
Enhancement of hydrodesulfurization of 4,6-dimethyldibenzothiophene catalyzed by CoMo catalysts supported on carbon-covered γ-Al2O3†
Feng
Cui‡
ab,
Guangci
Li‡
b,
Xuebing
Li
*b,
Mohong
Lu
a and
Mingshi
Li
*a
aJiangsu Key Laboratory of Advanced Catalytic Materials and Technology, School of Petrochemical Engineering, Changzhou University, Changzhou, 213164, PR China. E-mail: mingshili@cczu.edu.cn; Fax: +86 519 86330360; Tel: +86 519 86330360
bKey Laboratory of Biofuels, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, 266101, PR China. E-mail: lixb@qibebt.ac.cn; Fax: +86 532 80662757; Tel: +86 532 80662757
First published on 17th September 2014
Abstract
The carbon-covered aluminas were prepared by using different monocarboxylic acids as carbon sources to modify active alumina, and then were used as supports to prepare supported CoMo catalysts for hydrodesulfurization (HDS) of 4,6-dimethyldibenzothiophene (4,6-DMDBT). These monocarboxylic acid molecules can be readily converted to carbon species by thermal decomposition in a nitrogen atmosphere and deposited on an alumina surface. The carbon species can then effectively weaken the interaction between active metals and alumina, which improves the migration and growth of surface Mo species. This result further affected the morphology and orientation of surface sulfur species, that is, the slab length and stacking number of the MoS2 slabs, which closely relates to HDS activity. Since the surface Mo species supported on the alumina modified with acetic acid consisted of the MoS2 slabs with the shortest lengths, leading to the presence of more easily reducible sulfur species during the reaction, the corresponding catalyst exhibited the highest HDS activity for 4,6-DMDBT. In addition, the stacking numbers for all of the catalysts were relatively low, which hindered the adsorption of 4,6-DMDBT on the brim sites of the MoS2 stacks, and thus the HDS reaction mainly occurred through the direct desulfurization route.
1. Introduction
Active alumina-supported transition metal sulfide catalysts (CoMoS/γ-Al2O3, NiMoS/γ-Al2O3, etc.) have been extensively used for hydrodesulfurization (HDS) in the hydrotreating process for several decades.1,2 In recent years, many studies have focused on the production of ultra-low sulfur transport fuels via deep desulfurization3,4 because of the increasingly stringent environmental regulations. In order to achieve this purpose it is necessary to convert refractory sulfur compounds with steric hindrance, such as alkylated dibenzothiophenes.3–5In the 1980s, H. Topsøe and his co-workers found and identified the presence of the Co–Mo–S structure by several new in situ catalyst characterization techniques, and they thought that this structure was responsible for the improvement of the catalytic activity.6–8 From then on, the presence of this structure and its function were also confirmed and studied by other researchers. Although the Co–Mo–S structure was believed to supply HDS active sites, the morphology of the structure, including slab length and stacking number, can greatly affect the catalytic properties.9–11 It is accepted that the Co–Mo–S structure consisting of multilayered MoS2 slabs exhibits more activity than that consisting of monolayered MoS2 slabs.12 Thus, many efforts were made to develop feasible and facile methods to control the morphology of the Co–Mo–S structure and to obtain multilayered MoS2 active phases. The addition of organic chelating agents, such as citric acid and EDTA, is one such feasible method in the catalyst preparation, mostly for supported CoMo catalysts.13–15 The chelating agent molecules can interact with active metals to form chelate compounds, which inhibit the active metals from strongly interacting with the support surface. The weak interaction benefits the formation of multilayered MoS2 slabs.
