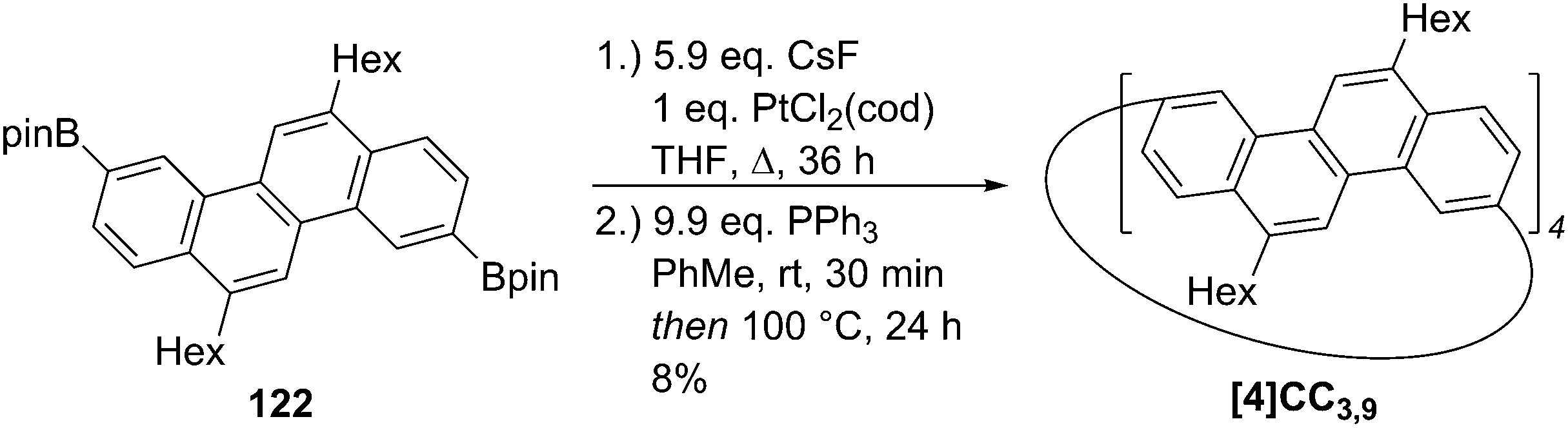
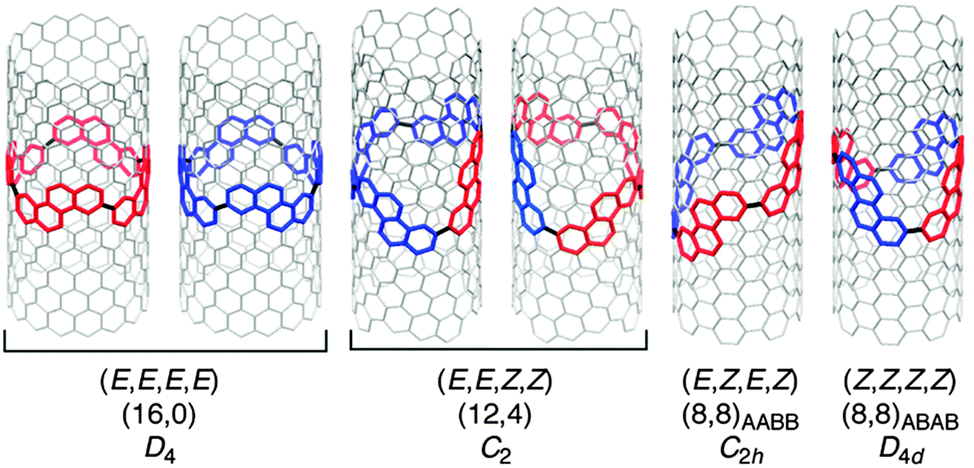
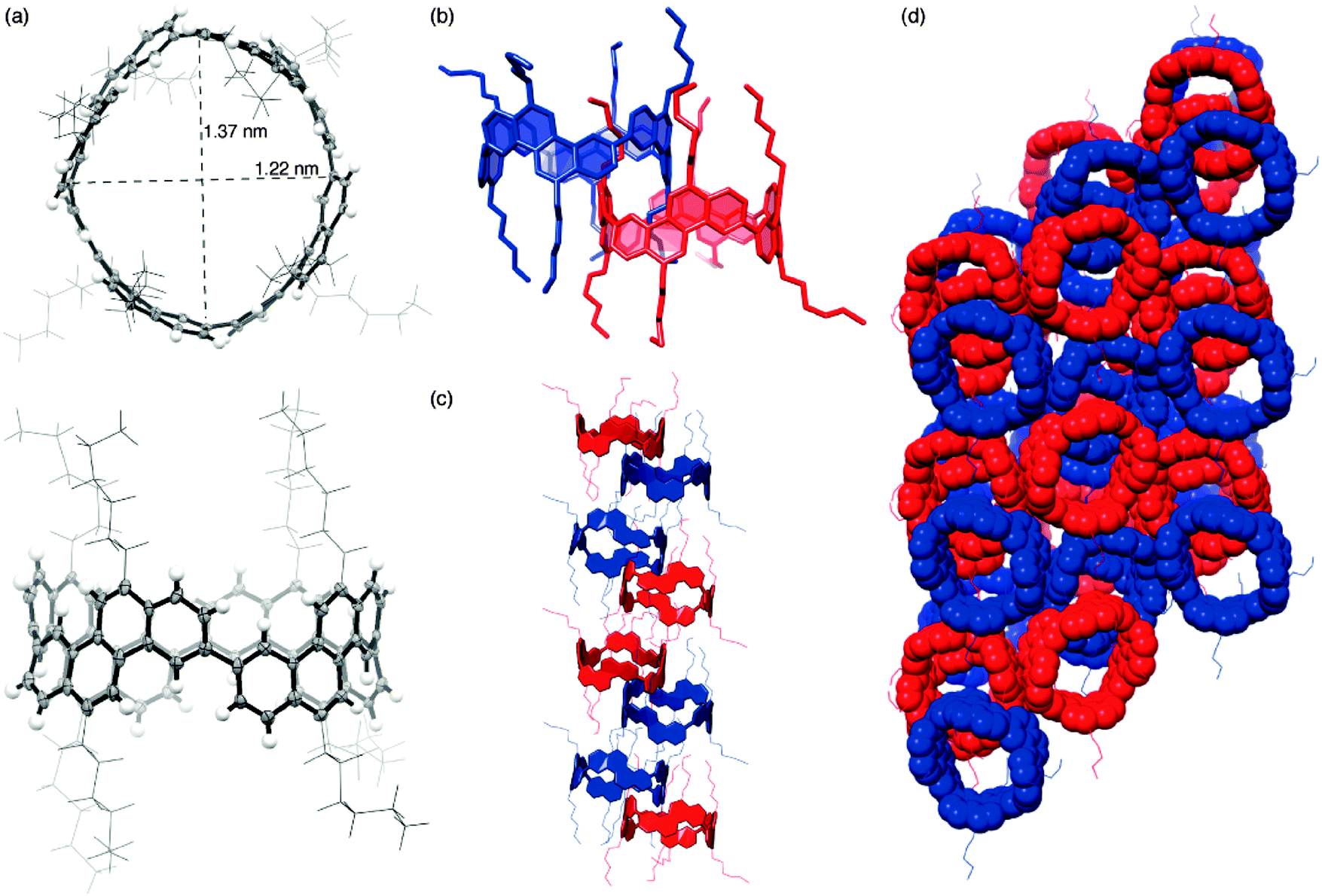
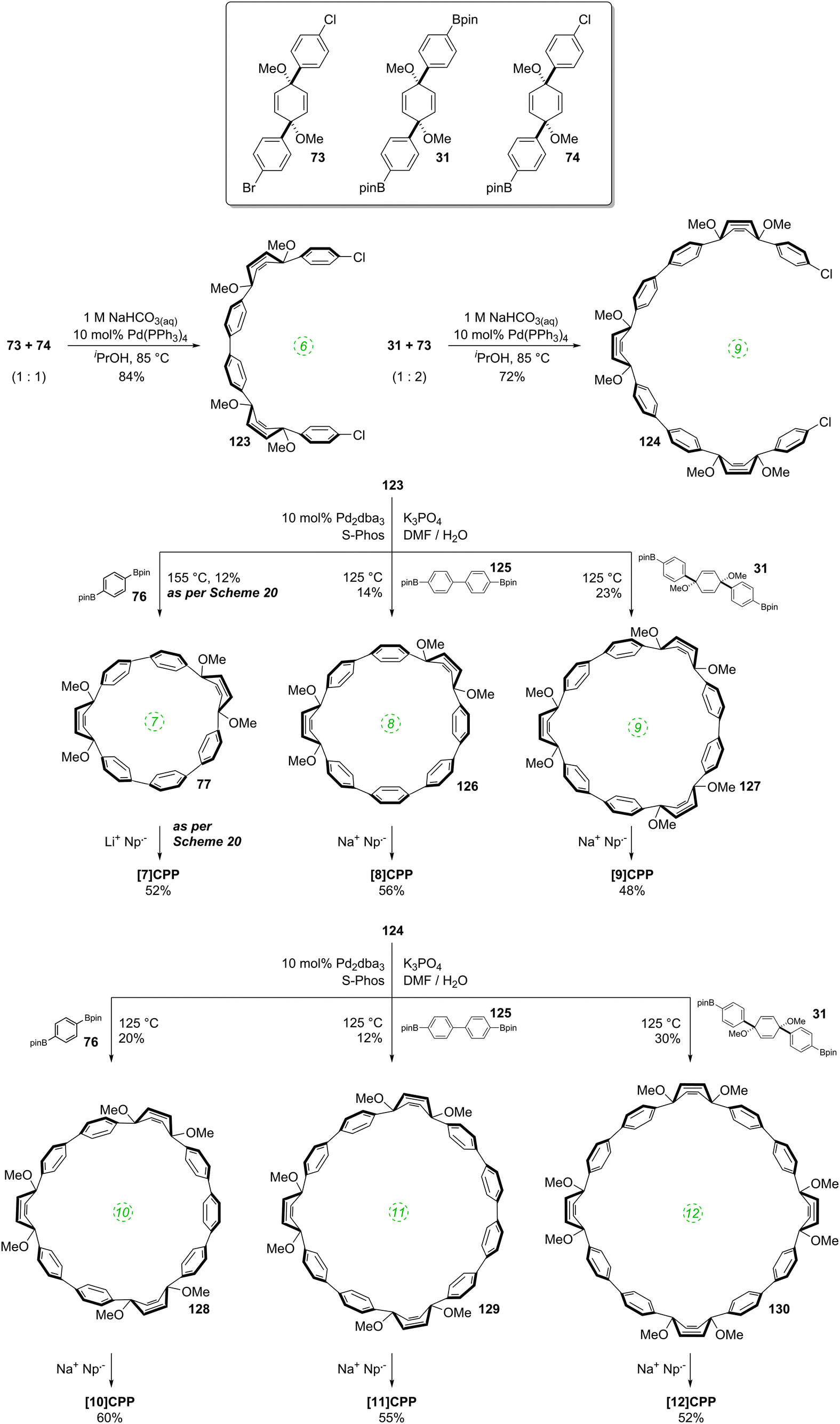
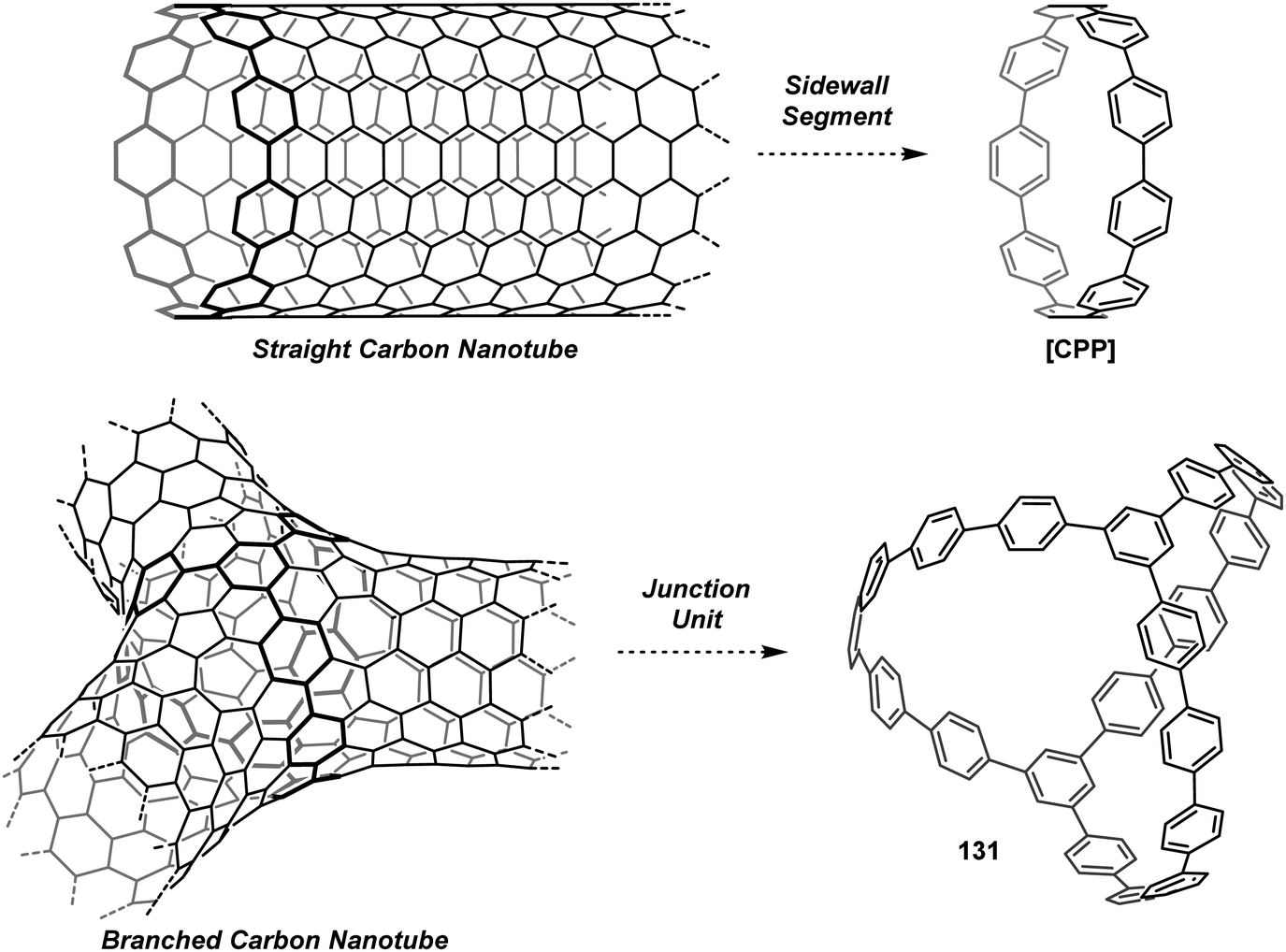
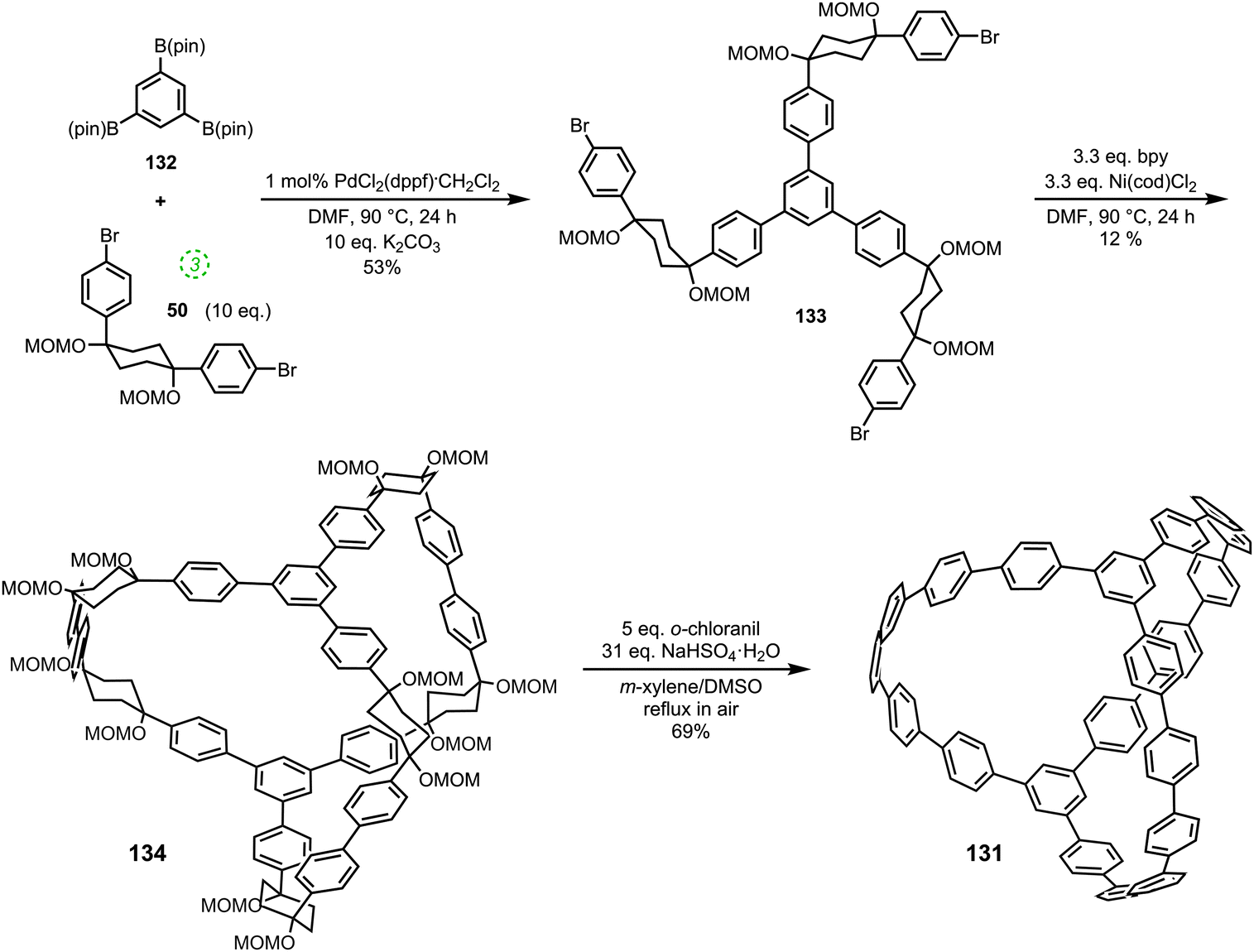
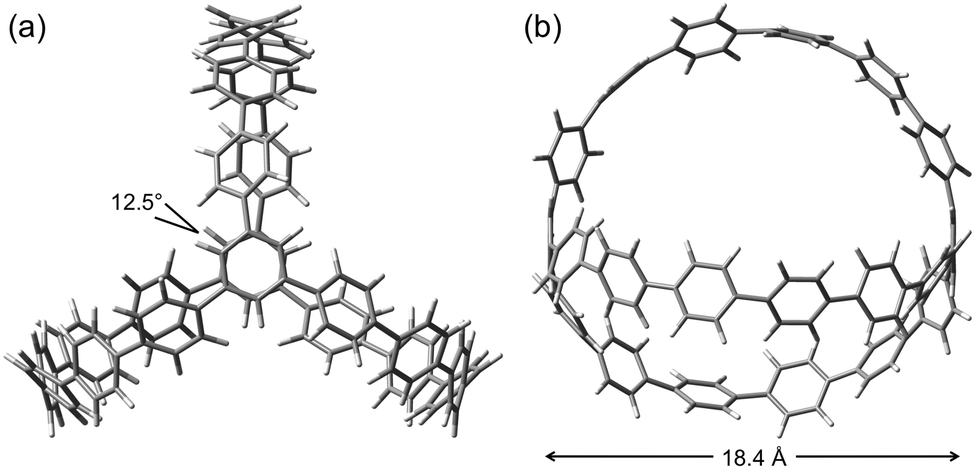
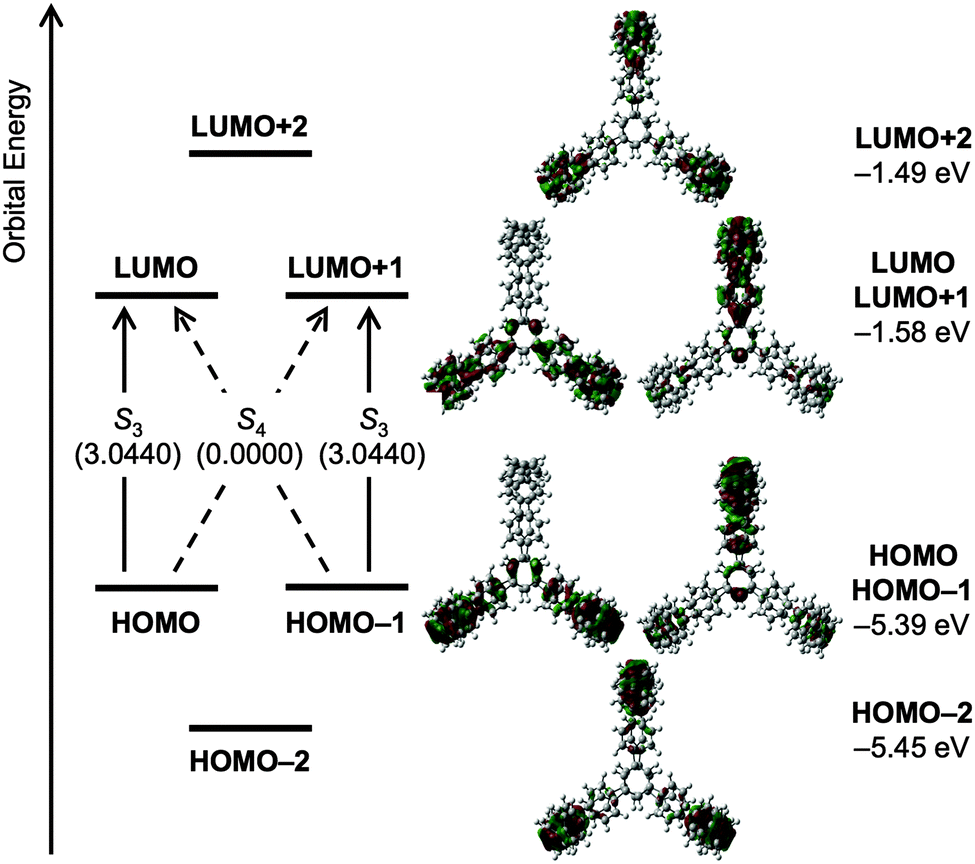
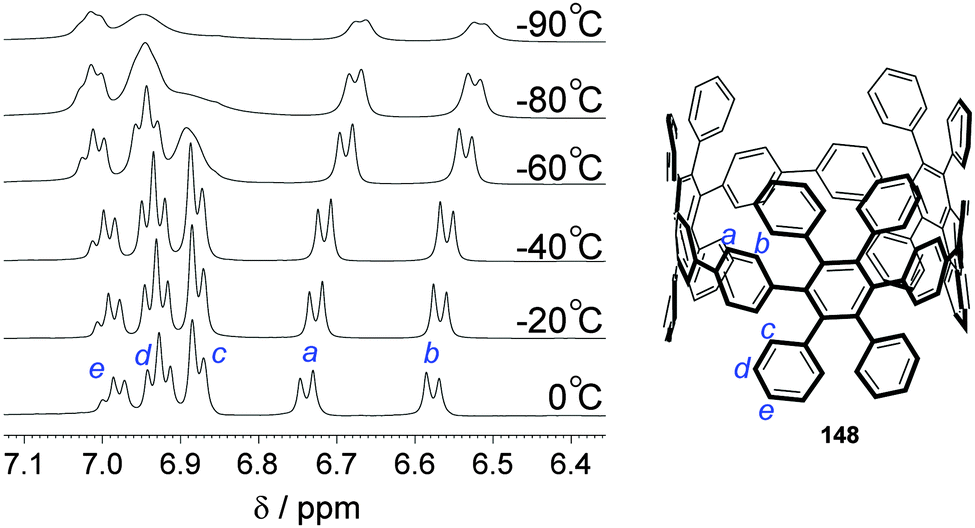
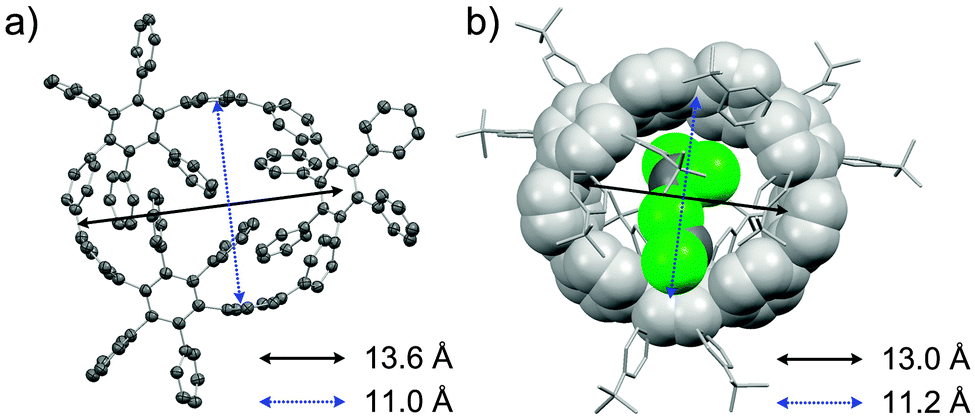
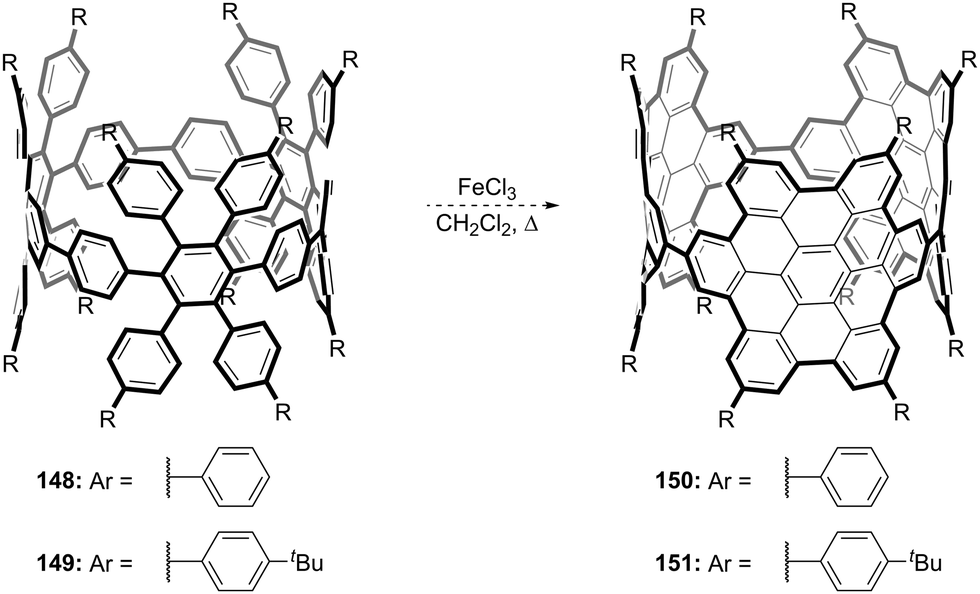
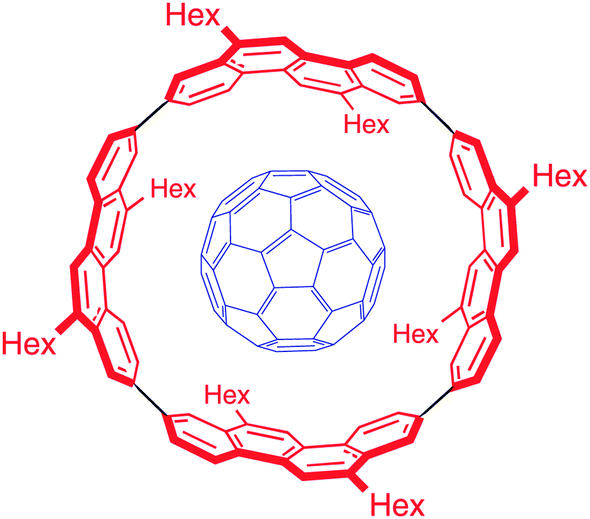
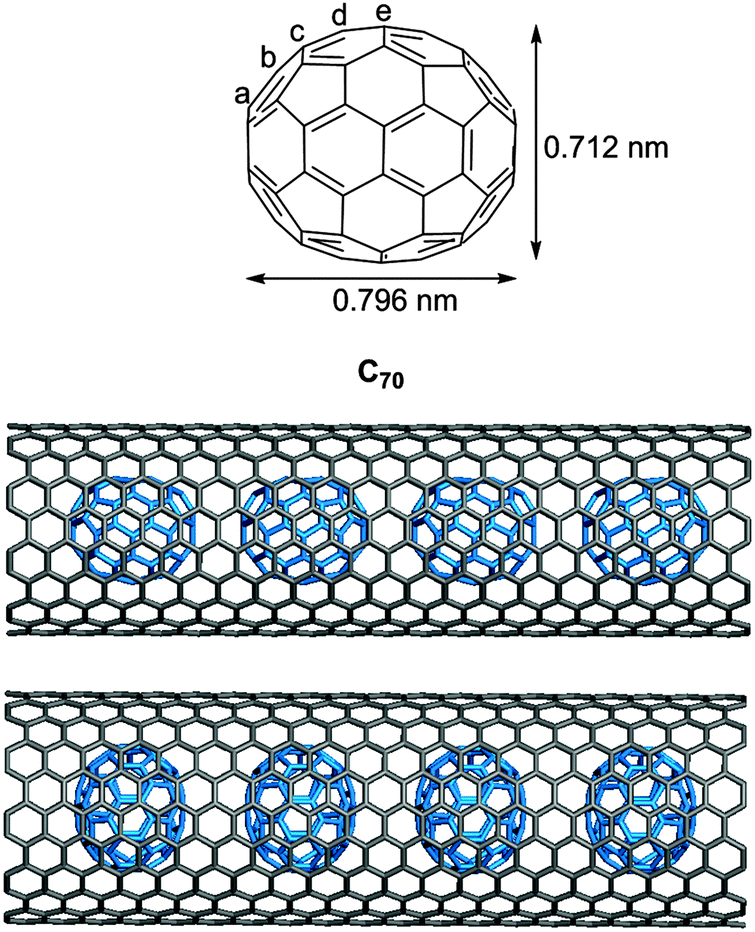
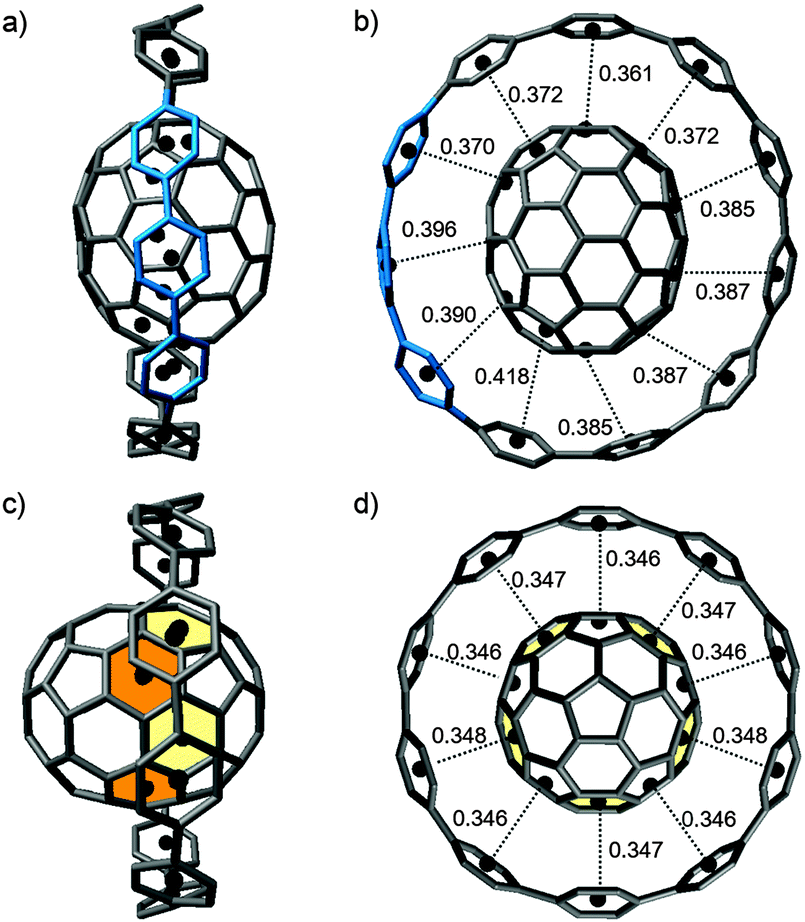
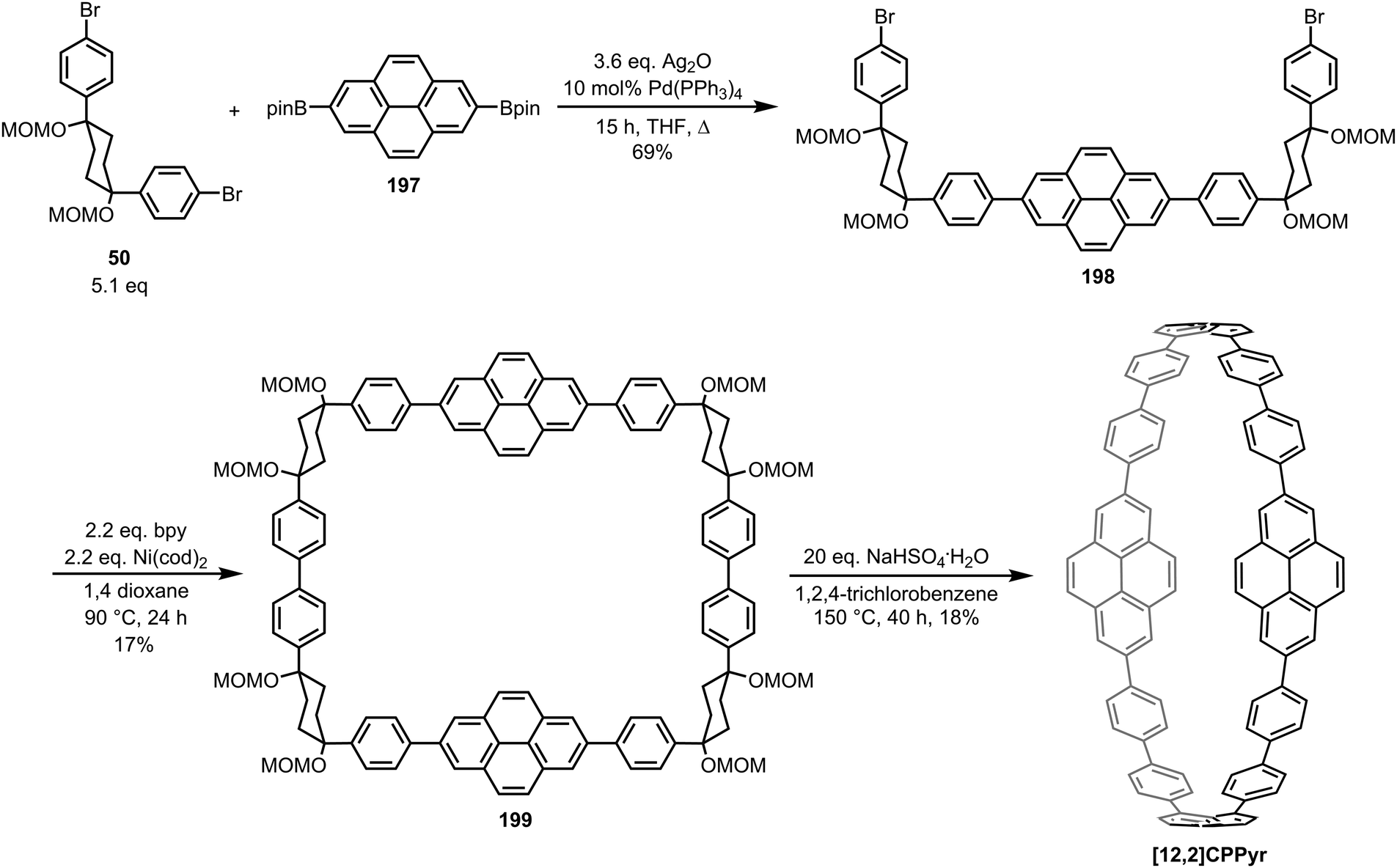
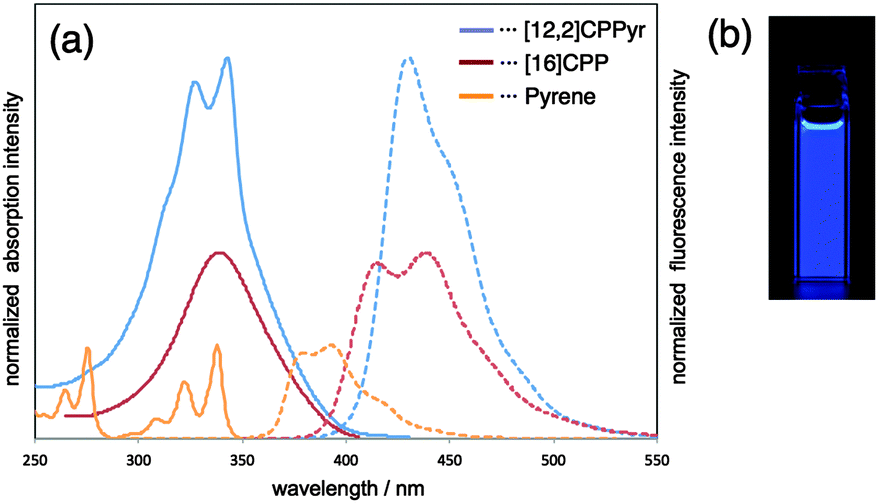
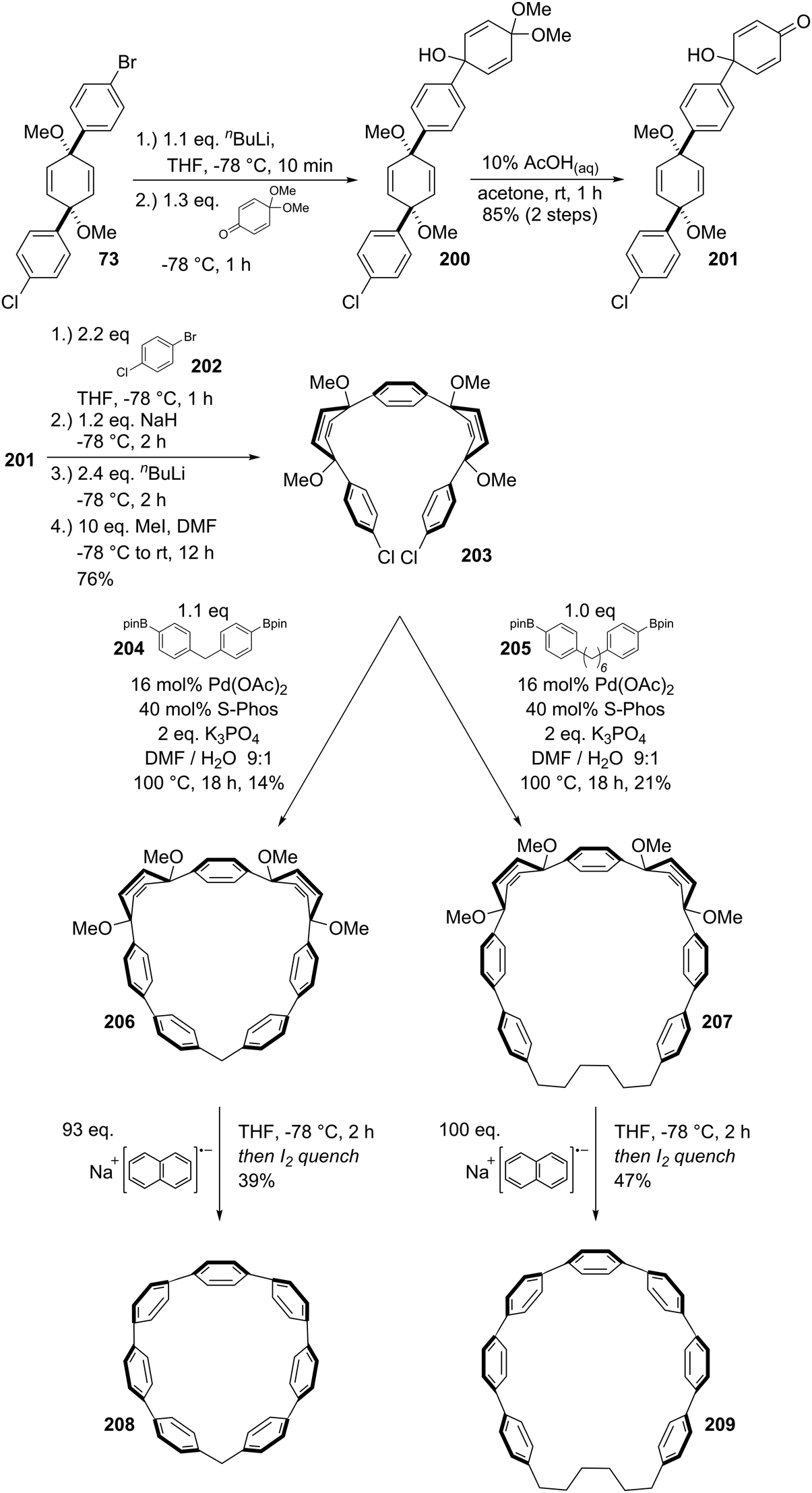
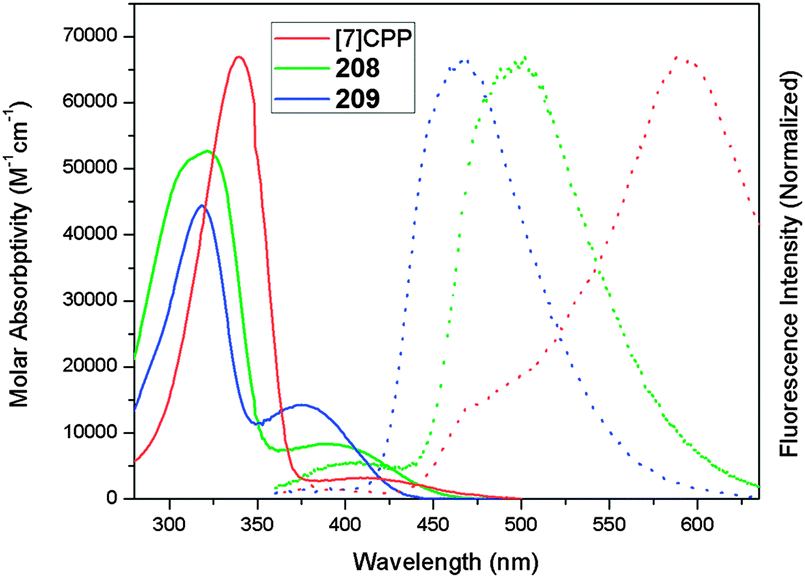
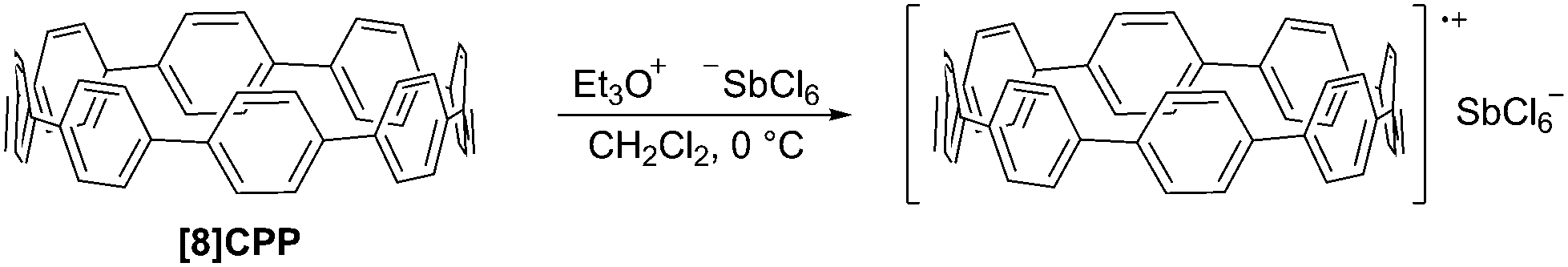DOI:
10.1039/C4CS00366G
(Review Article)
Chem. Soc. Rev., 2015,
44, 2221-2304
Cycloparaphenylenes and related nanohoops
Received
31st October 2014
First published on 4th March 2015
Abstract
The first synthesis of a cyclic oligophenylene possessing a radial π system was reported in 2008. In the short period that has elapsed since, there has been an ever-increasing level of interest in molecules of this type, as evidenced by the volume of publications in this area. This interest has been driven by the highly unusual properties of these molecules in comparison to their linear oligoarene analogues, as well as the diverse array of potential applications for them. Notably, CPPs and related structures were proposed as viable templates for the bottom-up synthesis of single-walled carbon nanotubes (SWCNTs), a proposition which has recently been realised. This review gives a comprehensive and strictly chronological (by date of first online publication) treatment of literature reports from the inception of the field, with emphasis on both synthesis and properties of CPPs and related nanohoops. (The scope of this review is restricted to molecules possessing a radial π system consisting entirely of subunits which are aromatic in isolation, e.g. CPPs, but not cycloparaphenyleneacetylenes or cyclopolyacetylenes).
Introduction
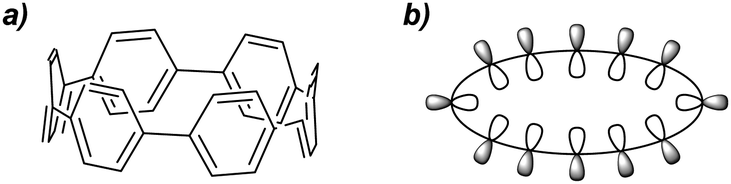
New undergraduate students of chemistry are taught that aromatic molecules are planar, by virtue of their π-systems. This is not universally true, however – biaryl structures such as BINOL are an obvious example of systems possessing non-planar π-systems due to the competing effects of orbital overlap and steric encumbrance. An extreme manifestation of non-planarity in a π-system may be found in the cycloparaphenylenes (CPPs), which possess radially cyclic π-systems (Fig. 1).
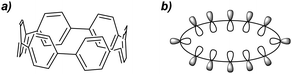 |
| | Fig. 1 (a) [6]Cycloparaphenylene ([6]CPP). (b) A schematic representation of the radial π system of a CPP. | |
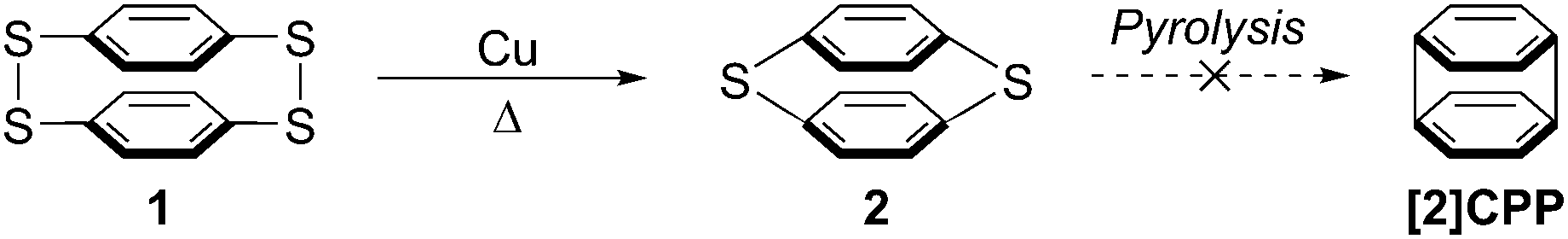
It is only in the last 7 years that CPPs have moved from theoretical curiosities to synthetically accessible molecules. However, a true chronological treatment of the field needs to start much earlier, with a report from Parekh and Guha dating from 1934.1 This describes an attempted synthesis of the smallest member of this class of compounds, [2]CPP, and also raises the possibility of synthesising [3]CPP. As shown in Scheme 1, these authors effected a Cu-mediated partial desulfurization of material they believed to be p,p′-diphenylenetetrasulfide 1, a [22]paracyclophane. This reportedly afforded the more strained [12]paracyclophane 2. However, they were not able to effect complete desulfurization to access [2]CPP. To the eye of a contemporary chemist, [2]CPP looks to be far too strained a structure to be viable, but nevertheless it is interesting to see CPP structures being considered so long ago.
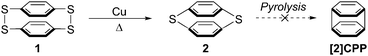 |
| | Scheme 1 Parekh and Guha's attempted synthesis of [2]CPP. | |
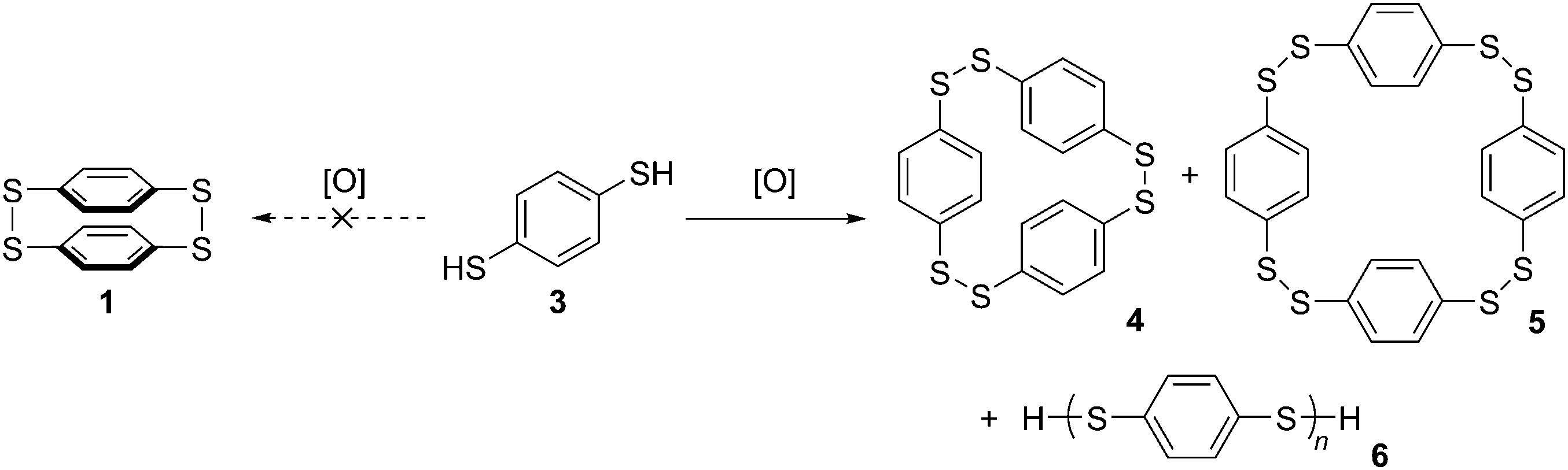
A caveat which must be noted concerning this report is that the authors characterised 1 and 2 primarily on the basis of elemental analysis, so the possibility they isolated a higher oligomer cannot be excluded. Indeed, they themselves acknowledged this possibility, and the fact that the substance they believed to be 1 was insoluble in all organic solvents lends credence to the idea it was in fact polymeric. To the best of our knowledge, structures 1 and 2 have never been isolated and characterized again subsequently. Parekh and Guha prepared 1 by oxidation of 1,4-benzenedithol 3, but it was later shown that even at high dilution this reaction affords only [23]paracyclophane 4 and/or [24]paracyclophane 5, as well as polymeric material 6 (Scheme 2).2,3
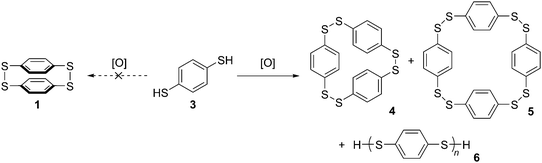 |
| | Scheme 2 Oxidation of 3 does not actually afford dimer 1. | |
Only many years later, in 1993, did CPP chemistry advance significantly further when Vögtle and co-workers published a paper entitled “On the way to macrocyclic paraphenylenes”.4 This was the culmination of a decade of efforts to access CPPs and although Vögtle was ultimately unsuccessful in synthesising CPPs, this seminal work nevertheless discloses several conceptually distinct approaches to CPPs, which undoubtedly paved the way for the many later successes this review describes.
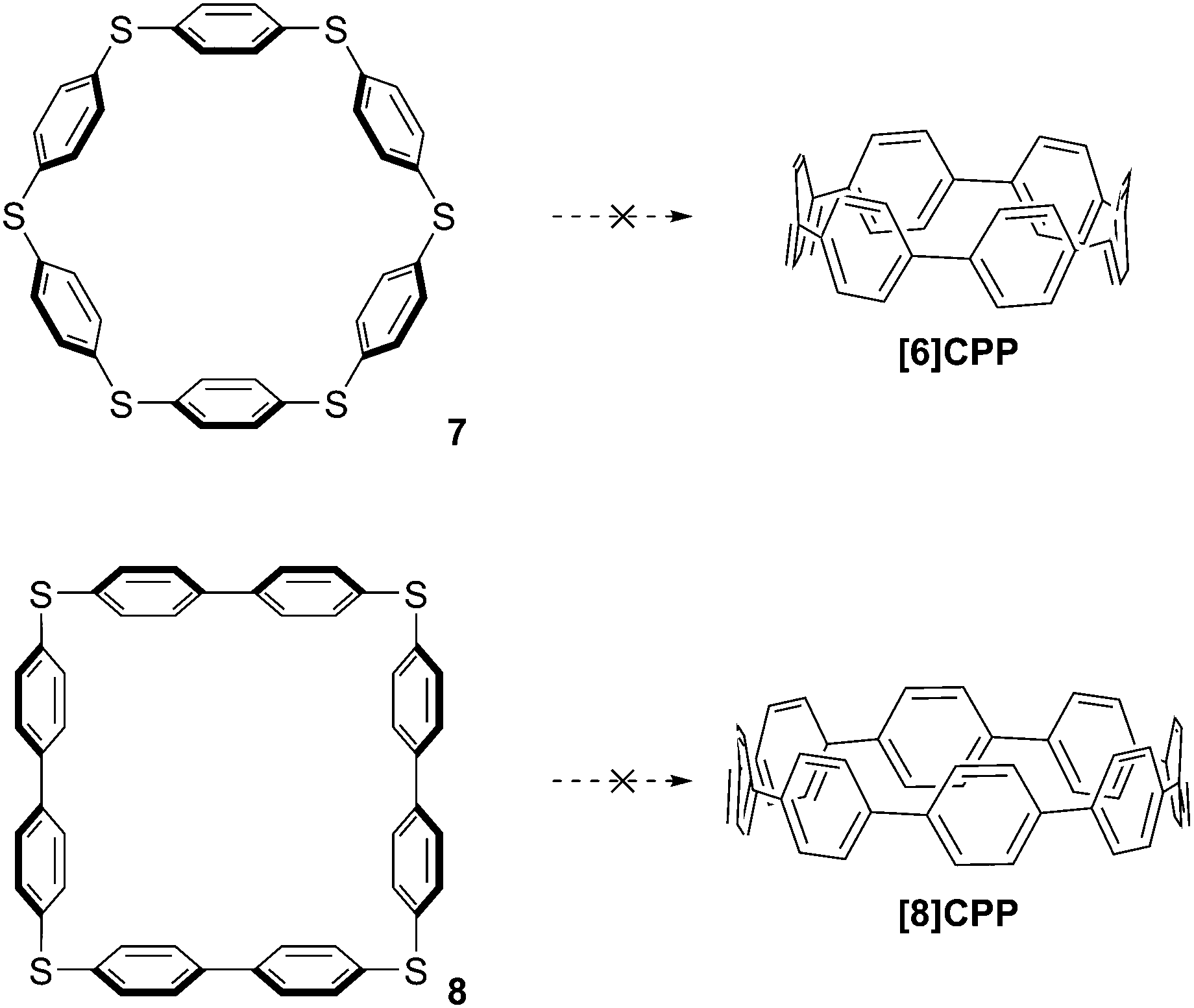
In the first instance, Vögtle attempted to establish a desulfurisation route to CPPs which bears some resemblance to Parekh and Guha's strategy, the key difference being that Vögtle targeted larger CPPs (specifically [6]CPP and [8]CPP). These are much more plausible targets than [2]CPP, but nevertheless still possess formidable strain energy due to the deformation of the π system away from planarity. The synthesis of Vögtle's precursors 7 and 8 is surprisingly concise and high-yielding,5 but unfortunately all attempts to transform them into CPPs were unsuccessful (Scheme 3).
