Alternating ring-opening copolymerization of styrene oxide and maleic anhydride using asymmetrical bis-Schiff-base metal(III) catalysts†
Received
18th August 2014
, Accepted 23rd September 2014
First published on 23rd September 2014
Abstract
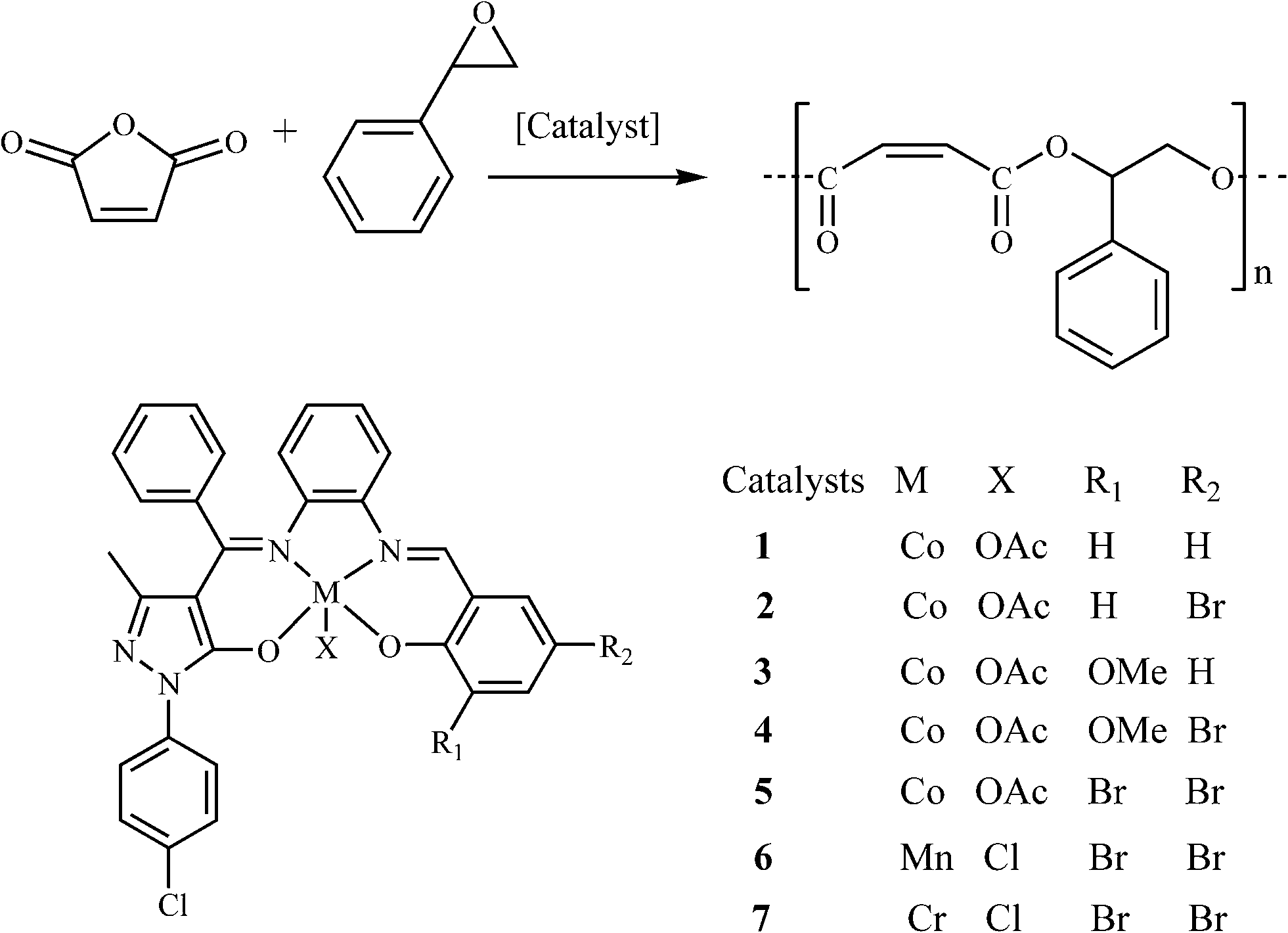
A ring-opening copolymerization of styrene oxide with maleic anhydride was performed by applying a series of asymmetrical bis-Schiff-base metal(III) catalysts. The chromium catalyst exhibited the best performance and therefore the catalyst (salen)CrCl (salen = 4-((Z)-(2-((E)-3,5-dibromo-2-hydroxybenzylideneamino)phenylimino)(phenyl)methyl)-1-(4-chlorophenyl)-3-methyl-1H-pyrazol-5(4H)-one) (7) was chosen for further studies. The investigations on the effect of different co-catalysts on the copolymerization of styrene oxide and maleic anhydride revealed that DMAP showed the highest activity, followed by PPN+Cl−, whereas Ph3P showed the lowest activity. 1H NMR and MALDI-TOF-MS spectra of the copolymers formed confirmed the alternating microstructures. The copolymerization of styrene oxide with maleic anhydride bearing a double bond in its structure was shown to be highly dependent on the polymerization condition, type of co-catalyst, monomer to catalyst ratio, temperature and time used in the copolymerization reaction. Applying chain transfer agents, alcohols, resulted in a decrease in molecular weight.
1. Introduction
Polyesters are generally produced by the step-growth condensation polymerization from diols, diacids or diesters, which is an energy intensive process requiring drastic conditions, such as high temperatures and high vacuum removal of small molecule byproducts to drive the reaction towards high conversion, and they require very accurate co-monomer stoichiometries to obtain high-molecular-weight polymers.1,2 Although the ring-opening polymerization of cyclic esters takes place under mild reactions without any byproducts, the availability of structurally diverse monomers restricts the scope of the polymer architecture.3 Alternatively, the ring-opening copolymerization of epoxides with cyclic anhydrides has the potential to produce a wide range of polymer backbone structures.4
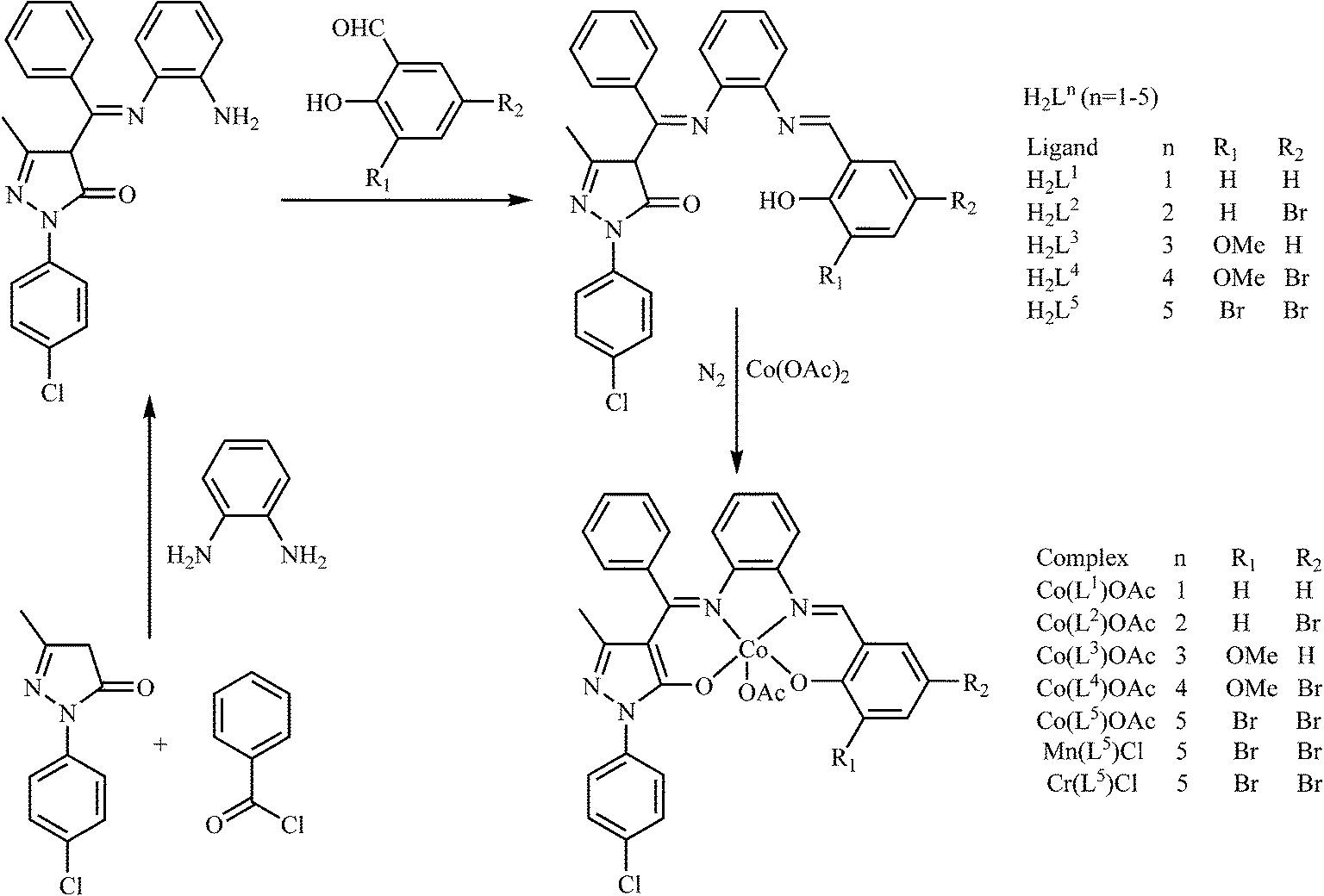
The catalytic coupling of epoxide and anhydride with inorganic salts or tertiary amines as catalysts was earlier reported by Inoue5 and Maeda,6 but the difficulty of obtaining high molecular weight alternating polyesters and the undesirable side reactions of epoxide homo-polymerization prevented its popularity for a considerable time. It was not until 2007 that Coates7 employed zinc 2-cyano-β-diketiminato complexes as catalysts for the solution copolymerization of vinyl cyclohexene oxide and diglycolic anhydride, affording high molecular weight copolymers (Mn = 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) with narrow molecular weight distributions. This attracted the attention of the scientists despite the lower catalytic activities and significant amounts of ether linkages for unsaturated anhydride–epoxide copolymerization. Since then, a series of metal–porphyrinato,8 metal–salen9 and manganese–corrole10 complexes have been reported to catalyze this copolymerization, several of which are capable of yielding high molecular weight polymers with low molecular weight distributions. However, for the copolymerization of unsaturated anhydride maleic anhydride (MA) and styrene oxide (SO), these catalysts displayed low activity (53% of SO conversion, Mn = 1420 g mol−1) and significant amount of ether linkages.11 As a matter of fact, the distinctive advantage of asymmetrical salen-type Schiff-base scaffolds12 or analogies13,14 endows the possibility of further fine-tuning their structures by introducing different electronic and steric effects. In the previous studies, we have found that asymmetrical salen-type Schiff-base catalysts are more efficient than symmetrical salen-type Schiff-base catalysts in catalyzing cyclohexene oxide and MA with regards to catalytic activity and selectivity.15–17 To the best of our knowledge, research on the copolymerization of electron-withdrawing SO and MA using the metal complexes based on asymmetrical salen-type Schiff-base ligands as the catalysts has not been reported. Herein, based on a series of new asymmetrical Schiff-base ligands H2Ln (n = 1–5) synthesized from the reaction of o-phenylenediamine, 1-(4-Cl-phenyl)-3-methyl-4-benzoyl-5-pyrazolone (Cl-PMBP), and different aldehyde derivatives, a series of new catalysts [Co(Ln)OAc] (n = 1, 1; n = 2, 2; n = 3, 3; n = 4, 4; n = 5, 5), [Mn(L5)Cl] (6) and [Cr(L5)Cl] (7) have been obtained by a two-step sequence. The ring-opening copolymerization behaviors of SO with MA in the presence of different co-catalysts bis(triphenylphosphine)iminium chloride (PPN+Cl−), 4-N,N-dimethylaminopyridine (DMAP) and triphenylphosphine (Ph3P) with a stipulated reaction procedure have been examined.