Generally, the properties of the support play a key role in the catalytic activity, stability and structure of the active phase of the catalyst.16,17 Although alumina is a commonly used commercial support due to its low cost, good thermal stability and volatile acidity,18 it can strongly interact with active metals and thus hinder their sulfurization and formation of multilayered MoS2 slabs. Therefore, another effective method to obtain multilayered MoS2 active phases is to replace alumina and use other supports that weakly interact with active metals. Carbon materials are preferable to alumina as supports because of their high surface areas and low acidities, which are favorable for the dispersion of active metals and the formation of more active multilayered MoS2 structures.19–21 Despite carbon-supported catalysts usually being more active than alumina-supported ones, their properties of poor mechanical strength and low bulk density limit their applications in HDS. In order to overcome these shortcomings and to combine the advantages of both materials, a new support, carbon-covered alumina, was developed, which exhibited superior performance in HDS reactions.22–24
To the best of our knowledge, most carbon-covered aluminas were prepared via pyrolysis of hydrocarbons. Because hydrocarbons cannot interact with the surface of alumina, the obtained carbon species may easily migrate and aggregate, resulting in a nonuniform distribution of the carbon layer on alumina. This may cause a negative effect on the dispersion of active metals. Recently, Li et al.25 used citric acid as a carbon source to prepare carbon-covered alumina; they found that citric acid molecules can react with the surface hydroxyl groups of alumina and then convert into carbon species. The well-dispersed carbon species isolated the active metals and facilitated the formation of active WS2 structures. However, the effect of the carbon species on the CoMo active component and the MoS2 structure has yet to be mentioned. Although González-Cortés26,27 and Green28 developed a urea-organic matrix method to prepare CoMoS HDS catalysts with high activity and found that the formed surface carbon materials may improve HDS activity, the carbon materials derived from thiophene, which were formed during the presulfurization of the catalysts, only interacted with Mo species and did not affect the alumina support directly. In order to investigate the effect of alumina supports modified with carbon on CoMoS catalyst activity, herein we report the preparation of CoMo catalysts supported on carbon-covered alumina supports, prepared by using different monocarboxylic acids as carbon sources. The changes in CoMo and MoS2 structures were investigated by XRD and HRTEM. The results revealed that the formed carbon species effectively weakened the interaction between the active metals and alumina, leading to a decrease in the MoS2 slab length and an increase in the number of easily reducible sulfur species, thus leading to an improvement in HDS activity.
2. Experimental
2.1 Chemicals
All chemicals were purchased from commercial suppliers and used without any purification: aluminium hydroxide xerogel (70 wt.% Al2O3, Yantai Henghui Chemical Engineering Co., Ltd., China), (NH4)6Mo7O24·4H2O, Co(NO3)2·6H2O, CS2 and decalin (Sinopharm, AR standard), 4,6-dimethyldibenzothiophene (Aladdin, 97%), n-dodecane (Aladdin, ≥99.5% GC standard).2.2 Preparation of carbon-covered γ-Al2O3 supports
Commercial aluminium hydroxide xerogel (AHx) was used as the alumina precursor. Typically, 20.0 g of AHx was mixed with aqueous nitric acid solution (20 wt.%) to a suitable viscosity and then extruded in a radial extruder with a 1.6 mm sieve. Then, the extrudates were dried in air at 80 °C overnight, calcinated at 600 °C for 4 h with a heating ramp of 2 °C min−1, and then crushed into particles with lengths of 3–5 mm. The obtained primary alumina support was labelled S-1.The carbon-covered supports were prepared by using different monocarboxylic acids to modify support S-1. Typically, 10.0 g of S-1 was added to a 150 ml conical flask containing 30.0 ml of 0.40 M formic acid solution, and the conical flask was then shaken at room temperature for 12 h. After that, S-1 was separated by filtration, washed with deionized water until neutral pH was obtained, and dried in air at 80 °C overnight. After calcination in nitrogen at 600 °C for 4 h, the carbon-covered support was obtained, and it was labelled S-2. When acetic acid, 1-propionic acid, or 1-butyric acid were used to replace formic acid as the carbon source, the corresponding alumina supports were labelled S-3, S-4 and S-5, respectively.