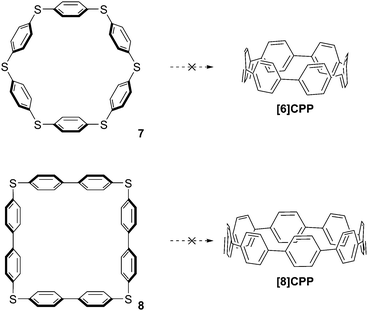 |
| | Scheme 3 Vögtle's attempted preparations of [6]CPP and [8]CPP by desulfurization. | |
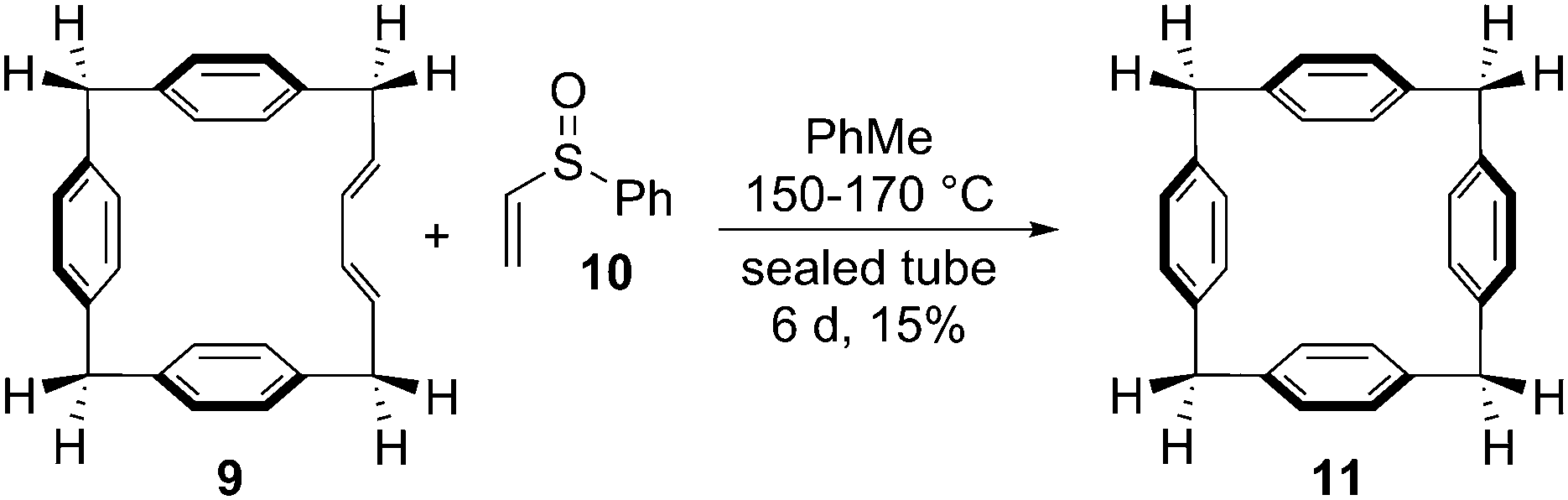
The Vögtle group next sought to access CPPs using Diels–Alder reactions to install aromatic rings in a pre-existing macrocycle. There was circumstantial precedent for this in Miyahara's synthesis of a [14]paracyclophane 11 from macrocyclic diene 9 and phenyl vinyl sulfoxide 10 (Scheme 4).6 In the event, Vögtle's approach differed somewhat in that macrocycles 14 and 17, prepared by Wittig cyclooligomerisation, contained enyne units as opposed to dienes (Scheme 5). All attempts to transform 14 or 17 into CPPs were reportedly unsuccessful.
 |
| | Scheme 4 Miyahara's synthesis of a [14]paracyclophane. | |
 |
| | Scheme 5 Vögtle's attempted synthesis of CPPs by Diels–Alder reaction. | |
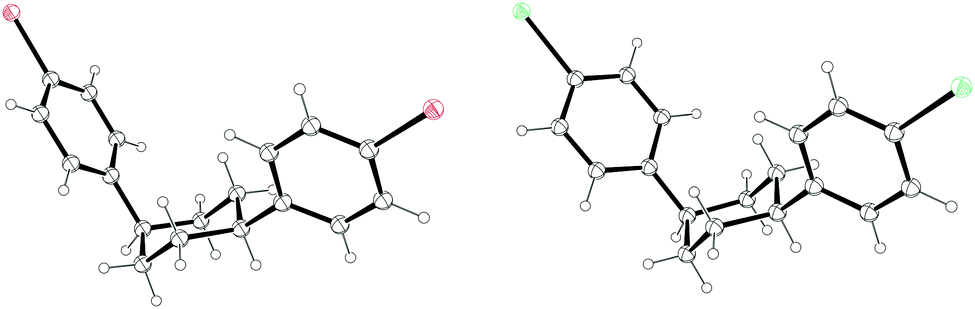
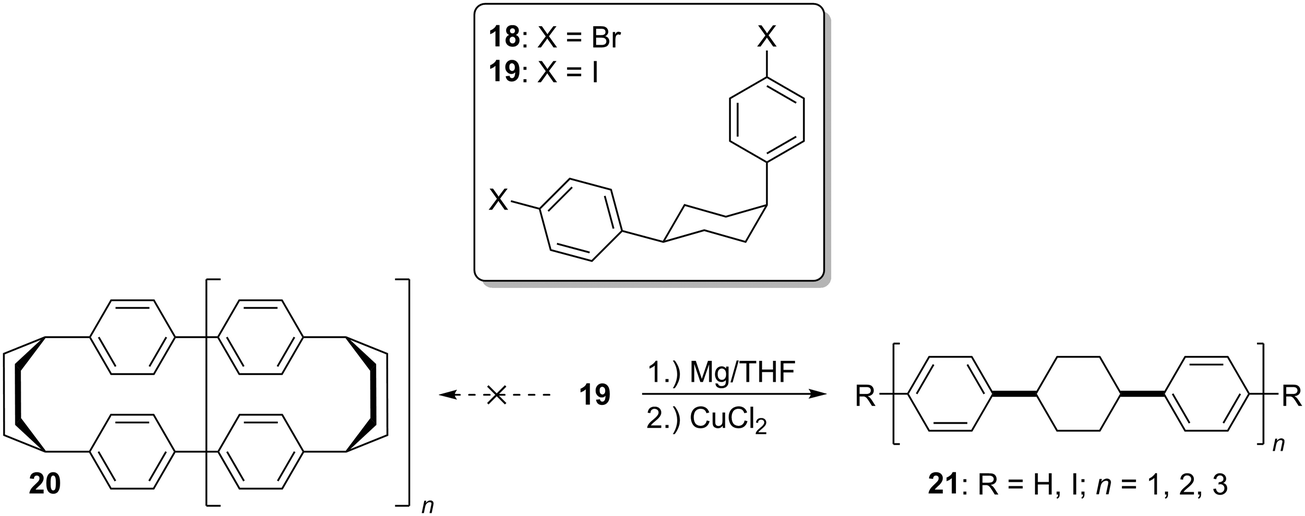
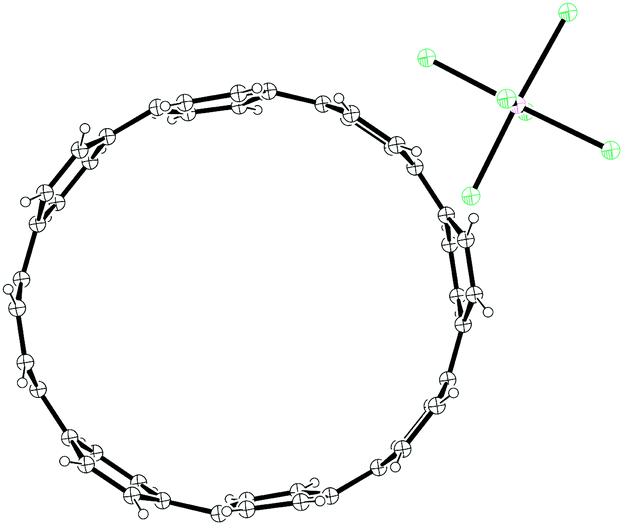
The third strategy explored by Vögtle, seemingly in the most detail, was that of assembling macrocycles containing cyclohexane rings as well as arenes. Such macrocycles would be expected to be far less strained than CPPs since they contain sp3 hybridised carbons and therefore would be much better able to accommodate the curvature of the macrocycle. Once such macrocycles had been formed, aromatisation of the cyclohexane rings would lead to CPPs, the increase in strain energy being offset by the gain in aromaticity. As a first attempt, 1,4-syn-diaryl cyclohexane building blocks 18 and 19 were synthesised. X-ray crystal structures of these suggested that these molecules adopted a conformation which would favour cyclooligomerisation, at least in the solid state (Fig. 2). However, macrocycle formation under conditions of Kharasch coupling was in fact unsuccessful, giving linear oligomers instead (Scheme 6).
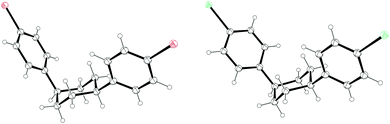 |
| | Fig. 2 ORTEP diagram of 1,4-syn-diaryl cyclohexanes 18 and 19, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. For 19, only one molecule of three in the unit cell is shown for clarity. | |
 |
| | Scheme 6 Failed cyclooligomerisation of 1,4-syn-diaryl cyclohexanes 18 and 19. | |
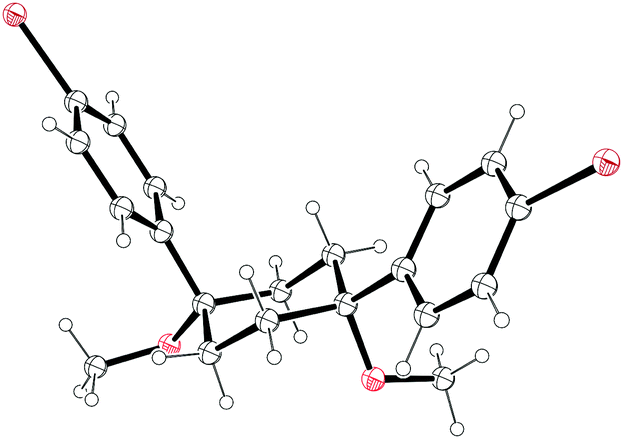
NMR data for 18 suggested that in contrast to the solid state, 18 was conformationally labile in solution, with the desired conformer for cyclisation probably being only minimally populated. On this basis, Vögtle and co-workers designed and synthesised a modified 1,4-syn-diaryl cyclohexane building block 22 that possessed additional substituents, the idea being that these would impart a degree of preorganisation to the structure, favouring in solution the conformer required for macrocycle formation (Fig. 3). Once again, X-ray crystallography indicated a solid state conformation compatible with cyclooligomerisation (Fig. 4), but once again this desired transformation was not successful. In an attempt to increase still further the preorganisation of the system, the authors synthesised bicyclohexyl derivative 23. In this prospective CPP precursor, the doubly spirocyclic ketal would serve to rigidify the cyclohexane rings, allowing the equatorially disposed phenyl rings to adopt a coplanar arrangement. This might favour dimerisation rather than the formation of higher oligomers. However, Vögtle does not actually report para-halogenation of 23 or attempts at macrocyclisation. By the same rationale, doubly spirocyclic thiirane 24 was targeted, but was not synthetically accessible.
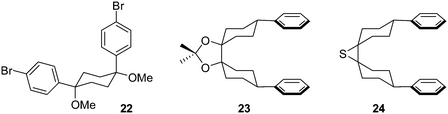 |
| | Fig. 3 Alternative building blocks for accessing macrocyclic CPP precursors targeted by the Vögtle group. | |
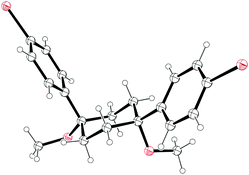 |
| | Fig. 4 ORTEP diagram of 1,4-syn-diaryl cyclohexane 22, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
The final strategy explored by Vögtle was accessing CPPs from macrocyclic precursors derived through cyclative McMurry coupling. As shown in Scheme 7, McMurry himself had reported the reductive cyclisation of diketone 25 (n = 1) to give cyclotetrakis(1,4-cyclohexylidene) 26 (n = 1).7 Vögtle extended this approach to a larger ring, assembling homologated diketone precursor 25 (n = 2) using chemistry analogous to that of McMurry, although 25 was of unexpectedly poor solubility. Reductive cyclisation gave cyclopentakis(1,4-cyclohexylidene) 26 (n = 2), but in a quantity sufficient only for characterisation by high resolution mass spectrometry; insufficient material was produced for elaboration towards [5]CPP. As a final point of note, in the concluding remarks of this report, Vögtle states that approaches to CPPs based on preorganised 1,4-disubstituted cyclohexane building blocks appear to be the most promising. This comment was very prescient, given the successes reported fifteen or more years later (vide infra).
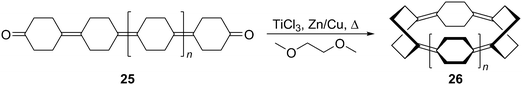 |
| | Scheme 7 Access to cyclooligo(1,4-cyclohexylidenes). | |
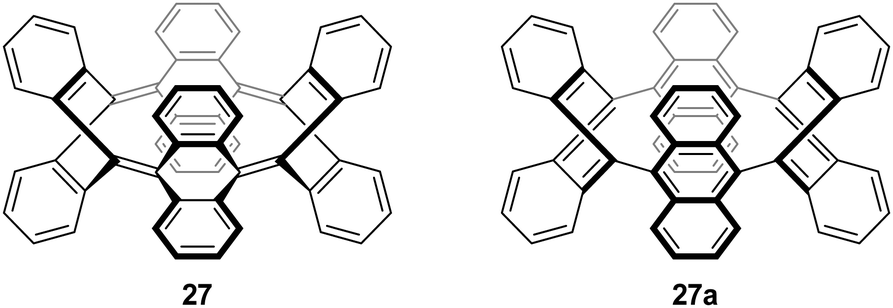
Subsequent to Vögtle's report, two further reports of particular relevance appeared. In 1996, Herges and co-workers reported the synthesis of a “picotube” 27 (so named by analogy with larger nanotubes).8,9 This annulated derivative of 26 (n = 1) can be conceived of as a [4]CPP derivative (Fig. 5), although it should be noted that X-ray crystallographic evidence (Fig. 6) indicates the picotube exists in the quinodimethane form (27), as opposed to its fully aromatic bond shift isomer (27a).
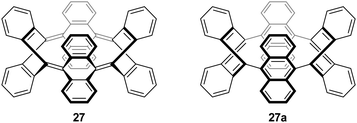 |
| | Fig. 5 Bond shift isomers of picotube 27. | |
 |
| | Fig. 6 ORTEP diagrams of picotube 27, showing ellipsoids at 30% probability. H atoms have been omitted for clarity, as have a molecule of MeCN and a molecule of CS2 in the unit cell. | |
In 2000, a computational study of selected CPPs was reported by Chandrasekhar and co-workers.10 This addressed the question of molecular structure and aromatic character and predicted [5]CPP and [6]CPP to have significantly different properties. As regards the relative stability of the quinoid and benzenoid (i.e. fully aromatic) forms (cf.27 and 27a), [5]CPP was predicted to have a quinoid structure, whereas [6]CPP was predicted to have a benzenoid structure. Later computational studies at different levels of theory instead predicted [5]CPP to be benzenoid and when [5]CPP was in fact synthesised fourteen years later,11–13 X-ray crystallographic analysis confirmed it to be benzenoid in nature on the basis of the observed bond lengths.13 Nevertheless, this early computational study identified the key trend of decreasing aromaticity with decreasing ring size.
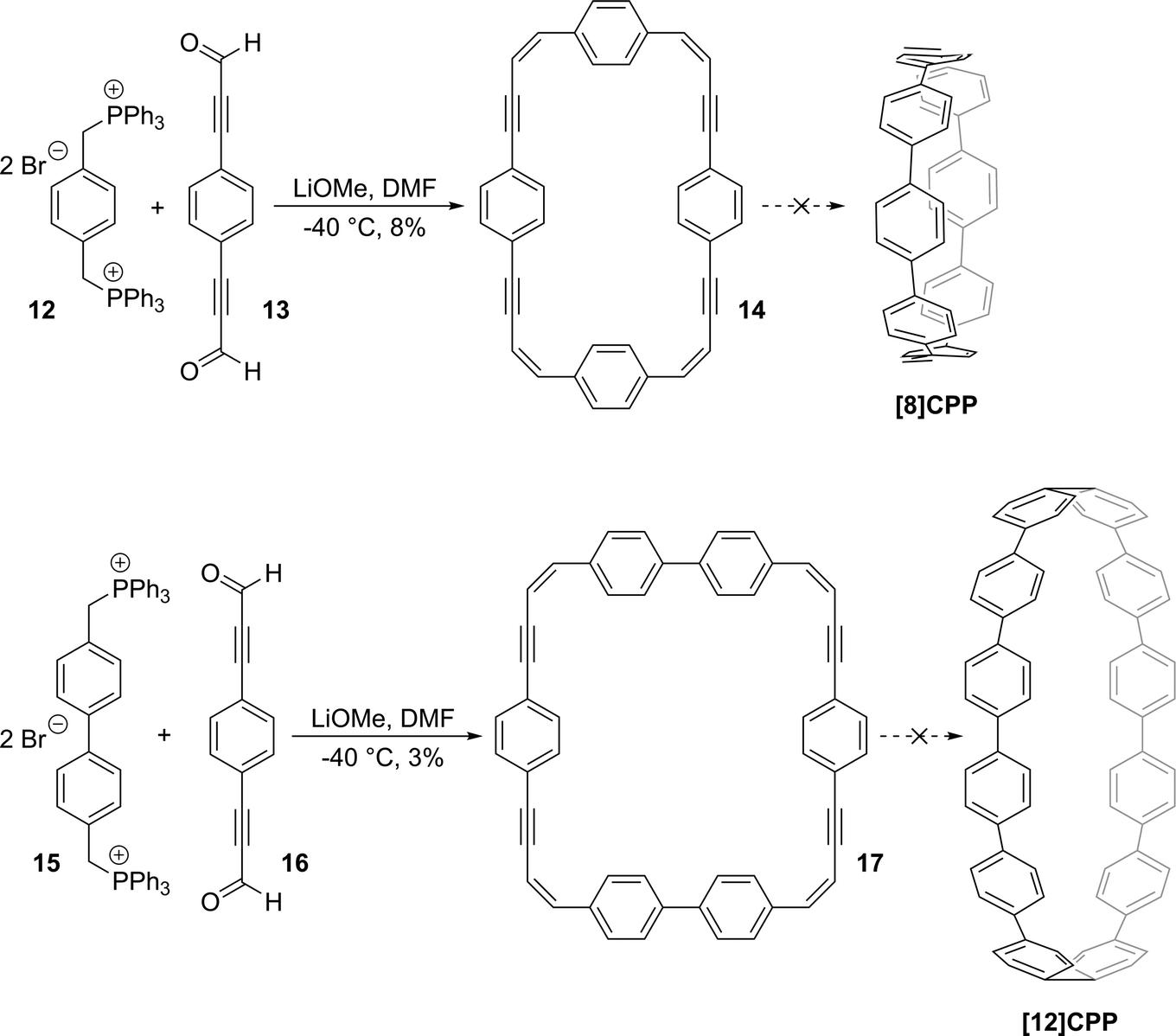
Subsequent to Chandrasekhar's study, no further papers specifically concerned with CPPs were published until 2008, when the first successful synthesis of a CPP was reported, although a review in 2006 provides a comprehensive overview of the more general field of “molecular loops and belts”, including some discussion of CPPs.14 Thus, the above section describes the state of the art at the point when CPPs moved from the computer screen to the round-bottom flask.
2008
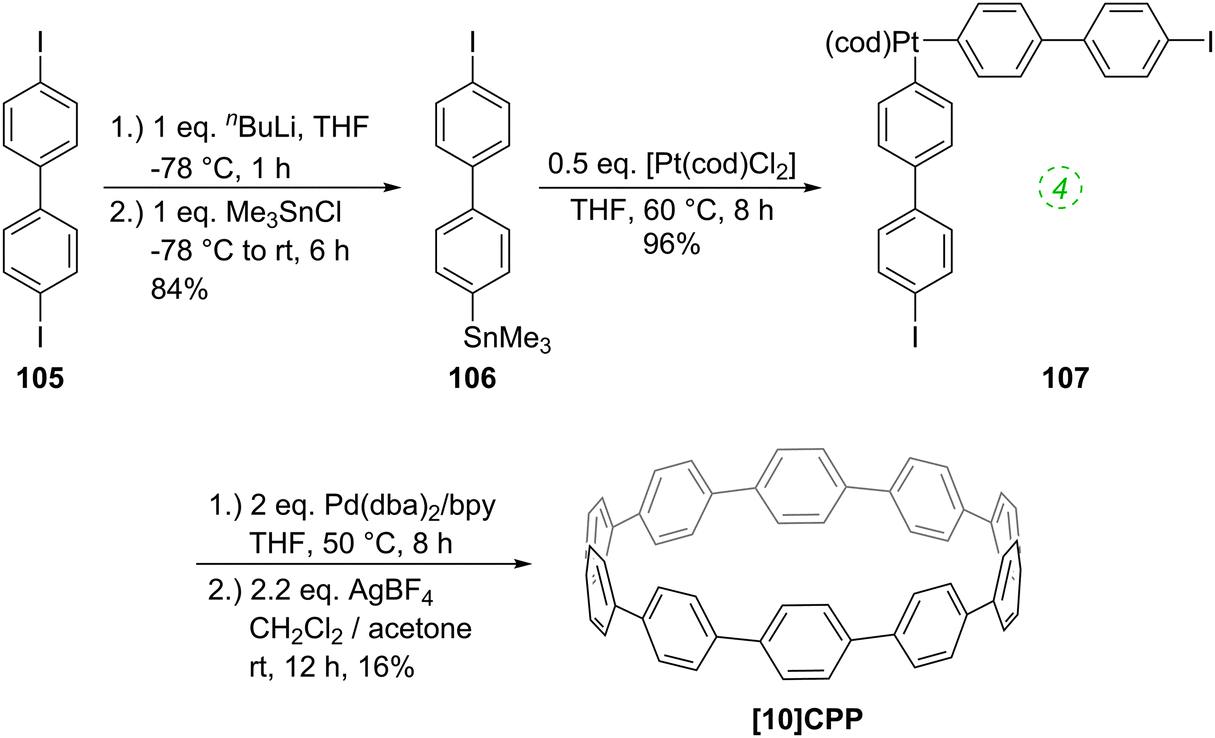
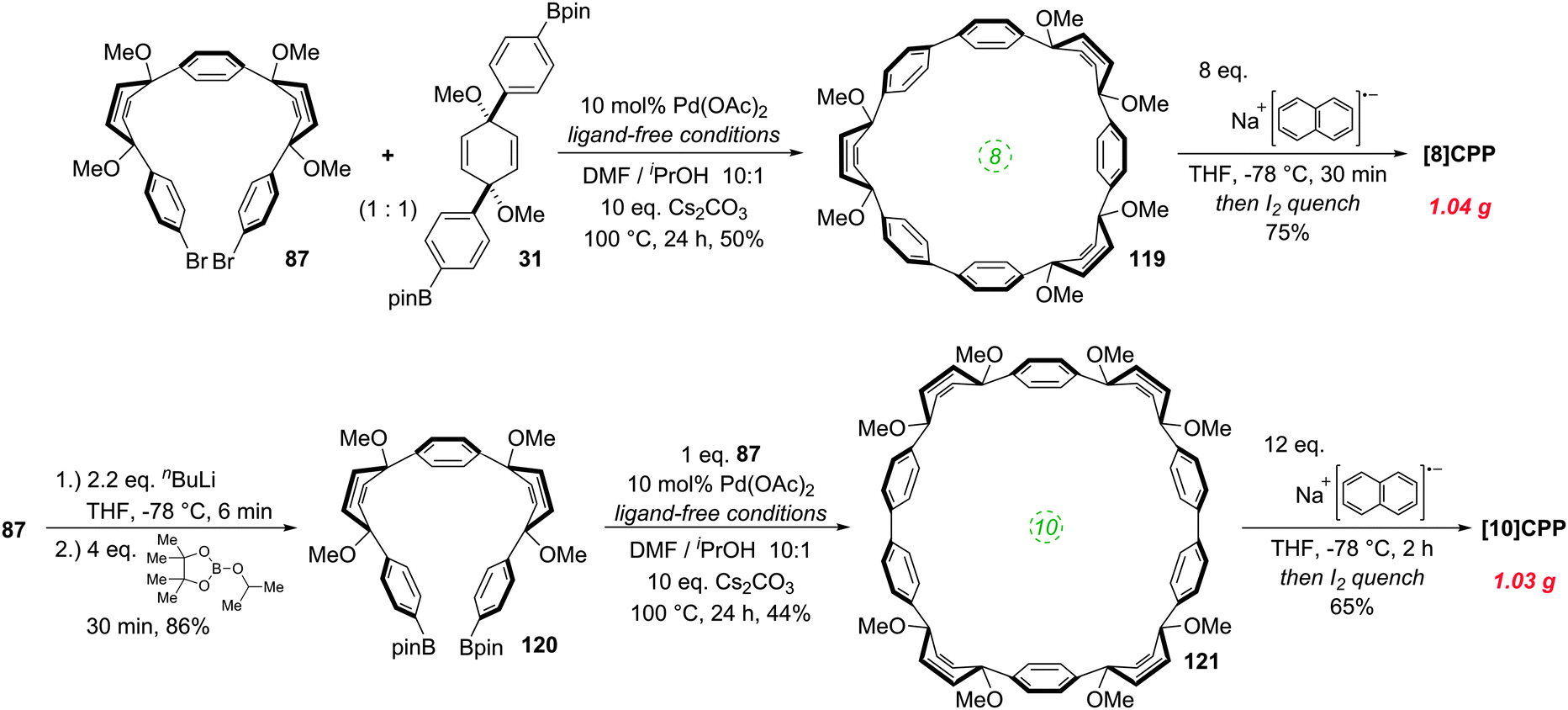
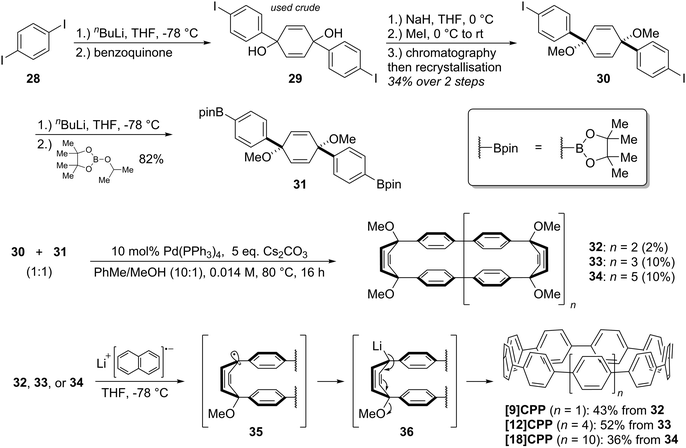
It was in 2008 that Bertozzi and co-workers published their landmark synthesis of [9]CPP, [12]CPP and [18]CPP.15 These three CPPs were synthesised from a common precursor and the approach has conceptual similarity with Vögtle's attempts to access CPPs using 1,4-syn-diaryl cyclohexane building blocks. However, a crucial difference is that the non-aromatic rings in the CPP precursors are not cyclohexanes but cyclohexa-1,4-dienes. The non-aromatic rings therefore contain two sp3 centres and four sp2 centres; this combination confers both curvature and also rigidity/preorganisation. Another difference is that the macrocyclisation is effected by means of Suzuki–Miyaura coupling, not Kharasch coupling. The synthetic route is depicted in detail in Scheme 8.
 |
| | Scheme 8 The first synthesis of CPPs, reported by Bertozzi and co-workers. | |
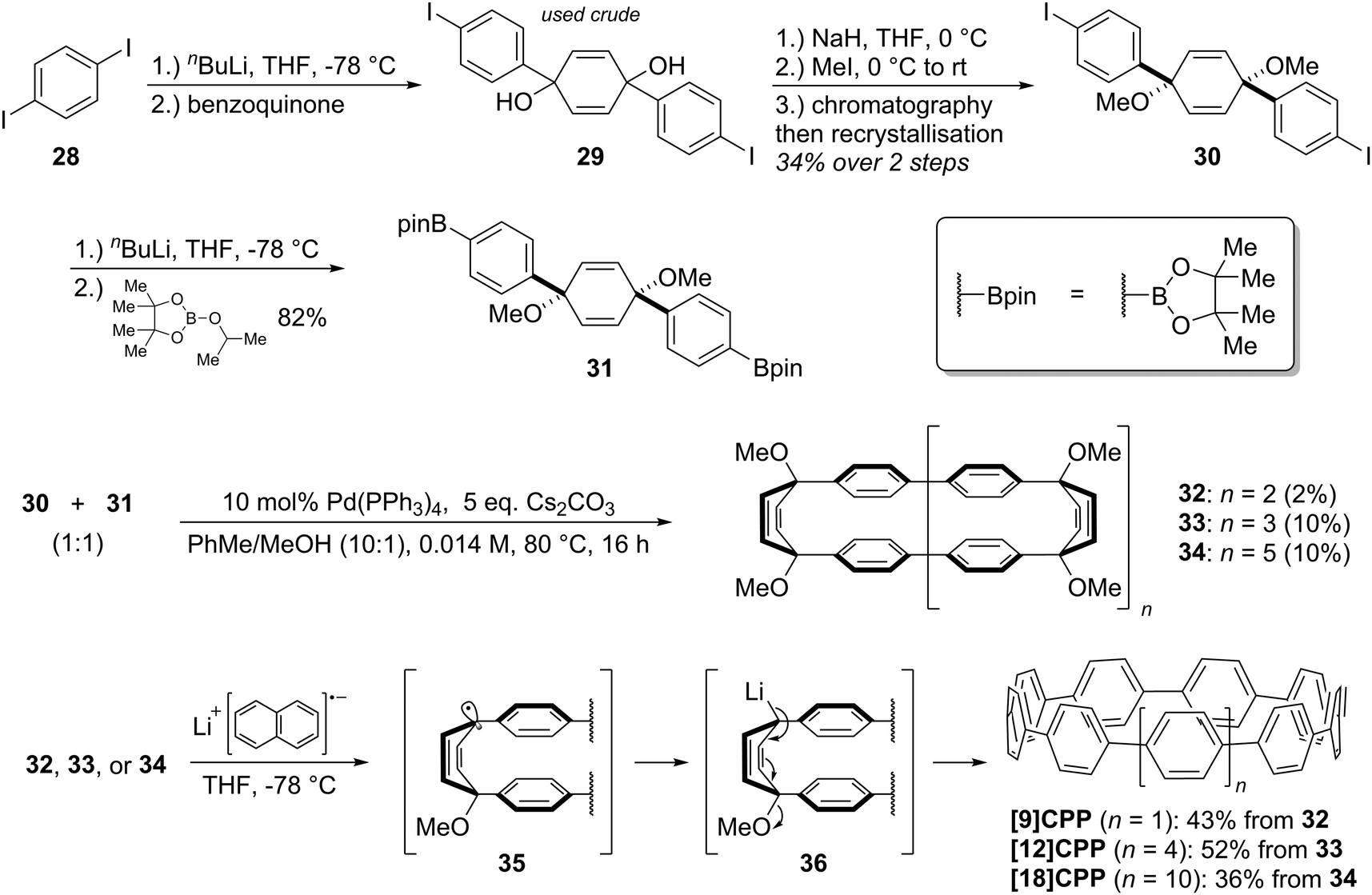
Monolithiation of 1,4-diiodobenzene 28 followed by addition to ≈0.5 equivalents of benzophenone gave diol 29 (syn/anti ratio not specified). This crude diol was methylated with methyl iodide, which gave pure syn bis(ether) 30 after chromatography and subsequent recrystallization (recrystallization alone being ineffective for purification). With one cross-coupling partner in hand, the authors next converted a portion of 30 to the corresponding bis(pinacolborane) 31 by double lithiation and addition to 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (ITDB), employing careful control of the addition times. With both difunctionalised precursors in hand, the authors undertook macrocyclisation by exposing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 30 and 31 to conditions for Suzuki–Miyaura cross-coupling. Formation of a direct 1:1 adduct of 30 and 31 (i.e. a precursor to [6]CPP) was not observed – instead higher oligomers formed. Thus, 33 (arising from the combination of 2 molecules of 30 and 2 of 31), as well as 34 (arising from 3 molecules of 30 and 3 of 31), were each formed in a yield of 10%. A smaller macrocycle, 32, was also isolated as an unexpected additional product (in a lower yield of 2%). Formation of 32 is surprising insofar as it must arise from a total of three molecules of 30 and/or 31. Any macrocyclisation of an odd number of molecules of 30 and/or 31 must necessarily involve at least one homocoupling event in addition to the expected Suzuki–Miyaura cross-couplings; in the case of 32, the authors ascribe its formation to a homocoupling of 31. Transformation of the cyclised precursors into the target CPPs was effected by single electron transfer reduction using lithium naphthalenide. It is proposed that the first one-electron reduction leads to loss of methoxide and formation of stabilised radical 35. A second one-electron reduction then converts this to lithiated intermediate 36, which eliminates a second equivalent of methoxide as shown to aromatise the ring.
:1 mixture of 30 and 31 to conditions for Suzuki–Miyaura cross-coupling. Formation of a direct 1:1 adduct of 30 and 31 (i.e. a precursor to [6]CPP) was not observed – instead higher oligomers formed. Thus, 33 (arising from the combination of 2 molecules of 30 and 2 of 31), as well as 34 (arising from 3 molecules of 30 and 3 of 31), were each formed in a yield of 10%. A smaller macrocycle, 32, was also isolated as an unexpected additional product (in a lower yield of 2%). Formation of 32 is surprising insofar as it must arise from a total of three molecules of 30 and/or 31. Any macrocyclisation of an odd number of molecules of 30 and/or 31 must necessarily involve at least one homocoupling event in addition to the expected Suzuki–Miyaura cross-couplings; in the case of 32, the authors ascribe its formation to a homocoupling of 31. Transformation of the cyclised precursors into the target CPPs was effected by single electron transfer reduction using lithium naphthalenide. It is proposed that the first one-electron reduction leads to loss of methoxide and formation of stabilised radical 35. A second one-electron reduction then converts this to lithiated intermediate 36, which eliminates a second equivalent of methoxide as shown to aromatise the ring.
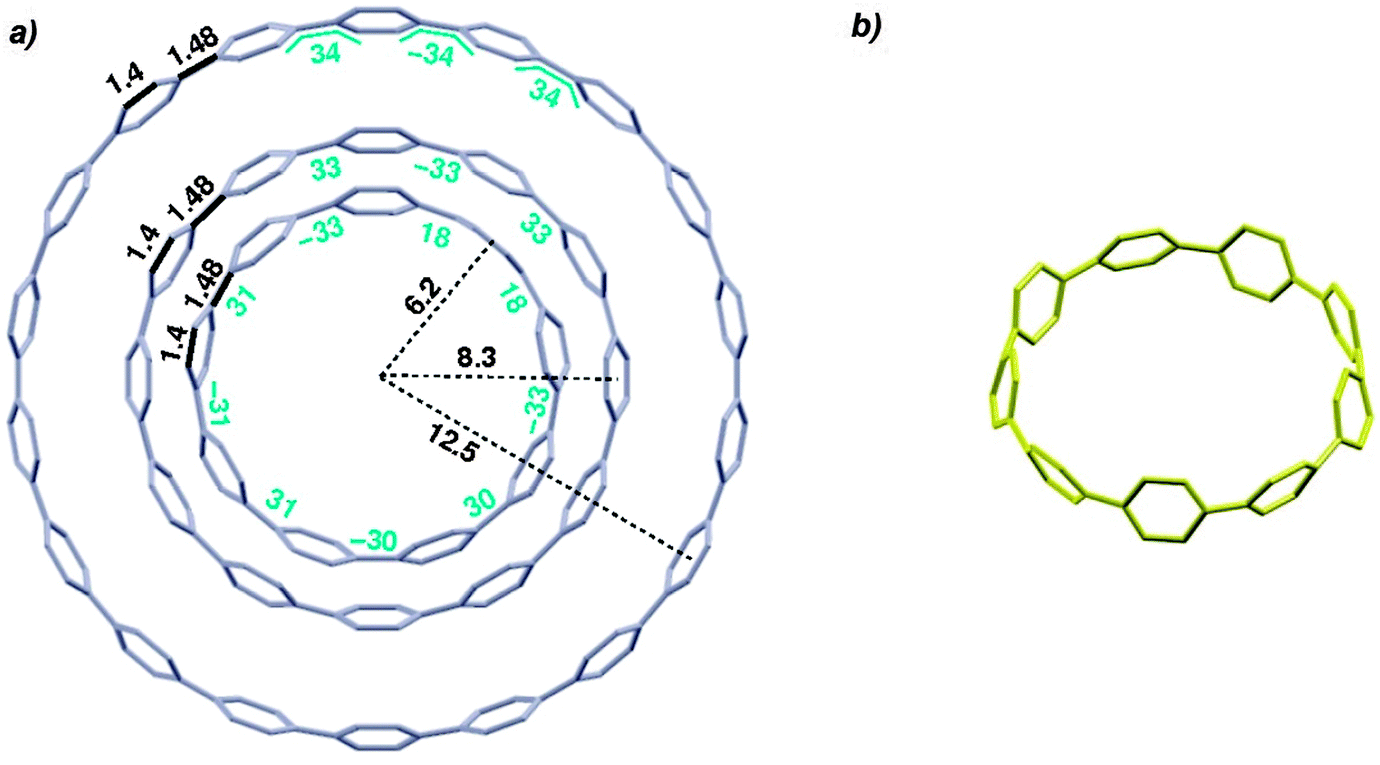
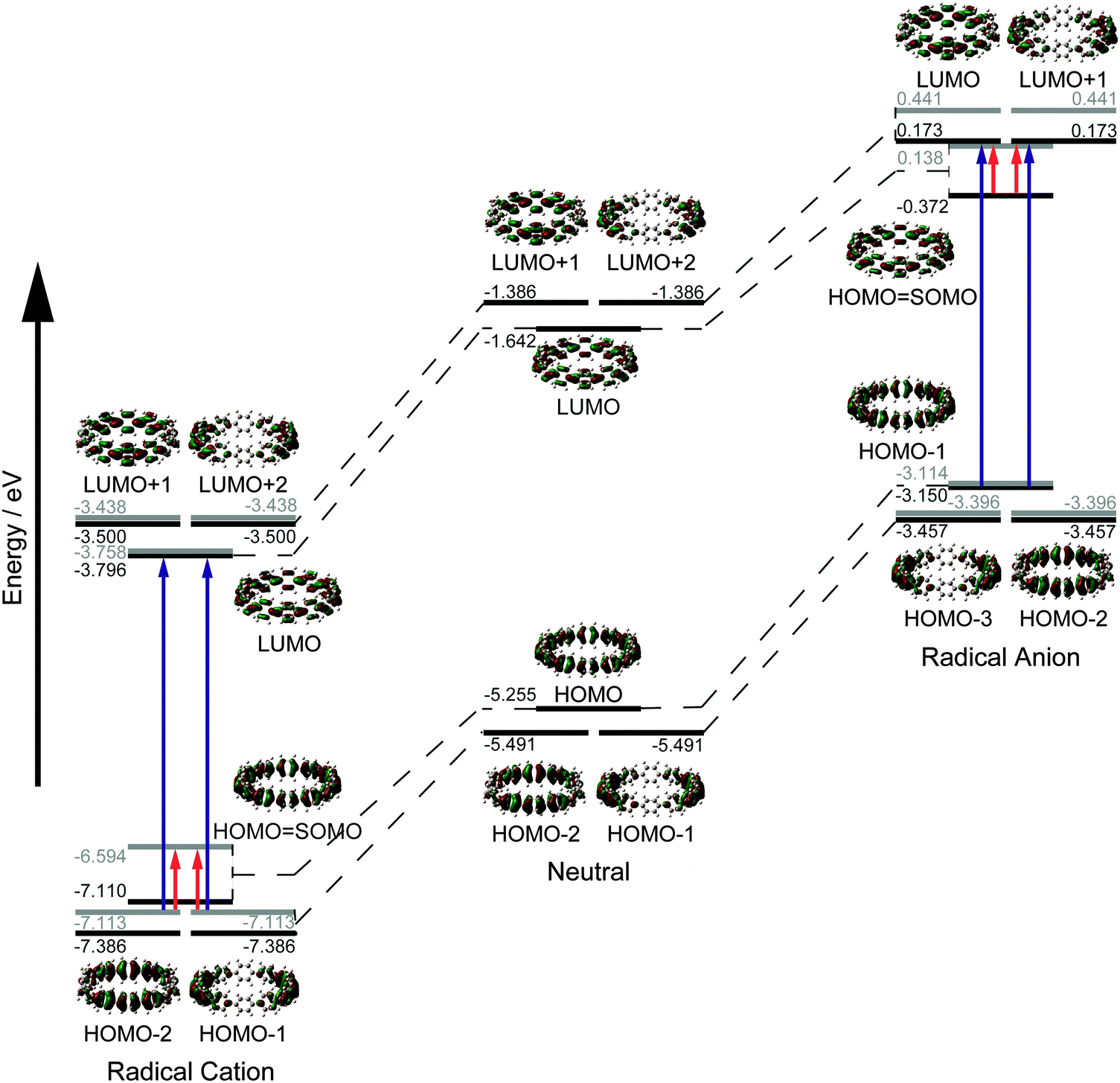
In addition to this synthetic milestone, Bertozzi and co-workers' communication also describes computational studies of the CPPs they had synthesised, as well as characterisation of the CPPs by various methods. Energy minimised structures of the three CPPs were calculated with DFT methods (Fig. 7a). An interesting aspect of CPP geometry is the dihedral angle between the phenyl rings, which was calculated to be non-zero in every case. While at first glance it might be assumed that CPPs would have dihedral angles of zero (and hence [n]CPP would have Cn symmetry), this is in fact not the case – occlusion of the aryl hydrogens disfavours conformers with dihedral angles of zero. Linear oligophenylenes provide a precedent for this, for example biphenyl, which has a dihedral angle between the two phenyl rings of around 45°.16,17 For the even-numbered CPPs (i.e. [12]CPP and [18]CPP), the lowest energy conformation was calculated to be an alternating staggered arrangement of aryl rings. However, such a conformation is not possible for any odd-numbered CPP; in the case of [9]CPP, the energy-minimised structure possesses a range of dihedral angles between 18° and 33° (Fig. 7b). The authors considered the alternative possibilities of a Möbius π system,18 arising from successive clockwise dihedral angles of (360–n) for [n]CPP, but discounted this as they were found to be appreciably higher in energy.
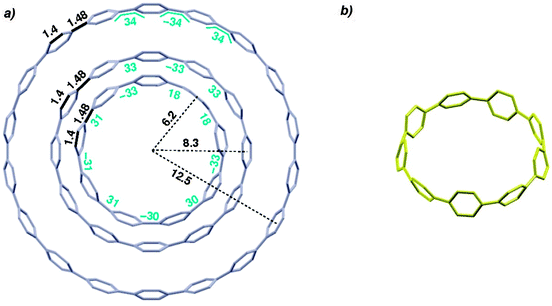 |
| | Fig. 7 (a) Bond lengths, dihedral angles and radii in energy minimised structures of [9]-, [12]- and [18]CPP. All lengths (black) and dihedral angles (cyan) are in angstroms and degrees, respectively. (b) Side-on view of the energy minimised structure of [9]CPP. Reprinted with permission from ref. 15. Copyright 2008 American Chemical Society. | |
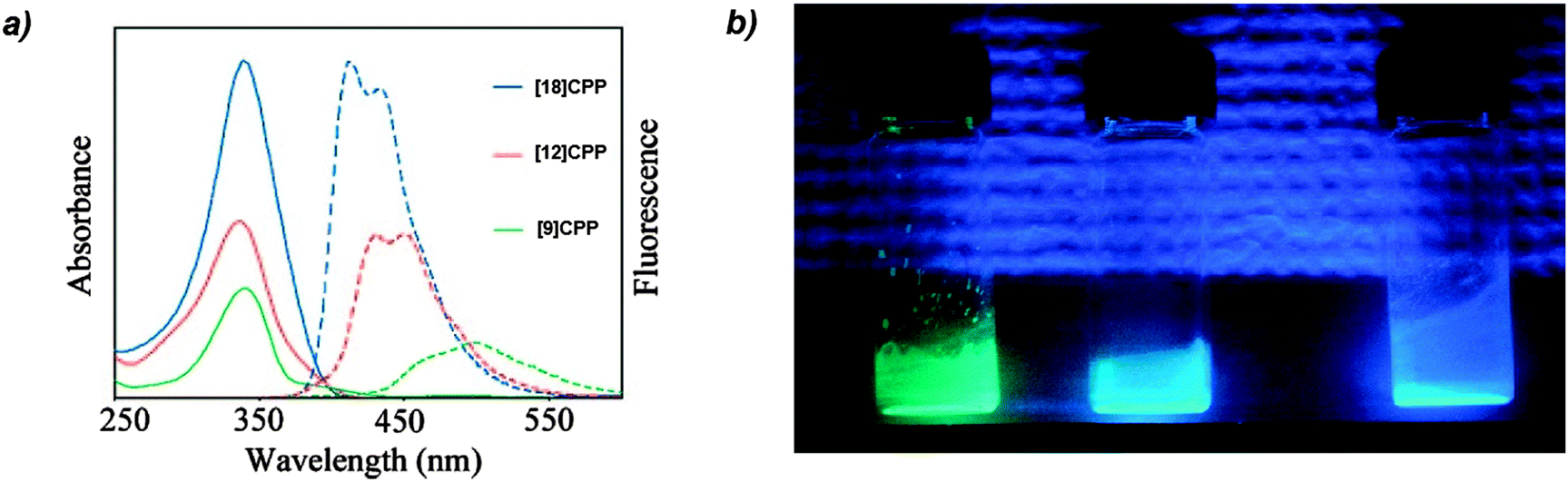
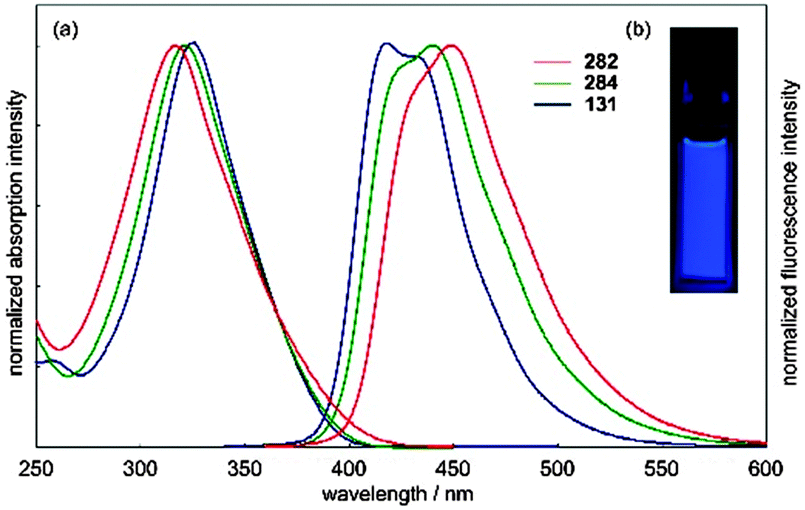
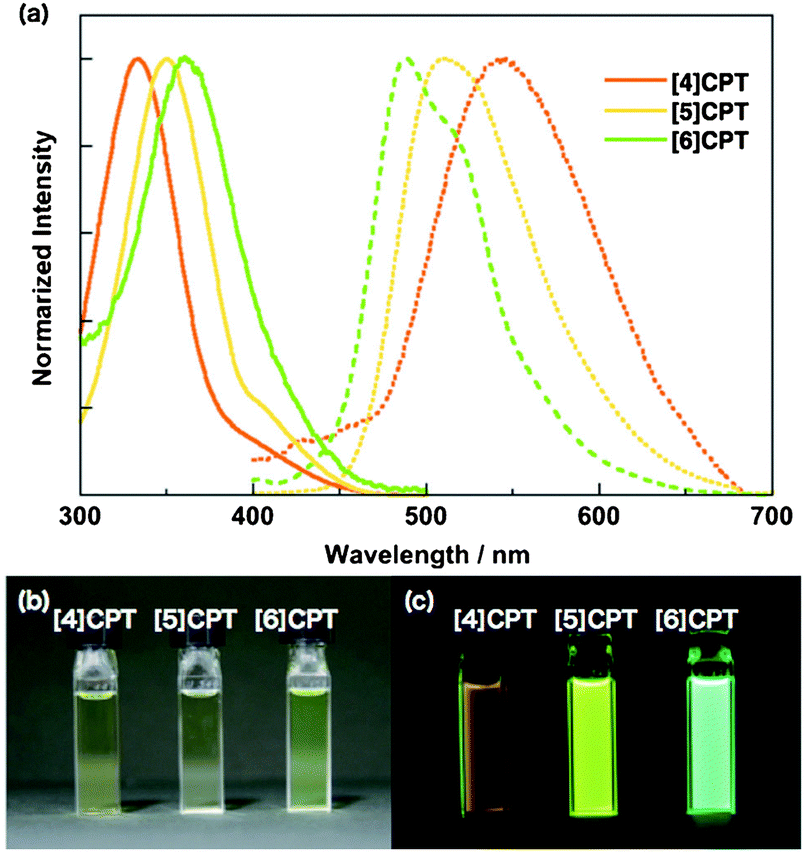
The absorbance and fluorescence spectra of the CPPs were found to have some surprising characteristics (Fig. 8). The absorption maxima were found to be independent of ring size, with λmax ≈ 340 nm in all three cases. This contrasts with the linear oligophenylene case, wherein increasing the number of phenylene units leads to an increase in λmax.19 All three CPPs were observed to be fluorescent, with the Stokes shift increasing as CPP ring size decreased. This result was rationalised through computation, with the large Stokes shifts being attributed to the extent of relaxation in the excited state. Specifically, in smaller rings, the increased ring strain and deviation from planarity allows for a greater reduction in the dihedral angles in the excited state.
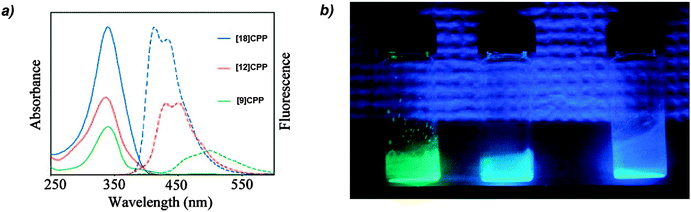 |
| | Fig. 8 (a) Absorbance (solid lines) and fluorescence (dashed lines) spectra of [9]-, [12]- and [18]CPP. (b) CPPs in the solid state, irradiated at 365 nm. Reprinted with permission from ref. 15. Copyright 2008 American Chemical Society. | |
2009
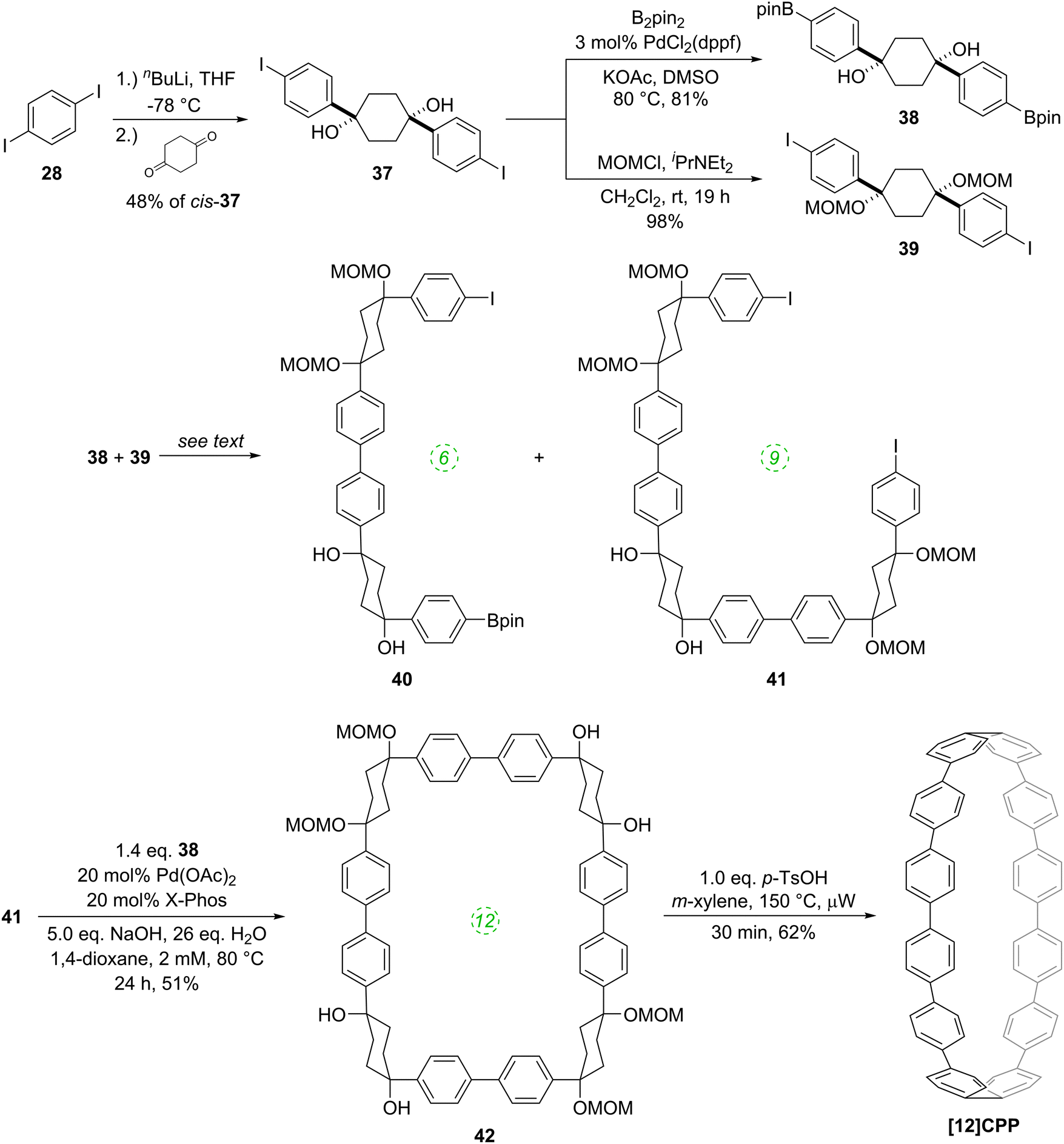
The significance of the Bertozzi group's work was rapidly acknowledged by the community at large, with a review published in early 2009 highlighting the potential applicability of the CPPs as starting materials for the “bottom-up” synthesis of carbon nanotubes.20 Shortly after this, Itami and co-workers published their first contribution (of many) to the field of CPPs, reporting a selective synthesis of [12]CPP.21 Itami's work has parallels with that of Bertozzi, insofar as the CPP is accessed through macrocyclisation of a precursor containing non-aromatic rings, followed by a final aromatisation step. Indeed, Itami's building blocks consist of 1,4-diphenylcyclohexanes, directly comparable to structures such as 22 explored by Vögtle. However, there are also key differences between Itami's work and the previous reports: Itami's modular approach to macrocycle construction permitted the selective formation of [12]CPP, with no other precursors to CPPs of different sizes being formed. Also, whereas Vögtle's attempts at cyclization employed a copper-mediated process, Itami's group exhaustively explored palladium-catalysed processes, eventually identifying conditions which permitted macrocycle formation in good yield. Furthermore, the final aromatisation step is oxidative, not reductive. The synthetic strategy is depicted in Scheme 9.
 |
| | Scheme 9 The synthesis of [12]CPP reported by Itami and co-workers. | |
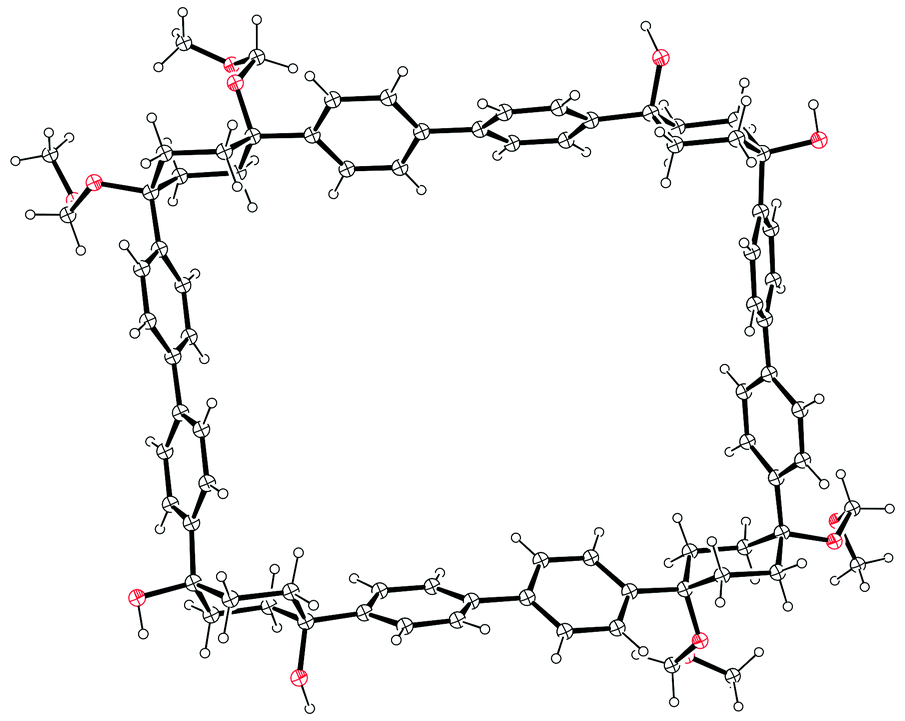
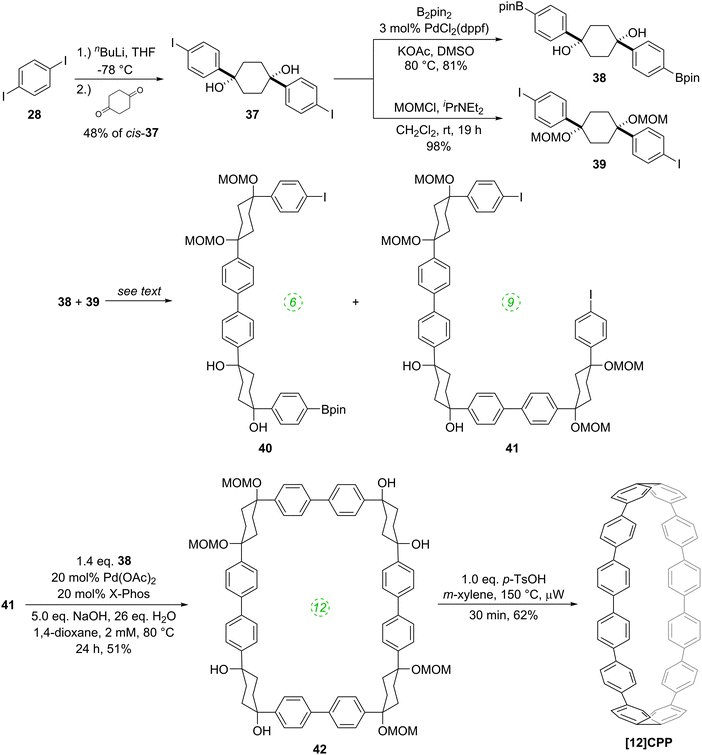
Monolithiation of 1,4-diiodobenzene 28 and addition of a twofold excess of this to cyclohexane-1,4-dione gave cyclohexa-1,4-diol 37, in which formation of the cis isomer was favoured in around a 4:1 ratio. Bis(iodide) 37 represents a point of divergence for the synthesis, with a portion of 37 undergoing Miyaura borylation to give diborylated building block 38, whereas a further portion was protected as the corresponding bis(methoxymethyl ether) 39. Itami specifically notes that this combination of building blocks bearing both free and protected hydroxyl functionalities greatly aided the purification of some of the later intermediates. The first strategy then employed by Itami to effect a union of these two building blocks was to carry out a Suzuki–Miyaura coupling of 38 and 39 in a 1:1 ratio, with the aim of synthesising 6-ring building block 40. In the event, even under optimised conditions using 10 mol% Pd(PtBu3)2 with 5 equivalents of NaOH and water, in toluene at 60 °C for 24 h, the desired 6-ring building block 40 was formed in only 18% – the major product was in fact 9-ring building block 41, formed in 27% yield. The corresponding reaction in 1,4-dioxane gave only oligomers. In view of these results, Itami's team altered their strategy to deliberately target the production of 9-ring building block 41. Thus, after further optimisation, it was found that treating 38 with a ten-fold excess of 39 in the presence of 10 mol% PdCl2(dppf) with 5 equivalents of NaOH and water, in 1,4-dioxane, with [38] = 8 mM, at 60 °C for 24 h, gave the 9-ring building block 41 in 81% yield (and with near-quantitative recovery of unreacted 39). With access to 41 secured, the next challenge was to couple this with a further equivalent of 38 to effect macrocyclisation, i.e. a tandem intermolecular-intramolecular dual Suzuki–Miyaura sequence. Unsurprisingly, this also required extensive optimisation, but it was eventually determined that treating 41 with a slight excess of 38 under the conditions shown in Scheme 9 gave cyclised [12]CPP precursor 42 in 51% yield; use of Buchwald's X-Phos ligand22 was crucial for obtaining this yield. It proved possible to obtain an X-ray crystal structure of 42 (Fig. 9), in which it can be see that the non-linearity of the macrocycle is entirely accommodated in the cyclohexyl rings – the biaryl motifs do not exhibit curvature. Finally, conversion of 42 into [12]CPP was accomplished using p-toluenesulfonic acid (pTSA) in meta-xylene. Microwave irradiation and a temperature of 150 °C were essential to obtain any [12]CPP at all. The mechanism of the final aromatisation step was not entirely clear – the action of the acid is presumably to effect MOM deprotection and elimination of water, but this would transform the cyclohexyl rings into cyclohexadienes – a further oxidative step would then be required to aromatise the ring. Itami himself noted that the identity of the oxidant was unclear.
