
Solvent-free oxidation of ethylbenzene over hierarchical flower-like core–shell structured Co-based mixed metal oxides with significantly enhanced catalytic performance†
Renfeng
Xie
,
Guoli
Fan
,
Lan
Yang
and
Feng
Li
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, P. O. BOX 98, Beijing, 100029, PR China. E-mail: lifeng@mail.buct.edu.cn; Fax: +8610 64425385; Tel: +8610 64451226
First published on 17th September 2014
Abstract
Herein, we reported the development of new and cost-effective cobalt-based metal oxide catalysts for the oxidation of ethylbenzene, which is considered to be of much importance for the production of high value-added raw materials. The heterogeneous Co-based catalyst system, hierarchical flower-like core–shell structured Co–Zn–Al mixed metal oxides supported on alumina (CoZnAl-MMO/Al2O3), was reproducibly prepared by a two-step process, which involved in situ growth of a two-dimensional Co–Zn–Al layered double hydroxide precursor on amorphous alumina microspheres followed by calcination. The materials were characterized by XRD, SEM, TEM, HRTEM, TPR, XPS and nitrogen adsorption–desorption measurement. The results revealed that CoZnAl-MMO/Al2O3 catalysts exhibited high dispersion of cobalt species due to well-developed three-dimensional flower-like CoZnAl-MMO platelets as well as the separating effect of the resulting ZnO phase. As-synthesized CoZnAl-MMO/Al2O3 catalysts were studied in the oxidation of ethylbenzene without the addition of any solvent and additive using tert-butyl hydroperoxide as the oxygen source and showed much higher catalytic activity and selectivity for acetophenone compared with the conventional supported Co-based catalyst prepared by incipient impregnation. Furthermore, such cost-effective CoZnAl-MMO/Al2O3 catalysts possessed high stability and could be reused at least three times without remarkable loss of the catalytic activity.
Introduction
The controlled partial oxidation of alkanes to produce desired products including alcohols, ketones and carboxylic acids, which are key building blocks in organic synthesis, is one of the most important industrial reactions. For instance, selective oxidation of ethylbenzene (EB) is of much importance for the production of high value-added acetophenone (AP),1,2 which is a useful raw material for the production of perfumes, pharmaceuticals, resins and alcohols. In this attractive field, numerous methods have been developed by using different oxidizing agents (e.g., molecular oxygen,3–5 hydrogen peroxide,6tert-butyl hydroperoxide (TBHP)7–9) and a variety of metal catalysts. As for the oxidation of EB, some homogeneous catalysts, such as metal acetylacetonates1 and metalloporphyrins,10,11 give excellent activity and selectivity.12 Despite their advantages, the problem of separation and recovery of homogenous catalysts, as well as high cost and easy deactivation, limits their large-scale practical industrial applications.Over the past decade, considerable attention has been drawn to the exploration of clean and environmentally friendly routes for selective oxidation of alkanes relying on a wide variety of heterogeneous catalyst systems, which can make the catalysts easy to recycle and regenerate. Recently, several heterogeneous catalysts, such as Mn-containing complexes supported on alumina/silica,13–15 supported Co-containing complexes,16–20 Mn-MCM-41,21,22 Cr-MCM-41,4 Co-HMS,23 M-APO-11 (M = Co, Mn and V),24 CeAPO-5 molecular sieves,25 hybrid metalloporphyrin@silica nanocomposite microspheres26 and Mn-, Cu-containing hydrotalcites,3,5,6,27 were reported for the oxidation of EB to AP. Among them, some catalysts have showed either good selectivity or good activity in the reaction, but the ability to achieve both excellent activity and high selectivity is more limited. Meanwhile, the practical application of some catalysts containing metal complexes is severely limited by their poor thermal stability under oxidative conditions.
Layered double hydroxides (LDHs, [M1−x2+Mx3+(OH)2]x+ [Ax/n]n−·mH2O) belong to a class of highly ordered two-dimensional (2D) anionic clay materials.28,29 Their structure is based on M2+(OH)6 octahedral units sharing edges to build M(OH)2 brucite-like layers in which trivalent cations can replace some of the divalent ions giving positively charged sheets, and the charge is balanced by intercalated anions in the hydrated interlayer regions.30,31 After calcination at intermediate temperatures (450–600 °C), LDHs are converted into highly active mixed metal oxides (MMO) with good thermal stability, large surface area, and high metal dispersion.32–35 Although some LDHs could catalyze the oxidation of EB,3,5,27 they exhibited moderate catalytic activity. Also, MMO derived from CuNiCr-LDHs could effectively catalyze the oxidation of EB with 60% conversion and 65% selectivity for AP at 70 °C for 8 h.9 The toxicity of such a type of Cr-containing catalysts, however, may cause severe environmental pollution.
Because of its excellent stability, good accessibility, high specific surface area, and excellent physical strength, alumina has been extensively used as a catalyst support. Although the dispersion of active metal components on alumina is often discussed in the literature, effective techniques to enhance metal dispersion are rarely reported. On the other hand, increasing environmental concerns with chemical production require a greener and energy-efficient process.36 Correspondingly, the use of neat substrates without any solvent and additive in the field of heterogeneous catalysis, which can save resources and energy, particularly is a subject of considerable interest from the standpoint of environmentally benign production.37,38 Therefore, in the present work, we synthesized a new heterogeneous catalyst system consisting of hierarchical flower-like core–shell structured CoZnAl-MMO supported on amorphous alumina (CoZnAl-MMO/Al2O3) by a two-step process, which involved in situ growth of 2D CoZnAl-LDH platelets on alumina microspheres and the following calcination. The catalytic performance of as-synthesized CoZnAl-MMO/Al2O3 catalysts was investigated in liquid-phase oxidation of EB using TBHP as an oxidant without the addition of any solvent and additive. To the best of our knowledge, there has been no report about cost-effective cobalt-based metal oxides with hierarchical flower-like microstructure for the oxidation of aromatic alkanes until now.