2.3 Preparation of CoMo catalysts
The catalysts were prepared by incipient wetness impregnation of the obtained supports with an aqueous solution containing (NH4)6Mo7O24·4H2O and Co(NO3)2·6H2O. The loading amounts of CoO and MoO3 were controlled to be 2.7 wt.% and 17.3 wt.%, respectively. The impregnated supports were then dried in air at 80 °C overnight, before calcination at 500 °C in nitrogen for 4 h. The CoMo catalysts were in turn labelled C-1, C-2, C-3, C-4 and C-5, according to their corresponding supports S-1, S-2, S-3, S-4 and S-5, respectively.2.4 Catalysts characterization
2.5 HDS activity tests
The CoMo catalysts were presulfurized in a high-pressure tubular microreactor before use. Typically, 2.0 g of catalyst was packed into a stainless steel tube and pre-treated at 200 °C for 3 h under flowing He, and the tube was then cooled to room temperature. Then, the sulfurizing agent (decalin solution containing 3 wt.% CS2) was pumped into the catalyst bed and the sulfurization was carried out at 300 °C and 2.0 MPa H2 for 8 h with a liquid hour space velocity (LHSV) of 2.0/h and H2/feed = 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
The HDS activity tests were carried out in a 100 ml stainless steel autoclave with mechanical stirring. Typically, 40 g of reactant (0.4 wt.% 4,6-DMDBT and 0.2 wt.% CS2 dissolved in decalin) was transferred into the reactor, accompanied with 0.4 g of the sulfurized catalyst. Prior to reaction, the reactor was purged three times with H2 to replace the air inside and then pressurized to 2.0 MPa at room temperature. The reaction was performed at 340 °C under stirring at a rate of 600 r min−1 for 6 h. The liquid product was collected by centrifugation and analyzed by an Agilent 7890 GC instrument equipped with a flame ionization detector (FID) and a HP-5 column.
3 Results and discussion
3.1 Characterization of carbon-covered γ-Al2O3 supports
XRD patterns of the primary and carbon-covered alumina supports are displayed in Fig. 1. All patterns can be well indexed to the γ-Al2O3 phase (JCPDS 00-050-0741), where two characteristic diffraction peaks at 45.9° and 66.7° are assigned to the (400) and (440) planes of γ-Al2O3, respectively. The similar diffraction patterns and comparable intensities of the diffraction peaks indicated that the crystalline structure of the primary alumina was not remarkably affected by the monocarboxylic acid solutions. Nevertheless, the colour of the supports became darker in order (from white to dark grey) with increasing carbon number for the monocarboxylic acids, which suggests significant changes in the surfaces of the supports. | ||
| Fig. 1 XRD patterns of the different alumina supports. | ||
All supports were characterized by means of TG analysis to investigate the quantities of surface hydroxyl groups and carbon species. As seen from Table 1, the hydroxyl group content of all carbon-covered supports was lower than that of S-1, implying that organic acid can interact with the surface hydroxyl groups of alumina and that the hydroxyl groups may be removed in the form of H2O. Among the supports, the hydroxyl group content was lowest for S-3, which may be related to the reactivity of the monocarboxylic acids with the hydroxyl groups, where acetic acid was most prone to react with hydroxyl groups. With the increase in the carbon number of the carboxylic acids, the quantity of carbon species deposited on alumina gradually increased. Because of the different reactivities of the monocarboxylic acids with hydroxyl groups, the carbon species content does not change linearly with the molecular weights of the monocarboxylic acids.
| Support | Hydroxyl group contenta (wt.%) | Carbon species contentb (wt.%) |
|---|---|---|
| a The hydroxyl group content was calculated by the weight loss of the supports from 600 to 1200 °C under nitrogen atmosphere. b The carbon species content was calculated by using the weight loss of the supports from 200 to 1200 °C under air atmosphere and subtracting the hydroxyl group content. | ||
| S-1 | 2.5 | 0 |
| S-2 | 2.1 | 3.6 |
| S-3 | 1.6 | 3.9 |
| S-4 | 1.7 | 4.2 |
| S-5 | 1.9 | 4.6 |
The recorded adsorption/desorption isotherms for all supports are shown in Fig. S1 (see ESI†); all of the isotherms were of type IV, which is characteristic of mesoporous materials. Their hysteresis loops at relative pressures (P/P0) above 0.43 were of type H3, which means that the pores were mainly derived from aggregates or agglomerates of alumina particles.29 The same type and similar shapes of the isotherms indicated that all the supports might possess similar pore structures. The textural properties (specific surface area, pore volume and average pore diameter) of the supports are listed in Table 2. The pore structures of the supports were not obviously affected after treatment with the monocarboxylic acids. Since the pores of the supports were formed by the stacking of alumina particles, the textural properties have a close relationship with the shape and size of the particles. Thus, it was concluded that the organic acids only changed the surface hydroxyl group content of alumina, without influencing the particle size.
