
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes
Martin
Oestreich
*,
Julia
Hermeke†
and
Jens
Mohr†
Institut für Chemie, Technische Universität Berlin, Straße des 17. Juni 115, D-10623 Berlin, Germany. E-mail: martin.oestreich@tu-berlin.de; Fax: +49 (0)30 314 28829; Tel: +49 (0)30 314 29721
First published on 13th February 2015
Abstract
The bond activation chemistry of B(C6F5)3 and related electron-deficient boranes is currently experiencing a renaissance due to the fascinating development of frustrated Lewis pairs (FLPs). B(C6F5)3's ability to catalytically activate Si–H bonds through η1 coordination opened the door to several unique reduction processes. The ground-breaking finding that the same family of fully or partially fluorinated boron Lewis acids allows for the related H–H bond activation, either alone or as a component of an FLP, brought considerable momentum into the area of transition-metal-free hydrogenation and, likewise, hydrosilylation. This review comprehensively summarises synthetic methods involving borane-catalysed Si–H and H–H bond activation. Systems corresponding to an FLP-type situation are not covered. Aside from the broad manifold of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bond reductions and CX/C–X defunctionalisations, dehydrogenative (oxidative) Si–H couplings are also included.
X bond reductions and CX/C–X defunctionalisations, dehydrogenative (oxidative) Si–H couplings are also included.
 Martin Oestreich | Martin Oestreich (born in 1971 in Pforzheim/Germany) is currently Professor of Organic Chemistry at the Technische Universität Berlin. He received his diploma degree with Paul Knochel (Marburg, 1996) and his doctoral degree with Dieter Hoppe (Münster, 1999). After a two-year postdoctoral stint with Larry E. Overman (Irvine, 1999–2001), he completed his habilitation with Reinhard Brückner (Freiburg, 2001–2005) and was appointed as Professor of Organic Chemistry at the Westfälische Wilhelms-Universität Münster (2006–2011). He also held visiting positions at Cardiff University in Wales (2005) and at The Australian National University in Canberra (2010). |
 Julia Hermeke | Julia Hermeke (born in 1986 in Neuruppin/Germany) studied chemistry at the Freie Universität Berlin (2006–2009) and at the University of Hong Kong (2009–2011). She spent internships with Bayer Schering Pharma AG in Berlin and with Evonik Degussa GmbH in Marl (2008). She obtained her bachelor's degree with Hans-Ulrich Reißig at the Freie Universität Berlin and her master's degree with Patrick H. Toy at the University of Hong Kong. Her master studies were supported by a scholarship of the Studienstiftung des deutschen Volkes (2009–2011). She is currently in her final phase of her doctoral degree in the group of Martin Oestreich at the Technische Universität Berlin. |
 Jens Mohr | Jens Mohr (born in 1987 in Otterndorf/Germany) studied chemistry at the Westfälische Wilhelms-Universität Münster (2007–2012) and spent a three-month research internship at Hoffmann-La Roche in Basel (2011). He obtained his bachelor's (2010) and master's degrees (2012) with Martin Oestreich. His master studies were supported by an Almuth Klemer fellowship from the Organisches-Chemisches Institut of the Westfälische Wilhelms-Universität Münster. He is currently pursuing graduate research in the group of Martin Oestreich at the Technische Universität Berlin. |
1. Introduction
The potent boron Lewis acid tris(pentafluorophenyl)borane [B(C6F5)3, Fig. 1] is one of those molecules that revealed its relevance to synthetic chemistry long after its discovery. Its preparation was described in the early 1960s1 but, without any obvious use, these reports laid dormant for decades with hardly any citations. The situation changed in the early 1990s when Marks and co-workers found that B(C6F5)3 and cognate electron-deficient boranes are excellent co-catalysts in metallocene-mediated alkene polymerization.2 B(C6F5)3's ability to act as catalyst in its own right was then discovered by Piers and co-workers in the late 1990s. These authors showed that B(C6F5)3 catalyses CX bond hydrosilylations3 (reduction) as well as dehydrogenative Si–O couplings4 (oxidation) by a counterintuitive mechanism. It was proposed largely on the basis of kinetic data that B(C6F5)3 activates the hydrosilane rather than the carbonyl group by an, at that time, unusual η1 coordination.5 Although these contributions were noticed by the community, B(C6F5)3 catalysis still continued to develop rather slowly.6 The advent of frustrated Lewis pairs (FLPs) in the mid 2000s finally moved B(C6F5)3 and related fully or partially fluorinated boranes into the limelight. Stephan's7 and also Erker's8 findings that Lewis pairs, either frustrated or formed reversibly, composed of those boron Lewis acids and a rather broad range of Lewis bases activate small molecules without the need for a transition metal were a significant advancement.9,10 The heterolytic splitting of dihydrogen and its application in transition-metal-free hydrogenation is particularly relevant and closely related to the Piers-type activation of the weaker Si–H bond (90 kcal mol−1 for Si–H versus 103 kcal mol−1 for H–H).11 H–H bond activation is in fact not even limited to FLP-like systems, and B(C6F5)3 alone will activate dihydrogen without the assistance of a deliberately added Lewis base.12 The Lewis basic reactant is enough to step in. The purpose of this review is to collect synthetic methods involving borane-catalysed Si–H and H–H bond activation in a single article. We explicitly excluded clear cases of FLP-type systems as these were comprehensively covered by several reviews in recent years.9,10
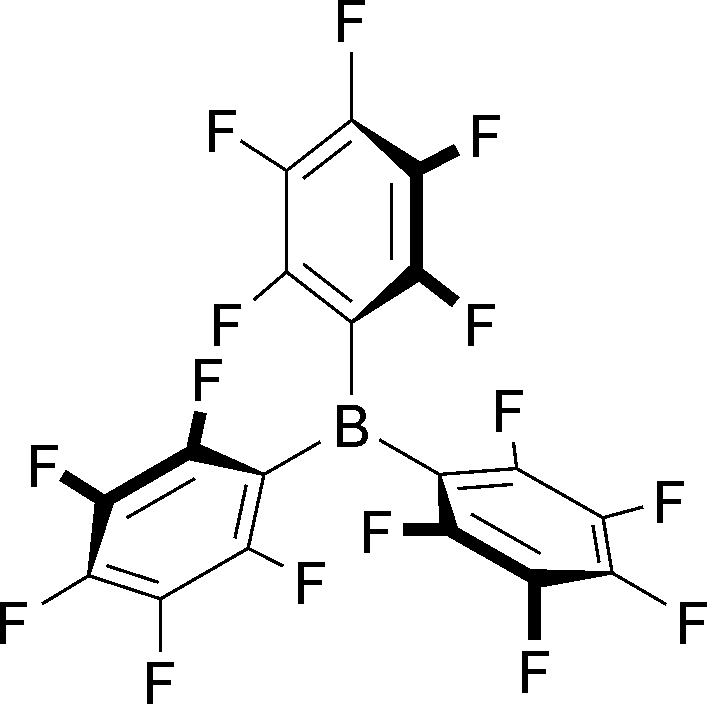 | ||
| Fig. 1 Tris(pentafluorophenyl)borane [B(C6F5)3]. | ||
2. Mechanistic aspects
The fascination with B(C6F5)3 catalysis began with the seminal discovery by Piers and co-workers that the B(C6F5)3-catalysed carbonyl hydrosilylation obeys a counterintuitive three-step mechanism (XO, Scheme 1).3,5 A series of significant experiments established that Lewis adduct II composed of B(C6F5)3 and carbonyl compound I is not a competent intermediate in the catalytic cycle. B(C6F5)3 was in fact shown to be largely present in its free form. Instead, it was proposed that B(C6F5)3 is involved in the activation of the Si–H bond of hydrosilane III through reversible η1 rather than η2 coordination, and the intermediacy of the unusual complex IV was suggested. The enhanced Lewis acidity of the silicon atom in IV would then facilitate the nucleophilic attack by the Lewis-basic oxygen atom of I. The hydride is transferred from the silicon to the boron atom in that step to afford ion pair VI, that is a borohydride along with a silylcarboxonium ion. The catalysis would come full circle by hydride transfer from the borohydride to the electrophilic carbon atom of the silylcarboxonium ion, thereby yielding silyl ether VII and releasing free B(C6F5)3. That unique mechanism was in accordance with kinetic analyses, NMR spectroscopic measurements and isotopic labelling experiments. Preliminary computational investigations lent further support to the existence of the hydrosilane–borane adduct IV.11 Later, Rendler and Oestreich verified the nature of the actual activation step for carbonyl compounds I with the aid of a silicon-stereogenic hydrosilane as a stereochemical probe.13,14 The Si–H bond activation was shown to proceed through the SN2–Si transition state V (XO).
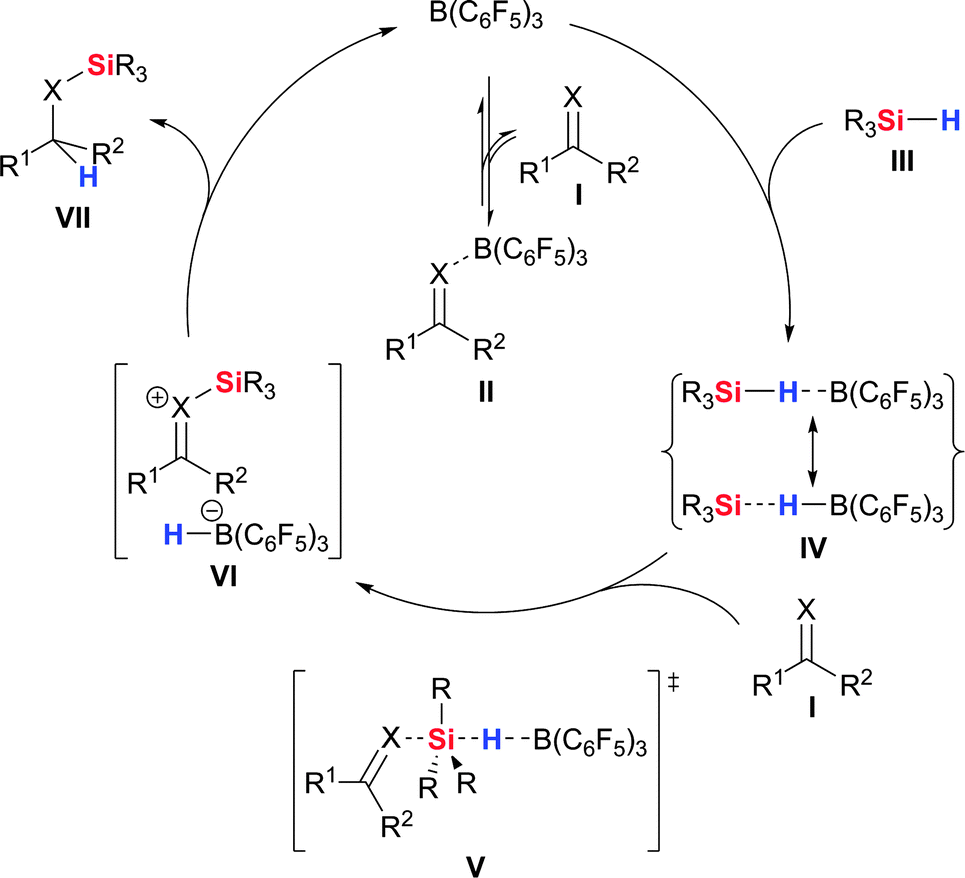 | ||
| Scheme 1 Mechanism of the B(C6F5)3-catalysed hydrosilylation of ketones (XO) and imines (XNR3). | ||
Recently, Sakata and Fujimoto further supported the experimentally observed mechanism by quantum-chemical calculations with acetone and Me3SiH as model compounds.15 Their results are in full agreement with the Piers–Oestreich mechanism.5,13 That work also included a comparison of B(C6F5)3 with BF3 as catalyst, and it was found that conventional carbonyl activation is operative in the latter case.16 The Si–H bond is cleaved in a four-membered transition state where the carbonyl oxygen atom accommodates both the boron atom and the weakly Lewis acidic silicon atom. The hydride is directly transferred from the silicon atom to the highly electrophilic carbonyl carbon atom. The fundamentally different activation modes exerted by BF3 (CO group) and B(C6F5)3 (Si–H bond) were mainly attributed to the electronic nature of their boron centres.
Just recently, Piers, Tuononen, and co-workers finally presented the long-awaited experimental evidence for the missing link of the catalytic cycle, that is the hydrosilane–borane adduct IV.17 These authors utilised antiaromatic 1,2,3-tris(pentafluorophenyl)-4,5,6,7-tetrafluoro-1-boraindene18 (1) as a more Lewis acidic model structure for B(C6F5)3 and were able to isolate its adduct with Et3SiH (2a) (3, Scheme 2). The careful study includes variable-temperature NMR spectroscopic analysis of the equilibrium of adduct formation and structural characterisation of 3 by X-ray crystallography. Quantum-chemical calculations were used to further probe the stability of 3 and to compare 3 with Et3SiH/B(C6F5)3 and the hypothetical adduct of Et3SiH (2a) and perfluoropentaphenylborole. The adduct Et3SiH/B(C6F5)3 was found to be weakest, and the higher stability of 3 was rationalized by a fine balance of steric and electronic effects rather than merely being the result of the higher Lewis acidity of 3 relative to B(C6F5)3. The molecular structure of 3 shows the anticipated Si–H–B bridge with the hydrogen atom remaining at the silicon atom. The Si–H distance is 1.51(2) Å and, hence, in the typical range of Si–H bonds (1.48 Å); the B–H distance of 1.46(2) Å is substantially longer than typical B–H bonds (1.14 Å). The Si–H–B bridge is not linear but significantly bent with a bond angle of 157°, a feature that it shares with related, less-bent [Si–H–Si]+ hydronium ions.19
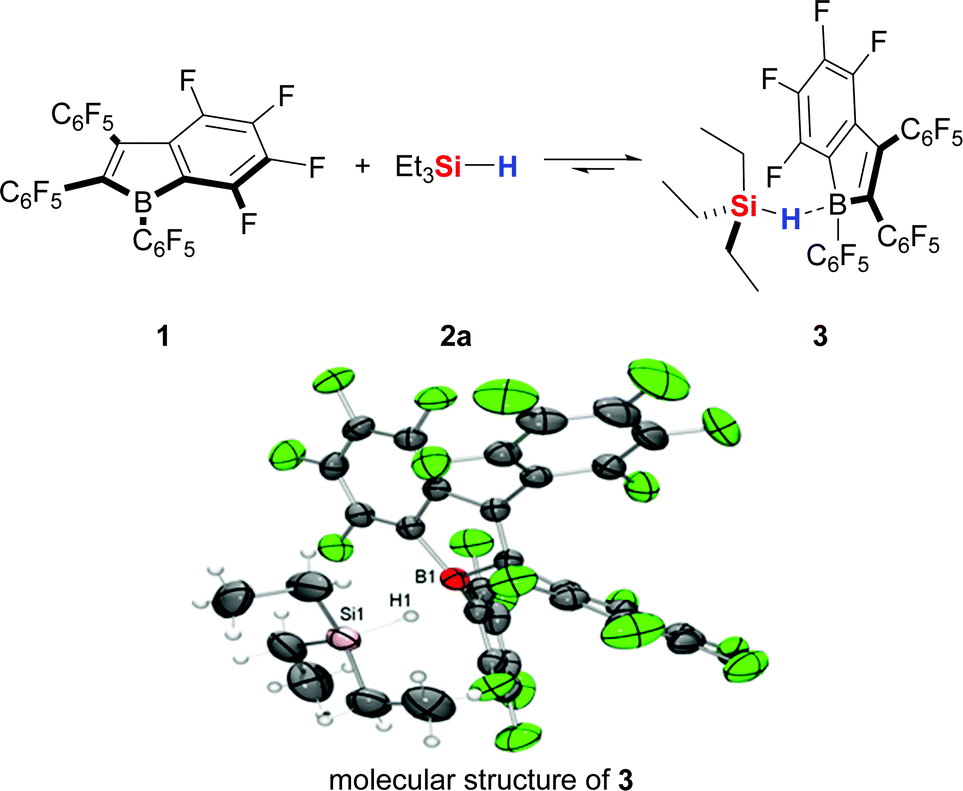 | ||
| Scheme 2 Adduct formation between Et3SiH (2a) and 1,2,3-tris(pentafluorophenyl)-4,5,6,7-tetrafluoro-1-boraindene (1) and its molecular structure 3. | ||
Regarding the imine hydrosilylation, the above basic mechanistic principles are still valid (XNR3, Scheme 1).20 Again, B(C6F5)3 dissociates from adduct II to form adduct IV with hydrosilane III. Imine I then acts as a nucleophile in the concerted displacement of the Si–H bond (IV → V → VI). However, there were doubts about the involvement of the silyliminium ion in the borohydride reduction step (VI → VII). Hog and Oestreich had tried to apply the aforementioned stereochemical probe13 to the hydrosilylation of imines but results were inconclusive.21 Initial attempts to determine the absolute configuration and the enantiomeric purity of the silicon-stereogenic hydrosilane reisolated after the hydrosilylation/Si–N cleavage sequence had failed. Also, the stereogenicity at the silicon atom had not induced any diastereoselectivity (dr = 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :26 in the corresponding carbonyl hydrosilylation13); the free amine was isolated in racemic form after hydrolysis. These findings led the authors to question the transformation of VI into VII, and to propose several alternative hydride transfer scenarios. Later, Mewald and Oestreich were able to show that the stereochemical information at the silicon atom is indeed lost in the catalytic cycle.22 It was speculated that dissociation of the silyliminium ion accounts for the racemization. However, the same authors demonstrated through matched–mismatched combinations of an axially chiral borane catalyst and a chiral hydrosilane reagent that the silyliminium ion is in fact participating in the borohydride reduction.22
:26 in the corresponding carbonyl hydrosilylation13); the free amine was isolated in racemic form after hydrolysis. These findings led the authors to question the transformation of VI into VII, and to propose several alternative hydride transfer scenarios. Later, Mewald and Oestreich were able to show that the stereochemical information at the silicon atom is indeed lost in the catalytic cycle.22 It was speculated that dissociation of the silyliminium ion accounts for the racemization. However, the same authors demonstrated through matched–mismatched combinations of an axially chiral borane catalyst and a chiral hydrosilane reagent that the silyliminium ion is in fact participating in the borohydride reduction.22
This confusion was finally solved by Oestreich and co-workers.23 A closer look at the imine hydrosilylation by NMR spectroscopy revealed the formation of unexpected intermediates that play a major role in the catalytic cycle (Scheme 3). The existence of free amine X and silylated enamine VIII in equimolar ratio indicated that a competing reaction pathway must be added to the established mechanistic picture. These intermediates emerge from the abstraction of a proton in the α-position of the silyliminium ion in VI (VI → VIII and IX). The resulting iminium ion in IX is reduced to the free amine X (IX → X). That free amine X is then transformed into the silylated amine VII by a two-step process: FLP-like and at that time unprecedented Si–H bond cleavage (X → XI)24 is followed by protonation of the silylated enamine VIII with the kinetically stable ion pair XI25 (XI → VII). The latter step also converts VIII back into the silyliminium ion VI. Based on these findings, the generally accepted mechanism must be expanded by another reaction pathway with another borohydride reduction step. The fact that there are two potentially stereoselectivity-determining steps hidden in the catalytic cycle must be considered for asymmetric variants.23
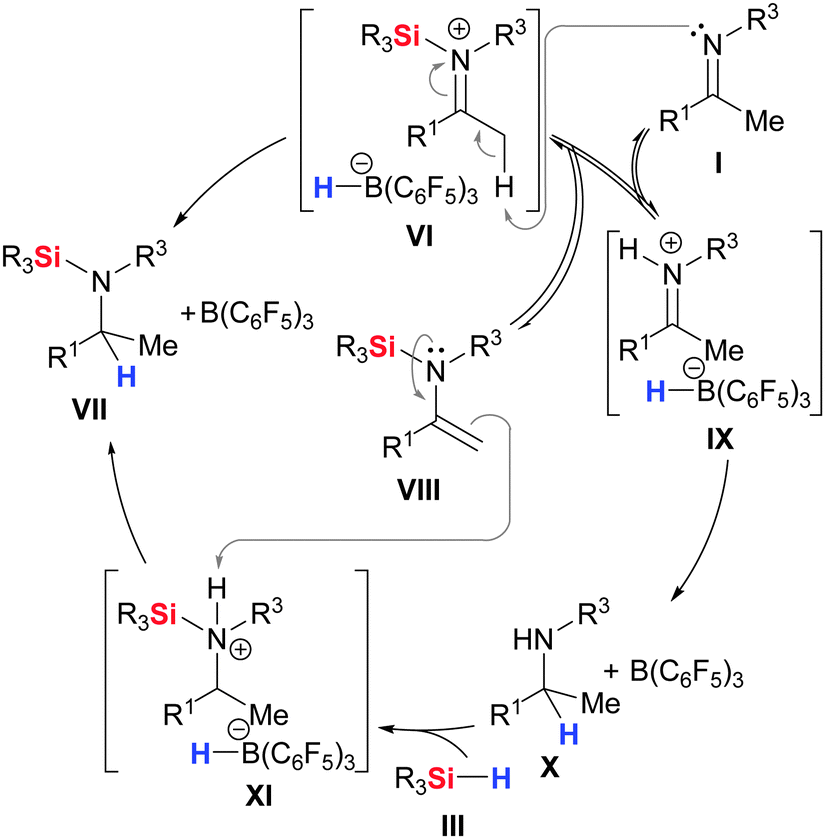 | ||
| Scheme 3 Competing pathways in B(C6F5)3-catalysed hydrosilylation of imines. | ||
The related hydrogenation is believed to pass through similar key intermediates as the hydrosilylation (cf.Scheme 1). Cooperative H–H activation by the Lewis acid catalyst and the Lewis basic substrate results in an ion pair composed of an iminium ion and a borohydride.26,27 The subsequent hydride transfer affords an amine–borane adduct that dissociates into the amine and the free borane catalyst. It must be noted though that there is evidence of an autocatalytic pathway where the amine rather than the imine is involved in the FLP-type heterolytic H–H splitting. Initially proposed by Klankermayer,28 this competing reaction was confirmed by quantum-chemical calculations by Pápai and co-workers.27c
3. Hydrosilylation and hydrogenation
3.1 Reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) X double bonds
X double bonds
As shown above, B(C6F5)3 is a suitable catalyst for the activation of the Si–H bond. The focus of this section is on hydrosilylation of CX bonds, initially introduced by Piers and co-workers.3,5 The ground-breaking discovery by Stephan and, independently, Klankermayer that the same catalyst also activates the H–H bond led to the development of several related hydrogenations, and these will be presented together with the hydrosilylations where appropriate. Piers and co-workers reported the B(C6F5)3-catalysed hydrosilylation of various aromatic and, later, aliphatic carbonyl compounds using Ph3SiH (2b) (4/5 → 7/8, Scheme 4).3,5 These catalyses are generally quite efficient, and it was shown in subsequent years that other electron-deficient boranes decorated with at least one C6FnH5−n group (n = 1–5) are also capable of promoting these reductions.29
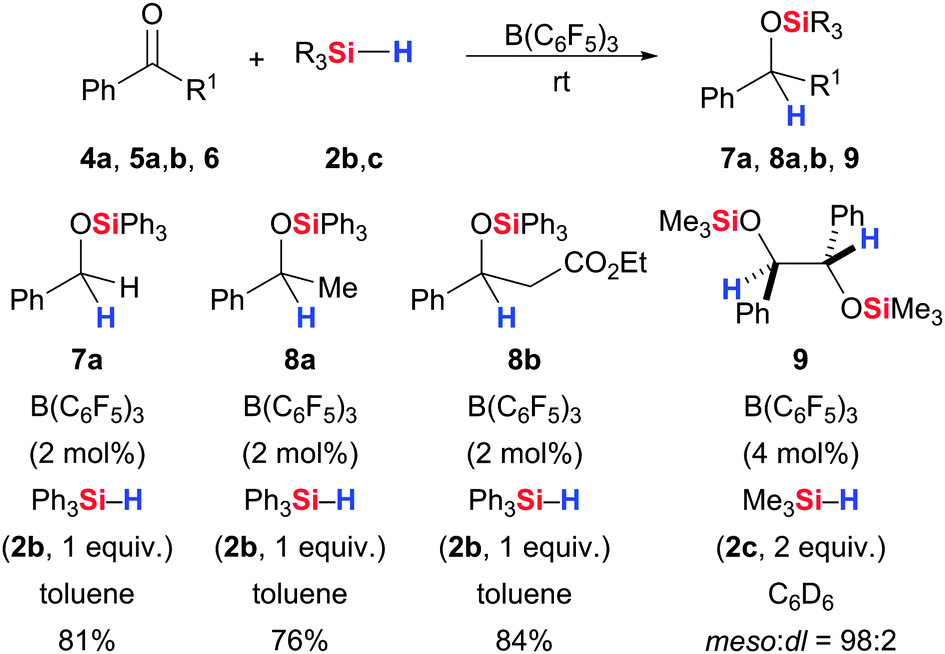 | ||
| Scheme 4 B(C6F5)3-catalysed hydrosilylation of aldehydes and ketones by Piers (examples 1–3) and Rosenberg (example 4). | ||
Rosenberg and co-workers elaborated the diastereoselective hydrosilylation of α-diketones where the steric bulk of the hydrosilane determines the relative configuration (syn/anti or meso/dl) of the 1,2-diol (6 → 9, Scheme 4).29e “Small” hydrosilanes and dihydrosilanes afforded meso whereas “large” hydrosilanes gave dl configuration.