000 g mol−1) with narrow molecular weight distributions. This attracted the attention of the scientists despite the lower catalytic activities and significant amounts of ether linkages for unsaturated anhydride–epoxide copolymerization. Since then, a series of metal–porphyrinato,8 metal–salen9 and manganese–corrole10 complexes have been reported to catalyze this copolymerization, several of which are capable of yielding high molecular weight polymers with low molecular weight distributions. However, for the copolymerization of unsaturated anhydride maleic anhydride (MA) and styrene oxide (SO), these catalysts displayed low activity (53% of SO conversion, Mn = 1420 g mol−1) and significant amount of ether linkages.11 As a matter of fact, the distinctive advantage of asymmetrical salen-type Schiff-base scaffolds12 or analogies13,14 endows the possibility of further fine-tuning their structures by introducing different electronic and steric effects. In the previous studies, we have found that asymmetrical salen-type Schiff-base catalysts are more efficient than symmetrical salen-type Schiff-base catalysts in catalyzing cyclohexene oxide and MA with regards to catalytic activity and selectivity.15–17 To the best of our knowledge, research on the copolymerization of electron-withdrawing SO and MA using the metal complexes based on asymmetrical salen-type Schiff-base ligands as the catalysts has not been reported. Herein, based on a series of new asymmetrical Schiff-base ligands H2Ln (n = 1–5) synthesized from the reaction of o-phenylenediamine, 1-(4-Cl-phenyl)-3-methyl-4-benzoyl-5-pyrazolone (Cl-PMBP), and different aldehyde derivatives, a series of new catalysts [Co(Ln)OAc] (n = 1, 1; n = 2, 2; n = 3, 3; n = 4, 4; n = 5, 5), [Mn(L5)Cl] (6) and [Cr(L5)Cl] (7) have been obtained by a two-step sequence. The ring-opening copolymerization behaviors of SO with MA in the presence of different co-catalysts bis(triphenylphosphine)iminium chloride (PPN+Cl−), 4-N,N-dimethylaminopyridine (DMAP) and triphenylphosphine (Ph3P) with a stipulated reaction procedure have been examined.
2. Experimental
General remarks: Tetrahydrofuran (THF) HPLC grade was purchased from Fisher Scientific and purified over basic alumina columns. Other solvents were used as received from Sigma Aldrich and stored over 3 Å activated molecular sieves. SO purchased from Sigma Aldrich, was dried over CaH2, distilled and stored under dry N2 prior to use. MA purchased from Alfa Aesar, was dried overnight under vacuum and sublimed twice under dry N2 prior to use. DMAP, Ph3P and PPN+Cl− were purchased from Fluka and used as received. All the manipulations of air and water sensitive compounds were carried out under dry N2 using the standard Schlenk line techniques. Elemental analyses were performed on a Perkin-Elmer 240C elemental analyzer. Infrared spectra were recorded on a Nicolet Nagna-IR 550 spectrophotometer in the region 4000–400 cm−1 using KBr pellets. 1H NMR spectra were recorded on a JEOL EX 400 spectrometer in CDCl3 or DMSO-d6 at room temperature. Gel permeation chromatography (GPC) analyses of the molecular weights (Mn and Mw) and molecular weight distribution (PDI = Mw/Mn) of the polymers were performed on a Waters 150C instrument in HPLC THF using polystyrene as standard. MALDI-TOF-MS analysis was performed on a Voyager DE-STR from Applied Biosystems equipped with a 337 nm nitrogen laser. An accelerating voltage of 25 kV was applied. Mass spectra of 1000 shots were accumulated. The polymer samples were dissolved in HPLC THF at a concentration of 1 mg mL−1. The cationization agent used was KCF3COO (Alfa Aesar, >99%) dissolved in HPLC THF at a concentration of 5 mg mL−1. The matrix used was DCTB (Alfa Aesar, DCTB = trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile) and was dissolved in HPLC grade THF at a concentration of 40 mg mL−1. The solution of matrix, salt and polymer were mixed in a volume ratio of 4:1:4, respectively. The mixed solution was transferred on a stainless steel MALDI target and left to dry. The spectra were recorded from the crude products. In-house developed software was used to characterize the polymers in detail, which allowed the elucidation of the individual chain structures, the chemical composition and topology of copolymer.18 Differential scanning calorimetry of polymer samples was performed on a TA Instruments Q1000 instrument equipped with a LNCS and automated sample. Typical DSC experiments were performed in crimped aluminum pans under nitrogen with a heating rate of 10 °C min−1 from −70 °C to 200 °C.