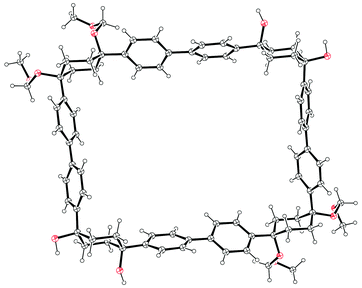 |
| | Fig. 9 ORTEP diagrams of [12]CPP precursor 42, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Molecules of hexane and methanol in the unit cell have been omitted for clarity. | |
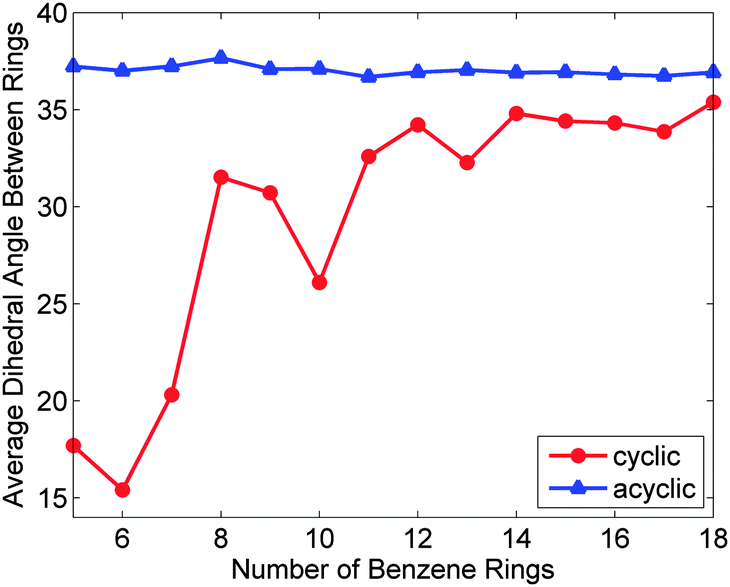
The third publication on CPPs in 2009 was a sole-author paper from Wong, which disclosed a comprehensive computational study.23 Time-dependent density functional theory was employed to model not only the CPPs synthesised by Bertozzi et al. ([9]CPP, [12]CPP and [18]CPP, Fig. 10), but actually every ring size between [5]CPP and [18]CPP. In addition, linear oligo(paraphenylenes) with 5–18 phenyl rings were also modelled for comparison. For many of the computed properties, the values for the CPPs were found to converge with those of their linear analogues with increasing ring size. For example, Fig. 11 shows the calculated average dihedral angles for both classes of molecules with varying numbers of phenyl rings. Whereas in the linear oligophenylene series (blue triangles) the average dihedral angle is essentially invariant with number of repeat units, for the corresponding CPPs (red circles), the average angle be seen to vary markedly for the smaller CPPs, before converging towards the linear value.
 |
| | Fig. 10 Energy minimised structures of [9]-, [12]- and [18]CPP. Reprinted with permission from ref. 23. Copyright 2009 American Chemical Society. | |
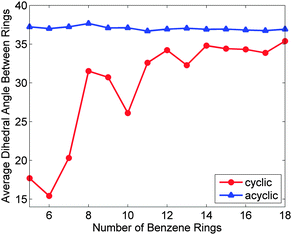 |
| | Fig. 11 Average dihedral angle between adjacent benzene rings as a function of paraphenylene size. As the number of benzene rings increases, the average dihedral angle for the cyclic paraphenylenes asymptotically approaches the acyclic dihedral angle of 37°. Reprinted with permission from ref. 23. Copyright 2009 American Chemical Society. | |
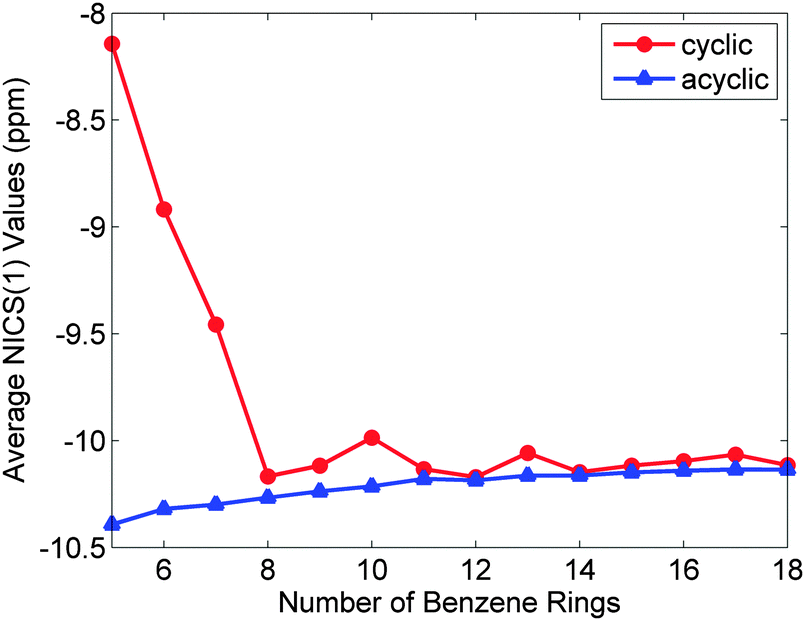
A key aspect of Wong's work is the rationalisation of the seemingly counter-intuitive trends in the optical properties, i.e. the fact that for CPPs, the larger the ring, the larger the lowest excitation energy, which is rather unexpected! In contrast, for the linear series, the greater the number of phenyl rings, the smaller the lowest excitation energy. Wong concluded that the principal differences in the absorption spectra of the linear oligomers and their CPP counterparts are attributable to various contributions to the electron–hole interaction energy. Thus, for CPPs, an electron–hole pair has a larger Coulombic attraction than in the linear case and this decreases more quickly than the quasiparticle band gap upon increasing ring size. The combined effect of these two trends is for the optical absorption gap to increase overall with increasing ring size. Finally, in order to attempt to quantify the aromaticity of the various CPPs, Wong calculated the NICS(1) (nucleus-independent chemical shift) values, a parameter that quantifies the π-orbital aromaticity, with more negative values indicating a greater degree of aromaticity, whereas more positive values denote more quinoidal character. (For reference, at the same level of theory, the calculated value for benzene is −11.5 ppm.) As shown in Fig. 12, for the larger CPPs, the calculated values are similar to those for the linear oligophenylenes, but for smaller CPPs (n < 8), aromaticity diminishes in a pronounced fashion as a function of diminishing ring size.
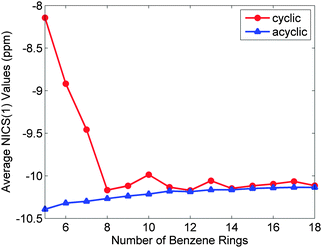 |
| | Fig. 12 Average NICS(1) values of phenyl rings in the cyclic and acyclic paraphenylenes as a function of size. More negative NICS(1) values correspond to an enhanced aromatic character of the system. All NICS(1) calculations were obtained at the PBE0/6-31G(d,p) level of theory. Reprinted with permission from ref. 23. Copyright 2009 American Chemical Society. | |
2010
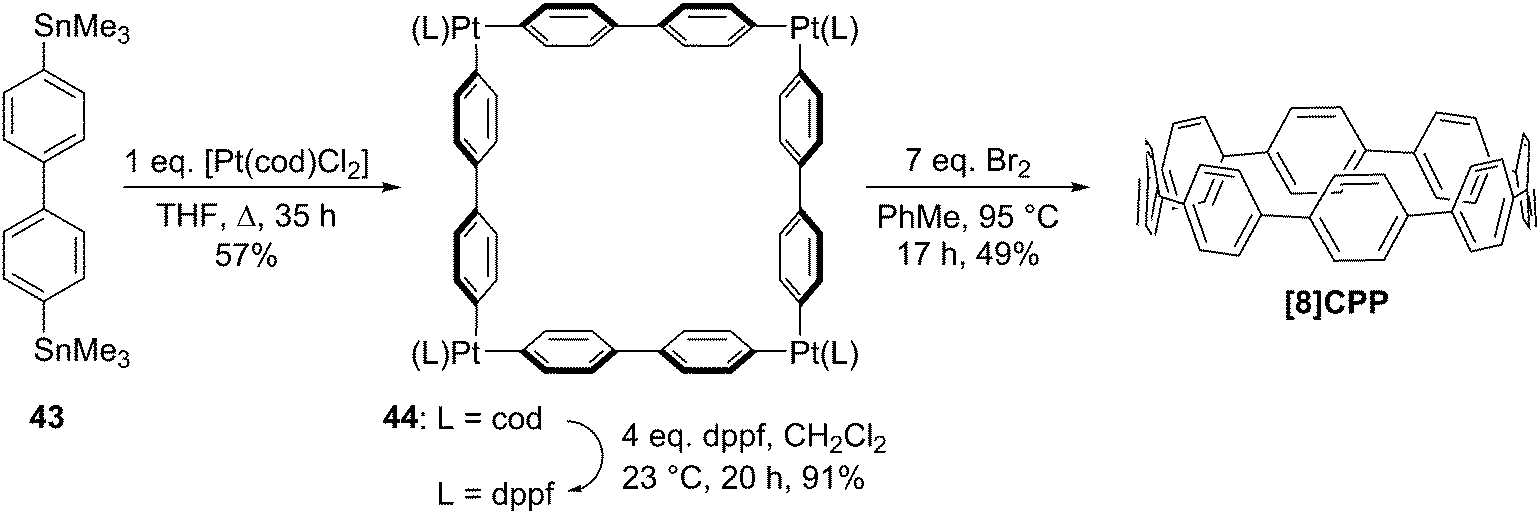
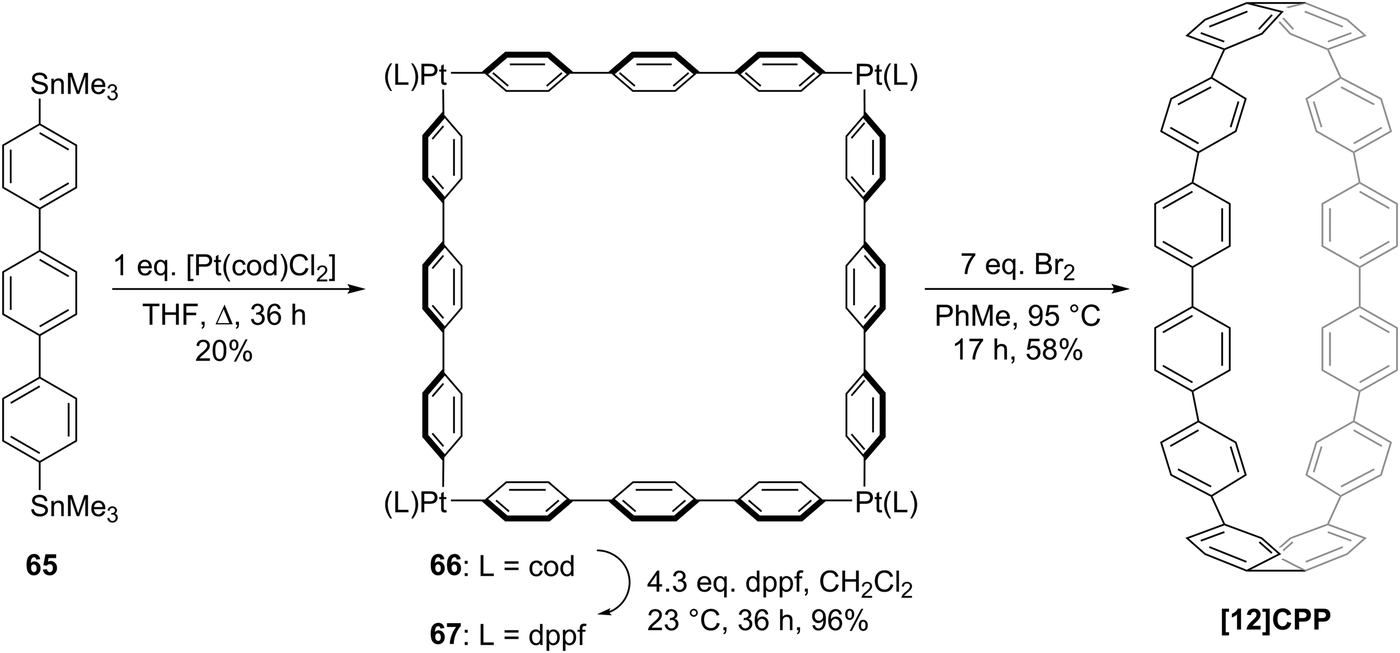
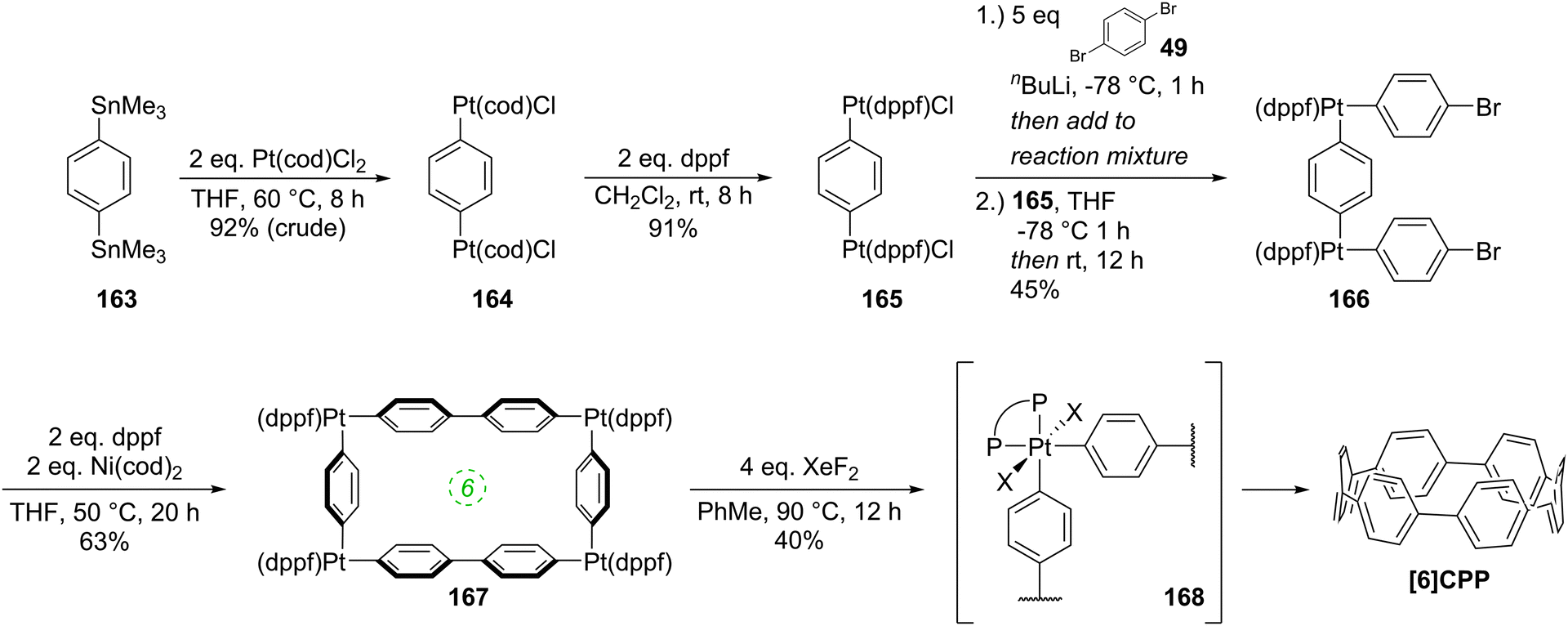
The following year began with a report from Yamago and co-workers of a third, conceptually original synthetic route to CPPs.24 The strategy relies on the formation of a tetraplatinum precursor to a CPP, which is essentially strain-free due to the square-planar geometry of Pt(II). Treatment with an oxidant then induces four-fold reductive elimination to the CPP. The synthesis is depicted in Scheme 10. A 4,4′-bis(stannylated) biphenyl precursor 43 was transmetallated with an equimolar amount of a Pt(II) source to give macrocycle 44 (L = cod). After a straightforward ligand exchange to give 44 (L = dppf), treatment with bromine as oxidant led presumably to formation of transient Pt(IV) species; these which are known to undergo reductive elimination forming either Ar–Ar bonds or Ar–Br bonds preferentially, depending on the choice of ligand.25 In this instance, four-fold Ar–Ar bond formation gave [8]CPP, at the time the smallest CPP to be reported.
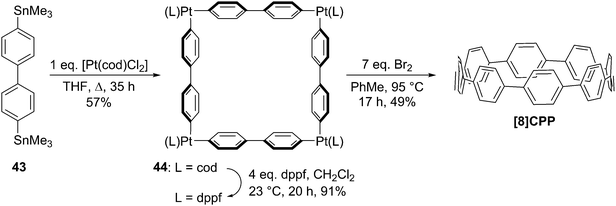 |
| | Scheme 10 The synthesis of [8]CPP reported by Yamago and co-workers. | |
While the use of a super-stoichiometric quantity of Pt has cost implications for scale-up, Yamago's route nevertheless is exceedingly concise and high yielding, giving [8]CPP in just 3 steps from 42 and 25% overall yield. Just as for Bertozzi and Itami, Yamago's approach to CPPs is also informed by the early work of Vögtle. In this case the idea of taking a precursor in which all rings are already aromatic and excising the linking atoms to access a CPP had already been explored by Vögtle (albeit unsuccessfully), using sulfur in the place of platinum, as shown in Scheme 3. Upon characterisation of [8]CPP, the Yamago group observed a continuation of the trend first reported by Bertozzi, that of smaller CPPs having larger Stokes shifts – approximately 200 nm in the case of [8]CPP.
The next paper on CPPs to be published specifically addressed the issue of the large Stokes shift. Sundholm and co-workers used time-dependent density functional theory, to model CPPs from [6]CPP to [11]CPP.26 They calculated the absorption and emission spectra using the BP86 GGA functional and in the case of [6]CPP, also using the B3LYP functional (which gave appreciably different results). The calculated absorption and emission maxima are not in agreement with those observed experimentally by Bertozzi and Jasti, and the authors provide a discussion of the possible reasons for this.
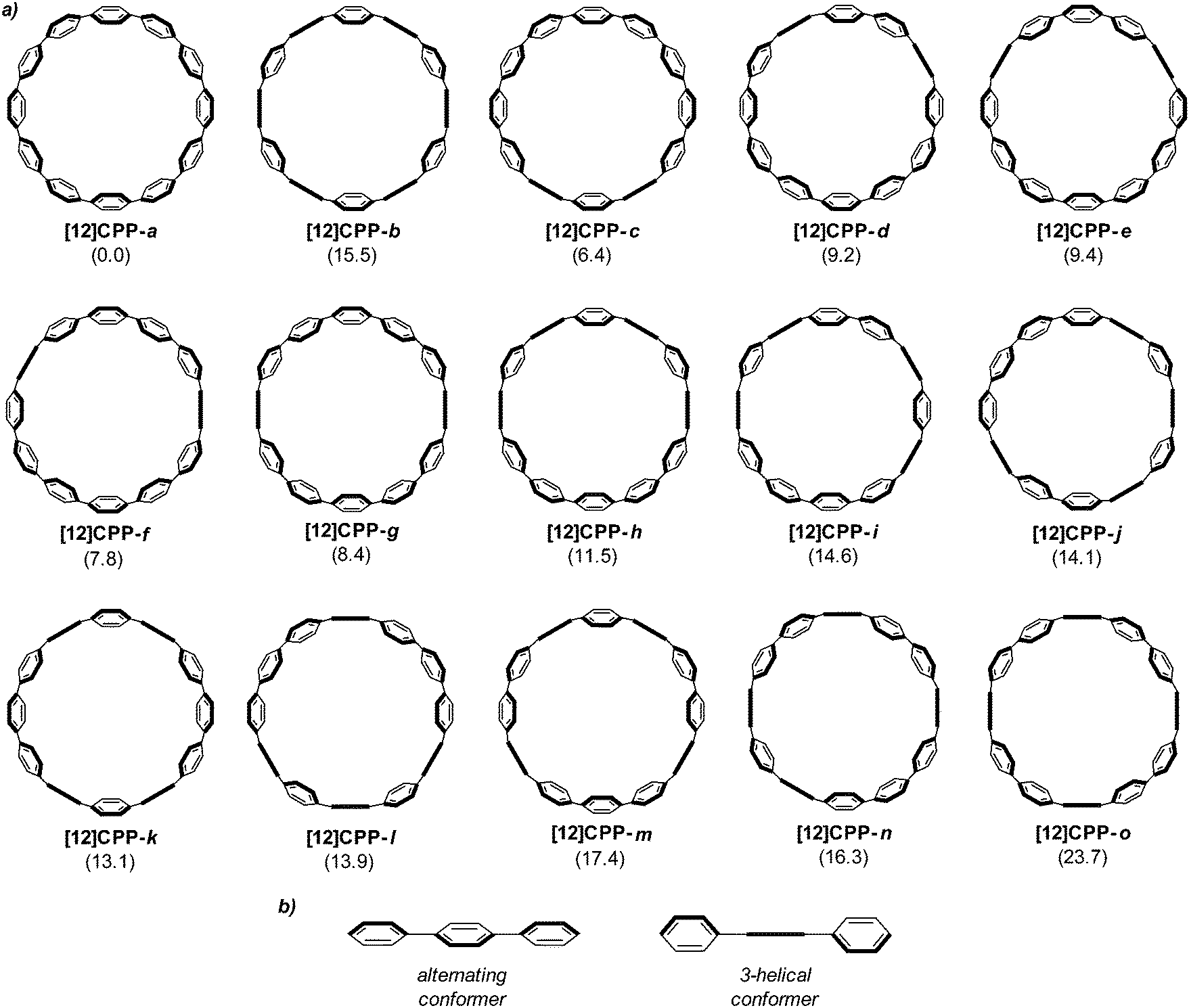
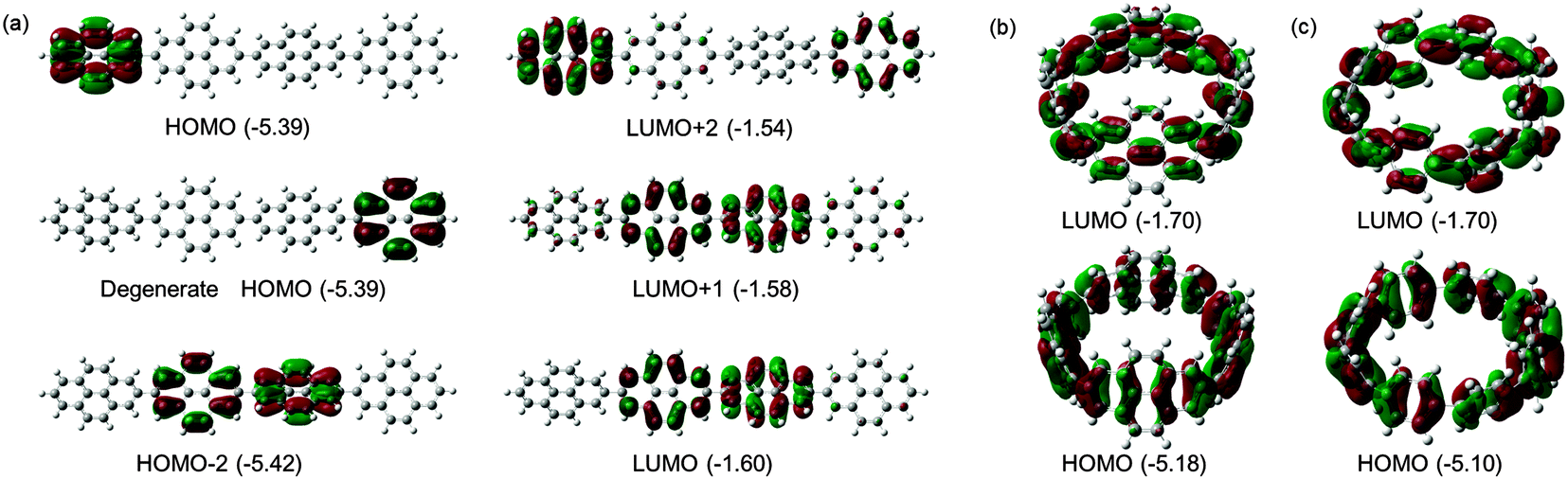
Computational studies on CPPs in fact outnumbered experimental studies in 2010, and the next one to be published, from Itami and co-workers, addressed the question of the strain energies of CPPs, as well as the related cycloparaphenyleneacetylenes (CPPAs).27 Itami noted that the three experimental studies reported at that point had all included computed strain energies for CPPs, which were different in all three reports. As such, further computational studies were merited and the Itami group began by carrying out a conformational search for [12]CPP (B3LYP/6-31G(d) level of theory). They identified 15 local minima for [12]CPP, which are shown in Fig. 13a. The conformer of lowest energy, [12]CPP-a (top left of Fig. 13a) is of D6d symmetry and consists of alternately twisted phenyl rings (dihedral angle ≈ 33°). Another conformer of high symmetry, [12]CPP-b, has D3d symmetry. These two conformers are cyclic extensions of two known stable conformations of p-terphenyl, the “alternating” and the “3-helical” conformers (Fig. 13b). The other minima identified for [12]CPP (c–o, Fig. 13a) are all combinations of these two p-terphenyl conformers. The calculated HOMO and LUMO for [12]CPP-a are shown in Fig. 14; the quinoidal nature of the LUMO may be clearly seen.
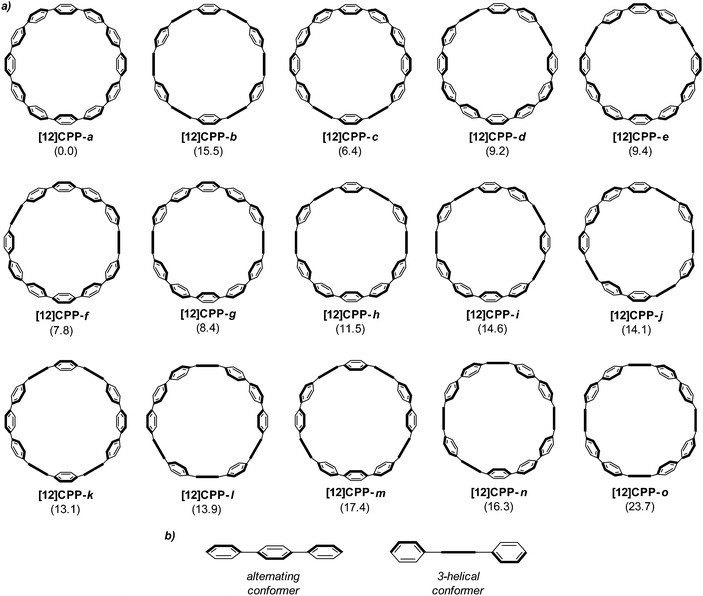 |
| | Fig. 13 (a) Conformations of [12]CPP (ΔG in KJ mol−1 relative to [12]CPP-a) and (b) two conformations of p-terphenyl. Reprinted with permission from ref. 27. Copyright 2010 American Chemical Society. | |
 |
| | Fig. 14 HOMO (bottom) and LUMO (top) of [12]CPP-a. Reprinted with permission from ref. 27. Copyright 2010 American Chemical Society. | |
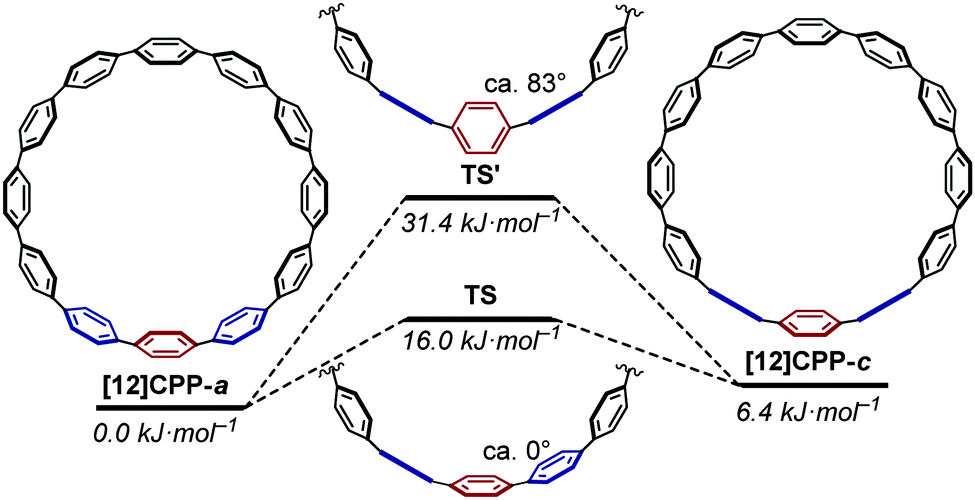
The barrier to interconversion between [12]CPP-a and [12]CPP-c (involving the rotation of a single phenyl ring) was studied. It was determined that interconversion via a transition state (TS) with the three relevant phenyl rings near-coplanar was lower in energy than the alternative transition state (TS') with the three phenyl rings near-perpendicular (Fig. 15).
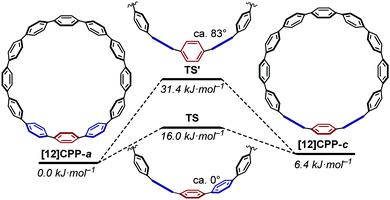 |
| | Fig. 15 Rotation of a phenyl ring through TS or TS′ (ΔG, based on [12]CPP-a). Reprinted with permission from ref. 27. Copyright 2010 American Chemical Society. | |
Itami et al. further studied CPPs of different sizes (n = 6 to 20) determining that for even-numbered CPPs (n = 6, 8, 10, etc.) the most stable conformers were all-alternating ones (analogous with [12]CPP-a), whereas for odd-numbered CPPs (n = 7, 9, 11, etc.) the most stable conformers were the ones with one three-helical motif in every instance. Finally, they also undertook a comparison of CPPs with CPPAs (cycloparaphenyleneacetylenes)28,29 – nanohoops consisting of alternating acetylene and p-phenylene units. They found that for any given diameter of nanohoop, a CPP was of higher energy than a comparably-sized CPPA.
The potential for CPPs and derivatives to be used as starting materials for the de novo synthesis of carbon nanotubes was highlighted by a second review in 2010, published by Jasti30 (the first author on the initial Bertozzi group paper on CPPs). This particular review was focused specifically on carbon nanotubes with discrete chirality. Very soon after, a publication from Taubert, Pichierri et al. followed, concerning magnetically induced currents in CPPs (n = 6 to 11).31 Based on computation, the authors predict that [6]CPP (with 4n π electrons) has a slight antiaromatic character overall, while [7]CPP (with 4n + 2π electrons) is aromatic, albeit only weakly, having a ring current susceptibility strength only about ¼ that of benzene. The authors also modelled lithium and magnesium complexes of CPPs, finding that Li2[6]CPP (a CPP dianion having 4n + 2π electrons) has a net ring current strength of 28 nA T−1 (being 2.4 times that of benzene). The possibility to compare these computational data to experimental data has not yet arisen, although a metal complex of a CPP polyanion has subsequently been reported (vide infra, Fig. 57).
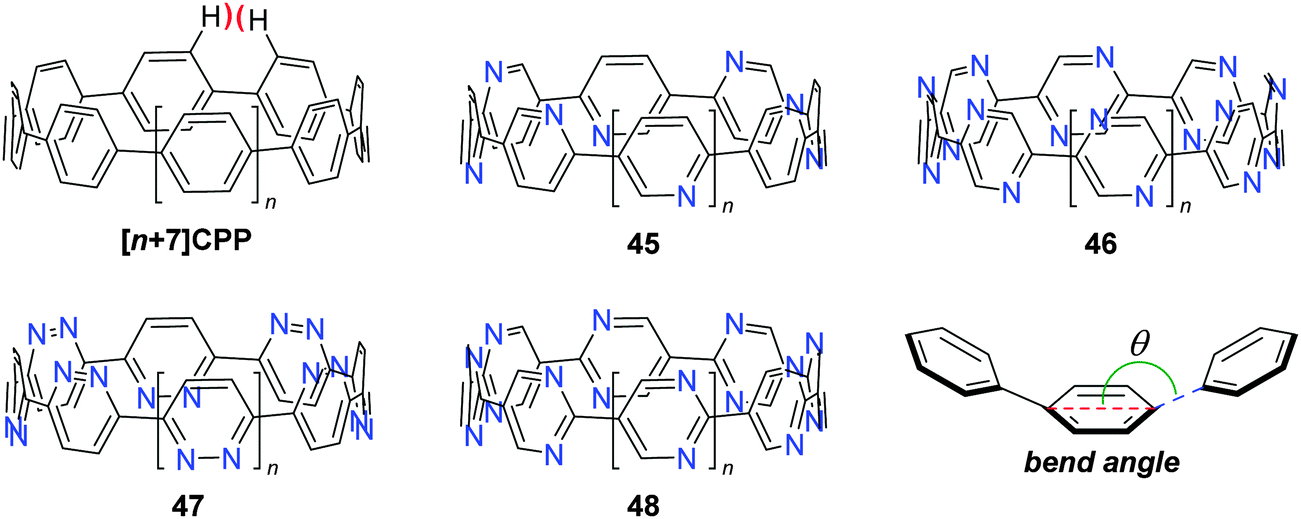
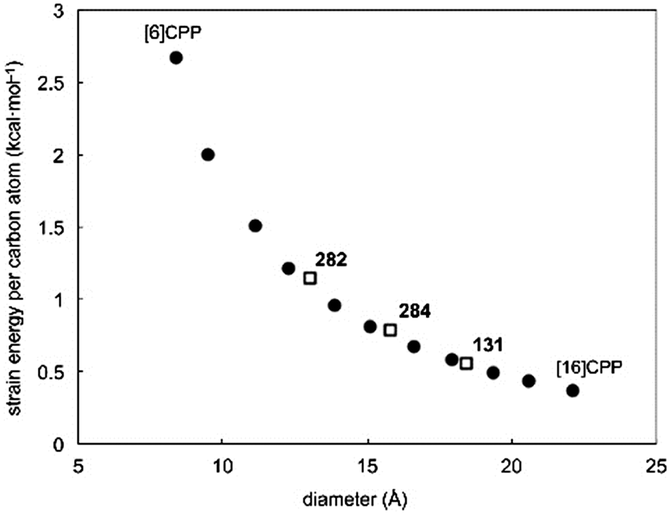
The fourth (and final) computational study disclosed in 2010 was carried out by the Bachrach group.32 This work compared the CPPs to various hypothetical aza-analogues, as shown in Fig. 16. In the first instance, CPPs themselves were modelled and results in good agreement with Itami's report27 were obtained. Bachrach considered the non-zero dihedral angles in the CPPs, arising from unfavourable interaction of the ortho,ortho′ hydrogens, and proposed analogues 45–48, in which such interactions should be diminished or removed entirely. Replacement of aryl hydrogens with nitrogen lone pairs ought to allow the dihedral angles to approach zero, giving an idealised “ribbon-like” structure. In the event, computations do indeed predict 46 and 48 (the pyrazinyl and pyrimidinyl analogues) to have dihedral angles <2°, as these structures are the ones entirely lacking in ortho,ortho′ hydrogen interactions. Another significant aspect of the work relates the overall strain energy of a CPP/aza-CPP to the bend angle of the aromatic ring (θ, Fig. 16), finding the two correlate extremely closely for any given series. Crucially, the more aromatic the individual repeating unit in the nanohoop, the greater the increase in strain energy upon deviation from a bend angle of 180°. Pyrimidine, having a lower aromatic stabilisation energy, is predicted to form nanohoops of appreciably less strain energy than the corresponding parent CPPs of the same size. Thus, of the various structures studied, Bachrach suggests 48 as a particularly appealing (and likely tractable) synthetic target. However, no syntheses of 45–48 have been reported to date.
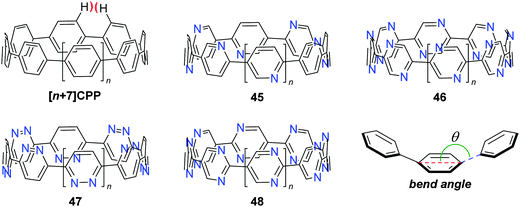 |
| | Fig. 16 Aza–CPPs modelled by the Bachrach group. | |
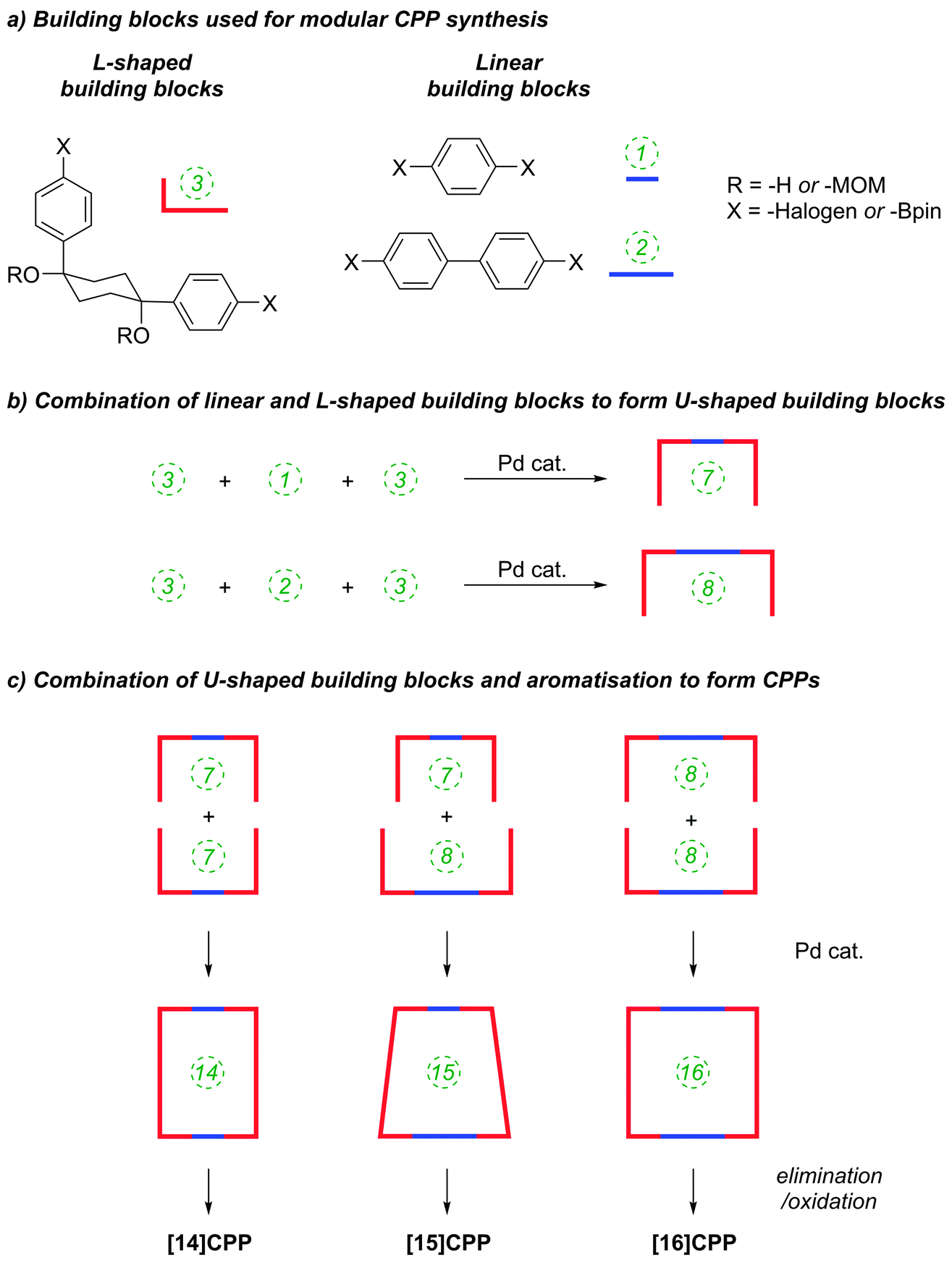
The Itami group published a second paper on CPP synthesis towards the end of 2010, describing selective syntheses of [14]CPP, [15]CPP and [16]CPP.33 The work is an expansion of the methodology described in their preceding paper21 (Scheme 9). Previously, Itami had accessed [12]CPP by combining four “L-shaped” building blocks each comprising three rings, where the 90° bend was accommodated in the cyclohexyl ring. This subsequent report described modular, selective syntheses of these larger CPPs by using the same kind of 1,4-diphenylcyclohexane L-shaped building blocks, in conjunction with “linear” building blocks. The same key reactions, when employed with different combinations of building blocks, gave rise to the different CPPs. As previously, all reactions to link the building blocks together (and to effect the macrocyclisation) were effected by means of Suzuki–Miyaura cross-coupling reactions. The strategy is outlined in schematic form in Scheme 11; the specific reactions used to reduce this strategy to practice are shown in Scheme 12.
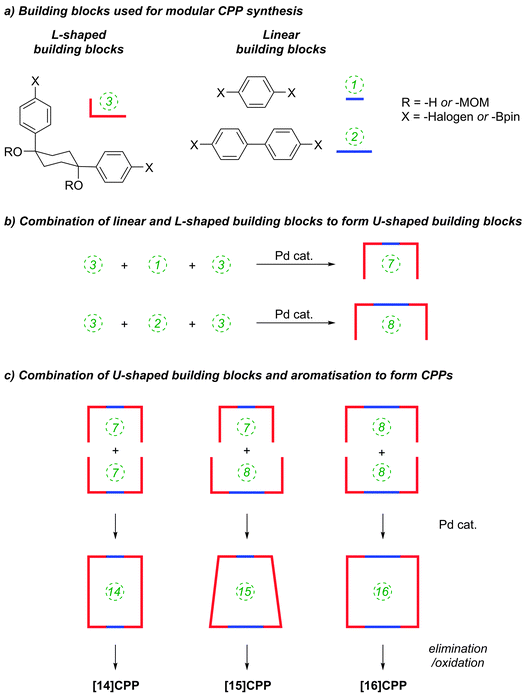 |
| | Scheme 11 Schematic representation of the modular strategy employed by Itami and co-workers. | |
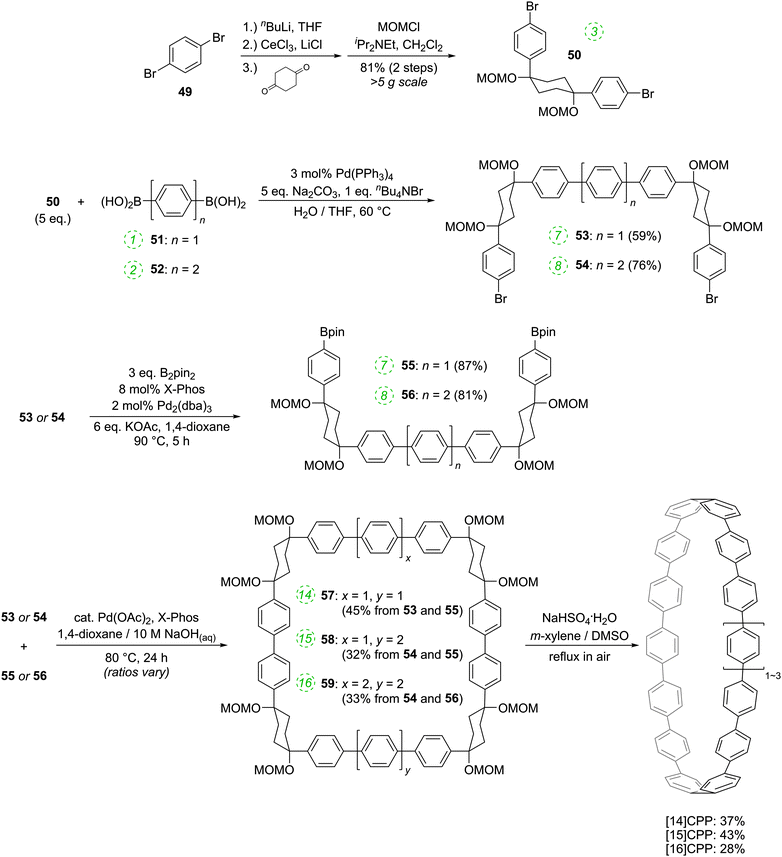 |
| | Scheme 12 Itami's route to [14]CPP, [15]CPP and [16]CPP. | |
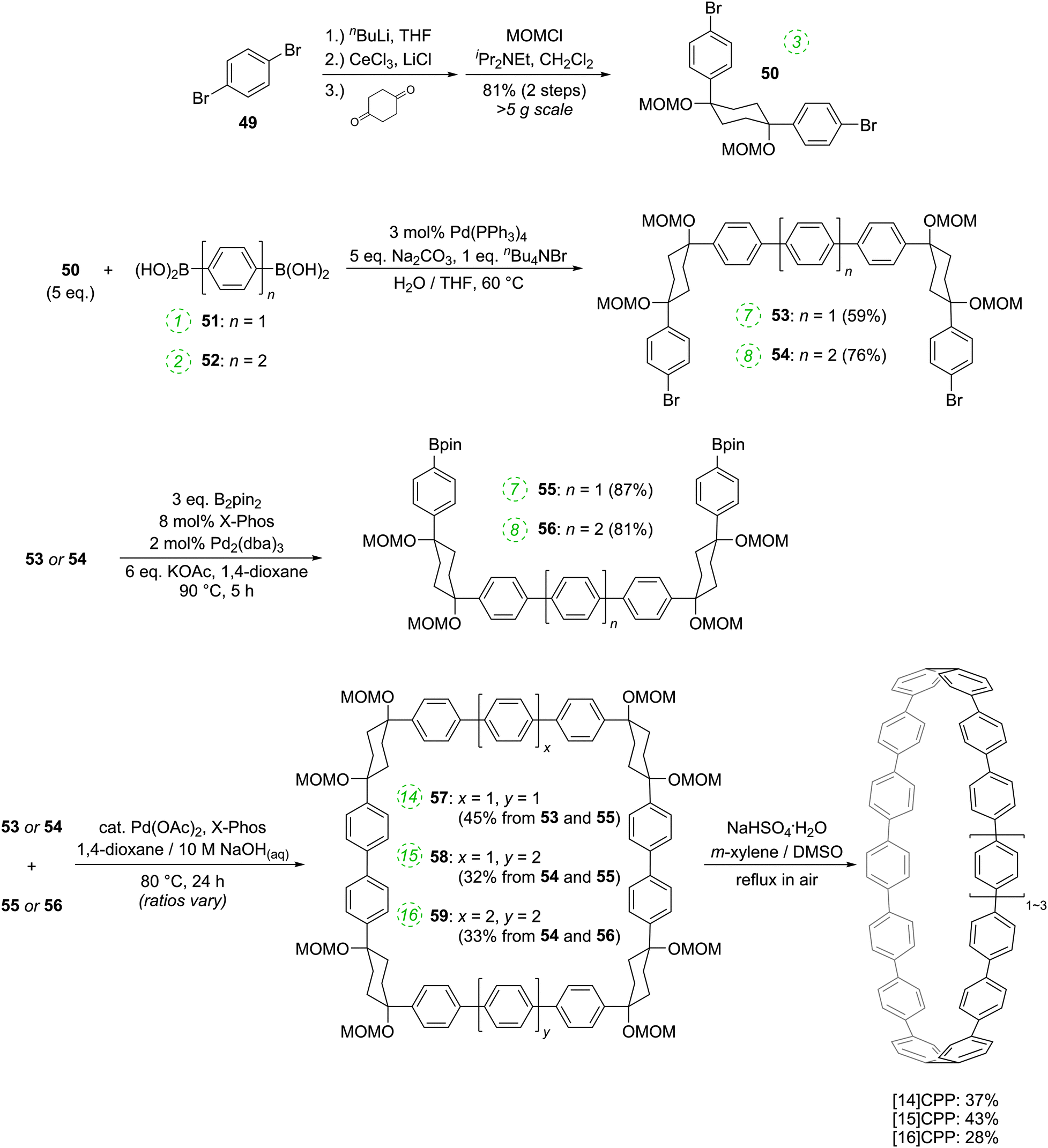
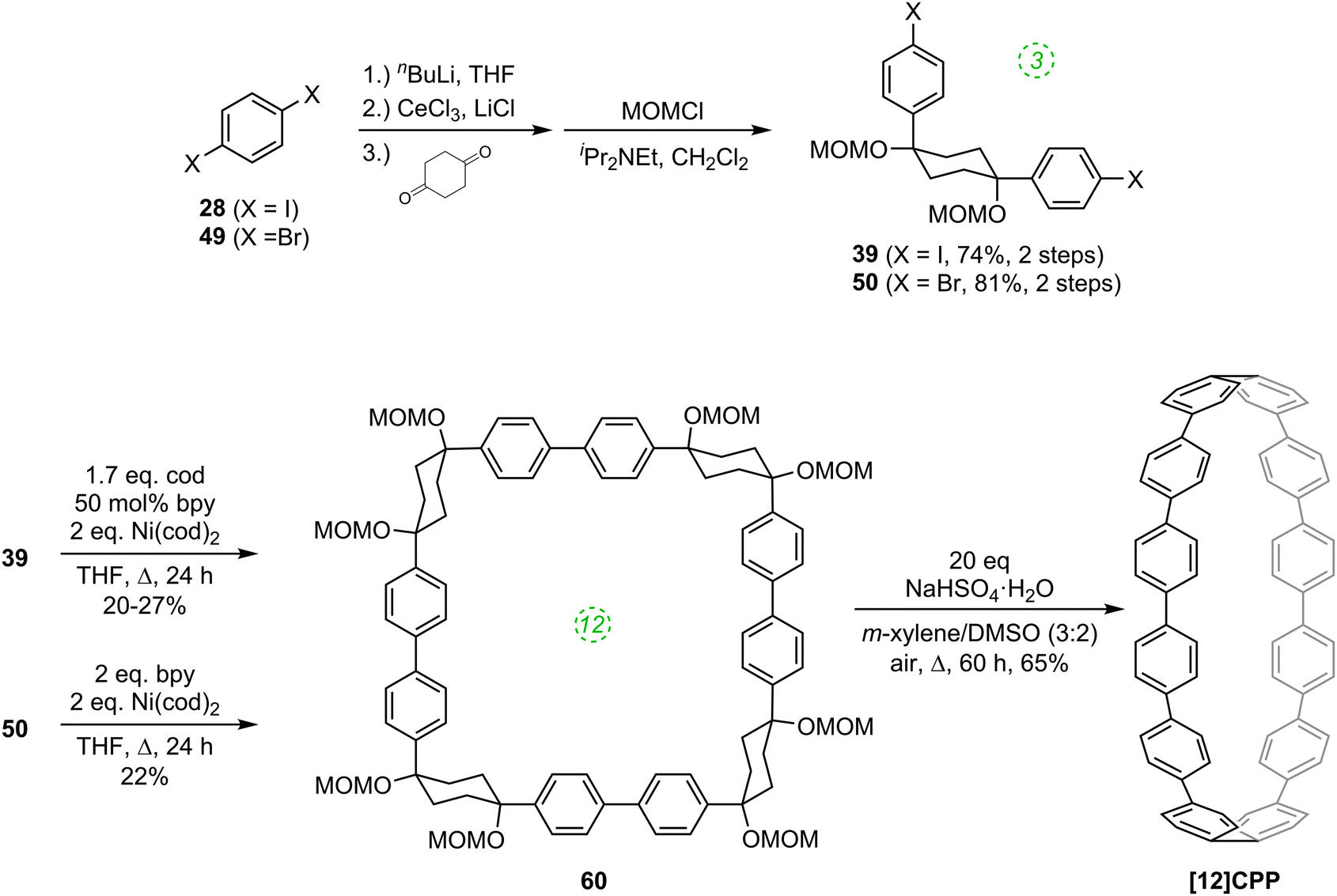
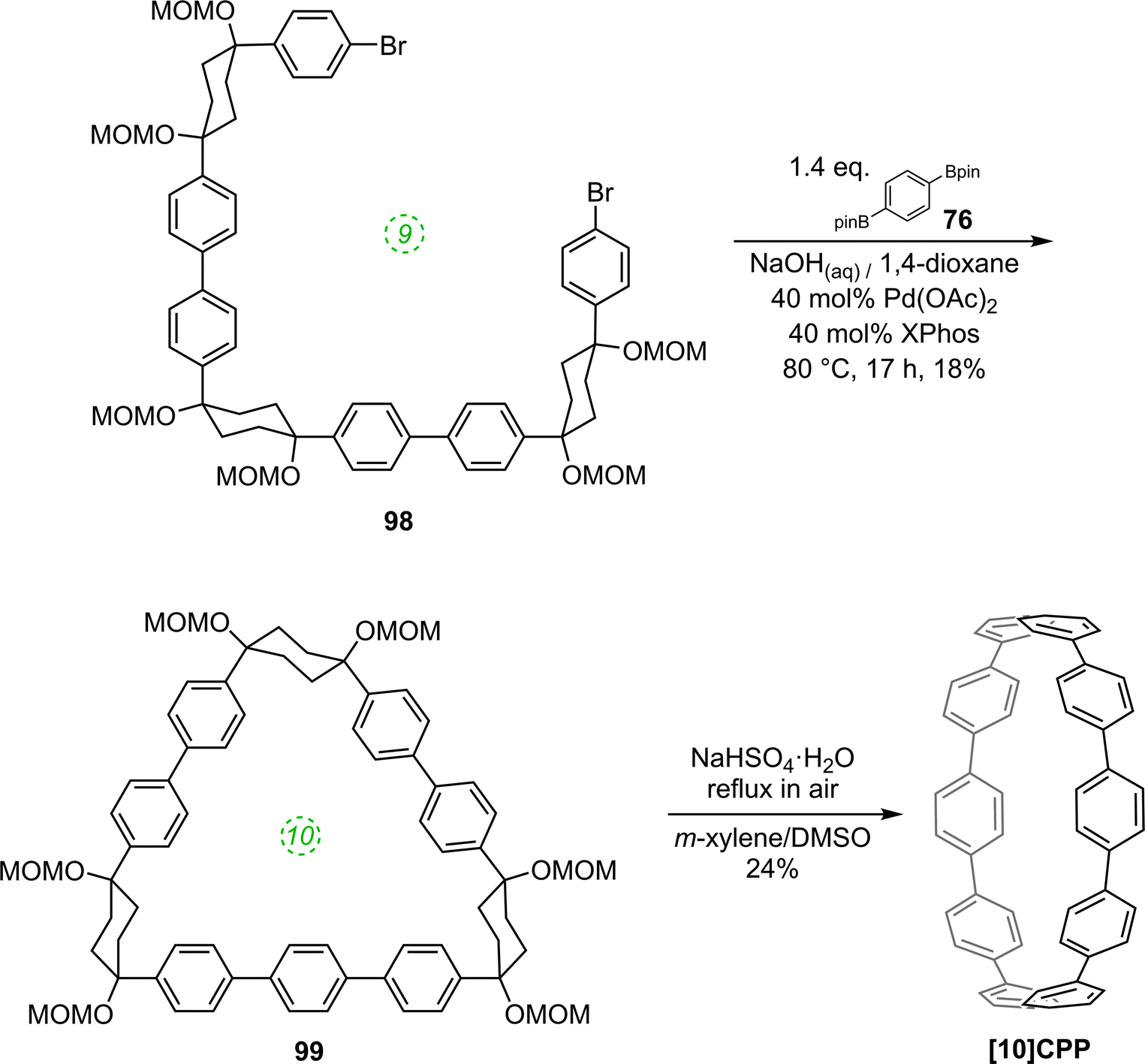
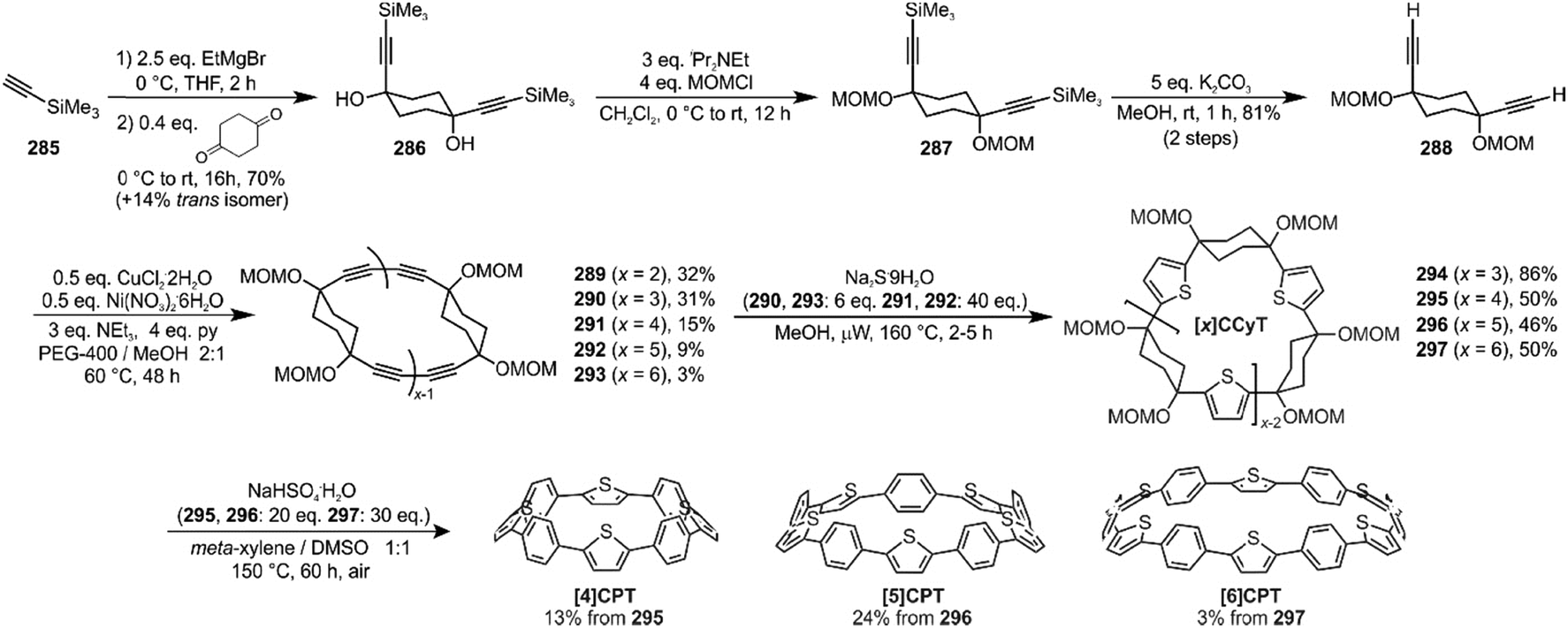
The route begins with the synthesis of dibromo 3-ring building block 50, in a manner analogous to the diiodo analogue 39 (Scheme 9); the optimised route to 50 has allowed its synthesis on a multigram scale. L-shaped building block 50 was then combined with bis(boronic acids) – either with 1-ring linear building block 51 or with 2-ring linear building block 52. Use of a five-fold excess of the dibromide ensured that the U-shaped building blocks 53 and 54 (comprising 7 or 8 rings, respectively) were formed in acceptable yield, still bearing two unreacted aryl bromides for further functionalisation. Miyaura borylation of 53 or 54 gave the corresponding complementary bis(boryl) building blocks 55 or 56, respectively. Finally, combination of a dibromide (53 or 54) with a bis(boryl) building block (55 or 56) under conditions of high dilution (2 mM dibromide) gave the macrocyclic CPP precursors 57–59. Theoretically, the [15]CPP precursor 58 could be formed either by combination of 54 and 55 or by combination of 53 and 56. However, Itami reports only the coupling of 54 and 55 – no comment is made on the viability or otherwise of a union of 53 and 56. The final aromatisation step required some optimisation – the conditions reported for the analogous aromatisation to give [12]CPP (Scheme 9, 42 → [12]CPP) were not applicable here. Thus, it was determined that use of conventional heating as opposed to microwave irradiation, with DMSO co-solvent added to the meta-xylene and a different acid catalyst (NaHSO4·H2O) was able to effect the desired eight-fold MOM deprotection/dehydration/oxidative aromatisation sequence to the final CPPs. It is explicitly stated that this transformation is carried out in air, which strongly suggests the oxidant required for the final step is in fact molecular oxygen.