The related B(C6F5)3-catalysed hydrogenation of carbonyl compounds remained elusive for many years although its feasibility had been proposed in a computational study by Nyhlén and Privalov a few years ago.30 Just recently, this gap was closed by the groups of Stephan31 and Ashley.32 Stephan and co-workers had already found that 1,1-diphenylethylene is reduced to the corresponding alkane when treated with dihydrogen in the presence of catalytic amounts of B(C6F5)3 and Et2O (10a → 11a, Scheme 5, upper).33 Inspired by this, the aforementioned groups were able to hydrogenate ketones as well as aldehydes to the corresponding alcohols with catalytic amounts of B(C6F5)3 in ethereal solvents (5/4 → 12/13, Scheme 5, lower). It was shown that not the carbonyl substrate but the solvent acts as the Lewis-base component in the heterolytic splitting of dihydrogen. Subsequent proton and hydride transfers generate the alcohol. Hence, the mechanism is different from the case calculated by Nyhlén and Privalov and resembles a situation with an external Lewis base rather than the catalytic cycle described in Scheme 1.
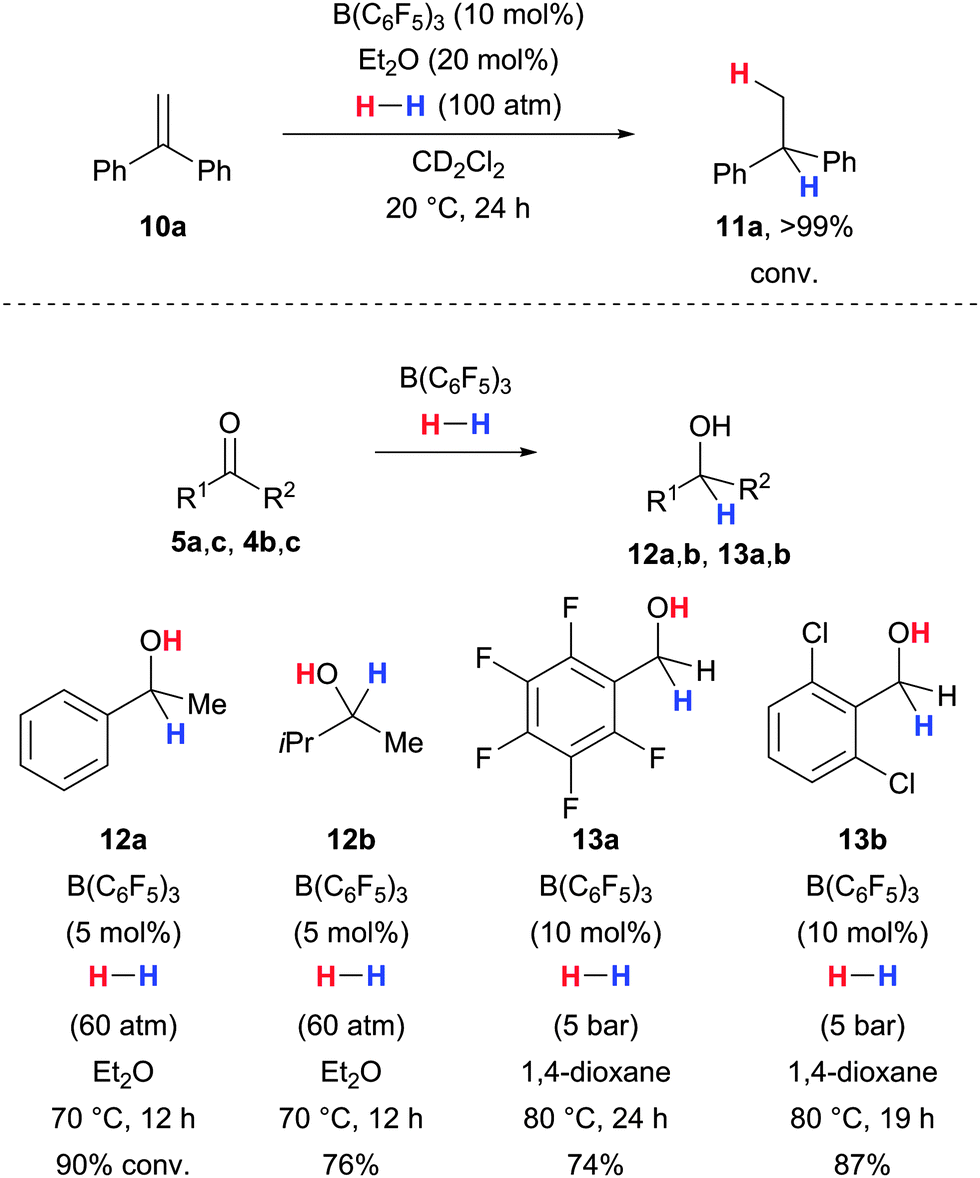 | ||
| Scheme 5 Hydrogenation of an alkene by Stephan (upper) and hydrogenations of carbonyl compounds using ether solvents as the Lewis base components by Stephan (lower, examples 1 and 2) and Ashley (examples 3 and 4). | ||
The attempt to extend this carbonyl reduction chemistry to the thioketone analogues was successfully accomplished by Rosenberg and co-workers.34 This substrate class participates in the B(C6F5)3 catalysis to form silyl thioethers with sterically accessible tertiary hydrosilanes and dihydrodisilanes such as HMe2SiSiMe2H (2e) (14 → 15, Scheme 6). The mechanism proposed by Rosenberg and co-workers is the same as that of the carbonyl hydrosilylation (cf.Scheme 1).5 However, these authors showed that formation of Lewis adduct II is less pronounced with thioketones than ketones. The hydrosilylation of benzophenone with Ph3SiH (2b) is nevertheless faster than that of the corresponding thioketone; the ketone is chemoselectively reduced in the presence of the thioketone. Several years later, Rosenberg and co-workers applied the same hydrosilylation to the preparation of polymers containing Si–S linkages next to (unreacted) Si–H bonds (14 + 2f → 15c, Scheme 6).35 The Si–Si bonds in the polyphenylsilane reactant remained untouched. The use of other catalysts for this postpolymerisation modification, no matter if ionic or radical, would lead to Si–Si bond cleavage. Not all Si–H bonds were converted into Si–S linkages but Rosenberg and co-workers were not able to estimate the degree of the Si–H substitution.
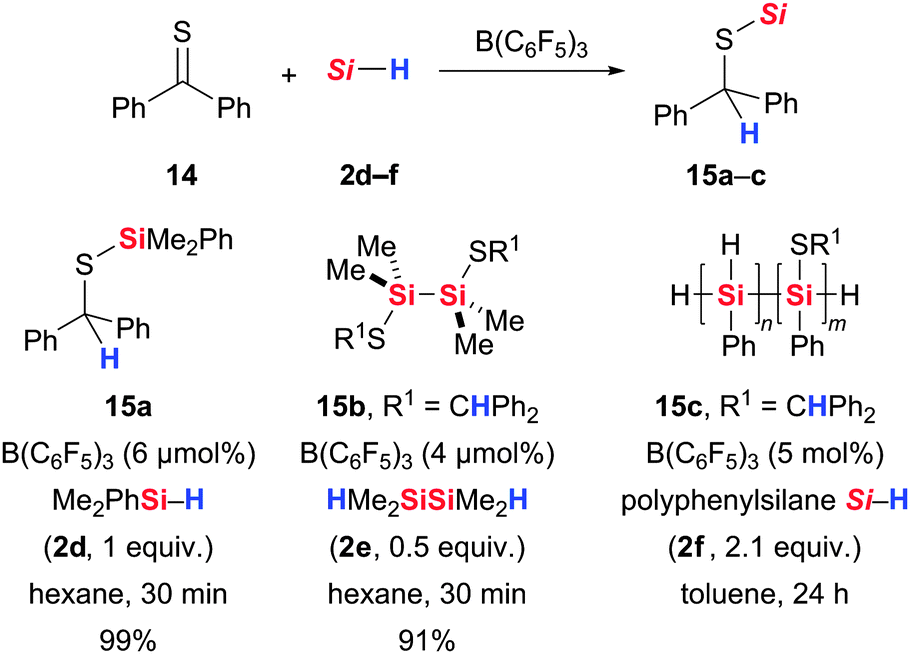 | ||
| Scheme 6 B(C6F5)3-catalysed hydrosilylation of thioketones by Rosenberg. | ||
Likewise, the B(C6F5)3-catalysed Si–H bond activation was applied to imines by Piers and co-workers (16/17 → 18/19 → 20/21, Scheme 7).20,36 Benzaldehyde-derived imines with representative substituents R3 at the imine nitrogen atom were systematically subjected to the standard procedure with Me2PhSiH (2d). As expected, the R3 group had a profound influence on the reaction rate, and SO2Ph and tBu were optimal requiring only 30 minutes at room temperature for completion. Imines derived from acetophenone, indan-1-one and benzophenone with benzyl protection were also successfully reduced.
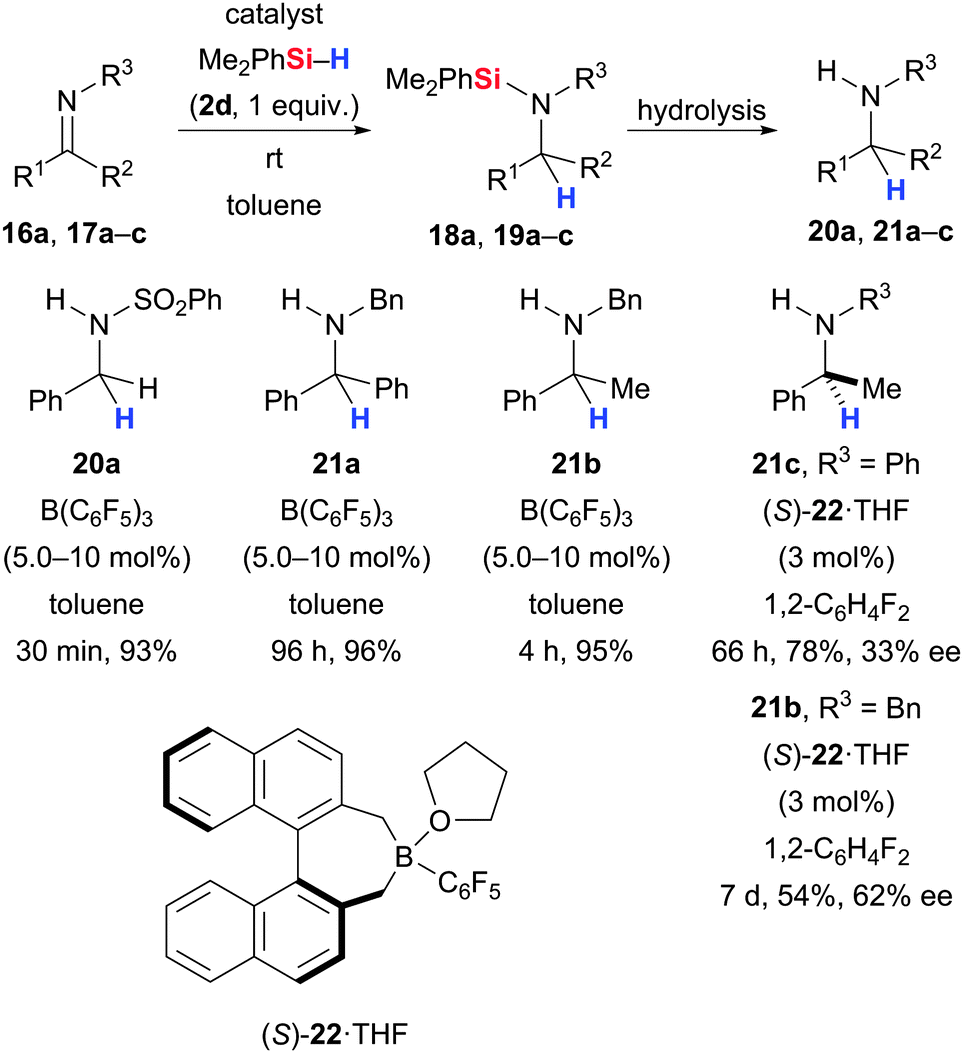 | ||
| Scheme 7 B(C6F5)3-catalysed hydrosilylation of imines. | ||
Mewald and Oestreich introduced the chiral electron-deficient borane (S)-22·THF29h to render the imine hydrosilylation enantioselective (17 → 19 → 21, Scheme 7).22 The level of enantioinduction was in fact promising but the refined mechanistic picture of this reaction illustrates that it will be extremely difficult to achieve high enantiocontrol due to competing enantioselectivity-determining hydride transfer steps (cf.Scheme 3).23 This was also illustrated by the work of Klankermayer and co-workers who utilised a chiral borane based on a functionalised camphor-derived backbone in the hydrosilylation of imines but could not detect any enantioinduction.36a Only in combination with an external phosphine Lewis base substantial enantioinduction (≤87% ee) was observed albeit yields were relatively low (not shown). Zhu and Du were able to reach higher yields by applying chiral binaphthyl-based boranes as catalysts but enantiomeric excesses did not exceed 82% ee (not shown).36b
The related B(C6F5)3-catalysed hydrogenation of imines was independently discovered by the groups of Stephan26 (16/17 → 20/21, Scheme 8) and Klankermayer.28 Aldehyde- as well as ketone-derived imines 16–17 were successfully reduced to the corresponding amines 20–21. Imine 16a bearing an electron-withdrawing protecting group reacted significantly slower than more basic imines 16b, 17d–17e. The group of Stephan also extended this method to diastereoselective hydrogenations of chiral imines (not shown).37
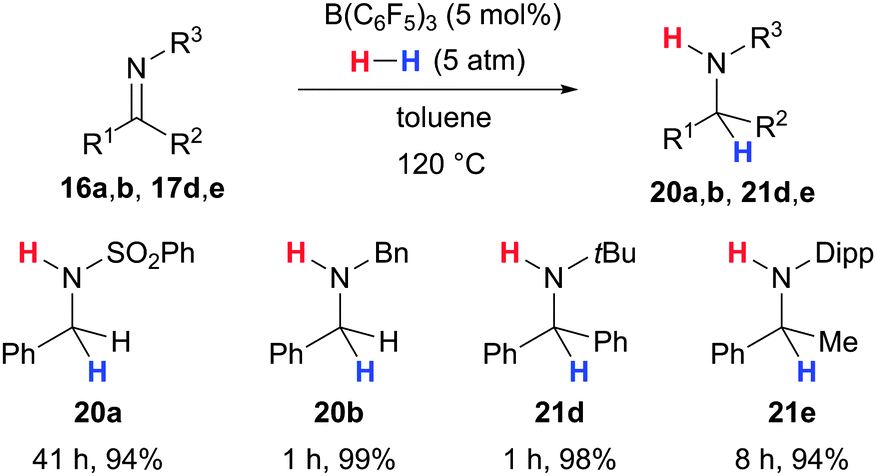 | ||
| Scheme 8 B(C6F5)3-catalysed hydrogenation of imines by Stephan. Dipp = 2,6-(Me2CH)C6H3. | ||
Ashley and co-workers were able to hydrogenate imines under milder reaction conditions (16c → 20c, Scheme 9).38 Similar to the hydrogenation of carbonyl compounds (cf.Scheme 5),31,32 the solvent d8-THF acts as the Lewis base in this system. To bypass its Lewis pair formation with the catalyst, the sterically more hindered B(C6F5)3 congener B(C6Cl5)(C6F5)2 was used.
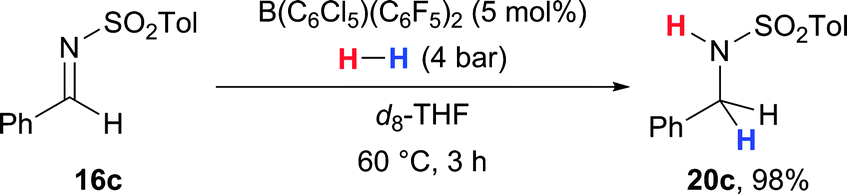 | ||
| Scheme 9 Hydrogenation of imines using the solvent as the Lewis base component by Ashley. | ||
Chen and Klankermayer had already observed 13% ee with (+)-α-pinene-derived B(C6F5)3-congener 2339a as chiral catalyst in the hydrogenation of phenyl-protected imine 17c (17c → 21c, Scheme 10).28 The same substrate 17c was reduced with high enantiomeric excess by Liu and Du five years later.40 The chiral borane 24a, prepared in situ by hydroboration with Piers' borane HB(C6F5)2,39 induced high enantioselectivity in the reduction of a variety of alkyl aryl ketimines, including substrates bearing additional Lewis basic sites (17 → 21, Scheme 10). However, meta-substituted substrates as well as diaryl and dialkyl ketimines showed poor enantioselectivities (not shown).
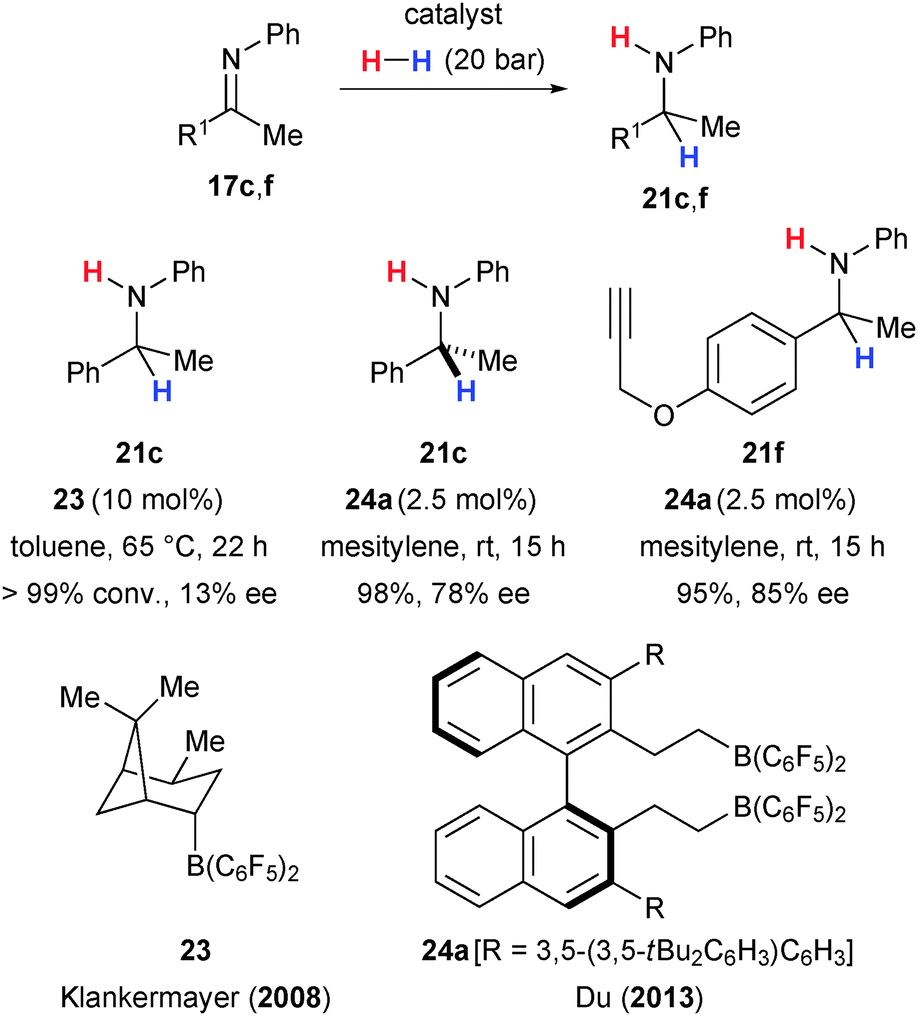 | ||
| Scheme 10 Enantioselective imine hydrogenations catalysed by chiral electron-deficient boranes. | ||
In addition to the direct hydrogenation of imines employing dihydrogen gas, methods for the transfer hydrogenation employing sacrificial dihydrogen sources were also developed. Stephan and co-workers treated various imines 16–17 with catalytic amounts of B(C6F5)3 in iPr2NH (25) (Scheme 11).41 A Meerwein–Pondorf–Verley-type reduction resulted in the formation of the corresponding amines 20–21. The excess of the dihydrogen source 25 is believed to shift the equilibrium toward the desired amine.