Results and discussion
Catalyst characterization
Fig. 1A depicts the powder XRD patterns of CoZnAl-LDH/Al2O3 catalyst precursors. It can be seen that all samples mainly present the characteristic (003), (006), and (012) peaks corresponding to the basal spacing and higher-order diffractions of NO3−-type hydrotalcite-like materials, similar to those typically reported in the literature.28 With the incorporation of Zn ions, the intensities of characteristic (003) and (006) diffraction peaks for the LDH phase are weakened, indicative of the decrease in the integrity of the layered structure. Meanwhile, when the [Zn2+]/([Co2+] + [Zn2+]) atomic ratio increases to 0.5, the impurity, zinc carbonate hydroxide (Zn4(CO3)(OH)6·H2O, JCPDS no. 03-0787), is also observed, as indicated by its characteristic diffraction peak at a 2θ value of about 13.0°.39 However, the diffractions related to the crystalline Al2O3 phase cannot be found in the patterns of samples, suggestive of its amorphous nature. The above results demonstrate that LDHs are successfully formed on the surface of Al2O3. | ||
| Fig. 1 XRD patterns of CoZnAl-LDH/Al2O3 precursors (A) and CoZnAl-MMO/Al2O3 (B). | ||
Fig. 1B shows the XRD patterns of calcined CoZnAl-LDH/Al2O3 samples. In the case of MMO-0/A sample, crystalline spinel phases, such as Co3O4 and/or CoAl2O4, can be observed. However, it is rather difficult to distinguish between these spinels due to their almost identical diffraction positions. Furthermore, some Co3+ cations can be replaced by Al3+ in spinels,40 and consequently, another type of spinel-like Co2+(Co3+, Al)2O4 phase may be present. With the increase in [Zn2+]/([Co2+] + [Zn2+]) atomic ratio to 0.5, the ZnO phase is obviously observed. In the case of MMO-0.75/A, only the ZnO phase is detected and no spinel phases are found, probably due to the high dispersion nature of cobalt-containing spinels. As for Im-MMO/A and MMO-0.5 comparison samples, the only indexed crystalline phases are Co-containing spinels (Fig. S1†).
The morphology of the representative MMO-0.5/A sample was characterized by FE-SEM. As shown in Fig. 2a, amorphous Al2O3 presents a uniform spherical morphology with an even surface and a mean diameter of about 3.0 μm. In the case of MMO-0.5/A, a large quantity of densely packed and interlaced nanoplatelets with visible edges and almost no ab-faces is distributed homogeneously over the Al2O3 support surface, and the crystallite size of nanoplatelets in the c-direction is about 10–20 nm. No agglomeration or sintering of adjacent platelets is observed. Moreover, it is interesting to note that the diameter of the resulting MMO-0.5/A increases to about 4.5 μm (Fig. 2b and c). A typical flower-like core–shell structure of the cracked MMO-0.5/A sample can be seen from the inset in Fig. 2c. Obviously the formed Al2O3 core is smaller than the pure Al2O3 microsphere due to the formation of the Al-containing CoZnAl-MMO shell. Elemental mapping indicates the homogeneous distributions of Co, Zn and Al species in the MMO-0.5/A sample (Fig. 3).
 | ||
| Fig. 2 FE-SEM images of samples: (a) Al2O3 microspheres; (b, c) MMO-0.5/A; (d) Im-MMO-0.5/A; (e) MMO-0.5. The inset in (c) shows a typical flower-like core–shell structure of cracked MMO-0.5/A. | ||
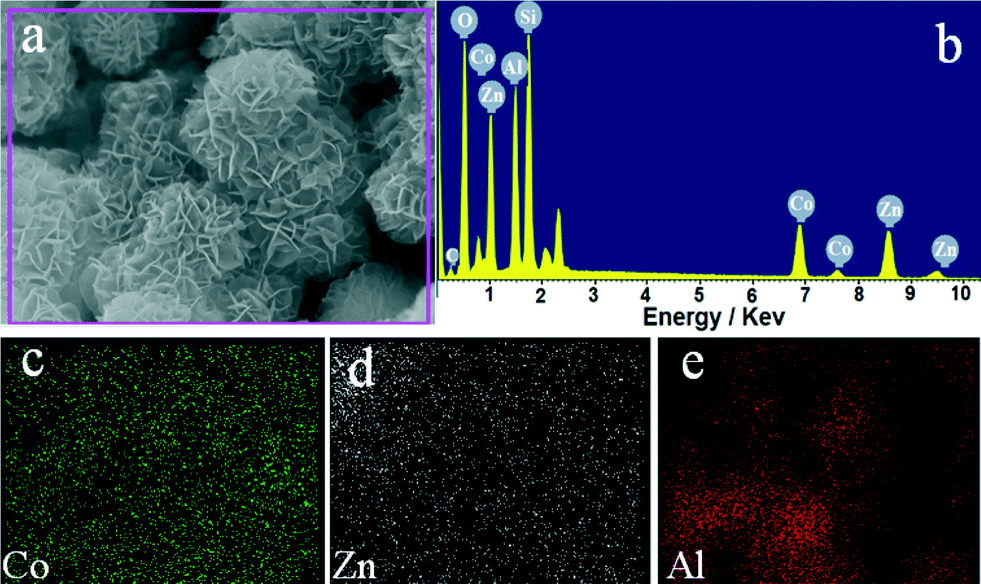 | ||
| Fig. 3 FE-SEM image (a), EDX spectrum (b) of the representative MMO-0.5/A sample and elemental mapping of Co (c), Zn (d), and Al (e). | ||
In addition, as for the anisotropic CoZnAl-MMO platelet in MMO-0.5/A, the size in the ab-direction is obviously larger than that in the c-direction. This is due to the fact that the initial CoZnAl-LDH crystallites all tend to grow in their ab-direction perpendicular to the Al2O3 substrate through a homogeneous nucleation mechanism41 in order to optimize their growth and avoid collisions between growing crystallites. It suggests that direct in situ growth of LDHs on the surface of Al2O3 facilitates attachment and stabilization of LDH particles, leading to the uniform distribution of CoZnAl-MMO platelets on the support and thus the formation of the hierarchical flower-like core–shell microstructure. In comparison, the FE-SEM image of Im-MMO-0.5/A depicts that some irregular aggregates composed of small particles are distributed randomly on the support (Fig. 2d). Also, unsupported MMO-0.5 presents the morphology of uniform hexagonal platelets with a mean lateral size of about 200 nm and a thickness of about 50 nm (Fig. 2e).