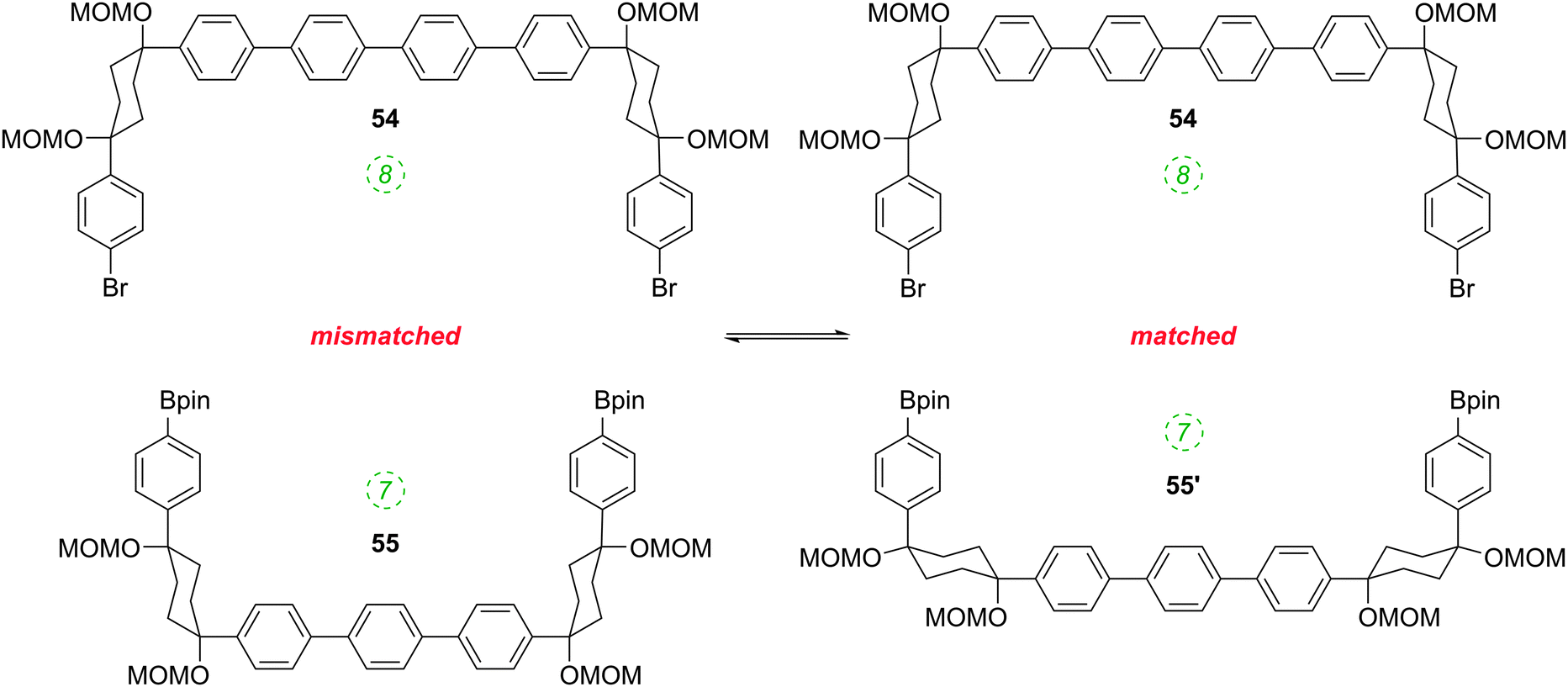
The formation of the 14-ring precursor 57 and the 16-ring precursor 59 both involve the union of 2 fragments of the same size. In contrast, formation of the 15-ring precursor 58 involves two fragments which at first glance are mismatched in terms of size (Scheme 11c, middle column). Nevertheless, 58 is formed in comparable yield to 57 and 59 and Itami attributes this to ring flipping in the smaller U-shaped building block (Scheme 13). Computational modelling supports this proposal and it seems that the change in building block “width” upon interconversions of the two dual ring-flip conformers (55 and 55′) is similar to the dimensions of an additional phenyl ring. Crucially, this allows for the targeted synthesis of odd-numbered CPPs.
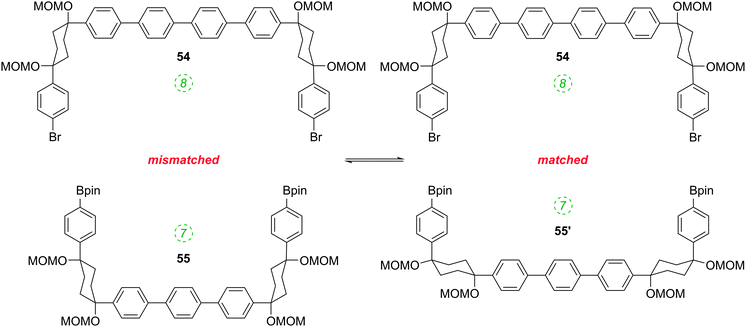 |
| | Scheme 13 Ring flipping allows for macrocyclisation to odd-numbered CPP precursors. | |
2011
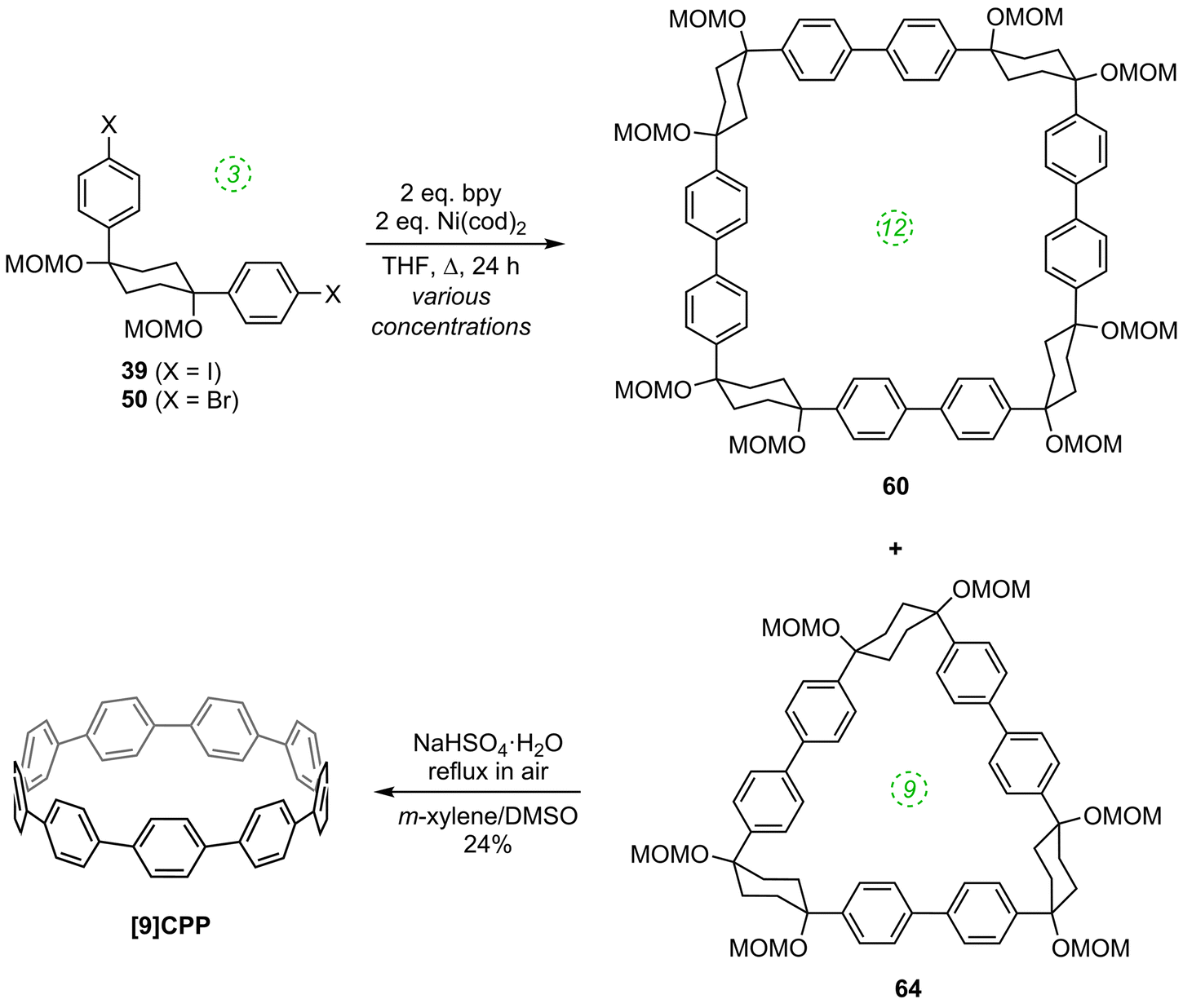
The first publication on CPPs in 2011 was again from the Itami group, who revisited the synthesis of [12]CPP, this time using a nickel-mediated macrocyclisation approach.34 The new synthesis was far more concise and high-yielding, and is shown in Scheme 14. L-shaped building blocks 39 or 50 were prepared as previously (on a 50 g scale), but rather than derivatise these to give a nucleophilic component for a cross-coupling (e.g. a boronic acid), they were instead directly cyclised to 12-ring macrocyclic CPP precursor 60, using super-stoichiometric quantities of nickel(0). Both this approach and the previous report from Bertozzi are so-called “shotgun” cyclisations, potentially capable of producing many cyclic oligomers of different sizes. However, whereas Bertozzi's approach yielded macrocycles with 9, 12 and 18 rings, Itami's Ni-mediated reaction was more selective. In this current paper, only the formation of [12]CPP precursor 60 was reported (although see also Scheme 16), in impressive yield given the number of individual couplings required for its formation. The reaction conditions required extensive optimisation and optimal conditions were found to be slightly different for the two different precursors 39 or 50. A total of 5 g of 60 was prepared; its transformation into [12]CPP (under the conditions established in Itami's preceding report) occurred in good yield and 0.5 g of [12]CPP was prepared. In terms of overall yield (11–13%), number of steps and cost of reagents, this new route to [12]CPP offered significant advantages over the previous ones.
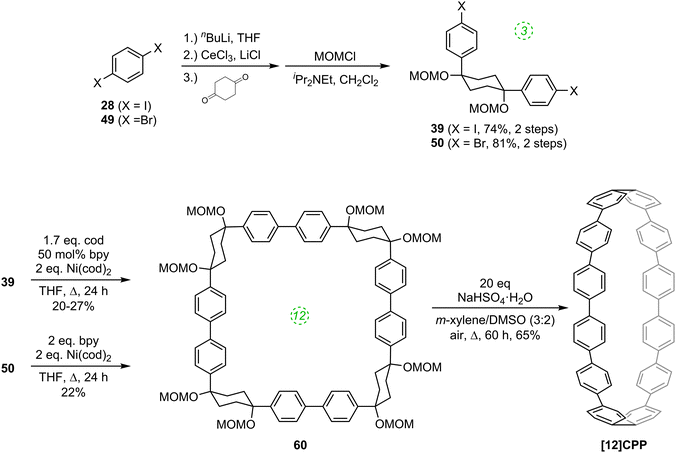 |
| | Scheme 14 Itami's nickel-mediated synthesis of [12]CPP. | |
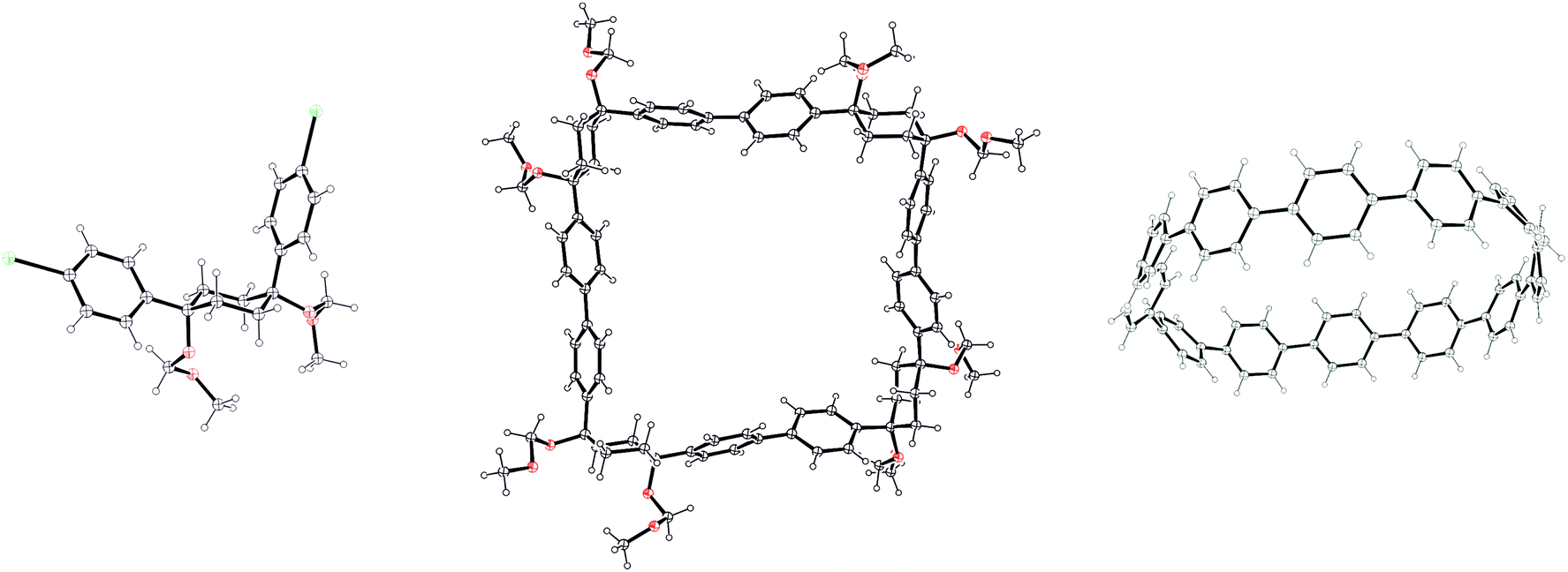
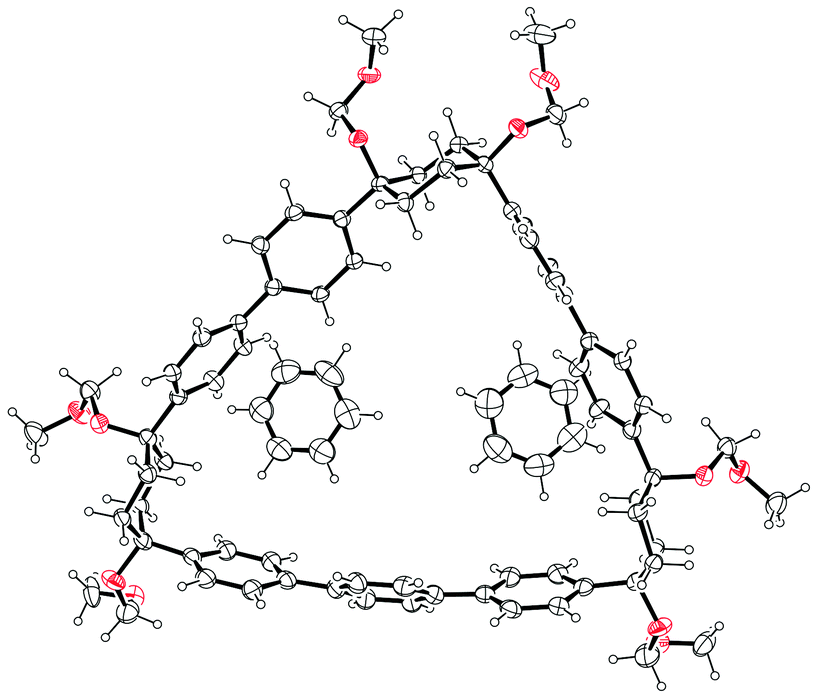
Itami's report also included X-ray crystal structures for 39, 60 and [12]CPP (the first time a structure had been obtained for a CPP). These are shown in Fig. 17. A possible reason for the selectivity observed in the cyclisation of 39 and 50 is hinted at by the X-ray structure for 39. It can be seen that the molecule is indeed L-shaped (in the solid state at least), with an “included” angle of ≈80° between the two aryl substituents on the cyclohexane chair. Assuming such a conformer predominates in solution, this angle is sufficiently close to the ideal value of 90° to favour formation of a tetramer. The X-ray structure of macrocycle 60 is surprising, insofar as it shows a “square” arrangement of its twelve constituent rings, whereas the analogue of 60 that lacks some of the MOM protecting groups, 42, adopted a “rectangular” conformation in the solid state (Fig. 9). In fact, slow interconversion of 60 on the NMR timescale between its square and rectangular conformers permitted the barrier to this interconversion to be calculated by a variable-temperature NMR study. Finally, the solid state structure of 60 is not the D6d conformer [12]CPP-a (Fig. 13) that was previously calculated to be the lowest energy conformer in the gas phase. Instead, the structure of 60 was closest to the D3d symmetry of conformer [12]CPP-b; this is attributed to crystal packing forces.
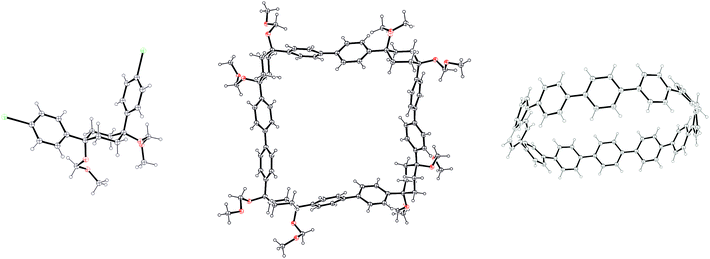 |
| | Fig. 17 ORTEP diagrams 39, 60 and [12]CPP, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Disordered atoms and solvent have been omitted for clarity. | |
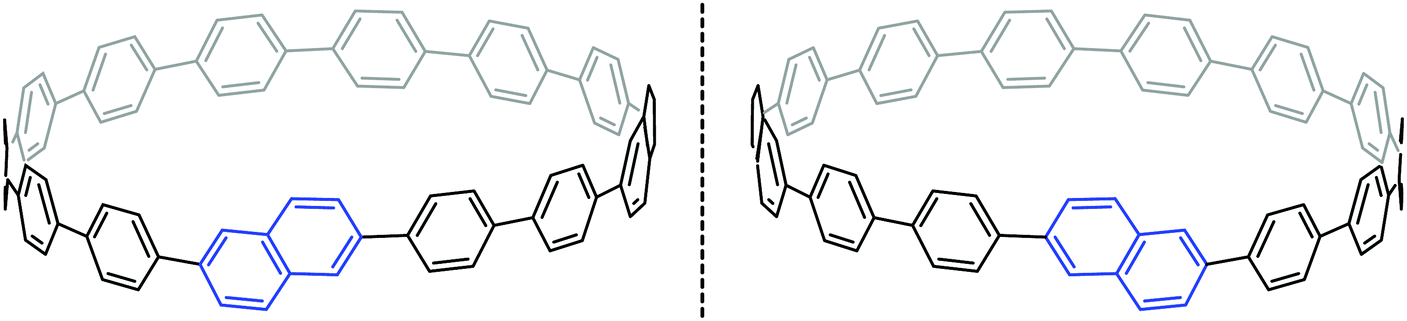
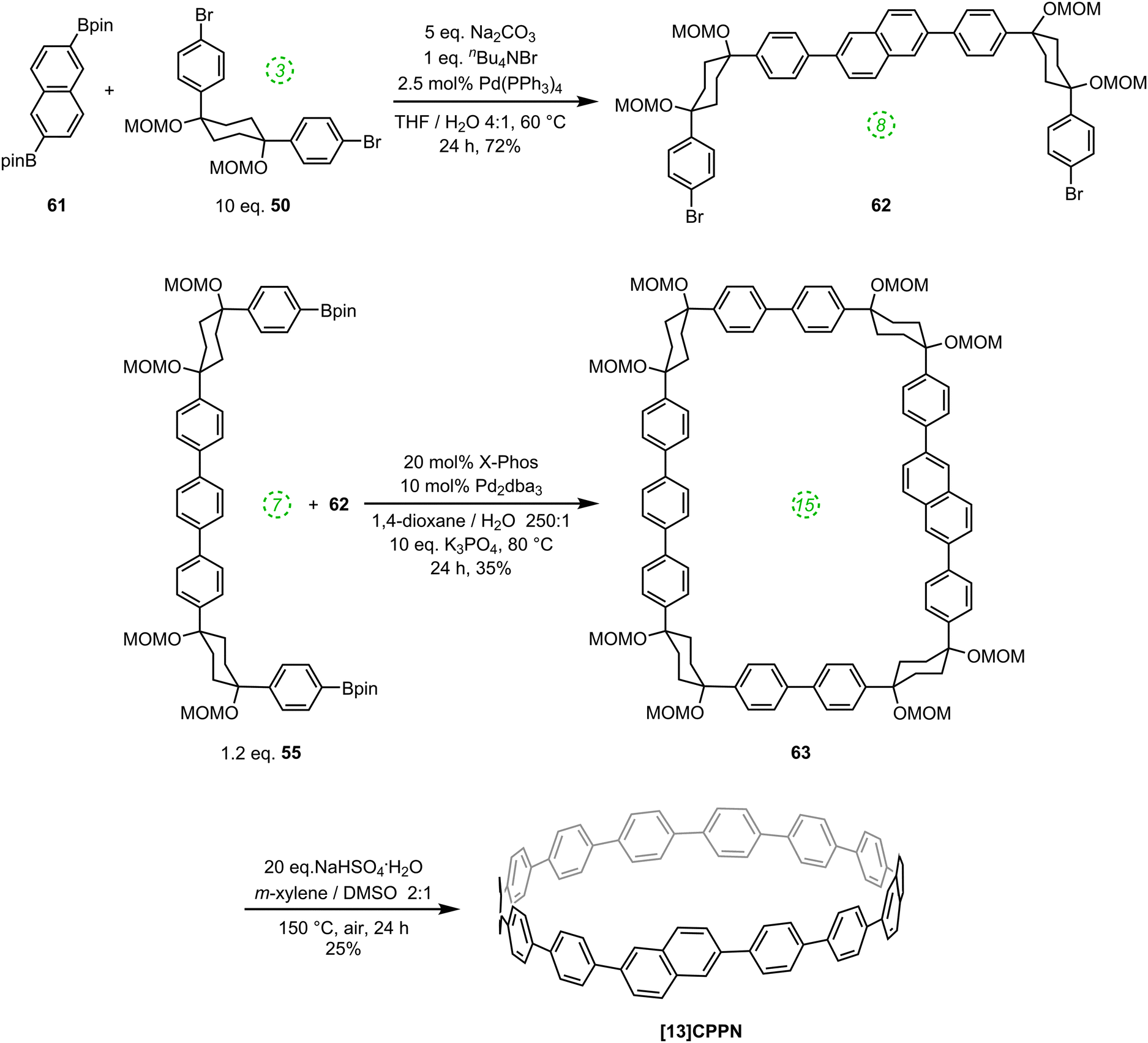
Another paper then followed from the Itami group in quick succession. This work described the first synthesis of a chiral CPP derivative using a variant of the methodology described above.35 A 2,6-naphthalenediyl motif was incorporated into a CPP structure, to give cyclo[13]paraphenylene-2,6-naphthylene, abbreviated [13]CPPN. (Note that the “13” refers to the number of phenylene units, not the total number of rings in the nanohoop.) The chirality arises not from the inclusion of a tetrahedral atom in the structure; rather, the molecule exhibits helical chirality (Fig. 18), with racemisation occurring by the rotation of the naphthyl unit around the two C–C bonds that connect it to the adjacent phenyl rings.
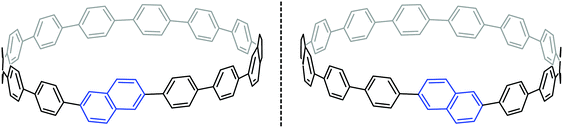 |
| | Fig. 18 Enantiomers of [13]CPPN. | |
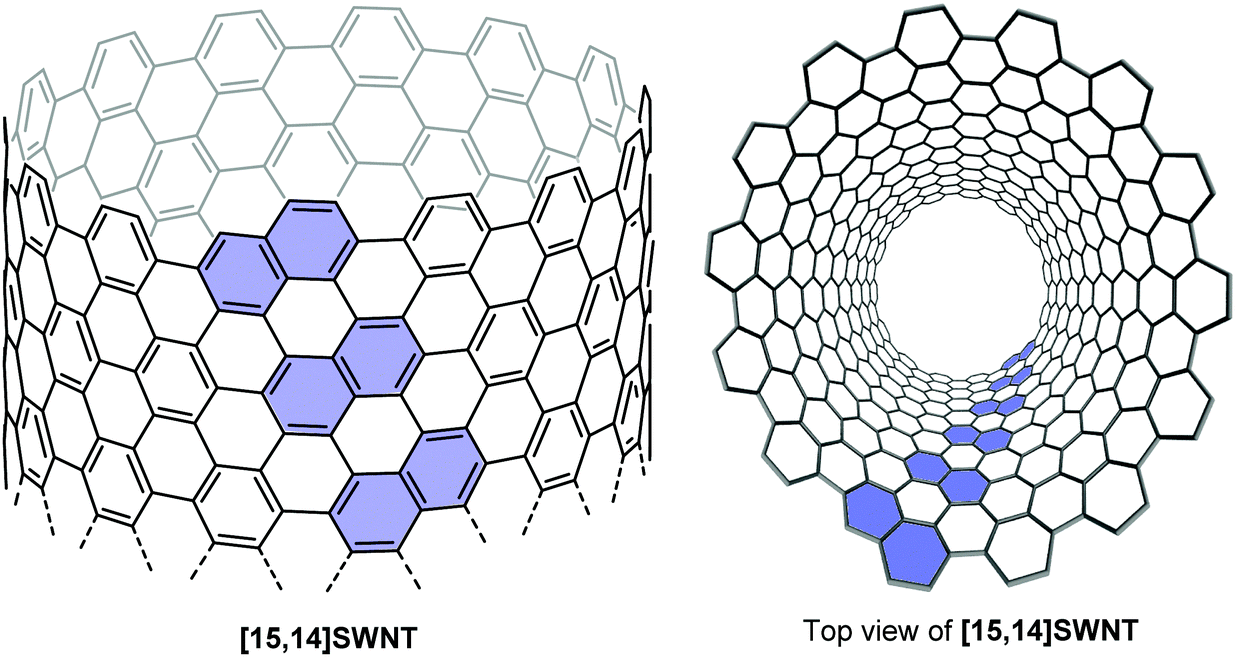
Such helically chiral nanorings could potentially be used as templates for the bottom-up synthesis of chiral carbon nanotubes. In this specific case, [13]CPPN can be envisaged as a template for a [15,14]SWCNT (single-walled carbon nanotube), as shown in Fig. 19. The synthesis of [13]CPPN is shown in Scheme 15. L-shaped building block 50 was combined with bifunctional naphthalene 61 to give U-shaped building block 62. This in turn was combined with the previously synthesised 55 to give macrocycle 63. The final deprotection/dehydration/aromatisation sequence was a little lower-yielding than previously, but nevertheless furnished the desired [13]CPPN.
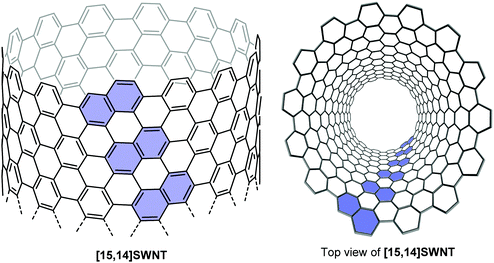 |
| | Fig. 19 [13]CPPN as a repeating unit for a chiral nanotube. Reprinted with permission from ref. 35. Copyright 2011 American Chemical Society. | |
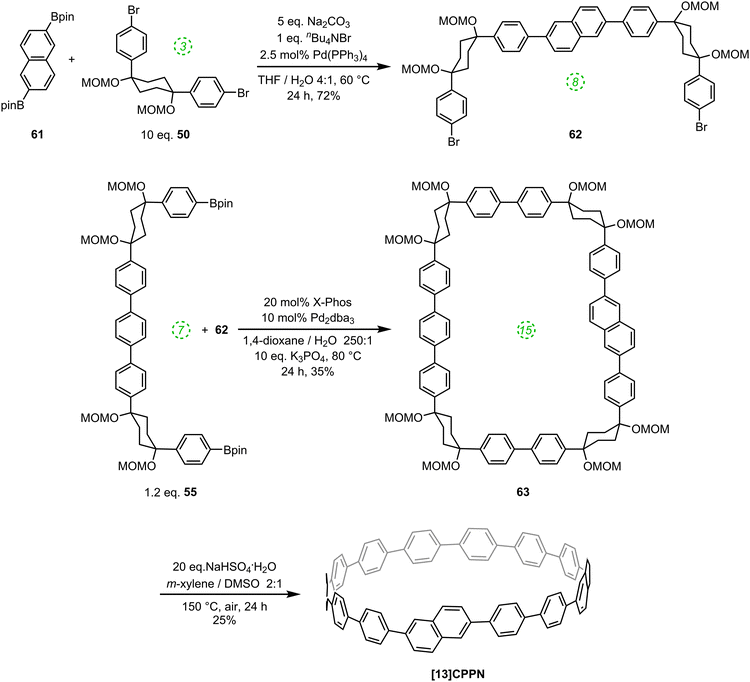 |
| | Scheme 15 Itami's synthesis of [13]CPPN. | |
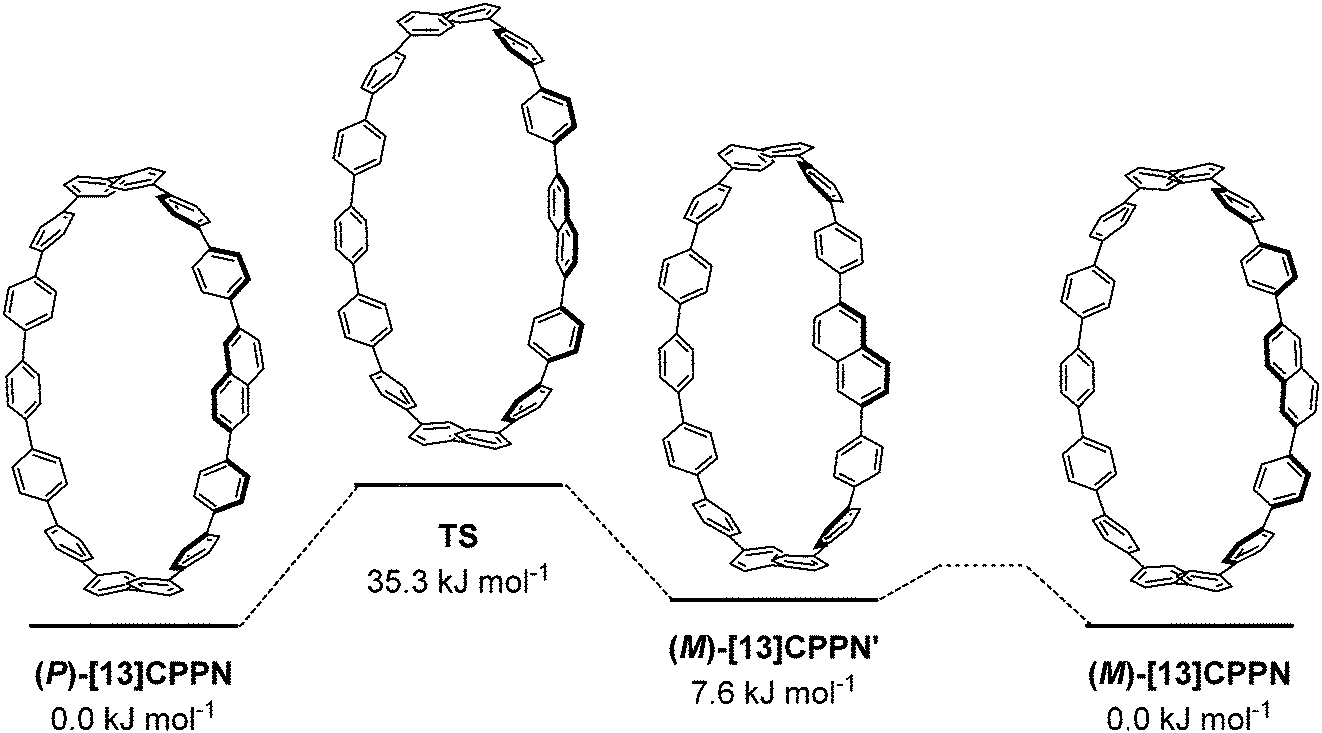
Itami and co-workers carried out computational modelling to determine the barrier to racemisation for [13]CPPN, as shown in Fig. 20. The (P)-enantiomer of [13]CPPN shown on the left has an “all-alternating” conformation of the phenyl and naphthyl rings; this was found to be the lowest energy of the various minima identified. In the transition state for its interconversion to (M)-[13]CPPN, the dihedral angles between the naphthyl unit and both adjacent phenyl rings are both ≈90°. The calculated barrier to interconversion of 35.3 kJ mol−1 implies that [13]CPPN racemises rapidly at room temperature. Calculations on other hypothetical chiral CPP derivatives were also reported.
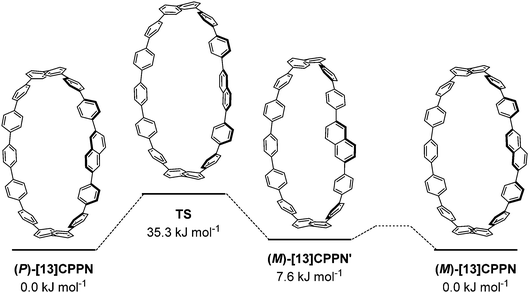 |
| | Fig. 20 Calculated barrier to racemisation for [13]CPPN. Reprinted with permission from ref. 35. Copyright 2011 American Chemical Society. | |
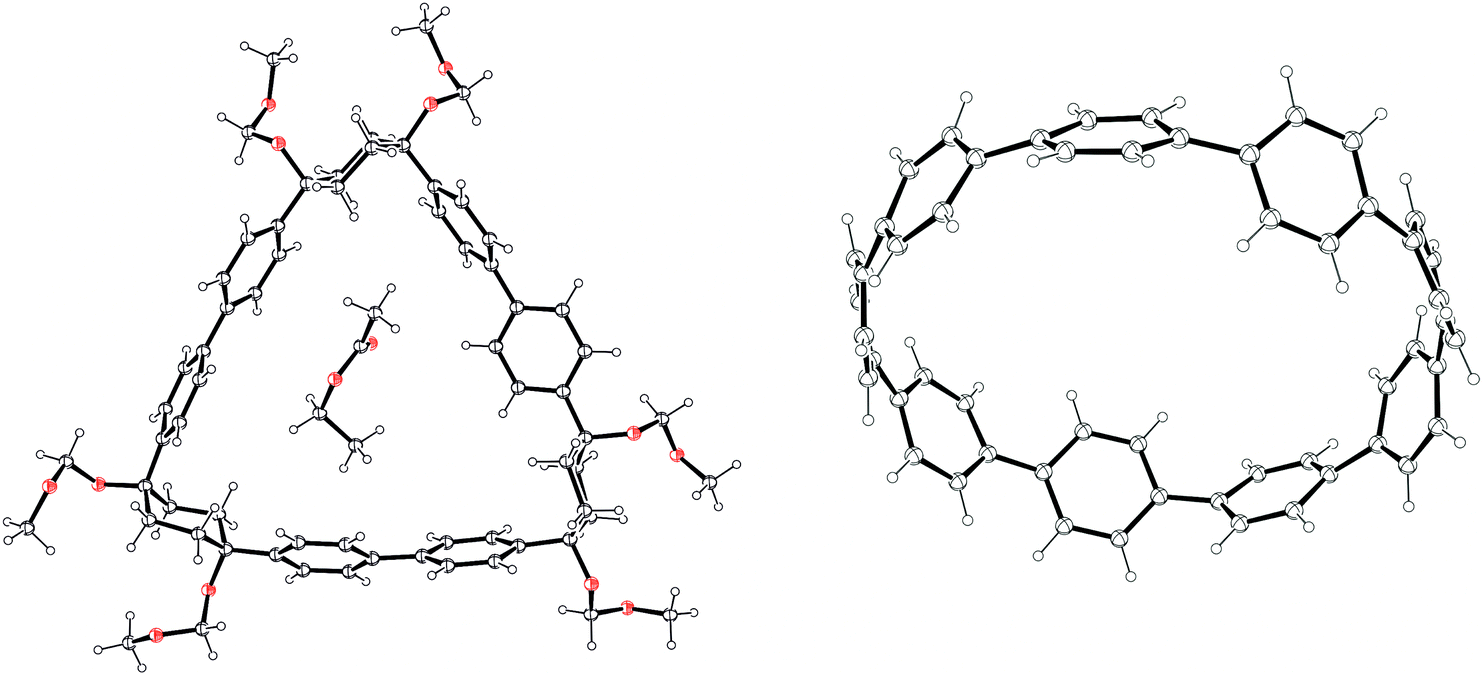
The third paper in quick succession from Itami in 201136 is an extension of the nickel-mediated methodology for formation of [12]CPP they had reported three months previously (Scheme 14). Subsequent to that earlier disclosure, the Itami group discovered that a byproduct from the nickel-mediated shotgun cyclisation, that they had assumed was a linear oligomer, was in fact cyclic trimer 64 (Scheme 16), as determined by X-ray crystallography. It proved possible to convert 64 into [9]CPP as previously, albeit in lower yield, which was attributed to the higher strain energy of [9]CPP compared to [12]CPP. An X-ray structure for the end product was also acquired. A further exploration of the cyclisation conditions determined that both tetramer 60 and trimer 64 were formed regardless of the choice of L-shaped building block; the highest yield of trimer 64 was 32%.
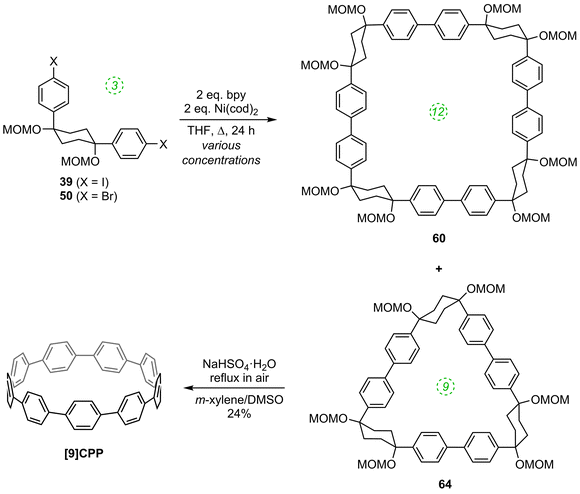 |
| | Scheme 16 Itami's synthesis of [9]CPP. | |
The crystal structures for 64 and [9]CPP are shown in Fig. 21. Trimer 64 comprises cyclohexyl rings with an “included” angle of ≈70°. Computational modelling showed the strain energy of 64 to be 33 kJ mol−1, appreciably higher than that for 60, which was calculated to be close to strain-free (7 kJ mol−1). The crystal structure of [9]CPP was slightly distorted into an ellipsoidal conformation, in contrast to the crystal structure for [12]CPP. This could be attributed to the incorporation of two molecules of THF solvent upon crystallisation. A comparison of bond lengths for the [9]CPP and [12]CPP X-ray structures showed the mean Cipso–Cipso bond length to be slightly shorter for [9]CPP than [12]CPP; the same trend was observed for the mean Cortho–Cortho bond length. This was taken to be indicative of slightly increased quinoidal character in the smaller CPP.
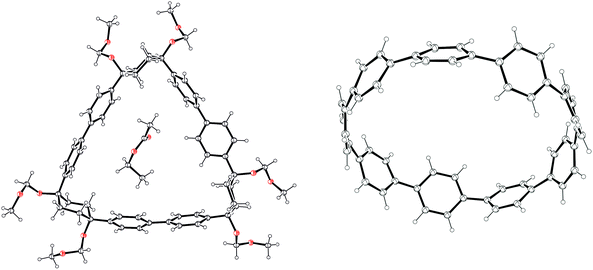 |
| | Fig. 21 ORTEP diagrams 64·EtOAc and [9]CPP, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Disordered solvent has been omitted from the structure of [9]CPP for clarity. | |
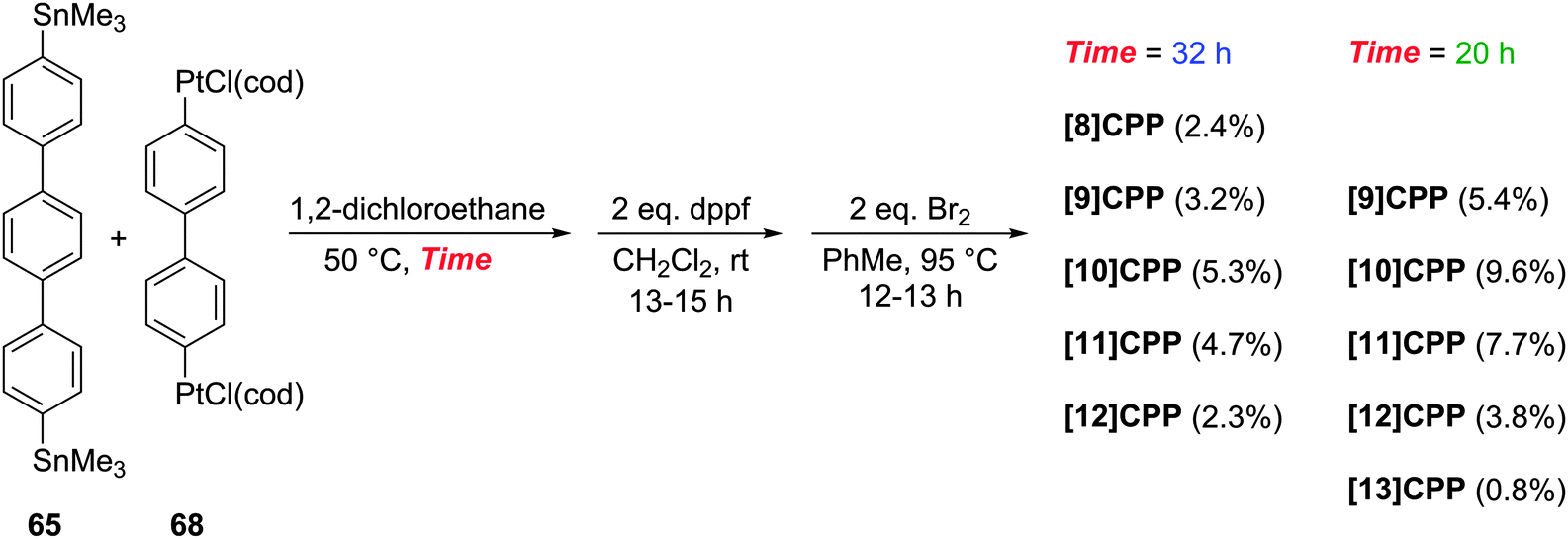
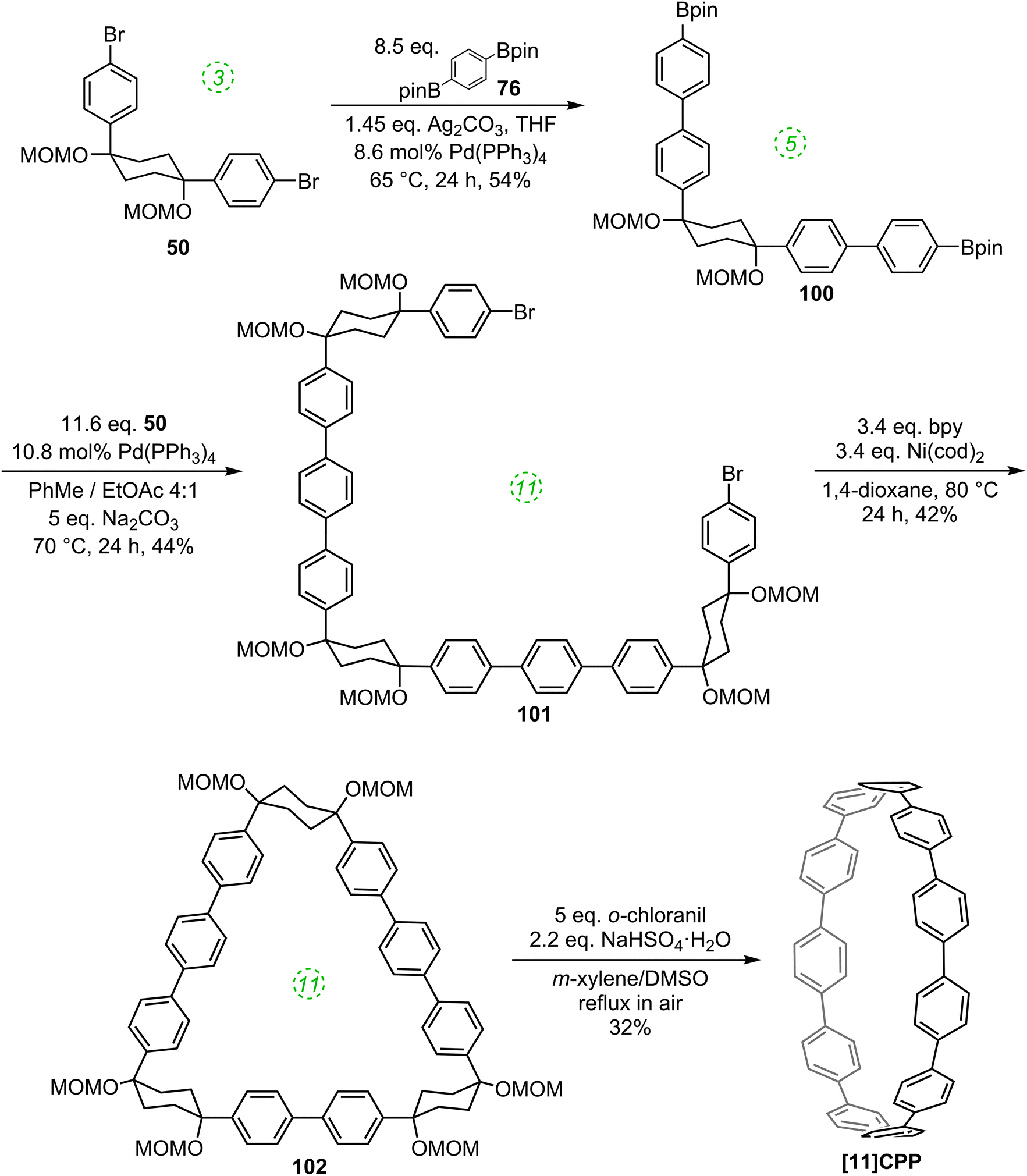
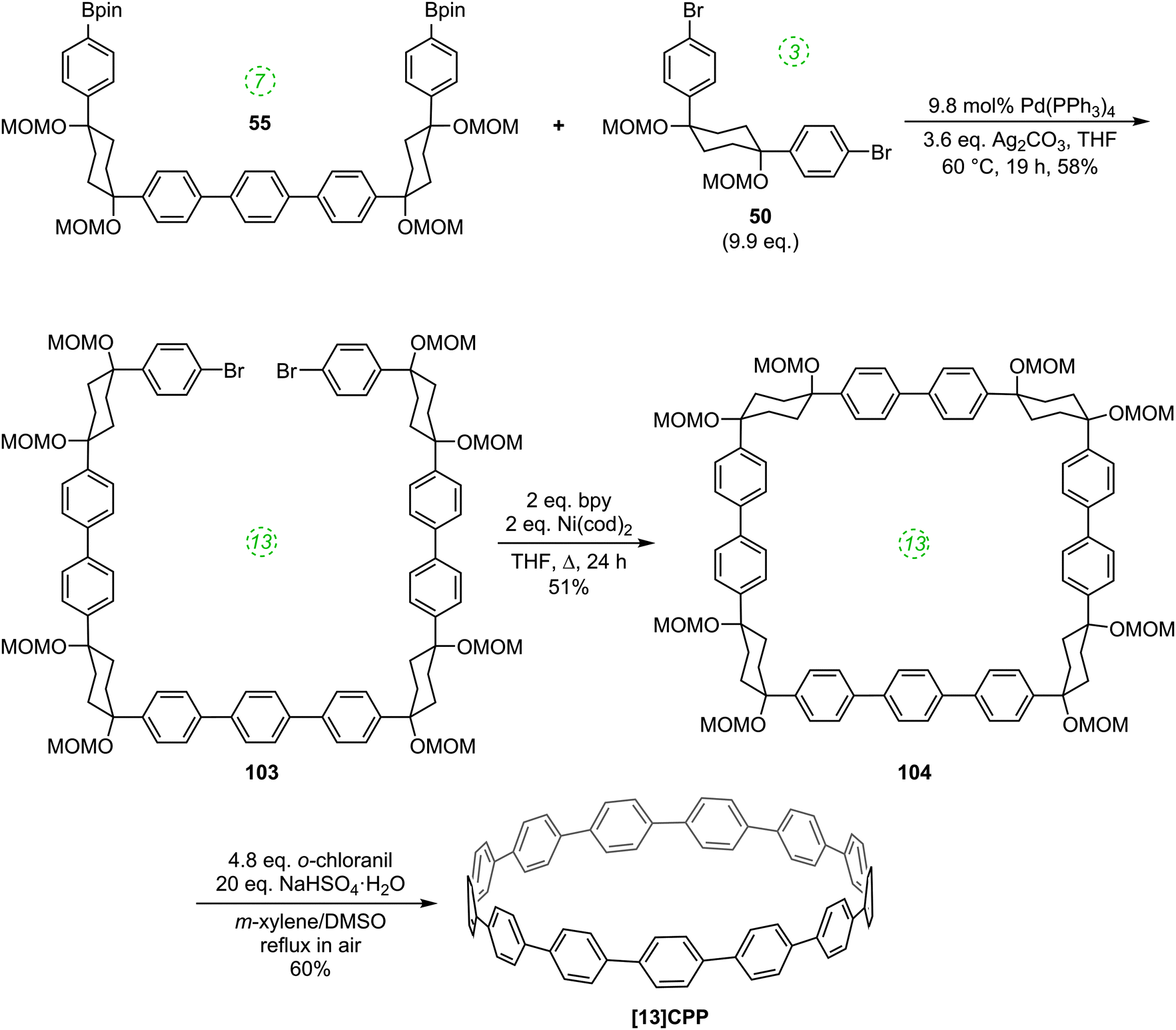
Yamago followed up his 2010 communication on the synthesis of [8]CPP with a full account in 2011 which extended the platinum-based methodology to effect syntheses of all CPPs from [8]CPP to [13]CPP.37 In the same way that a selective synthesis of [8]CPP was possible using a biphenyl building block (Scheme 10), so a selective synthesis of [12]CPP proved to be possible using a terphenyl building block 65 (Scheme 17).
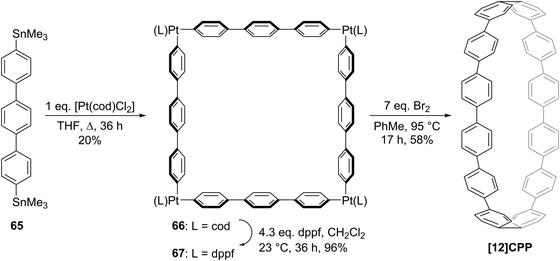 |
| | Scheme 17 Yamago's platinum-mediated synthesis of [12]CPP. | |
The Yamago group next attempted a selective synthesis of [10]CPP by combining bis(stannyl)terphenyl building block 65 with bis(platinum)biphenyl building block 68 (Scheme 18). Ligand exchange with dppf was then effected without isolation of the intermediate. Oxidation with elemental bromine gave rise to not only the expected [10]CPP, but in fact all CPPs from [8]CPP to [13]CPP! In addition, it was found that simply varying the reaction time for the first step profoundly affected the product distribution. The fact that multiple CPPs had been produced was obvious from the crude 1H-NMR of the reaction mixture for the final step. As had been noted in previous publications, all known CPPs exhibit a single resonance in their 1H-NMR spectra, due to fast interconversion of CPP conformers on the NMR timescale. However, chemical shift varies with size of the CPP (the resonance moves downfield with increasing ring size, except for [5]- and [6]CPP). Thus, the presence of several singlets in the aryl region was indicative of multiple CPPs having been formed. Separation of the CPPs was achieved using gel permeation chromatography. The formation of multiple CPPs was attributed to the transmetallation between platinum and aryl stannanes being a reversible process (which had in fact been reported previously38). It is notable that the process provides access to odd-numbered CPPs. Whereas any even-numbered CPP could be accessed from a rectangular tetraplatinum precursor devoid of ring strain (cf.44 and 67), access to an odd-numbered CPP requires a tetraplatinum precursor where the C–Pt–C bond angles must necessarily be distorted away from 90°. As such, formation of odd-numbered CPPs might be expected to be disfavoured, but examination of the product ratios in Scheme 18 does not suggest this is the case. Most surprising of all is the formation of [13]CPP – with the building blocks employed by Yamago, this could not have arisen from a tetraplatinum precursor and instead must have arisen from a pentaplatinum or hexaplatinum precursor. It should be noted that Yamago's work constituted the first report of a synthesis of [10], [11] or [13]CPP.
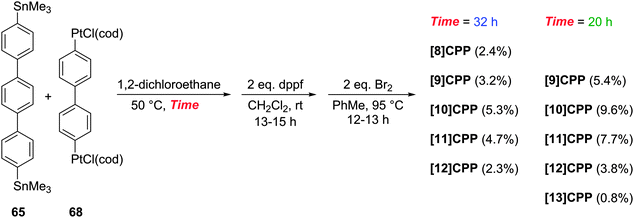 |
| | Scheme 18 Yamago's platinum-mediated synthesis of [8–13]CPP. | |
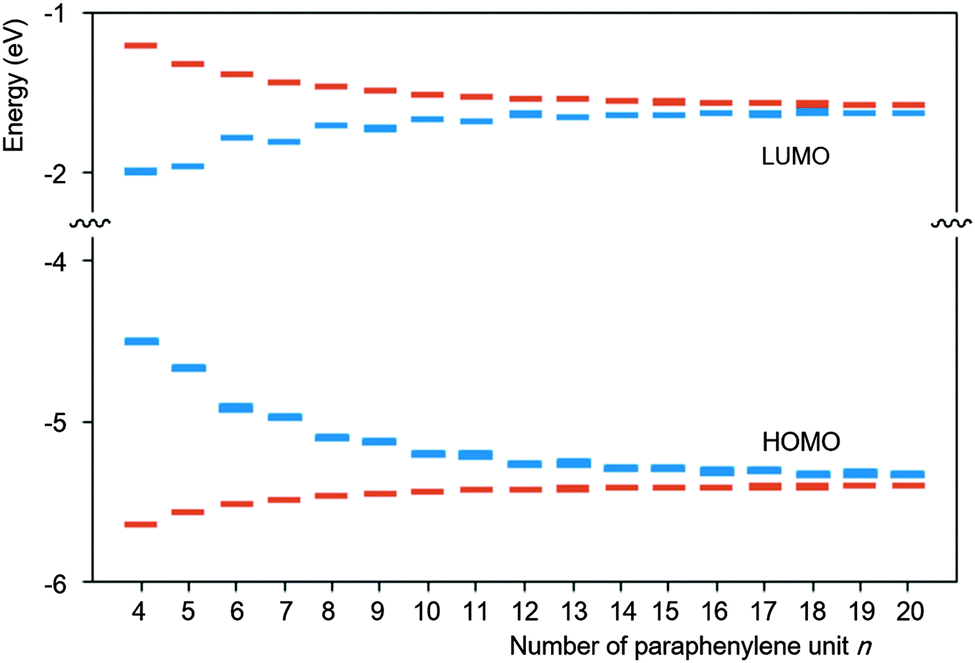
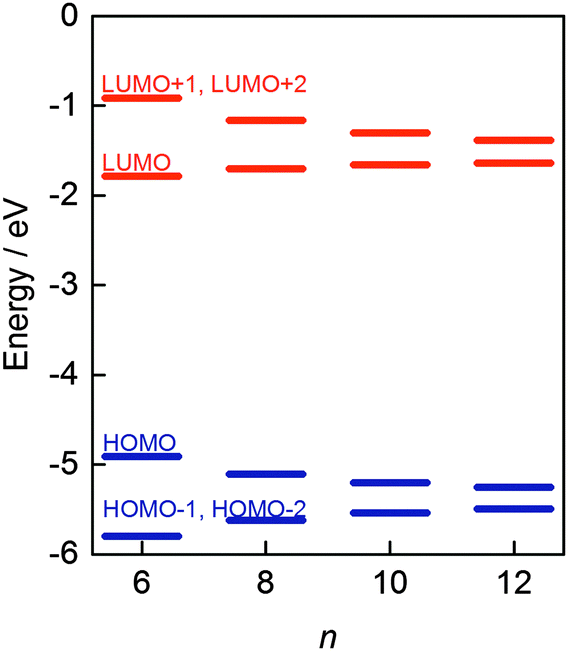
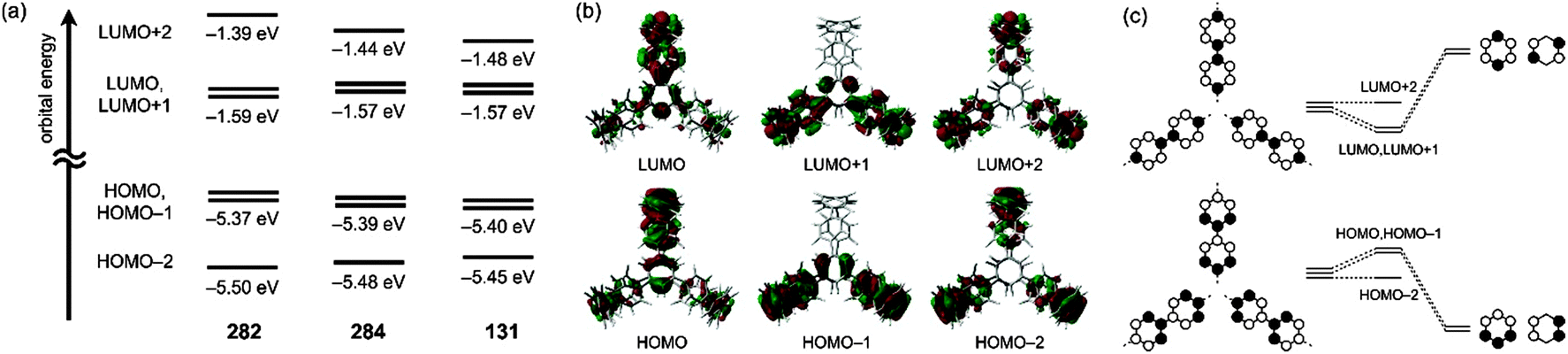
Yamago and co-workers undertook extensive computational modelling and experimental characterisation of their library of CPPs. Strain energies, as well as HOMO and LUMO energies were computed for all CPPs from [4]CPP to [20]CPP; the calculated strain energies were in close agreement with those calculated by Bachrach.32 The calculated HOMO and LUMO energy levels are represented pictorially in Fig. 22. As the size of the CPP gets smaller, the energy of the HOMO increases and the energy of the LUMO increases, which is the opposite of the trend observed for linear oligophenylenes. Yamago ascribes this divergent behaviour to the ring-size dependence of the aromaticity of the phenylene units. As suggested previously by Wong's calculated NICS(1) values and Itami's X-ray data for [9] and [12]CPP, smaller CPPs have diminished aromatic character and increased quinoid character. The trend in CPP HOMO and LUMO energies is not entirely smooth, however, with a small but noticeable offset between energies for even-numbered and odd-numbered CPPs, as had been previously noted by Bachrach. Yamago rationalises this as being due to the conformational differences between the two types of CPP.