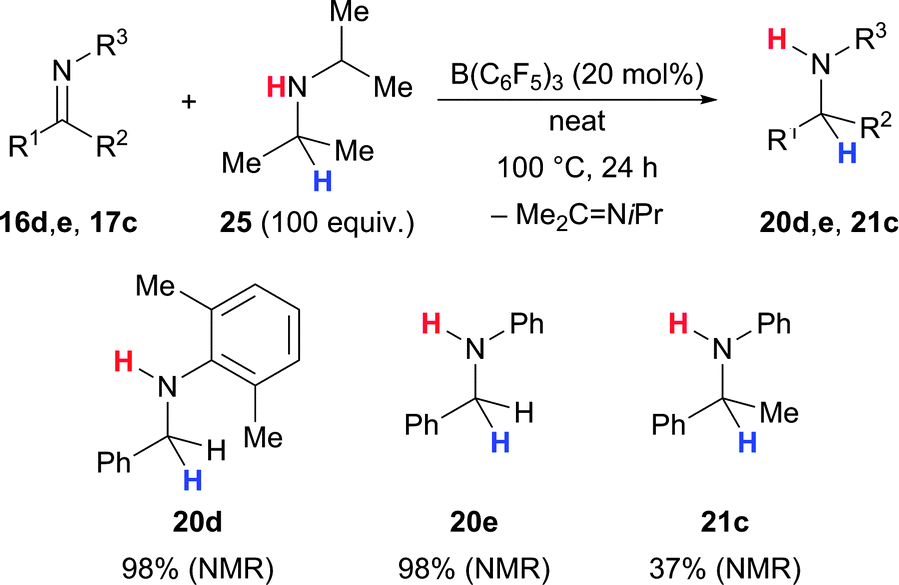 | ||
| Scheme 11 B(C6F5)3-catalysed Meerwein–Pondorf–Verley-type transfer hydrogenation of imines by Stephan. | ||
Chatterjee and Oestreich adopted a different approach toward imine reduction by employing cyclohexa-1,4-diene 26 as a dihydrogen surrogate (16/17 + 26 → 20/21, Scheme 12).42 The net reaction is a transfer hydrogenation that was believed to be initiated by B(C6F5)3-mediated dihydrogen release from 26 followed by the pathways (including autocatalysis)26–28 of the B(C6F5)3-catalysed imine hydrogenation. However, monitoring the reaction by 1H NMR spectroscopic analysis suggested a different scenario that again involves hydride abstraction from 26 by B(C6F5)3 (ref. 43) but no immediate dihydrogen formation. The high-energy Wheland intermediate is a strong Brønsted acid that protonates the imine substrate to arrive directly at the iminium ion–borohydride ion pair. Various tosyl-protected aldimines, e.g., 16f and 16g, were successfully hydrogenated. Ketimines were less reactive but different phenyl-protected imines, e.g., 17h, or imine 17g bearing the removable para-methoxyphenyl (PMP) group were reduced in excellent yield.
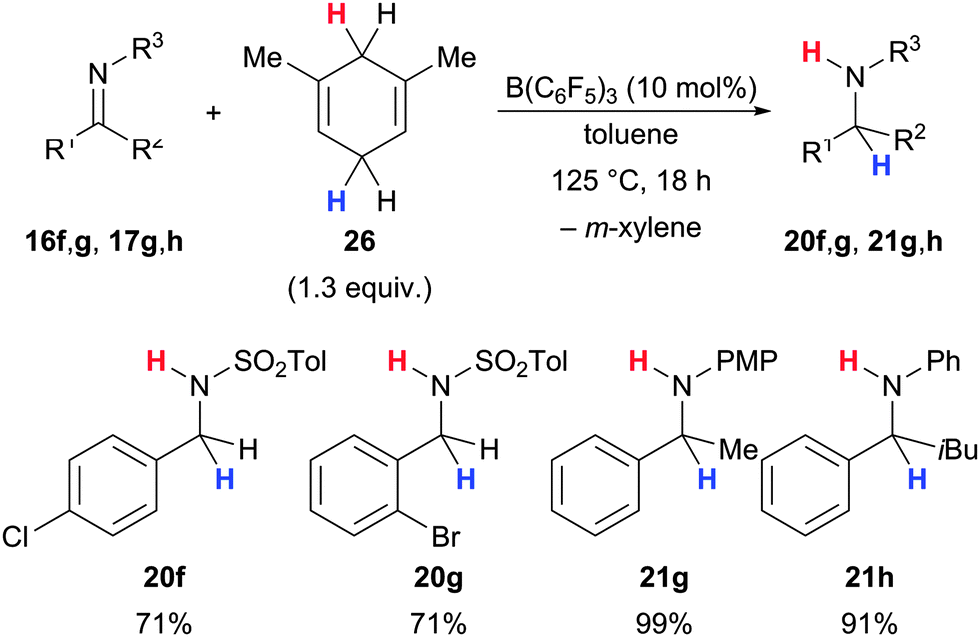 | ||
| Scheme 12 Transfer hydrogenation of imines by Oestreich. | ||
Recently, Mohr and Oestreich applied the B(C6F5)3-catalysed hydrogenation approach to the reduction of oxime ethers (Scheme 13).44 Decoration of the basic oxygen atom with sterically demanding substituents (tBu or SiiPr3) enabled the chemoselective reduction to the corresponding oxygen-protected hydroxylamines with leaving the weak N–O bond intact (27 → 28). Although the method was largely unaffected by the electronic and steric properties of the substituents at the oxime carbon atom, it remained limited to ketone-derived oxime ethers.
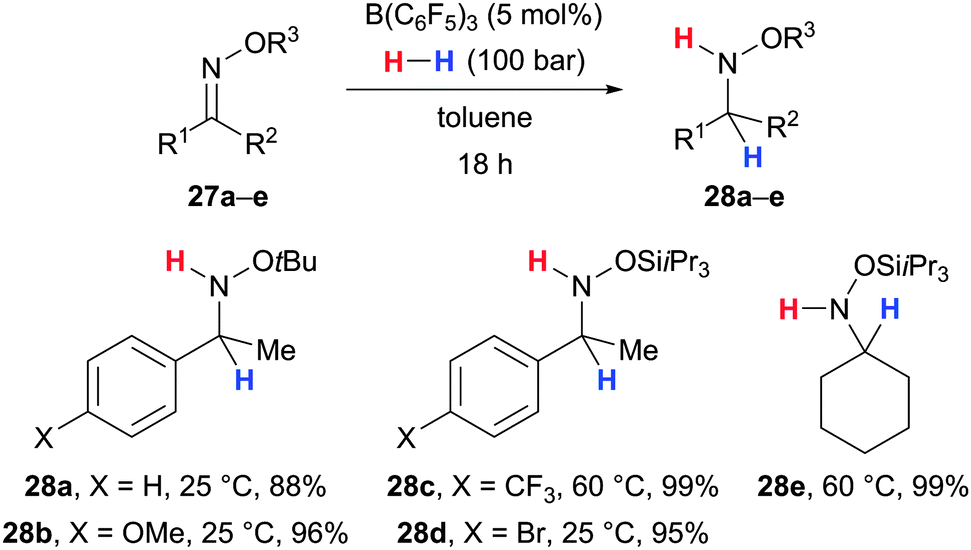 | ||
| Scheme 13 B(C6F5)3-catalysed hydrogenation of oxime ethers by Oestreich. | ||
Another well-investigated transformation is the hydrosilylation of alkenes, tracing back to the pioneering work of Gevorgyan and co-workers (10 → 29, Scheme 14).45 The reaction follows the usual pattern of Si–H bond activation by B(C6F5)3 with the π-basic alkene acting as nucleophile. A β-silycarbenium ion is formed as an intermediate, and the reaction is terminated by hydride transfer from the borohydride. Using various hydrosilanes (excluding sterically hindered ones such as iPr3SiH), excellent yields were obtained with substituted styrenes (including polymerisable indene), aliphatic alkenes, cyclohexene as well as properly protected oxygen-containing alkenes. The addition across the C–C double bond is trans-selective since the hydride attack occurs from the less hindered face; e.g., 1-methylcyclohex-1-ene gives good cis relative configuration (10e → 29d).
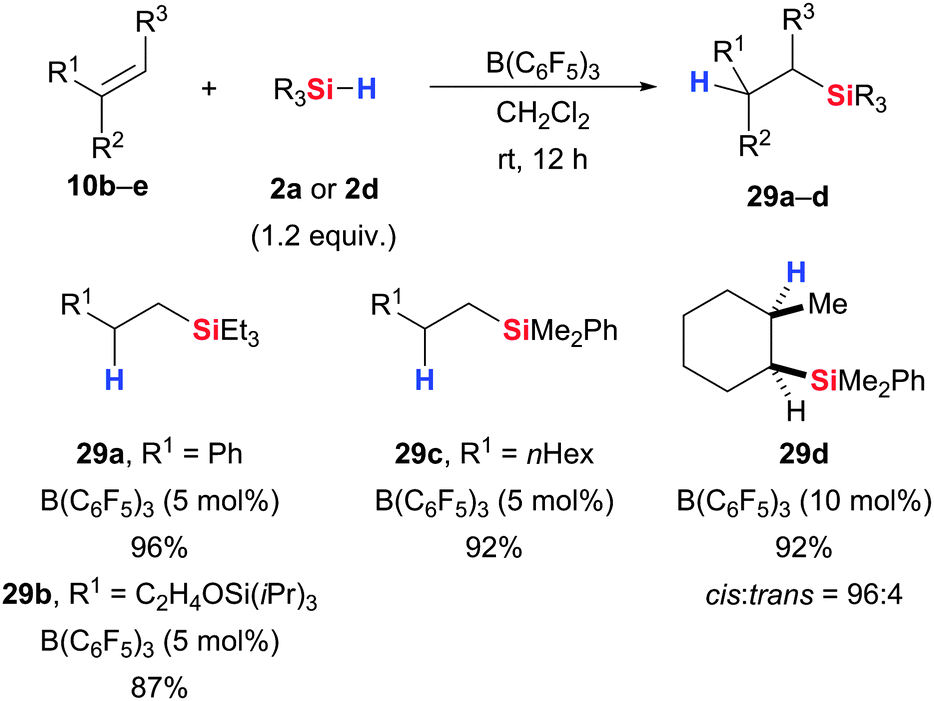 | ||
| Scheme 14 B(C6F5)3-catalysed hydrosilylation of alkenes by Gevorgyan. | ||
Recently, Simonneau and Oestreich introduced the new strategy of ionic transfer hydrosilylation where the hydrosilane is formed in situ by B(C6F5)3-promoted decomposition of easy-to-handle 3-silylated cyclohexa-1,4-dienes (10 + 30 → 29, Scheme 15).46 This approach is particularly beneficial in the case of gaseous hydrosilanes, e.g., highly flammable and explosive Me3SiH (b.p. 6.7 °C). As described for the B(C6F5)3-catalysed transfer hydrogenation of imines (cf.Scheme 12),42 the catalysis is initiated by abstraction of one of the bis(allylic) hydrides from the hydrosilane surrogate,43 forming a silicon-stabilised Wheland complex with the borohydride as counteranion. We note here that the Wheland intermediate might also be described as a benzene-stabilised silicon cation. That ion pair readily transforms into Me3SiH and benzene. Applied to alkenes, the in situ-generated Me3SiH engages in the above B(C6F5)3-catalysed hydrosilylation (cf.Scheme 14).45a Simonneau and Oestreich demonstrated the broad scope of this transfer hydrosilylation for terminal and internal alkenes (Scheme 15).46 For instance, oct-1-ene (10d → 29e) reacted with complete anti-Markovnikov selectivity, norbornene (10f → 29f) underwent exo-selective hydrosilylation and 1-methylcyclohex-1-ene (10e → 29g) featured cis-selective addition across its trisubstituted C–C double bond. Conversely, 2-methylindene shows opposite regioselectivity due to benzylic stabilisation of the carbenium ion, thereby overruling tertiary carbenium ion formation as well as steric factors.
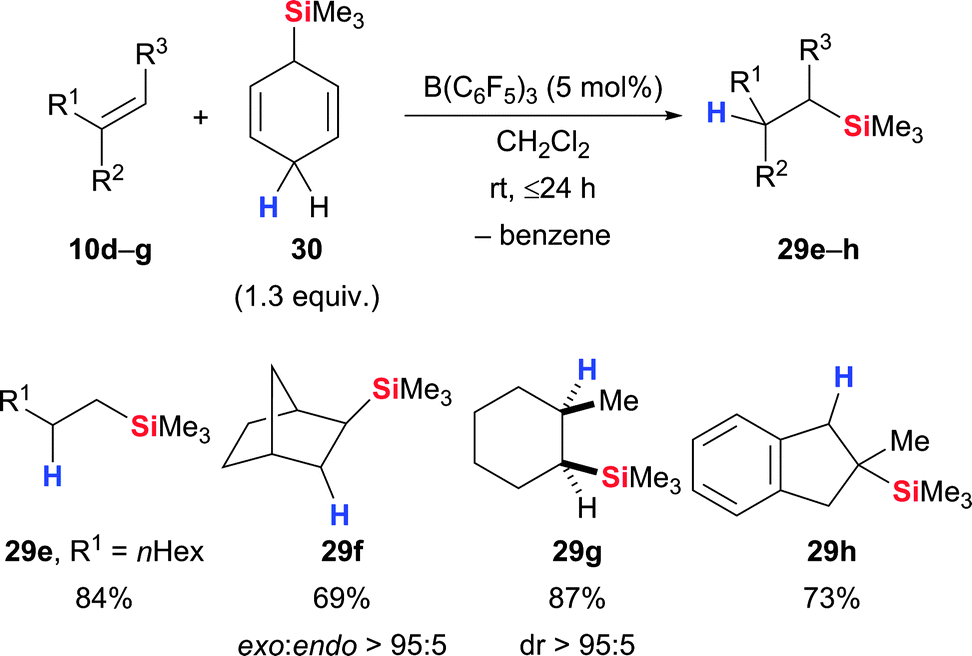 | ||
| Scheme 15 B(C6F5)3-catalysed transfer hydrosilylation of alkenes by Oestreich. | ||
Piers and co-workers extended the hydrosilylation of carbonyl functions to α,β-unsaturated acceptors (Scheme 16, upper).47 The chemoselectivity is usually excellent with enones, favoring 1,4- over 1,2-reduction (31a,b → 32a,b). Not surprisingly, 1,2-reduction becomes competitive with increased steric bulk at the β-carbon atom but in cases where the 1,2-pathway also suffers from steric congestion the conjugate reduction still wins (31a → 32a). However, predominant 1,2-hydrosilylation is seen with enals (31c → 32c). These 1,4-hydrosilylations afford silyl enol ethers that participate in another hydrosilylation to yield β-silylated silyl ethers (32d → 33, Scheme 16, lower).48 The diastereoselectivity of the addition of the Si–H bond across the C–C double bond is trans as a result of the sterically controlled borohydride reduction of the intermediate silylcarboxonium ion (cf. mechanistic discussion of the alkene hydrosilylation); the silyloxy and silyl groups are in a cis relationship.
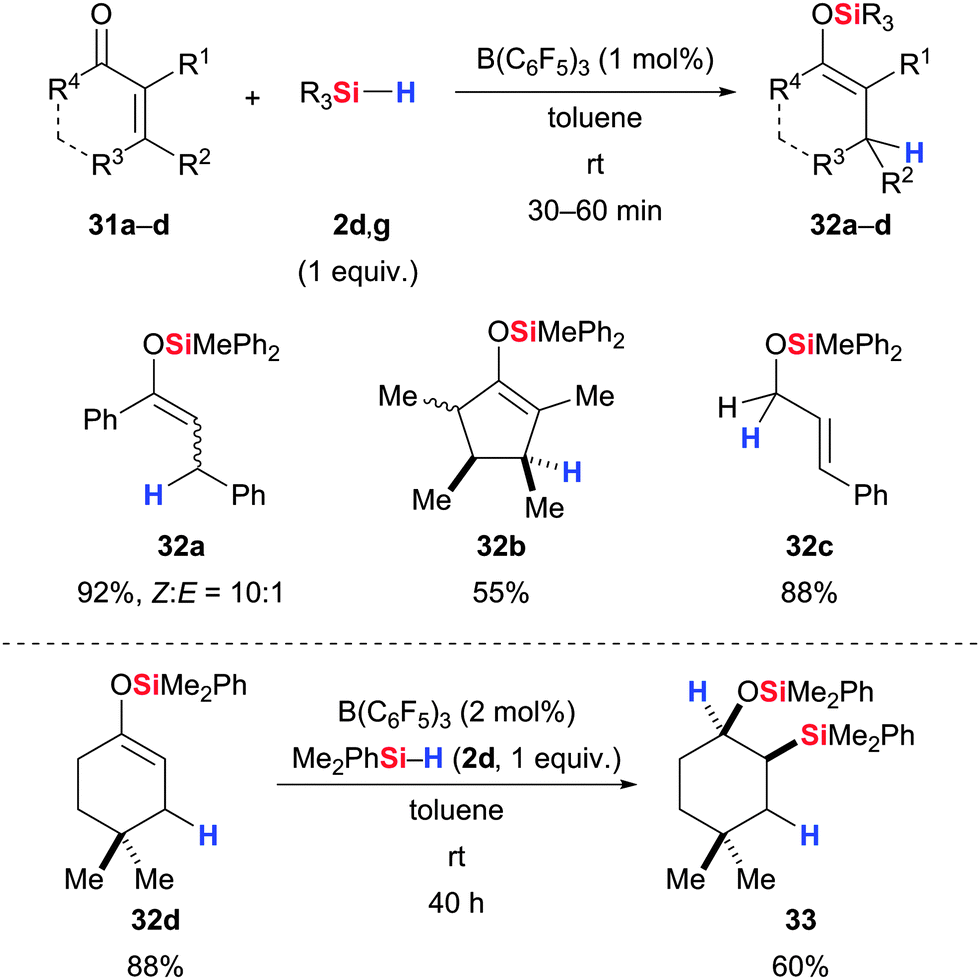 | ||
| Scheme 16 B(C6F5)3-catalysed hydrosilylation of enones, enals, and silyl enol ethers by Piers. | ||
Chandrasekhar and co-workers later employed polymethylhydrosiloxane (PMHS, 2h) in this reduction (10 → 11Scheme 17).49 The PMHS–B(C6F5)3 system allowed for highly chemo- and regioselective reduction of various conjugated alkenes in excellent yields and was superior to other PMHS–Lewis acid combinations. The advantages of PMHS (2h), such as being inexpensive, environmentally friendly as well as air- and moisture-stable, make the procedure a practical alternative to the existing protocol.