Further, the TEM and HRTEM images of representative MMO-0.5/A (Fig. 4a–c) undoubtedly confirm a well-defined flower-like core–shell microstructure, where the amorphous Al2O3 core is coated by the vertically oriented and interlaced platelet-like MMO thin flakes. In addition, an HRTEM image of a single nanoflake depicts the lattice fringes with interplanar spacings of about 2.86 Å and 2.45 Å (Fig. 4d), corresponding to the (220) and (311) planes of the Co3O4 spinel phase, respectively.
 | ||
| Fig. 4 TEM (a, b, c) and HRTEM (d) images of the representative MMO-0.5/A sample. | ||
To investigate the textural properties of Al2O3 caused by loading CoZnAl-MMO, N2 adsorption–desorption isotherms of samples were measured (Fig. 5A). All of the isotherms exhibit intermediate isotherms between type II (absence of a plateau at high P/P0) and type IV category (low N2 adsorption at low P/P0),42,43 with a large hysteresis loop in the relative pressure range of 0.45 to 1.0, indicating the presence of capillary condensation in the mesopore structure. According to the IUPAC classification,44 all samples present a mixture hysteresis loop of H2 and H3 types with an abrupt closure at a relative pressure of 0.5 due to the tensile strength effect, suggesting the presence of slit-shaped and ink-bottle pores in the whole mesopore network associated with the aggregates of plate-like particles on the surface of hierarchical core–shell microstructures. Moreover, CoZnAl-MMO/Al2O3 samples exhibit a narrow pore size distribution ranging from 2 to 5 nm (Fig. 5B). Compared with Zn-free MMO-0/A, Zn-containing samples have a larger specific surface area and total pore volume (Table 1). The results demonstrate that part of the pores of Al2O3 support can be blocked by the in situ formed well-developed 2D LDH microcrystallines. As for the Im-MMO-0.5/A comparison sample (Fig. S2†), the much smaller surface area and pore volume may be the result of the absence of surface flower-like micro-nanostructures as well as aggregation of particles.
 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms (A) and the pore size distributions (B) of MMO-0/A (a), MMO-0.25/A (b), MMO-0.5/A (c), and MMO-0.75/A (d). | ||
It is well known that the oxidation states of active metal species play an important role in catalytic activity in oxidation reactions. H2-TPR measurements were carried out to determine different forms of oxides in MMO-x/A samples. As shown in Fig. 6, the reduction of MMO-x/A samples strongly depends on the Co content. In the case of MMO-0/A, because the shapes of reduction profiles are not symmetrical and reduction peaks possess shoulders, the broad peak is fitted to four peaks by deconvoluting the original profiles with the elevated temperature. The first small peak at about 340 °C is assigned to the reduction of surface Co3+ species;45 the second peak at about 390 °C represents the reduction of Co3+ to Co2+ species in the bulk phase; the third peak at about 458 °C is associated with the further reduction of Co2+ to Co0 species; the fourth small peak at about 515 °C is ascribed to the reduction of large cobalt-containing spinel particles. Interestingly, it is noted that the reduction temperature for MMO-x/A samples presents an increasing trend with the introduction of Zn due to the formation of (Co2+, Zn)Al2O4 or (Co2+, Zn) (Co3+, Al)2O4 spinel-like phases. In the case of CoZnAl-LDH/Al2O3-derived samples, Co2+ and Co3+ ions in spinel phases can interact with neighboring groups through the Co–O bonds polarized by Al3+ or Zn2+ ions in metal oxides formed through the decomposition of the LDH phase, thus hindering the reduction of cobalt species. Furthermore, the interlaced platelet-like microstructure is helpful to resist aggregation and sintering of MMO particles. For example, in the case of MMO-0.5/A, the peak temperatures related to reduction of Co3+ to Co2+ and Co2+ to Co0 increase to about 507 and 590 °C, respectively. Also, the peak areas are much smaller than those for MMO-0/A in spite of the absence of small amounts of highly dispersed Co species and large oxide crystallites. Such increased reduction difficulty for MMO-x/A samples with a lower cobalt content may be attributed to the higher dispersion of Co species due to the separating effect of the ZnO phase around active Co-containing species, which inhibits H2 approach to Co2+/Co3+ species and thus delays their reduction.42 Considering the similar aluminum content in these samples, it appears that the addition of Zn plays an important role in the improvement of cobalt dispersion. Meanwhile, Zn present in the LDH-derived samples is responsible for their high surface areas (Table 1).
 | ||
| Fig. 6 H2-TPR profiles of MMO-0/A (a), MMO-0.25/A (b), MMO-0.5/A (c), and MMO-0.75/A (d). | ||
In comparison, Im-MMO-0.5/A (Fig. S3†) presents a typical reduction behavior of Co3O4.46 However, the reduction temperatures are higher than those of Co3O4, which is ascribed to the presence of a smaller amount of Co3O4 particles weakly interacting with support as well as the aggregation of Co3O4 particles. In contrast, the reduction of unsupported MMO-0.5 involves several complex steps covering a wide range from 250 to 750 °C, which correspond to the reduction of different superficial Co species. The low-temperature reduction from 250 to 500 °C is associated with the reduction of surface Co-containing spinel particles, because hydrogen is easily able to access and reduce these superficial cobalt specials.47,48 The high-temperature reduction above 500 °C, which occurs at much higher temperatures than those for MMO-x/A samples, is assigned to the reduction of large Co-containing spinel particles. The higher area of high-temperature peaks than that of low-temperature peaks should be mainly due to severe aggregation of the spinel particles.
XPS analyses were performed on the MMO-0.5/A sample (Fig. 7). Core levels of Co 2p, Zn 2p, Al 2p and O 1s can be identified from the XPS survey spectra. It can be seen that the XPS of the Co 2p 3/2 region can be fitted into three contributions. The first intense peak with a binding energy (BE) value of about 779.9 eV is assigned to Co 2p3/2 for Co3+ cations.49,50 The presence of Co2+ species can be confirmed by two peaks at 781.6 and 786.8 eV for Co 2p3/2 and its shake-up satellite, respectively, due to the fact that the low-spin Co3+ cation only leads to much weaker satellite features than does high-spin Co2+ with unpaired valence 3d electron orbitals.51 The BE value of the Zn 2p3/2 peak is about 1021.5 eV, indicative of the presence of Zn2+ species.52 The fine spectrum of Al 2p shows a peak at around 74.2 eV, which is related to the Al3+ species bonded to oxygen.53 From the O 1s spectrum, it is seen that there are two fitted peaks representing two different types of surface oxygen species. There is general agreement between the literature and the present results such that the intense peak with a lower BE at around 530.5 eV is characteristic of lattice oxygen bound to metal cations of the spinel structure, while the small peak with a higher BE at 532.2 eV belongs most likely to surface oxygen,54 including mainly oxygen species of hydroxyl groups.55 The aforementioned results are in good consistency with the XRD and TPR results.