2.1 Preparation of 1-(4-Cl-phenyl)-3-methyl-4-benzoyl-2-pyrazolin-5-one
The 1,4-dioxane solution (25 mL) of 1-(4-Cl-phenyl)-3-methyl-5-pyrazolone (1.04 g, 5 mmol) was stirred with calcium hydroxide (0.9 g, 1 mmol) and barium hydroxide (0.9 g, 1 mmol) under room temperature for 30 min. A solution of benzoyl chloride (0.70 mL, 5.5 mmol) in absolute 1,4-dioxane (5 mL) was slowly added, and the resulting mixture was refluxed under nitrogen atmosphere for 24 h. After cooling to room temperature, the reaction mixture was poured into a 40 mL solution of ice-cold hydrochloric acid [HCl:H2O = 3:7 (v/v)], and the resulting yellow precipitate was filtered and recrystallized from acetone and distilled water. Yield: 1.3 g (82%). 1H NMR (400 MHz, DMSO-d6, ppm): δ 7.87 (d, 2H, J = 8.4 Hz, ArH), 7.63 (d, 2H, J = 7.6 Hz, ArH), 7.60 (d, 1H, J = 6.8 Hz, ArH), 7.54–7.51 (m, 2H, ArH), 7.44 (d, 2H, J = 8.4 Hz, ArH), 2.09 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 195.3; 160.4; 148.4; 139.8; 137.1; 133.6; 132.0; 129.8; 129.7; 129.6; 129.5; 128.9; 128.8; 121.7; 121.6; 106.9; 16.0.
2.2 Synthesis of the half-unit Schiff-base precursor HL0
(HL0 = 4-((2-aminophenylimino)(phenyl)methyl)-3-methyl-1-(4-Cl-phenyl)-pyrazol-5-one)
To a solution of 1-(4-Cl-phenyl)-3-methyl-4-benzoyl-2-pyrazolin-5-one (312.8 mg, 1 mmol) in absolute EtOH (7 mL), a solution of 1,2-diaminobenzene (118.8 mg, 1.1 mmol) in absolute EtOH (3 mL) was slowly added, and the resulted mixture was refluxed for 6 h, After cooling to room temperature, the insoluble precipitate was filtered off, and recrystallized with absolute EtOH to give an brown microcrystalline product. Yield: 298.4 mg (74%). Anal. calcd for C23H19N4OCl: C, 68.57; H, 4.75; N, 13.91. Found: C, 68.55; H, 4.80; N, 13.87. FT-IR (KBr, cm−1): 3324 (w), 3054 (m), 1624 (s), 1578 (vs), 1535 (w), 1495 (s), 1387 (s), 1321 (w), 1207 (m), 1146 (w), 1090 (w), 1049 (w), 1007 (w), 835 (m), 745 (w), 702 (w), 596 (w). 1H NMR (400 MHz, DMSO-d6, ppm): δ 12.43 (s, 1H, –OH), 8.03 (d, 1H, J = 7.6 Hz, ArH), 7.37 (d, 6H, J = 6.0 Hz, ArH), 7.32 (d, 2H, J = 7.6 Hz, ArH), 6.96–6.92 (m, 1H, ArH), 6.68 (d, 1H, J = 8.4 Hz, ArH), 6.59 (d, 1H, J = 7.6 Hz, ArH), 6.45 (d, 1H, J = 8.0 Hz, ArH), 3.94 (s, 2H, –NH2), 2.09 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 170.6; 160.3; 150.1; 144.8; 142.6; 138.8; 132.9; 131.8; 131.1; 129.6; 129.5; 129.3; 129.2; 128.9; 128.8; 128.3; 128.2; 121.7; 121.6; 120.2; 118.6; 108.3; 16.0.
2.3 Synthesis of series of asymmetric bis-Schiff-base ligand H2Ln (n = 1–5)
For H2L1: to a solution of HL0 (80.6 mg, 0.2 mmol) in absolute CHCl3 (5 mL), a solution of salicylaldehyde (21.0 μL, 0.2 mmol) in absolute EtOH (5 mL) was slowly added, and the resultant mixture was refluxed for 6 h. After cooling to room temperature, the insoluble precipitate was filtered off, and washed with cold absolute EtOH (10 mL) three times to yield the bright yellow microcrystalline product of H2L1. Yield: 80.8 mg (80%). Anal. calcd for C30H23ClN4O2: C, 71.01; H 4.54; N, 11.05. Found: C, 70.97; H, 4.62; N, 11.01. FT-IR (KBr, cm−1): 3329 (b), 1640 (m), 1614 (vs), 1570 (s), 1536 (m), 1491 (s), 1418 (w), 1385 (s), 1275 (m), 1233 (w), 1204 (m), 1171 (w), 1140 (w), 1086 (w), 1051 (m), 1007 (m), 937 (w), 914 (w), 895 (w), 829 (w), 756 (m), 700 (m), 681 (w), 627 (w), 586 (w), 557 (w), 519 (w), 503 (w), 440 (w). 1H NMR (400 MHz, DMSO-d6, ppm): δ 12.88 (s, 1H, –OH), 12.02 (s, 1H, –OH), 8.95 (s, 1H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 8.06 (d, 2H, J = 8.8 Hz, ArH), 7.94 (d, 1H, J = 6.0 Hz, ArH), 7.53–7.41 (m, 10H, ArH), 7.25 (t, 1H, J = 7.2 Hz, ArH), 7.06–6.97 (m, 2H, ArH), 6.82 (d, 1H, J = 7.2 Hz, ArH), 1.44 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 167.1; 164.9; 163.6; 161.3; 157.2; 149.5; 149.4; 137.7; 134.0; 132.5; 132.1; 131.6; 130.9; 129.8; 129.6; 129.2; 129.0; 128.9; 128.8; 128.6; 128.5; 123.5; 123.4; 122.6; 122.3; 121.6; 118.5; 116.1; 107.8; 16.0.
N), 8.06 (d, 2H, J = 8.8 Hz, ArH), 7.94 (d, 1H, J = 6.0 Hz, ArH), 7.53–7.41 (m, 10H, ArH), 7.25 (t, 1H, J = 7.2 Hz, ArH), 7.06–6.97 (m, 2H, ArH), 6.82 (d, 1H, J = 7.2 Hz, ArH), 1.44 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 167.1; 164.9; 163.6; 161.3; 157.2; 149.5; 149.4; 137.7; 134.0; 132.5; 132.1; 131.6; 130.9; 129.8; 129.6; 129.2; 129.0; 128.9; 128.8; 128.6; 128.5; 123.5; 123.4; 122.6; 122.3; 121.6; 118.5; 116.1; 107.8; 16.0.
For H2L2: the bright yellow asymmetric bis-Schiff-base ligand H2L2 solid product was prepared in the same way as H2L1 except that 5-bromo-salicylaldehyde (42.0 mg, 0.2 mmol) was used instead of salicylaldehyde (21.0 μL, 0.2 mmol). Yield: 91.7 mg (78%). Anal. calcd for C30H22BrClN4O2: C, 61.45; H, 3.76; N, 9.56. Found: C, 61.42; H, 3.83; N, 9.54. FT-IR (KBr, cm−1): 3352 (b), 1634 (m), 1613 (m), 1566 (s), 1533 (m), 1491 (vs), 1464 (m), 1379 (s), 1313 (m), 1279 (m), 1254 (m), 1213 (m), 1142 (w), 1123 (w), 1088 (w), 1049 (w), 1007 (m), 972 (w), 864 (w), 831 (m), 777 (m), 754 (w), 733 (m), 704 (m), 631 (w), 613 (w), 588 (w), 548 (w), 505 (w), 482 (w), 455 (w), 426 (w). 1H NMR (400 MHz, DMSO-d6, ppm): δ 13.12 (s, 1H, –OH), 11.55 (s, 1H, –OH), 8.95 (s, 1H, –CN), 8.34 (s, 1H, ArH), 8.13 (d, 2H, J = 8.8 Hz, ArH), 7.58–7.46 (m, 8H, ArH), 7.41–7.39 (d, 1H, J = 8.0 Hz, ArH), 7.20 (t, 1H, J = 7.6 Hz, ArH), 6.98 (t, 2H, J = 8.4 Hz, ArH), 6.58 (d, 1H, J = 8.0 Hz, ArH), 1.44 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 167.4; 165.1; 163.1; 163.0; 156.5; 149.5; 149.4; 139.5; 135.8; 134.3; 134.2; 131.1; 129.3; 129.2; 129.1; 129.0; 128.9; 128.8; 128.7; 128.6; 128.5; 128.4; 123.5; 123.4; 123.3; 122.7; 118.5; 115.8; 107.7; 16.3.