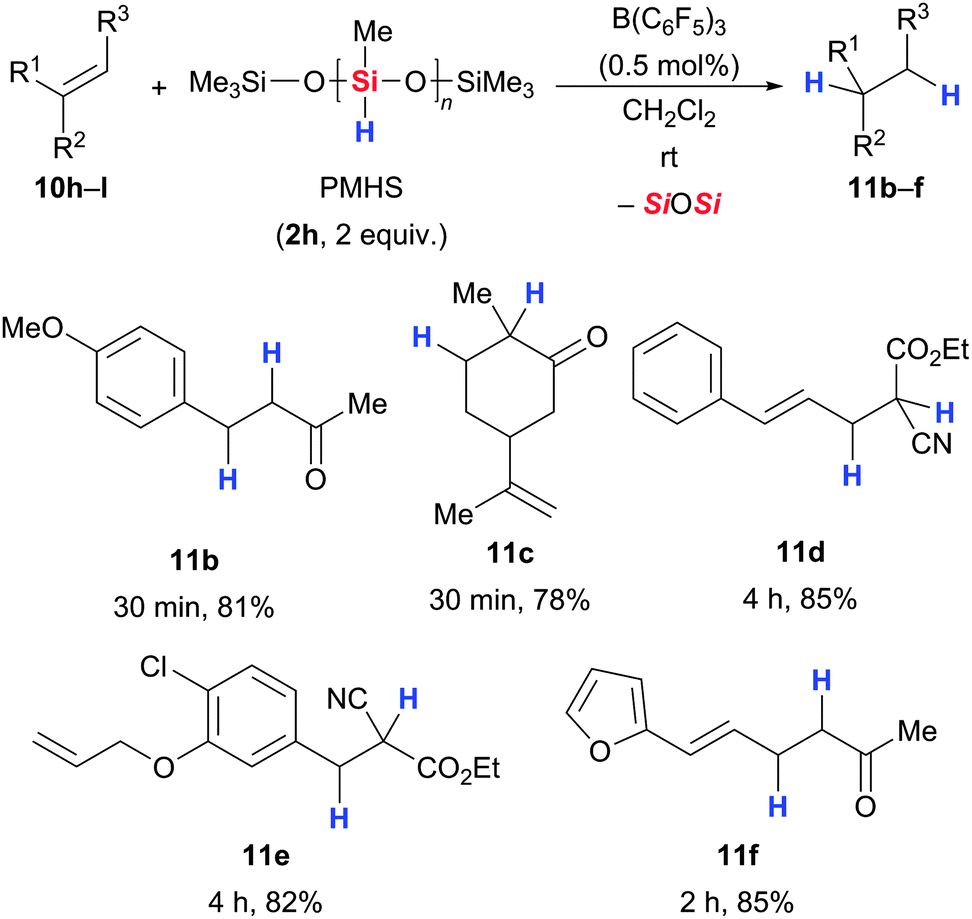 | ||
| Scheme 17 B(C6F5)3-catalysed hydrosilylation of α,β-unsaturated acceptors employing PMHS (2h) by Chandrasekhar. | ||
Similar to the hydrosilylation of silyl enol ethers (Scheme 16, lower), Tan and Zhang accomplished the reduction of enamines to afford the corresponding tertiary amines in excellent yields (34 → 35, Scheme 18).50 Only two examples have been reported to date but the related FLP-catalysed enamine hydrogenation is a popular application of this transformation.51
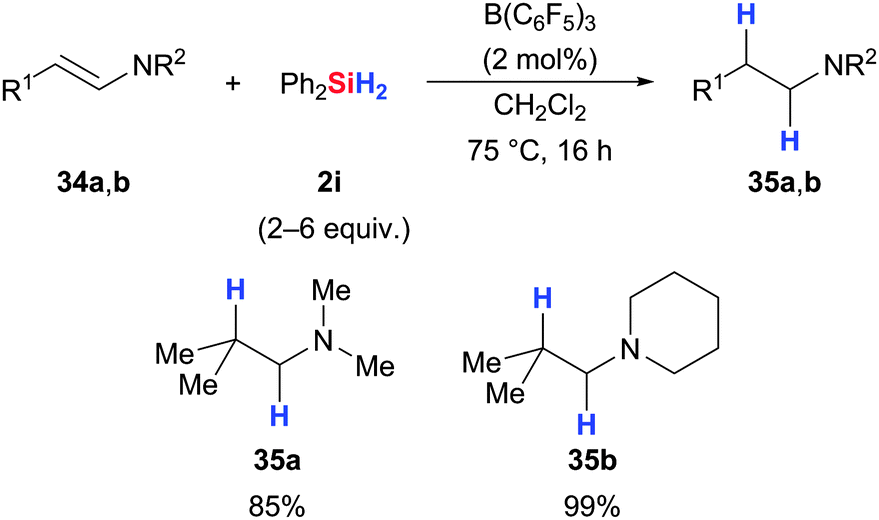 | ||
| Scheme 18 B(C6F5)3-catalysed hydrosilylation of enamines by Zhang. | ||
3.2 Reduction of carboxyl groups to the aldehyde oxidation level
The pioneering work of Piers and co-workers on carbonyl hydrosilylation also included carboxyl compounds as substrates (36 → 37 → 4, Scheme 19).3,5 Initially limited to aryl esters,3 the authors later extended the scope to (functionalised) aliphatic esters.5 Hydrolysis of the acetal 37 then affords the aldehyde 4. It is important to note that overstoichiometric amounts of the hydrosilane result in further degradation of the acetal 37, yielding complex mixtures of various oxidation states.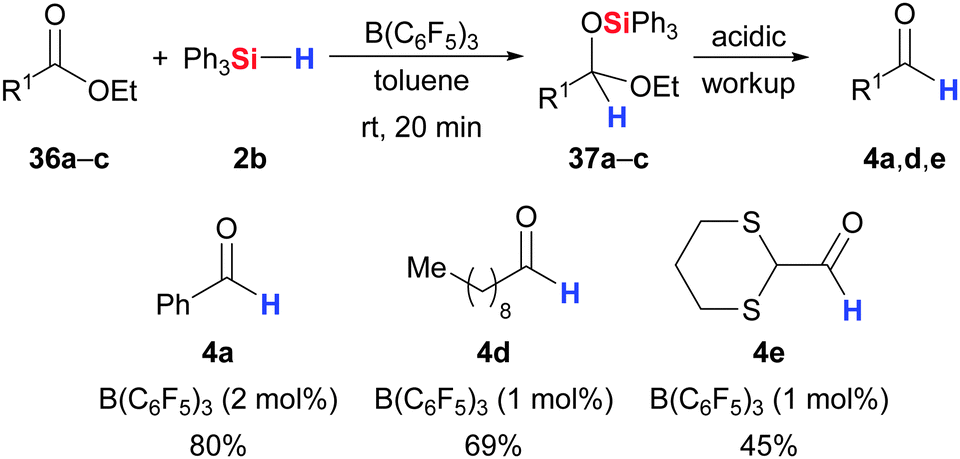 | ||
| Scheme 19 B(C6F5)3-catalysed hydrosilylation of carboxylic esters to acetals by Piers. | ||
The related hydrosilylation of carboxylic acids was recently elaborated by Brookhart and co-workers (38 → 39 → 4, Scheme 20).52,53 Again, the acetal is easily hydrolysed to the aldehyde (39 → 4). Acetal formation was particularly effective with bulky tertiary hydrosilanes whereas smaller tertiary as well as secondary hydrosilanes led to overreduction. For example, hydrosilylation of phenylpropanoic acid with TMDS (1,1,3,3-tetramethyldisiloxane) cleanly produced fully reduced n-propylbenzene (not shown). The substrate scope ranges from linear and branched aliphatic to aromatic carboxylic acids with excellent tolerance of unsaturation (double and triple bonds). The difference between this and Piers' carboxyl hydrosilylation is that the present transformation begins with a B(C6F5)3-catalysed dehydrogenative Si–O coupling (see Section 7) that converts the free carboxylic acid into the corresponding silyl ester.
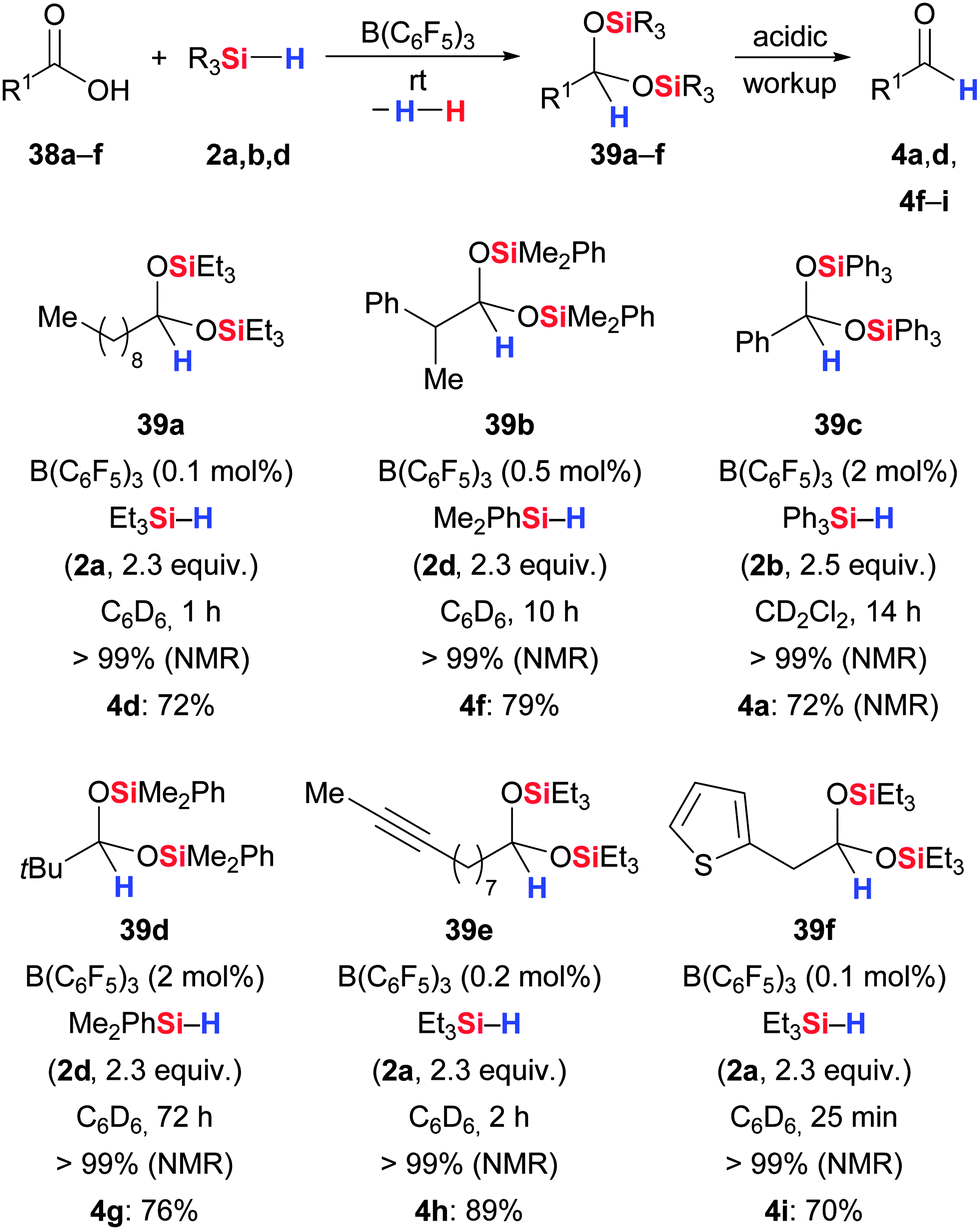 | ||
| Scheme 20 B(C6F5)3-catalysed hydrosilylation of carboxylic acids to acetals by Brookhart. | ||
3.3 Deoxygenation
Another attractive application of the B(C6F5)3-catalyzed hydrosilylation was introduced by Gevorgyan, Yamamoto, and co-workers.54,55 These authors showed that alcohols, carbonyl as well as carboxyl compounds are fully reduced to the hydrocarbon at room temperature when treated with excess Et3SiH (2a) in the presence of catalytic amounts of B(C6F5)3 (Schemes 21 and 23). By this, primary alcohols are converted into the corresponding alkane (13 → 40, Scheme 21). However, deoxygenation of secondary and tertiary alcohols failed. These alcohols undergo the initial dehydrogenative Si–O coupling (cf.Scheme 40) but the generated silyl ethers are too sterically hindered to react further with Et3SiH–B(C6F5)3 adduct. Chemoselective formation of silyl ethers from primary alcohols is possible with equimolar amounts of Et3SiH (2a). Nimmagadda and McRae further advanced this methodology by utilizing less hindered nBuSiH3 and Et2SiH2 (2j), respectively.56 By this, secondary and tertiary alcohols were now susceptible to deoxygenation (not shown).57,58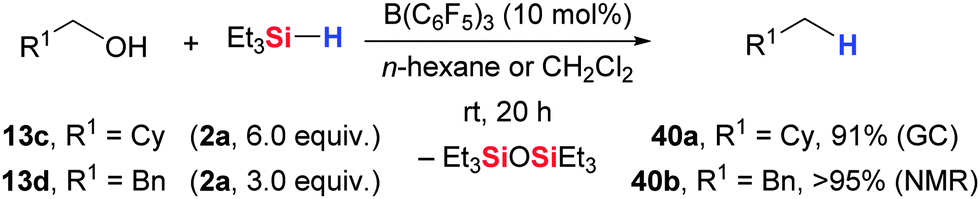 | ||
| Scheme 21 B(C6F5)3-catalysed deoxygenation of alcohols. | ||
An intriguing application of this deoxygenation was reported by Gagné and co-workers by converting monosaccharides and polysaccharides into mixtures of saturated and unsaturated hydrocarbons, i.e., alkanes and alkenes, respectively, with Et2SiH2 (2j) in the presence of catalytic amounts of B(C6F5)3 (41 → 40, Scheme 22).59
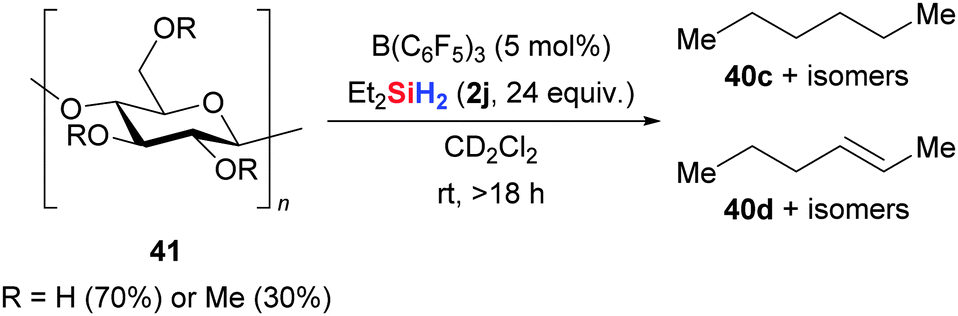 | ||
| Scheme 22 B(C6F5)3-catalysed deoxygenation of carbohydrates by Gagné. | ||
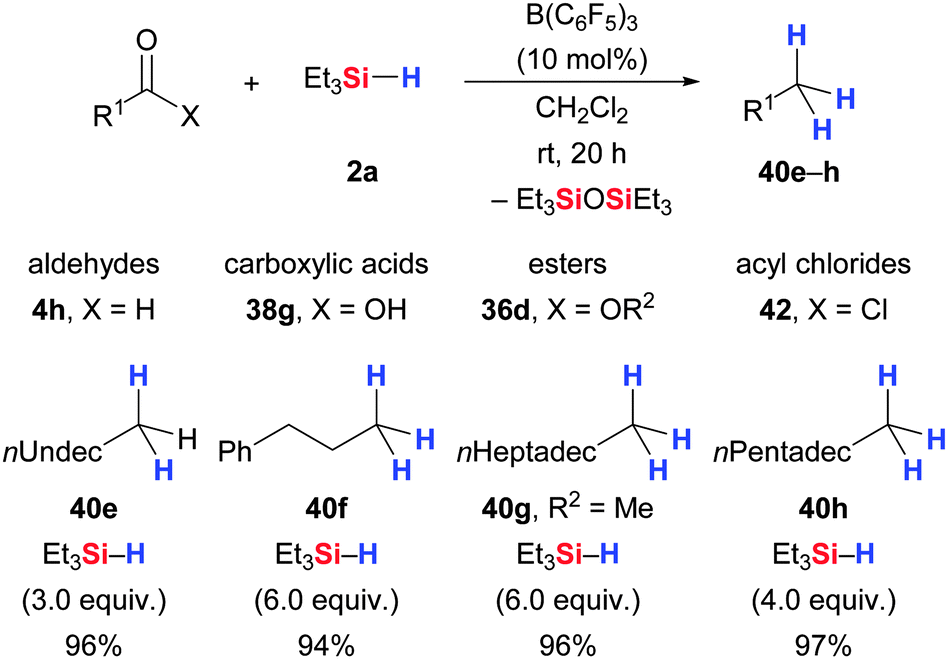 | ||
| Scheme 23 B(C6F5)3-catalysed deoxygenation of various carbonyl and carboxyl compounds by Gevorgyan and Yamamoto. | ||
Defunctionalization of carbonyl and carboxyl compounds works equally well with excess Et3SiH (2a) in the presence of B(C6F5)3 (Scheme 23).59 For example, n-dodecanal is converted into n-dodecane in high yield (4h → 40e). Similarly, the exhaustive reduction of aliphatic carboxylic acids (38g → 40f) as well as esters (36d → 40g) and acyl chlorides (42 → 40h) derived thereof proceeded smoothly at ambient temperature. Conversely, partial reduction was seen with aromatic carbonyl and carboxyl compounds, affording the corresponding TES-protected benzylic alcohols (not shown). Moreover, the authors indicate in the final paragraph of their publication that ketones, acetals and nitriles were also amenable to this procedure but no examples were reported. Tan and Zhang later showed that α,β-unsaturated carboxyl compounds, e.g., cinnamic acid (43), undergo exhaustive reduction with Ph2SiH2 (2i) (43 → 40f, Scheme 24).50 Their study also included an example of an enol ether that was reduced to the corresponding hydrocarbon (44 → 40f).50
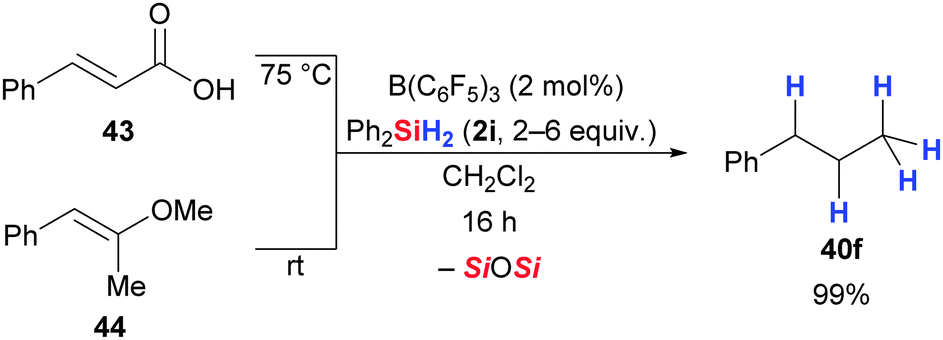 | ||
| Scheme 24 B(C6F5)3-catalysed exhaustive reduction of an α,β-unsaturated carboxylic acid and an enol ether. | ||
One year later, Chandrasekhar and co-workers found that PMHS (2h) can also be used as reductant in B(C6F5)3-catalysed carbonyl deoxygenations.59b,60,61 Various aromatic and aliphatic carbonyl groups were rapidly reduced to methylene groups (5 → 40, Scheme 25). For example, benzophenone was cleanly defunctionalised to furnish diphenylmethane (5d → 40i). The new hydrosilane–borane combination displays excellent scope. Alkyl and aryl halides remained untouched, and unsaturation in form of alkenes was tolerated as well (5e → 40j). Remarkably, even carboxylic esters did not react (5f → 40k). Rosenberg and co-workers demonstrated that defunctionalisation by B(C6F5)3-catalysed hydrosilylation is also applicable to thioketones (Scheme 26).35 Thiobenzophenone underwent rapid desulfurisation with PhSiH3 (2k) or Ph2SiH2 (2i) at room temperature to yield diphenylmethane quantitatively (14 → 40i).
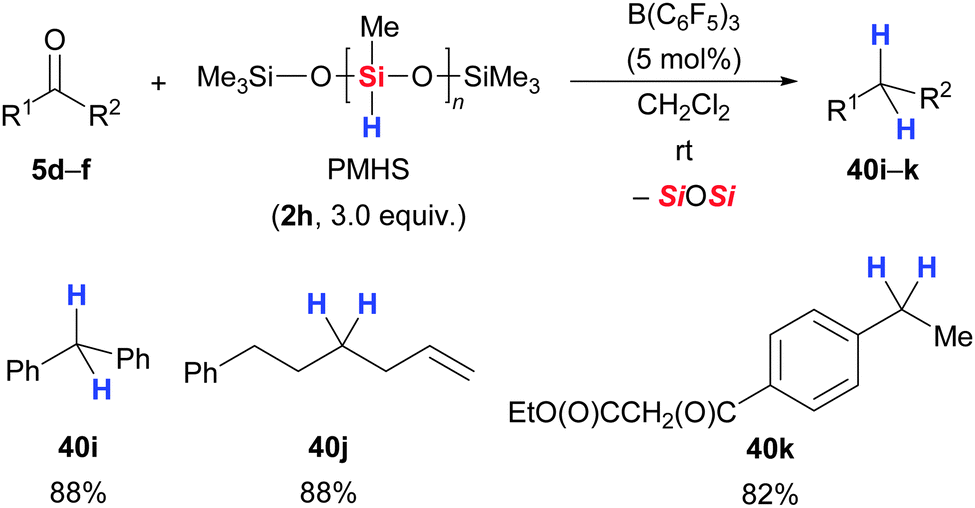 | ||
| Scheme 25 B(C6F5)3-catalysed deoxygenation of various carbonyl compounds using PMHS by Chandrasekhar. | ||
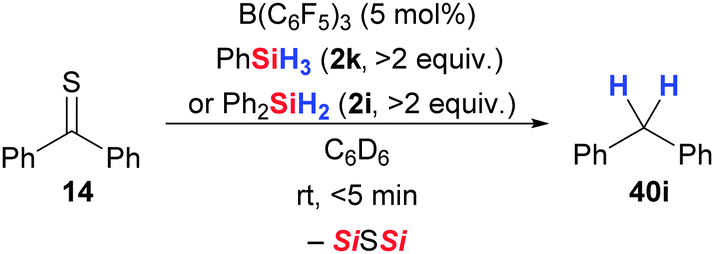 | ||
| Scheme 26 B(C6F5)3-catalysed desulfurisation of a thioketone by Rosenberg. | ||
Chandrasekhar and co-workers reported that the reduction of benzamide to benzylamine with the PMHS/B(C6F5)3 reducing agent failed (45 → 46, not shown).59b In contrast, Tan and Zhang50 as well as Blondiaux and Cantat62 identified a system that allows for the deoxygenation of primary, secondary and tertiary amides. Also, Tan and Zhang mentioned that the reduction of benzamide did not occur at temperatures as high as 120 °C, probably due to conjugation of the amide group with the phenyl ring.50 Blondiaux and Cantat recently accomplished the challenging reduction of primary amides, with one example being benzamide. The “trick” is to protect one of the N–H bonds with Me3SiCl, followed by B(C6F5)3-catalysed hydrosilylation with TMDS (not shown).62
Tan and Zhang used various phenyl-substituted hydrosilanes in their amide reduction;50 secondary and tertiary amides were reduced to the corresponding amines in good yields but N-phenylamides were limited to three examples: N-phenylacetamide, 1-methyl-2-indolinone and 3,4-dihydro-2(1H)-quinolinone (not shown). The deoxygenation of a broad range of aliphatic and aromatic amides was later presented by the groups of Cantat62 (45 → 46, Scheme 27) and Adronov63 (not shown). Cantat and co-workers utilised cost-efficient, non-toxic and air-stable PMHS (2h) and TMDS (2l) as reducing agents. It is important to note that high temperatures are required to thermally cleave the relatively stable amide–B(C6F5)3 adduct. Interestingly, in α,β-unsaturated amides 1,4-reduction proceeds faster than the deoxygenation (not shown).