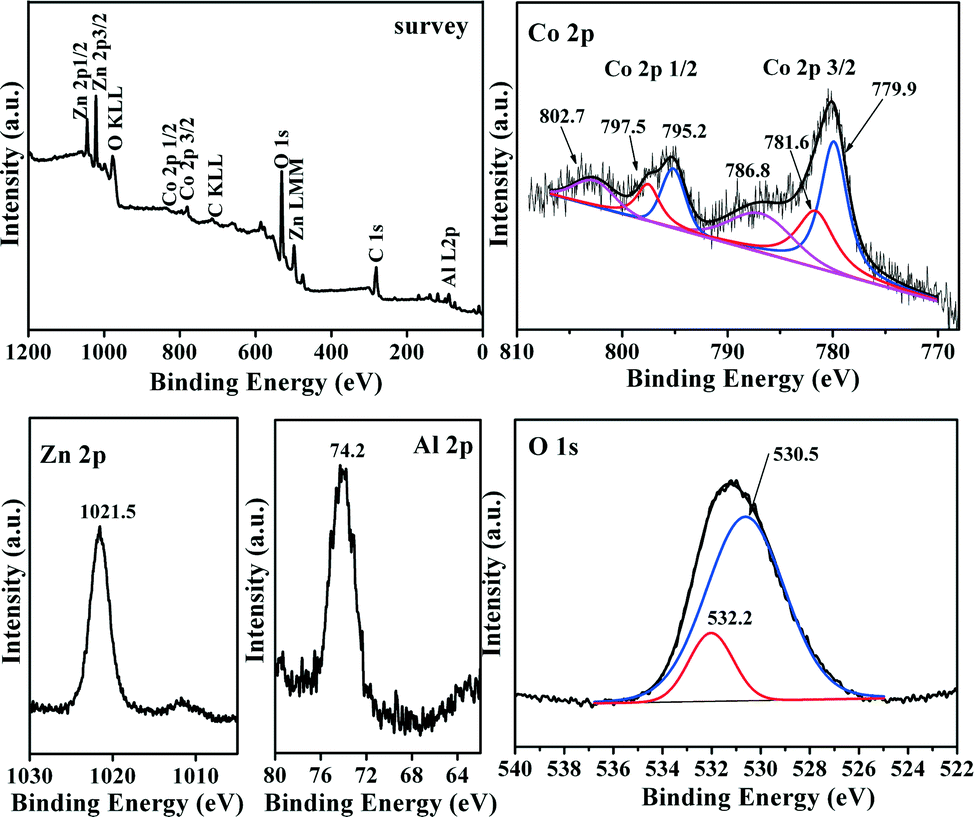 | ||
| Fig. 7 XPS spectra of the survey, Co 2p, Zn 2p, Al 2p, and O 1s of MMO-0.5/A sample. | ||
Catalytic activity of MMO-x/A catalysts
The aerobic oxidation of EB catalyzed by hierarchical flower-like core–shell MMO-x/A catalysts with TBHP was investigated. The products are mainly AP, benzaldehyde (BZ) and benzoic acid (BA), and no oxidation products of the aromatic ring are found. Fig. 8 presents the reaction pathway that proceeds during EB oxidation. | ||
| Fig. 8 Reaction pathway for the oxidation of EB. | ||
The activities of MMO-0.5/A, Im-MMO-0.5/A, MMO-0.5 and blank for the oxidation of EB were measured. As shown in Fig. 9, only 5.2% EB conversion is obtained after 12 h in the absence of the catalyst. In contrast, MMO-0.5/A shows much higher activity for selective oxidation of EB than Im-MMO-0.5/A or MMO-0.5, although MMO-0.5/A has a similar or much lower amount of Co loading.
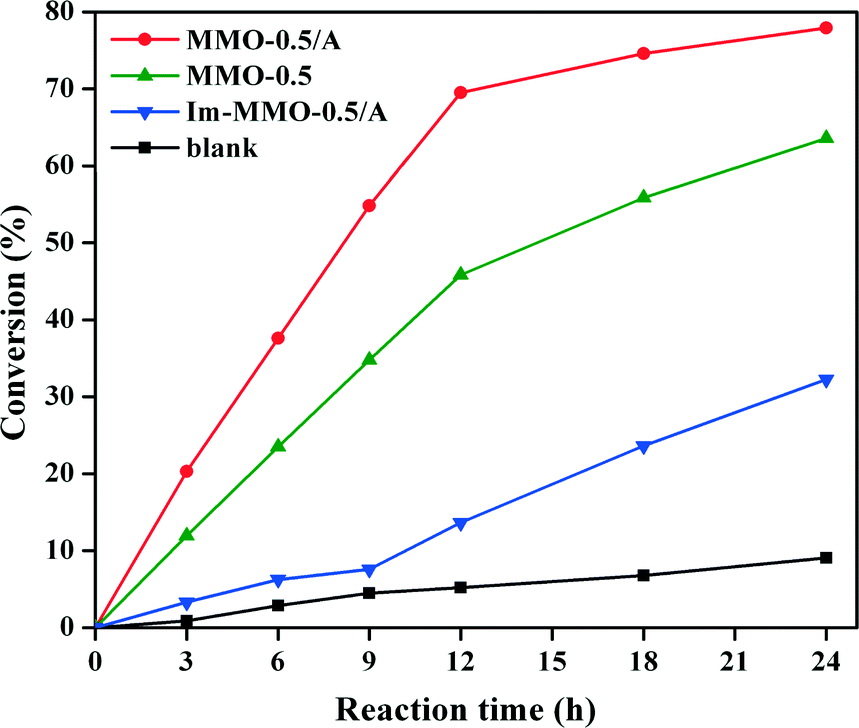 | ||
Fig. 9 Comparison of the catalytic performance of MMO-0.5/A, Im-MMO-0.5/A, and MMO-0.5 for oxidation of EB. Reaction conditions: EB, 10 mmol; catalyst, 0.1 g; reaction temperature, 120 °C; EB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TBHP molar ratio, 1:3. :TBHP molar ratio, 1:3. | ||
The reaction results over different MMO-x/A catalysts are summarized in Table 2. It is found that MMO-x/A catalysts exhibit a remarkable difference in the catalytic performance, and both the conversion of EB and the selectivity for AP increase with the decrease in Co content when the [Zn2+]/([Zn2+] + [Co2+]) molar ratio is lower than 0.5. On the contrary, both the conversion and the selectivity for AP decrease with the decrease in Co content when the [Zn2+]/([Zn2+] + [Co2+]) molar ratio is higher than 0.5. For the MMO-0.5/A catalyst, the maximum conversion of EB reaches 69.5% and the selectivity for AP is about 80.4%. In addition, it is worth noting that after 12 h of reaction, the turnover number (TON) value of MMO-0.5/A is 62.0, which is about 5 times higher than those observed for Im-MMO-0.5/A and MMO-0.5 comparison catalysts.