For H2L3: the orange asymmetric bis-Schiff-base ligand H2L3 solid product was prepared in the same way as H2L1 except that o-vanillin (30.5 mg, 0.2 mmol) was used instead of salicylaldehyde (21.0 μL, 0.2 mmol). Yield: 82.4 mg (77%). Anal. calcd for C31H25ClN4O3: C, 69.27; H, 4.66; N 10.43. Found: C, 69.25; H, 4.72; N, 10.41. FT-IR (KBr, cm−1): 3356 (b), 1634 (m), 1613 (s), 1568 (s), 1537 (m), 1491 (vs), 1462 (m), 1418 (m), 1379 (m), 1325 (m), 1281 (m), 1254 (m), 1142 (w), 1123 (w), 1088 (w), 1049 (m), 1007 (m), 972 (m), 934 (w), 866 (w), 831 (m), 777 (m), 735 (m), 704 (m), 613 (w), 586 (w), 546 (w), 505 (w), 482 (w), 457 (w), 430 (w). 1H NMR (400 MHz, DMSO-d6, ppm): δ 12.81 (s, 1H, –OH), 12.04 (s, 1H, –OH), 8.94 (s, 1H, –CN), 8.06 (d, 2H, J = 8.4 Hz, ArH), 7.51–7.41 (m, 9H, ArH), 7.25 (t, 1H, J = 7.2 Hz, ArH), 7.16 (d, 1H, J = 7.6 Hz, ArH), 7.05 (t, 1H, J = 7.6 Hz, ArH), 6.95 (t, 1H, J = 8.0 Hz, ArH), 6.86 (d, 1H, J = 8.0 Hz, ArH), 3.80 (s, 3H, –OCH3), 1.45 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 167.8; 165.6; 160.3; 156.5; 152.4; 150.1; 145.8; 145.7; 139.5; 134.0; 131.1; 129.9; 129.4; 129.3; 129.2; 129.1; 128.9; 128.8; 128.7; 128.6; 123.7; 123.6; 123.3; 123.2; 122.9; 122.5; 119.6; 118.2; 101.3; 56.3; 16.1.
For H2L4: the bright yellow asymmetric bis-Schiff-base ligand H2L4 solid product was prepared in the same way as H2L1 except that 5-bromo-2-hydroxy-3-methoxy-benzaldehyde (48.0 mg, 0.2 mmol) was used instead of salicylaldehyde (21.0 μL, 0.2 mmol). Yield: 100.2 mg (81%). Anal. calcd for C31H24BrClN4O3: C, 60.39; H, 3.90; N, 9.09. Found: C, 60.35; H, 3.97; N, 9.04. FT-IR (KBr, cm−1): 3354 (b), 1632 (m), 1613 (s), 1568 (s), 1535 (m), 1491 (vs), 1466 (m), 1445 (m), 1420 (m), 1377 (m), 1325 (m), 1273 (m), 1250 (m), 1190 (w), 1144 (w), 1090 (w), 1049 (m), 1009 (m), 976 (m), 864 (m), 833 (m), 800 (w), 772 (m), 748 (m), 706 (m), 679 (w), 611 (w), 584 (w), 559 (w), 546 (w), 507 (w), 480 (w), 457 (w), 424 (w). 1H NMR (400 MHz, DMSO-d6, ppm): δ 13.04 (s, 1H, –OH), 11.39 (s, 1H, –OH), 8.95 (s, 1H, –CN), 8.12 (d, 2H, J = 9.2 Hz, ArH), 7.91 (d, 1H, J = 2.4 Hz, ArH), 7.54–7.42 (m, 8H, ArH), 7.29 (d, 1H, J = 2.4 Hz, ArH), 7.20 (t, 1H, J = 7.2 Hz, ArH), 6.99 (t, 1H, J = 7.6 Hz, ArH), 6.63 (t, 1H, J = 4.8 Hz, ArH), 3.86 (s, 3H, –OCH3), 1.43 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 167.3; 165.6; 160.2; 156.5; 154.7; 150.1; 145.8; 145.7; 139.5; 135.1; 133.2; 130.9; 129.4; 129.3; 129.2; 129.1; 128.9; 128.8; 128.7; 128.6; 127.4; 123.7; 123.6; 123.2; 123.1; 121.8; 121.4; 116.8; 107.3; 56.3; 16.2.
For H2L5: the bright yellow asymmetric bis-Schiff-base ligand H2L5 solid product was prepared in the same way as H2L1 except that 3,5-dibromo-2-hydroxybenzaldehyde (56.0 mg, 0.2 mmol) was used instead of salicylaldehyde (21.0 μL, 0.2 mmol). Yield: 103.0 mg (78%). Anal. calcd for C30H21Br2ClN4O2: C, 54.15; H, 3.16; N, 8.42. Found: C, 54.13; H, 3.24; N, 8.39. FT-IR (KBr, cm−1): 3337 (b), 1636 (m), 1613 (s), 1570 (s), 1535 (m), 1491 (vs), 1443 (m), 1383 (s), 1323 (m), 1296 (m), 1213 (m), 1150 (w), 1090 (w), 1049 (m), 1009 (m), 972 (w), 939 (w), 887 (w), 866 (w), 829 (m), 775 (w), 741 (m), 706 (w), 687 (w), 633 (w), 611 (w), 588 (w), 565 (w), 502 (w), 463 (w). 1H NMR (400 MHz, DMSO-d6, ppm): δ 13.04 (s, 1H, –OH), 12.94 (s, 1H, –OH), 8.94 (s, 1H, –CN), 8.08–8.00 (m, 4H, ArH), 7.51–7.43 (m, 8H, ArH), 7.27 (t, 1H, J = 7.6 Hz, ArH), 7.07 (t, 1H, J = 8.0 Hz, ArH), 6.78 (d, 1H, J = 8.0 Hz, ArH), 1.46 (s, 3H, –CH3). 13C NMR (400 MHz, DMSO-d6, ppm): δ 170.6; 160.3; 160.1; 159.3; 149.4; 144.8; 144.7; 139.5; 138.8; 133.9; 133.0; 131.8; 131.0; 129.6; 129.5; 129.3; 129.2; 128.9; 128.8; 128.7; 128.6; 124.5; 124.4; 123.9; 122.6; 122.5; 118.3; 116.2; 108.4; 16.3.
2.4.1 General procedure for the preparation of the corresponding Co(III)-bis-Schiff-base complexes [Co(Ln)(OAc)] (n = 1–5, 1–5).
The synthetic procedures and characterizations for complexes 1–5 can be found in the ESI.†
2.4.2 General procedure for the preparation of the corresponding Mn(III)-bis-Schiff-base complexes [Mn(L5)Cl] (6).
For [Mn(L5)(Cl)] (6): solid H2L5 (99.7 mg, 0.15 mmol) and anhydride Mn(OAc)2 (76.1 mg, 0.15 mmol) were added to a flame dried Schlenk flask charged with a Teflon-coated stir bar. Under an atmosphere of dry N2, absolute CHCl3 (5 ml) and MeOH (5 ml) were injected and the resultant mixture was stirred at ambient temperature for 6 h. The stopper of the flask was removed, lithium chloride (38.0 mg, 0.90 mmol) was added and the resultant reddish brown solution was stirred at room temperature for 10 h while exposed to dry air. The final solution was filtered and the clear filtrate was left to stand at room temperature for several days to give the dark brown polycrystalline solid product of 6. Yield: 129.0 mg, 57%. Anal. calc for C30H19Cl2Br2MnN4O2: C, 47.80; H, 2.52; N, 7.44. Found: C, 47.76; H, 2.64; N, 7.39. FT-IR (KBr, cm−1): 3443 (b), 2928 (w), 1634 (vs), 1597 (s), 1559 (s), 1524 (m), 1497 (m), 1464 (s), 1427 (s), 1381 (m), 1355 (w), 1339 (w), 1313 (w), 1285 (w), 1215 (s), 1090 (m), 1061 (m), 1013 (m), 873 (w), 854 (w), 829 (w), 789 (w), 756 (w), 721 (m), 604 (m), 582 (w), 550 (m), 519 (w), 490 (w). ESI-MS (in THF) m/z 754.15 [M − H]+; 717.71 [M − Cl]+.