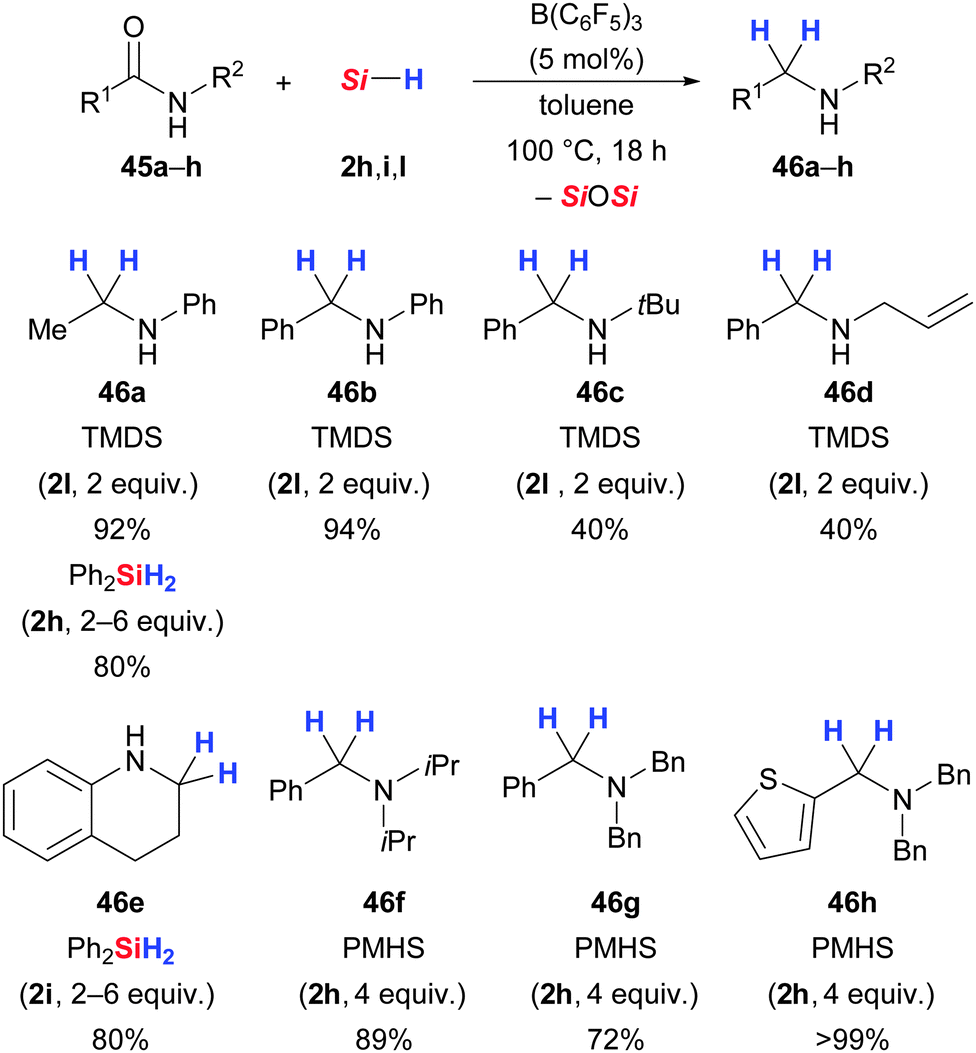 | ||
| Scheme 27 B(C6F5)3-catalysed amide-to-amine reduction by Cantat. | ||
Tan and Zhang presented one example of an aryl isocyanate reduction to afford the corresponding aniline in quantitative yield (47 → 46i, Scheme 28).50
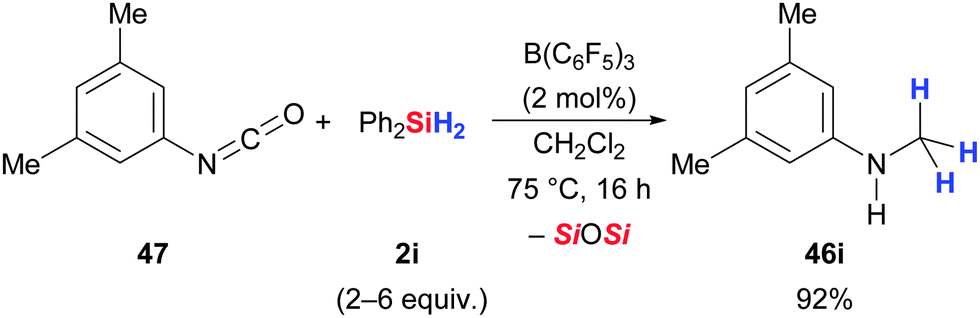 | ||
| Scheme 28 B(C6F5)3-catalysed deoxygenation of an isocyanate by Zhang. | ||
3.4 Reduction of heterocycles
Tan and Zhang had also included an electron-rich heterocycle, namely indole, into their broad screening of substrates for the B(C6F5)3-catalysed hydrosilylation.50,64 Indole is reminiscent of the enamine motif that is indeed reduced to amines under the typical reaction setup (cf.Scheme 18). Consequently, free indole and N-methyl indole participate nicely in the reduction with Ph2SiH2 (2i) to yield the correspondings indolines (48a,b → 49a,b, Scheme 29). Likewise, the hydrogenation of N-methyl protected indoles works equally well (48b,c → 49b,c, Scheme 29).37c Mechanistic investigations concerning Si–H and H–H bond activation in the presence of heteroarenes were undertaken by Ingleson and co-workers and these revealed a complex situation with multiple competing pathways that eventually lead to the reduced compounds (not shown).65 In their N-silylation studies, Paradies and co-workers also observed reduction of indoles to indolines (cf.Scheme 43).24b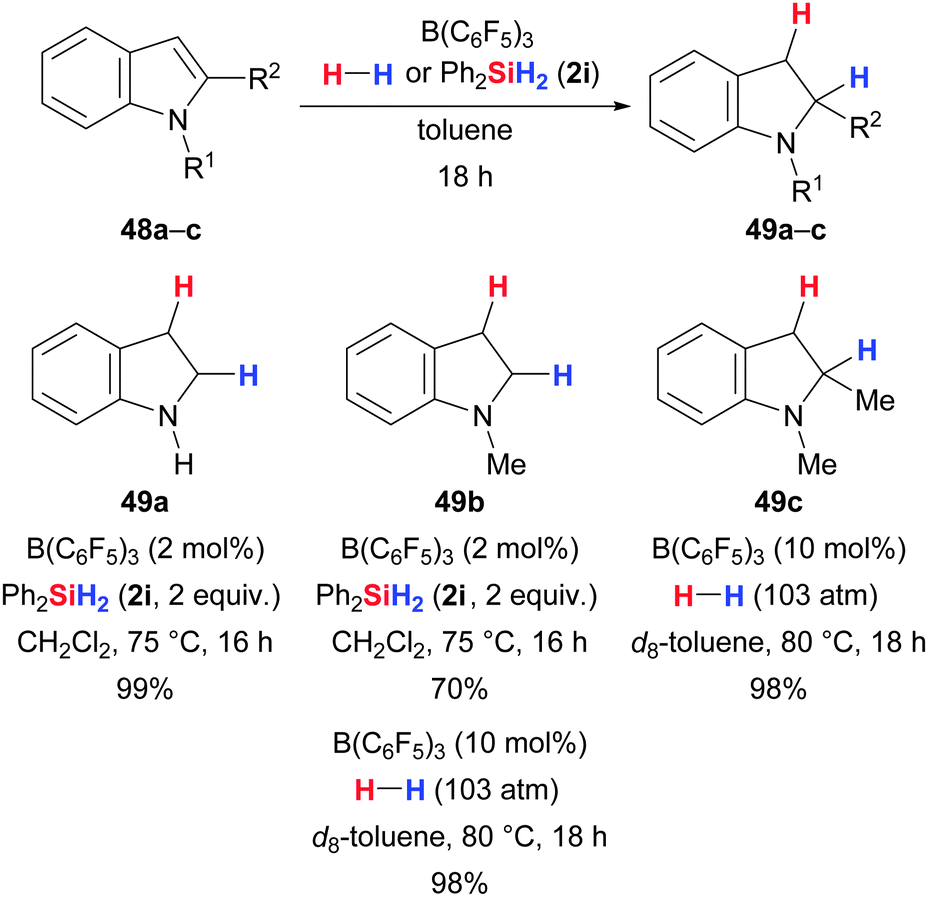 | ||
| Scheme 29 B(C6F5)3-catalysed reduction of indoles by Zhang (with hydrosilanes) and Stephan (with dihydrogen). | ||
Moreover, imine-like heteroarenes are also reduced with dihydrogen in the presence of electron-deficient boranes (Scheme 30).66 Examples include the hydrogenation of acridine (50 → 53), various quinolines (51 → 54) and 1,10-phenanthroline (52 → 55). Interestingly, 2-phenylquinoline was reduced to 1,2,3,4-tetrahydro-2-phenylquinoline with dihydrogen (51a → 54a) but gave 1,4-reduction exclusively (51a → 56) when treated with Et3SiH (2a) in the presence of B(C6F5)3.66 The substrate scope of the quinoline hydrogenation was later extended by Soós and co-workers.67 The use of a sterically more demanding and slightly less Lewis acidic catalyst, MesB(C6F4H)2, prevented Lewis pair formation with the substrates, thereby even enabling the reduction of the free quinoline (51b,c → 54b,c).
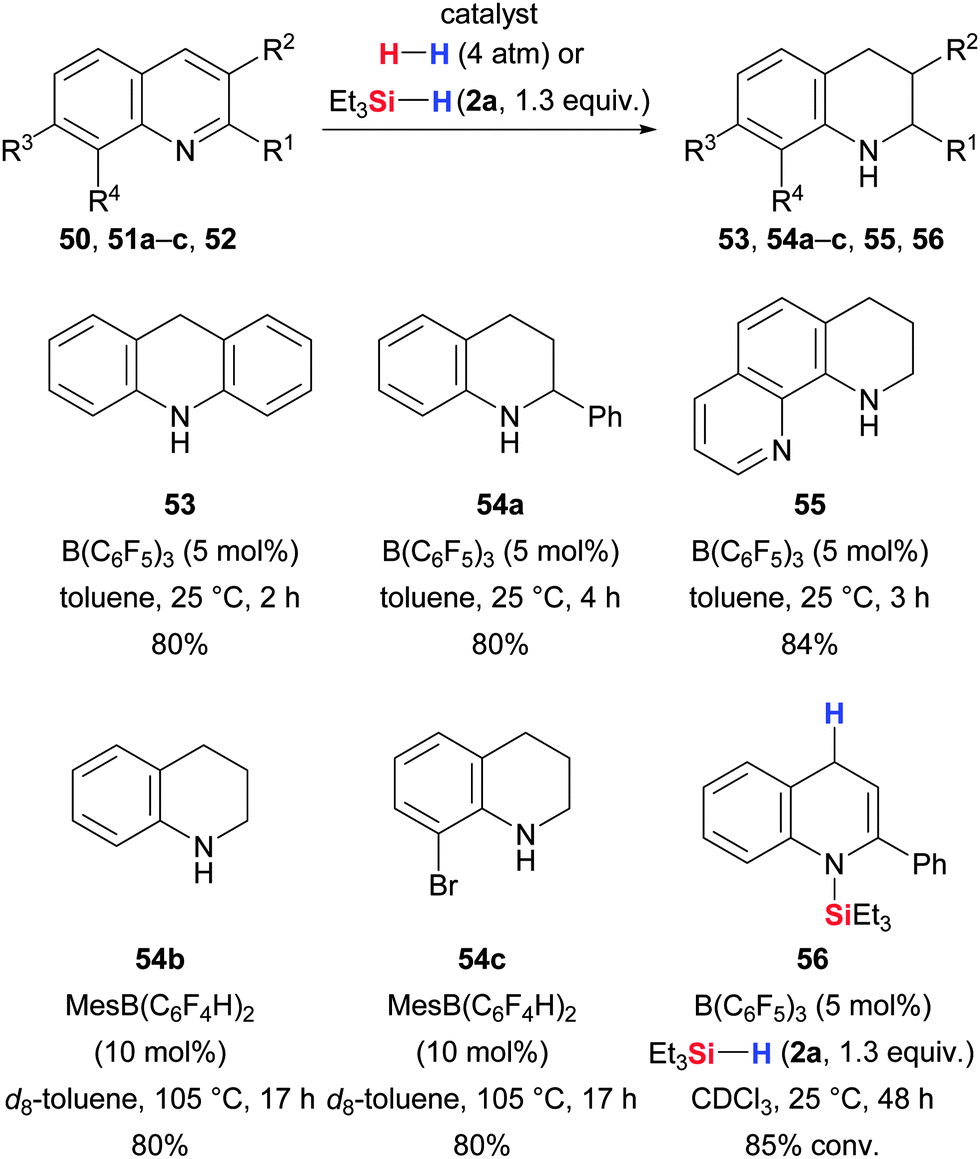 | ||
| Scheme 30 Catalytic reduction of heterocycles by Stephan (with B(C6F5)3) and Soós (with MesB(C6F4H)2). | ||
When quinolines are treated with an excess of dihydrosilane 2j in the presence of B(C6F5)3, 3-silylated 1,2,3,4-tetrahydroquinolines are formed as illustrated by the group of Park and Chang (51 → 57, Scheme 31, upper).68 Mechanistic as well as computational studies revealed that an initial 1,4-reduction is followed by regioselective hydrosilylation of the thus formed N-silylated enamine.69 For substrates bearing substituents in the 2-position, excellent diastereoselectivities were achieved (51f → 57c). In the case of isoquinolines, reduction products with silylation in β-position with respect to the nitrogen atom were obtained (58 → 59, Scheme 31, lower).
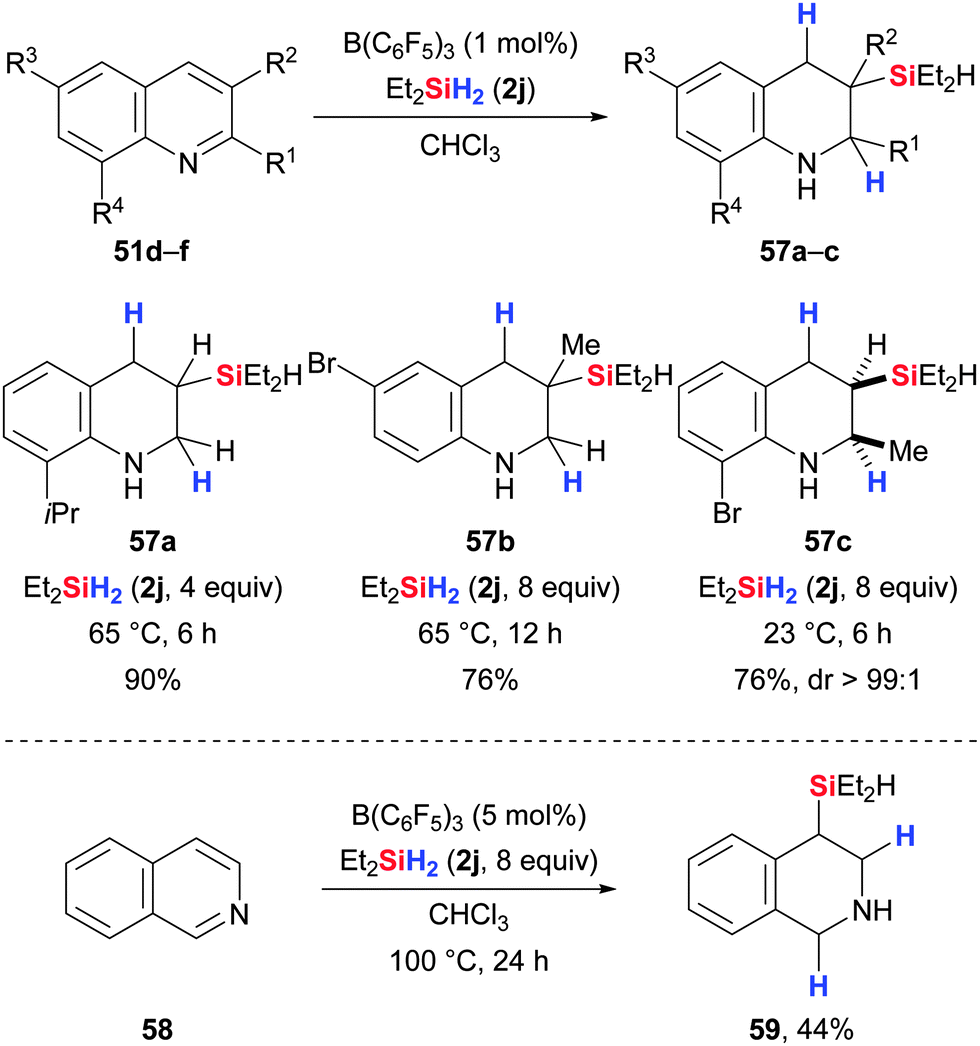 | ||
| Scheme 31 B(C6F5)3-catalysed reduction of quinolines (upper) and isoquinolines (lower) by Park and Chang. | ||
A stereoselective hydrogenation method for 2,3-disubstituted quinoxalines was developed by Zhang and Du (60 → 61, Scheme 32).70 Employing B(C6F5)3 as catalyst resulted in the diastereoselective formation of cis-2,3-disubstituted 1,2,3,4-tetrahydroquinoxalines whereas the use of chiral borane 24b afforded the same target molecules in enantioselective fashion.
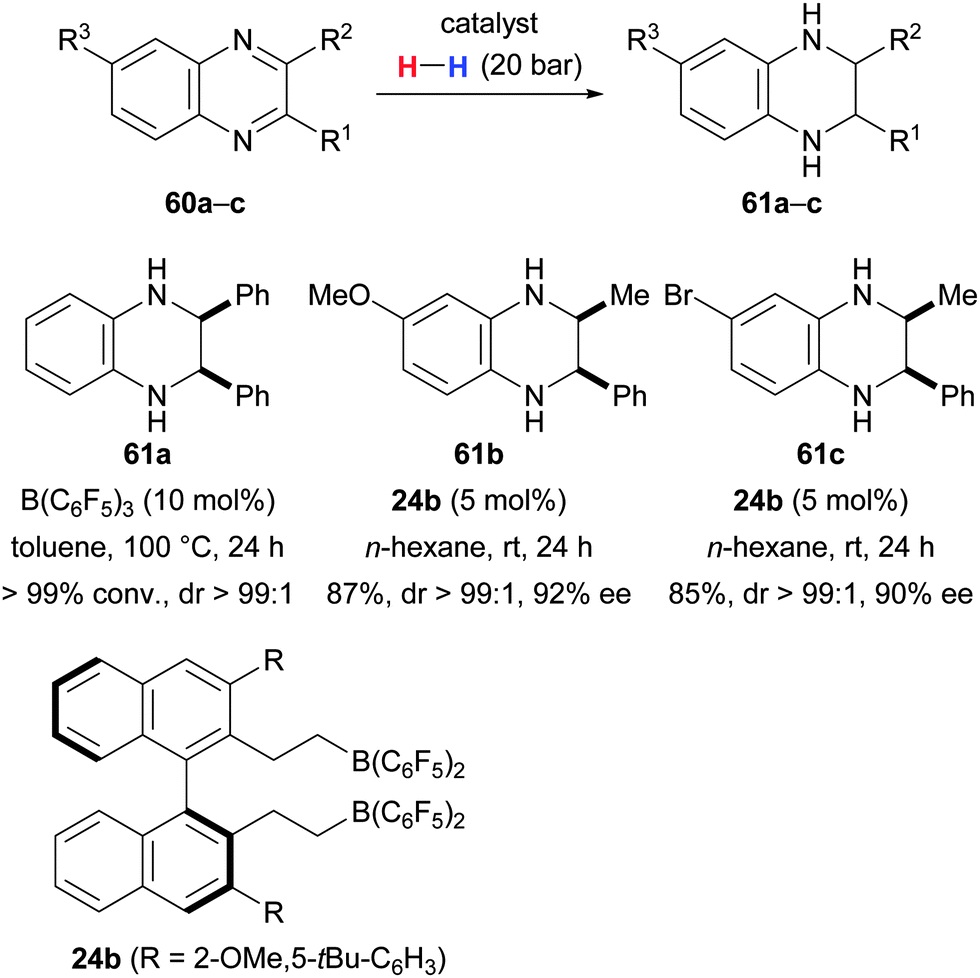 | ||
| Scheme 32 Stereocontrolled reduction of quinoxalines by Du. | ||
A B(C6F5)3-mediated exhaustive reduction of anilines71 and a few heteroarenes72 under hydrogen atmosphere to afford the hydridoborate salts was discovered by Stephan and co-workers (62a/63a/50 → 64a–c, Scheme 33). Although stoichiometric amounts of B(C6F5)3 are required, multiple turnovers are achieved before the final H–H bond activation by the formed amine and B(C6F5)3 produces the hydridoborate salts.