| Catalysts | Cob (wt%) | Conversion (%) | Selectivity (mol%) | TON (mol mol−1) | ||
|---|---|---|---|---|---|---|
| AP | BZ | BA | ||||
|
a Reaction conditions: EB, 10 mmol; catalyst, 0.1 g; reaction temperature, 120 °C; EB:TBHP molar ratio, 1:3; reaction time, 12 h.
b Measurement by ICP-AES.
c Other product: 1-phenethyl alcohol.
|
||||||
| Blank | — | 5.2 | 43.3 | 36.9 | 19.8 | — |
| MMO-0/A | 12.8 | 52.1 | 76.4 | 18.3 | 5.3 | 24.0 |
| MMO-0.25/A | 9.5 | 54.8 | 79.1 | 12.9 | 8.0 | 34.0 |
| MMO-0.5/A | 6.6 | 69.5 | 80.4 | 12.1 | 7.5 | 62.0 |
| MMO-0.75/A | 2.1 | 22.6 | 60.1 | 29.6 | 10.3 | 63.4 |
| Im-MMO-0.5/Ac | 6.7 | 13.6 | 23.6 | 45.6 | 12.4 | 12.0 |
| MMO-0.5 | 25.9 | 45.8 | 51.6 | 38.5 | 9.9 | 10.4 |
In addition, compared with other catalysts reported in the previous literature,7,8,12,13,18–20,56–62 the as-synthesized MMO-0.5/A catalyst behaves in a comparable way or even better in most cases, in spite of different reaction conditions. To the best of our knowledge, this is the best result for the oxidation of EB to AP over Co-based metal oxide catalysts.
A number of research studies have revealed that ZnO can function as a physical spacer among Cu nanoparticles to achieve a high copper surface area.63,64 However, large ZnO particles can be formed under high calcination temperatures, which results in reduced activity.65 In our case, a similar result is obtained with the addition of zinc. Interestingly, note that the introduction of an appropriate amount of zinc into MMO-x/A greatly promotes the oxidation of EB to AP. Therefore, high conversion and selectivity for AP are achieved over MMO-0.5/A. In the present heterogeneous catalyst system, in situ immobilization of Co-containing metal oxides onto the surface of alumina can greatly enhance the activity and selectivity. The remarkably enhanced catalytic performance for MMO-x/A catalysts should be ascribed to the specific 3D core–shell flower-like architecture with enhanced surface area, suitable mesopore distribution and high metal dispersion, which can offer more adsorption sites and active reaction centers and allow substrates to diffuse freely on the MMO platelets. With the increase in the [Zn2+]/([Zn2+] + [Co2+]) molar ratio to 0.5, the catalytic activity is enhanced gradually due to the increase in the metal dispersion in spite of the decrease in the active Co content. However, in the case of MMO-0.75/A, the less Co loading amount (about 2.1 wt%) leads to a dramatic decrease in the conversion of EB, although Co species possess higher dispersion as evidenced by the above TPR results.
Table 3 shows the effects of different reaction conditions on the solvent-free oxidation of EB over the representative MMO-0.5/A catalyst. It is well known that increasing the amount of the catalyst in batch reaction processing will increase the conversion due to more active sites provided by catalysts. However, the above positive effect is not always to be utilized if the selectivity for the product is considered. As shown in Table 3, (tests 1, 2 and 3), the conversion of EB over MMO-0.5/A improves with a catalyst dosage of 0.05 to 0.2 g. After an increase in selectivity for AP in the cases of lower catalyst dosages, the selectivity decreases from 80.4% to 69.6% with increasing catalyst dosage from 0.1 to 0.2 g. This is because with the increasing amount of active sites, higher conversion leads to a relatively lower concentration of EB in the reaction solution. However, the excess Co active sites are prone to adsorb AP easily. Therefore, AP may further be converted to BZ and BA, leading to the decrease in the selectivity for AP. On increasing the ratio of EB:TBHP from 1:2 to 1:3 (Table 3, tests 2 and 4), the conversion of EB increases from 38.8% to 69.5%. However, further increase in the molar ratio of EB:TBHP does not lead to enhanced conversion (Table 3, test 5). In contrast, the conversion of EB slightly decreases to 67.6%. This result is consistent with the observation by Singh et al.24 As the reaction time is increased from 6 to 24 h at 120 °C (Table 3, tests 2, 6 and 7), the EB conversion and the selectivity for AP increase from 37.6% to 77.9% and 54.8% to 81.6%, respectively, while the selectivity for BZ and BA decreases. As shown in Table 3 (tests 2, 8, 9 and 10), elevating the reaction temperature from 80 to 120 °C results in a continuous increase in the conversion of EB and the selectivity for AP because the rate of reaction can be enhanced with the reaction temperature. Further increase in the reaction temperature from 120 to 140 °C slightly improves the conversion of EB. Inversely, the selectivity for AP displays a decrease. This is because high temperature is beneficial to heterolytic cleavage of C–C bond, thus enhancing the rate of the formation of BZ with elevated temperatures.