2.4.3 General procedure for the preparation of the corresponding Cr(III)-bis-Schiff-base complexes [Cr(L5)Cl] (7).
For [Cr(L1)Cl] (7): solid H2L5 (199.4 mg, 0.3 mmol) and anhydrous CrCl2 (37.0 mg, 0.3 mmol) were added to a flame dried Schlenk flask charged with a Teflon-coated stir bar. Under an atmosphere of dry N2, absolute THF (10 mL) was injected and the resultant mixture was stirred at ambient temperature for 24 h. The stopper of the flask was removed, and the resultant brown solution was stirred at room temperature for another 24 h while exposed to dry air. After the reaction solution was poured into diethyl ether (50 mL), the organic layer was washed with aqueous saturated NH4Cl (3 × 30 mL) and brine (3 × 30 mL) followed by drying with anhydrous Na2SO4. After filtration to remove solid impurities and drying agent, toluene (20 mL) was added into the filtrate. The final clear solution was left to stand at room temperature for several days to obtain the brown polycrystalline solid product of 7. Yield: 136.0 mg (60%). Anal. calcd for C30H19Cl2Br2CrN4O2: C, 47.99; H, 2.53; N, 7.46. Found: C, 47.89; H, 2.64; N, 7.40. FT-IR (KBr, cm−1): 3057 (w), 2924 (m), 2854 (m), 1627 (s), 1563 (s), 1528 (m), 1462 (vs), 1447 (s), 1427 (s), 1381 (m), 1313 (m), 1216 (w), 1152 (m), 1055 (m), 1014 (w), 843 (w), 790 (w), 750 (s), 712 (w), 690 (w), 594 (w), 548 (w), 514 (w). ESI-MS (in THF) m/z 751.21 [M − H]+; 714.77 [M − Cl]+.
2.5 Structure determination
The single crystals of complexes [Co(L3)(OAc)] (3) and [Co(L4)(OAc)] (4) of suitable dimensions were mounted onto thin glass fibers. All the intensity data were collected on a Bruker SMART CCD diffractometer (Mo-Kα radiation and λ = 0.71073 Å) in Φ and ω scan modes. Structures were solved by Direct methods followed by difference Fourier syntheses, and then refined by full-matrix least-squares techniques against F2 using SHELXTL-97.19 All other non-hydrogen atoms were refined with anisotropic thermal parameters. Absorption corrections were applied using SADABS.20 All hydrogen atoms were placed in calculated positions and isotropically refined using a riding model.
2.6 Copolymerization procedures of SO and MA
The copolymerization between SO and MA was performed in both bulk and solution (toluene) with a stipulated molar ratio of SO:MA:catalyst:cocatalyst for the selected reaction time.
2.6.1 Representative copolymerization procedure in bulk.
In the standard Schlenk vacuum line system, SO (2.5 mmol), MA (2.5 mmol), catalyst 7 (0.01 mmol) and co-catalyst DMAP (0.01 mmol) were placed in a vial equipped with a small stir bar. The vial was sealed with a Teflon lined cap, and placed in an aluminum heat block preheated to the desired temperature of 110 °C. After the allotted reaction time of 150 min, the vial was removed from the heat block and a small aliquot was removed for crude 1H NMR analysis to determine monomer conversion. The viscous reaction mixture was then dissolved in a minimum amount of CH2Cl2, and precipitated with an excess of anhydrous diethyl ether. The polymer was collected and dried in vacuum to give a white solid product.
2.6.2 Representative copolymerization procedure in solution.
In the standard Schlenk vacuum line system, SO (2.5 mmol), MA (2.5 mmol), catalyst 7 (0.01 mmol) and co-catalyst DMAP (0.01 mmol) were placed in a vial equipped with a small stir bar. Then absolute toluene (1 ml) was injected into the vial. The vial was sealed with a Teflon lined cap, and placed in an aluminum heat block preheated to the desired temperature of 110 °C. After the allotted reaction time of 300 min, the vial was removed from the heat block and a small aliquot was removed for crude 1H NMR analysis to determine monomer conversion. The viscous reaction mixture was then dissolved in a minimum amount of CH2Cl2, and precipitated with an excess of anhydrous diethyl ether. The polymer was collected and dried in vacuum to give a white solid product.
2.6.3 Representative characterization of copolymers from SO and MA.
Copolymer (SO–MA): FT-IR (KBr, cm−1): 3316 (w), 2956 (w), 2874 (w), 2649 (w), 1962 (w), 1732 (vs), 1649 (m), 1574 (w), 1535 (w), 1496 (w), 1453 (m), 1401 (m), 1350 (w), 1207 (s), 1162 (s), 1078 (w), 1026 (w), 1001 (w), 915 (w), 818 (w), 760 (m), 699 (m), 637 (w), 525 (w). 1H NMR (400 MHz, DMSO-d6): 7.32 (s, 5H, ArH), 6.44 (d, 2H, J = 7.2 Hz, –CHCH–), 6.03–5.94 (m, 1H, –CH–), 4.40–4.10 (m, 2H, –CH2–).
3. Results and discussion
3.1 Synthesis of the novel asymmetrical bis-Schiff-base ligands and corresponding complexes
As shown in Scheme 1, we synthesized five novel asymmetrical bis-Schiff-base ligands H2Ln (n = 1–5). The asymmetrical bis-Schiff-base ligands were synthesized by a three-step procedure, wherein we first prepared 1-(4-Cl-phenyl)-3-methyl-4-benzoyl-2-pyrazolin-5-one by the reaction of 1-(4-Cl-phenyl)-3-methyl-5-pyrazolone with benzoyl chloride in 1:1 molar ratio in absolute 1,4-dioxane. Then HL0 was obtained by the reaction of 1-(4-Cl-phenyl)-3-methyl-4-benzoyl-5-pyrazolone with o-phenylenediamine in either 1:1 or 2:1 molar ratio in absolute EtOH. When the monoamine precursor HL0 was allowed to react with one of the aldehyde derivatives (salicylaldehyde, 5-bromo-salicylaldehyde, o-vanillin or 5-bromo-2-hydroxy-3-methoxy-benzaldehyde, 3,5-dibromo-2-hydroxy-benzaldehyde) in 1:1 molar ratio, a series of new asymmetrical bis-Schiff-base ligands H2Ln (n = 1–5) were obtained in good yields, respectively. In addition to these ligands, the corresponding Co(III) complexes (1–5), Mn(III) complex 6 and Cr(III) complex 7 were synthesized. All of the synthesized ligands and complexes were well characterized by elemental analyses, FT-IR and 1H NMR (ESI†).
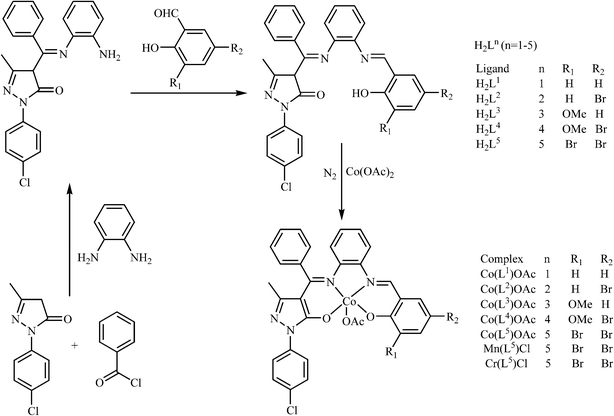 |
| | Scheme 1 Synthesis of ligands H2Ln and complexes 1–7. | |
3.2 X-ray single-crystal structures of complexes 3 and 4
The molecular structures of complexes [Co(L3)(OAc)(H2O)]·H2O ([3(H2O)]·H2O) and [Co(L4)(OAc)(H2O)]·EtOH ([4(H2O)]·EtOH) were determined by X-ray single-crystal diffraction analyses. Crystallographic data for the two complexes are presented in Table 1, and selected lengths and angles are given in Table 2.