| Samples | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|
| S-1 | 265 | 0.71 | 10.7 |
| S-2 | 273 | 0.75 | 10.9 |
| S-3 | 266 | 0.77 | 11.5 |
| S-4 | 262 | 0.75 | 11.4 |
| S-5 | 259 | 0.69 | 10.7 |
3.2 Characterization of CoMo catalysts
Fig. 2 shows the XRD patterns of fresh CoMo catalysts in the oxidised state. For C-1, in addition to the diffraction peaks of γ-Al2O3, diffraction peaks at 23.5°, 24.4° and 30.7° which correspond to the Al2(MoO4)3 phase (JCPDS 01-084-1652) can be observed, without any observation of diffraction peaks corresponding to the MoO3 phase. This result suggests that most of the MoO3 interacted with alumina and transformed into Al2(MoO4)3 on the surface of the support during the calcination, implying a strong interaction between Mo and the support. Although the diffraction peak at 26.5° corresponding to the CoMoO4 phase (JCPDS 00-021-0868) was present, its intensity was very weak, indicating a low crystallinity. Because of the low loading content of CoO, its diffraction peaks were not observed. When the support was treated with formic acid, the intensity of the peak corresponding to the CoMoO4 phase increased remarkably and the diffraction peak corresponding to MoO3 appeared. Combined with the result in Table 1, it was believed that the carbon species deposited on the alumina surface could reduce the interaction between MoO3 and the support, favoring the migration and aggregation of Mo species and the formation of the CoMoO4 phase. Therefore, as the carbon species content increased, the intensity of the peaks corresponding to Al2(MoO4)3 decreased and that of the peak corresponding to the CoMoO4 phase was enhanced. | ||
| Fig. 2 XRD patterns of the fresh CoMo catalysts. | ||
Although the weak interaction of MoO3 with the support accelerated the aggregation of MoO3, the interaction of CoO with MoO3 to form the CoMoO4 phase was inhibited when the support was treated with 1-propionic acid or 1-butyric acid. This may be owing to the deposition of the carbon species further weakening the interaction forces such that the MoO3 particles become larger, and the growth rate of MoO3 becomes more rapid than that of the formation of the CoMoO4 phase. In addition, large MoO3 particles were reduced to MoO2 by surface carbon species during calcination. As a result, the intensity of the peaks corresponding to Al2(MoO4)3 further decreased and the MoO2 phase (JCPDS 00-032-0671) was predominant on the support surface.
Prior to HDS reaction, fresh catalysts need to be presulfurized; their XRD patterns are shown in Fig. 3. It appears that the diffraction peaks corresponding to the Al2(MoO4)3, MoO3, and CoMoO4 phases disappeared after presulfurization of the catalysts, indicating the high degree of sulfurization. In spite of this, in each pattern there was no characteristic diffraction peak corresponding to MoS2, implying that the formed MoS2 may possess a poorly crystalline structure and that its particles are too small to be detected by XRD. In addition, because of the presence of Co, the CoMoS phase may possibly be present as small sized particles, which also cannot be detected. It was observed that in comparison to MoO3, the conversion of MoO2 to MoS2 was more difficult at low temperature, and thus diffraction peaks corresponding to MoO2 were still observed for the surfaces of sulfurized C-4 and C-5.
 | ||
| Fig. 3 XRD patterns of the sulfurized CoMo catalysts. | ||
Fig. 4 presents the TPR patterns of the sulfurized CoMo catalysts. All show two distinct peaks at 160–400 °C; the first peak at 198 °C can be assigned to the reduction of surface sulfur atoms that are weakly bonded to Mo,30 while the second peak at 315 °C can be assigned to the partial reduction of the small MoS2 crystallites.31 For all patterns, the reduction temperatures of the two peaks did not notably change, which suggests that treatment with monocarboxylic acids did not affect the reducibility of the surface sulfur species. The hydrogen consumption, however, was different, and the area of the first peak decreased in the order C-3 > C-4 > C-5 > C-2 > C-1, which means that the amount of easily reducible surface sulfur species decreased correspondingly. This result is in good agreement with that from the XRD results, showing that the more CoMoO4 phase, the easier the reduction of the formed sulfur species during sulfurization. It was believed that reducing the surface sulfur atoms at low temperature could create coordinatively unsaturated sites (CUS), that is, anionic sulfur vacancies, which were reported to be responsible for the HDS activity.12 From this viewpoint, it is thought that C-3 should exhibit the highest HDS activity among all of the catalysts.