| Test | Mass of catalyst (g) | EB:TBHP (mol per mol) |
Reaction time (h) | Temperature (°C) | Conversion (%) | Selectivity (mol%) | ||
|---|---|---|---|---|---|---|---|---|
| AP | BZ | BA | ||||||
| a Reaction conditions: EB, 10 mmol; catalyst, MMO-0.5/A. | ||||||||
| 1 | 0.05 | 1:3 |
12 | 120 | 39.5 | 63.5 | 27.3 | 9.2 |
| 2 | 0.1 | 1:3 |
12 | 120 | 69.5 | 80.4 | 12.1 | 7.5 |
| 3 | 0.2 | 1:3 |
12 | 120 | 72.3 | 69.6 | 20.1 | 10.3 |
| 4 | 0.1 | 1:2 |
12 | 120 | 38.8 | 66.4 | 28.8 | 4.8 |
| 5 | 0.1 | 1:4 |
12 | 120 | 67.6 | 76.5 | 17.3 | 6.2 |
| 6 | 0.1 | 1:3 |
6 | 120 | 37.6 | 54.8 | 32.4 | 12.8 |
| 7 | 0.1 | 1:3 |
24 | 120 | 77.9 | 81.6 | 14.0 | 4.4 |
| 8 | 0.1 | 1:3 |
12 | 80 | 20.7 | 73.1 | 14.6 | 12.3 |
| 9 | 0.1 | 1:3 |
12 | 100 | 41.3 | 73.5 | 19.7 | 8.8 |
| 10 | 0.1 | 1:3 |
12 | 140 | 70.7 | 64.4 | 29.6 | 6.0 |
Recovery and recyclability are two of the most attractive hallmarks of heterogeneous catalysts. Before reuse of the catalyst, MMO-0.5/A can be easily recovered from the reaction mixture by simple centrifugation, washing with ethanol and acetone, and then drying at 80 °C. Subsequently, the recycling performance of MMO-0.5/A was evaluated under identical reaction conditions. As shown in Fig. 10, MMO-0.5/A can maintain its excellent catalytic activity in the oxidation of EB (reaching an AP yield of 53.9%) at 120 °C for 12 h after recycling three times. The selectivity for AP is close to 80% in all cases, and the conversion of EB only decreases by about 1.0%. Elemental analysis by ICP-AES further reveals that the Co leaching loss is only about 0.8 wt% of the total cobalt after four consecutive cycles, suggestive of the good stability of the catalyst. This can be explained by the fact that in situ growth of LDH precursors on the Al2O3 support is beneficial to form a strong interaction between active species and support matrix and thus keep a remarkable stability for the oxidation of EB. In addition, a further reaction of test 2 in Table 3 was performed. After a reaction time of 2 h, the reaction was hot filtered to remove the solid catalyst. Then, the reaction was allowed to continue under the same conditions. Only a trace amount of the desired product could be identified under the same conditions after another reaction for 12 h. The above results strongly suggest that the present reaction proceeds mostly on the heterogeneous surface.
 | ||
| Fig. 10 Reproducibility of MMO-0.5/A in the oxidation of EB. Reaction conditions: EB, 10 mmol; catalyst, 0.1 g; reaction temperature, 120 °C; EB:TBHP molar ratio, 1:3; reaction time, 12 h. | ||
In general, the current study can be more advantageous over previous studies because of the two following merits. Firstly, the synthesis of CoZnAl-LDH precursors was performed by an in situ growth route, which resulted in excellent stability of the resultant core–shell MMO-x/A catalysts due to the strong adhesion of flower-like Co-containing metal oxide micro/nanostructures on the support. Secondly, a uniform distribution of multi-metal species in the platelet-like CoZnAl-LDH precursor facilitated high dispersion of active Co species, as well as strong metal–support interactions, thus leading to significantly enhanced catalytic activity in the oxidation of EB.
Conclusions
In summary, Co-containing LDH microcrystallites were in situ grown on the surface of microspherical alumina. The resulting Al2O3-supported CoZnAl-MMO presented a hierarchical flower-like core–shell microstructure due to well-developed 2D platelet-like LDHs grown on the support surface. Compared with the conventional catalyst prepared by incipient impregnation and the bulk CoZnAl-MMO catalyst, Al2O3-supported CoZnAl-MMO possessed higher metal dispersion due to the homogeneous distribution of MMO nanoplatelets as well as the stronger interaction between active species and matrix. In particular, the introduction of an appropriate amount of Zn into LDH precursors could greatly improve the dispersion of active Co species and thus catalytic performance in solvent-free liquid-phase oxidation of EB with TBHP as an oxidant to produce AP. Increasing the reaction temperature, reaction time and catalyst dosage could improve the conversion of EB by accelerating the rate of oxidation, but a higher reaction temperature and catalyst dosage could lead to the decrease in the selectivity for AP due to the enhanced deep oxidation. Moreover, this catalyst possessed attractive stability and could retain high catalytic activity after recycling three times. Owing to their intrinsic properties such as high surface area, chemical stability and low cost, it is expected that such a type of highly-dispersed and stable hierarchical flower-like core–shell Co-based metal oxide catalysts may work as an alternative, which are probably appropriate in the practical oxidation of alkylaromatics.Experimental
Preparation of amorphous Al2O3 microspheres
In a typical synthesis, Al2(SO4)3·18H2O (0.014 mol), CO(NH2)2 (0.042 mol), and sodium tartrate (0.0035 mol) were dissolved in distilled water (200 mL) and ethanol (80 mL) to form a mixed solution and then the solution was transferred into an autoclave, followed by hydrothermal treatment at 165 °C for 1 h. The white precipitate was separated by centrifugation and washed with distilled water and alcohol several times. The product was dried in a vacuum oven at 80 °C for 8 h. Finally, amorphous Al2O3 microspheres were obtained by calcination of the above precipitation at 500 °C for 6 h.Preparation of CoZnAl-MMO/Al2O3 samples
In the synthesis of Al2O3-supported CoZnAl-LDHs, Co(NO3)2·6H2O, Zn(NO3)2·3H2O, and NH4NO3 (0.0375 mol) with different [Zn2+]/([Zn2+] + [Co2+]) molar ratios of x (x = 0, 0.25, 0.5 and 0.75) were dissolved in 280 mL of deionized water to obtain a clear solution with a total metal cation concentration of 0.3 M. The resulting solution was added to a four-necked flask with 0.4 g of Al2O3, and the resulting slurry was transferred to an autoclave and aged at 100 °C for 12 h, thoroughly washed with deionized water until the pH reached 7, and dried at 60 °C for 12 h in air. The obtained CoZnAl-LDH/Al2O3 precursors were denoted as L-x/A. L-x/A was calcined in static air at 500 °C for 6 h, and the calcined product was denoted as MMO-x/A.For comparison, Al2O3 (0.4 g) was added to 1.0 mL of Co(NO3)2·6H2O and Zn(NO3)2·3H2O solution (0.7 M) and then impregnated for 12 h. The resulting supported catalyst precursor was obtained after drying at 60 °C for 12 h, and calcined in static air at 500 °C for 6 h, and the calcined product was denoted as Im-MMO-0.5/A. In addition, CoZnAl-LDH with a Co/Zn/Al molar ratio of 2:2:1 was prepared by our previously reported separate nucleation and aging step method.66 The CoZnAl-LDH precursor was calcined in static air at 500 °C for 6 h, and the calcined product is denoted as MMO-0.5.