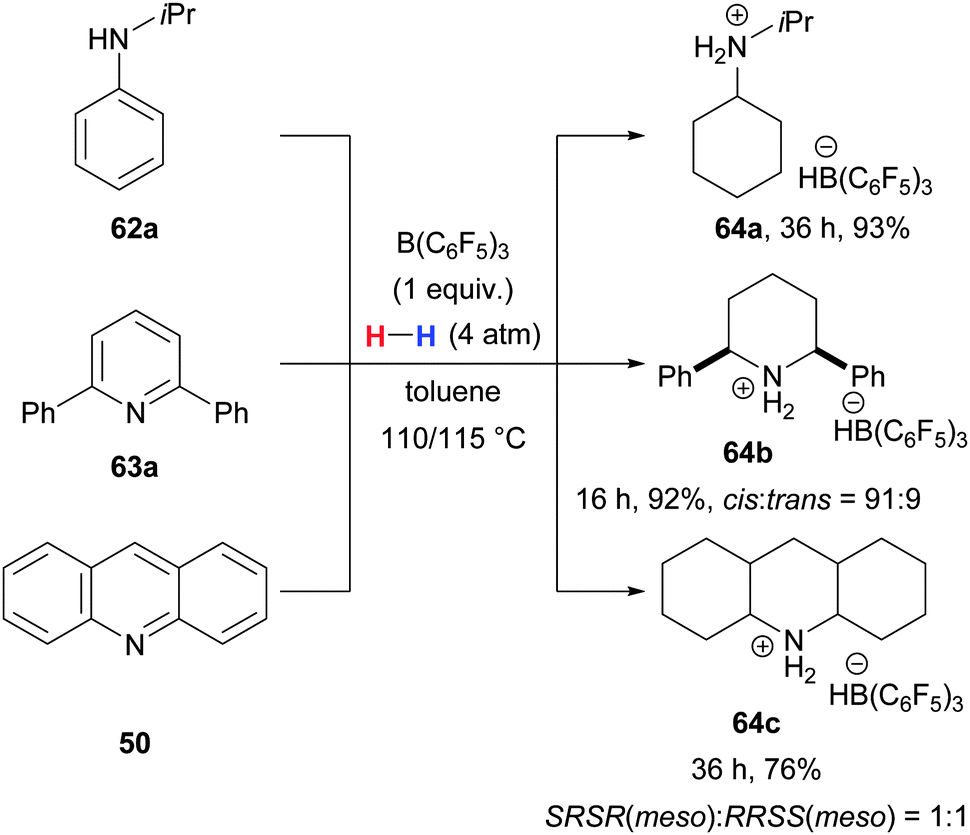 | ||
| Scheme 33 B(C6F5)3-catalysed reduction of anilines and heteroarenes to hydridoborate salts by Stephan. | ||
Shortly thereafter, Liu and Du developed a truly catalytic method to reduce pyridines to piperidines with dihydrogen (63 → 66, Scheme 34).73 These authors found that Piers' borane HB(C6F5)2 (ref. 39b) was able to catalyse this reaction but its hydroboration products with electron-poor alkenes showed an even higher catalytic activity. In situ-formed borane 65 was used to promote the exhaustive reduction of pyridines with various alkyl or aryl substitution patterns with high levels of diastereoselection. 2-Bromopyridines were dehalogenated (not shown), and selective hydrogenation of one pyridine core was observed for bipyridine 63f.
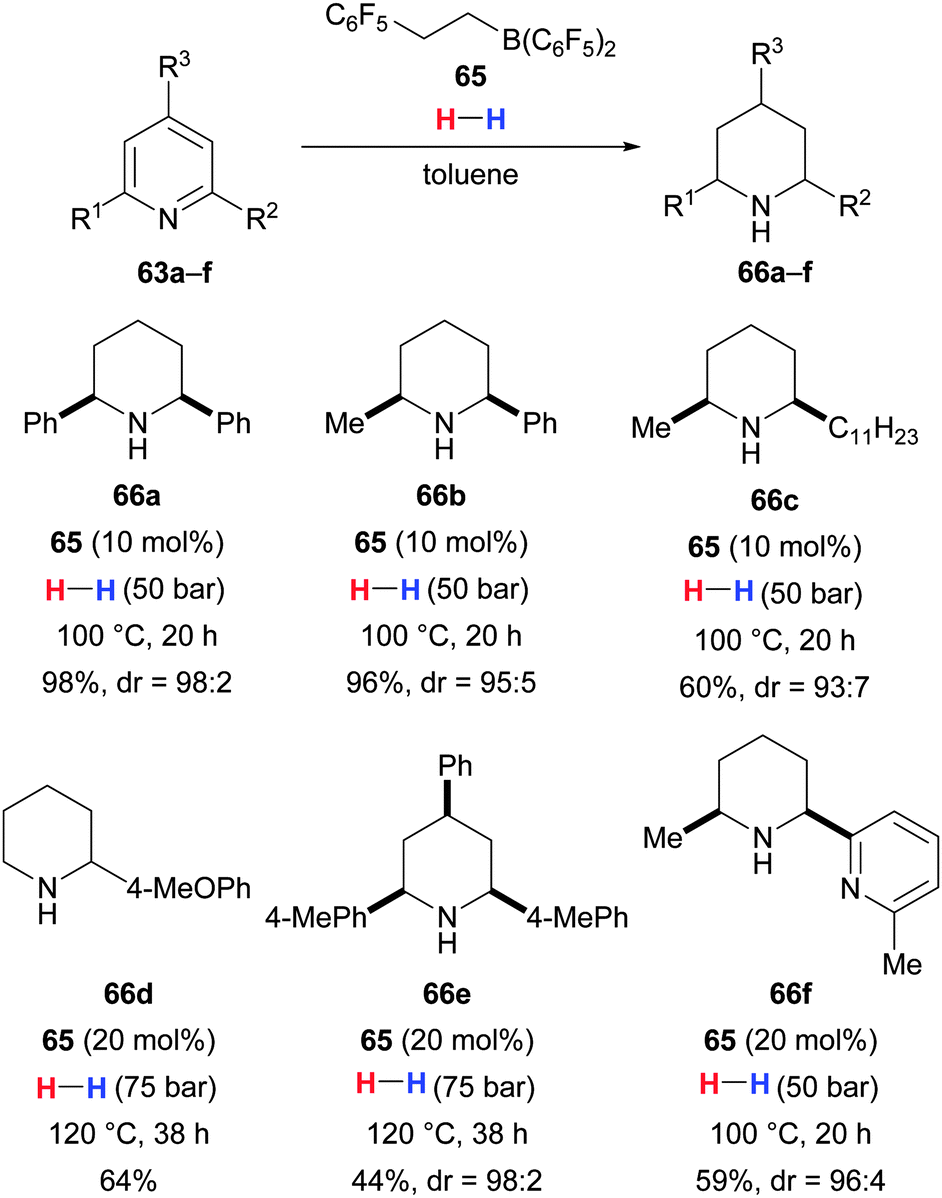 | ||
| Scheme 34 Reduction of pyridines by Du. | ||
4. Transformations of ethers
4.1 Cleavage of dialkyl and aryl alkyl ethers
Gevorgyan, Yamamoto and co-workers had mentioned that Et3SiH (2a) with B(C6F5)3 reduces ethers to alkanes under mild conditions (cf. Section 3.3).54,55 Aryl methyl ethers yielded the corresponding silyl ethers and methane quantitatively (e.g., 67a → 8c, Scheme 35). Likewise, 2,3-dihydrobenzofuran was ring-opened with Si–O and C–H bond formations (67b → 8d). Primary ethers with linear alkyl chains, cyclic ethers such as tetrahydropyran (67c → 7b) as well as secondary ethers such as iPr2O were fully reduced to the respective hydrocarbons. Tertiary ethers did not react.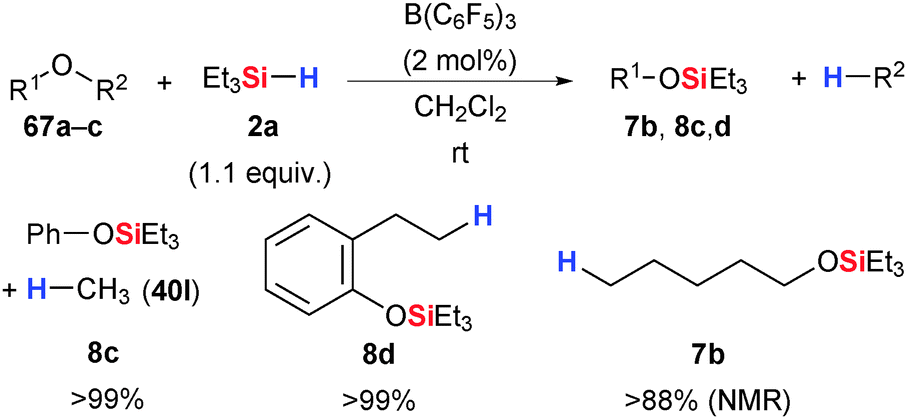 | ||
| Scheme 35 B(C6F5)3-catalysed ether cleavage by Gevorgyan and Yamamoto. | ||
Njardarson and co-workers were inspired by this work and extended this mild ether cleavage to unsaturated cyclic ethers.74 Their contribution is focused on 2,5-dihydrofurans and 3,6-2H-dihydropyrans (Scheme 36). The C–O cleavage in 2,5-dihydrofurans proceeded cleanly in high yield (68 → 13) but thermodynamically controlled alkene migration was seen in few cases. Homoallylic alcohols were obtained from 3,6-2H-dihydropyrans, and scrambling of the double bond geometry occurred with selected substrates (68c → 13g). The selective incorporation of deuterium in the allylic position was demonstrated with Et3SiD (68d → 13h, Scheme 36).
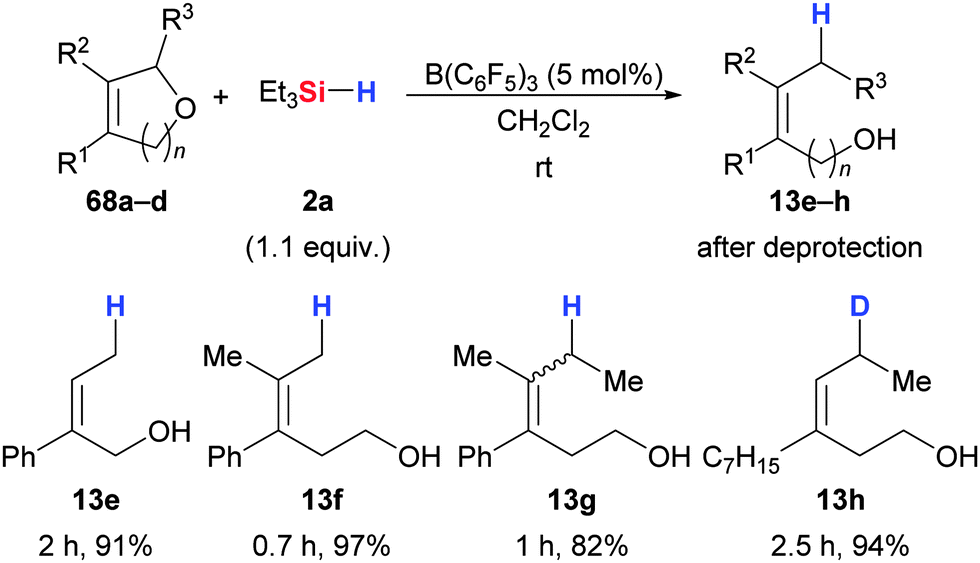 | ||
| Scheme 36 B(C6F5)3-catalysed cleavage of cyclic ethers by Njardarson. | ||
4.2 Cleavage of alkyl silyl ethers (Piers–Rubinsztajn reaction)
Alkyl silyl ethers, i.e., alkoxysilanes, convert into disiloxanes when reacted with hydrosilanes in the presence of catalytic amounts of B(C6F5)3. This dehydrocarbonative condensation is commonly referred to as the Piers–Rubinsztajn reaction but might also be called a transetherification. This transformation is ideal for the rapid formation of 3D silicon structures for silicon-based polymers.Chojnowski and co-workers described the preparation of disiloxanes along with hydrocarbons from alkoxysilanes for the first time (7 → 69, Scheme 37, upper).75 Their goal was to examine the mechanism of this reaction by means of kinetic and UV spectroscopic measurements. The reaction of MePh2SiH (2g) with Me3SiOnOct (7c) was used as model reaction. Their kinetic analysis showed that the reaction rate is proportional to the catalyst concentration, and the reaction is 1st order in both reactants. The catalyst, B(C6F5)3, is mainly available in free form because Lewis pair formation with the alkoxysilane 7 or disiloxanes 69 is relatively weak. The proposed mechanism passes through a complex of the hydrosilane, B(C6F5)3 and the alkoxysilane (cf.V, Scheme 1). Hydride transfer from the silicon to the boron atom generates an oxonium ion intermediate that can react with the borohydride in three different ways, that is hydride attack at either of the three groups attached to the oxonium ion oxygen atom. Out of these three options, the cleavage of the C–O bond will irreversibly form the disiloxane and the alkane. Breaking either of the Si–O bonds results in the backward reaction or a hydrosilane/alkoxysilane metathesis.
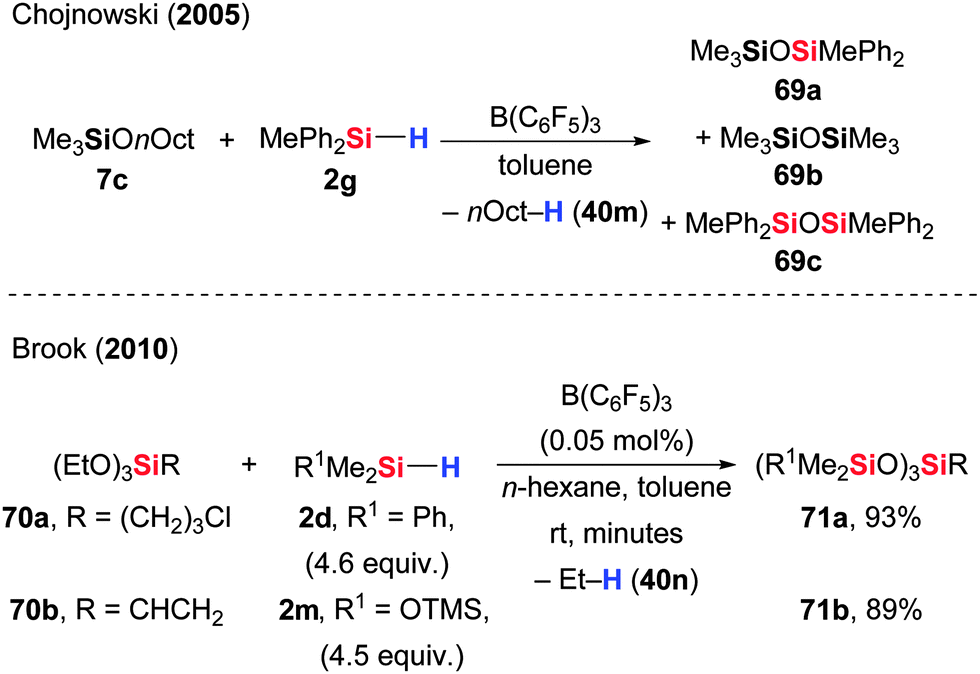 | ||
| Scheme 37 B(C6F5)3-catalysed dehydrocarbonative condensation of alkoxysilanes and hydrosilanes: the Piers–Rubinsztajn reaction. | ||
Brook and co-workers became interested in the Piers–Rubinsztajn reaction for the preparation of silicon-based polymers and described the synthesis of oligosilicone building blocks from functionalized trialkoxysilanes (70 → 71, Scheme 37, lower).76,77 For example, ω-haloalkyl- and vinyl-substituted trialkoxysilanes were compatible with the reaction conditions of the dehydrocarbonative condensation. Neither hydrodehalogenation nor hydrosilylation of the alkenyl group occurred, thereby allowing for the generation of alkenyl-modified silicones.
5. Transetherification and deoxygenation of phosphonic and phosphinic esters
Denis and co-workers investigated the B(C6F5)3-catalysed C–O/Si–O metathesis (cf. Section 4) as well as the deoxygenation (cf. Section 3.3) of phosphonic esters (Scheme 38, upper).78 The choice of hydrosilane determines the chemoselectivity. With tertiary hydrosilanes, phosphonic alkyl esters are converted into the corresponding silyl esters in high yields (72 → 73). Conversely, exhaustive reduction to free phosphines was achieved with excess of dihydro- or trihydrosilanes (72 → 74). In contrast, phosphinic alkyl esters are deoxygenated to the free phosphines already with one equivalent of dihydro- or trihydrosilane (75 → 76, Scheme 38, lower). The conversion to their corresponding silyl esters was not reported.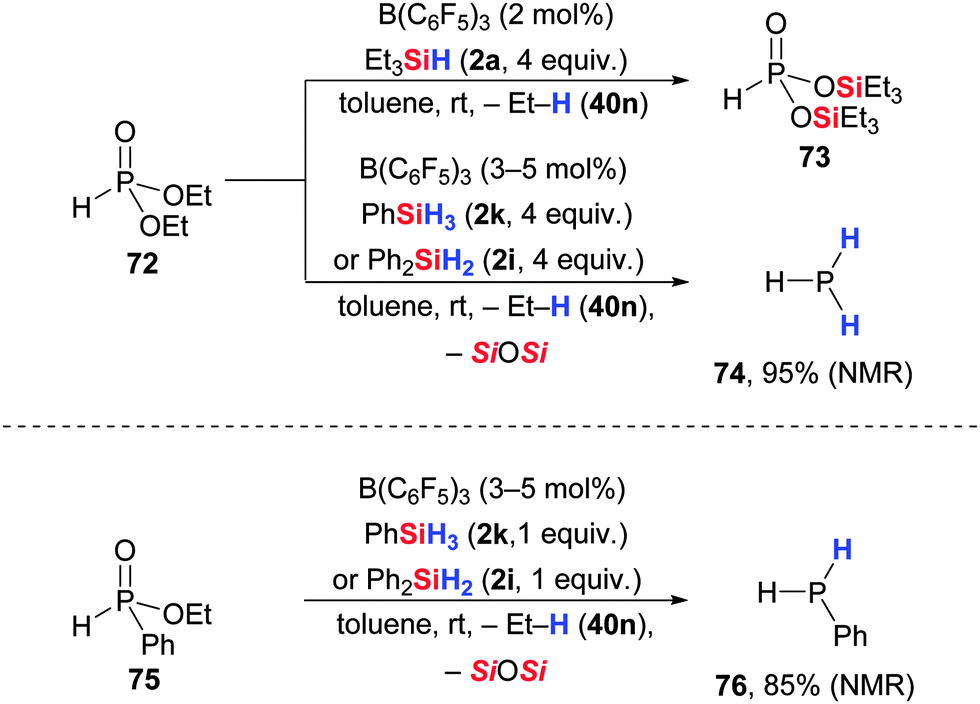 | ||
| Scheme 38 B(C6F5)3-catalysed reaction of phosphonic (upper) and phosphinic esters (lower) with hydrosilanes. | ||
6. Hydrodefluorination with hydrosilanes
Caputo and Stephan introduced a simple protocol for the activation of C(sp3)–F bonds (Scheme 39).79 The combination of Et3SiH (2a) and catalytic amounts of B(C6F5)3 was shown to convert (α-oxygenated) alkyl fluorides into the corresponding alkanes along with the formation of Et3SiF (78). Quantitative yields were obtained for primary and tertiary fluorides within a few minutes (77 → 40). However, ether 77d required 18 h at 60 °C to afford hydrodefluorinated 40r in 72% yield; the CH(CF3)2 group remained intact.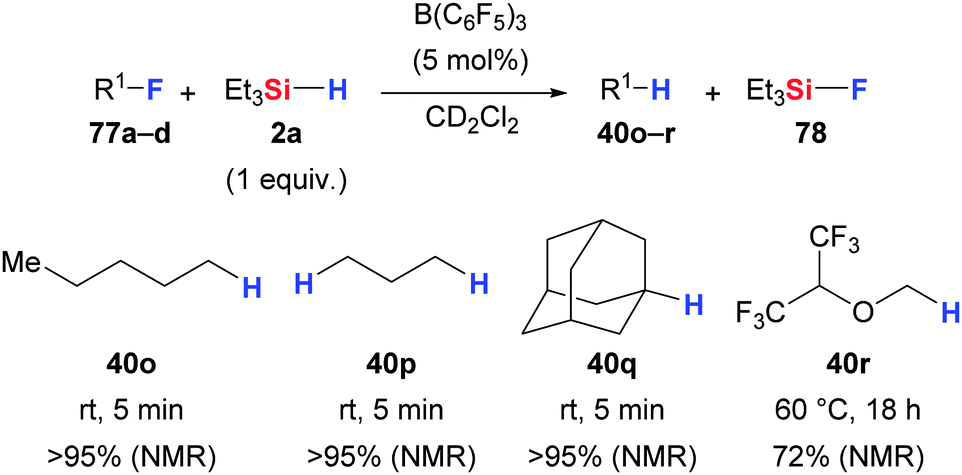 | ||
| Scheme 39 B(C6F5)3-catalysed hydrodefluorination of (α-heteroatom-substituted) alkyl fluorides by Stephan. | ||
7. Dehydrogenative Si–X couplings
The dehydrogenative coupling of alcohols,80–82 thiols35,80a or amines24 and hydrosilanes is, besides the hydrosilylation of CX bonds, another major application of catalysis with B(C6F5)3. These oxidations are generally high-yielding, releasing dihydrogen as the sole byproduct.
The methodology was introduced by Piers and co-workers, and their initial report was focused on the dehydrogenative silylation of primary, secondary, tertiary and phenolic hydroxy groups to afford the corresponding silyl ethers (13/12 → 7/8, Scheme 40).4 Interestingly, secondary and tertiary react faster than primary alcohols (1 h for 12c → 8eversus 24 h for 13i → 7d) but shorter reaction times are possible with higher catalyst loadings or at elevated temperatures. The functional-group tolerance of the method is excellent. Shortly thereafter, Gevorgyan, Yamamoto and co-workers reported similar observations in connection with their protocol for the deoxygenation of alcohols with Et3SiH/B(C6F5)3 (cf.Scheme 21).54 Primary alcohols were defunctionalised after the Si–O coupling whereas the reaction stopped at this stage with the bulkier secondary and tertiary alcohols as well as phenols, even in the presence of excess of hydrosilane. Kawakami and co-workers applied Sommer's essentially enantiopure silicon-stereogenic hydrosilane 2n (>99% ee) to this dehydrogenative Si–O coupling and observed inversion of the stereochemistry at the silicon atom in (R)-7f with hardly any erosion of the enantiomeric excess (95% ee).14 The stereochemical course at the silicon atom is in agreement with the general mechanistic picture (cf.V, Scheme 1). The hydroxy group attacks the silicon atom in the hydrosilane–borane adduct from the backside.