 | ||
| Fig. 4 TPR patterns of the sulfurized CoMo catalysts. | ||
Representative TEM micrographs of the sulfurized catalysts before the HDS reaction are shown in Fig. 5, in which the black fringes with a spacing of about 0.62 nm were indexed to the (002) basal planes of MoS2 crystallites. Considering that the morphology and orientation of the CoMoS phase are closely related with the HDS activity of catalysts,32,33 the distributions of the slab lengths and stacking numbers of the MoS2 slabs were measured for all the catalysts, and the results are shown in Fig. 6 and 7. Compared with the other catalysts, C-1 had longer slabs that mostly exceeded 2 nm, which indicated that the Mo species can migrate and aggregate easily on the surface of S-1. When the carbon species were deposited on the supports, the lengths of the MoS2 slabs decreased, mainly to 2–4 nm. It was therefore concluded that the surface carbon species may play a role of “isolating agents”. When the monocarboxylic acid molecules come into contact with the alumina support, they readily interact with the surface hydroxyl groups and the fatty acid chains become fixed on the support surface through C–O–Al bonds. After calcination in nitrogen, these fatty acid chains decomposed and converted into carbon species with high C/H ratios. The carbon species are well dispersed on the support surface like “carbon stacks”, preventing Mo species from aggregation and growth. When the support was treated with acetic acid, the average length of the MoS2 slabs was calculated to be 2.3 nm and was found to be the shortest. If the carbon species content increased further, the obtained “carbon stacks” would become larger and some adjacent stacks might connect with each other, which would hinder the effective inhibition of the migration of the Mo species. This is the reason why the average slab length of C-4 or C-5 is longer than that of C-3.
 | ||
| Fig. 5 TEM images of the sulfurized catalysts: (a) C-1, (b) C-2, (c) C-3, (d) C-4 and (e) C-5. | ||
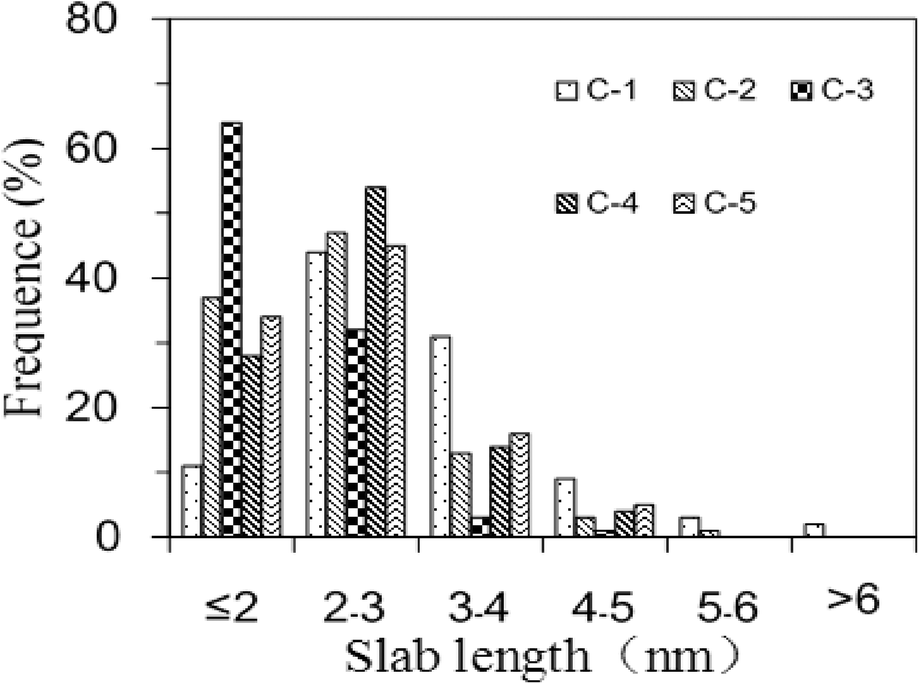 | ||
| Fig. 6 Slab length distributions of the MoS2 slabs on the sulfurized catalysts. | ||
 | ||
| Fig. 7 Stacking distributions of the MoS2 slabs on the sulfurized catalysts. | ||
In terms of the stacking numbers of the MoS2 slabs, apart from for C-5, the MoS2 slabs of the catalysts mainly consisted of 2–4 layers. As the content of carbon species increased, the average stacking number gradually decreased (from 2.6 for C-2 to 1.7 for C-5). According to the XRD results in Fig. 2, the addition of carbon species effectively weakens the interaction of the Mo species with the support. However, many literature reports34–36 have demonstrated that a weak interaction force is favorable for the formation of MoS2 slabs with high stacking numbers, which is contrary to the result obtained here. This contradiction may be attributed to the reduction of Mo species. As discussed above, weak interaction forces promote the growth of Mo species into large particles and these particles would consist of MoO3 crystallites. If these particles could be successfully sulfurized, the formed MoS2 slabs might have a high stacking number. Prior to sulfurization, however, most MoO3 particles were reduced to MoO2 by surface carbon species during calcination. The formed MoO2 cannot be sulfurized easily under the present conditions (see Fig. 3). As a result, the stacking number of the MoS2 slabs, which are derived from the unreduced MoO3, decreased in order. For the shorter slab lengths with similar stacking numbers, there are more brim and edge sites of the MoS2 phase on the catalyst surface, which may lead to the TPR results observed in Fig. 4.