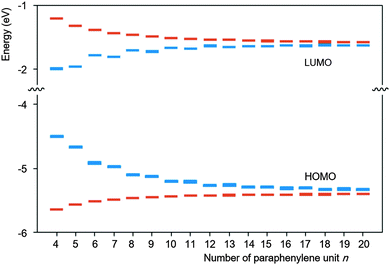 |
| | Fig. 22 Calculated HOMO and LUMO energies of CPPs (blue) and oligoparaphenylenes (red). Reprinted with permission from ref. 37. Copyright 2011 American Chemical Society. | |
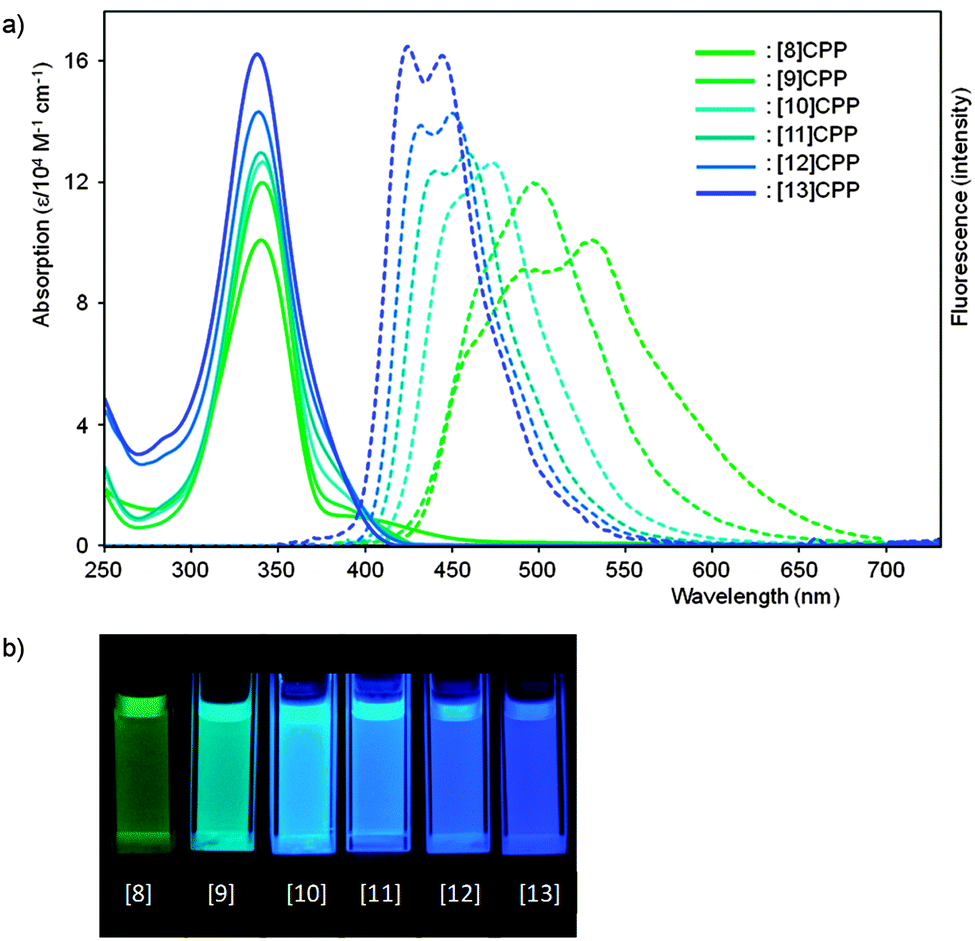
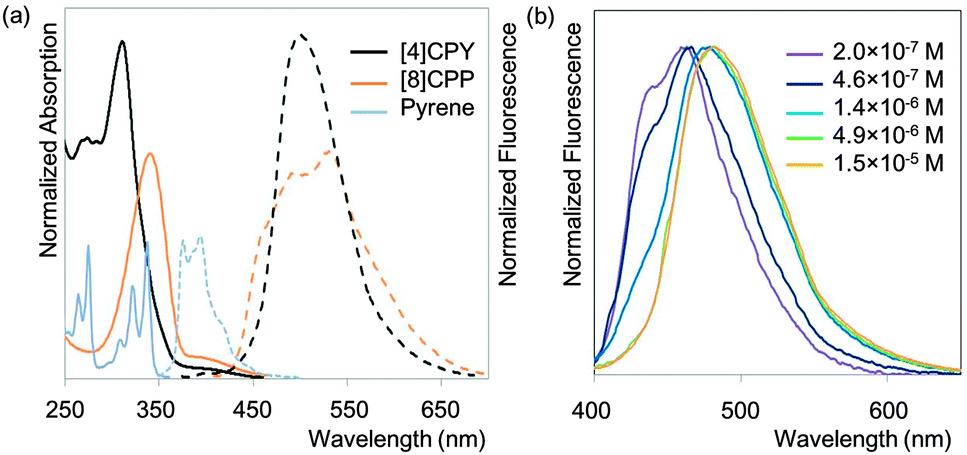
The observed absorbance and fluorescence spectra are shown in Fig. 23. The absorption maxima for the different CPPs are essentially invariant, at around 340 nm. This is in agreement with Bertozzi's observations, but is not what would be expected on the basis of the calculated HOMO/LUMO levels in Fig. 22. This disagreement was investigated using time-dependent density functional theory (TD-DFT), which revealed that in all cases the HOMO → LUMO transition is actually forbidden, with minimal or no oscillator strength. The observed absorption is in fact due to several other transitions, specifically HOMO − 2 → LUMO, HOMO − 1 → LUMO, HOMO → LUMO + 1 and HOMO → LUMO + 2. As regards the trend in the fluorescence spectra, the trend for an increase in Stokes shift with smaller rings is again observed, for which Yamago offers the same rationale as Bertozzi and Sundholm.
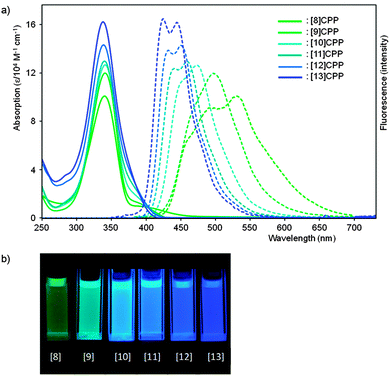 |
| | Fig. 23 (a) Absorbance (solid lines) and fluorescence (dashed lines) spectra for [8–13]CPPs. (b) Fluorescence emission of [8–13]CPPs. Reprinted with permission from ref. 37. Copyright 2011 American Chemical Society. | |
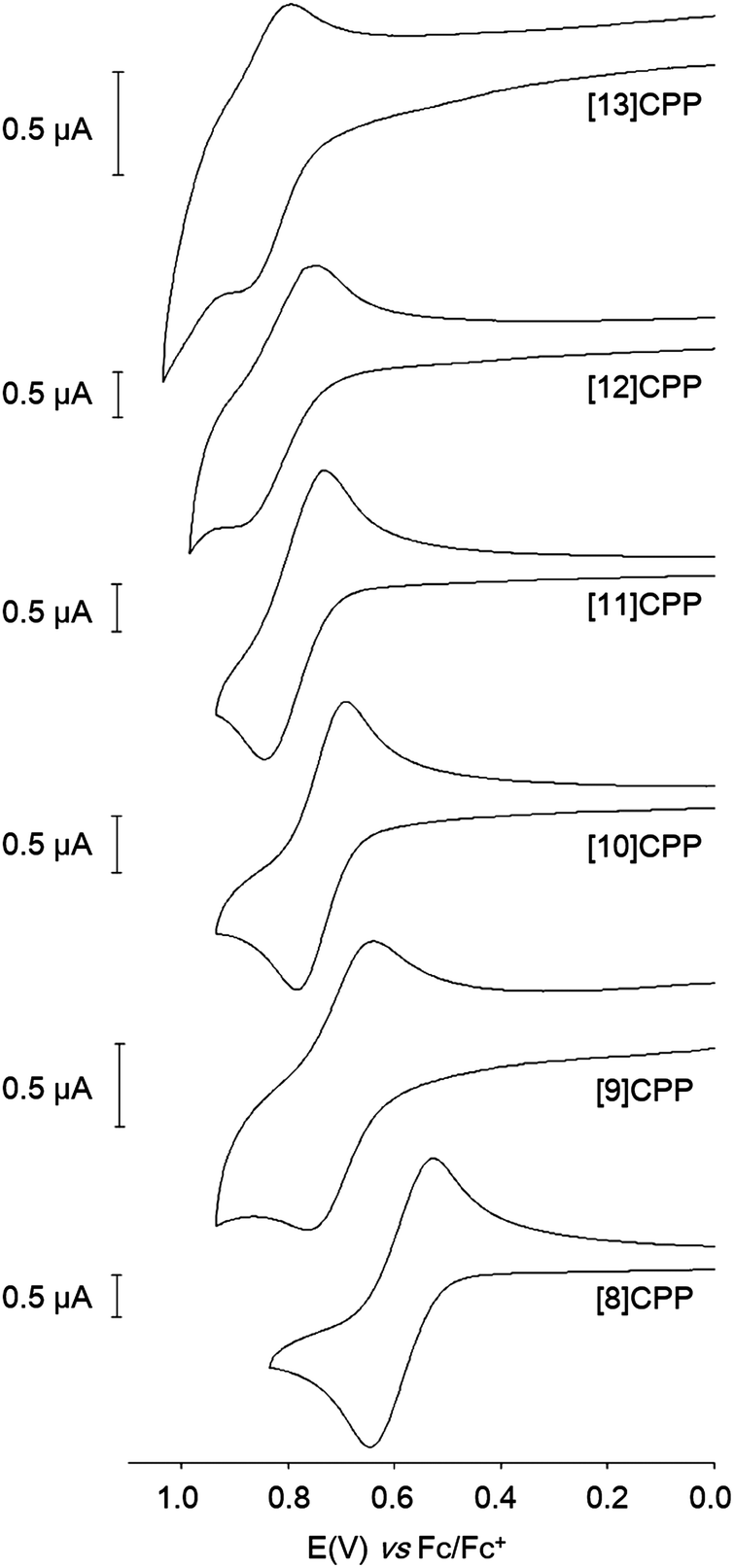
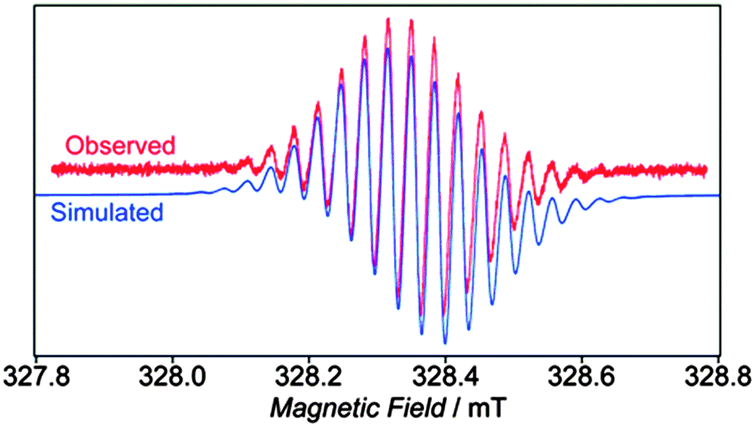
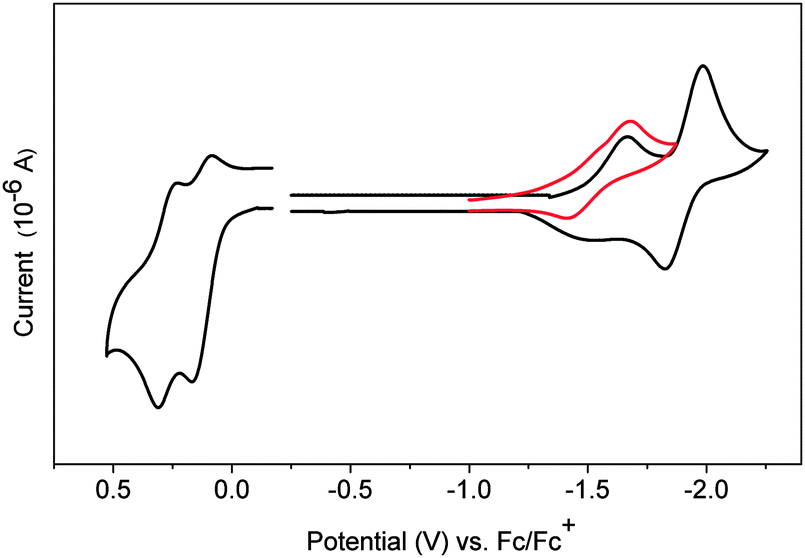
Finally, Yamago's paper is the first to describe an electrochemical study of CPPs. Cyclic voltammograms for [8] to [13]CPP are reproduced in Fig. 24. Every CPP showed a reversible oxidation wave, implying the CPP radical cations formed are stable under the conditions of the experiment. Oxidation potential correlates with CPP size, with larger CPPs having a higher oxidation potential, as expected from the calculated HOMO energies in Fig. 22.
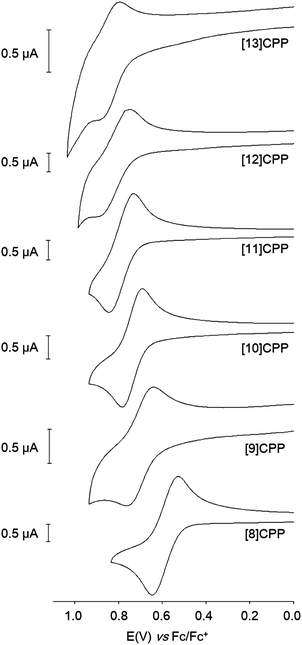 |
| | Fig. 24 Cyclic voltammograms for [8] to [13]CPP in 1,1,2,2-tetrachloroethane with 0.1 M Bu4NPF6. Reprinted with permission from ref. 37. Copyright 2011 American Chemical Society. | |
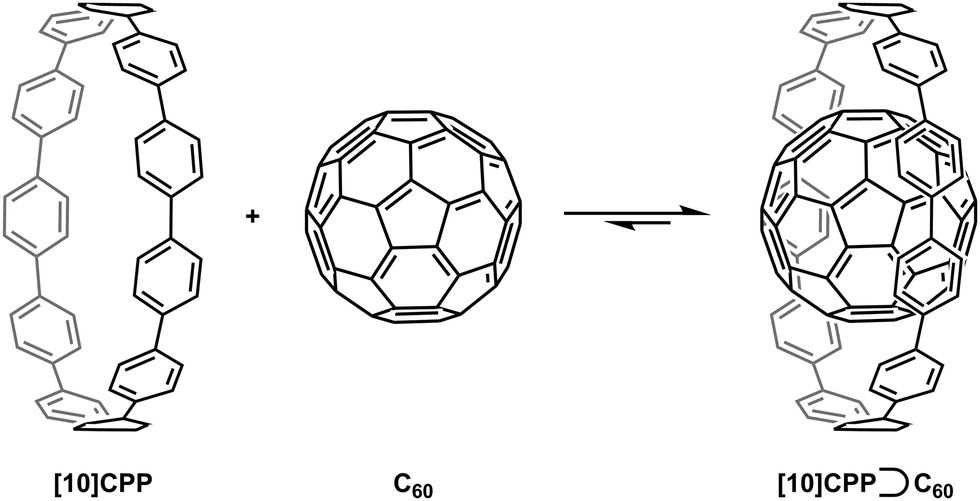
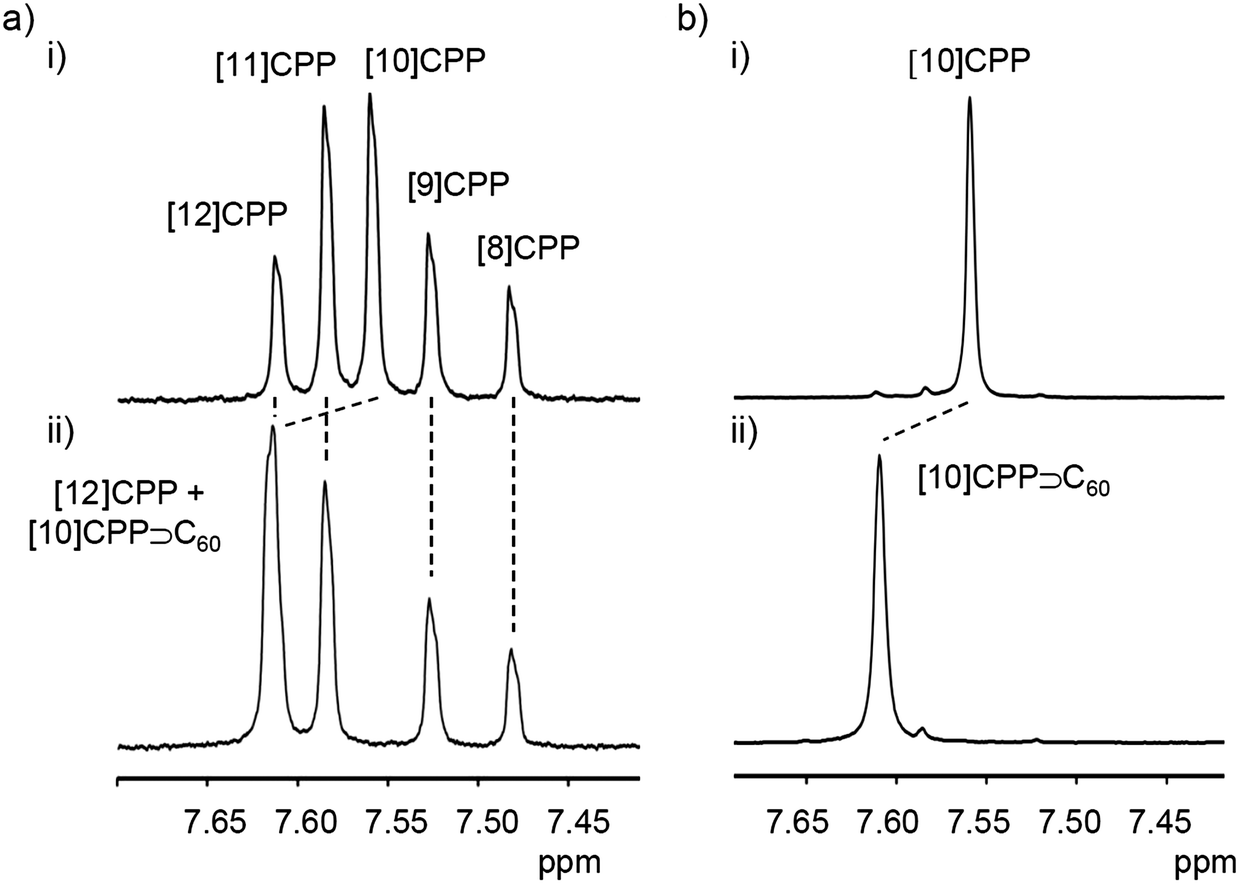
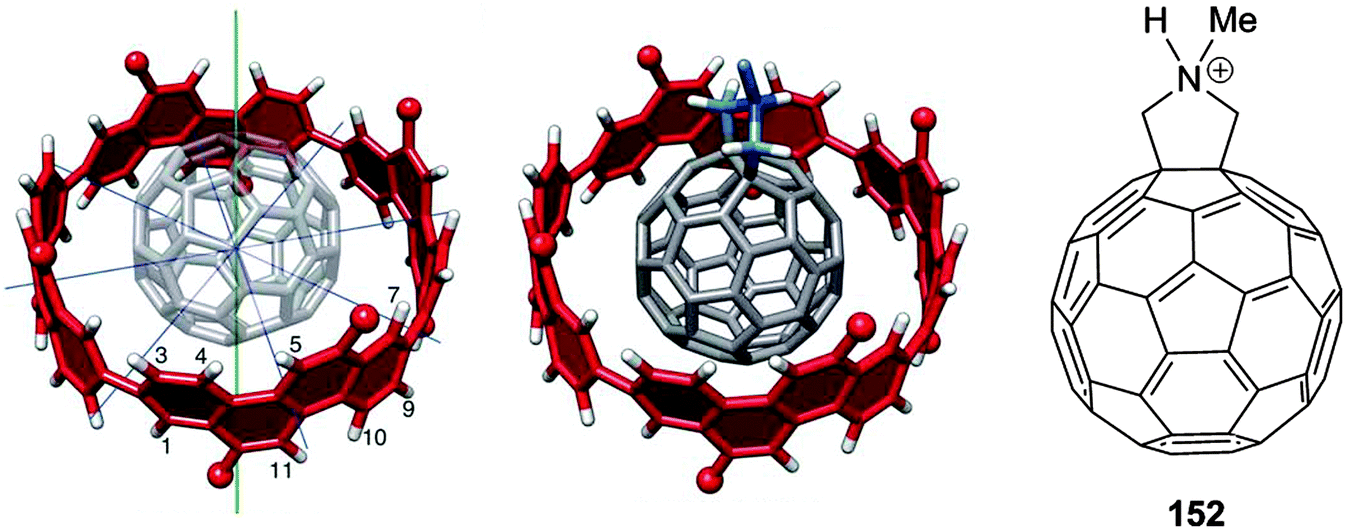
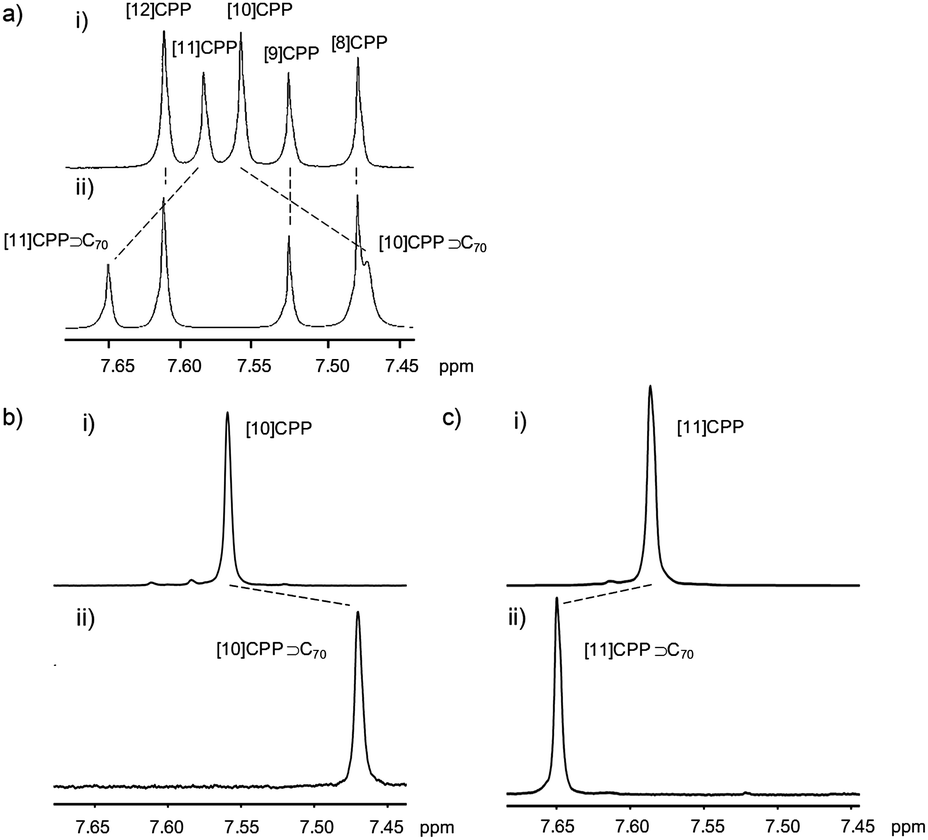
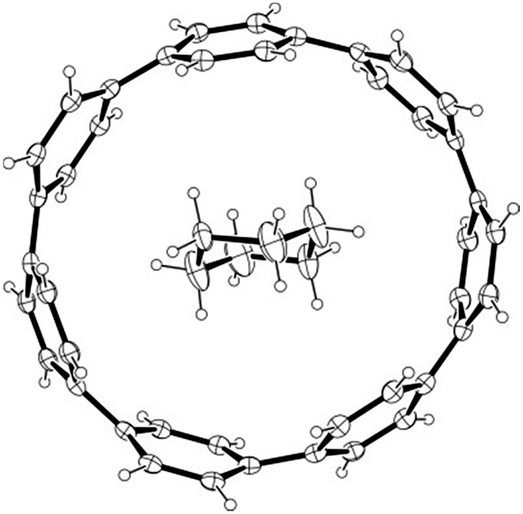
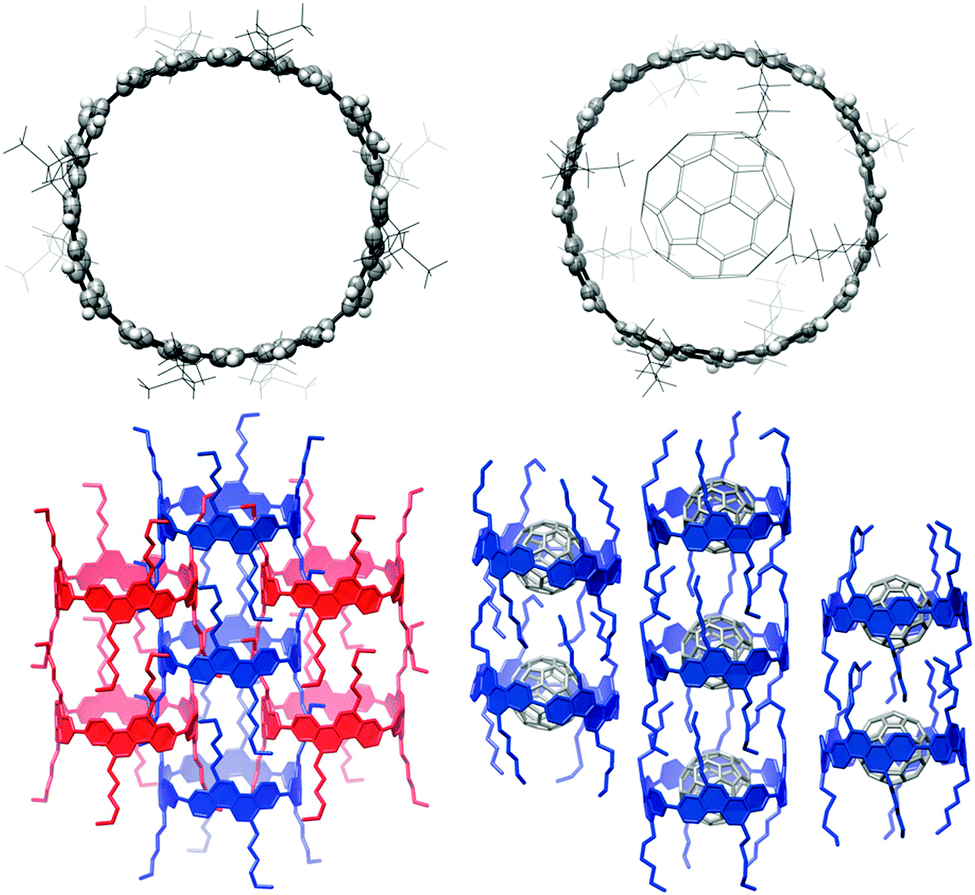
A second paper from Yamago then appeared some two months later on the host–guest chemistry of CPPs.39 The Yamago group were able to determine that [10]CPP was an effective host for buckminsterfullerene (C60), as shown in Scheme 19. The interaction between these two species was first detected by 1H-NMR spectroscopy. As mentioned above, the chemical shift for the aryl singlet in CPPs varies with ring size. Thus, when C60 was added to a mixture of [8]–[12]CPP, the resonance for [10]CPP was observed to shift upfield (Fig. 25a), whereas the resonances for the other CPPs did not change. The same shift was also observed upon addition of C60 to a solution of [10]CPP alone (Fig. 25b).
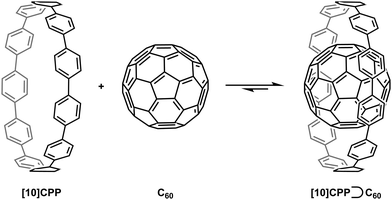 |
| | Scheme 19 Selective encapsulation of C60 by [10]CPP. | |
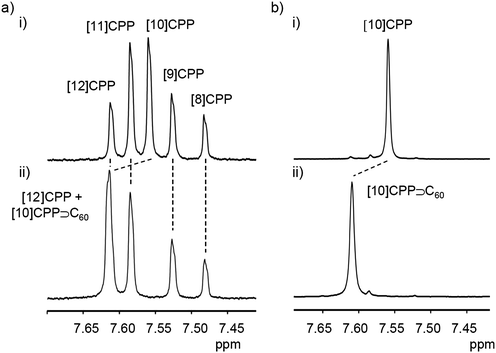 |
| | Fig. 25
1H-NMR spectra in CDCl3 at room temperature of (a) [8]–[12]CPPs before (i) and after (ii) the addition of C60 and (b) isolated [10]CPP before (i) and after (ii) the addition of C60. Reprinted with permission from ref. 39. Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
The 1:1 stoichiometry of the complex was confirmed with a Job's plot and the binding constant in ortho-dichlorobenzene was determined to be Ka = (6.0 ± 0.2) × 103 L−1 mol. C60 was observed to quench the fluorescence of [10]CPP – fluorescence quenching experiments in toluene determined the Stern–Völmer constant to be KSV = (4.34 ± 0.04) × 106 L−1 mol and the binding constant to be Ka = (2.79 ± 0.03) × 106 L−1 mol (much larger than in ortho-dichlorobenzene). Thus, it was determined that encapsulation of C60 by [10]CPP results in a stabilisation of around 38 kJ mol−1, around two orders of magnitude greater than the previously reported value for encapsulation of C60 by [6]CPPA (cycloparaphenyleneacetylene).
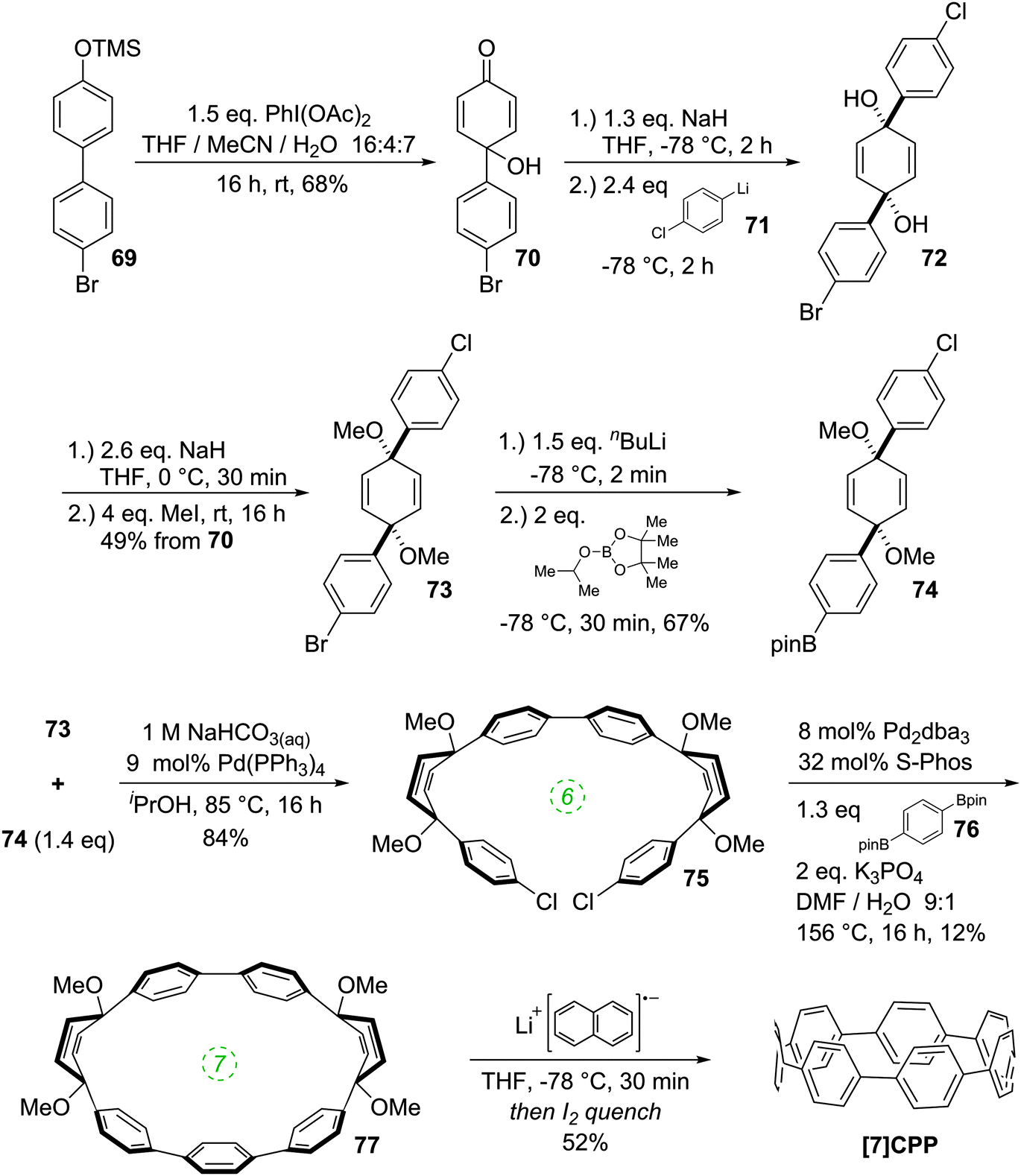
A 2011 publication from Jasti and co-workers reported the first synthesis of [7]CPP, the smallest CPP described at that point.40 They employed an approach which was a variant of that employed in the original 2008 disclosure of the first CPP syntheses. As shown in Scheme 20, the key to Jasti's strategy is the exploitation of the enhanced reactivity of aryl bromides with respect to aryl chlorides. Dearomatisation of biphenyl 69 with hypervalent iodine gave hydroxyketone 70. To ketone 70 was added aryllithium 71, the preparation of which proceeded by preferential lithium–halogen exchange of a bromide in the presence of a chloride. The deprotonation of 70 with sodium hydride prior to addition of 71 favoured the formation of the desired syn product 72. This was methylated to give 73, which is an analogue of 30, but with two differentiated halides. A further lithium–halogen exchange on 73 (again selective for the aryl bromide), followed by reaction with ITDB gave 74. Combination of 73 and 74 by Suzuki–Miyaura cross-coupling under comparatively mild conditions gave 6-ring linear precursor 75, with the two aryl chlorides remaining untouched. The key dual intramolecular/intermolecular Suzuki–Miyaura macrocyclisation step was extremely demanding. Not only would the hitherto inert aryl chlorides need to be functionalised, but computational modelling suggested that the target CPP precursor 77 possessed 67 kJ mol−1 of strain energy, quite aside from the strain energy of [7]CPP itself. In the event, use of Buchwald's S-Phos ligand under forcing conditions was able to provide 77 in 12% yield, the byproducts of the reaction being linear oligomers. Finally, reductive aromatisation with lithium naphthalenide afforded [7]CPP. An X-ray crystal structure was obtained for CPP precursor 77, and is shown in Fig. 26.
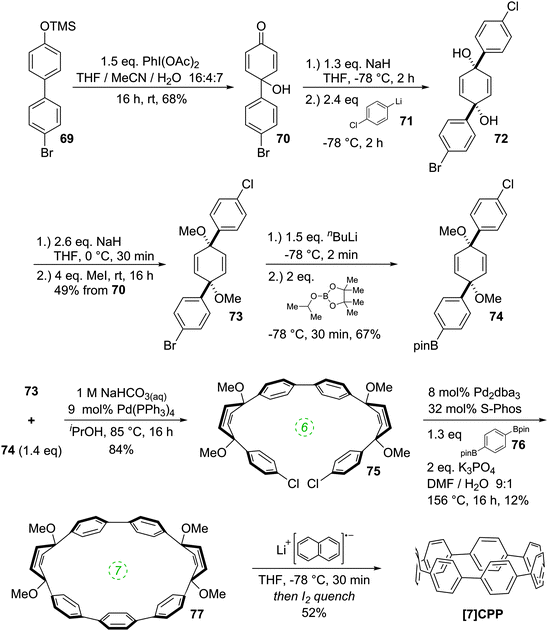 |
| | Scheme 20 Jasti's synthesis of [7]CPP. | |
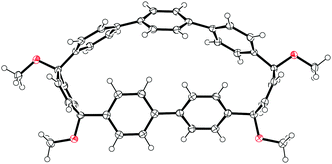 |
| | Fig. 26 ORTEP diagram of 77, showing ellipsoids at 50% probability. H atoms are shown as spheres of arbitrary radius. | |
[7]CPP exhibited an absorption maximum at 339 nm, in agreement with the previously observed trend that λmax does not vary with CPP ring size. The fluorescence spectrum of [7]CPP has a maximum at 592 nm, making [7]CPP an orange emitter, and continuing the trend of smaller CPPs having larger Stokes shifts. However, unlike the larger CPPs, [7]CPP had a very low quantum yield of only 0.007, implying a rather forbidden transition (for comparison, the quantum yield for [12]CPP was measured as being 0.81).
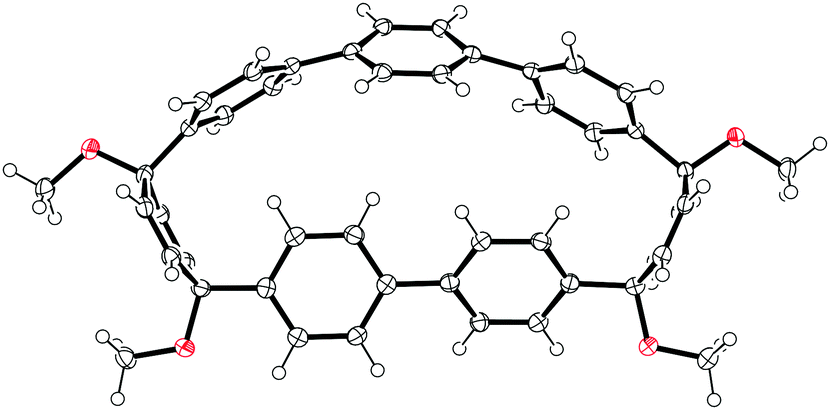
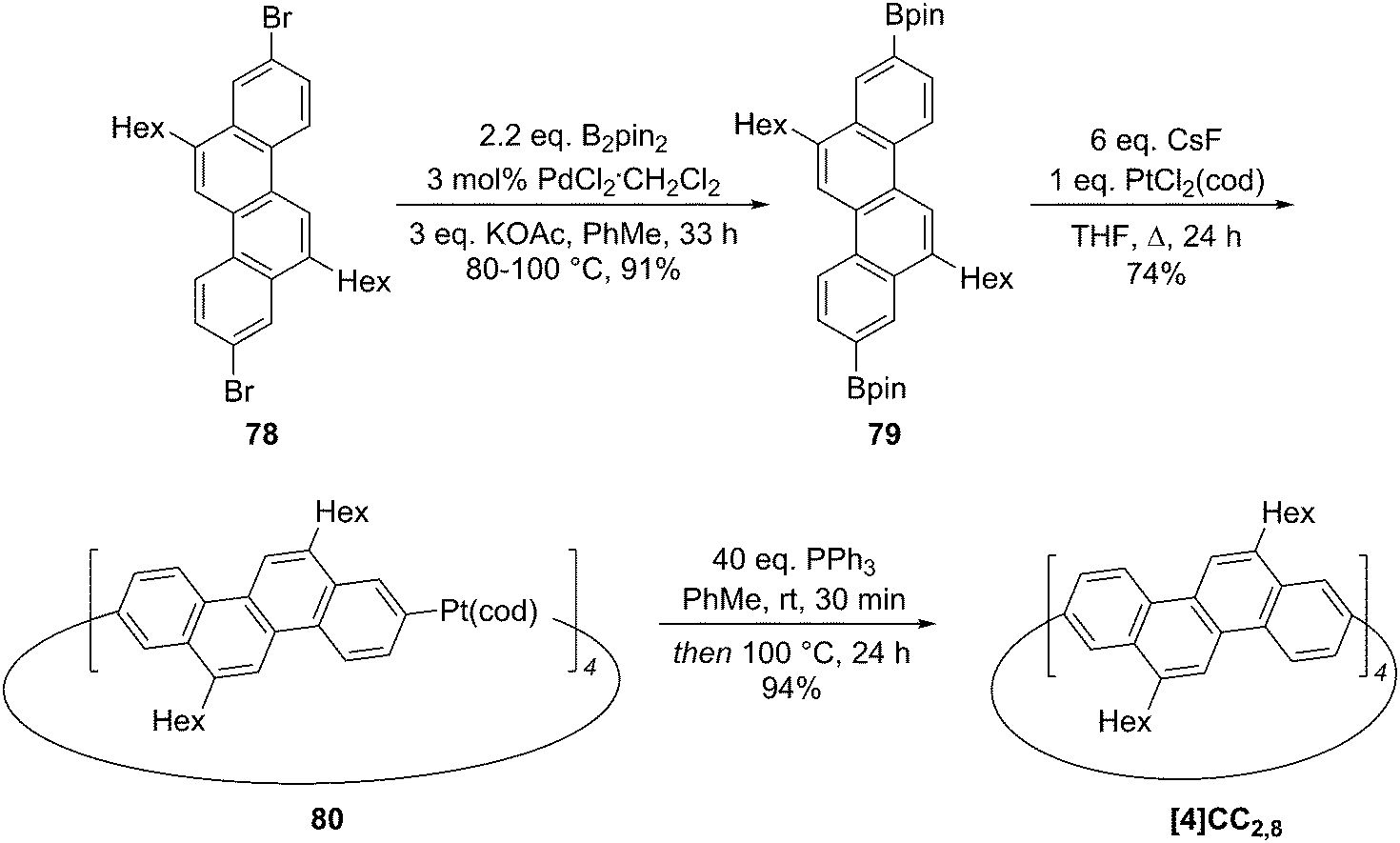
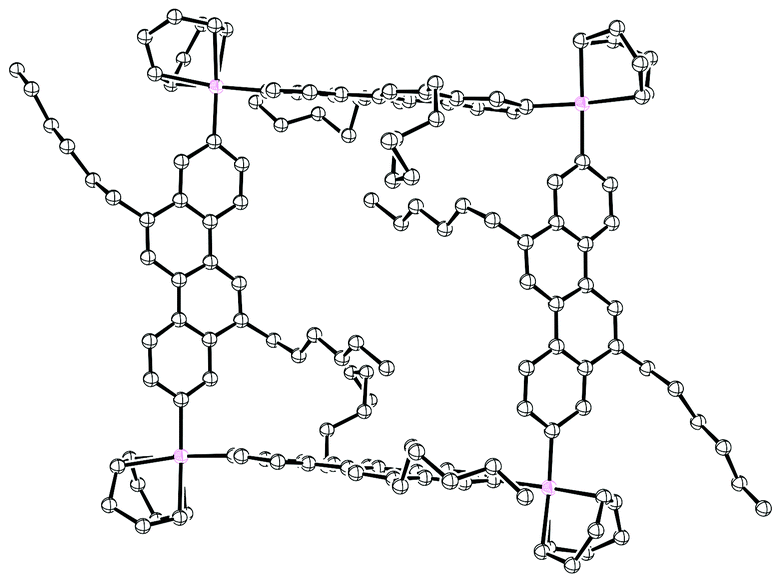
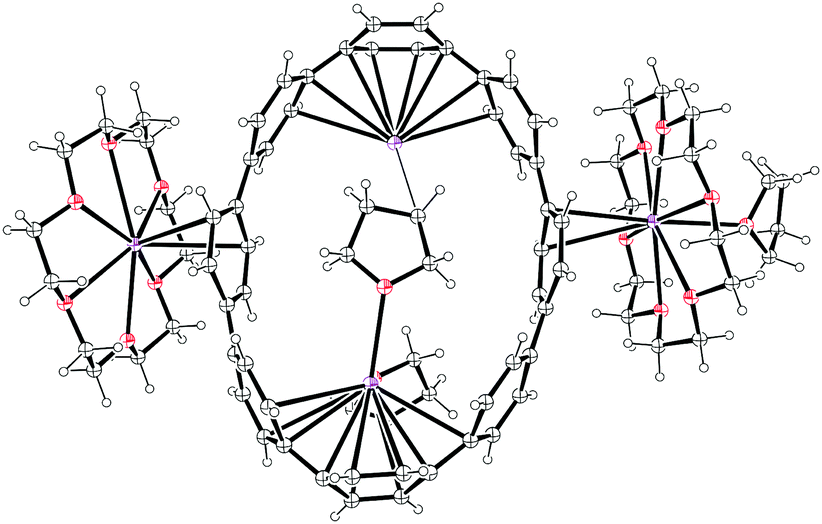
2011 saw the first contribution to the field from the Isobe group, who had previously published extensively on macrocyclic aromatic compounds, which lacked a radial π system as found in CPPs (such work therefore falls outside the scope of this review). Isobe now reported the synthesis of all stereoisomers of [4]cyclo-2,8-chrysenylene (“[4]CC2,8”) using a variant of the platinum-mediated methodology developed by Yamago.41 The synthetic route is shown in Scheme 21. Miyaura borylation of 2,8-dibromochrysene 78 gave bis(borylated) building block 79. This boron-containing linear fragment then underwent transmetallation with a platinum source, in a manner analogous to Yamago's tin-containing monomers, to give tetraplatinum complex 80, for which an X-ray crystal structure was obtained (Fig. 27). Whereas Yamago had effected reductive elimination from 43 and 66via a ligand exchange and oxidation with elemental bromine, Isobe reports that treatment of 80 with an excess of triphenylphosphine leads directly to the product of four-fold reductive elimination, [4]CC2,8. (Yamago had reported use of triphenylphosphine to be unsuccessful in his CPP syntheses).
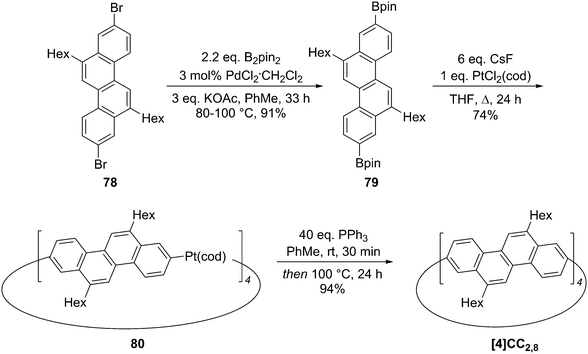 |
| | Scheme 21 Isobe's synthesis of [4]CC2,8 stereoisomers. | |
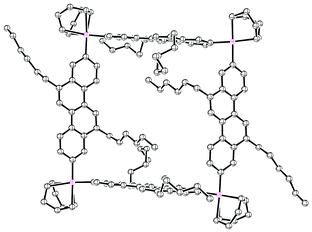 |
| | Fig. 27 ORTEP diagram of 80, showing ellipsoids at 50% probability. A second molecule of 80 in the unit cell has been omitted for clarity, as have solvent molecules and all hydrogens. | |
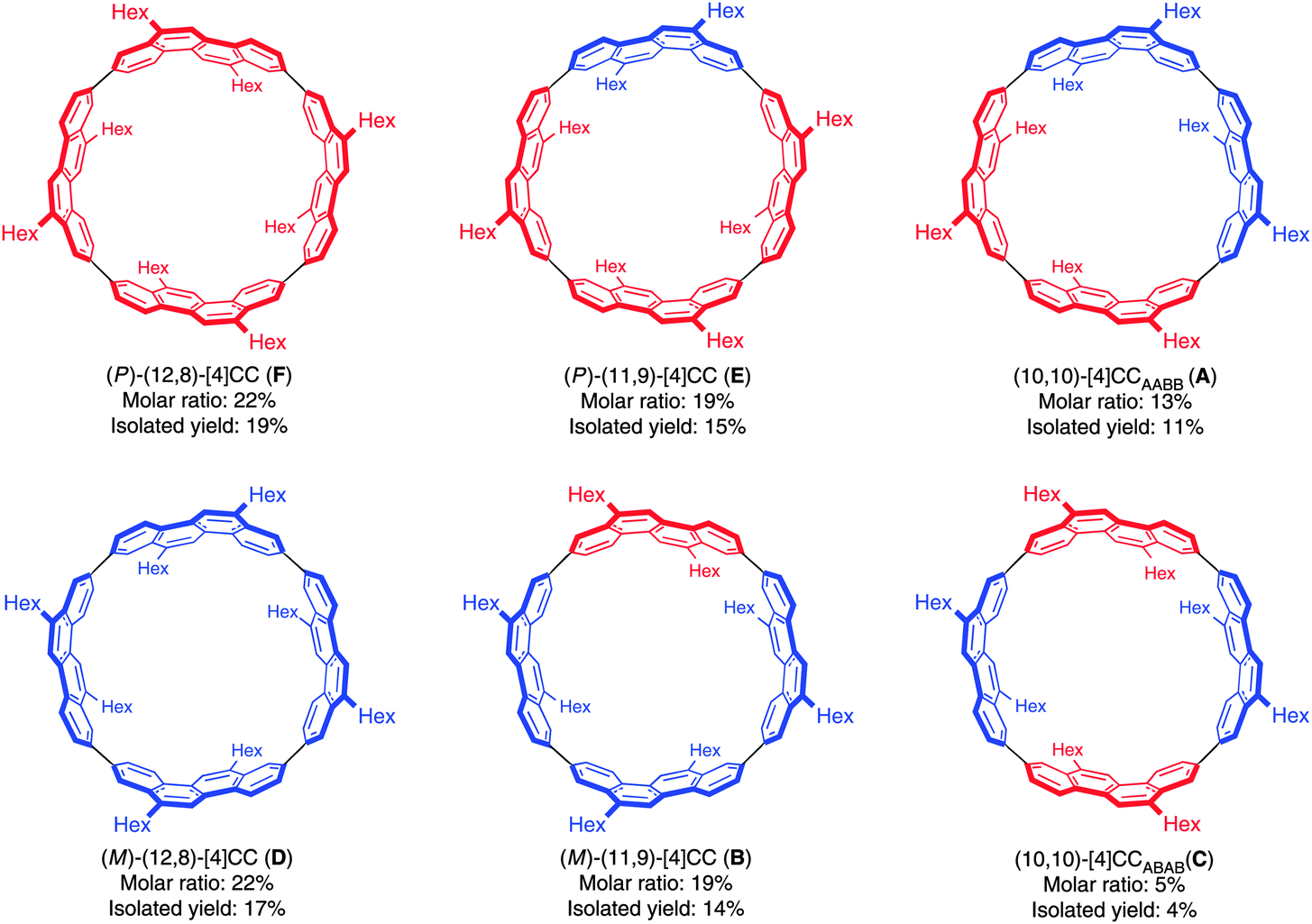
Just as for Itami's [13]CPPN (Fig. 18), Isobe's [4]CC2,8 is capable of exhibiting stereoisomerism. However, whereas [13]CPPN possesses only a single 2,6-naphthyl unit (and hence can exist as two enantiomers), [4]CC2,8 possesses four repeating units of C2 symmetry (2,8-chrysylene units) and so the situation is more complex. In fact, six distinct isomers are possible for [4]CC2,8, which are shown in Fig. 28. The six isomers are designated A to F. Structures D and F are a pair of enantiomers, with their helical chirality indicated as (M) and (P) respectively. Similarly, B and E are a pair of enantiomers, whereas A and C are achiral. The two numbers in brackets for each isomer are that isomer's chiral index.
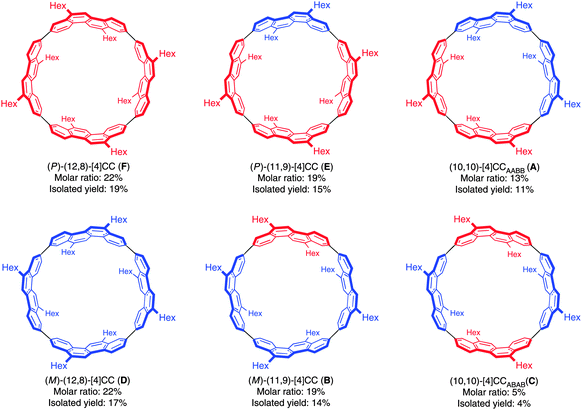 |
| | Fig. 28 Stereoisomers A–F of [4]CC2,8. The two possible chrysenylene orientations are shown in red and blue. Adapted with permission from ref. 41. Copyright 2011 Nature Publishing Group. | |
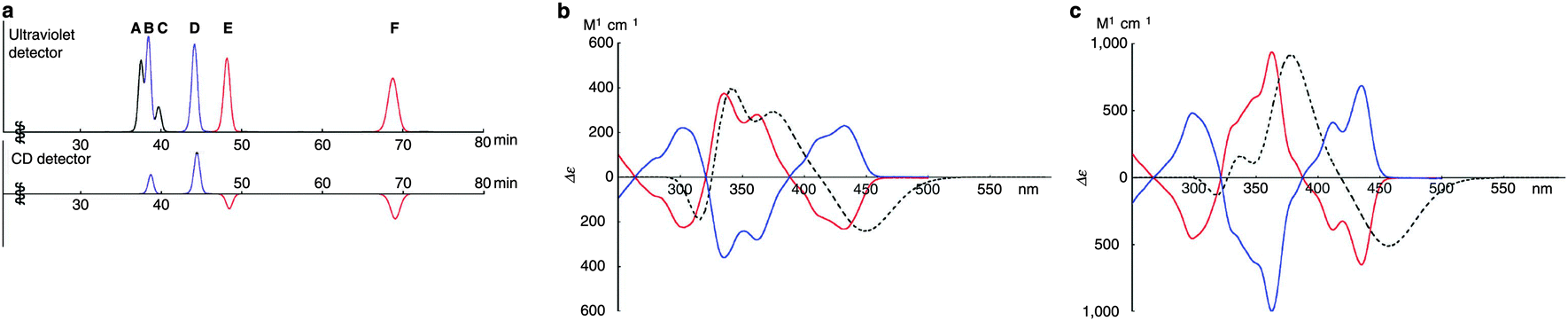
A key difference between Itami's [13]CPPN and Isobe's [4]CC2,8 concerns ease of racemisation by rotation of the naphthylenyl or chrysenylenyl units. [13]CPPN racemises spontaneously at room temperature by free rotation of the naphthylenyl unit. It is also the case that the chrysenylenyl units in tetraplatinum complex 80 are freely rotating, as determined from the apparent simplicity of its NMR spectrum. However, in contrast, interconversion of the six isomers of [4]CC2,8 is not rapid at room temperature and they are in fact separable by preparative HPLC, using a chiral column of cholesterylated silica (the designations A to F refer to their order of elution). Assignment of the six isomers was carried out by means of NMR and circular dichroism (CD) spectroscopy. As shown in Fig. 29, of the six peaks observed in the HPLC trace, two were not detected by the CD detector, and were therefore assigned as the achiral isomers A and C. Distinguishing between these two was possible based on their 13C-NMR spectra: A has C2h symmetry and 18 aromatic carbon environments, whereas C has D4d symmetry and only 9 aromatic carbon environments. For the four remaining isomers, comparison of their CD spectra established that B and E were enantiomeric, as were D and F. Distinguishing these further again required inspection of the 13C-NMR spectra: B and E have C2 symmetry and 36 aromatic carbon environments, whereas D and F have D4 symmetry and only 9 aromatic carbon environments. Finally, assigning the absolute configurations of B, D, E and F required prediction of their CD spectra with TD-DFT methods and comparison with experiment.
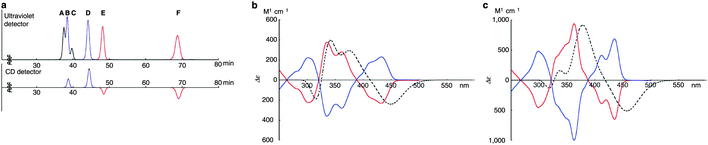 |
| | Fig. 29 (a) HPLC trace for [4]CC2,8. The upper trace shows absorbance at 300 nm and the lower trace shows CD detection at 420 nm. (b) CD spectra for (12,8)-[4]CC2,8: blue line – spectrum for B; red line – spectrum for E; dashed line – computed spectrum for truncated analogue of E. (c) CD spectra for (11,9)-[4]CC2,8: blue line – spectrum for D; red line – spectrum for F; dashed line – computed spectrum for truncated analogue of F. Adapted with permission from ref. 41. Copyright 2011 Nature Publishing Group. | |
The Isobe group's work potentially provides small molecule templates for the bottom-synthesis of chiral carbon nanotubes. Towards this aim, the group also report having carried out preliminary experiments attempting the asymmetric synthesis of [4]CC2,8. Thus, when the transformation of 80 to [4]CC2,8 was carried out in the presence of cholesteryl stearate, the products were enriched in the (P) isomers, with E being formed in 11% e.e. and F being formed in 17% e.e.
The day after Isobe's report, the Wong group published another computational study, this time focusing on chiral CPP derivatives such as [13]CPPN (Scheme 15).42 Wong et al. focused their studies on the excited states of CPPNs, CPPAs (in this case meaning cycloparaphenylene-2,6-anthracenylenes, although the same abbreviation has been used elsewhere for cycloparaphenylene-acetylenes) and CPPTs (cycloparaphenylene-2,8-tetracenylenes). Of these, they identified CPPAs as being unique insofar as they exhibit large photoinduced transitions, as a result of the symmetry-breaking effects of introduction of the anthracenylene unit, in combination with a good alignment of band gaps for the anthracene and the phenylene backbone.
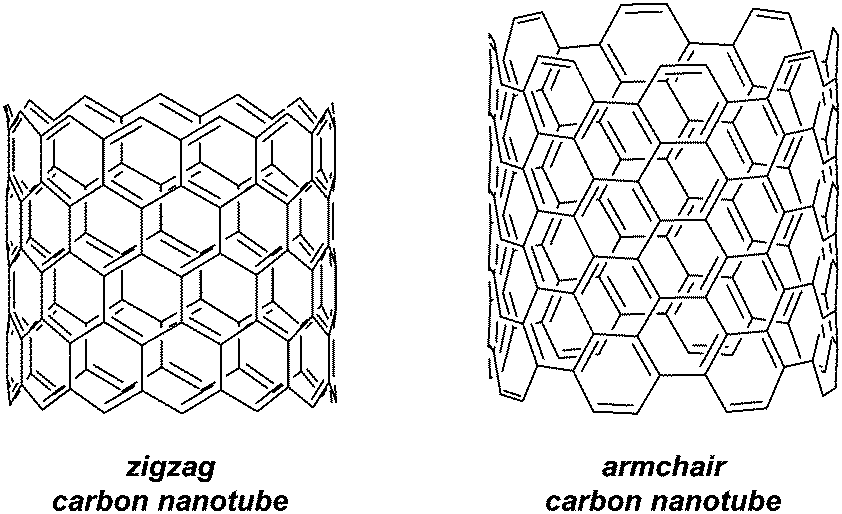
In surveying the literature from 2011, mention should also be made of a publication of from the Jasti group, which is of indirect relevance to CPPs.43 The group carried out a computational study on [5.7]ncyclacenes, which could constitute a heterojunction between “zigzag” and “armchair” regions of a carbon nanotube (Fig. 30). It is specifically armchair carbon nanotubes for which CPPs are potential precursors; the two types of nanotube have significantly different characteristics (armchair SWNTs are metallic, whereas zigzag SWNTs are semiconducting).
 |
| | Fig. 30 Armchair and zigzag nanotube fragments. | |
2012
The year began with a report from Jasti et al. which again broke the record for synthesis of the smallest known CPP.44 The key tactic used in this synthesis of [6]CPP was to employ the oxidative dearomatisation with hypervalent iodine that had been used in the group's prior synthesis of [7]CPP, but to utilise it twice. The synthetic route is shown in Scheme 22. Thus, 69 is dearomatised to hydroxyketone 70, as per the synthesis of [7]CPP, but 70 is instead treated with a lithiated two-ring building block 81 to give 4-ring fragment 82 in a syn-selective reaction. After methylation to give 83 and desilylation to give 84, the second dearomatisation furnishes hydroxyketone 85 without interfering with the functionality elsewhere in the molecule. A second syn-selective addition of a lithiated aryl (in this case formed from 49), followed by methylation, gave 5-ring precursor 87. Ring closure is effected by the same dual intermolecular/intramolecular Suzuki–Miyaura strategy employed for the synthesis of [7]CPP, albeit under different conditions. The low yield for this step is once again attributed to appreciable ring strain in the CPP precursor product 88. Finally, reductive aromatisation gave [6]CPP. Yamago had previously calculated37 that [6]CPP has a formidable strain energy of 407 kJ mol−1 (cf. 357, 307, 280 and 247 kJ mol−1 for [7]–[10]CPP, respectively). Crystal structures were obtained both for precursor 88 and for [6]CPP itself (Fig. 31). An unusual aspect of [6]CPP concerns its crystal packing – as shown in Fig. 32, it assembles into a linear alignment in the solid state, akin to a supramolecular nanotube. Nanosized channels through the crystal lattice are clearly visible. (In contrast, the previously acquired structures for [9] and [12]CPP exhibited a herringbone packing structure).