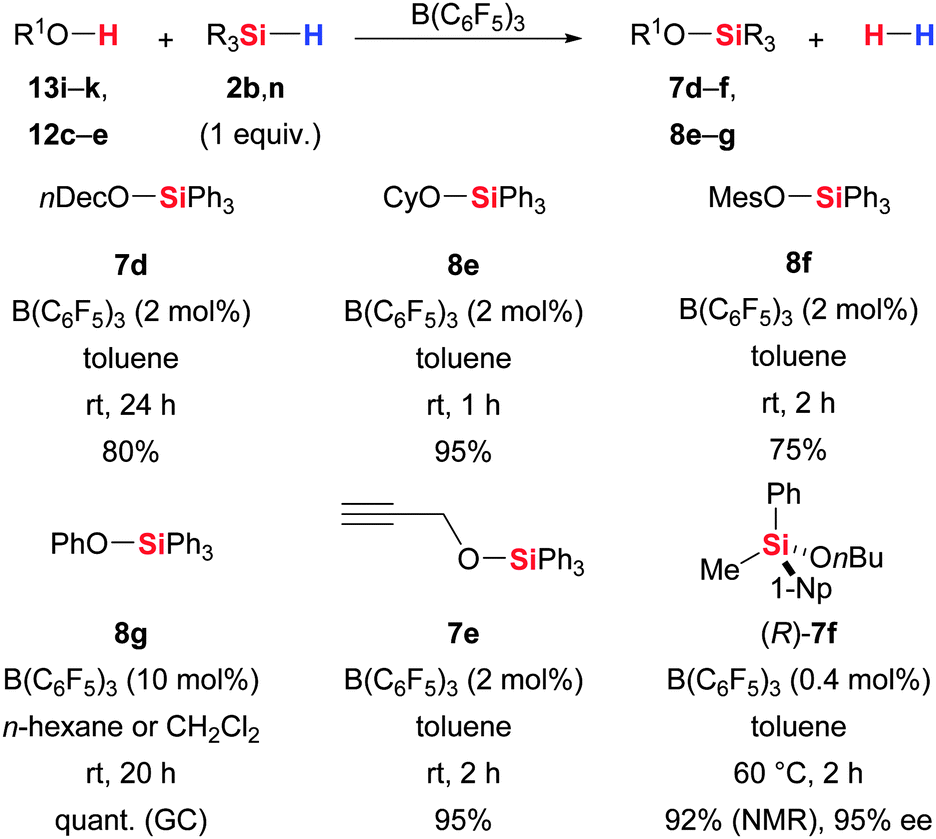 | ||
| Scheme 40 B(C6F5)3-catalysed dehydrogenative Si–O couplings. | ||
Dussault and co-workers merged the B(C6F5)3-catalysed dehydrogenative Si–O coupling with the alkene hydrosilylation (cf.Scheme 14) promoted by the same catalyst into a one-pot procedure (Scheme 41).81 This domino process allows for the synthesis of cyclic siloxanes in regio- and stereoselective fashion. For example, chemoselective alcohol silylation of 12f with Ph2SiH2 (2i) generated alkoxysilane 8h that engaged in a rapid intramolecular hydrosilylation of the tethered alkene to yield siloxane 29i.
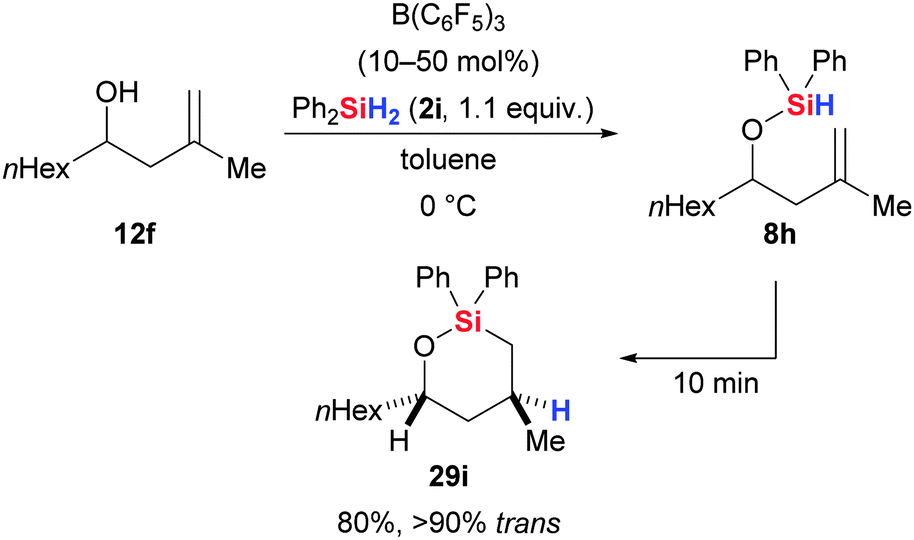 | ||
| Scheme 41 B(C6F5)3-catalysed dehydrogenative Si–O coupling followed by intramolecular alkene hydrosilylation. | ||
The potential of this methodology was further tested in the dehydrogenative coupling of aliphatic and aromatic thiols (79/80 → 15/81, Scheme 42).35,80a It was Rosenberg and co-workers to elaborate the sulfur chemistry based on Piers' seminal work. These authors demonstrated that tertiary hydrosilane 2d as well as symmetrically substituted dihydrodisilane 2e and polyphenylsilane 2f are applicable to the Si–S coupling; the Si–Si bonds of the latter reactants were stable in the presence of B(C6F5)3 (cf. Rosenberg's reduction of thioketones, Scheme 6). Reactions were generally fast, giving near-quantitative yields within minutes at room temperature. The clean coupling of HMe2SiSiMe2H (2e) and 1,2-benzenedithiol is particularly noteworthy as no acyclic oligomeric byproducts were formed (80 → 81).
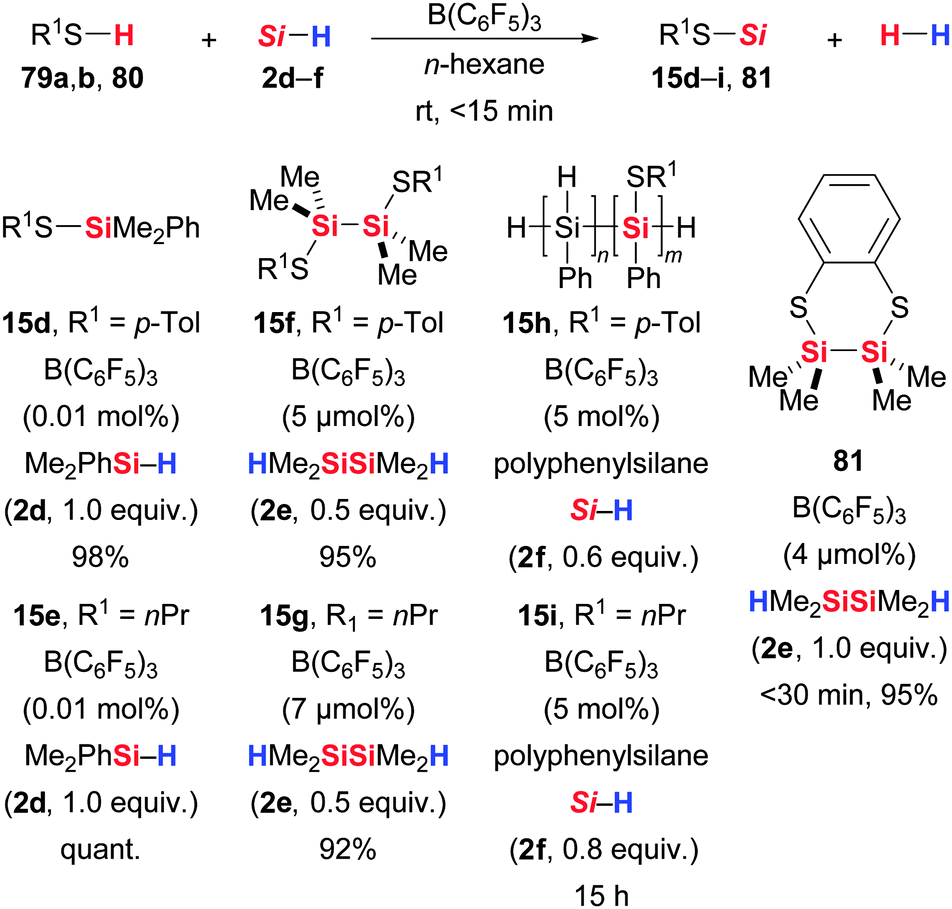 | ||
| Scheme 42 B(C6F5)3-catalysed dehydrogenative Si–S couplings. | ||
The related dehydrogenative Si–N coupling had long been neglected. It was introduced by Oestreich and co-workers as part of their mechanistic investigation on the B(C6F5)3-catalysed imine hydrosilylation (X → VII, Scheme 3)23 but the test reaction of Me2PhSiH (2d) and 1-phenylethylamine was rather slow (21c → 19c, Scheme 43). Shortly thereafter, Paradies and co-workers turned this into a useful methodology (21/49/62/82/83 → 19/87/84/85/86, Scheme 43).24b Anilines 62 (including carbazole 82) in any form were amenable to this B(C6F5)3 catalysis at elevated temperature. However, pyrroles were not participating and indoles were cleanly converted into the N-silylated indolines (cf.Scheme 29 for indole-to-indoline reduction). It is worth mentioning that these authors propose that anilines and carbazoles react by the same mechanism established for the Si–O coupling whereas indoles undergo silylation of the nitrogen atom followed by a rearrangement/reduction sequence to yield indolines.
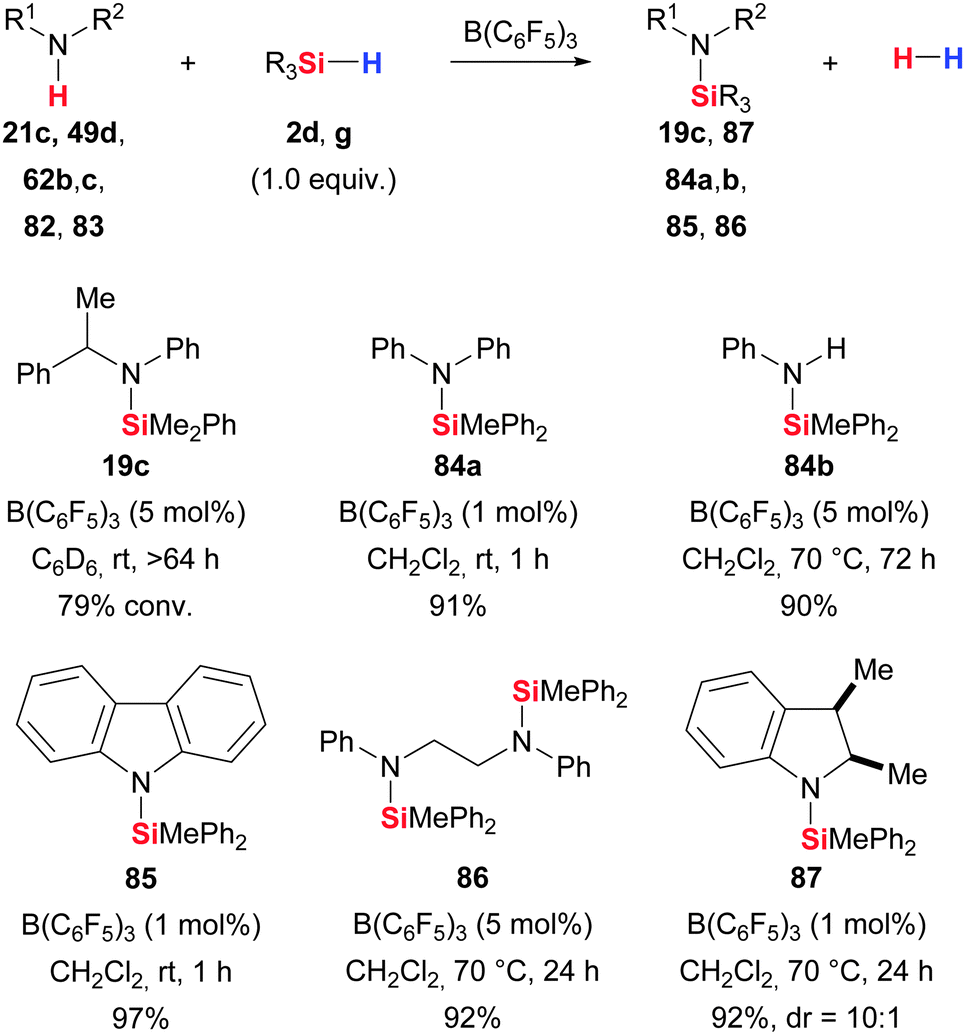 | ||
| Scheme 43 B(C6F5)3-catalysed dehydrogenative Si–N couplings. | ||
8. Intramolecular sila-Friedel–Crafts-type reactions
Curless and Ingleson developed a method to perform intramolecular sila-Friedel–Crafts-type reactions catalytic in B(C6F5)3 (Scheme 44).83 Conventional activation of the Si–H bond in biphenyl-substituted hydrosilane 2o with B(C6F5)3 followed by intramolecular attack of the ortho arene at the silicon atom results in a silicon-stabilised Wheland intermediate that rearomatises by deprotonation with 2,6-dichloropyridine to furnish silole 88. The borohydride and the protonated pyridine base liberate dihydrogen as byproduct (Scheme 44, upper). A one-pot hydrosilylation/dehydrosilylation sequence starting from alkyne 89via vinylsilane 2p to access silaindene 90 was also reported (Scheme 44, lower). The initiation of this sequence constitutes the first example of a B(C6F5)3-catalysed hydrosilylation of an alkyne.46c Moderate trans-selectivity in the hydrosilylation event (resulting in the Z-configured vinylsilane) was observed.84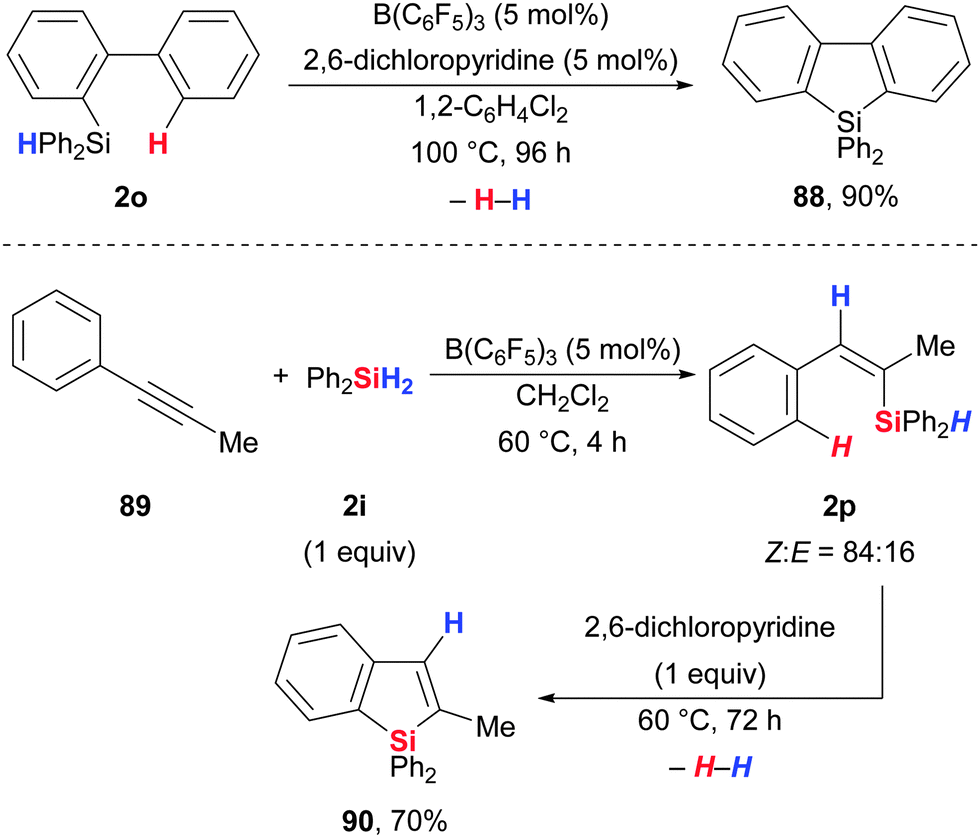 | ||
| Scheme 44 B(C6F5)3-catalysed sila-Friedel–Crafts-type reactions by Ingleson. | ||
9. Short summary
It is evident from the enormous progress in recent years that catalysis with electron-deficient boranes is “alive and well”. Different from previous decades, the potential of these Lewis acids to contribute substantially to transition-metal-free catalysis matches the current zeitgeist of sustainable chemistry. B(C6F5)3-catalysed hydrosilylation and even more so hydrogenation are fundamental accomplishments. Of course, there are still problems, e.g., the limited functional group tolerance due to the strong Lewis acidity of the catalysts. Newly designed boranes where their Lewis acidity is finely balanced with their ability to mediate the bond activation will be required to address this issue. The same applies to the development of chiral catalysts for enantioselective variants. Again, the synthesis of such boron Lewis acids will be challenging but the work of Liu and Du nicely demonstrates that relatively simple solutions are indeed available.40 We strongly believe that B(C6F5)3 as well as FLP chemistry will continue to grow and that there is much more to be discovered.Acknowledgements
This research was supported by the Cluster of Excellence Unifying Concepts in Catalysis of the Deutsche Forschungsgemeinschaft (EXC 314/2). Dr Sebastian Rendler (Freiburg/Münster) and Dr Marius Mewald (Münster/Berlin) deserve particular mention for their enthusiasm and commitment. M.O. is indebted to the Einstein Foundation (Berlin) for an endowed professorship.Notes and references
- (a) A. G. Massey, A. J. Park and F. G. A. Stone, Proc. Chem. Soc., 1963, 212 CAS; (b) A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS; (c) A. G. Massey and A. J. Park, J. Organomet. Chem., 1966, 5, 218–225 CrossRef CAS.
- For early examples, see: (a) X. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1991, 113, 3623–3625 CrossRef CAS; (b) X. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10015–10031 CrossRef CAS; (c) B. Temme, G. Erker, J. Karl, H. Luftmann, R. Fröhlich and S. Kotila, Angew. Chem., Int. Ed. Engl., 1995, 34, 1755–1757 CrossRef CAS; (d) Y.-X. Chen, C. L. Stern, S. Yang and T. J. Marks, J. Am. Chem. Soc., 1996, 118, 12451–12452 CrossRef CAS ; for an overview, see: ; (e) E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887–4892 CrossRef CAS PubMed.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- For overviews, see: (a) T. Robert and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 5216–5218 CrossRef CAS PubMed; (b) G. Erker, Dalton Trans., 2005, 1883–1890 RSC; (c) W. E. Piers, Adv. Organomet. Chem., 2004, 52, 1–76 CrossRef; (d) K. Ishihara and H. Yamamoto, Eur. J. Org. Chem., 1999, 527–538 CrossRef CAS; (e) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC.
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (b) P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed; (c) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- (a) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC; (b) P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543–7546 CrossRef CAS PubMed; (c) H. Wang, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966–5968 RSC.
- For overviews of frustrated Lewis pair chemistry, see: (a) D. W. Stephan, Acc. Chem. Res. DOI:10.1021/ar500375j; (b) M. Alcarazo, Synlett, 2014, 1519–1520 CrossRef; (c) D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC; (d) Top. Curr. Chem., ed. G. Erker and D. W. Stephan, Springer, Berlin, Heidelberg, 2013, vol. 332 Search PubMed; (e) Top. Curr. Chem., ed. G. Erker and D. W. Stephan, Springer, Berlin, Heidelberg, 2013, vol. 334 Search PubMed; (f) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CAS; (g) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 RSC; (h) A. L. Kenward and W. E. Piers, Angew. Chem., Int. Ed., 2008, 47, 38–41 CrossRef CAS PubMed.
- For an overview of applications of H–H and Si–H bond activations catalysed by electron-deficient boranes as well as frustrated Lewis pairs, see: X. Feng and H. Du, Tetrahedron Lett., 2014, 55, 6959–6964 CrossRef CAS.
- For a comparison of Si–H and H–H bond activations catalysed by electron-deficient boranes, see: W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed.
- For reviews of hydrogenation reactions catalysed by electron-deficient boranes or frustrated Lewis pairs, see: (a) L. Shi and Y.-G. Zhou, ChemCatChem, 2015, 7, 54–56 CrossRef CAS; (b) Y. Liu and H. Du, Acta Chim. Sin., 2014, 72, 771–777 CrossRef CAS; (c) L. J. Hounjet and D. W. Stephan, Org. Process Res. Dev., 2014, 18, 385–391 CrossRef CAS; (d) J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552–3557 CrossRef CAS PubMed; (e) J. Paradies, Synlett, 2013, 777–780 CrossRef CAS; (f) D. W. Stephan, Org. Biomol. Chem., 2012, 10, 5740–5746 RSC; (g) D. W. Stephan, Chem. Commun., 2010, 46, 8526–8533 RSC; (h) D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC.
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed.
- For a previous attempt to verify this step by using a silicon-stereogenic silane as stereochemical probe, see: S. Shinke, T. Tsuchimoto and Y. Kawakami, Silicon Chem., 2007, 3, 243–249 CrossRef CAS.
- K. Sakata and H. Fujimoto, J. Org. Chem., 2013, 78, 12505–12512 CrossRef CAS PubMed.
- For reductions of carbonyl compounds with hydrosilanes and BF3·OEt2, see: (a) M. P. Doyle, C. T. West, S. J. Donnelly and C. C. McOsker, J. Organomet. Chem., 1976, 117, 129–140 CrossRef CAS; (b) J. L. Fry, M. Orfanopoulos, M. G. Adlington, W. R. Dittman, Jr. and S. B. Silverman, J. Org. Chem., 1978, 43, 374–375 CrossRef CAS.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- A. Y. Houghton, V. A. Karttunen, W. E. Piers and H. M. Tuononen, Chem. Commun., 2014, 50, 1295–1298 RSC.