3.3 HDS activity
The results of the HDS of 4,6-DMDBT are listed in Table 3, in which C-3 exhibited the highest catalytic activity. This result is in accordance with TPR data, showing that the catalyst with more easily reducible sulfur species reveals relatively higher HDS activity. An appropriate amount of carbon species deposited on the support improves the formation of CUS, while excess carbon species lead to the reduction of MoO3 to MoO2 and thus decrease the number of MoS2 slabs. Thus, the conversion first increased and then decreased along the series of the different catalysts.| Catalysts | Relative conversion of 4,6-DMDBT | Reaction rate (10−8 mol g−1cat s−1) | ||
|---|---|---|---|---|
| 320 °C | 340 °C | 320 °C | 340 °C | |
| a Reaction conditions: 4,6-DMDBT (0.16 g), decalin (40 g), catalyst (0.4 g), and stirring at 600 r min−1 for 6 h. | ||||
| C-1 | 1.00 | 1.20 | 1.24 | 1.49 |
| C-2 | 1.11 | 1.34 | 1.37 | 1.66 |
| C-3 | 1.26 | 1.52 | 1.56 | 1.89 |
| C-4 | 1.20 | 1.43 | 1.48 | 1.77 |
| C-5 | 1.16 | 1.37 | 1.44 | 1.70 |
It was accepted that the HDS of 4,6-DMDBT underwent two parallel routes involving direct desulfurization (DDS) and hydrodesulfurization (HYD). The DDS route proceeded via chemisorption of the sulfur atom in the molecule on the CUS of MoS2 slabs (edge sites) followed by hydrogen transfer and sulfur elimination, while the HYD route proceeded via adsorption of aromatic rings in the molecule on the MoS2 stack (brim sites) followed by saturation of the aromatic rings and sulfur elimination.37,38 For all of the catalysts, the selectivities to 3,3′-dimethylbiphenyl were higher than 98%, indicating that the HDS reaction mostly underwent via the DDS route and that this route was not impacted markedly. This result is unusual because generally the HYD route is predominant relative to the DDS route for 4,6-DMDBT hydrodesulfurization. Herein, two possible reasons for this deviation are proposed: (i) the low stacking numbers of the MoS2 slabs. Among all of the catalysts, C-2 had the highest average stacking number (2.6 layers), implying that the average height of the MoS2 stack was about 1.7 nm (spacing 0.64 nm × 2.6). Because the molecular size of 4,6-DMDBT is approximately 0.95 nm × 0.8 nm, the low stacking numbers may hinder the adsorption of 4,6-DMDBT on the brim sites of the MoS2 stacks and thus affect the HYD route. (ii) The low sulfurization degree of CoO. According to the literature,24,26,27,39 the presulfurization temperature of CoMo catalysts is usually 400 °C or higher, which benefits sulfurization of active components. In this work, however, this temperature was 300 °C. Low sulfurization temperatures may inhibit the formation of Co sulfide, while this species is responsible for the hydrogenation of aromatic rings. Thus, very little hydrogenated products were generated. However, the specific reason needs to be affirmed by further experiments in the future.