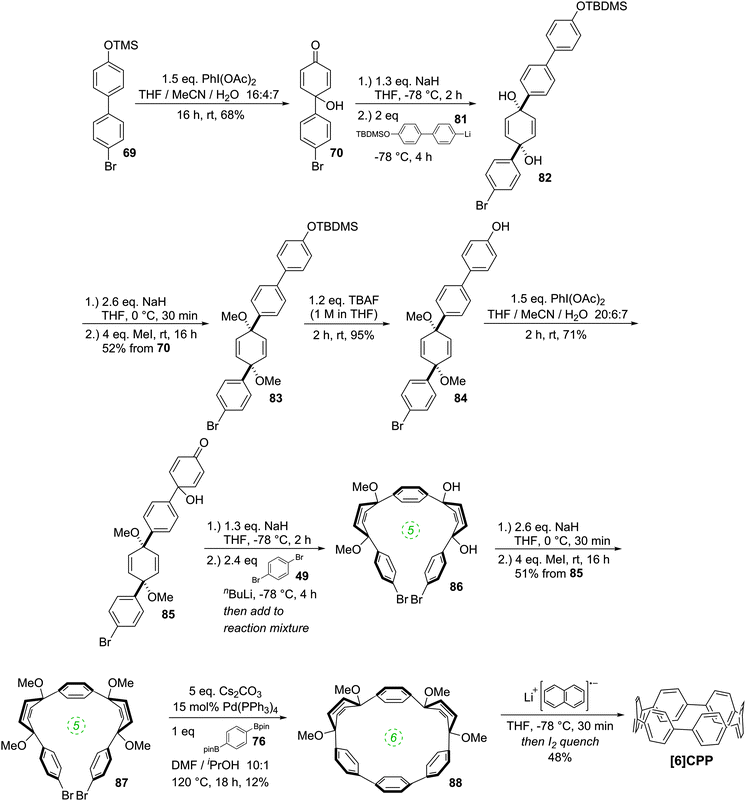 |
| | Scheme 22 Jasti's synthesis of [6]CPP. | |
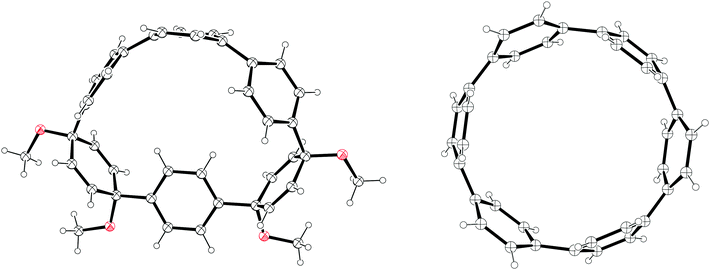 |
| | Fig. 31 ORTEP diagram of 88 and [6]CPP, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. A second, disordered molecule of 88 in the unit cell has been omitted for clarity. | |
 |
| | Fig. 32 Crystal packing of [6]CPP, 3 × 3 × 3 unit cells. Hydrogen atoms have been omitted for clarity. | |
Upon characterisation of [6]CPP, it was found to exhibit several characteristics which were not in keeping with trends observed for the larger CPPs. For example, while [6]CPP had an absorption maximum at 338 nm (essentially the same as larger CPPs), it was not fluorescent. This represents the continuation of a trend insofar as the quantum yield reported for [7]CPP was very low in contrast to larger CPPs. The chemical shift in the 1H-NMR spectrum of [6]CPP is also anomalous. Thus far, a correlation had been observed between CPP ring size and chemical shift, i.e. the smaller the ring, the more upfield the resonance. However, the signal for [6]CPP is in fact shifted 0.16 ppm downfield relative to [7]CPP. Finally, [6]CPP was also characterised electrochemically and found to have a half-wave oxidation potential of 0.44 V.
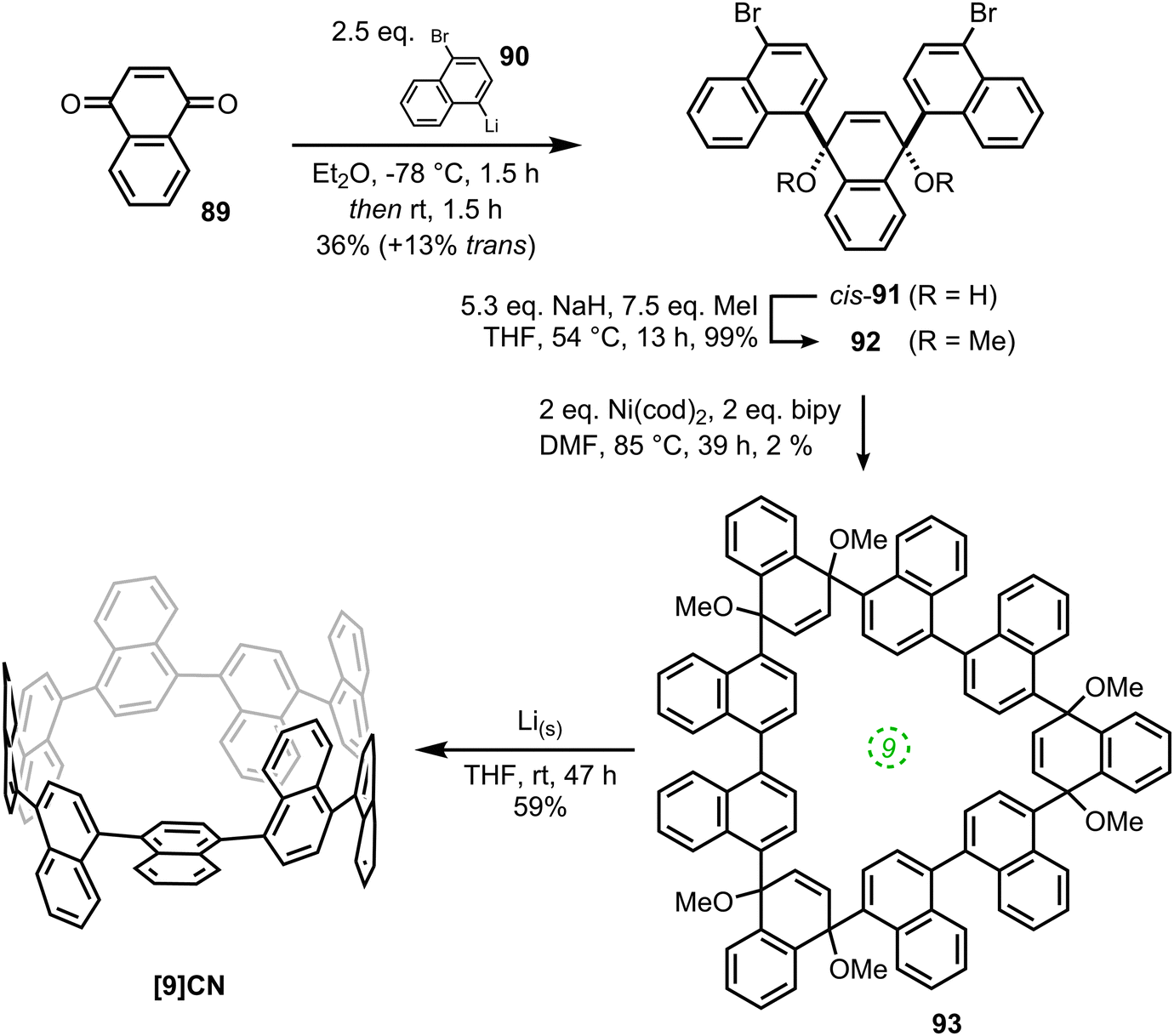
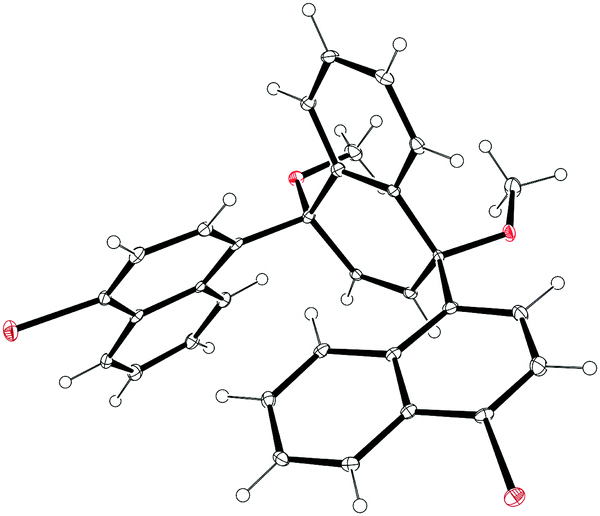
The second paper published in 2012 was from the Itami group and concerned the synthesis of a multiply-annulated CPP derivative, specifically [9]cyclo-1,4-naphthylene (“[9]CN”).45 They employed the nickel-mediated “shotgun” approach which had previously allowed access to [12]CPP (Scheme 14) and [9]CPP (Scheme 16). The synthetic approach is shown in Scheme 23. Treatment of naphthoquinone 89 with an excess of lithiated naphthalene 90 gave linear precursor 91, with the desired syn diastereomer favoured by 11:4. Methylation afforded 92, for which a crystal structure was obtained (Fig. 33). From this structure, it was determined that the “included” angle was ≈71°, i.e. smaller than for the non-annulated analogue 39. On this basis, it was predicted that the “shotgun” cyclisation would favour formation of a trimer over a tetramer. Further in support of this expectation, modelling of cyclic trimer 93 suggested it had near-negligible strain energy (6 kJ mol−1). In the event, 93 was indeed the only cyclic oligomer formed, although the yield was very low (2%), with formation of linear oligomers predominating. Careful optimisation of the reaction concentrations and the conditions for purification were required even to achieve this isolated yield. With 93 in hand, the Itami group attempted its reductive elimination to [9]CN, but the use of lithium naphthalenide failed in this case. It was eventually determined that the use of granular lithium metal allowed for the production of [9]CN and its isolation in 59% yield.
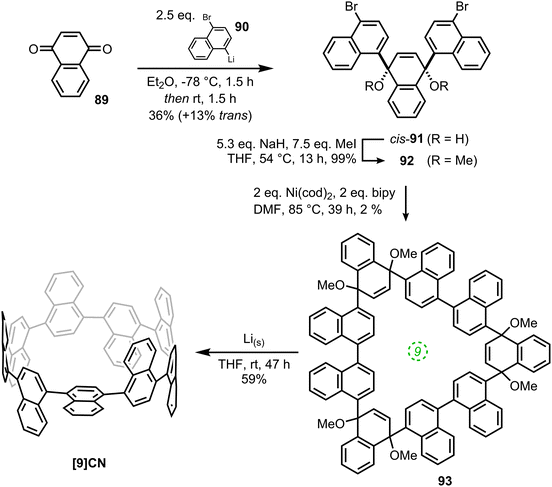 |
| | Scheme 23 Itami's synthesis of [9]CN. Adapted with permission from ref. 45. Copyright 2012 American Chemical Society. | |
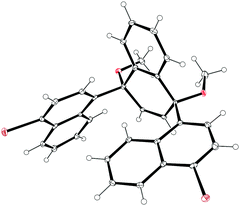 |
| | Fig. 33 ORTEP diagram of 92, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
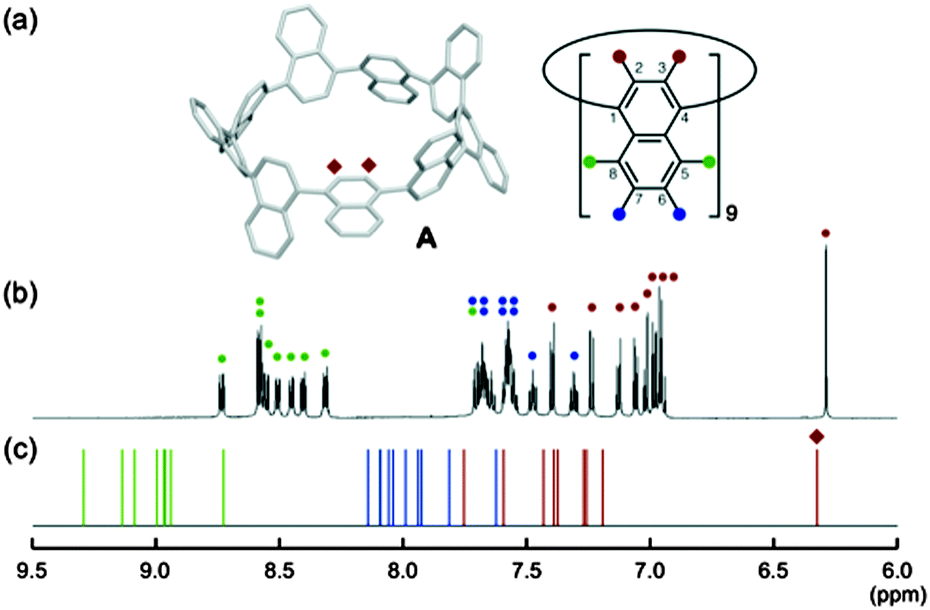
The NMR spectrum of [9]CN is markedly different from that of the CPPs – rather than a singlet, the spectrum comprises many overlapping resonances, indicative of a structure of low symmetry and of slow interconversion of conformers on the NMR timescale. The spectrum is shown in Fig. 34. When heated to 150 °C in DMSO-d6, the spectrum coalesced to three broad singlets, corresponding to the three environments shown as red, green and blue circles in Fig. 34a, indicating fast interconversion of conformers. However, at room temperature, the molecule adopts a structure of C2 symmetry as shown in Fig. 34a – as this is an odd-numbered nanoring, a simple conformation of alternately oriented rings is not possible and a 3-helical motif (cf.Fig. 13b) must be incorporated. The central naphthyl unit of this motif projects two hydrogens into the centre of the nanoring and these account for the unusually upfield singlet which is observed (Fig. 34c, red diamond). Overall, 27 discrete environments are present for the 54 hydrogens in [9]CN. Concerning absorption and emission spectra, [9]CN exhibited λmax = 378 nm (i.e. a longer wavelength than [9]CPP), with several other maxima. [9]CN was fluorescent, with λem = 491 nm, almost identical to the value for [9]CPP.
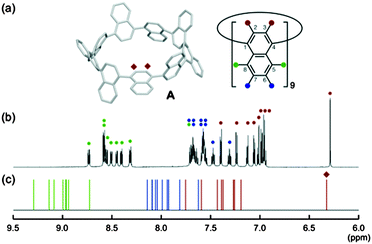 |
| | Fig. 34 (a) Energy minimised structure of [9]CN. (b) 1H-NMR spectrum of [9]CN in THF-d8. (c) Predicted 1H-NMR chemical shifts of [9]CN calculated at the B3LYP/6-11+G(2d,p)//B3LYP/6-31G(d) level. Reproduced with permission from ref. 45. Copyright 2012 American Chemical Society. | |
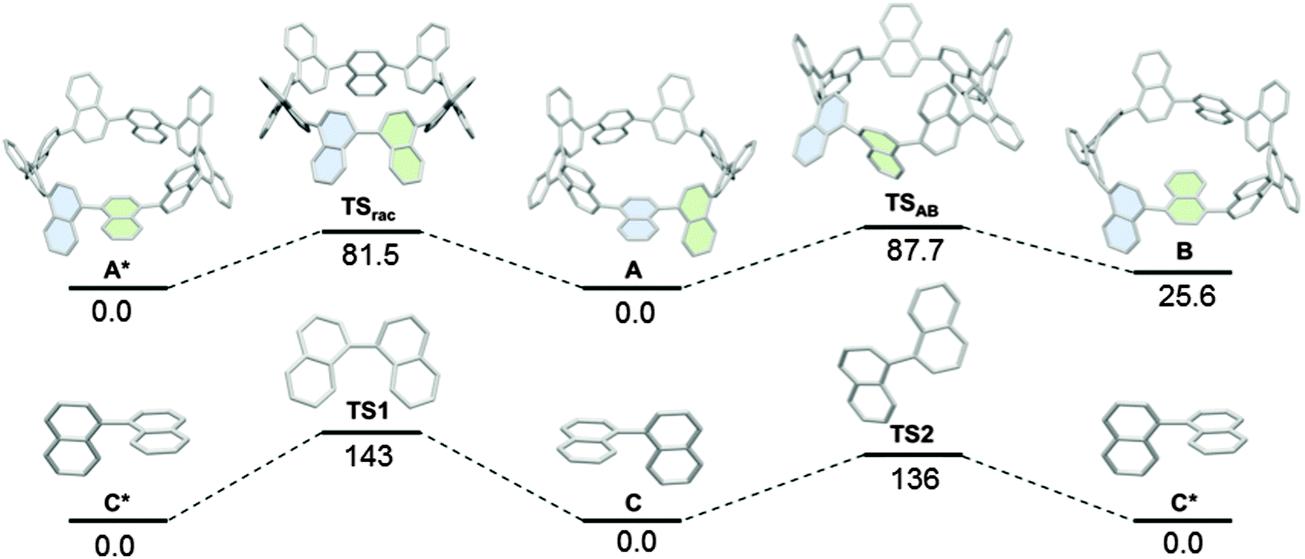
Considering the conformer A in Fig. 34a, it can be seen that [9]CN is in fact a chiral molecule. Computer modelling was undertaken by the Itami group to study the interconversion of enantiomers; racemisation of 1,1′-binaphthyl was studied at the same level of theory for comparison (Fig. 35). Thus, the barrier to racemisation from [9]CN-A to its enantiomer [9]CN-A*via the transition state TSrac was calculated to be 81.5 kJ mol−1, which perhaps surprisingly is lower than the corresponding barrier for racemisation of 1,1′-binaphthyl. Itami ascribes this to the ring strain present in [9]CN.
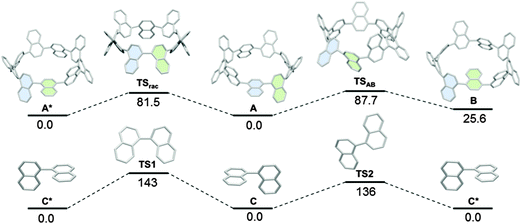 |
| | Fig. 35 Rotation pathways of (top) [9]CN and (bottom) 1,1′-binaphthyl. Values are relative Gibbs free energies (ΔG) in kJ mol−1 at 298.15 K and 1 atm calculated at the B3LYP/6-31G(d) level. The symbol * represents the enantiomer of the corresponding structure. Adapted with permission from ref. 45. Copyright 2012 American Chemical Society. | |
Shortly after Itami's [9]CN publication, Jasti and Sisto published a commentary on their synthesis of [7]CPP, setting it in the context of the work that had come before.46 There then followed a theoretical study from Irle, Morokuma and co-workers on the mechanism of “bottom-up” SWCNT growth from a CPP template.47 The authors studied a mechanism of SWCNT growth from a CPP that proceeds by addition of ethynyl radicals, and find this process to be more energetically favourable than the alternative of a Diels–Alder based growth mechanism as proposed previously by Scott.48,49 They state that the ˙C2H radicals in fact have two roles in the SCWNT elongation process, both abstracting hydrogen from the growing SCWNT and also providing the source of carbon for its growth.
Itami published an account in early 2012 of his group's efforts towards controlled synthesis of SCWNTs and nanographenes,50 which was quickly followed by a paper from the Isobe group on the kinetics of isomerisation of the isomers of [4]CC2,8 that they had reported the previous year (Fig. 28).51 As shown in Fig. 36, when pure [4]CC2,8-F was heated to 80 °C in toluene, interconversion between [4]CC2,8 isomers occurred, with equilibrium being reached after ≈50 h. It can be seen that isomer [4]CC2,8-E was formed first, but the quantity of this then diminished after ≈4 h, as other isomers were also formed. This strongly suggests that [4]CC2,8-F interconverts into [4]CC2,8-E in the first instance and then on to the other isomers. This initial interconversion involved the rotation of only one of the chrysene units in [4]CC2,8, which suggests the units rotate one by one. Isobe's group were also able to derive various thermodynamic parameters for the equilibration process. On the same day, Nakano and co-workers published a computational study contrasting CPPs with linear oligophenylenes, as well as cyclic and linear acenes.52 They employed long-range corrected spin-unrestricted density functional theory (LC-UDFT) to investigate the diradical character of the species under study, finding that CPPs and linear oligophenylenes have closed shell configurations, whereas both linear and cyclic acenes have singlet diradical character. The group also calculated the various third-order non-linear optical polarisabilities.
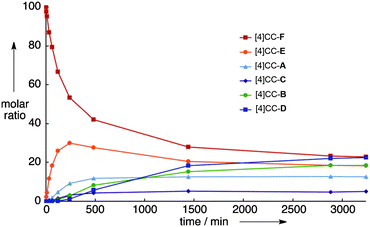 |
| | Fig. 36 Time-course analysis of isomerization of [4]CC2,8-F in toluene at 80 °C. Adapted with permission from ref. 51. Copyright 2012 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim. | |
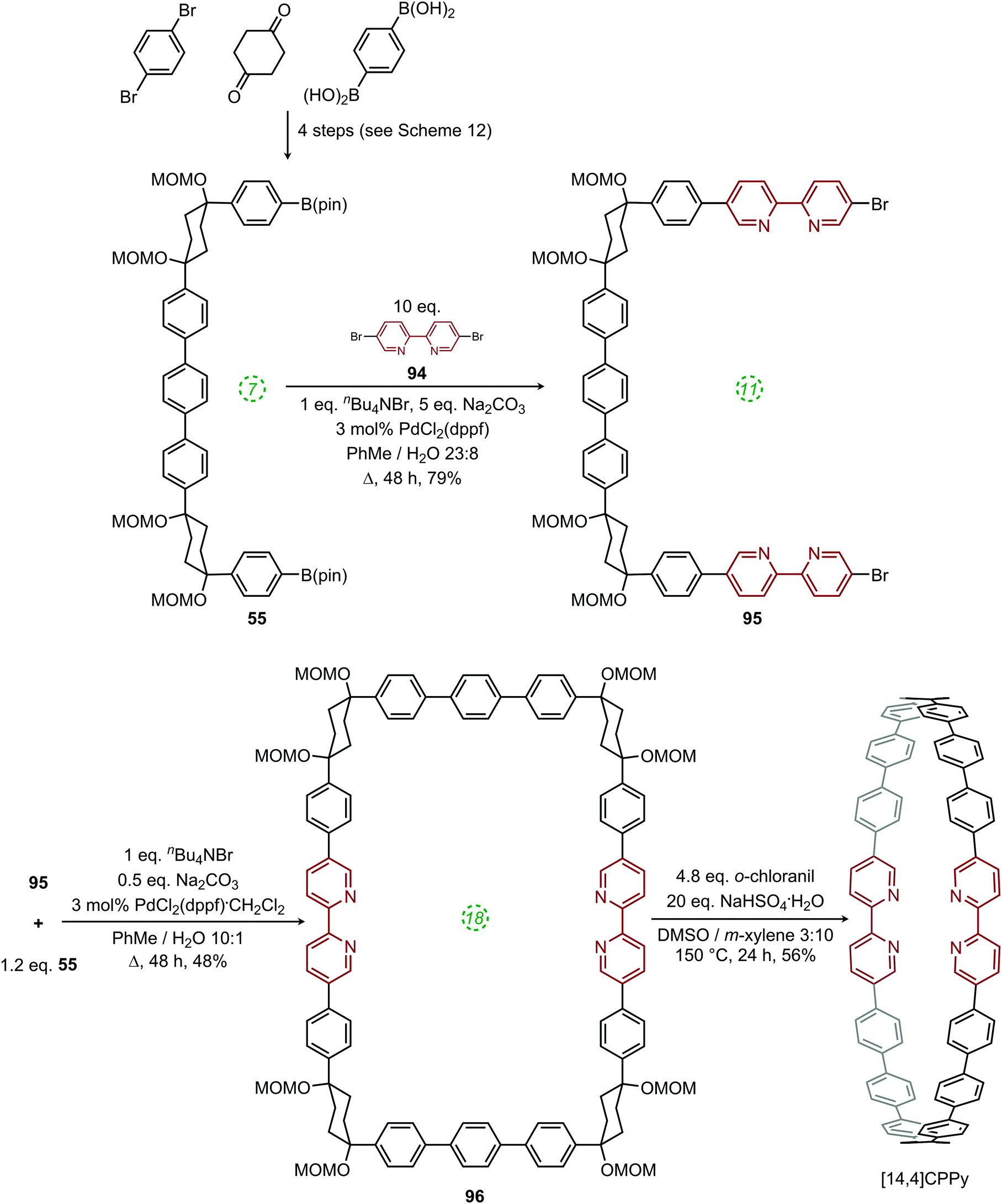
A week later, Itami published the first report of a heteroatom-containing CPP derivative.53 The molecule in question, cyclo[14]paraphenylene[4]2,5-pyridylidene, was abbreviated as “[14,4]CPPy” and its synthesis is shown in Scheme 24. Previously reported U-shaped building block 55 was elaborated with two equivalents of 5,5′-dibromo-2,2′-bipyridyl 94 to give extended U-shaped building block 95. The dual intermolecular/intramolecular Suzuki–Miyaura macrocyclisation of 95 with a further equivalent of 55 gave CPPy precursor 96. Finally, oxidative aromatisation gave [14,4]CPPy; in a change from Itami's previous reports, this last step involved the explicit addition of an oxidant (ortho-chloranil). With [14,4]CPPy in hand, the group set about the characterisation of this “tetraaza-CPP” and its absorbance and fluorescence spectra are shown in Fig. 37.
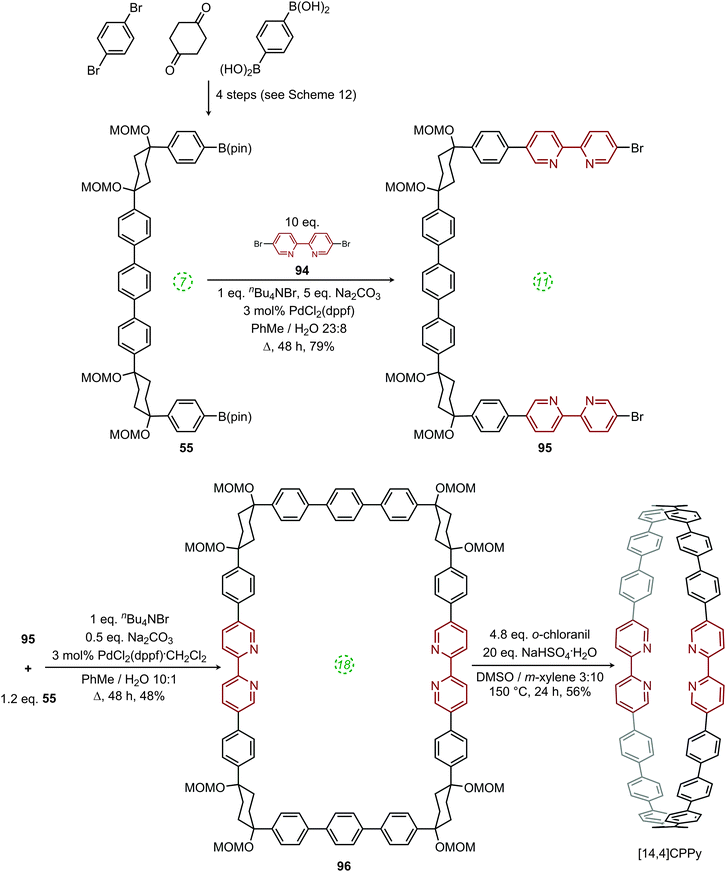 |
| | Scheme 24 Itami's synthesis of [14,4]CPPy. | |
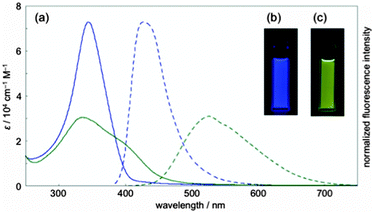 |
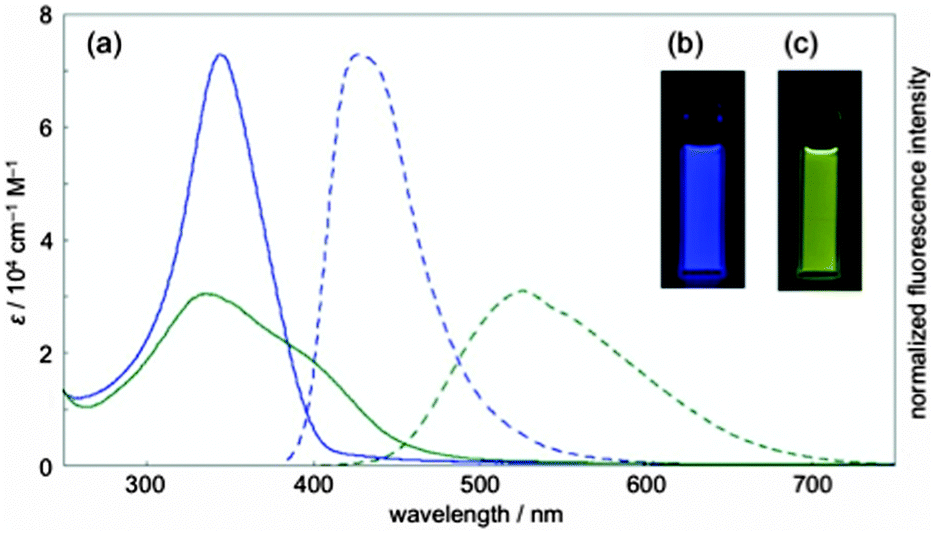
| | Fig. 37 (a) UV/vis absorption (solid lines) and fluorescence spectra (broken lines) of dichloromethane solution of [14,4]CPPy (blue lines) and after adding HCl (green lines). (b) Fluorescence of [14,4]CPPy. (c) Fluorescence of [14,4]CPPy + HCl. Reproduced with permission from ref. 53. Copyright 2012 American Chemical Society. | |
[14,4]CPPy has λmax = 344 nm and overall has an absorption spectrum very similar to the larger CPPs. In its fluorescence spectrum, an emission was observed at λem = 427, similar to the behaviour of [18]CPP. However, the presence of the heteroatoms in [14,4]CPPy has the potential to modulate significantly its properties and the Itami group found that protonation with HCl greatly altered the optical properties, inducing a large bathochromic shift (see Fig. 37). Finally, in preliminary experiments, Itami and co-workers were able to demonstrate the use of [14,4]CPPy as a transition metal ligand, forming a 1:2 complex with palladium(II).
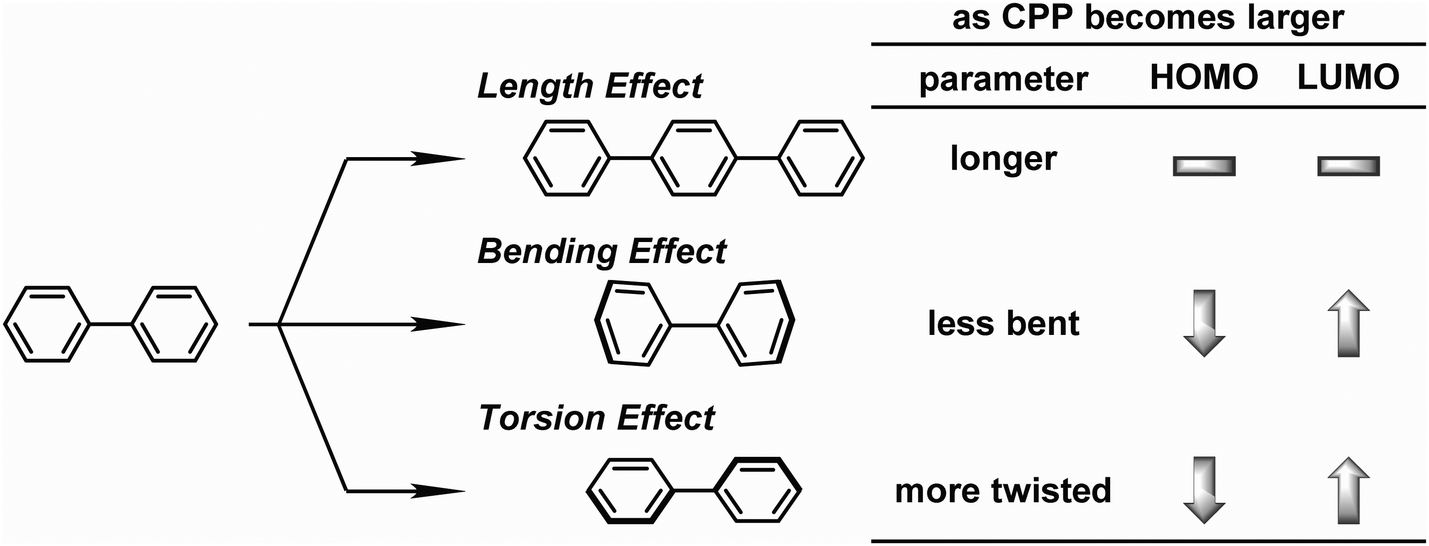
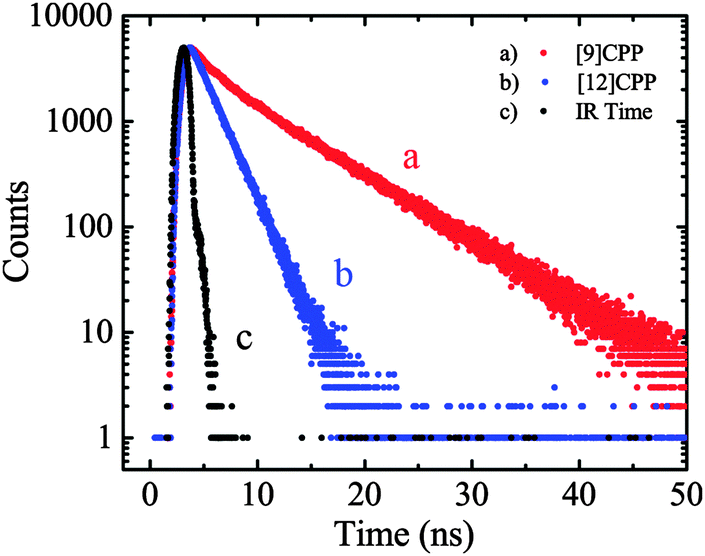
On the same day the above study was published, Itami also published the results of a collaborative study with Yamaguchi, Irle et al. on the photophysical properties of CPPs.54 This study combined experimental and theoretical approaches and many new data were presented. Although the invariance of λmax for the most intense absorption had previously been noted, the group also studied the longest-wavelength absorption maxima. These correspond to the forbidden HOMO → LUMO transition and are therefore of much lower intensity than the most intense absorption, (and so are not readily visible in Fig. 23a). They found that these maxima are blue-shifted with increasing the ring size (from 395 nm for [9]CPP to 365 nm for [16]CPP), as are the emission maxima in the fluorescence spectra (from 494 nm for [9]CPP to 438 nm for [16]CPP). Quantum yields are experimentally determined for [9], [12], [14], [15] and [16]CPP and are high in all cases (0.73 ≤ ΦF ≤ 0.90), in contrast to Jastis data for [6] and [7]CPP. These values increase slightly in a polymer matrix, but are greatly lowered in the solid state. Fluorescence lifetimes are also reported: τS = 10.6 ns for [9]CPP and 2.2 ns for [12]CPP. As regards the various structural factors which influence the energies of the HOMO and LUMO, the key findings of this work are summarised in Fig. 38.
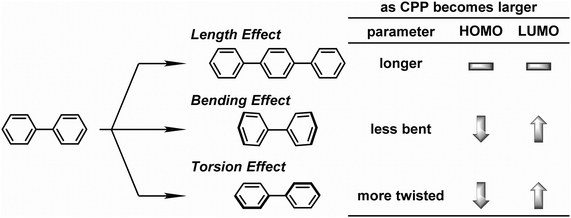 |
| | Fig. 38 Length, bending, and torsion effects in HOMO–LUMO energies of CPPs. Adapted with permission from ref. 54. Copyright 2012 Royal Society of Chemistry. | |
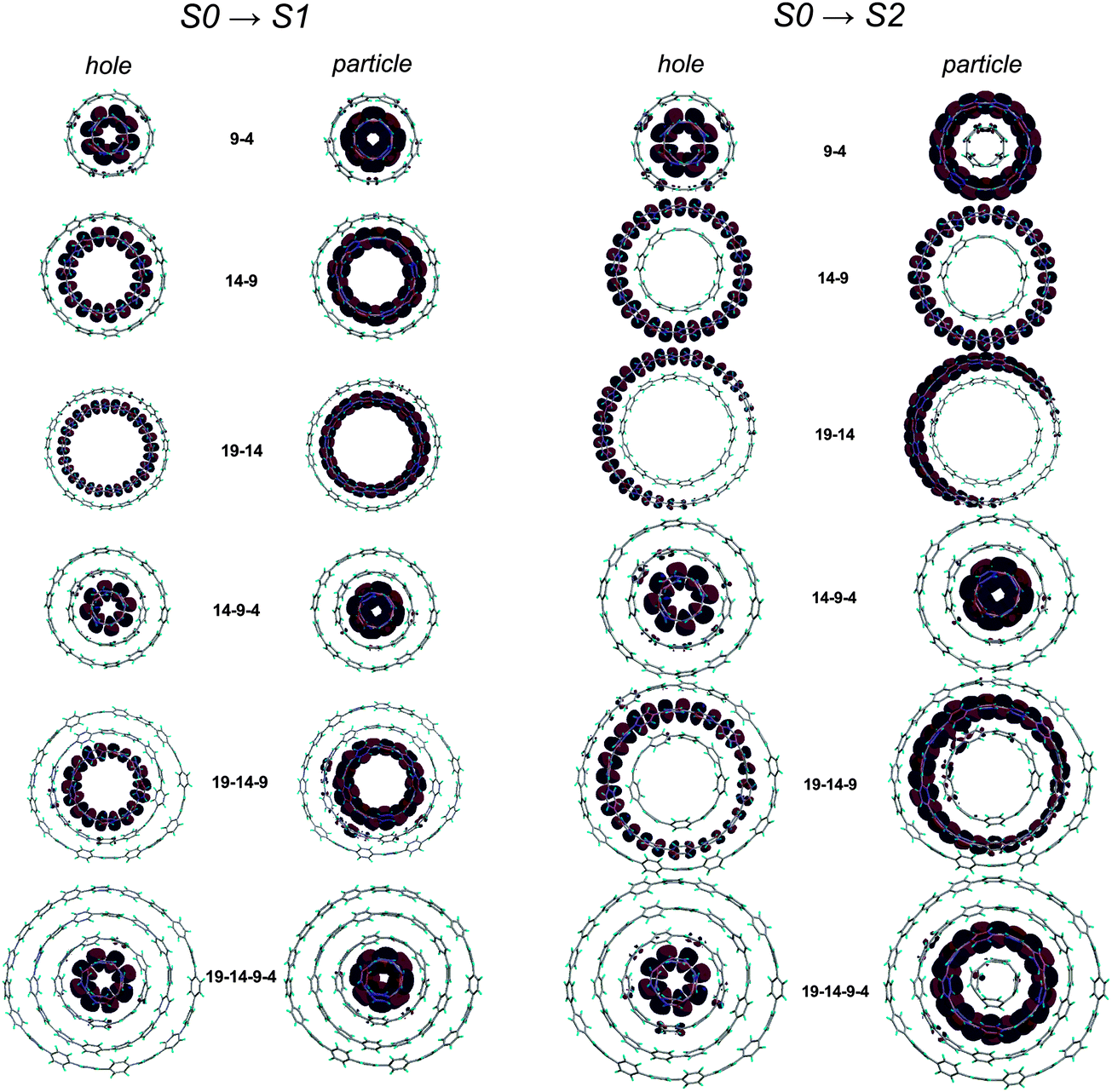
A computational study by Fomine and co-workers examined the intriguing possibility of forming “Russian doll” supramolecular complexes of two, three or four differently sized CPPs.55 Simple geometrical considerations suggested that an inclusion complex would ideally be formed between two CPPs differing in size by five phenylene units (it is stated this is a universal rule, regardless of specific ring size). Thus, the complexes to be studied were chosen to be combinations of [4], [9], [14] and [19]CPP. Their electronic structures of were computed at the M06-2X/6-31G* level of theory. The dominant natural transition orbital pairs for the S0 → S1 and S0 → S2 transitions are shown in Fig. 39. Fomine calculates that significant binding energies can exist, and can be >280 kJ mol−1 for complexes containing [14] and [19]CPP. While [4]CPP remains unknown (and may well remain so, given its calculated strain energy of >600 kJ mol−1!), experimental validation of the proposed inclusion complexes involving [9], [14] and [19]CPP ought to be entirely viable; a recent report by López Navarette, Baonza and Casado does in fact present indirect evidence for such an encapsulation (vide infra, Fig. 95).
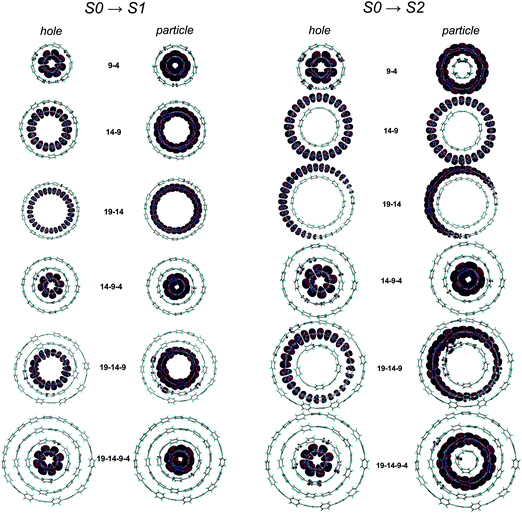 |
| | Fig. 39 The dominant natural transition orbital pairs for S0 → S1 (left half) and S0 → S2 (right half) transitions in Russian doll complexes. In each case, the “hole” is on the left, the “particle” on the right. Adapted with permission from ref. 55. Copyright 2012 Springer-Verlag. | |
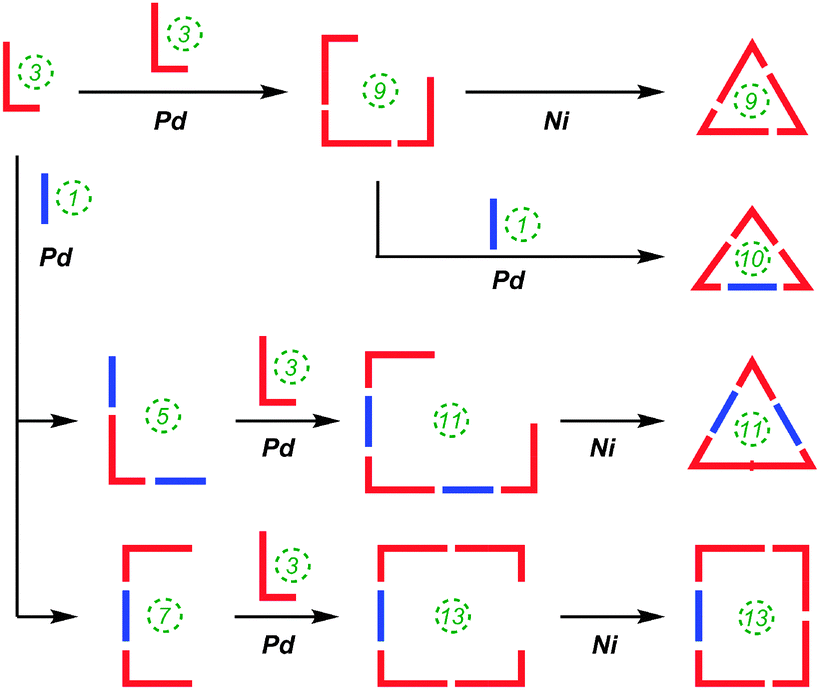
Also in 2012, Itami reported on the successful combination of his two previously described methodologies (i.e. Pd-catalysed and Ni-mediated couplings) to effect selective syntheses of [9], [10], [11] and [13]CPP using hybrid methodology.56 The versatility of this approach is that it allows for many more possible combinations of Itami's previously-reported building blocks, given that the Pd-catalysed couplings join a nucleophilic and an electrophilic functionality, whereas the Ni-mediated couplings join two electrophilic fucnctionalities. The approach is represented schematically in Scheme 25.
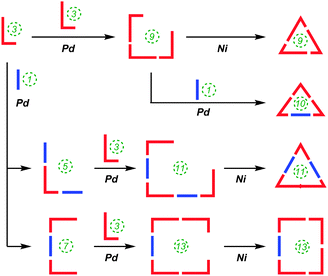 |
| | Scheme 25 Schematic representation of Itami's hybrid Pd/Ni approach to [9]–[11] and [13]CPP. | |
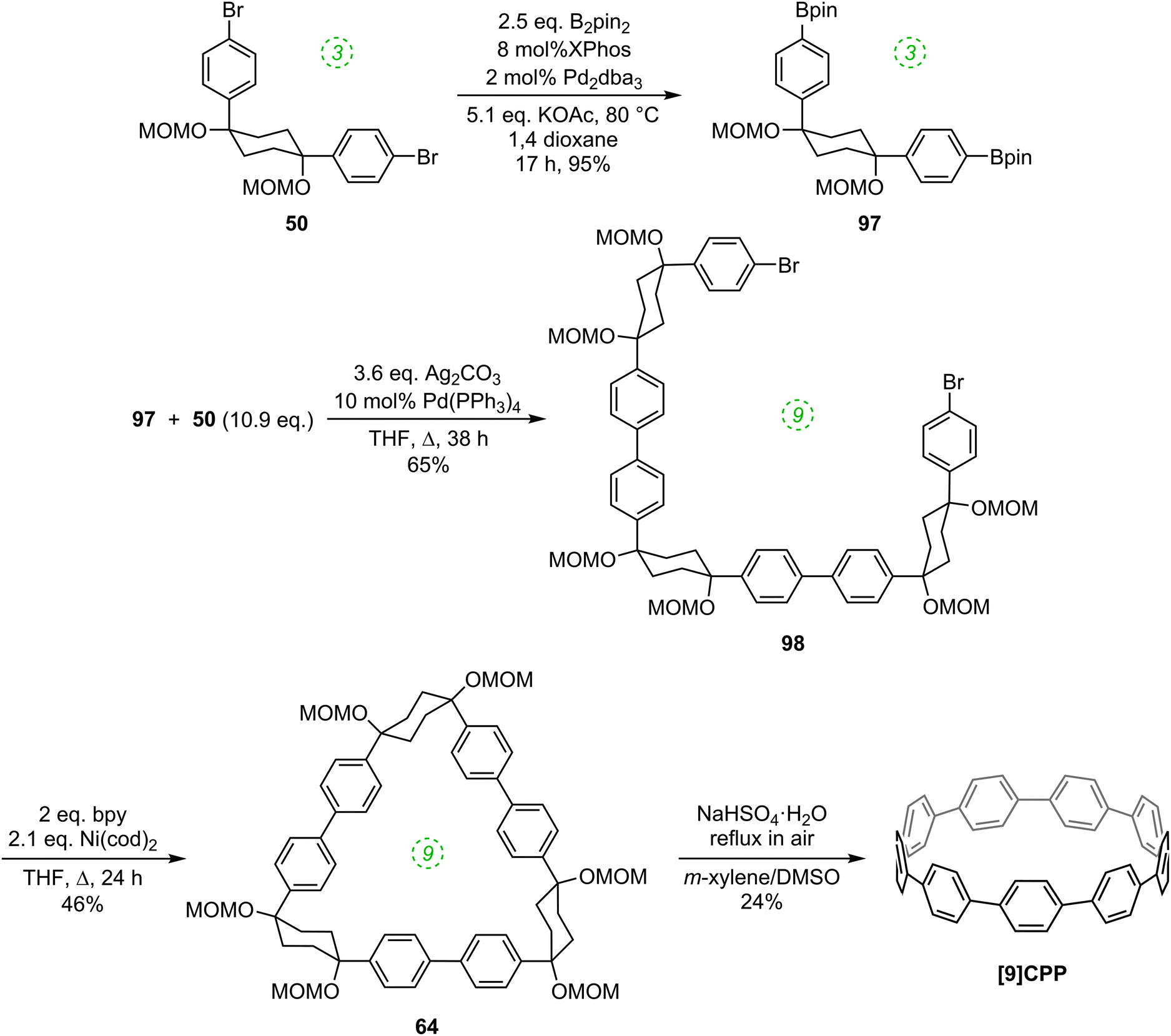
Thus, whereas the nickel-only “shotgun” approach to [9]CPP (Scheme 16) also yielded the [12]CPP precursor 60, the hybrid approach allows for a selective approach to [9]CPP only, as shown in Scheme 26. Previously known L-shaped building block 50 underwent Miyaura borylation to give difunctionalised Bpin building block 97. These two building blocks were then combined, with 50 in a large excess, to furnish acyclic 9-ring building block 98 by means of two intermolecular Suzuki–Miyaura couplings, with unreacted 50 being recovered in good yield. The reductive nickel-mediated coupling was then employed to cyclise 98 cleanly to give known macrocycle 64 only, whose transformation to [9]CPP had previously been demonstrated.
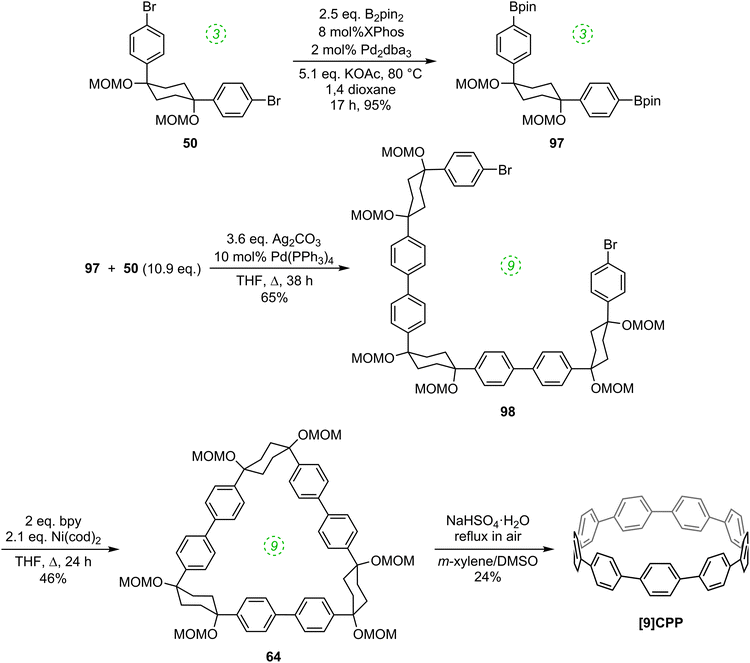 |
| | Scheme 26 Itami's selective hybrid synthesis of [9]CPP. | |
A nice illustration of the versatility of this hybrid approach is given in Scheme 27, where only a very minor modification to the approach to [9]CPP instead allows the selective synthesis of [10]CPP (for the first time). Thus, acyclic 9-ring building block 98 was combined with 1-ring fragment 76via the dual intermolecular/intramolecular Suzuki–Miyaura process to give macrocyclic 99. A crystal structure was obtained for 99, which is shown in Fig. 40. Oxidative aromatisation of 99 gave [10]CPP as expected. In a similar vein, syntheses of [11]CPP (Scheme 28) and [13]CPP (Scheme 29) were also effected using previously known building blocks; both of these CPP ring sizes were previously unknown. In the same publication, the Itami group also reported alternative syntheses of [14] and [16]CPP utilising nickel-mediated couplings, whereas their previous routes (Scheme 12) employed only palladium-catalysed couplings. In all, following the publication of this report, routes to all CPPs from [6]CPP to [16]CPP, as well as [18]CPP had been established.
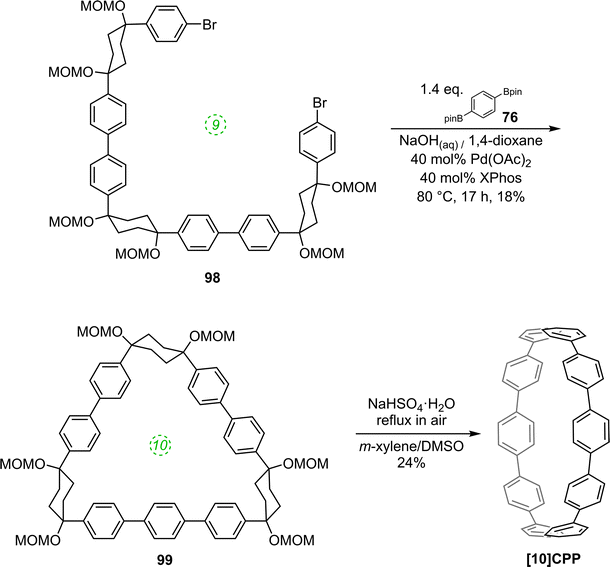 |
| | Scheme 27 Itami's selective hybrid synthesis of [10]CPP. | |
 |
| | Fig. 40 ORTEP diagram of 99·2C6H6, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
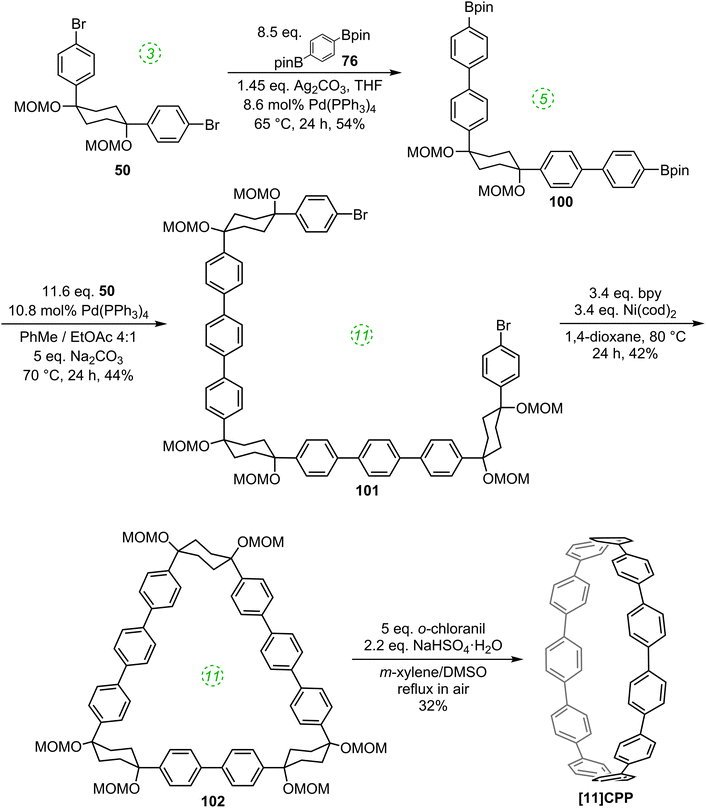 |
| | Scheme 28 Itami's selective hybrid synthesis of [11]CPP. | |
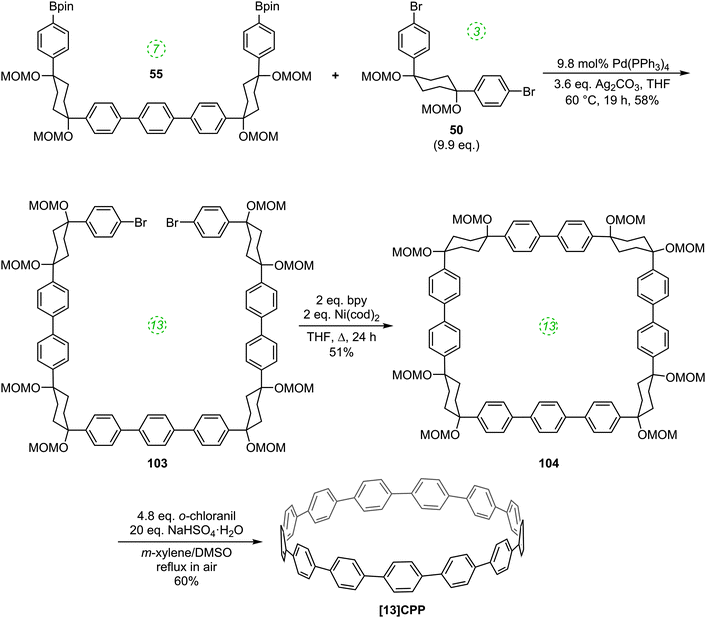 |
| | Scheme 29 Itami's selective hybrid synthesis of [13]CPP. | |
A short time after the above disclosure, Itami published a personal account of his group's endeavours in the field of CPPs.57 This in turn was followed by a report from Yamago of a selective (and unexpected) synthesis of [10]CPP.58 Building on their previous successes with square-planar platinum(II) complexes, the Yamago group sought to exploit a palladium-mediated coupling reaction reported by Osakada.59 As shown in Scheme 30, from 4,4′-diiodobiphenyl 105, it proved possible to prepare monostannyl building block 106via a selective single lithium–halogen exchange reaction. Upon treatment with half an equivalent of a platinum(II) source, 2:1 complex 107 was formed in good yield by transmetallation. Complex 107 may be thought of as a 4-ring L-shaped building block for CPP synthesis and its oligomerisation ought therefore to give rise ultimately to [8]CPP (by cyclodimerisation), [12]CPP (by trimerisation) or [16]CPP (by tetramerisation), etc. Surprisingly, however, applying Osakada's conditions for reductive biaryl formation with palladium(0) furnished [10]CPP (and no other CPPs). Yamago advances a mechanism to account for this unexpected selectivity, in which an acyclic trimer of 107 (which would possess 12 phenylene units) undergoes cyclisation with concomitant loss of a biphenyl unit, giving a macrocyclic 10-ring triplatinum complex which then undergoes reductive elimination to [10]CPP. In addition to the surprising selectivity of this reaction, it is also noteworthy that the reductive elimination of platinum to give the CPP product occurs spontaneously, whereas in Yamago's previous reports, addition of an oxidant was required to induce such a reductive elimination. The Yamago group also attempted cyclooligomerisation of 107 using bis(cyclooctadiene)nickel(0), which gave a mixture of [8], [10], [12] and [16]CPP, more in line with their initial expectations, albeit in lower yield than the palladium-mediated process. The Yamago group were also able to obtain an X-ray crystal structure of [10]CPP, which is shown in Fig. 41.
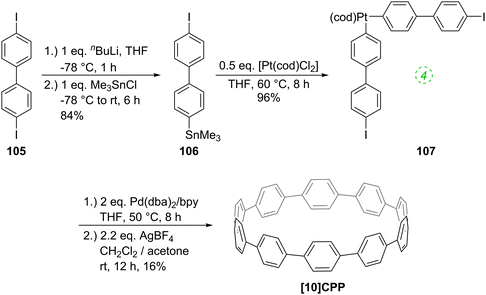 |
| | Scheme 30 Yamago's serendipitous synthesis of [10]CPP. | |
 |
| | Fig. 41 ORTEP diagram of [10]CPP˙nhexane, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
Also in 2012, Jasti extended the chemistry of CPPs to derivatives bearing aryl substituents.60 The rationale for wanting to do so was that such molecules could potentially serve as precursors for the synthesis of ultrashort SWCNTs via an oxidative cyclisation approach. The target of Jasti's study, 108 (Fig. 42) was of interest as a simplified model of 109, which upon controlled oxidation could conceivably furnish ultrashort SWCNT 110. The synthesis of 108 exploits Jasti's previously developed methodology, although with some interesting variations.
 |
| | Fig. 42 Targets related to SWCNTs: (a) [12]CPP. (b) Tetraphenyl-[12]CPP. (c) Strategy for accessing an ultrashort SWCNT. Reproduced with permission from ref. 60. Copyright 2012 American Chemical Society. | |
As shown in Scheme 31, the route commences with arylboronic acid 111. A four-fold Suzuki–Miyaura coupling with tetrabromoquinone 112 gave tetraarylquinone 113. This in turn was treated with para-iodophenyllithium 114 to afford cyclohexane-1,4-diol 115. Notably, the reaction was rather selective for the desired syn isomer of the product, even without the prior deprotonation of the hydroxyls using sodium hydride (as was required in other cases, cf.Schemes 20 and 22). Permethylation of 115 gave three-ring building block 116, which was then incorporated into a macrocyclisation sequence analogous to those Jasti had developed previously. Reaction with an excess of differentially functionalised building block 74 gave 9-ring acyclic precursor 117, with the aryl chloride functionalities remaining unchanged under the comparatively mild conditions employed.