- (a) S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767–769 RSC; (b) S. J. Connelly, W. Kaminsky and D. M. Heinekey, Organometallics, 2013, 32, 7478–7481 CrossRef CAS.
- J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS PubMed.
- D. T. Hog and M. Oestreich, Eur. J. Org. Chem., 2009, 5047–5056 CrossRef CAS.
- M. Mewald and M. Oestreich, Chem. – Eur. J., 2012, 18, 14079–14084 CrossRef CAS PubMed.
- J. Hermeke, M. Mewald and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 17537–17546 CrossRef CAS PubMed.
- For dehydrogenative silylation of amines, see: (a) ref. 23 ; (b) L. Greb, S. Tamke and J. Paradies, Chem. Commun., 2014, 50, 2318–2320 RSC.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS PubMed.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701–1703 RSC . This work also contains one example of a B(C6F5)3-catalysed reductive aziridine ring opening.
- Computational models for the mode of dihydrogen activation by borane–imine and related borane–phosphine systems have been extensively discussed: (a) T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef CAS PubMed; (b) Y. Guo and S. Li, Inorg. Chem., 2008, 47, 6212–6219 CrossRef CAS PubMed; (c) T. A. Rokob, A. Hamza, A. Stirling and I. Pápai, J. Am. Chem. Soc., 2009, 131, 2029–2036 CrossRef CAS PubMed; (d) A. Hamza, A. Stirling, T. A. Rokob and I. Pápai, Int. J. Quantum Chem., 2009, 109, 2416–2425 CrossRef CAS; (e) B. Schirmer and S. Grimme, Chem. Commun., 2010, 46, 7942–7944 RSC; (f) S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS PubMed; (g) I. Bakó, A. Stirling, S. Bálint and I. Pápai, Dalton Trans., 2012, 41, 9023–9025 RSC; (h) D. M. Camaioni, B. Ginovska-Pangovska, G. K. Schenter, S. M. Kathmann and T. Autrey, J. Phys. Chem. A, 2012, 116, 7228–7237 CrossRef CAS PubMed; (i) T. A. Rokob, I. Bakó, A. Stirling, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2013, 135, 4425–4437 CrossRef CAS PubMed; (j) L. L. Zeonjuk, N. Vankova, A. Mavrandonakis, T. Heine, G.-V. Röschenthaler and J. Eicher, Chem. – Eur. J., 2013, 19, 17413–17424 CrossRef CAS PubMed; (k) H. Zaher, A. E. Ashley, M. Irwin, A. L. Thompson, M. J. Gutmann, T. Krämer and D. O'Hare, Chem. Commun., 2013, 49, 9755–9757 RSC; (l) M. Pu and T. Privalov, ChemPhysChem, 2014, 15, 2936–2944 CrossRef CAS PubMed; (m) M. Pu and T. Privalov, ChemPhysChem, 2014, 15, 3714–3719 CrossRef CAS PubMed.
- D. Chen and J. Klankermayer, Chem. Commun., 2008, 2130–2131 RSC.
- For further examples of hydrosilylation of carbonyls catalysed by electron-deficient boranes, see: ref. 14; (a) V. Gevorgyan, M. Rubin, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS PubMed; (b) R. Roesler, B. J. N. Har and W. E. Piers, Organometallics, 2002, 21, 4300–4302 CrossRef CAS; (c) N. Asao, T. Ohishi, K. Sato and Y. Yamamoto, Tetrahedron, 2002, 58, 8195–8203 CrossRef CAS; (d) G. B. Bajracharya, T. Nogami, T. Jin, K. Matsuda, V. Gevorgyan and Y. Yamamoto, Synthesis, 2004, 308–311 CAS; (e) M. K. Skjel, A. Y. Houghton, A. E. Kirby, D. J. Harrison, R. McDonald and L. Rosenberg, Org. Lett., 2010, 12, 376–379 CrossRef CAS PubMed; (f) P. Bach, A. Albright and K. K. Laali, Eur. J. Org. Chem., 2009, 1961–1966 CrossRef CAS; (g) K. Benda, W. Regenhardt, E. Schaumann and G. Adiwidjaja, Eur. J. Org. Chem., 2009, 1016–1021 CrossRef CAS ; hydrosilylation of acetophenone using a chiral binaphthyl-based borane, but with no enantioinduction: ; (h) M. Mewald, R. Fröhlich and M. Oestreich, Chem. – Eur. J., 2011, 17, 9406–9414 CrossRef CAS PubMed ; hydrosilylation of acetophenone using a B(C6F5)3 congener: ; (i) J. Mohr, M. Durmaz, E. Irran and M. Oestreich, Organometallics, 2014, 33, 1108–1111 CrossRef CAS ; for a catalytic hydrosilylation of oxalic acid, see: ; (j) E. Feghali, O. Jacquet, P. Thuéry and T. Cantat, Catal. Sci. Technol., 2014, 4, 2230–2234 RSC.
- J. Nyhlén and T. Privalov, Dalton Trans., 2009, 5780–5786 RSC.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 7492–7495 CrossRef CAS PubMed.
- D. J. Harrison, R. McDonald and L. Rosenberg, Organometallics, 2005, 24, 1398–1400 CrossRef CAS.
- P. T. K. Lee, M. K. Skjel and L. Rosenberg, Organometallics, 2013, 32, 1575–1578 CrossRef CAS.
- For further examples of hydrosilylation of imines catalysed by electron-deficient boranes, see: ref. 21; (a) D. Chen, V. Leich, F. Pan and J. Klankermayer, Chem. – Eur. J., 2012, 18, 5184–5187 CrossRef CAS PubMed ; hydrosilylation by using a chiral binaphthyl-based borane: ref. 22 and 23; (b) X. Zhu and H. Du, Org. Biomol. Chem., 2015, 13, 1013–1016 RSC ; hydrosilylation by using a B(C6F5)3 congener: ref. 29i.
- (a) Z. M. Heiden and D. W. Stephan, Chem. Commun., 2011, 47, 5729–5731 RSC (diastereoselective); for further examples of imine hydrogenations catalysed by electron-deficient boranes, see: ; (b) C. Jiang, O. Blacque and H. Berke, Chem. Commun., 2009, 5518–5520 RSC; (c) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338–12348 CrossRef CAS PubMed; (d) J. W. Thomson, J. A. Hatnean, J. J. Hastie, A. Pasternak, D. W. Stephan and P. A. Chase, Org. Process Res. Dev., 2013, 17, 1287–1292 CrossRef CAS.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2014, 53, 10218–10222 CrossRef CAS PubMed . Hydrogenation of several heteroarenes and alkenes are also included in this work.
- Borane 23 was first prepared by Piers and co-workers: (a) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492–5503 CrossRef CAS; (b) D. J. Parks, R. E. von H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809–811 CrossRef CAS.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed.
- J. M. Farrell, Z. M. Heiden and D. W. Stephan, Organometallics, 2011, 30, 4497–4500 CrossRef CAS.
- I. Chatterjee and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 1965–1968 CrossRef CAS PubMed.
- For a similar abstraction of hydrides from Hantzsch esters by B(C6F5)3, see: (a) J. D. Webb, V. S. Laberge, S. J. Geier, D. W. Stephan and C. M. Crudden, Chem. – Eur. J., 2010, 16, 4895–4902 CrossRef CAS PubMed ; for an abstraction of hydride from an N-silyl dihydropyridine by B(C6F5)3, see: ; (b) D. V. Gutsulyak, A. van der Est and G. I. Nikonov, Angew. Chem., Int. Ed., 2011, 50, 1384–1387 CrossRef CAS PubMed.
- J. Mohr and M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 13278–13281 CrossRef CAS PubMed.
- (a) M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS PubMed; (b) for hydrostannation of unsaturated hydrocarbons with tributyltin hydride formed in situ from tributylchlorostannane and Et3SiH, see: V. Gevorgyan, J.-X. Liu and Y. Yamamoto, Chem. Commun., 1998, 37–38 RSC.
- (a) A. Simonneau and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 11905–11907 CrossRef CAS PubMed ; for a quantum-chemical treatment, see: ; (b) K. Sakata and H. Fujimoto, Organometallics, 2015, 34, 236–241 CrossRef CAS ; for broad investigation of transfer hydrosilylation, see: ; (c) S. Keess, A. Simonneau and M. Oestreich, Organometallics DOI:10.1021/om501284a.
- J. M. Blackwell, D. J. Morrison and W. E. Piers, Tetrahedron, 2002, 58, 8247–8254 CrossRef CAS.
- For the defunctionalisation of enol ethers, see Scheme 24.
- S. Chandrasekhar, G. Chandrashekar, M. S. Reddy and P. Srihari, Org. Biomol. Chem., 2006, 4, 1650–1652 CAS.
- M. Tan and Y. Zhang, Tetrahedron Lett., 2009, 50, 4912–4915 CrossRef CAS.
- Ref. 8b; (a) V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskelä, T. Repo, P. Pyykkö and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117–14119 CrossRef CAS PubMed; (b) G. Erős, H. Mehdi, I. Pápai, T. A. Rokob, P. Király, G. Tárkányi and T. Soós, Angew. Chem., Int. Ed., 2010, 49, 6559–6563 CrossRef PubMed; (c) S. Schwendemann, T. A. Tumay, K. V. Axenov, I. Peuser, G. Kehr, R. Fröhlich and G. Erker, Organometallics, 2010, 29, 1067–1069 CrossRef CAS; (d) S. Schwendemann, R. Fröhlich, G. Kehr and G. Erker, Chem. Sci., 2011, 2, 1842–1849 RSC; (e) G. Erős, K. Nagy, H. Mehdi, I. Pápai, P. Nagy, P. Király, G. Tárkányi and T. Soós, Chem. – Eur. J., 2012, 18, 574–585 CrossRef PubMed; (f) K. Chernichenko, M. Nieger, M. Leskelä and T. Repo, Dalton Trans., 2012, 41, 9029–9032 RSC.
- D. Bézier, S. Park and M. Brookhart, Org. Lett., 2013, 15, 496–499 CrossRef PubMed.
- Partial reductions of carboxylic acids to silyl ethers were already mentioned by Gevorgyan and Yamamoto in ref. 29a.
- V. Gevorgyan, J.-X. Liu, M. Rubin, S. Benson and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 8919–8922 CrossRef CAS.
- For further examples of ether cleavages, see: (a) V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS PubMed; (b) J. Heo, T. Kang, S. G. Jang, D. S. Hwang, J. M. Spruell, K. L. Killops, J. H. Waite and C. J. Hawker, J. Am. Chem. Soc., 2012, 134, 20139–20145 CrossRef CAS PubMed ; for an example of ether cleavages for formation of polymers, see: ref. 29b; for examples of B(C6F5)3-assisted intrachain ring-opening polymerization, see: ; (c) I. Perez-Baena, F. Barroso-Bujans, U. Gasser, A. Arbe, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 775–779 CrossRef CAS ; for examples of B(C6F5)3-catalysed ether cleavages in natural product syntheses, see: ; (d) D. F. Taber and S. C. Malcolm, J. Org. Chem., 2001, 66, 944–953 CrossRef CAS PubMed; (e) D. F. Taber, Q. Jiang, B. Chen, W. Zhang and C. L. Campbell, J. Org. Chem., 2002, 67, 4821–4827 CrossRef CAS PubMed; (f) J. R. Vyvyan, J. M. Oaksmith, B. W. Parks and E. M. Peterson, Tetrahedron Lett., 2005, 46, 2457–2460 CrossRef CAS; (g) N. A. McGrath, E. S. Bartlett, S. Sittihan and J. T. Njardarson, Angew. Chem., Int. Ed., 2009, 48, 8543–8546 CrossRef CAS PubMed ; for an application in lignin models, see: ; (h) E. Feghali and T. Cantat, Chem. Commun., 2014, 50, 862–865 RSC.
- R. D. Nimmagadda and C. McRae, Tetrahedron Lett., 2006, 47, 5755–5758 CrossRef CAS.
- They successfully applied their methodology by the use of n-butylhydrosilane (nBuSiH3) or diethylhydrosilane (2j) also to ketones and aldehydes.
- For a reductive dehydroxylation of Baylis–Hillman adducts using PMHS (2h) and catalytic B(C6F5)3, see: S. Chandrasekhar, G. Chandrashekar, K. Vijeender and M. S. Reddy, Tetrahedron Lett., 2006, 47, 3475–3478 CrossRef CAS.
- (a) L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 1646–1649 CrossRef CAS PubMed ; for further examples of deoxygenation processes catalysed by electron-deficient boranes, see: ref. 29a, d and 56; (b) S. Chandrasekhar, C. R. Reddy and B. N. Babu, J. Org. Chem., 2002, 67, 9080–9082 CrossRef CAS PubMed ; for a catalytic deoxygenation of oxalic acid, see: ref. 29j.
- For B(C6F5)3-catalyzed reductive alkylation of alkoxybenzenes using aldehyde as alkylating agents in the presence of PMHS (2h), see: S. Chandrasekhar, S. Khatun, G. Rajesh and C. R. Reddy, Tetrahedron Lett., 2009, 50, 6693–6697 CrossRef CAS.
- For a B(C6F5)3-catalysed reductive coupling of carbonyl compounds with alkoxysilanes in the presence of PMHS (2h), see: S. Chandrasekhar, G. Chandrashekar, B. Nagendra Babu, K. Vijeender and K. Venkatram Reddy, Tetrahedron Lett., 2004, 45, 5497–5499 CrossRef CAS.
- E. Blondiaux and T. Cantat, Chem. Commun., 2014, 50, 9349–9352 RSC.
- R. C. Chadwick, V. Kardelis, P. Lim and A. Adronov, J. Org. Chem., 2014, 79, 7728–7733 CrossRef CAS PubMed.
- For a further example, see: G. T. Beng, R. Meesala, M. N. Mordi and S. M. Mansor, Synth. Commun., 2014, 44, 1291–1295 CrossRef CAS.
- L. D. Curless, E. R. Clark, J. J. Dunsford and M. J. Ingleson, Chem. Commun., 2014, 50, 5270–5272 RSC.
- S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884–4886 RSC.
- G. Erős, K. Nagy, H. Mehdi, I. Pápai, P. Nagy, P. Király, G. Tárkányi and T. Soós, Chem. – Eur. J., 2012, 18, 574–585 CrossRef PubMed.
- N. Gandhamsetty, S. Joung, S.-W. Park, S. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 16780–16783 CrossRef CAS PubMed.
- J. Hermeke, H. F. T. Klare and M. Oestreich, Chem. – Eur. J., 2014, 20, 9250–9254 CrossRef CAS PubMed.
- Z. Zhang and H. Du, Angew. Chem., Int. Ed., 2015, 54, 623–626 CAS.
- T. Mahdi, Z. M. Heiden, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 4088–4091 CrossRef CAS PubMed.
- (a) T. Mahdi, J. N. del Castillo and D. W. Stephan, Organometallics, 2013, 32, 1971–1978 CrossRef CAS; (b) for a similar B(C6F5)3-mediated hydrogenation of carbodiimides, see: M. H. Holthausen, M. Colussi and D. W. Stephan, Chem. – Eur. J., 2015, 21, 2193–2199 CrossRef CAS PubMed.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 12968–12971 CrossRef CAS PubMed.
- D. J. Mack, B. Guo and J. T. Njardarson, Chem. Commun., 2012, 48, 7844–7846 RSC.
- J. Chojnowski, S. Rubinsztajn, J. A. Cella, W. Fortuniak, M. Cypryk, J. Kurjata and K. Kaźmierski, Organometallics, 2005, 24, 6077–6084 CrossRef CAS.
- J. B. Grande, D. B. Thompson, F. Gonzaga and M. A. Brook, Chem. Commun., 2010, 46, 4988–4990 RSC.
- Examples for the synthesis of silicon-based polymers with siloxanes: (a) S. Rubinsztajn and J. A. Cella, Polym. Prepr., 2004, 45, 635–636 CAS; (b) S. Rubinsztajn and J. A. Cella, Macromolecules, 2005, 38, 1061–1063 CrossRef CAS; (c) J. Chojnowski, W. Fortuniak, J. Kurjata, S. Rubinsztajn and J. A. Cella, Macromolecules, 2006, 39, 3802–3807 CrossRef CAS; (d) C. Longuet, C. Joly-Duhamel and F. Ganachaud, Macromol. Chem. Phys., 2007, 208, 1883–1892 CrossRef CAS; (e) C. Xunjun, C. Yingde, Y. Guoqiang and L. Liewen, J. Appl. Polym. Sci., 2007, 106, 1007–1013 CrossRef; (f) J. Chojnowski, S. Rubinsztajn, W. Fortuniak and J. Kurjata, J. Inorg. Organomet. Polym. Mater., 2007, 17, 173–187 CrossRef CAS; (g) D. B. Thompson and M. A. Brook, J. Am. Chem. Soc., 2008, 130, 32–33 CrossRef CAS PubMed; (h) J. Cella and S. Rubinsztajn, Macromolecules, 2008, 41, 6965–6971 CrossRef CAS; (i) J. Chojnowski, S. Rubinsztajn, W. Fortuniak and J. Kurjata, Macromolecules, 2008, 41, 7352–7358 CrossRef CAS; (j) J. Kurjata, W. Fortuniak, S. Rubinsztajn and J. Chojnowski, Eur. Polym. J., 2009, 45, 3372–3379 CrossRef CAS; (k) J. B. Grande, F. Gonzaga and M. A. Brook, Dalton Trans., 2010, 39, 9369–9378 RSC; (l) D. J. Keddie, J. B. Grande, F. Gonzaga, M. A. Brook and T. R. Dargaville, Org. Lett., 2011, 13, 6006–6009 CrossRef CAS PubMed; (m) B. A. Kamino, J. B. Grande, M. A. Brook and T. P. Bender, Org. Lett., 2011, 13, 154–157 CrossRef CAS PubMed; (n) F. Gonzaga, J. B. Grande and M. A. Brook, Chem. – Eur. J., 2012, 18, 1536–1541 CrossRef CAS PubMed; (o) J. B. Grande, A. S. Fawcett, A. J. McLaughlin, F. Gonzaga, T. P. Bender and M. A. Brook, Polymer, 2012, 53, 3135–3142 CrossRef CAS; (p) M. J. Gretton, B. A. Kamino, M. A. Brook and T. P. Bender, Macromolecules, 2012, 45, 723–728 CrossRef CAS; (q) J. B. Grande, T. Urlich, T. Dickie and M. A. Brook, Polym. Chem., 2014, 5, 6728–6739 RSC ; for hydride transfer ring-opening polymerization of cyclic siloxanes catalysed by B(C6F5)3, see: ; (r) J. Chojnowski, J. Kurjata, W. Fortuniak, S. Rubinsztajn and B. Trzebicka, Macromolecules, 2012, 45, 2654–2661 CrossRef CAS ; for the synthesis of triarylamine–siloxanes using the Piers–Rubinsztajn reaction, see: ; (s) B. A. Kamino, B. Mills, C. Reali, M. J. Gretton, M. A. Brook and T. P. Bender, J. Org. Chem., 2012, 77, 1663–1674 CrossRef CAS PubMed; (t) M. J. Gretton, B. A. Kamino and T. P. Bender, Macromol. Symp., 2013, 324, 82–94 CrossRef CAS ; for a polymerisation of phenylsilane catalysed by B(C6F5)3 to form branched polysilanes, see: ; (u) A. Feigl, I. Chiorescu, K. Deller, S. U. H. Heidsieck, M. R. Buchner, V. Karttunen, A. Bockholt, A. Genest, N. Rösch and B. Rieger, Chem. – Eur. J., 2013, 19, 12526–12536 CrossRef CAS PubMed ; for an overview, see: ; (v) M. A. Brook, J. B. Grande and F. Ganachaud, in Adv. Polym. Sci., ed. A. M. Muzafarov, Springer, Berlin, Heidelberg, 2011, vol. 235, pp. 161–183 Search PubMed.
- J.-M. Denis, H. Forintos, H. Szelke and G. Keglevich, Tetrahedron Lett., 2002, 43, 5569–5571 CrossRef CAS.
- C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27–30 CrossRef CAS.
- For further examples of dehydrogenative silylation of alcohols, see: ref. 4, 14, 29a, h–j, 54, 55a, b and 56; (a) D. J. Harrison, D. R. Edwards, R. McDonald and L. Rosenberg, Dalton Trans., 2008, 3401–3411 RSC; (b) T. Strašák, J. Sýkora, M. Lamač, J. Kubišta, M. Horáček, R. Gyepes and J. Pinkas, Organometallics, 2013, 32, 4122–4129 CrossRef; (c) A. Simonneau, J. Friebel and M. Oestreich, Eur. J. Org. Chem., 2014, 2077–2083 CrossRef CAS.
- R. Shchepin, C. Xu and P. Dussault, Org. Lett., 2010, 12, 4772–4775 CrossRef CAS PubMed.
- Examples for dehydrogenative silylation of alcohols for the synthesis of polymers: (a) D. Zhou and Y. Kawakami, Macromolecules, 2005, 38, 6902–6908 CrossRef CAS; (b) Z. Zhang, L. J. Lyons, J. J. Jin, K. Amine and R. West, Chem. Mater., 2005, 17, 5646–5650 CrossRef CAS; (c) L. Xue and Y. Kawakami, Polym. J., 2007, 39, 379–388 CrossRef CAS; (d) M. A. Hoque, Y. Kakihana, S. Shinke and Y. Kawakami, Macromolecules, 2009, 42, 3309–3315 CrossRef CAS; (e) Z. Zhang, J. Dong, R. West and K. Amine, J. Power Sources, 2010, 195, 6062–6068 CrossRef CAS; (f) N. Moitra, S. Ichii, T. Kamei, K. Kanamori, Y. Zhu, K. Takeda, K. Nakanishi and T. Shimada, J. Am. Chem. Soc., 2014, 136, 11570–11573 CrossRef CAS PubMed.
- L. D. Curless and M. J. Ingleson, Organometallics, 2014, 33, 7241–7246 CrossRef CAS.
- For trans-selective hydrosilylations of alkynes using phosphonium salts, see: (a) M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308–18310 CrossRef PubMed; (b) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef CAS PubMed.
Footnote |
| † J.H. and J.M. contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |