Y addition (Fig. 11). It was also confirmed that the steam reforming reaction recovered the catalytic activity that was deactivated by the oxidative treatment (Fig. 12b).
Steam reforming reaction is an endothermic reaction.32 The fact that the outlet temperature was lower compared to the inlet temperature during the three-way catalysis above 673 K demonstrated that the endothermic reaction was taking place during the three-way catalysis (Fig. S6 in the ESI†). Rh/Zr–Y–La–O showed high steam reforming activity (Fig. 11) and correspondingly the recovery of the catalyst was confirmed. (Fig. 12b). Based on these findings, it is reasonable to conclude that the recovery of the catalyst by the steam reforming reaction took place under the actual three-way catalysis conditions. Therefore, the recovery function of Rh/Zr–Y–La–O can be regarded as “self-regeneration”. Overall, Rh/Zr–Y–La–O showed excellent performance, even after the aging treatment.
5. Conclusions
High activity of the fresh and aged Rh/Zr–Y–La–O in the three-way catalytic processes was identified in this study. The effects of Y addition to the supports were also investigated in detail. Yttrium addition to Rh/Zr–La–O increased the oxidation of rhodium compared with that to Rh/Zr–La–O; moreover, the reducibility of rhodium was also enhanced, as indicated by CO-TPR. Rh/Zr–Y–La–O showed high steam reforming activity, comparable to that of Rh/Zr–La–O. Thus, the regeneration of Rh was likely pronounced for Rh/Zr–Y–La–O. In the case of Rh/Zr–Y–O, rhodium particles were fully oxidized after the oxidation treatment. Yttrium addition to ZrO2 dramatically decreased the steam reforming activity. Rhodium particles on ZrO2 and Zr–Y–O are readily oxidized during reaction, leading to low three-way catalytic activity.After the aging treatment, Rh/Zr–Y–La–O showed higher rhodium dispersion than the other tested catalysts. This is likely because of the relatively high surface area of the support after the aging treatment and stabilization of ZrO2 by the addition of Y. Excellent steam reforming activity was maintained for Rh/Zr–Y–La–O after the aging treatment. The high steam reforming activity regenerates deactivated oxidized Rh, providing the best three-way catalytic activity. These results demonstrate the importance of the catalyst support in designing three-way catalysts. Our findings highlight the potential for designing and developing effective three-way catalysts with high tolerance to oxidative reaction conditions in recently developed vehicles and engines.
Notes and references
- H. Shinjoh, M. Hatanaka, Y. Nagai, T. Tanabe, N. Takahashi, T. Yoshida and Y. Miyake, Top. Catal., 2009, 52, 1967 CrossRef CAS.
- J. R. Theis and R. W. McCabe, Catal. Today, 2012, 184, 262 CrossRef CAS PubMed.
- R. Burch, P. K. Loader and N. A. Cruise, Appl. Catal., A, 1996, 147, 375 CrossRef CAS.
- K. Dohmae, Y. Nagai, T. Tanabe, A. Suzuki, Y. Inada and M. Nomura, Surf. Interface Anal., 2008, 40, 1751 CrossRef CAS.
- C. P. Hwang, C. T. Yeh and Q. M. Zhu, Catal. Today, 1999, 51, 93 CrossRef CAS.
- M. Zimowska, J. B. Wagner, J. Dziedzic, J. Camra, B. Borzeccka-Prokop and M. Najbar, Chem. Phys. Lett., 2006, 417, 137 CrossRef CAS PubMed.
- R. W. McCabe, R. K. Usmen, K. Ober and H. S. Gandhi, J. Catal., 1995, 151, 385 CrossRef CAS.
- M. Uenishi, H. Tanaka, M. Taniguchi, I. Tan, Y. Nishihata, J. I. Mizuki and T. Kobayashi, Catal. Commun., 2008, 9, 311 CrossRef CAS PubMed.
- S. I. Matsumoto, Catal. Today, 2004, 90, 183 CrossRef CAS PubMed.
- H. Muraki and G. Zhang, Catal. Today, 2000, 63, 337 CrossRef CAS.
- H. S. Gandhi, G. W. Graham and R. W. McCabe, J. Catal., 2003, 216, 433 CrossRef CAS.
- M. Shelef and R. W. McCabe, Catal. Today, 2000, 62, 35 CrossRef CAS.
- G. C. Koltsakis and A. M. Stamatelos, Prog. Energy Combust. Sci., 1997, 23, 1 CrossRef CAS.
- M. Shelef and G. W. Graham, Catal. Rev.: Sci. Eng., 1994, 36, 433 CAS.
- Z. WengSieh, R. Gronsky and A. T. Bell, J. Catal., 1997, 170, 62 CrossRef CAS.
- D. J. C. Yates and E. B. Prestridge, J. Catal., 1987, 106, 549 CrossRef CAS.
- M. A. Newton, Chem. Soc. Rev., 2008, 37, 2644 RSC.
- M. A. Newton, B. Jyoti, A. J. Dent, S. G. Fiddy and J. Evans, Chem. Commun., 2004, 2382 RSC.
- T. Tanabe, A. Morikawa, M. Hatanaka, N. Takahashi, Y. Nagai, A. Sato, O. Kuno, H. Suzuki and H. Shinjoh, Catal. Today, 2012, 184, 219 CrossRef CAS PubMed.
- R. Burch and P. K. Loader, Appl. Catal., A, 1996, 143, 317 CrossRef CAS.
- H. Kawabata, Y. Koda, H. Sumida, M. Shigetsu, A. Takami and K. Inumaru, Chem. Commun., 2013, 49, 4015 RSC.
- H. Kawabata, Y. Koda, H. Sumida, M. Shigetsu, A. Takami and K. Inumaru, Catal. Sci. Technol., 2014, 4, 697 CAS.
- H. G. Scott, J. Mater. Sci., 1975, 10, 1527 CrossRef CAS.
- C. Dujardin, A. S. Mamede, E. Payen, B. Sombret, J. P. Huvenne and P. Granger, Top. Catal., 2004, 30–1, 347 CrossRef.
- T. Chafik, A. M. Efstathiou and X. E. Verykios, J. Phys. Chem. B, 1997, 101, 7968 CrossRef CAS.
- M. Haneda, K. Shinoda, A. Nagane, O. Houshito, H. Takagi, Y. Nakahara, K. Hiroe, T. Fujitani and H. Hamada, J. Catal., 2008, 259, 223 CrossRef CAS PubMed.
- E. Rogemond, N. Essayem, R. Frety, V. Perrichon, M. Primet, M. Chevrier, C. Gauthier and F. Mathis, J. Catal., 1999, 186, 414 CrossRef CAS.
- J. Barbier-Jr and D. Duprez, Appl. Catal., B, 1994, 4, 105 CrossRef.
- J. P. Breen, R. Burch and H. M. Coleman, Appl. Catal., B, 2002, 39, 65 CrossRef CAS.
- Supported Metals in Catalysis, ed. J. A. Anderson and M. Fernandez Garcia, ICP, London, 2005, p. 140 Search PubMed.
- Shokubai-kouza, Catalyst design, ed. The Catalysis Society of Japan, ISBN: 4061914057, Kodan-sha, 1985, vol. 5, p. 141 Search PubMed.
- M. E. Domine, E. E. Iojoiu, T. Davidian, N. Guilhaume and C. Mirodatos, Catal. Today, 2008, 133, 565 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CO conversion, EDX analysis data, XRD patterns of the Rh 111 diffraction peak and XPS spectra are provided. See DOI: 10.1039/c4cy01032a |
| This journal is © The Royal Society of Chemistry 2015 |