4 Conclusion
In this work, different monocarboxylic acids were selected as carbon sources to prepare carbon-covered alumina supports by wetness impregnation followed by calcination in nitrogen. The formed carbon species deposited on the support surface decreased the surface hydroxyl group content of alumina while the textural properties were not obviously changed. The carbon species effectively weakened the interaction between active metals and alumina, which improved the migration of surface Mo species and the formation of CoMoO4 species. Further increase in carbon species content may have resulted in the growth of MoO3, which then reacted with the carbon species to form large sized MoO2 that was then difficult to sulfurize to MoS2. An optimum amount of carbon species can play the role of “isolating agents”, which inhibit the growth of Mo species and lead to the generation of subsequent MoS2 slabs with short lengths. These MoS2 slabs generate more coordinatively unsaturated sites after reduction by hydrogen at low temperature, and thus C-3 (the CoMo catalyst supported on the carbon-covered γ-Al2O3 support prepared by modification with acetic acid solutions) exhibited the highest HDS activity for 4,6-DMDBT.Acknowledgements
Financial support from the National Natural Science Foundation, China (no. U1162203) is gratefully acknowledged. We also thank Dr. Y. P. Li (the College of Chemical Engineering, China University of Petroleum, Qingdao, P. R. China) for HRTEM characterizations.Notes and references
- H. Topsøe, B. S. Clausen and F. E. Massoth, Hydrotreating Catalysis—Science and Technology, Springer, Berlin, 1996, vol. 11, p. 162 Search PubMed.
- C. S. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- B. C. Gates and H. Topsøe, Polyhedron, 1997, 16, 3213–3217 CrossRef CAS.
- K. G. Knudsen, B. C. Cooper and H. Topsøe, Appl. Catal., A, 1999, 189, 205–215 CrossRef CAS.
- M. Breysse, G. Djega-Mariadassou, S. Pessayre, C. Geantet, M. Vrinat, G. Perot and M. Lemaire, Catal. Today, 2003, 84, 129–138 CrossRef CAS.
- H. Topsøe, B. S. Clausen, R. Candia, C. Wivel and S. Mørup, J. Catal., 1981, 68, 433–452 CrossRef.
- C. Wivel, R. Candia, B. S. Clausen, S. Mørup and H. Topsøe, J. Catal., 1981, 68, 453–463 CrossRef CAS.
- N.-Y. Topsøe and H. Topsøe, J. Catal., 1983, 84, 386–401 CrossRef.
- A. Stanislaus, A. Marafi and M. S. Rana, Catal. Today, 2010, 153, 1–68 CrossRef CAS PubMed.
- V. M. Kogan and P. A. Nikulshin, Catal. Today, 2010, 149, 224–231 CrossRef CAS PubMed.
- S. Eijsbouts, L. C. A. van den Oetelaar and R. R. van Puijenbroek, J. Catal., 2005, 229, 352–364 CrossRef CAS PubMed.
- H. Topsøe, Appl. Catal., A, 2007, 322, 3–8 CrossRef PubMed.
- T. Kubota, N. Rinaldi, K. Okumura, T. Honma, S. Hirayama and Y. Okamoto, Appl. Catal., A, 2010, 373, 214–221 CrossRef CAS PubMed.
- A. V. Pashigreva, G. A. Bukhtiyarova, O. V. Klimov, Y. A. Chesalov, G. S. Litvak and A. S. Noskov, Catal. Today, 2010, 149, 19–27 CrossRef CAS PubMed.