Characterization
X-ray diffraction (XRD) data of the samples were collected at room temperature using a Rigaku D/Max-RB diffractometer with a graphite-filtered Cu Kα source (λ = 0.15418 nm).Elemental analysis was performed using a Shimadzu ICPS-7500 inductively coupled plasma atomic emission spectroscope (ICP-AES) after the samples were dissolved in dilute hydrochloric acid (1:1).
The morphology of the samples was investigated using a field emission scanning electron microscope (FE-SEM, Zeiss Supra 55 with an accelerating voltage of 20 kV) which was combined with energy-dispersive X-ray spectroscopy (EDX) performed using an Oxford Instruments INCA analyzer for determination of the metal composition.
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were carried out using a JEM-2100F electron microscope (JEOL, Japan) operated at an accelerating voltage of 200 kV.
N2 adsorption–desorption isotherms of the samples were obtained with a Micromeritics ASAP 2020 sorptometer apparatus at −196 °C. All samples were outgassed prior to analysis at 200 °C before adsorption measurements. The total specific surface areas were evaluated by the multipoint Brunauer–Emmett–Teller (BET) method. The mesopore size distribution and average pore diameter were determined by the Barrett–Joyner–Halenda (BJH) method applied to adsorption isotherms.
X-ray photoelectron spectroscopy (XPS) spectra were recorded using a VG ESCALAB 2201 XL spectrometer with monochromatic Al Kα X-ray radiation (1486.6 eV photons). Binding energies were calibrated based on the graphite C 1s peak at 284.5 eV.
The reduction behavior of calcined samples was studied by hydrogen temperature-programmed reduction (H2-TPR) using a Micromeritics ChemiSorb 2720 instrument. A 0.1 g sample, which was placed in a quartz U-tube reactor, was degassed at 200 °C for 2 h under argon flow (40 mL min−1). Then TPR was conducted in a stream of 10% (v/v) H2/Ar (50 mL min−1) with a heating rate of 5 °C min−1 up to 1000 °C. The effluent gas was analyzed by a thermal conductivity detector (TCD).
Selective oxidation of EB
The oxidation of EB was conducted in a glass reactor equipped with a condenser and magnetic stirrer. Typically, EB (10 mmol) and a certain amount of catalyst were added, followed by 70% aqueous TBHP with a desired EB/TBHP molar ratio. The reaction was performed at a certain temperature for a certain period with continuous magnetic stirring at 1000 rpm. The catalysts were separated by centrifugation. The yield of products was analyzed using an Agilent (GC-7890B) gas chromatograph with an HP-5 column and FID detector. The products formed in the reactions were further characterized by GC-MS analysis. The TON value was calculated based on the moles of EB converted per mole of Co. In addition, leaching of the metal during the course of the reaction was verified by resubmission of the filtrate for further reaction under the same reaction conditions. In the reusability tests, the catalysts were removed from the reaction mixtures, washed with ethyl alcohol and acetone, and dried at 80 °C for 12 h. Then, fresh EB was employed in the repeated tests.Acknowledgements
We gratefully acknowledge the financial support from the 973 Program (2011CBA00506), the National Natural Science Foundation of China, the Program for Changjiang Scholars and Innovative Research Team in University (IRT1205), and a project from the Beijing Engineering Center for Hierarchical Catalysts.Notes and references
- R. Alcántara, L. Canoira, P. G. Joao, J. M. Santos and I. Vázquez, Appl. Catal., A, 2000, 203, 259 CrossRef.
- M. Ghiaci, Z. Sadeghi, M. E. Sedaghat, H. Karimi-Maleh, J. Safaei and A. Gil, Appl. Catal., A, 2010, 381, 121 CrossRef CAS PubMed.
- V. R. Choudhary, J. R. Indurkar and V. S. Narkhede, J. Catal., 2004, 227, 257 CrossRef CAS PubMed.
- S. E. Dapurkar, H. Kawanami, T. Yokoyama and Y. Ikushima, Catal. Commun., 2009, 10, 1025 CrossRef CAS PubMed.
- S. K. Jana, Y. Kubota and T. J. Tatsumi, J. Catal., 2007, 247, 214 CrossRef CAS PubMed.
- K. Bahranowski, R. Dula and M. Gasior, Appl. Clay Sci., 2001, 18, 93 CrossRef CAS.
- S. Vetrivel and A. Pandurangan, J. Mol. Catal. A: Chem., 2004, 217, 165 CrossRef CAS PubMed.
- K. George and S. Sugunan, Catal. Commun., 2008, 9, 149 CrossRef PubMed.
- S. Vetrivel and A. Pandurangan, Ind. Eng. Chem. Res., 2005, 44, 692 CrossRef CAS.
- X. G. Li, J. Wang and R. He, Chin. Chem. Lett., 2007, 18, 1053 CrossRef CAS PubMed.
- C. Guo, Q. Peng, Q. Liu and G. Jiang, J. Mol. Catal. A: Chem., 2003, 192, 295 CrossRef CAS.
- C. Lu, Z. Fu, Y. Liu, F. Liu, Y. Wu, J. Qin, X. He and D. Yin, J. Mol. Catal. A: Chem., 2010, 331, 106 CrossRef CAS PubMed.
- M. Arshadi and M. Ghiaci, Appl. Catal., A, 2011, 399, 75 CrossRef CAS PubMed.
- D. Habibi and A. R. Faraji, Appl. Surf. Sci., 2013, 276, 487 CrossRef CAS PubMed.
- D. Habibi, A. R. Faraji, M. Arshadi, H. Veisi and A. Gil, J. Mol. Catal. A: Chem., 2014, 382, 41 CrossRef CAS PubMed.
- M. Arshadia, M. Ghiacia, A. A. Ensafia, H. Karimi-Maleh and Steven L. Suib, J. Mol. Catal. A: Chem., 2011, 338, 71 Search PubMed.
- D. Habibi, A. R. Faraji, M. Arshadi, S. Heydari and A. Gil, Appl. Catal., A, 2013, 466, 282 CrossRef CAS PubMed.
- R. Wang, B. Gao and W. Jiao, Appl. Surf. Sci., 2009, 255, 4109 CrossRef CAS PubMed.
- K. O. Xavier, J. Chacko and K. K. M. Yusuff, Appl. Catal., A, 2004, 258, 251 Search PubMed.
- K. O. Xavier, J. Chacko and K. K. M. Yusuff, J. Mol. Catal. A: Chem., 2002, 178, 275 CrossRef CAS.