Table 1 Crystal data and structure refinement for complexes [3(H2O)]·H2O and [4(H2O)]·EtOH
| Compound |
[3(H2O)]·H2O |
[4(H2O)]·EtOH |
| Formula |
C33H30ClCoN4O7 |
C35H33BrCoClN4O7 |
| Fw |
688.99 |
795.94 |
| Cryst syst |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
|
a, Å |
10.418(12) |
12.326(15) |
|
b, Å |
12.073(14) |
12.739(15) |
|
c, Å |
13.296(16) |
13.021(16) |
|
α, deg |
90.683(2) |
62.534(2) |
|
β, deg |
110.550(2) |
82.794(2) |
|
γ, deg |
96.916(2) |
79.008(2) |
|
V, Å3 |
1551.7(3) |
1779.1(4) |
|
Z
|
2 |
2 |
|
D
calc, g cm−3 |
1.475 |
1.486 |
| Cryst size, mm3 |
0.35 × 0.27 × 0.20 |
0.29 × 0.23 × 0.21 |
| Temp, K |
296(2) |
296(2) |
|
F(000) |
712 |
812 |
|
μ, mm−1 |
0.695 |
1.733 |
|
θ range, deg |
2.11–27.21 |
1.82–26.06 |
| Reflns measd |
8839 |
9352 |
| Reflns used |
6387 |
6701 |
| Params |
417 |
443 |
|
R (I > 2σ(I)) |
R
1 = 0.0512 |
R
1 = 0.0553 |
|
W
R
2 = 0.1486 |
W
R
2 = 0.1547 |
|
R (all data) |
R
1 = 0.0708 |
R
1 = 0.0923 |
|
W
R
2 = 0.1700 |
W
R
2 = 0.1834 |
|
S
|
1.054 |
0.878 |
Table 2 Selected bond lengths (Å) and bond angles (°) for [3(H2O)]·H2O and [4(H2O)]·EtOH
| [3(H2O)]·(H2O) |
[4(H2O)]·EtOH |
| Co(1)–N(3) 1.930(3) |
Co(1)–N(3) 1.920(4) |
| Co(1)–N(4) 1.883(3) |
Co(1)–N(4) 1.880(3) |
| Co(1)–O(1) 1.955(2) |
Co(1)–O(1) 1.925(3) |
| Co(1)–O(2) 1.904(2) |
Co(1)–O(2) 1.872(3) |
| Co(1)–O(5) 1.885(2) |
Co(1)–O(3) 1.882(3) |
| Co(1)–O(6) 1.971(2) |
Co(1)–O(6) 1.956(3) |
| N(3)–Co(1)–N(4) 84.69(10) |
N(3)–Co(1)–N(4) 85.71(15) |
| N(3)–Co(1)–O(1) 94.02(10) |
N(3)–Co(1)–O(1) 95.00(13) |
| N(3)–Co(1)–O(2) 178.46(10) |
N(3)–Co(1)–O(2) 178.00(14) |
| N(3)–Co(1)–O(5) 93.00(10) |
N(3)–Co(1)–O(3) 92.71(14) |
| N(3)–Co(1)–O(6) 91.18(10) |
N(3)–Co(1)–O(6) 89.57(14) |
| N(4)–Co(1)–O(2) 94.14(10) |
N(4)–Co(1)–O(2) 94.50(14) |
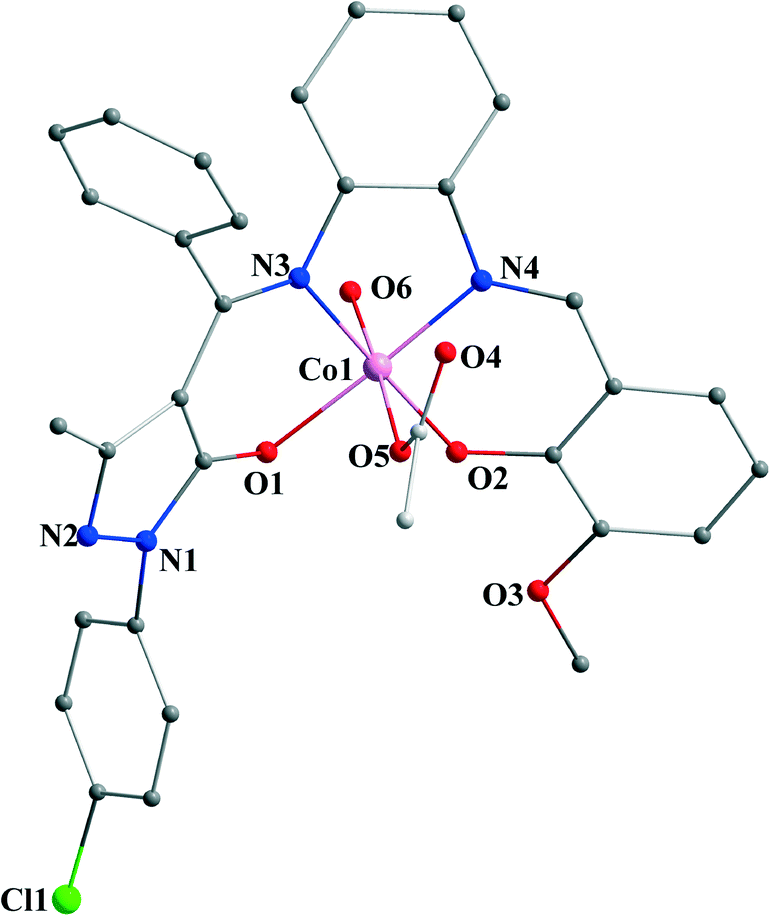
Complex [3(H2O)]·H2O crystallizes in the triclinic space group of P, and its structural unit is composed of one mononuclear [Co(L3)(OAc)(H2O)] framework and one solvated H2O molecule. As shown in Fig. 1, the central Co3+ ion (Co1) lies in a six-coordinated environment and adopts a slightly distorted octahedral geometry, where two N atoms (N3 and N4) and two O atoms (O1 and O2) from the asymmetrical bis-Schiff-base ligand H2L3 form an equatorial plane, two other O atoms (O5 from the coordinated OAc− anion and O6 from the coordinated H2O) occupy the two axial positions of the coordination polyhedron. The distortion to the octahedron is primarily caused by the non-planar conformation of the equatorial planes with the mean deviations of 0.08–0.16 Å and the dihedral angle of 29.7 between the two stable delocalized six-membered (CoNCCCO) chelate rings and two N–Co–O bite angles (N3–Co1–O1 and N4–Co1–O2) of 94.02(10) ° and 94.14(10) °, respectively. The existence of coordinated OAc− anion at the axial position suggested the formation of the oxidized Co(III) complex 3. The solvated H2O molecule is not bound to the monomer framework and no interaction with the host structure is observed.
 |
| | Fig. 1 Perspective drawing of [3(H2O)]·H2O. H atoms and solvated molecules are omitted for clarity. | |
Complex [4(H2O)]·EtOH crystallizes in the triclinic space group of P, and its structural unit is composed of one mononuclear [Co(L4)(OAc)(H2O)] framework and one solvated EtOH molecule. As shown in Fig. 2, the central Co3+ ion (Co1) lies in a six-coordinated environment and adopts a slightly distorted octahedral geometry, where two N atoms (N3 and N4) and two O atoms (O1 and O2) from the asymmetrical bis-Schiff-base ligand H2L4 form an equatorial plane, two other O atoms (O3 from the coordinated OAc− anion and O6 from the coordinated H2O) occupy the two axial positions of the coordination polyhedron. The distortion to the octahedron is primarily caused by the no-planar conformation of the equatorial planes with the mean deviations of 0.08–0.14 Å and the dihedral angle of 25.9° between the two stable delocalized six-membered (CoNCCCO) chelate rings and two N–Co–O bite angles (N3–Co1–O1 and N4–Co1–O2) of 95.00(13)° and 94.50(14)°, respectively. The existence of coordinated OAc− anion at the axial position suggested the formation of the oxidized Co(III) complex 4. The solvated EtOH molecular is not bound to the monomer framework and no interaction with the host structure is observed.
 |
| | Fig. 2 Perspective drawing of [4(H2O)]·EtOH. H atoms and solvated molecules are omitted for clarity. | |
3.3 Alternating copolymerization studies of SO and MA catalyzed by complexes 1–7
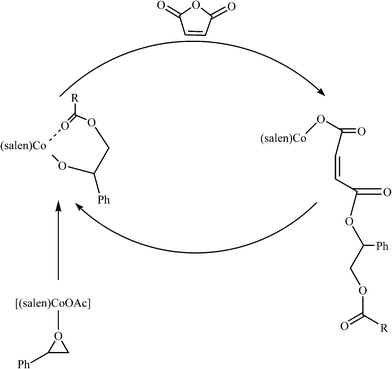
As shown in Scheme 2, the bulk or solution copolymerization behavior of SO and MA monomers employing complexes 1–7 as catalysts in the presence of co-catalyst (DMAP, Ph3P or PPN+Cl−) are examined, and the results are summarized in Table 3. In comparison, the blank experiments in bulk copolymerization without catalyst and co-catalyst (entry 1 in Table 3) show no copolymers obtained. Simultaneously, co-catalyst DMAP alone (entry 2 in Table 3) or catalyst 7 alone (entry 3 in Table 3) are not very active (SO conversions 11–14%); thus, lower activity can be observed, and the lower molecular weight of copolymers (Mn = 1000–1600 g mol−1) obtained contain a high percentage of ether linkages (55–63%). In contrast, the incorporation of one of complexes 1–7 as the catalyst and DMAP as the co-catalyst (other entries in Table 3) not only accelerates the copolymerization, but also significantly stimulates the formation of ester linkages. Based on the experiments, we propose that a copolymer was generated via the mechanism proposed in Scheme 3. The cobalt(III) complex acts as a Lewis acid and activates an epoxide for ring-opening to form a cobalt(III) alkoxide, then the cobalt(III) alkoxide reacts with anhydride to form a polymer.
 |
| | Scheme 2 Synthesis of polyesters from SO and MA using 1–7 as catalysts. | |
Table 3 Copolymerization of SO and MA catalyzed by catalysts 1–5
| Entry |
Monomer |
Method |
Cat |
Co-cat |
Molar ratio |
Time |
T
|
Conv.a |
Ethera |
M
n
|
PDIb |
T
g
|
|
|
M1 + M2 |
|
|
|
M1:M2:Cat:Co-cat |
min |
°C |
% |
% |
|
|
|
|
Determined by 1H NMR.