- L. Peña, D. Valencia and T. Klimova, Appl. Catal., B, 2014, 147, 879–887 CrossRef PubMed.
- M. Breysse, C. Geantet, P. Afanasiev, J. Blanchard and M. Vrinat, Catal. Today, 2008, 130, 3–13 CrossRef CAS PubMed.
- E. Furimsky and F. E. Massoth, Catal. Today, 1999, 52, 381–495 CrossRef CAS.
- K. Hellgardt and D. Chadwick, Ind. Eng. Chem. Res., 1998, 37, 405–411 CrossRef CAS.
- M. Kouzu, Y. Kuriki, F. Hamdy, K. Sakanishi, Y. Sugimoto and I. Saito, Appl. Catal., A, 2004, 265, 61–67 CrossRef CAS PubMed.
- H. Farag, I. Mochida and K. Sakanishi, Appl. Catal., A, 2000, 194, 147–157 CrossRef.
- F. Severino, J. Laine and A. Lopez-Agudo, J. Catal., 2000, 189, 244–246 CrossRef CAS.
- P. M. Boorman, K. Chong, R. A. Kydd and J. M. Lewis, J. Catal., 1991, 128, 537–550 CrossRef CAS.
- J. P. R. Vissers, F. P. M. Mercx, S. M. A. M. Bouwens, V. H. J. de Beer and R. Prins, J. Catal., 1988, 114, 291–302 CrossRef CAS.
- P. A. Nikulshin, V. A. Salnikov, A. V. Mozhaev, P. P. Minaev, V. M. Kogan and A. A. Pimerzin, J. Catal., 2014, 309, 386–396 Search PubMed.
- H. Li, M. Li, Y. Chu, F. Liu and H. Nie, Appl. Catal., A, 2011, 403, 75–82 CrossRef CAS PubMed.
- S. L. González-Cortés, T.-C. Xiao, P. M. F. J. Costa, B. Fontal and M. L. H. Green, Appl. Catal., A, 2004, 270, 209–222 CrossRef PubMed.
- S. L. González-Cortés, T.-C. Xiao, T.-W. Lin and M. L. H. Green, Appl. Catal., A, 2006, 302, 264–273 CrossRef PubMed.
- M. L. H. Green and T.-C. Xiao, WO 2004000456, 2003.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207–219 CrossRef CAS.
- P. Afanasiev, Appl. Catal., A, 2006, 303, 110–115 CrossRef CAS PubMed.
- B. Scheffer, N. J. J. Dekker, P. J. Mangnus and J. A. Moulijn, J. Catal., 1990, 121, 31–46 CrossRef CAS.
- E. J. M. Hensen, P. J. Kooyman, Y. van der Meer, A. M. van der Kraan, V. H. J. de Beer, J. A. R. van Veen and R. A. van Santen, J. Catal., 2001, 199, 224–235 CrossRef CAS.
- Y. Araki, K. Honna and H. Shimada, J. Catal., 2002, 207, 361–370 CrossRef CAS.
- L. Qu, W. Zhang, P. J. Kooyman and R. Prins, J. Catal., 2003, 215, 7–13 CrossRef CAS.
- L. Ding, Z. Zhang, Y. Zheng, Z. Ring and J. Chen, Appl. Catal., A, 2006, 301, 241–250 CrossRef PubMed.
- U. Usman, M. Takaki, T. Kubota and Y. Okamoto, Appl. Catal., A, 2005, 286, 148–154 CrossRef CAS PubMed.
- F. Bataille, J. Lemberton, P. Michaud, G. Pérot, M. Vrinat, M. Lemaire, E. Schulz, M. Breysse and S. Kasztelan, J. Catal., 2000, 191, 409–422 CrossRef CAS.
- J. Lauritsen, M. Nyberg, J. Nørskov, B. Clausen, H. Topsøe, E. Lægsgaard and F. Besenbacher, J. Catal., 2004, 224, 94–106 CrossRef CAS PubMed.
- O. Y. Gutiérrez and T. Klimova, J. Catal., 2011, 281, 50–62 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy00814f |
| ‡ These authors contributed equally to this work and should be considered as co-first authors. |
| This journal is © The Royal Society of Chemistry 2015 |