- K. M. Parida and S. Soumya Dash, J. Mol. Catal. A: Chem., 2009, 306, 54 CrossRef CAS PubMed.
- S. Vetrival and A. Pandurangan, Appl. Catal., A, 2004, 264, 243 CrossRef PubMed.
- S. S. Bhoware, S. Shylesh, K. R. Kamble and A. P. Singh, J. Mol. Catal. A: Chem., 2006, 255, 123 CrossRef CAS PubMed.
- P. S. Singh, K. Kosuge, V. Ramaswamy and B. S. Rao, Appl. Catal., A, 1999, 177, 149 CrossRef CAS.
- S. Devika, M. Palanichamy and V. Murugesan, Appl. Catal., A, 2011, 407, 76 CrossRef CAS PubMed.
- D.-H. Shen, L.-T. Ji, Z.-G. Liu, W.-B. Sheng and C.-C. Guo, J. Mol. Catal. A: Chem., 2013, 379, 15 CrossRef CAS PubMed.
- B. Monteiro, S. Gago, S. S. Balula, A. A. Valente, I. S. Goncalves and M. Pillinger, J. Mol. Catal. A: Chem., 2009, 312, 23 CrossRef CAS PubMed.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- P. S. Braterman, Z. P. Xu and F. Yarberry, in Handbook of Layered Materials, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, Marcel Dekker, New York, 2004, p. 373 Search PubMed.
- D. G. Evans and R. C. T. Slade, Struct. Bonding, 2006, 119, 1 CAS.
- F. Li and X. Duan, Struct. Bonding, 2006, 119, 193 CrossRef CAS.
- D. Tichit and B. Coq, CATTECH, 2003, 7, 206 CrossRef CAS.
- D. Tichit, D. Lutic, B. Coq, R. Dur and R. Teissier, J. Catal., 2003, 219, 167 CrossRef CAS.
- F. Li, X. Jiang, D. G. Evans and X. Duan, J. Porous Mater., 2005, 12, 55 CrossRef CAS.
- F. Kovanda, T. Rojka, J. Dobesova, V. Machovic, P. Bezdicka, L. Obalova, K. Jiratova and T. Grygar, J. Solid State Chem., 2006, 179, 812 CrossRef CAS PubMed.
- M. R. Schwarzman and M. P. Wilson, Science, 2009, 326, 1065 CrossRef CAS PubMed.
- H. Zhao, J. C. Zhou, H. Luo, C. Y. Zeng, D. H. Li and Y. J. Liu, Catal. Lett., 2006, 108, 49 CrossRef CAS.
- V. R. Choudhary and D. K. Dumbre, Catal. Commun., 2011, 13, 82 CrossRef CAS PubMed.
- N. Kanari, D. Mishra, I. Gaballah and B. Dupré, Thermochim. Acta, 2004, 410, 93 CrossRef CAS.
- J. Pérez-Ramírez, G. Mul, F. Kapteijn and J. A. Moulijn, J. Mater. Chem., 2001, 11, 2529 RSC.
- H. Chen, F. Zhang, S. Fu and X. Duan, Adv. Mater., 2006, 18, 3089 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1 CrossRef CAS.
- S. Abelló and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2006, 96, 102 CrossRef PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- H. K. Lin, H. C. Chiu, H. C. Tsai, S. H. Chien and C. B. Wang, Catal. Lett., 2003, 88, 3 CrossRef.
- J. Zheng, J. J. Yu, J. Cheng, T. C. Xiao, M. O. Jones, Z. P. Hao and P. P. Edwards, Fuel Process. Technol., 2010, 91, 97 CrossRef PubMed.
- L. Chmielarz, P. Kustrowski, A. Rafalska-Lasocha and R. Dziembaj, Thermochim. Acta, 2002, 395, 225 CrossRef.
- P. Arnoldy and J. A. Moulijn, J. Catal., 1985, 93, 38 CrossRef CAS.
- M. P. Hyman and J. M. Vohs, Surf. Sci., 2011, 605, 383 CrossRef CAS PubMed.
- E. Martono and J. M. Vohs, J. Catal., 2012, 291, 79 CrossRef CAS PubMed.
- C. Altavilia and E. Ciliberto, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 309 CrossRef PubMed.
- G. Schoen, J. Electron Spectrosc. Relat. Phenom., 1973, 2, 75 CrossRef.
- N. M. Figueiredo, N. J. M. Carvalho and A. Cavaleiro, Appl. Surf. Sci., 2011, 257, 5793 CrossRef CAS PubMed.
- C. Yoon and D. L. Cocke, J. Catal., 1988, 113, 267 CrossRef CAS.
- M. Muhler, R. Schlogl and G. Ertl, J. Catal., 1992, 138, 413 CrossRef CAS.
- R. Jothiramalingam, B. Viswanathan and T. K. Varadarajan, J. Mol. Catal. A: Chem., 2006, 252, 49 CrossRef CAS PubMed.
- S. K. Jana, P. Wu and T. Tatsumi, J. Catal., 2006, 240, 268 CrossRef CAS PubMed.
- T. Radhika and S. Sugunan, Catal. Commun., 2007, 8, 150 CrossRef CAS PubMed.
- S. S. Bhoware and A. P. Singh, J. Mol. Catal. A: Chem., 2007, 266, 118 CrossRef CAS PubMed.
- J.-Y. Qi, H.-X. Ma, X.-J. Li, Z.-Y. Zhou, M. C. K. Choi, A. S. C. Chan and Q.-Y. Yang, Chem. Commun., 2003, 1294 RSC.
- G. S. Kumar, M. Palanichamy, M. Hartmann and V. Murugesan, Catal. Commun., 2007, 8, 493 CrossRef CAS PubMed.
- S. L. H. Rebelo, M. M. Q. Simões, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mol. Catal. A: Chem., 2003, 201, 9 CrossRef CAS.
- T. Shishido, Y. Yamamoto, H. Morioka and K. Takehira, Appl. Catal., A, 2004, 263, 249 CrossRef CAS PubMed.
- H. H. Tseng, H. Y. Lin, Y. F. Kuo and Y. T. Su, Chem. Eng. J., 2010, 160, 13 CrossRef CAS PubMed.
- C. Hariharan, Appl. Catal., A, 2006, 304, 55 CrossRef CAS PubMed.
- Y. Zhao, F. Li, R. Zhang, D. G. Evans and X. Duan, Chem. Mater., 2002, 14, 4286 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental results. See DOI: 10.1039/c4cy00744a |
| This journal is © The Royal Society of Chemistry 2015 |