Determined by GPC.
Determined by DSC.
|
| 1 |
SO + MA |
Bulk |
— |
— |
250:250:0:0 |
150 |
110 |
— |
— |
— |
— |
— |
| 2 |
SO + MA |
Bulk |
— |
DMAP |
250:250:0:1 |
150 |
110 |
14 |
55 |
1000 |
1.56 |
25 |
| 3 |
SO + MA |
Bulk |
7
|
— |
250:250:1:0 |
150 |
110 |
11 |
63 |
1600 |
1.37 |
27 |
| 4 |
SO + MA |
Bulk |
1
|
DMAP |
250:250:1:1 |
150 |
110 |
89 |
6 |
2600 |
1.11 |
22 |
| 5 |
SO + MA |
Bulk |
2
|
DMAP |
250:250:1:1 |
150 |
110 |
92 |
11 |
3000 |
1.28 |
27 |
| 6 |
SO + MA |
Bulk |
3
|
DMAP |
250:250:1:1 |
150 |
110 |
83 |
<1 |
3300 |
1.24 |
29 |
| 7 |
SO + MA |
Bulk |
4
|
DMAP |
250:250:1:1 |
150 |
110 |
90 |
<1 |
3400 |
1.26 |
32 |
| 8 |
SO + MA |
Bulk |
5
|
DMAP |
250:250:1:1 |
150 |
110 |
92 |
5 |
3800 |
1.15 |
35 |
| 9 |
SO + MA |
Bulk |
6
|
DMAP |
250:250:1:1 |
150 |
110 |
95 |
<1 |
4000 |
1.12 |
35 |
| 10 |
SO + MA |
Bulk |
7
|
DMAP |
250:250:1:1 |
150 |
110 |
98 |
<1 |
4700 |
1.09 |
35 |
 |
| | Scheme 3 Proposed mechanism for the copolymerization of SO and MA (R = alkyl or polymer). | |
3.3.1 Effect of catalyst structure of complexes 1–7 on catalytic copolymerization of SO and MA.
To study the effect of the electronic and steric environment of a series of asymmetrical bis-Schiff-base complexes [Co(Ln)(OAc)] (n = 1–5, 1–5) on the catalytic performance, the copolymerization (entries 4–8 in Table 3) of SO and MA are carried out in bulk with DMAP as the co-catalyst under the stipulated molar ratio (250:250:1:1) of SO:MA:catalyst:co-catalyst at polymerization temperature of 110 °C for 150 min. As expected, catalyst 1 gives significantly higher activity of SO (89% of SO conversion) and better selectivity (6% of ether content) in the presence of co-catalyst DMAP. Interestingly, the utilization of the electron-withdrawing Br substituent para to the phenoxide group in catalyst 2 (R1 = Br, R2 = H), when compared to catalyst 1 (R1 = H, R2 = H), gives relatively higher reactivity (92% of SO conversion), which is possibly due to the increase of electrophilicity of Co3+ ion in 2 due to the delocalization of electron conjugation. Conversely, the electron-donating MeO substituent ortho to the phenoxide group in catalyst 3 (R1 = H, R2 = MeO) has a negative effect on the catalytic activity (83% of SO conversion). With regards to the effect of both the electron-withdrawing Br substituent at the para orientation and the electron-donating MeO substituent at the ortho orientation to the phenoxide group in catalyst 4 (R1 = Br, R2 = MeO) or two electron-withdrawing Br substituents at the para and ortho orientations to the phenoxide group in catalyst 5 (R1 = Br, R2 = Br), a similar trend in catalytic activity is observed, in which catalyst 5 proves to be more active (92% of SO conversion) for SO–MA copolymerization, while the activity (90% of SO conversion) of catalyst 4 is almost identical to that of catalyst 1. To study the effect of the metal of the catalyst 5 on the catalytic performance, we decided to vary the metal in the catalyst 5. Of the three catalysts, the chromium-based catalyst 7 exhibited the best performance under the applied conditions, followed by the manganese-based catalyst 6. The cobalt-based catalyst 5 showed the least activity under the applied conditions. The polymer produced by 7/DMAP appears to be an almost perfectly alternating CHO–MA copolymer (<1% of the ether content shown in Fig. 3), which is in agreement with the MALDI-TOF-MS spectrum (Fig. 4) of the product, where an m/z interval of 218 between the consecutive peaks corresponding to the addition of a [SO + MA] repeating unit (entry 10 in Table 3) is observed. These are obscured in the MALDI-TOF-MS spectrum by the intrinsically charged DMAP-end capped chains. The glass transition temperatures of the copolymers were determined by differential scanning calorimetry analysis. These data are summarized in Table 3. Specifically, the glass transition temperature found for the polyesters formed from SO and MA with catalysts 1–7 were 22, 27, 29, 32, 35, 35, and 35 °C, respectively. There is a slight fluctuation in the Tgs of the copolymers, which is most probably the result of the low molecular weight of the copolymers and possibly the inclusion of the traces of monomer.
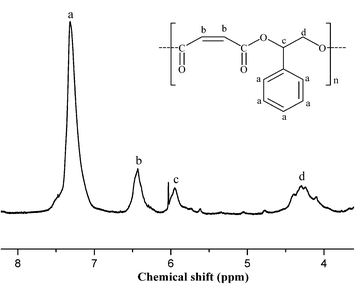 |
| | Fig. 3 Representative 1H NMR spectrum of SO–MA copolymer, Table 3, entry 10. | |
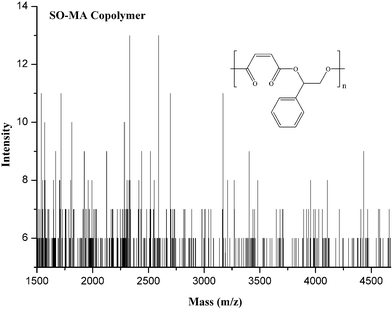 |
| | Fig. 4 Representative MALDI-TOF-MS spectrum of SO–MA copolymer, Table 3, entry 10. | |
3.3.2 Effect of copolymerization procedures of catalyst 7 on the catalytic copolymerization of SO and MA.
Because the chromium-based catalyst 7 proved to be the most active of all, catalyst 7 was chosen for further studies with different copolymerization procedures. The results are listed in Table 4. The typical solution polymerization (entry 1 in Table 4) from 7/DMAP under the stipulated molar ratio (250:250:1:1) of SO:MA:catalyst:co-catalyst at the polymerization temperature of 110 °C for 300 min also produces the expected perfectly alternating SO–MA copolymer, and also leads to both higher SO conversion (95%) and the largest copolymer Mn (5200 g mol−1, PDI = 1.07) than those in bulk as the general feature.12a It is evident that the asymmetrical catalyst 7 is better than similar catalyst described in the literature for SO–MA copolymerization (Mn = 1420 g mol−1, PDI = 1.53).11 To study the effect of co-catalysts on catalytic behavior, two other types of co-catalysts including Ph3P and PPN+Cl− are compared in combination with the catalyst 7 for the solution ring-opening copolymerization of SO–MA (2–3 in Table 4). Compared with the effective catalysis of DMAP, no copolymer was obtained with Ph3P as co-catalyst, and this can be attributed to the large cone angle of three steric phosphines in Ph3P considerably lowering its nucleophilicity. Onium salt PPN+Cl− had shown considerable higher activity as a co-catalyst than Ph3P. In agreement with the common feature of the variation of catalyst and co-catalyst concentration on the catalytic activity,12,13 for DMAP, there is a slight decrease in the reactivity with increasing or decreasing catalyst and co-catalyst concentration (entries 4–5 in Table 4). The polymer molecular weight (2900–3700 g mol−1) also decreased due to the chain transfer effect of excess of DMAP or the insufficient reaction of less DMAP.13b Considering the effect of the reaction temperature on the polymerization behavior (entries 6–7 in Table 4), a lower temperature (90 °C) results in lower polymerization rate (56% of CHO conversion) and lower polymer molecular weight (Mn = 2400 g mol−1). It indicates that a lower reaction temperature shows negative influence on the reaction rate constant under the consistent conditions. According to Arrhenius equation, an elevated temperature favors most of the reactions to a certain degree because their activation energy is usually positive.21 It appears that higher temperature is more desirable for ring-opening copolymerization of SO–MA, but the active decline of the catalyst 7 is caused by higher temperatures (130 °C). The effect of polymerization time on the catalytic activity is similar to that of the polymerization temperature. The catalytic activity increases at first from 150 min to 300 min, then decreases from 300 min to 450 min, and the peak appears at 300 min. The relatively lower polymer molecular weight (Mn = 2400 g mol−1) and a larger PDI (1.33) than those (Mn = 5200 g mol−1 and PDI = 1.07) at the reaction time of 300 min from the same 7/DMAP system along with the higher ether content (20%) of poly(ester-co-ether) are observed for the longest reaction time of 450 min, which should also be due to the chain transfer effect of DMAP with the longest reaction time. Compared to the run without chain transfer agents (Table 4, entry 1), the addition of benzyl alcohol (Table 4, entry 10) or n-butyl alcohol (Table 4, entry 11) as chain transfer agents in all the cases resulted in the expected reduction of molecular weight (Mn = 3000–3100 g mol−1 and PDI = 1.53–1.56), whereas the conversions remained comparable. We assumed that the chelating effects of the chain transfer agents on the copolymerization of SO and MA might deactivate the catalyst, but it plays only a limited role in this catalyst system.
Table 4 Copolymerization of SO and MA catalyzed by catalyst 7 from different polymerization procedures
| Entry |
Method |
Co-cat |
Molar ratio |
Time |
T
|
Conv.a |
Ethera |
M
n
|
PDIb |
T
g
|
| M1:M2:Cat:Co-cat |
min |
°C |
% |
% |
|
Determined by 1H NMR.
Determined by GPC.
Determined by DSC.
Reaction conditions: solution, [SO]:[MA]:[7]:[DMAP]:[benzyl alcohol] = 250:250:1:1:10.
Reaction conditions: solution, [SO]:[MA]:[7]:[DMAP]:[n-butyl alcohol] = 250:250:1:1:10.
|
| 1 |
Solution |
DMAP |
250:250:1:1 |
300 |
110 |
95 |
<1 |
5200 |
1.07 |
35 |
| 2 |
Solution |
Ph3P |
250:250:1:1 |
300 |
110 |
— |
— |
— |
— |
— |
| 3 |
Solution |
PPN+Cl− |
250:250:1:1 |
300 |
110 |
86 |
7 |
4300 |
1.16 |
34 |
| 4 |
Solution |
DMAP |
150:150:1:1 |
300 |
110 |
90 |
11 |
3700 |
1.38 |
33 |
| 5 |
Solution |
DMAP |
500:500:1:1 |
300 |
110 |
72 |
13 |
2900 |
1.24 |
32 |
| 6 |
Solution |
DMAP |
250:250:1:1 |
300 |
90 |
56 |
18 |
2400 |
1.14 |
20 |
| 7 |
Solution |
DMAP |
250:250:1:1 |
300 |
130 |
71 |
12 |
4500 |
1.33 |
33 |
| 8 |
Solution |
DMAP |
250:250:1:1 |
150 |
110 |
82 |
6 |
2700 |
1.41 |
33 |
| 9 |
Solution |
DMAP |
250:250:1:1 |
450 |
110 |
97 |
20 |
2400 |
1.33 |
33 |
| 10d |
Solution |
DMAP |
250:250:1:1 |
300 |
110 |
84 |
15 |
3100 |
1.56 |
23 |
| 11e |
Solution |
DMAP |
250:250:1:1 |
300 |
110 |
82 |
19 |
3000 |
1.53 |
31 |
4. Conclusion
In conclusion, we have significantly demonstrated the higher activity of asymmetrical Schiff-base catalysts 1–7 compared to the similar catalysts described in the literature for the ring-opening copolymerization of SO with MA. Among the seven catalysts applied, the chromium catalyst 7 proved to be the most active. The MALDI-TOF-MS spectrum of the copolymer formed by 7/DMAP showed a completely alternating microstructure. Of three co-catalysts tested, DMAP exhibited the highest activity, PPN+Cl− showed a relatively higher activity than Ph3P, which exhibited a considerably low activity. The variation of the monomer to catalyst ratio, polymerization time and polymerization temperature affected the epoxide–anhydride copolymerization in a similar way as observed for the corresponding SO–MA copolymerization. Upon investigating the role of chain transfer agents in the alternating ring-opening copolymerization of SO with MA, it was found that chain transfer agents resulted in the reduction of molecular weight, whereas the conversions remained comparable. Further studies that are focused on increasing the efficiency and expanding the monomer scope with the asymmetrical Schiff-base metal catalysts are currently in progress.
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation (21373160), the Scientific Research Program Foundation of Shaanxi Provincial Education Department (2013JK0701) and the Scientific Research Culture Foundation of Xi'an University of Science and Technology (201117).
References
- M. J.-L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC.
- J. Luston and F. Vass, Adv. Polym. Sci., 1984, 56, 92–132 CrossRef.
- J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2(586) DOI:10.1038/ncomms1596.
-
(a) T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef CAS;
(b) T. Aida, K. Sanuki and S. Inoue, Macromolecules, 1985, 18, 1049–1055 CrossRef CAS;
(c) S. Asano, T. Aida and S. Inoue, Macromolecules, 1985, 18, 2057–2061 CrossRef CAS;
(d) T. Aida, Y. Maekawa, S. Asano and S. Inoue, Macromolecules, 1988, 21, 1195–1202 CrossRef CAS.
- Y. Maeda, A. Nakayama, N. Kawasaki, K. Hayashi, S. Aiba and N. Yamamoto, Polymer, 1997, 38, 4719–4725 CrossRef CAS.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
-
(a) A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed;
(b) A. Bernard, C. Chatterjee and M. H. Chisholm, Polymer, 2013, 54, 2639–2646 CrossRef CAS PubMed;
(c) N. D. Harrold, Y. Li and M. H. Chisholm, Macromolecules, 2013, 46, 692–698 CrossRef CAS;
(d) S. Huijser, E. H. Nejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
-
(a) E. H. Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef CAS;
(b) E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS;
(c) D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- C. Robert, T. Ohkawara and K. Nozaki, Chem. – Eur. J., 2014, 20, 4789–4795 CrossRef CAS PubMed.
- E. H. Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC.
- P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2012, 256, 953–1114 CrossRef CAS PubMed.
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2008, 252, 1871–1995 CrossRef CAS PubMed.
-
(a) S. Huijser, B. B. P. Staal, J. Huang, R. Duchateau and C. E. Koning, Biomacromolecules, 2006, 7, 2465–2469 CrossRef CAS PubMed;
(b) S. Huijser, B. B. P. Staal, J. Huang, R. Duchateau and C. E. Koning, Angew. Chem., Int. Ed., 2006, 45, 4104–4108 CrossRef CAS PubMed.
- D.-F. Liu, L.-Y. Wu, W.-X. Feng, X.-M. Zhang, J. Wu, L.-Q. Zhu, D.-D. Fan, X.-Q. Lü and Q. Shi, J. Mol. Catal. A: Chem., 2014, 382, 136–145 CrossRef CAS PubMed.
- J. Wu, D.-F. Liu, L.-Y. Wu, X.-M. Zhang, L.-Q. Zhu, D.-D. Fan, X.-Q. Lü and Q. Shi, J. Organomet. Chem., 2014, 749, 302–311 CrossRef CAS PubMed.
- L.-Q. Zhu, D.-F. Liu, L.-Y. Wu, W.-X. Feng, X.-M. Zhang, J. Wu, D.-D. Fan, X.-Q. Lü, R. Lu and Q. Shi, Inorg. Chem. Commun., 2013, 37, 182–185 CrossRef CAS PubMed.
-
(a) S. Huijser, B. B. P. Staal, J. Huang, R. Duchateau and C. E. Koning, Biomacromolecules, 2006, 7, 2465–2469 CrossRef CAS PubMed;
(b) S. Huijser, B. B. P. Staal, J. Huang, R. Duchateau and C. E. Koning, Angew. Chem., Int., Ed., 2006, 45, 4104–4108 CrossRef CAS PubMed.
-
G. M. Sheldrick, SHELXL-97: Program for Crystal Structure Refinement, Göttingen, Germany, 1997 Search PubMed.
-
G. M. Sheldrick, SADABS, University of Göttingen, 1996 Search PubMed.
- S.-B. Tan, J.-J. Li and Z.-C. Zhang, Macromolecules, 2011, 44, 7911–7916 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic files of complexes [3(H2O)]·H2O and [4(H2O)]·EtOH in CIF format, CCDC 1013801 for [3(H2O)]·H2O, 1013802 for [4(H2O)]·EtOH. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cy01064g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.