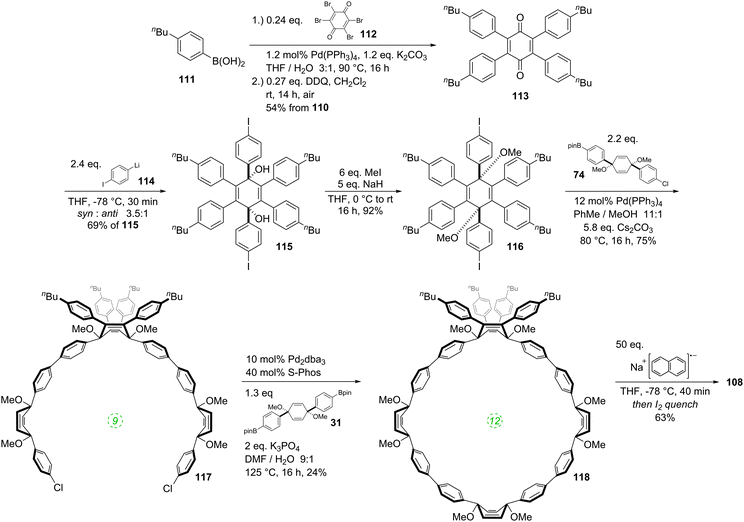 |
| | Scheme 31 Jasti's synthesis of tetraphenyl-[12]CPP 107. | |
Prior to attempting the synthesis of macrocycle 118, Jasti and co-workers were concerned that the presence of the heavily substituted cyclohexadiene ring might bias the conformational equilibrium away from the reactive conformer required. In the event, however, use of 3-ring nucleophilic building block 31 under the more forcing conditions for Suzuki–Miyaura coupling developed previously gave the desired CPP precursor 118 in a superior yield to those obtained for 77 or 88 (Jasti's precursors to [7]CPP and [6]CPP respectively). This is most likely due to the lower ring strain that would be present in 118. Sodium naphthalenide then effected reductive aromatisation of 118 to give the target tetraphenyl-[12]CPP 108. This is a slight departure from previously reported conditions for such a reductive aromatisation, where lithium naphthalenide (Schemes 8, 20 and 22) or granular lithium metal (Scheme 23) were used; clearly the optimal reductant varies with the CPP (or derivative) in question. In its UV/vis absorption spectrum, 108 exhibited an absorption maximum at 328 nm, not dissimilar to [12]CPP itself, but a second maximum at 240 nm was also observed, which Jasti ascribes to the phenyl substitution. 108 is fluorescent, with a Stokes shift close to that of [12]CPP and an identical quantum yield. The use of n-butyl substituents in the structure of 108 is not commented on by Jasti, but it is reasonable to surmise that they were included to avoid problems with solubility.
Jasti followed the above paper with a report on the large-scale synthesis of [8]CPP and [10]CPP.61 The study was motivated by the fact that all previous reported syntheses had furnished milligram quantities of CPPs; minimising the cost of the reagents used was also a major imperative. The new routes employed building blocks Jasti had reported previously, namely 5-ring dibromide 87 (used in the synthesis of [6]CPP, Scheme 22) and 3-ring diborylated building block 31 (used in the original CPP syntheses of Bertozzi and Jasti, Scheme 8). The Jasti group were able to optimise further the synthesis of 87, such that it could be prepared in 30% overall yield; they prepared >20 g of 87 in one batch. Combination of 87 and 31 gave 8-ring macrocycle 119, reductive aromatisation of which using sodium naphthalenide gave over a gram of [8]CPP in a single batch (Scheme 32). In addition, transformation of 87 into doubly nucleophilic 5-ring building block 120 was effected in good yield. Combination of 87 and 120 in a 1:1 ratio gave macrocyclic 10-ring CPP precursor 121, which underwent reductive aromatisation to give [10]CPP (albeit in not quite as good a yield), again furnishing over a gram of material in a single batch.
 |
| | Scheme 32 Jasti's gram-scale syntheses of [8] and [10]CPP. | |
With such quantities of material in hand, the Jasti group were able to secure X-ray crystal structures of both [8] and [10]CPP (Yamago's structure of [10]CPP, Fig. 41, had not been published at the time the Jasti group were carrying out this work). The structure obtained for [8]CPP is shown in Fig. 43. Excitingly, the group were also able to secure a structure for the inclusion complex [10]CPP⊃C60 (Unit cell, Fig. 44; packing structure, Fig. 45). It is insightful to contrast the structures of complexed and free [10]CPP. In the former, the average dihedral angle between adjacent phenylene units is 28.5 ± 2.5°, whereas for free [10]CPP, it is 27.3 ± 11.7°. The value for [10]CPP is affected by a single ring exhibiting dihedral angles of 47.1° and 48.6°; no such ring is present in the structure of [10]CPP⊃C60. Jasti attributes this difference to the efficient π–π interactions present in [10]CPP⊃C60 (which are presumably increased with diminishing dihedral angles). Nevertheless, an important caveat is that both Jasti and Yamago's structures of [10]CPP are solvated (which may influence the dihedral angles), whereas the structure of [10]CPP⊃C60 is not.
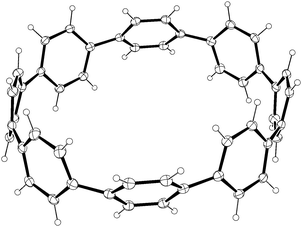 |
| | Fig. 43 ORTEP diagram of [8]CPP, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
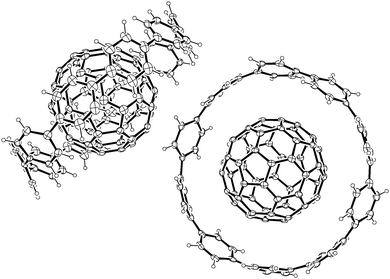 |
| | Fig. 44 ORTEP diagram of [10]CPP⊃C60, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
 |
| | Fig. 45 Crystal packing of [10]CPP⊃C60, 3 × 3 × 3 unit cells. Hydrogen atoms have been omitted for clarity. | |
Isobe's second publication of 2012 in the field of CPP derivatives concerned a synthesis of [4]cyclo-3,9-chrysenylene (“[4]CC3,9”).62 At first glance, the key concept behind this work appears comparatively straightforward, namely taking the methodology Isobe had previously described for the synthesis of [4]CC2,8 (Scheme 21) and modifying the points of attachment of the chrysenylene unit. However, both the realisation of this synthesis, as well as the properties of the products, differed appreciably from Isobe's previous reports. Whereas use of 2,8-diboryl building block 79 had previously given tetraplatinum complex 80 in good yield (Scheme 21), in the current case, using isomeric 3,9-diboryl building block 122, comparable reaction conditions furnished only a complex mixture, from which no discrete species could be isolated and identified. Accordingly, the Isobe group took the crude mixture of products and subjected it to the conditions known to effect reductive elimination (Scheme 33). Whilst this did produce the desired [4]CC3,9, the yield of only 8% over two steps is in marked contrast to the ease of synthesis of [4]CC2,8. Another key difference between this work and the previously reported access to [4]CC2,8 is that whereas all six possible isomers of [4]CC2,8 were isolated, only two isomers (an enantiomeric pair) of [4]CC3,9 were obtained in the present case.
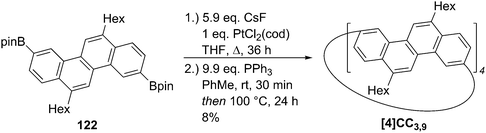 |
| | Scheme 33 Isobe's synthesis of [4]CC3,9. | |
The six possible isomers of [4]CC3,9 are depicted in Fig. 46. Purification of the crude mixture by preparative chiral HPLC gave two different fractions, which were shown to be enantiomeric by their NMR and CD spectra. Furthermore, the presence of nine aryl 13C-NMR resonances and four aryl 1H-NMR resonances implied D4 symmetry and the products were therefore identified as (+) and (−)-(16,0)-[4]CC3,9 (the two structures on the left of Fig. 46).
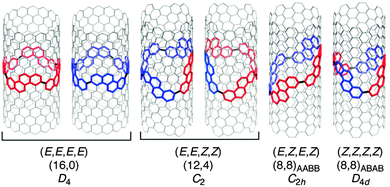 |
| | Fig. 46 Possible isomers of [4]CC3,9 embedded in SWCNTs. Two possible chrysenylene orientations are shown in red and blue. Configurations at the single-bond linkage are denoted as “E” or “Z”; chiral indices and point symmetry are also shown for each structure. Reproduced with permission from ref. 62. Copyright 2012 American Chemical Society. | |
In the case of [4]CC2,8, Isobe had previously shown that interconversion of isomers was possible at elevated temperature. In contrast, (−)-(16,0)-[4]CC3,9 did not undergo any isomerisation even after eight weeks at 200 °C; computer modelling failed to locate any transition states for isomerisation processes. Isobe and co-workers were able to crystallise (16,0)-[4]CC3,9 as the racemate and to obtain an X-ray crystal structure (Fig. 47). The structure consists of an “entangled supramolecular assembly”, whereby hexyl side-chains of one enantiomer occupy the central cavity of the other enantiomer, a so-called “thread-in-bead” structure.
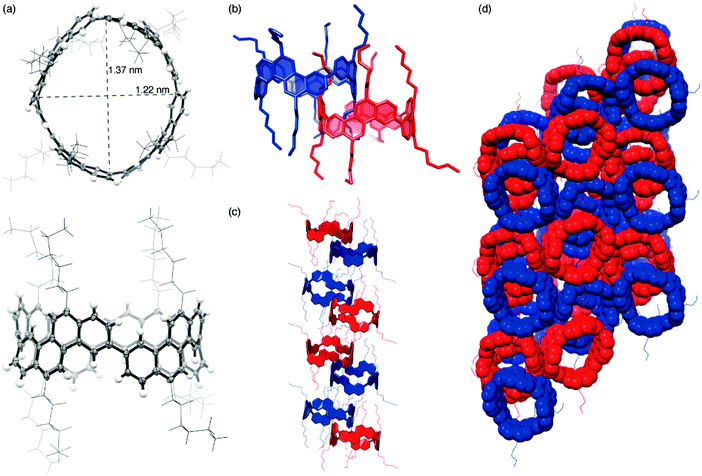 |
| | Fig. 47 Molecular and packing structures of (16,0)-[4]CC3,9. (a) Molecular structures of (+)-(16,0)-[4]CC3,9. Hexyl substituents are shown in wireframe; chrysenylenes are shown as ORTEP plots with thermal ellipsoids at 50% probability. Top and side views are shown. (b) An interwoven pair of enantiomers viewed from side. (c) Columnar array of the paired enantiomers. The pairs are alternately stacked, and four pairs are shown. (d) Packing structure of the columns viewed along the columnar direction. (+)- and (−)-isomers are shown in red and blue, respectively. Reproduced with permission from ref. 62. Copyright 2012 American Chemical Society. | |
Later in 2012, Jasti published a report of selective syntheses of all CPPs from [7]CPP to [12]CPP.63 The approach is highly modular and consolidates the various methodologies his group had developed prior to this point. As shown in Scheme 34, combination of the three previously reported building blocks 31, 73 and 74 allowed concise access to two key building blocks, 6-ring dichloride 123 and 9-ring dichloride 124. Both of these may then be combined with either a 1-ring (76), 2-ring (125) or 3-ring (31) building block under essentially the same conditions (use of the S-Phos ligand) to effect the dual intermolecular/intramolecular Suzuki–Miyaura macrocyclisation. This in turn gave rise to a series of macrocyclic precursors (77, 126–130), all of which underwent reductive aromatisation to give the full set of CPPs from [7]CPP to [12]CPP.
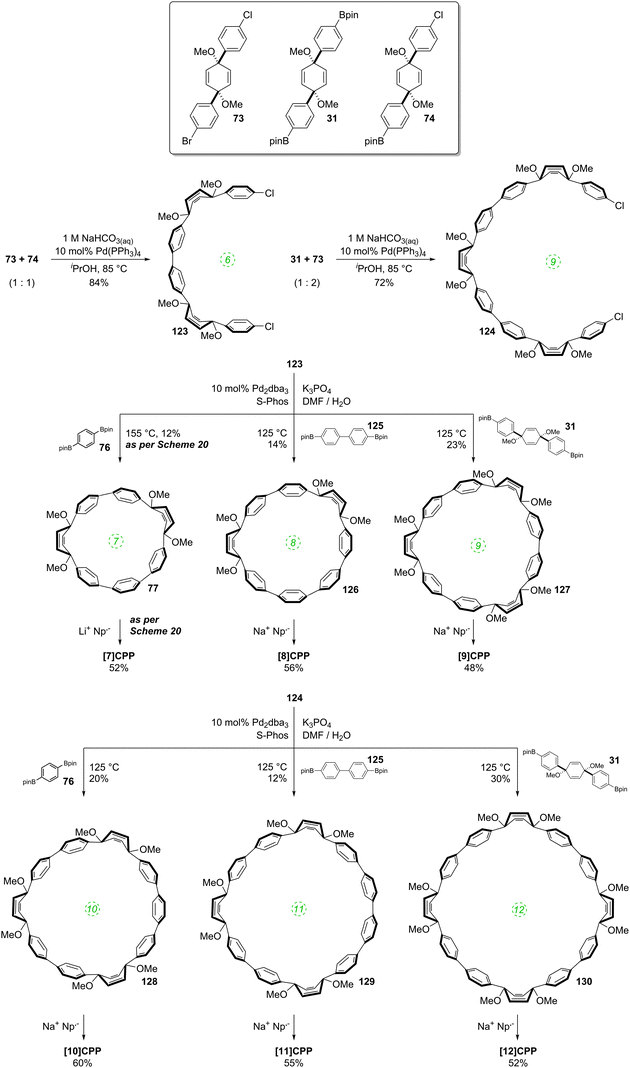 |
| | Scheme 34 Jasti's modular syntheses of [7] to [12]CPP. | |
In August, Irle, Morokuma and co-workers published a full paper describing computational studies on mechanisms of SWCNT growth from CPP templates.64 This work followed from their previous disclosure on this topic,47 and in this case gave consideration to the effects of nanotube diameter and chirality on the rate of nanotube growth. Their computations found that for the ethynyl radical-mediated SWCNT growth mechanism they had previously proposed, the chirality of the growing nanotube had a profound effect on the rate, whereas the diameter did not influence the rate. Upon modelling the alternative Diels–Alder SWCNT growth mechanism, they found that in this case the rate was highly dependent on nanotube diameter. They also found that that the ability of a SWCNT to avoid defect formation during growth is an intrinsic quality of the SWCNT edge.
Also in August, Kamada, Itami and co-workers reported the synthesis of a so-called “carbon nanocage”, a 3D variant of a CPP which could act as a Y-branching unit in SWCNTs.65 The relationship between the target molecule 131 and a SWCNT junction is shown in Fig. 48. Synthetic access to 131 relied on Itami's previously developed palladium and nickel coupling strategies, as shown in Scheme 35.
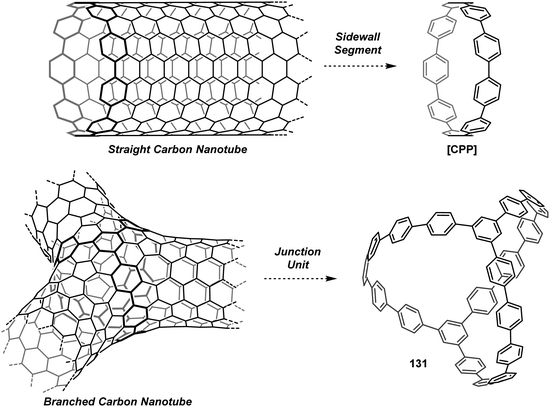 |
| | Fig. 48 A representative CPP and a carbon nanocage as segments of straight and branched SWCNTs. Reproduced with permission from ref. 65. Copyright 2013 Royal Society of Chemistry. | |
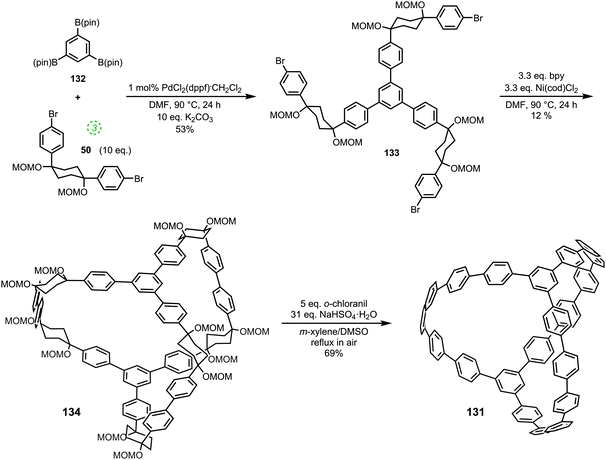 |
| | Scheme 35 Synthesis of 131. | |
Computational modelling of 131 determined the ground state to have D3 symmetry; the optimised structure is shown in Fig. 49. The two nodal phenyl rings are offset by an angle of 12.5° and the cavity diameter is 18.4 Å, which is intermediate between the values for [13] and [14]CPP. The strain energy of 131 was calculated to be 67.2 kJ mol−1, very close to the value for [9]CPP. In its absorption spectrum, 131 exhibited λmax = 325 nm; 131 was fluorescent with emission maxima at 418 and 431 nm and ΦF = 0.87. The calculated frontier orbitals for 131 are shown in Fig. 50. In contrast to the CPPs themselves, the HOMO → LUMO transition for 131 is symmetry-allowed. A two-photon absorption for 131 is also discussed.
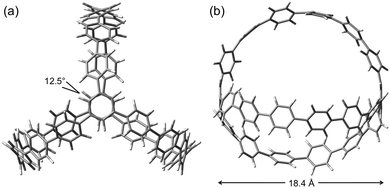 |
| | Fig. 49 Optimised structure of 131 (B3LYP/6-31G(d)). Reproduced with permission from ref. 65. Copyright 2013 Royal Society of Chemistry. | |
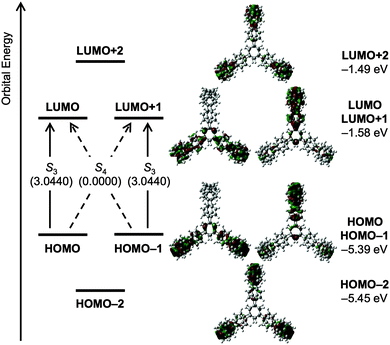 |
| | Fig. 50 Energy diagrams and pictorial representations of the frontier MOs of 131 calculated at the B3LYP/6-31G(d) level of theory. Excitation energies were computed by TD-DFT at the same level. Values in parentheses represent oscillator strengths (f). Reproduced with permission from ref. 65. Copyright 2013 Royal Society of Chemistry. | |
The issue of the unusual photophysical properties of CPPs was the subject of a group of studies toward the end of 2012. First of all, Majima and co-workers studied the fluorescence properties of CPPs,66 determining not only the Stokes shifts and quantum yields as previously, but also the fluorescence lifetimes of various CPPs, using the fluorescence upconversion method. They found upon decreasing the CPP ring size, as well as larger Stokes shifts and lower quantum yields, the CPPs also exhibited longer fluorescence lifetimes. This last trend is the same as that which is observed for linear oligoparaphenylenes. Shortly after, Kanemitsu also published on the photophysical properties of CPPs.67 This study employed one-photon and two-photon photoluminescence excitation spectra to determine the transition energy of the (optically forbidden) HOMO–LUMO gap. The dependence of the HOMO–LUMO gap with CPP ring size was found to be identical to that of the photoluminescence energy. The third study, from Irle and co-workers, attempted to rationalise thoroughly the relationship between CPP ring size and Stokes shift.68 They found that excitation from the S0 state to the S2 and S3 states induces Jahn–Teller distortion of the CPP shape from circular to oval. They identified large vibrational amplitudes in smaller CPPs, leading to greater Stokes shifts, whereas larger CPPs are more rigid. They also note that symmetry rules are violated to a greater extent in smaller CPPs and that this likely also contributes to the observed Stokes shifts.
In late 2012, Jasti published two papers on consecutive days, a review on routes to CPPs,69 followed by the first report of the synthesis of CPP dimers.70 Jasti's group targeted dimers of [8]CPP with a linking arylene between the two CPP rings. Two different linkers (para-phenylene and 1,5-naphthylene) were used, giving access to two dimers 135 and 136, both of which constitute more extensive fragments of a SWCNT than a single CPP alone (Fig. 51). Key to accessing these dimers was the modification of Jasti's previous methodology to introduce an additional functional handle onto one of the non-aromatic rings of the macrocyclic CPP precursor. As shown in Scheme 36, the handle for further functionalization (an alkenyl bromide) is present from the very first step: bromoquinone monoketal 137 undergoes attack by aryllithium species 114, followed by deprotection to give hydroxyketone 138. This is then subjected to deprotonation to form the corresponding alkoxide, addition of another equivalent of the same aryllithium 114 and exhaustive methylation in a one-pot procedure to provide 3-ring L-shaped building block 139 (simply the brominated analogue of the building block 30 used in the very first CPP synthesis). This is coupled under ligand-free conditions with 5-ring diboryl building block 120 to provide 8-ring macrocycle 140 (itself the bromo analogue of 119). Notably the alkenyl bromide remained unreacted during the macrocyclisation, but use of different conditions allow a two-fold Suzuki–Miyaura reaction with a diboryl building block: coupling of para-phenylene linker 76 with two equivalents of 140 gave precursor 141, which underwent the usual reductive aromatisation to give target para-phenylene-linked [8]CPP dimer 135. Analogously, coupling of 1,5-naphthylene linker 142 with two equivalents of 140 gave precursor 143, which in turn gave 1,5-naphthylene-linked [8]CPP dimer 136.
 |
| | Fig. 51 Dimers of [8]CPP synthesised by Jasti. Reproduced with permission from ref. 70. Copyright 2012 American Chemical Society. | |
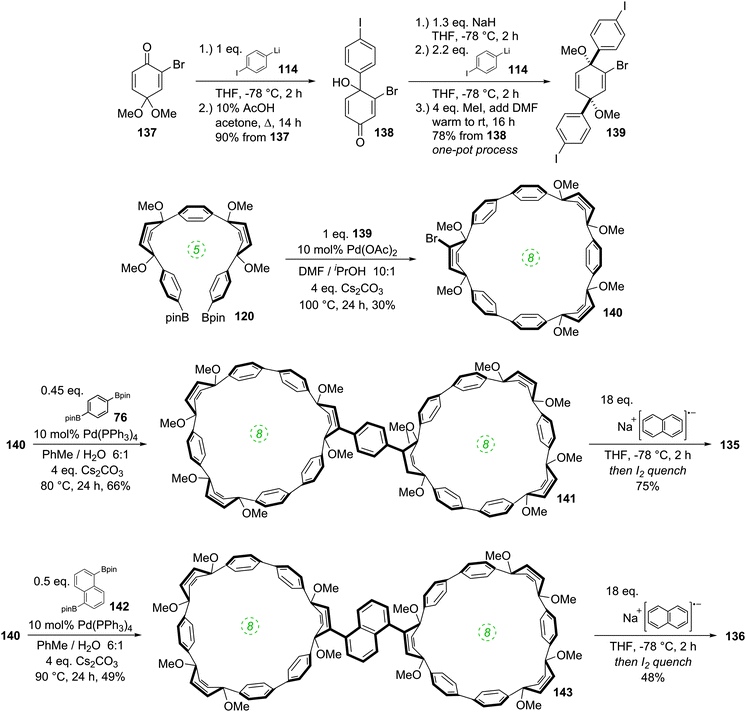 |
| | Scheme 36 Synthetic route to dimers of [8]CPP. | |
An alternative endgame to the synthesis in Scheme 36 can be envisaged: reductive aromatisation of 140 might plausibly furnish bromo-[8]CPP, which then could be cross-coupled with 76 or 142 to reach the same desired CPP dimers. Jasti makes no comment as to the plausibility of such an approach and the stability or otherwise of the C–Br bond in 140 under the single electron transfer conditions remains to be established. The photophysical characteristics of the dimers were probed and it was found that both 135 and 136 have both absorption and emission maxima that are extremely close to those of the monomeric [8]CPP. Interestingly, though, whereas the extinction coefficient for 135 is similar to [8]CPP, the 1,5-naphthylene-linked dimer 136 has an extinction coefficient which is nearly doubled. Attempts at growing X-ray quality crystals were hampered by the insolubility of 135 and 136; a preliminary structure was obtained for 135 which showed the two CPP rings to be trans oriented (vide infra), but this could not be refined for publication. Extensive computational studies on the new dimers were carried out, including modelling the reaction coordinate for the isomerisation between the cis and trans conformers of 135 (Fig. 52). In contrast to the solid state, computation predicts the cis conformer to be more stable in the gas phase due to the increased van der Waals interactions between the two CPP rings. It is interesting to note that the possibility of a directly-linked [8]CPP dimer (i.e. no intermediate arylene linker), while not discussed in the main text of Jasti and co-workers' article, has nevertheless been modelled and the results included in the ESI. Some time later, Itami and co-workers were in fact able to synthesise such a “true” dimer of [10]CPP (vide infra, Scheme 55).
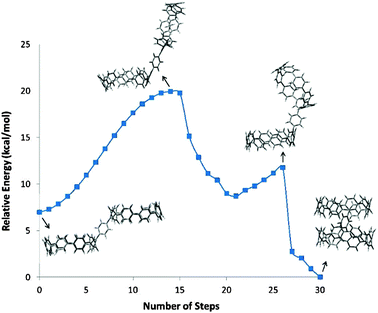 |
| | Fig. 52 Potential energy curve (B3LYP-D/6-31G(d,p)) of the trans to cis transition for 135. Reproduced with permission from ref. 70. Copyright 2012 American Chemical Society. | |
The final publication in 2012 came from Nishiuchi and Müllen, who sought to put into practice the idea of synthesising an ultrashort SWCNT by oxidative cyclisation of a polyarylated CPP precursor, an idea they had been exploring contemporaneously with the Jasti group (cf.Fig. 42).71 They targeted structures of general type 109 (Fig. 42), albeit with two unsubstituted phenylene linkers between the tetraarylated units instead of one. They did so using methodology inspired by the work of Itami and Jasti, but with significant additions of their own (Scheme 37).
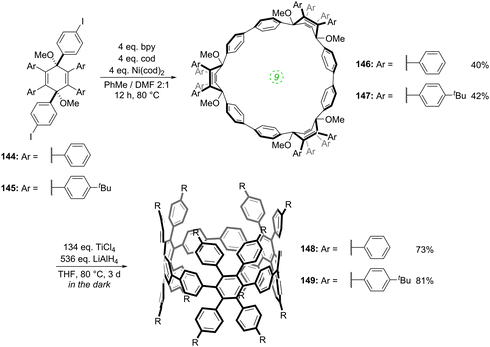 |
| | Scheme 37 Nishiuchi and Müllen's route to polyarylated CPPs. | |
L-shaped building blocks 144 and 145 were prepared using a route analogous to that devised by Jasti, before their nickel-mediated “shotgun” cyclooligomerisation to 146 and 147 in a similar fashion to the work of Itami. Mindful of Itami's previous syntheses of 60 and 64 (Scheme 16), Nishiuchi and Müllen anticipated the formation of both trimers and tetramers of 144 and 145, but in the event, only cyclotrimers 146 and 147 were obtained. They ascribe this result to the fact that 144 and 145 have been predicted computationally to have quite small “included” angles of ≈50°. Reductive aromatisation was then undertaken, but instead of the sodium naphthalenide which had become the Jasti group's reagent of choice by this time, Nishiuchi and Müllen employed low-valent titanium, which gave 148 and 149 in good yield. Structure optimisation of 148 (B3LYP/6-31G*) predicted a structure of C3 symmetry in which the CPP core was highly twisted, with dihedral angles of 51.2° and 78.7°. Crucially, such a twisted structure would not be expected to exhibit the extended π conjugation of the parent [9]CPP. The free rotation (or otherwise) of the phenylene units has implications for the plausibility of desired oxidative cyclisation of 148/149 to ultrashort SWCNT fragments. Thus, this rotation was investigated in a variable temperature NMR study (Fig. 53). The signals of interest (for protons labelled “a” and “b”) are observed as two doublets at 0 °C, whereas if rotation around the phenylene–phenylene linkage were slow on the NMR timescale, the protons protruding into the central cavity would be observed at a different chemical shift from those on the outside (i.e. four distinct resonances). Upon cooling, even to −90 °C, only a broadening effect is observed: “a” and “b” are still observed as only two resonances, implying fast rotation of the phenylene units even down to this low temperature. Crystal structures were obtained for both 148 and 149 (Fig. 54). In contrast to the computed gas phase structures, in the solid state, for 148, six of the twelve aryl substituents are oriented into the central cavity. In the case of 149, solvent molecules occupy the cavity, as the steric bulk of the tert-butyl groups precludes the orientation of aryl substituents directly into the cavity. Both structures are of C1 symmetry, with an oval distortion of the [9]CPP core. Strain energies were calculated and determined to be 393 kJ mol−1 for 148, which is appreciably higher than for the parent [9]CPP (274 kJ mol−1) and closer to the value for [6]CPP (402 kJ mol−1).
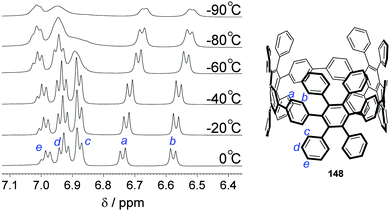 |
| | Fig. 53 Variable-temperature 1H-NMR spectra of 148 (500 MHz, CD2Cl2). Reproduced with permission from ref. 71. Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
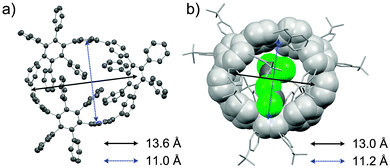 |
| | Fig. 54 Crystal structures of (a) 148 (ORTEP plot) and (b) 149 (the CPP core and the CH2Cl2 solvent are represented by a space-filling model and aryl substituents are represented as sticks). Hydrogens are omitted for clarity. Reproduced with permission from ref. 71. Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Oxidative cyclisation was then attempted for the CPP derivatives (Scheme 38). Of various oxidative conditions, it was established that treatment with iron(III) chloride in refluxing dichloromethane was optimal. In the case of 148, desired product 150 could not be identified in the crude reaction mixture; instead mass spectrometric evidence pointed to the presence of partially cyclised and chlorinated products. In the case of 149, the presence of the tert-butyl groups seemed to facilitate the oxidative cyclisation, perhaps due to their electron donating nature. Thus, 149 gave rise to a crude reaction mixture in which mass spectrometric signals corresponding to 151 could be observed, as well as a monochlorinated derivative of 151. However, signals corresponding to [151 + 2H]+, [151 + 4H]+ and [151 + 6H]+ were also observed (i.e. partial cyclisation products) and to date 151 has not been isolated in pure form. Use of more forcing conditions only lead to an increase in the formation of chlorinated side products. Nevertheless, this disclosure is significant in terms of advancing the field of de novo synthesis of SWCNTs.
 |
| | Scheme 38 Nishiuchi and Müllen's oxidative cyclisation of polyarylated CPPs to give model ultrashort SWCNTs. | |
2013
The year began with a publication from the Isobe group who demonstrated the exploitation of a CPP derivative to form a “molecular bearing”.72 Bearings are defined as consisting of two parts: an outer bearing shell and a “journal” which may roll freely within the shell. For their outer shell, Isobe and co-workers opted to use their previously reported [4]CC2,8, and for the journal, they employed a molecule of C60 (Fig. 55).
 |
| | Fig. 55 Structure of (P)-(12,8)-[4]CC2,8⊃C60, Isobe and co-workers' “molecular bearing”. | |
For the construction of an effective bearing, two criteria must be met. Firstly, the journal must be held tightly within the bearing in order to prevent its run-out from the bore. Secondly, however, despite the first constraint, the journal must be able to roll freely within the bore. Of course, the formation of an inclusion complex between [10]CPP and C60 had previously been reported (Scheme 19 and Fig. 25 and 44), but this system did not fulfil the first criterion – the association constant of log Ka = 3.8 was insufficient to stop the journal from running out of the bore on the NMR timescale. The Isobe group hypothesised that a [4]CC bearing shell would exhibit a higher Ka value, as the hindered rotation of the chrysenylene units (which they had previously demonstrated41) should impart a greater degree of (desirable) preorganisation to the structure of the bore. This indeed proved to be the case, and the association constant was calculated to be log Ka = 9.6 ± 0.2, higher than for any other supramolecular complex with C60 at that point. The fact that the journal did not run out from the bore was demonstrated by NMR. Thus, a 2:1 mixture of (P)-(12,8)-[4]CC2,8 and C60 showed two distinct sets of 1H-NMR resonances – one for the bore containing the C60 journal and one for the empty bore. (In contrast, for mixtures of [10]CPP and C60, a single averaged resonance was observed, indicative of exchange on the NMR timescale.) The other isomers of [4]CC2,8 (Fig. 28) exhibited slightly lower association constants, whereas (−)-(16,0)-[4]CC3,9 (Fig. 47) had an appreciably lower association constant of log Ka = 4.3 ± 0.0; Isobe attributes this weaker interaction to a smaller (sub-optimal) cavity size for (−)-(16,0)-[4]CC3,9 compared to the various isomers of [4]CC2,8. The alkyl side chains were not considered significant for binding.
To prove that the second criterion for a functioning bearing (unrestricted rolling of the journal in the bore) was also being met for (P)-(12,8)-[4]CC2,8⊃C60, the Isobe group again resorted to NMR data. As can be seen in Fig. 56 (left), an empty molecule of (P)-(12,8)-[4]CC2,8 possesses four distinct proton environments, due to its D4 symmetry. Four distinct resonances are indeed observed in the 1H-NMR spectrum: H1/H7 (singlet), H3/H9 (doublet), H4/H10 (doublet) and H5/H11 (singlet). In the case of the inclusion complex (P)-(12,8)-[4]CC2,8⊃C60, if the C60 guest were static, the complex would be of lower symmetry, giving rise to more inequivalent proton enviroments and more resonances in the 1H-NMR spectrum. However, the four resonances of (P)-(12,8)-[4]CC2,8 were not split upon formation of (P)-(12,8)-[4]CC2,8⊃C60, indicating that the C60 journal is freely rotating in the bore on the NMR timescale. This was still the case even at −60 °C.
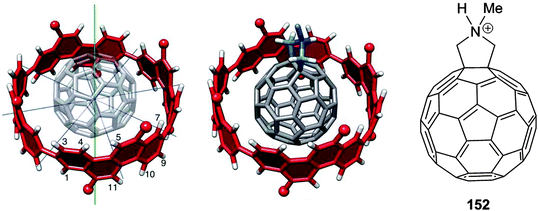 |
| | Fig. 56 Symmetry considerations for (P)-(12,8)-[4]CC2,8⊃C60 (left) and (P)-(12,8)-[4]CC2,8⊃152. Reproduced with permission from ref. 72. Copyright 2013 Royal Society of Chemistry. | |
Finally, the Isobe group explored the possibility of adding a “shaft” to the journal, by using C60 derivative 152 (Fig. 56, middle and right). Thus, N-methylfulleropyrrolidine was added to (P)-(12,8)-[4]CC2,8 (and protonated with trifluoroacetic acid to rigidify the “shaft” pyrrolidine ring and maintain the CS symmetry of the journal). If this bearing were static, the point group would be C1, but if the journal were free to rotate around the shaft, the point group would be C4. The observation of eight resonances in the 1H-NMR spectrum confirms the latter scenario, i.e. free rotation around the shaft means that the four chrysenylene units remain equivalent, but the “north” and “south” sides of the bore (i.e. the side with the shaft and the side without) are now inequivalent.
The second report of 2013 was a communication from Fujitsuka, Majima and co-workers describing the characterisation of CPPs by Raman spectroscopy.73 Examining [6] and [8]–[12]CPP, they were able to assign both inter-ring and intra-ring νCC stretches, as well as δCH in-plane bends. The wavenumbers for the νCC stretches were found to vary with CPP ring size, which the authors take as further evidence for the increasing levels of quinoidal character in the smaller CPPs. Jasti's first publication of 2013, was a review on molecular belts,74 quickly followed by a paper in collaboration with Petrukhina and co-workers, which describes the synthesis and characterisation of a CPP tetraanion.75 Upon exposure of [8]CPP to elemental potassium and 18-crown-6 in THF for 3 weeks, a solvated tetrapotassium complex was obtained, the X-ray crystal structure of which is shown in Fig. 57. The absorption spectrum exhibits λmax = 600 nm, significantly shifted from the λmax of neutral [8]CPP (340 nm). The crystal structure reveals a striking deviation from the neutral structure, in which the tetraanion adopts an ellipsoidal structure. It is able effectively to coordinate metal ions on both the inner and outer faces and the authors speculate that the ability to alter the shape of a nanobelt through redox processes may find application in host–guest chemistry.
 |
| | Fig. 57 ORTEP diagram of [{K(THF)([18]crown-6)+}{K+(K(THF)2+)([8]CPP4−)}-{K([18]crown-6)+}], showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
Isobe's second publication of 2013 concerned the synthesis of a “π-lengthened” belt-persistent cycloarylene, which in this case utilised an anthanthrenylene unit in place of a chrysenylene unit.76 The synthesis of this repeating unit is notable, in that it uses a readily available pigment 153 as the starting material (Scheme 39). Double nucleophilic addition and reductive aromatisation gave dibromides 154 and 155 with two different side chains. Palladium catalysed reductive removal of the halides (followed by hydrogenation for the hexynyl variant 157) gave substrates 156 and 158 for iridium catalysed C–H borylation. This was exceedingly regioselective, giving products 159 and 160 respectively. Such borylations are known to be sensitive to steric hindrance and Isobe's prior work afforded some precedent for the observed regiochemical outcome.77 Formation of tetraplatinum complexes 161 and 162 and their subsequent four-fold reductive elimination proceeded as previously, although more forcing conditions were employed in the last step.
 |
| | Scheme 39 Isobe's synthesis of [4]CA2,8 variants. | |
Production of the two [4]CA2,8 variants bearing different side-chains had unexpectedly divergent outcomes – the 6,12-bis[(triisopropylsilyl)ethynyl]-[4]CA2,8 was formed in only 2% yield, but as a racemic mixture of just a single diastereomer, (±)-(12,8)-[4]CA2,8. In contrast, the variant with the less sterically demanding side chains, 6,12-di-n-hexyl-[4]CA2,8, was formed in a higher 16% yield, as a mixture of all six possible stereoisomers (cf.Fig. 28). These were separated and identified by their CD and NMR spectra, as per Isobe's previous work (cf.Fig. 29). An X-ray crystal structure of 6,12-bis[(triisopropylsilyl)ethynyl]-(±)-(12,8)-[4]CA2,8 was obtained and is shown in Fig. 58. Isobe and co-workers probed the interconversion of the various stereoisomers of 6,12-di-n-hexyl-[4]CA2,8 as they had done previously for [4]CC2,8 (cf.Fig. 36). They were able to determine that 6,12-di-n-hexyl-[4]CA2,8 underwent isomerisation by the sequential rotation of one anthanthrenylene unit after another, as for [4]CC2,8 and that the enthaplic barrier to rotation for an anthanthrenylene unit was ΔH‡ ≈ +88 kJ mol−1, which surprisingly is ≈21 kJ mol−1 lower than the barrier to rotation of a chyrsenylene unit in [4]CC2,8.
 |
| | Fig. 58 ORTEP diagram of 6,12-bis[(triisopropylsilyl)ethynyl]-(P)-(12,8)-[4]CA2,8, showing ellipsoids at 30% probability. Hydrogens and isopropyl groups have been omitted for clarity. | |
It was in 2013 that another milestone in CPP chemistry was reached, when the idea of “bottom-up” synthesis of carbon nanotubes from a CPP template was demonstrated by Itami and co-workers.78 The experimental setup used by the Itami group to realise this feat is shown in Fig. 59a. A C-plane sapphire wafer which had been spin-coated with a solution of [12]CPP in toluene was heated to 500 °C and exposed to gaseous ethanol for 15 mins at a pressure of ≈1 Torr. Analysis of the wafer by transmission electron microscopy confirmed the formation of carbon nanotubes. While the nanotubes were not entirely uniform in diameter (and multiwalled structures were observed in addition to the targeted SWCNTs), the distribution of nanotube diameters (Fig. 59b, as determined from Raman spectroscopic data) showed a modal value of ≈1.6 nm, i.e. close to the diameter of [12]CPP (1.7 nm). Significantly, repetition of the experiment using [9]CPP as the template also led to nanotube formation (not shown), but in this case the distribution of nanotube diameters showed a smaller modal value of ≈1.3 nm, i.e. close to the diameter of [9]CPP (1.2 nm).
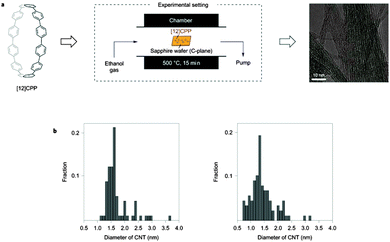 |
| | Fig. 59 (a) Schematic representation of experimental setup for Itami's synthesis of carbon nanotubes from CPPs; TEM image of nanotubes synthesised from [12]CPP. (b) Nanotube diameter distribution histograms for [12]CPP (left) and [9]CPP (right). Reproduced with permission from ref. 78. Copyright 2013 Nature Publishing Group. | |
Prior to Itami's work, two possible mechanisms for nanotube growth from a CPP template had been discussed in the literature. Scott has previously proposed a mechanism proceeding via an acetylenic Diels–Alder reaction, followed by oxidative aromatisation (Fig. 60, blue).48,49 Alternatively, the studies of Irle and Morokuma had focused on the possibility of a mechanism proceeding via an ethynyl radical47,64 (Fig. 60, green). The Itami group studied the stability of [12]CPP, exposing it to various temperatures for 15 mins, then acquiring infrared and fluorescence spectra. These indicated that major structural changes occurred at 450–500 °C and higher, which they ascribe to linear oligophenylene decomposition products arising from C–C bond cleavage. Given the similar bond dissociation energies for aryl C–C and C–H bonds, Itami speculates as to the possibility of a third mechanism for SWCNT growth, that proceeds via homolytic C–H bond scission at these elevated temperatures to give an sp2 carbon-centred radical (Fig. 60, red). This would then react with an unsaturated C2 species derived from ethanol to effect SWCNT growth. On the other hand, non-productive pathways such as dimerization of this radical can be envisioned, which could lead to CPP aggregates which in turn could act as templates for multi-walled nanotubes, branched structures, etc. While further optimisation is needed to improve the uniformity of the nanotubes produced by this method, this first demonstration of the viability of truly “bottom-up” SWCNT synthesis from CPPs is nevertheless a major landmark.
 |
| | Fig. 60 Possible mechanisms for CPP-initiated SWCNT growth. Reproduced with permission from ref. 78. Copyright 2013 Nature Publishing Group. | |
Yamago's first publication of 2013 described an evolution of his “platinum squares” approach to CPPs.79 The approach involves the synthesis of U-shaped diplatinum complexes and then combining these to access the by now familiar tetraplatinum CPP precursors using a nickel-mediated coupling (as had been exploited previously in the context of CPPs). Yamago demonstrated the applicability of his methodology to the synthesis of various even-numbered CPPs, but most significant of these was a new synthesis of [6]CPP, which is shown in Scheme 40. U-shaped building block 166 is prepared by the previously-established procedures before a Ni-mediated “shotgun” cyclisation selectively provides tetraplatinum complex 167. In this instance, the square planar geometry of platinum(II) ensures that dimerisation prevails over any other oligomerisation. Interestingly, the desired four-fold reductive elimination from 167 to [6]CPP could not be effected under the standard conditions. Treatment of 167 with bromine gave only para-dibromobenzene and para-dibromoterphenyl. Yamago hypothesised that these side-products arose by formation of platinum(IV) intermediate 168 (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Br), from which reductive elimination to form C–Br bonds was occurring in addition to the desired reductive elimination to form C–C bonds. To circumvent this problem, the Yamago group examined reagents that effect oxidative fluorination (since reductive elimination to form C–F bonds is known to be have a higher activation energy than formation of C–Br bonds). Thus, treatment of 167 with xenon difluoride gave [6]CPP in 40% yield, presumably via the intermediacy of 168 (XF). Other fluorine-containing reagents were effective, such as silver(I) fluoride and tetrabutylammonium fluoride; the latter result is somewhat surprising given that TBAF is a source of fluoride anions but not an oxidising agent. By this new procedure, Yamago was able to produce [6]CPP in an overall yield of 8.9% from commercially available starting materials.
Br), from which reductive elimination to form C–Br bonds was occurring in addition to the desired reductive elimination to form C–C bonds. To circumvent this problem, the Yamago group examined reagents that effect oxidative fluorination (since reductive elimination to form C–F bonds is known to be have a higher activation energy than formation of C–Br bonds). Thus, treatment of 167 with xenon difluoride gave [6]CPP in 40% yield, presumably via the intermediacy of 168 (XF). Other fluorine-containing reagents were effective, such as silver(I) fluoride and tetrabutylammonium fluoride; the latter result is somewhat surprising given that TBAF is a source of fluoride anions but not an oxidising agent. By this new procedure, Yamago was able to produce [6]CPP in an overall yield of 8.9% from commercially available starting materials.
 |
| | Scheme 40 Yamago's hybrid synthesis of [6]CPP. | |
Isobe's next publication returned to the concept of a molecular bearing (cf.Fig. 55) and this time focused on the evaluation of a number of different fullerene-derived journals.80 Association constants were measured for each different journal and were found to vary by several orders of magnitude depending on the substituents on/in the fullerene. Isobe's findings are represented on a logarithmic scale in Fig. 61.
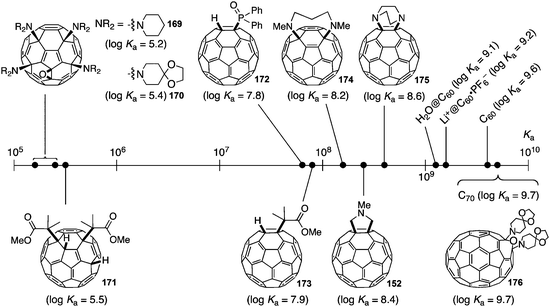 |
| | Fig. 61 Association constants for various fullerene derivatives with (P)-(12,8)-[4]CC2,8, as determined by Isobe and co-workers. Adapted with permission from ref. 80. Copyright 2013 American Chemical Society. | |
Several interesting trends are evident. The previously described (Fig. 55) molecular bearing, (P)-(12,8)-[4]CC2,8⊃C60, is shown on the right of Fig. 61, with a comparatively high association constant of logKa = 9.6. Similarly, previously described (Fig. 55) molecular bearing (P)-(12,8)-[4]CC2,8⊃152, where the journal contains a shaft, is shown to have a somewhat lower association constant of log Ka = 8.4. Isobe and co-workers now report data for several new fullerene-derived journals bearing shafts, 169–175. It can be seen that for all of these the association constants are appreciably lower, down to some four orders of magnitude lower than for (P)-(12,8)-[4]CC2,8⊃C60. Crudely, it may be seen that the association constant diminishes with increasing size of the shaft motif. However, even for the bearing with the lowest association constant, (P)-(12,8)-[4]CC2,8⊃169, the journal still rotates freely and does not run out of the bore, as shown by DOSY NMR. Another interesting aspect of this work is that Isobe et al. have studied endohedral fullerenes as journals (H2O⊂C60 and Li+⊂C60·PF6−), which in both cases form bearings with slightly lower association constants than for the bearing with an “empty” C60 journal. Finally, and perhaps most significantly, the group also examined C70 as a journal, finding it to have a higher association constant than for C60. Crucially, when a C70 derivative bearing a shaft moiety (176) was used, the association constant did not diminish, which may be ascribable to the oval shape of C70 and the fact that the shaft will consequently be further from the bore.
Having reported reduction of a CPP to its tetraanion earlier in the year, Jasti next published the results of a study on the oxidation of a CPP.81 Treatment of [8]CPP with triethyloxonium hexachloroantimonate led to the formation of a stable, isolable [8]CPP radical cation (Scheme 41). The [8]CPP radical cation reportedly exhibited absorption maxima well into the infra-red region. Jasti et al. also claimed that [8]CPP˙+ was capable of forming a charge-resonance dimer, ([8]CPP)2˙+, with a second molecule of [8]CPP. Upon titration of neutral [8]CPP into a solution of [8]CPP˙+, a new absorption was observed at 1747 nm, which was attributed to the delocalisation of the positive charge throughout both CPP moieties. The binding constant of the dimer was determined to be 1.15 ± 0.03 × 104 M−1, which is appreciably higher than the binding constants for charge-resonance dimers of planar aromatic systems. Computational modelling was carried out which was able to rationalise the observed absorbance of [8]CPP˙+ on the basis of the level of the SOMO and other orbitals. (Note that a later report from Yamago cast doubt on the identity of the species characterised in this study, vide infra, Fig. 117.)
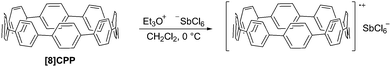 |
| | Scheme 41 Jasti's synthesis of {[8]CPP+}SbCl6−. | |
The first theoretical study to appear on the bottom-up synthesis of SWCNTs from CPPs since Itami's realisation of this concept was again from Irle and Morokuma.82 In this study they concentrated on the effect of reaction temperature, simulating the growth of an SWCNT from [6]CPP at 300, 500 and 800 K, using acetylene and acetylenyl radical as growth agents. With the latter, they predict the highest growth rate at 500 K; higher temperatures lead to increased defect formation (through the undesired formation of a new pentagon as opposed to the desired hexagon upon incorporation of the new C2 fragment). From these results they predict the existence of an optimum temperature for SWCNT growth. Notably, through determination of the various pathways through which pentagonal defects may be introduced, they observed that introduction of one defect renders the introduction of additional defects more likely, especially at higher temperature.
The theme of host–guest chemistry was returned to by Yamago, who, having previously studied the interaction between C60 and CPPs,39 now turned his attention to C70.83 The same approach was employed as in the Yamago group's previous study, namely adding C70 to a mixture of CPPs of various sizes and observing supramolecular interactions on the basis of changes in 1H-NMR chemical shifts. Whereas the spheroidal C60 has Ih symmetry, the ellipsoidal C70 has D5h symmetry (Fig. 62, top), which raises the possibility of its being accommodated in a SWCNT (or indeed in a CPP) in more than one possible orientation (Fig. 62, bottom).
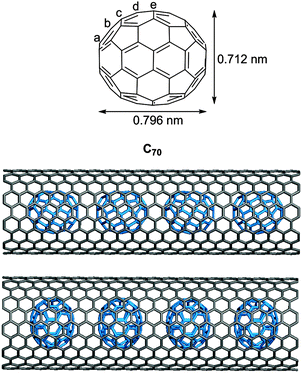 |
| | Fig. 62 Dimensions of C70 (top) and C70–SWCNT peapods in “lying” (middle) and “standing” (bottom) orientations. Adapted with permission from ref. 83. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
When the NMR experiment was carried out on a mixture of CPPs, both [10] and [11]CPP were observed to interact with C70 in solution (Fig. 63a); this interaction was reproducible with pure [10]CPP (Fig. 63b) and [11]CPP (Fig. 63c) also. Job's plots confirmed the stoichiometry of the interactions to be 1:1 in both cases. The supramolecular complexes were modelled computationally at the M06-2X/6-31G* level of theory. Computation predicts that the two orientations shown in Fig. 62 will also be observed in the CPP complexes. Thus, the larger [11]CPP will accommodate C70 in a “standing” orientation (Fig. 64a and b), whereas the smaller [10]CPP will accommodate C70 in a “lying” orientation (Fig. 64c and d). Notably, a significant “induced fit” is predicted for [11]CPP⊃C70, with the nanoring distorting away from its circular conformation to an oval. Also, for [10]CPP⊃C70, it is predicted that the nanoring host will not be oriented exactly over the equator of its C70 guest.
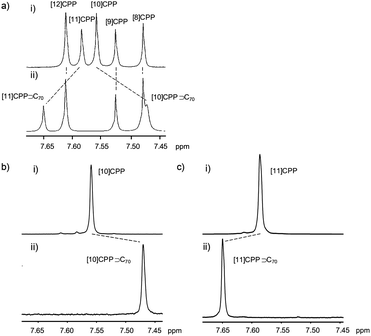 |
| | Fig. 63
1H-NMR spectra (CDCl3, rt) of (a) a mixture of [8]–[12]CPPs (i) before and (ii) after the addition of solid C70; (b) isolated [10]CPP; and (c) [11]CPP (i) before and (ii) after the addition of solid C70. Adapted with permission from ref. 83. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
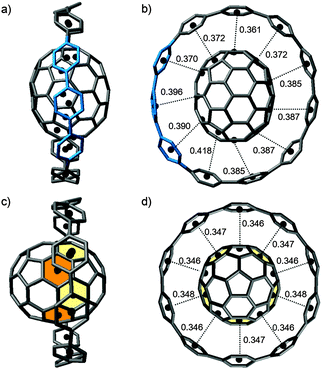 |
| | Fig. 64 Two orthogonal views of (a, b) [11]CPP⊃C70 and (c, d) [10]CPP⊃C70, as calculated by DFT. Adapted with permission from ref. 83. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
In order to try and gain some experimental evidence for the computed orientations of the C70 guests, the Yamago group examined the interaction of [10] and [11]CPP with a functionalised C70 guest, 177 (Fig. 65). It was predicted that the protruding diester motif in 177 would be deleterious in terms of accommodation of 177 in a nanoring in a “standing” orientation, but would have less of an effect on a “lying” orientation. The relevant inclusion complexes were formed and their association constants determined. As predicted, [11]CPP⊃177 had an association constant 13% smaller than [11]CPP⊃C70 (due to the diester interfering with the “standing” orientation), whereas the association constants for [10]CPP⊃177 and [10]CPP⊃C70 were almost equivalent. Further evidence in support of the computed orientations of the guests was obtained in the form of X-ray crystal structures of [11]CPP⊃C70 and [10]CPP⊃177 (Fig. 66).
 |
| | Fig. 65 Derivatised C70 guest 177. | |
 |
| | Fig. 66 ORTEP diagram of [10]CPP⊃177 (right) and [11]CPP⊃C70 (left), showing ellipsoids at 30% probability. Hydrogens and solvent molecules have been omitted for clarity. | |
The crystallographic studies confirm that the “induced fit” of C70 into [11]CPP that was predicted computationally is also observed experimentally in the solid state. Yamago speculates that a similar induced fit may well also occur upon incorporation of molecules of C70 into a (11,11)-SWCNT.
The subject of the Raman spectra of CPPs was returned to for the second time in 2013 by Swan and co-workers.84 They acquired experimental spectra for the same CPPs as studied earlier by Fujitsuka, Majima and co-workers,73 with the exception of [6]CPP. However, they predicted spectra computationally for every CPP from [4] to [20]CPP. Whereas Fujitsuka, Majima and co-workers had used Raman spectra to probe the degree of quinoidal character of various CPPs as the major focus of their study, Swan et al. were interested in a broader investigation and assignment of the various active Raman modes for CPPs. They found that some of the observed modes were common to both CPPs and SWCNTs (e.g. G-band, D-band, certain radial modes), but that most were absent in SWCNTs and unique to CPPs. They also found that all the active Raman modes were influenced by CPP ring size – for the larger CPPs, the values converged asymptotically with those of linear poly-para-phenylenes.
The Swager group published for the first time in the field of CPPs, having entered the area due to their interest in the bottom-up synthesis of SWCNTs.85 As a first attempt, they synthesised [12]CPP and attempted to homologate it by effecting Diels–Alder reactions, as proposed previously by Scott48,49 (Fig. 60, blue). However, they report being unsuccessful despite trying numerous combinations of dienophiles, Lewis acid activators and oxidants. In light of this failure, they considered synthesising a CPP derivative bearing polycyclic aromatic hydrocarbon (PAH) repeating units as opposed to simple phenylene units. They hoped such a CPP derivative would be more reactive towards Diels–Alder homologation, since Scott has previously shown that bay region Diels–Alder reactivity varied for different PAHs in the order biphenyl < phenanthrene < perylene.49 Accordingly, a CPP derivative containing perylene motifs, 178, was targeted. However, the Swager group did not propose to access 178 from a precursor that already contained perylene units – they anticipated such a strategy might be problematic due to the lower ability of the rigid perylene unit to accommodate the ring strain of the radial π system. Instead, they proposed to access 178 from pre-cyclised CPP derivative 179, through oxidative cyclisation of its 1,1′-binaphthyl units (Scheme 42a).
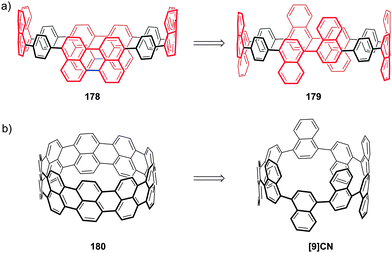 |
| | Scheme 42 Annulated CPP derivative 179 targeted by Swager. Adapted with permission from ref. 85. Copyright 2013 Georg Thieme Verlag. | |
Swager's nanohoop 179 is not dissimilar to [9]CN, previously reported by Itami.45 However, the Scholl reaction (the reaction with which Swager hoped to achieve the oxidative cyclisation to give perylenes) is known be highly susceptible to substrate structure, with undesired rearrangements often taking place (vide infra, Scheme 51). So, whereas access to 180 could well be envisaged from Itami's [9]CN as shown (Scheme 42b), the Swager group instead targeted 179, which they considered to be a “safer” option – i.e. unlike in [9]CN, the binaphthyl units in 179 are “isolated” by interstitial phenylene units, so would give isolated perylene units that should be less prone to unwanted rearrangement. Swager's group effected the synthesis of 179 using the methodology previously described by Itami (Scheme 43). With nanohoop 179 in hand, Swager reports that efforts to carry out its oxidation to 178 are underway.
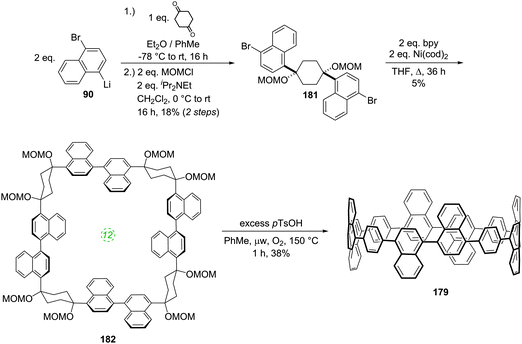 |
| | Scheme 43 Swager's synthesis of annulated [12]CPP derivative 179. | |
Yamago reported a significant extension of his platinum-based methodology, synthesising a 3D ball-like π-conjugated molecule from a hexaplatinum precursor.86 The approach was inspired in part by the work of Fujita, who had shown that treatment of tris(4-pyridyl) ligand 183 with a palladium(II) salt led to self-assembly of octahedral-shaped hexapalladium complex 184 (Scheme 44). Extending this idea to an all-carbon ligand framework and use of platinum instead of palladium, the Yamago group synthesised trifunctional building blocks 186 and 187. When these were combined in a 1:1 ratio, transmetallation led to formation of hexaplatinum complex 188. After a straightforward ligand exchange to give 189, treatment with xenon difluoride (cf.Scheme 40) gave unprecedented ball-like molecule 190. Computational modelling of 190 identified six energy minima of various symmetries, of which the D2 conformer was identified as being of lowest energy (Fig. 67). Interconversion between these structures is achieved by rotation of the aryl–aryl bonds; this was a fast process on the NMR timescale, with only a simple, averaged spectrum (3 resonances in the 1H-NMR spectrum) being observed even at temperatures as low as –80 °C. An X-ray crystal structure of 190 was also obtained (Fig. 68), in which the cavity of the molecule was occupied by disordered hexane solvent molecules. In contrast to the gas-phase computational predictions, in the solid state 190 adopts a structure closer to the S4 conformer, most likely due to crystal packing interactions.
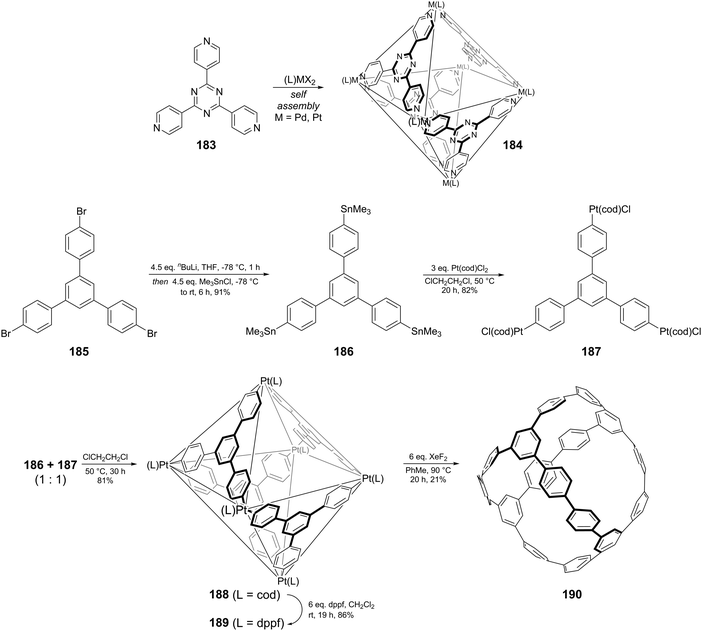 |
| | Scheme 44 Yamago's synthesis of ball-like π-conjugated 190. Adapted with permission from ref. 86. Copyright 2013 Nature Publishing Group. | |
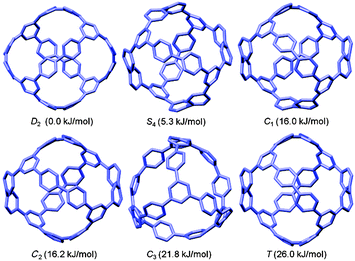 |
| | Fig. 67 computed conformers of 190 (B3LYP/6-31G*). Adapted with permission from ref. 86. Copyright 2013 Nature Publishing Group. | |
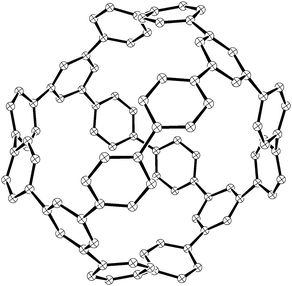 |
| | Fig. 68 ORTEP diagram of 190, showing ellipsoids at 30% probability. Hydrogens and solvent molecules have been omitted for clarity. | |
19th November 2013 saw the publication of three papers on CPP chemistry. The Yamago group reported the preparation and characterisation of both a CPP radical cation and a CPP dication.87 These were prepared chemically (electrochemical methods having been unsuccessful), by the first combination of [8]CPP with one or five equivalents of NOSbF6, respectively. Both the radical cation, [8]CPP˙+ SbF6− and the dication, [8]CPP2+ (SbF6−)2 were isolable solids that were stable for a matter of weeks at −30 °C under nitrogen. Both these novel species exhibited UV/vis/IR absorption spectra that were significantly different from that of [8]CPP (Fig. 69a). For both of the oxidised species, the shapes of the absorption bands were invariant with concentration, suggesting that both the radical cation and the dication exist as monomers in solution. Both exhibited large bathochromic shifts and in the case of the radical cation, the absorption band in the near infra-red region extended up to around 2300 nm. TD-DFT calculations (Fig. 69b) suggested that for the radical cation, the long-wavelength absorption is due to a transition from the degenerate HOMOs to the SOMO, whereas for the dication, the long-wavelength absorption may be ascribed to a transition from the degenerate HOMOs to the LUMO. Some discrepancies between data in this report and those in Jasti's preceding report81 were returned to in a later study (vide infra, Fig. 117).
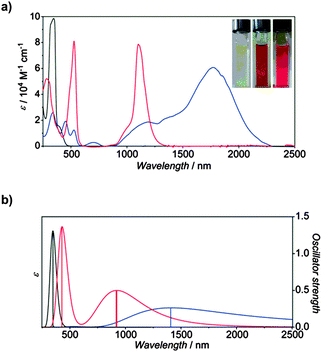 |
| | Fig. 69 (a) UV/vis/IR spectra of [8]CPP (black), [8]CPP˙+ SbF6− (blue) and [8]CPP2+ (SbF6−)2 (red) in CH2Cl2. Photographs of solutions of [8]CPP (left), [8]CPP˙+ SbF6− (centre), and [8]CPP2+ (SbF6−)2 (right) in CH2Cl2 under ambient light are also shown. (b) UV/vis/IR spectra obtained by TD-DFT calculations at the (U)B3LYP/6-31G(d) level of theory. Reproduced with permission from ref. 87. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
The radical cation [8]CPP˙+ SbF6− was studied by ESR spectroscopy and showed a symmetrically split multiplet with a 1H hyperfine coupling constant of 0.034 mT, indicative of complete delocalisation of the spin over all the phenylene units. The experimental spectrum was almost exactly superimposable on a theoretical spectrum generated on the assumption of equivalence of all H atoms in the structure (Fig. 70). The (closed-shell) dication [8]CPP2+ (SbF6−)2 was studied by NMR as opposed to ESR, and exhibited a single resonance in the 1H-NMR spectrum at δ = 5.24 ppm (far upfield from the value for [8]CPP), which indicates the equivalence of all the phenylene units, on the NMR timescale, at least.
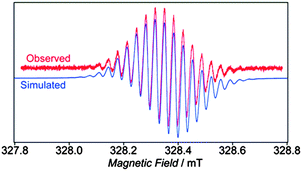 |
| | Fig. 70 Observed (red) and simulated (blue) ESR spectra of [8]CPP˙+ SbF6− at 20 °C. Reproduced with permission from ref. 87. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
An X-ray crystal-structure of the dication (as the hexachloroantimonate salt) was obtained (Fig. 71), which has some striking characteristics. Most notably, all phenylene–phenylene linkages are nearly coplanar, with dihedral angles in the range 3.6(4)° to 6.6(5)°, far lower than for neutral [8]CPP (which has dihedral angles up to 41.4(7)°). Also, the Cipso–Cipso bond length is shortened compared to neutral [8]CPP (by 2.3%), as is the Cortho–Cortho bond length (by 0.9%); in contrast, the Cipso–Cortho bond length is lengthened compared to neutral [8]CPP (by 0.3%). Taken together the above structural features provide compelling evidence for enhanced quinoidal character in the dication with respect to neutral [8]CPP.
 |
| | Fig. 71 ORTEP diagram of [8]CPP2+ (SbCl6−)2, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Disorder and solvent molecules have been omitted for clarity. | |
On the same day, Itami published two back-to-back papers in Chem. Commun. The first of these88 describes a divergent synthesis of [7]- and [8]CPP. The methodology described previously by the Itami group employed L-shaped building blocks containing three rings, such as 39 and 50. These were then cyclised to macrocyclic CPP precursors containing at least three such building blocks. As such, the smallest CPP accessible by this approach was 3 × 3 = [9]CPP. Therefore, new methodology was required to access smaller CPPs, and to this end Itami and co-workers developed the use of a smaller two-ring L-shaped precursor. The synthetic route is shown in Scheme 45. Key 2-ring building block 192 was accessed from 49 in 4 steps. Dual lithium–halogen exchange on 3-ring building block 50 and addition to two equivalents of 192 gave acyclic 7-ring intermediate 193. After MOM protection, this could be directly cyclised using the nickel-mediated procedure to give 7-ring macrocycle 195, or alternatively the dual intramolecular/intermolecular Suzuki–Miyaura sequence employing bifunctional boronic acid 51 gave 8-ring macrocycle 196. Both macrocycles underwent elimination/oxidation without the need for an external oxidant to afford [7]CPP and [8]CPP. The Itami group were able for the first time to obtain an X-ray crystal structure of [7]CPP, as shown in Fig. 72. A slight distortion away from a circular geometry is observable, but this was attributed merely to the presence of an included molecule of cyclohexane.
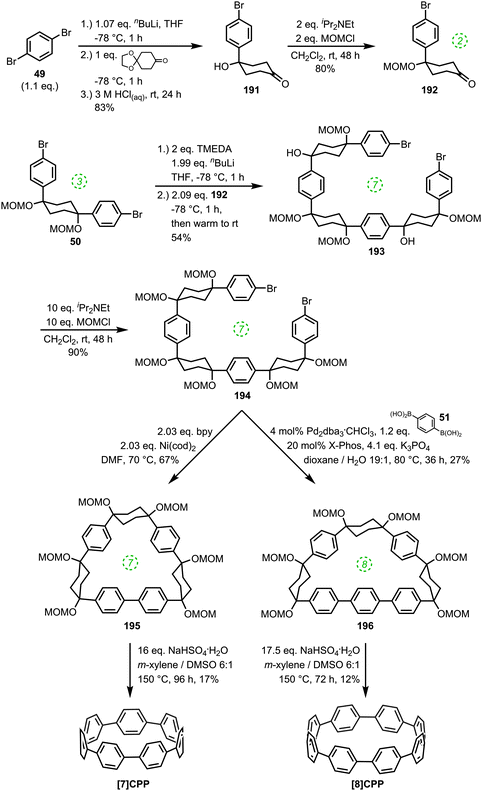 |
| | Scheme 45 Itami's synthesis of [7]- and [8]CPP. | |
 |
| | Fig. 72 ORTEP diagram of [7]CPP, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
The second of Itami's two back-to-back papers concerned the synthesis of the first pyrene-containing nanoring.89 The target structure, cyclo[12]-paraphenylene[2]-2,7-pyrenylene (denoted as [12,2]CPPyr by Itami), was assembled using Itami's by now well-established methodology employing L-shaped building blocks, nickel-mediated cyclisation and elimination/oxidation, as shown in Scheme 46. Certain modifications to the established methodology have been employed, such as use of silver oxide in the Suzuki–Miyaura coupling. Additionally, Itami explicitly notes that formation of 199 necessitated use of high dilution conditions (1 mM) to suppress formation of oligomers. Also, 1,2,4-trichlorobenzene is not a solvent Itami had reported previously for the elimination/oxidation step, although no comment is made as to whether or not the previous m-xylene/DMSO conditions failed in this instance.
 |
| | Scheme 46 Itami's synthesis of [12,2]CPPyr. | |
The absorption and emission spectra of [12,2]CPPyr are shown in Fig. 73. Several differences from the spectroscopic characteristics of CPPs are evident. [12,2]CPPyr has appreciably higher extinction coefficients than a CPP of similar diameter, [16]CPP, but a roughly comparable Stokes shift. The fluorescence quantum yield was calculated to be Φf = 0.21, which is significantly lower than for large CPPs, but the experimentally determined fluorescence lifetime (τs = 25.6 ns) is longer than for CPPs; pyrenes are known to have long fluorescence lifetimes.
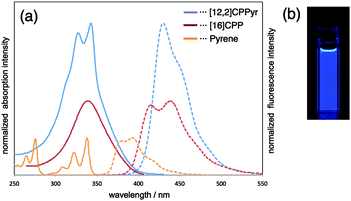 |
| | Fig. 73 (a) UV-vis absorption (solid lines) and fluorescence (broken lines) spectra of dichloromethane solution of [12,2]CPPyr (blue line), chloroform solution of [16]CPP (red line) and chloroform solution of pyrene (orange line). (b) Fluorescence of [12,2]CPPyr. Reproduced with permission from ref. 89. Copyright 2013 Royal Society of Chemistry. | |
The final CPP-related paper of 2013 came from the Jasti group, who synthesised two cleverly designed CPP derivatives (208 and 209) in order to delineate exactly which structural characteristics of the CPPs give rise to their unusual properties.90 As shown in Scheme 47, 5-ring acyclic dichloride 203 (analogous to dibromide 87, cf.Scheme 22) was synthesised using Jasti's by now well-developed methodology. This was then cyclised via a dual intermolecular/intramolecular Suzuki–Miyaura protocol employing bis(boryl) fragments 204 or 205, containing one or six sp3-hybridised carbons respectively. The resultant macrocycles 206 and 207 underwent oxidation/aromatisation to give CPP derivatives 208 and 209 which possess interrupted conjugation with respect to the parent CPPs. The first of these, 208, is closely analogous with [7]CPP and would be expected to possess similar ring strain, etc., but with the key difference that the presence of the single sp3 carbon means the π system is not fully radially conjugated. 208 therefore serves as a useful tool molecule to attempt to delineate the effects of radial conjugation from the other properties of CPPs (ring strain, non-planar phenylene units, torsion angles, etc.). In contrast, 209 will possess appreciably less ring strain than 208 by virtue of its longer aliphatic tether.
 |
| | Scheme 47 Jasti's synthesis of CPP analogues with interrupted conjugation. | |
In the first instance, 208 and 209 were characterised by cyclic voltammetry and compared to [7]CPP and a linear alkyl-substituted hepta-para-phenylene that had previously been reported.91 It was found that 208 exhibited a quasi-reversible peak ascribable to a reduction wave, but that the observed half-wave potential (−2.74 V, vs. FC/FC+) was appreciably altered from the value for [7]CPP (−2.57 V). Furthermore, oxidation of 208 was shown to be irreversible, having an onset potential at 0.63 V. The observed half-wave potential for the reduction wave in 209 (−2.75 V) was very close to that for 208, but the onset potential for the oxidation wave was 0.71 V. For comparison purposes, the onset potential for oxidation of the linear hepta-para-phenylene was 1.0 V. To put these results in context, the Jasti group carried out computational modelling which predicted 208 to have a very similar average torsional angles and a very similar degree of deformation of the phenylene rings from linearity with respect to [7]CPP. Thus, the main difference between [7]CPP and 208 is the interrupted conjugation; on the basis of the electrochemical data, therefore, it is clearly the uninterrupted radial π conjugation in the CPPs which plays the major role in narrowing the band gap by lowering the LUMO and raising the HOMO.
The absorption and emission spectra for 208 and 209 were acquired and are shown in Fig. 74, in comparison with those of [7]CPP. Several trends are evident. The Stokes shift for 208 is smaller than for [7]CPP, and that for 209 is smaller again; the quantum yields for 208 and 209 (Φf = 0.23 and 0.25 respectively) are much higher than that of [7]CPP (Φf = 0.006). Also of note, a second absorption maximum is observable (red-shifted with respect to the global maximum), which corresponds to the HOMO–LUMO transition. This is forbidden for CPPs, but becomes more significant in 208 and 209 due to the aliphatic tether breaking the symmetry of the molecules. The trends that the Jasti group have discerned will likely be of use in the design of CPP derivatives having targeted properties. It should be noted that shortly beforehand, Bodwell et al. had reported a study examining the characteristics of teropyrene analogues of 209, i.e. multiply-annulated analogues of 209, from the perspective of bottom-up SWCNT synthesis.92
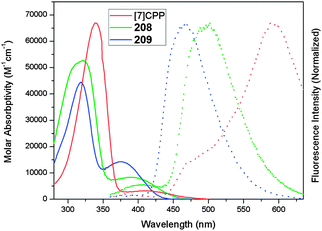 |
| | Fig. 74 UV/vis absorption and fluorescence spectra for [7]CPP, 208, and 209. Reproduced with permission from ref. 90. Copyright 2014 American Chemical Society. | |
2014
The first publication in 2014 saw the helically chiral (12,8)-[4]CC2,8 described by Isobe find a practical application in asymmetric catalysis.93 This collaborative work between Isobe and Soai demonstrated the applicability of enantiopure (12,8)-[4]CC2,8 in an asymmetric autocatalytic process, namely Soai's addition of di-iso-propylzinc to 2-tert-butylethynylpyrimidine-5-carbaldehyde 210.94 As shown in Scheme 48, addition of 4 equivalents of di-iso-propylzinc to 210 in the presence of 25 mol% of (12,8)-[4]CC2,8 was then followed by a second addition of larger quantities of 210 (16 equiv.) and di-iso-propylzinc (32 equiv.), giving secondary alcohol 211 in 83–86% yield and 91% e.e. (with (P)-(12,8)-[4]CC2,8 giving (R)-211 and (M)-(12,8)-[4]CC2,8 giving (S)-211). An additional two rounds of chiral amplification afforded 211 in >99.5% e.e. As 210 and the zinc reagent are both achiral, it follows that the chirality of 211 is ultimately derived from the chirality of the added nanohoop.
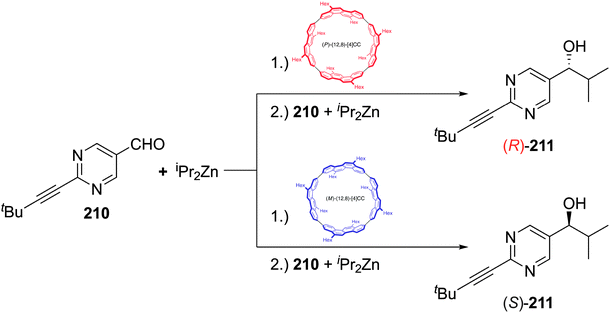 |
| | Scheme 48 Isobe's [4]CC2,8 used as a chiral initiator in Soai's autocatalytic addition of di-iso-propylzinc to 210. | |
On the same day that Yamago published a personal account of his work on CPPs,95 Müllen and Nishiuchi published a second study which extended their work of 2012 (see 148 and 149, Scheme 37), this time synthesising ultrashort SWCNT precursors that bear additional phenyl substituents and/or are of a larger diameter than 148 and 149.96 As shown in Scheme 49, L-shaped precursors 212 and 213 (elongated analogues of 145, Scheme 37) underwent nickel-mediated shotgun cyclisation to give cyclic oligomers 214 and 215. These in turn underwent reduction/aromatisation with sodium naphthalenide to afford multiply substituted CPP derivatives 216 and 217 (dodecaarylated analogues of [15]CPP and [21]CPP respectively). In contrast, however, when L-shaped precursor 145 was adorned with extra phenyl substituents to give 218, this did not undergo the desired nickel-mediated cyclisation. Instead, access to 219 necessitated the use of cuprate chemistry. Application of the reduction/aromatisation protocol to 219 furnished 220 (an octadecaarylated analogue of [9]CPP).
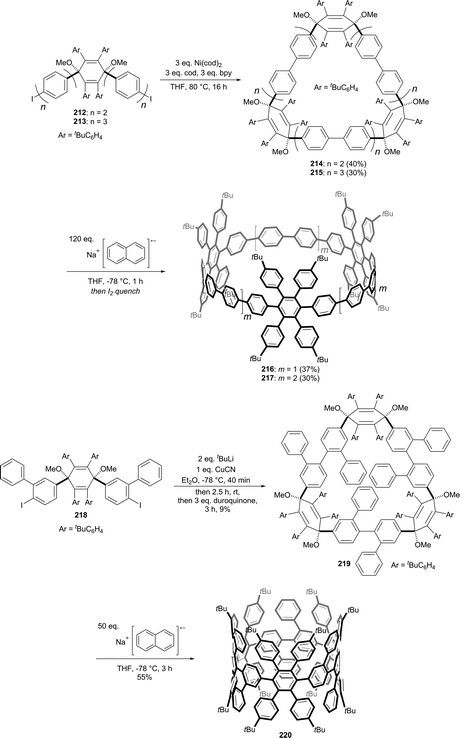 |
| | Scheme 49 Nishiuchi & Müllen's syntheses of ultrashort SWCNT precursors. | |
An X-ray crystal structure of 220 was obtained and is shown in Fig. 75. A distortion of the CPP core towards an ellipsoidal geometry is observed, but arguably more significant are the dihedral angles between adjacent phenylene rings, which are observed to be between 42° and 84°. Such a high dihedral angle (almost perpendicular!) had never been reported previously for a CPP or derivative. As well as 216, 217 and 220, Nishiuchi and Müllen also synthesised an analogue of 220 having a larger diameter, as shown in Scheme 50. Thus, 218 was doubly terphenylated to give 221, which underwent copper-mediated macrocyclisation and reduction/aromatisation to give 222 (a 30-fold arylated analogue of [15]CPP). Absorption and emission spectra were recorded for all the novel CPP analogues and are shown in Fig. 76.
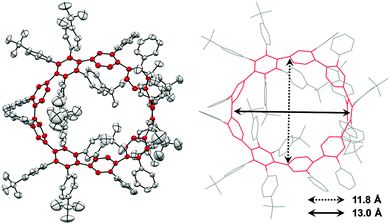 |
| | Fig. 75 X-ray crystal structure of 220. ORTEP drawing (left) and wireframe model (right). Ellipsoids at 50% probability. Hydrogen atoms and solvent molecules have been omitted for clarity. The CPP rings are depicted in red. Reproduced with permission from ref. 96. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
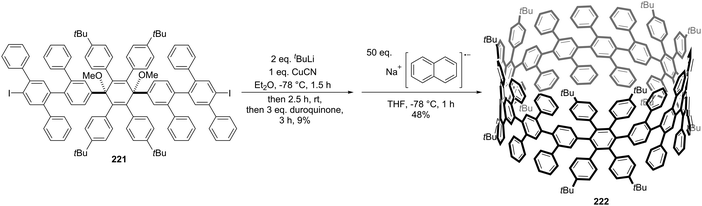 |
| | Scheme 50 Nishiuchi & Müllen's synthesis of ultrashort SWCNT precursor 222. | |
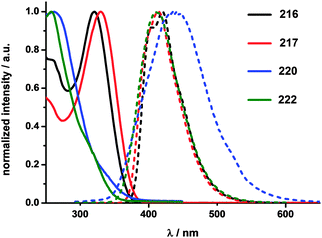 |
| | Fig. 76 UV/vis absorption (solid lines) and fluorescence (dashed lines) spectra for 216 (CH2Cl2), 217 (CH2Cl2), 220 (hexanes) and 222 (hexanes). Reproduced with permission from ref. 96. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
When Nishiuchi and Müllen attempted the cyclodehydrogenation of the CPP derivatives, they found that the more extensively arylated variants, 220 and 222, gave mixtures of dehydrogenated products under all conditions tried, indicating only partial formation of cyclic graphitic structures. Incomplete reaction was not a problem for 216 or 217, however – instead over-oxidation was observed. Thus, upon treatment of 216 or 217 with FeCl3, a comparatively clean process (which did not involve any chlorination) occurred, giving rise not to the expected products 223 and 224, but instead to those lacking an extra 2, 4 or even 6 hydrogens by mass spectrometry. In the case of 217, exhaustive NMR analysis identified the product missing two hydrogens to be 225, in which a 1,2-phenyl shift had occurred (Scheme 51). This unexpected shift and additional dehydrogenation was ascribed to relief of strain in the macrocyclic ring, since in model studies, the corresponding cyclodehydrogenation of a linear analogue of 216/217 proceeded as expected, without such a shift. Nishiuchi and Müllen point out that cyclodehydrogenation of the CPP derivative with the largest diameter (217) was the cleanest process, and that future efforts are therefore best targeted towards such larger CPP derivatives.
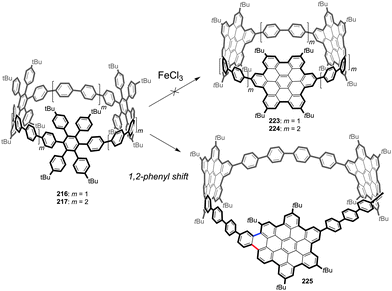 |
| | Scheme 51 Unexpected 1,2-phenyl shift upon oxidation of 217 gives 225. The bond which has migrated is shown in red and the additional bond which accounts for the loss of two further hydrogens is shown in blue. | |
The record set by Jasti in 2012 for the synthesis of the smallest CPP ([6]CPP) fell on 27th January 2014, when Yamago reported the first synthesis of [5]CPP.12 In order to overcome the intimidating strain energy of this structure (calculated to be 491 kJ mol−1),37 the Yamago group adopted a hybrid approach that incorporated concepts from both their own methodology (reductive elimination from a metal centre) and that of Jasti (reduction/aromatisation of a 1,4-cyclohexadiene after macrocyclisation). Yamago's synthesis is shown in Scheme 52. 4-Ring cyclohexadiene building block 82 was synthesised from 70 as previously described by Jasti (cf.Scheme 22). Protecting group manipulation gave 226, which underwent oxidative dearomatisation to give 227. An extra phenylene unit was added to this via addition of an aryllithium, which was followed by global silylation to give 5-ring macrocyclisation precursor 228. Ring closure was effected in this case using the nickel-mediated method, as opposed to the platinum-mediated method more commonly employed by Yamago's group. The resultant macrocycle 229 was formed in good yield; following global desilylation, a reduction/aromatisation sequence mediated by tin(II) chloride gave [5]CPP for the first time. Yamago specifically notes the importance of carrying out this final transformation under neutral conditions, as opposed to the acidic conditions more commonly used with SnCl2.
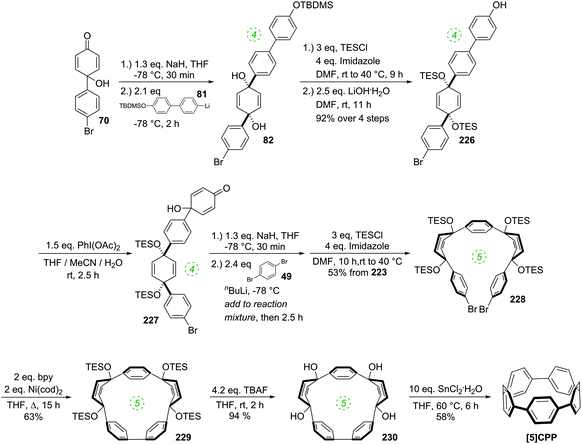 |
| | Scheme 52 Yamago's synthesis of [5]CPP. | |
[5]CPP is a dark purple solid which Yamago reports to be stable in air and soluble in many common solvents. It is non-fluorescent, which is keeping with the observations for larger CPPs ([8]CPP and larger – fluorescent; [7]CPP – very low Φf; [6]CPP non-fluorescent). The absorbance spectrum of [5]CPP is shown in Fig. 77, along with the computed oscillator strengths obtained by TD-DFT. The absorption at λmax = 335 nm is observed at a very similar wavelength to the larger CPPs; the weak absorption at λmax = 502 nm corresponds to the forbidden HOMO–LUMO transition. [5]CPP was also analysed electrochemically and the cyclic voltammogram obtained is shown in Fig. 78. Two chemically reversible oxidation waves are observed (scanning in tetrachloroethane), likely corresponding to formation of a radical cation and dication, (comparable to the oxidation products of [8]CPP studied previously by Jasti81 and Yamago87). In the negative direction (scanning in THF), two pseudoreversible reduction waves were observed, but evidence of the instability of the reduced species was discerned. The HOMO–LUMO gap was calculated by 1.69 eV based on the electrochemical data, but the discrepancy between this value and the value predicted computationally (2.71 eV) is significant, which Yamago states may be due to inaccuracies in the potentials measured in THF.
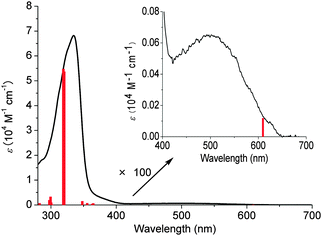 |
| | Fig. 77 UV-vis spectrum of [5]CPP in CHCl3 along with the oscillator strengths (red bars) obtained by TD-DFT calculations. Reproduced with permission from ref. 12. Copyright 2014 American Chemical Society. | |
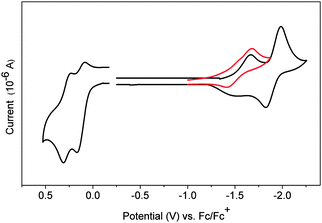 |
| | Fig. 78 Cyclic voltammograms of [5]CPP in C2H2Cl4 (for oxidation) or THF (for reduction) containing 0.1 mol L−1 Bu4NPF6 at room temperature at a scan rate of 0.1 V s−1 (for oxidation) or 0.05 V s−1 (for reduction). The red curve represents the voltammogram obtained upon scanning potential between −1.0 and −1.87 V. Reproduced with permission from ref. 12. Copyright 2014 American Chemical Society. | |
Quite aside for setting a record for the smallest CPP to be synthesised, Yamago points out several aspects of this work which are potentially highly significant. [5]CPP is a constituent fragment of C60 and as it is computed to have a similarly narrow HOMO–LUMO gap, it may well find applications in organic electronics. Also, the work showcases improved synthetic methodology which may find wider application. Specifically, it seems that carrying out the final reduction/aromatisation sequence on a hydroxyl-bearing precursor, as opposed to a methoxy-bearing precursor, is more facile. In this case it has permitted the use of a much milder reductant (SnCl2) than the dissolving metal conditions normally required.
Kamat and co-workers reported an extensive study on the excited state singlet and triplet characteristics of two representative CPPs ([9]CPP and [12]CPP).97 In both cases, rate constants have been determined for the three processes operative following excitation to the S1 state (i.e. radiative recombination, non-radiative recombination and inter-system crossing to the T1 state, represented diagrammatically in Fig. 79). Multiple techniques were employed to derive these values. For example, the data obtained from time-correlated-single photon counting (Fig. 80) clearly show [9]CPP to have a greater emission lifetime than [12]CPP. Kamat and co-workers were ultimately able to derive the following data: for [9]CPP, kr = 8.7 × 107 s−1, knr = 6.8 × 107 s−1, kisc = 4.4 × 107 s−1, τs = 5.3 × 10−9 s and τT = 6.7 × 10−5 s. In contrast, for [12]CPP, the values were kr = 4.4 × 108 s−1, knr = 2.1 × 107 s−1, kisc = 6.8 × 107 s−1, τs = 1.9 × 10−9 s and τT = 1.1 × 10−4 s.
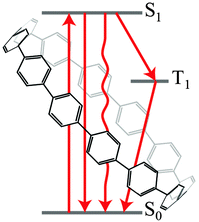 |
| | Fig. 79 Different fates of the S1 excited state of CPPs. Reproduced with permission from ref. 97. Copyright 2014 American Chemical Society. | |
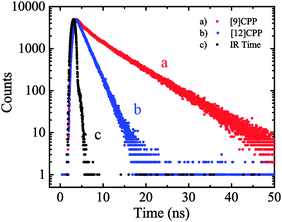 |
| | Fig. 80 Emission lifetime of (a) [9]CPP and (b) [12]CPP as recorded by TCSPC. Trace (c) represents the instrument response time (IRT). Reproduced with permission from ref. 97. Copyright 2014 American Chemical Society. | |
Itami's first publication of 2014 recounts the results of a collaboration with Sinohara on the purification of endohedral metallofullerenes using a CPP.98 Previous reports had described formation of a host–guest complex between C60 and [10]CPP (Scheme 19), between C70 and [10]- or [11]CPP (Fig. 63) and between various C60 or C70 derivatives and (12,8)-[4]CC2,8 (Fig. 61). In most cases, however, the guests had been empty fullerenes. Itami and Sinohara set about devising a system to effect the selective extraction of endohedral metallofullerenes from arc-processed raw soot, which is especially challenging, since it requires a system that can discriminate not only between fullerenes of different sizes but also between an endohedral metallofullerene and the corresponding empty fullerenes. Previously, such purifications had been achieved by multistage preparative HPLC, but this could only ever furnish tiny quantities and required prohibitive volumes of solvent.
Itami and Sinohara reasoned that since endohedral metallofullerenes comprise a positively charged metal ion encapsulated in a negatively charged fullerene, they would likely exhibit stronger intermolecular interactions with a π-conjugated host than the corresponding empty fullerenes. A CPP would be an appropriate host, and what is more, choice of a particular size of CPP could also effect discrimination based on the size of fullerene(s). The concept is represented schematically in Fig. 81.
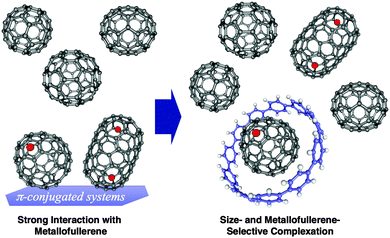 |
| | Fig. 81 Strategy for the size- and metallofullerene-selective complexation and extraction with a CPP. Reproduced with permission from ref. 98. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
In the first instance, NMR experiments indicated that [11]CPP appeared to be a good host for Lu2⊂C82, with the 1H-NMR resonance corresponding to [11]CPP shifting and broadening upon addition of the guest, whereas no change to the spectrum was observed when [12]CPP was used. UV/vis/IR titration experiments with [11]CPP and Gd⊂C82 showed evidence for interaction between [11]CPP and the fullerene cage, but the invariance of a characteristic absorption of Gd⊂C82 at 966 nm indicates there are no charge transfer interactions between [11]CPP and Gd⊂C82. A Job's plot confirmed the stoichiometry of the complex to be 1:1.
Fluorescence quenching experiments were carried out (Fig. 82), by which means the binding constants for various endohedral metallofullerenes were calculated (Gd⊂C82, Ka = 1.8 ± 0.1 × 106 M−1; Tm⊂C82, Ka = 1.8 ± 0.2 × 106 M−1; Lu2⊂C82, Ka = 1.8 ± 0.2 × 106 M−1; Sc3N⊂C80, Ka = 0.72 ± 0.05 × 106 M−1). To use such complexation to effect purification of an endohedral metallofullerene, Itami and Sinohara exploited the differences in solubility between the complex and its individual components. Thus, in toluene, they found that Gd@C82⊂[11]CPP had an appreciably lower solubility than the CPP or fullerene alone. When they treated a toluene solution of raw soot (which included Gd⊂C82) with [11]CPP, a precipitate formed, which was isolated by filtration. Mass spectrometric data showed the filtrate to be almost completely depleted of Gd⊂C82, whereas the precipitate was greatly enriched in Gd@C82⊂[11]CPP. This work therefore provides the basis for a non-chromatographic purification of endohedral metallofullerenes, a highly desirable and hitherto unrealised goal.
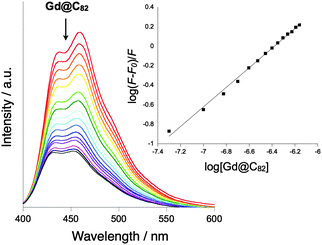 |
| | Fig. 82 Fluorescence spectra of [11]CPP (5.0 × 10−7 M, λexc = 370 nm) in toluene in the presence of various amounts of Gd⊂C82 (0–5.0 × 10−7 M) at 25 °C. Reproduced with permission from ref. 98. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Wegner and co-workers disclosed an inventive synthesis of annulated [8]CPP derivatives that relies on a rhodium-catalysed [2+2+2] cycloaddition.99 Wegner's synthesis employed the –MOM protected cyclohexane-1,4-diol building blocks that have been a common theme in Itami's work, in combination with monoprotected bis(acetylenes). As shown in Scheme 53, known building block 39 was appended to the terminal alkyne motif in 231, 232 or 233 using a Sonogashira reaction. After fluoride-mediated deprotection of the other alkyne, the resultant ω-iodoacetylenes 237–239 underwent cyclodimerisation under the very same Sonogashira reaction conditions to give 240–242 respectively. Each of these macrocycles then underwent a dual rhodium-catalysed [2+2+2] cycloaddition with an introduced alkyne 243 to afford cyclobis(terphenyl) macrocycles; this transformation constitutes a ring contraction from a 36-membered ring to a 32-membered ring in each instance. A combination of the three precursors (240–242) with various introduced (symmetrical) alkynes gave in total nine ring-contracted macrocycles, 244–252. Of these, four were subjected to elimination/oxidative aromatisation conditions to give annulated [8]CPP derivatives 253–256 (Scheme 53).
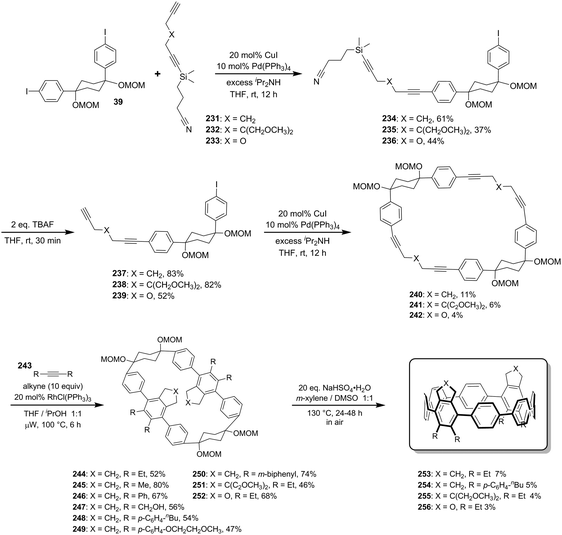 |
| | Scheme 53 Wegner's synthesis of annulated [8]CPP derivatives 253–256. | |
Although the yields for the final CPP-forming step were low, the Wegner group were able to isolate sufficient material to characterise the new [8]CPP derivatives. In the case of 256, an X-ray crystal structure was obtained, which is shown in Fig. 83; UV-vis absorption spectra and fluorescence spectra for 253–256 are shown in Fig. 84.
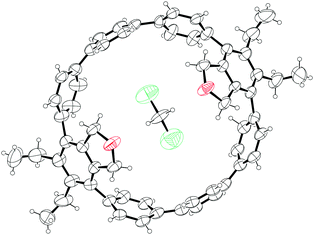 |
| | Fig. 83 ORTEP diagram of 256·CH2Cl2, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
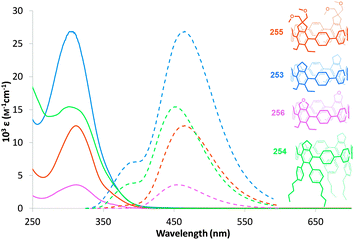 |
| | Fig. 84 Absorption (solid lines) and fluorescence (dashed lines) spectra for 253–256. Adapted with permission from ref. 99. Copyright 2014 American Chemical Society. | |
The annulated CPP derivatives 253–256 show markedly different optical properties from the parent [8]CPP. Specifically, they exhibit absorption maxima between 301 and 312 nm, appreciably shorter wavelengths than the value for [8]CPP of 340 nm (which is actually very similar for all CPPs). Extinction coefficients are also lower than for [8]CPP and fluorescence emission maxima (451–465 nm for 253–256) are all observed at shorter wavelengths than for [8]CPP (540 nm). All of these effects are attributed to distortions in the CPP ring induced by the extra substitution. Computation predicts larger dihedral angles between adjacent phenylene units due to the annulation and indeed, the X-ray structure of 256 shows a dihedral angle in the solid state of 53.8° between the annulated phenylene and its adjacent ring. This is higher than any dihedral angle in the parent [8]CPP. A strength of Wegner's route to CPP derivatives is the use of a late-stage diversification, allowing access to multiple CPP derivatives from a common precursor. In closing, it should be noted that an alternative route to the annulated [8]CPPs, namely using the [2+2+2] cycloaddition as the very final step, was reportedly unsuccessful. The Wegner group have also reported on how the [2+2+2] rhodium-catalysed alkyne cyclotrimerisation could be diverted down a different reaction pathway as a result of ring strain.100
In March, Jasti published a synthesis of [5]CPP in which a palladium catalysed boronate homocoupling was employed as the key macrocyclisation step (Scheme 54).13 It should be noted at the outset that although this paper was published several weeks after Yamago's report on [5]CPP, Jasti's manuscript was submitted for publication five days prior to the submission of Yamago's manuscript. Jasti's route to [5]CPP is distinct from Yamago's, starting as it does from 5-ring acyclic building block 120 that was previously employed to access [10]CPP (Scheme 32). Macrocyclised 257 was first observed by the Jasti group as a very minor byproduct in the reaction of 120 with 87 to give 121 (Scheme 32). Realising that 257 potentially offered access to [5]CPP, the group set about optimising conditions for the deliberate formation of 257, finding that use of high dilution and an equivalent of potassium fluoride effected boronate homocoupling to give 257 in 52% yield. Based on Jasti's previous work, dissolving metal reduction of 257 was expected to furnish [5]CPP. However, when 257 was treated with excess sodium naphthalenide at −78 °C, a stable dianion was formed (by reductive cleavage of two of the possible four C–O bonds) which did not react further to give [5]CPP. If the temperature was raised, decomposition occurred. Instead, if the dianion was quenched with methanol, reduced macrocycle 258 could be isolated in good yield. As 258 is at the same oxidation state as the target CPP, treatment with a base instead of a reductant was found to effect smooth elimination of two equivalents of methanol, giving [5]CPP in 69% yield. The concise nature of the synthesis, in conjunction with the high yields in each step, allowed the Jasti group to prepare 195 mg of [5]CPP. In addition, X-ray crystal structures were secured, not only for the novel intermediates 257 and 258, but also for [5]CPP itself (Fig. 85). The structure shows that the very high strain inherent in [5]CPP manifests itself partly in the form of a significant deviation away from planarity in the phenylene units. Despite this, the observed bond lengths indicate that benzenoid character is retained.
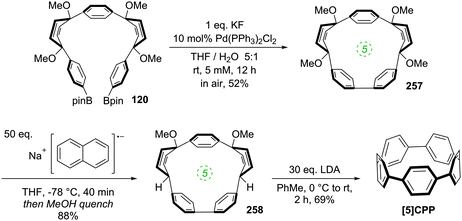 |
| | Scheme 54 Jasti's synthesis of [5]CPP. | |
 |
| | Fig. 85 ORTEP diagram of [5]CPP, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. | |
In March, Sato and co-workers published a study on [6]CPP in which the distortion away from a structure of D6h symmetry was considered in the context of pseudo-Jahn–Teller distortion.101 They employed vibronic coupling density analysis, determining that the instability of the D6h conformer primarily arises from the orbital vibronic couplings for the b1g vibrational mode (primarily coupling of the frontier orbitals and the σ-type occupied/unoccupied molecular orbitals).
Kanemitsu and co-workers disclosed their studies on exciton recombination dynamics in CPPs, which they studied using steady-state and time-resolved photoluminescence spectroscopy.102 They determined that the photoluminescence lifetime was dependent both on temperature and CPP ring size. As shown in Fig. 86, photoluminescence lifetime increases dramatically with CPP ring size at a constant temperature. Fig. 87 illustrates the effect of temperature for a given CPP, and a trend can be discerned here too, namely that an increase in temperature extends the photoluminescence lifetime. On the basis of the presented results, Kanemitsu concludes that excited state spreads out as the number of phenylene units increases (given the dependence of photoluminescence lifetime on CPP ring size). In addition, Kanemitsu concludes that the excitons in CPPs are delocalised and that their thermal distribution renders the photoluminescence lifetime temperature dependent.
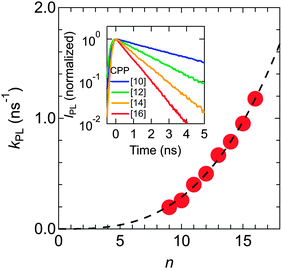 |
| | Fig. 86 The photoluminescence decay rate, kPL, at room temperature as a function of the number of benzene rings, n. The inset shows the photoluminescence decay curves of the [n]CPPs (n = 10, 12, 14, 16). Reproduced with permission from ref. 102. Copyright 2014 Royal Society of Chemistry. | |
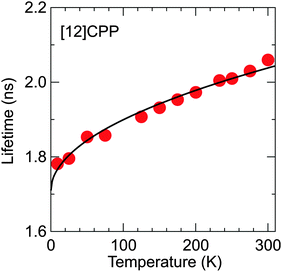 |
| | Fig. 87 The temperature dependence of the photoluminescence lifetime of [12]CPP embedded in a poly(methyl methacrylate) matrix. Reproduced with permission from ref. 102. Copyright 2014 Royal Society of Chemistry. | |
The Itami group reported the concise synthesis of a dimer of [10]CPP from a chloro-[10]CPP monomer, relying on their previously developed methodology (Scheme 55).103 Whereas the Jasti group had previously reported molecules containing two [8]CPP rings (135 and 136, Fig. 51), those were linked by phenylene or naphthylene linkers. In contrast, Itami's target (262) is the first true CPP dimer. Itami reasoned that 262 would be accessible by nickel-mediated dimerisation of chloro-[10]CPP 261. The idea of accessing 261 by direct chlorination of [10]CPP was considered but not pursued, as it would likely result in overhalogenation giving a mixture of mono, di, tri-halogenated CPPs, as well as unreacted CPP, the separation of which would be exceedingly difficult. Instead, Itami sought to synthesise a [10]CPP precursor that already possessed a chloro substituent prior to aromatisation. To this end, building block 98 (prepared previously by the Itami group in their “9+1” synthesis of [10]CPP, see Schemes 26 and 27) was coupled with bis(boryl) 1-ring fragment 259 (a monochloro analogue of 76) via the dual intermolecular/intramolecular Suzuki–Miyaura protocol to give macrocycle 260. This in turn underwent the elimination/aromatisation sequence to give 261. Final dimerisation using the nickel-mediated protocol proceeded without any issue to give [10]CPP dimer 262.
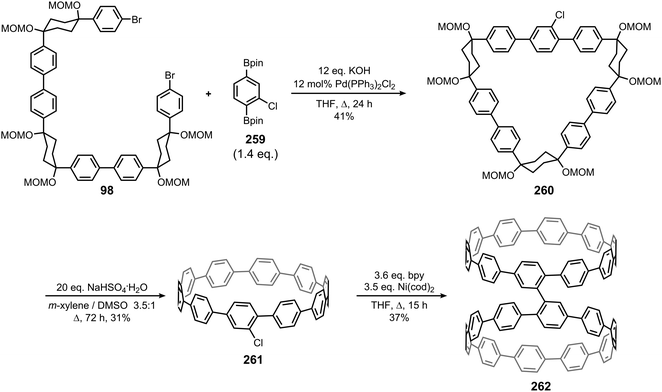 |
| | Scheme 55 Itami's synthesis of [10]CPP dimer 262. | |
Both 261 and 262 exhibited extremely similar absorption and fluorescence spectra to [10]CPP. As regards the conformation of 262, DFT modelling identified several local minima, which can be subdivided into “open” and “closed” conformations. As shown in Fig. 88, The global minimum is a “closed”-type conformer, with all identified “open” conformers lying at least 21.3 kJ mol−1 higher in energy. The lowest energy TS for interconversions between open and closed conformers is located around 37 kJ mol−1 higher in energy than the global minimum, as shown in Fig. 88. Itami comments that 262 is potentially a highly appropriate precursor for accessing a short SWCNT fragment oxidatively, although no studies to that effect are described.
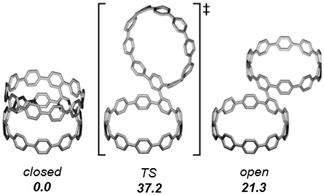 |
| | Fig. 88 Conformations of 262 and relative Gibbs free energies (in kJ mol−1). Adapted with permission from ref. 103. Copyright 2014 American Chemical Society. | |
Wang and co-workers reported the synthesis of a triply-annulated [9]CPP derivative that made use of a p-quinone Diels–Alder reaction to access the necessary 3-ring building block.104 Thus, as shown in Scheme 56, 1,4-diaryl diene 263 underwent a Diels–Alder cycloaddition to afford 264 (which exists in its diketo form in the solid state, as shown by X-ray crystallography). Methylation afforded 265, which underwent the nickel-mediated “shotgun” macrocyclisation, giving both cyclodimers 266 and cyclotrimers 267. For 266, the syn and anti isomers were separable, and the structure of syn-266 was established unambiguously by X-ray crystallography (Fig. 89). In contrast, for cyclotrimer 267, separation of the syn and anti isomers was not possible.
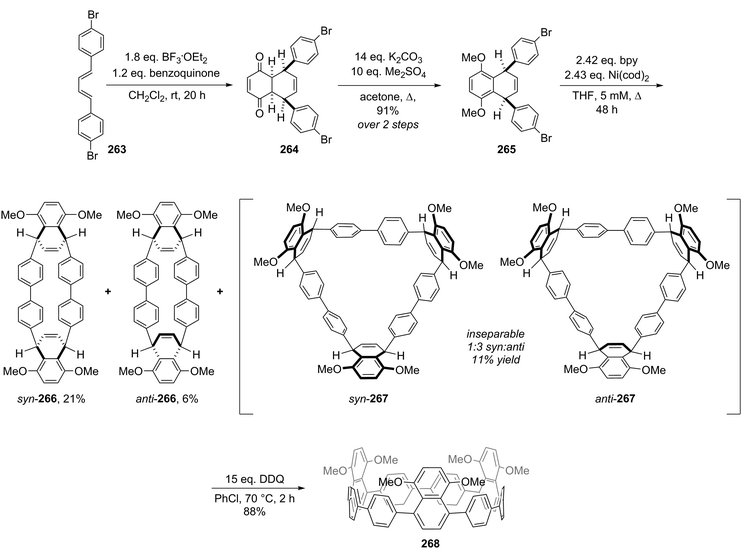 |
| | Scheme 56 Wang's synthesis of triannulated [9]CPP derivative 268. | |
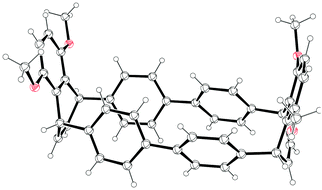 |
| | Fig. 89 ORTEP diagram of syn-266, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Only one of three molecules in the unit cell is shown for clarity. | |
The final aromatisation step in Wang's synthesis uses a different approach to that of Itami or Jasti. The non-aromatic rings in precursors 266 and 267 can be considered to be (benzannulated)1,4-cyclohexadienes, and are therefore more similar to Jasti's 1,4-cyclohexadiene-containing precursors as opposed to Itami's fully saturated cyclohexane-containing precursors. On the other hand, whereas Jasti's precursors are aromatised under reducing conditions (by reductive cleavage of the sp3 C–O bond), Wang's cyclohexadienes do not have any such C–O bond and are instead aromatised under oxidative conditions (more akin to Itami's approach). In the event, the Wang group were not able to aromatise either isomer of cyclodimer 266, most likely due to the high strain energy which would be present in the expected doubly benzannulated [6]CPP product. However, the isomeric mixture of cyclotrimers 267 underwent oxidative aromatisation in good yield upon treatment with DDQ, giving triannulated [9]CPP derivative 268. Wang notes that the mixture of two isomers gave rise to the single product 268, which exhibited only a simple NMR spectrum, indicative of rapid interconversion of atropisomers. As a final point of note, the conditions required for the aromatisation of 267 (DDQ, 70 °C, 2 h) were appreciably milder than those typically used by Itami, perhaps because Wang's precursor required only a single oxidation of each non-aryl ring. In contrast, Itami's precursors (e.g.199, Scheme 46) require two eliminations (of the –OMOM groups) and oxidation per cyclohexyl ring in order to aromatise.
Isobe returned to the concept of the molecular bearing in 2014, reporting detailed X-ray crystallographic and solid state NMR studies on his (12,8)-[4]CC2,8⊃C60 system105 (see Fig. 55). The solid state MAS (magic angle spinning) NMR data for (12,8)-[4]CC2,8⊃C60, consisted of a single resonance for C60, shifted upfield by 3 ppm relative to its resonance in the absence of the nanobelt host. This implies rapid rolling of the journal in the bore on the NMR timescale, since if the journal were static, the various carbons of C60 would be rendered inequivalent and multiple resonances would have been observed. Thus, the journal rolls freely in the bore, even in the solid state. The single resonance for C60 in (12,8)-[4]CC2,8⊃C60 was observed at a range of temperatures (from −30 °C to +70 °C). More dramatic evidence for the free rolling of the C60 journal in the solid state came from the crystallographic studies. Such free rolling gives rise to a large degree of disorder in the structure of (12,8)-[4]CC2,8⊃C60, which hampered attempts to acquire useful diffraction data using in-house X-ray facilities, but use of synchrotron X-ray beamlines allowed for atomic resolution structures to be obtained for the empty host, (±)-(12,8)-[4]CC2,8 (Fig. 90, left) and for (M)-(12,8)-[4]CC2,8⊃C60 (Fig. 90, right).
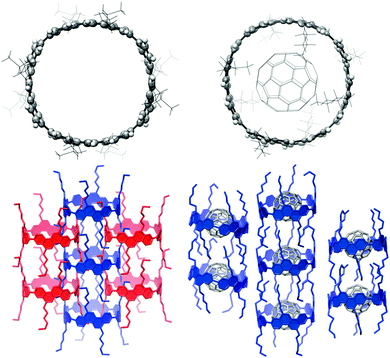 |
| | Fig. 90 Molecular structures from synchrotron X-ray diffraction analysis of a single crystal. For molecular structures viewed from the top, the hexyl chains and C60 are shown as wireframe diagrams, and the chrysenylenes are shown in ORTEP diagrams with thermal ellipsoids at the 30% level. For the packing structures viewed from the side, the (P)- and (M)-structures are coloured in red and blue, respectively. Solvent molecules with disorder are omitted for clarity. (left) Structures of (±)-(12,8)-[4]CC2,8. For the molecular structure, the (M)-structure is shown. Disordered hexyl chains are found, and one representative structure is shown. (right) Structures of (M)-(12,8)-[4]CC2,8⊃C60. One of the representative structures for four disordered C60 molecules (25% occupancy) and hexyl chains are shown. Reproduced with permission from ref. 105. Copyright 2014 National Academy of Sciences, USA. | |
A noteworthy attribute of the crystal structure of (M)-(12,8)-[4]CC2,8⊃C60 is the presence of disordered C60 molecules – four disordered structures were identified (25% occupancy). Isobe notes that such severe disorder may be indicative of only a very small energy difference between these structures, which would be a prerequisite for free journal rotation in the solid state. To gain further insight into the structure of (M)-(12,8)-[4]CC2,8⊃C60, the Hirschfeld surfaces for the C60 guest (and also for the host) were calculated.106,107 The coloured representations of this Hirshfeld surface shown in Fig. 91 are coloured according to curvedness, shape index and de (distance to external atoms). Significantly, the “curvedness” surface is uniformly red in the equatorial region (the region in close proximity to the [4]CC nanobelt host). The lack of nodes in this region indicates a lack of geometric inflection, again indicative of free rotation of the bearing in the solid state. In contrast, Fig. 92 shows the Hirshfeld surface calculated by Isobe from Jasti's previously reported crystal structure of [10]CPP⊃C60 – inflection points can be clearly seen in the equatorial region (yellow lines). This signifies that C60 does not freely rotate in the [10]CPP host in the solid state. The authors also point out the Hirshfeld surface for (M)-(12,8)-[4]CC2,8⊃C60, coloured for shape index (Fig. 91, third from left), shows green lines in a helical pattern, as a consequence of the helical chirality of the nanobelt. Finally, Isobe comments that the dynamic motion in the solid state uncovered here for the first time is very encouraging for the development of friction-free molecular machines.
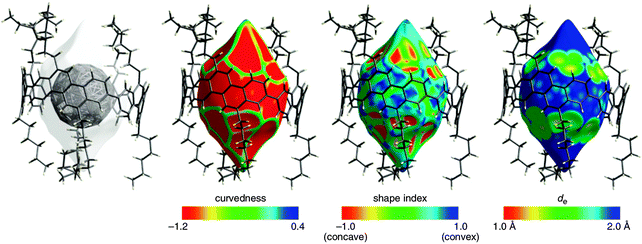 |
| | Fig. 91 Hirshfeld surface of (M)-(12,8)-[4]CC2,8⊃C60 with disordered structures. Solvent molecules with disorders are omitted for clarity. Curvedness, shape index, and de (distance from external atoms) are mapped in colours over the Hirshfeld surfaces. Reproduced with permission from ref. 105. Copyright 2014 National Academy of Sciences, USA. | |
 |
| | Fig. 92 Hirshfeld surface of [10]CPP⊃C60. The yellow nodal lines in the curvedness mapping shows the presence of dividing geometric inflection and thus the polygonal shape of the [10]CPP host. Reproduced with permission from ref. 105. Copyright 2014 National Academy of Sciences, USA. | |
Yamago described the application of his platinum squares methodology to the synthesis of a cyclic tetramer of pyrene, [4]CPY.108 Whereas Itami had previously synthesised a pyrene-containing nanoring (designated “[12,2]CPPyr”, see Scheme 46), Yamago's [4]CPY consists solely of pyrene units, with no interspersed phenylene units. Yamago's synthesis commences with pyrene itself, but its oxidation state changes over the course of the synthesis. Thus, reduction, bromination and metallation of pyrene (not shown) afford distannyl building block 269. As shown in Scheme 57, this readily forms the corresponding tetraplatinum complex 270. In this instance, the cod ligand is not exchanged for a dppf ligand, as was the case in Yamago's previous syntheses. Reductive elimination of platinum from 270 could not be induced cleanly by treatment with bromine, but instead it was found that excess triphenylphosphine furnished the desired [4]CHPY in good yield. Finally, [4]CHPY underwent oxidative aromatisation over palladium on carbon at high temperature, giving [4]CPY in near-quantitative yield.
 |
| | Scheme 57 Yamago's synthesis of [4]CHPY and [4]CPY. | |
Computational modelling (Fig. 93) had predicted that [4]CPY would be electronically very similar to [8]CPP, but very different from a linear oligopyrene, in which the individual pyrene units are electronically isolated. [4]CHPY was characterised electrochemically and its oxidation potential was found to be 0.56 V (vs. Fc/Fc+) by differential pulse voltammetry, almost identical to the value for [8]CPP.
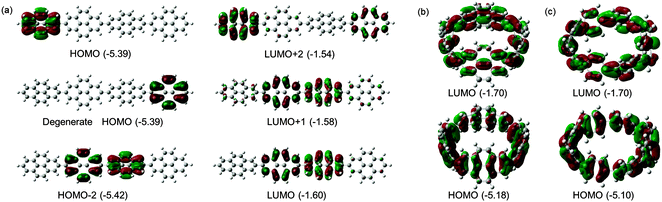 |
| | Fig. 93 HOMO and LUMO of (a) tetra-2,7-pyrene, (b) [4]CPY, and (c) [8]CPP. Orbital energies (in eV) calculated at the B3LYP/6-31G* level of theory are shown in parentheses. Reproduced with permission from ref. 108. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
[4]CPY exhibited absorption and fluorescence spectra (Fig. 94a) which are certainly more similar to those of [8]CPP than pyrene, although some key differences are evident, such as an absorption maximum at shorter wavelength (311 nm). Interestingly, [4]CPY showed concentration-dependent fluorescence (Fig. 94b), suggestive of the formation of intermolecular excimers.
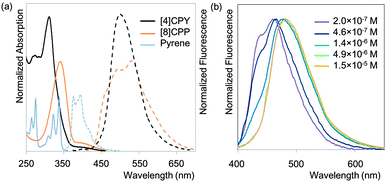 |
| | Fig. 94 (a) UV/visible absorption and fluorescence spectra of the solutions of [4]CPY, [8]CPP and pyrene in CHCl3. (b) Concentration-dependent fluorescence of [4]CPY in CHCl3. The fluorescence spectra were obtained by exciting the sample at λ = 370 nm. Reproduced with permission from ref. 108. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
López Navarrete, Baonza, Casado and co-workers reported a study that utilised Raman infrared spectroscopy to probe several aspects of CPP chemistry.109 In the first instance, the group paid particular attention to the radial breathing mode in the Raman IR spectra of the CPPs under study ([6]- to [12]CPP), as this has previously been used to discern information about the diameter of SWCNTs. For CPPs, a clear inverse relationship was demonstrated between the frequency of this particular mode and the diameter of the CPPs. A second key finding of the work concerned the effect of mechanical stress on CPPs. Application of an external pressure has a profound effect on the Raman spectra of CPPs, as shown in Fig. 95. It can be seen that for both [6]CPP and [12]CPP, the spectrum under pressure (pink line) is appreciably different from that beforehand (dotted line). Upon release of pressure, the spectrum for the sample [12]CPP essentially returns to that observed before the pressure was applied. On the other hand, the application of pressure to [6]CPP appears to induce a permanent deformation, since the spectrum after the release of pressure (solid black line) bears only partial resemblance to the spectrum before the pressure was applied.
 |
| | Fig. 95 785 nm Raman spectra of: (a) [6]CPP and (b) [12]CPP, before (dotted line), with an applied pressure around 6 GPa (pink line) and after the pressure release (solid line). Inserted schemes represent (from left to right) the molecular models before applied pressure, with applied pressure, and after release. Reproduced with permission from ref. 109. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
In an interesting extension of the above experiment, a 1:1 mixture of [6]CPP and [12]CPP was also subjected to high pressure, to determine if a [12]CPP⊃[6]CPP inclusion complex could be formed. It was anticipated that such a complex would protect the inner [6]CPP ring from deformation (a similar effect having been reported previously for double-walled carbon nanotubes). In the event, only very small changes were observed in the Raman spectrum, suggesting that effective encapsulation of [6]CPP had prevented its collapse. This result represents the first experimental evidence for the kind of CPP⊃CPP encapsulation first posited by Fomine (cf.Fig. 39).
Finally, the [10]CPP⊃C60 inclusion complex was studied under high pressure, and the results that were obtained are highly suggestive of the formation of a charge transfer complex under these conditions. Fig. 96 shows the spectra for [10]CPP⊃C60, both in solution and at high pressure. For comparison, the spectra of neutral C60, the C60 radical anion, neutral [10]CPP and the [10]CPP radical cation are also presented. It can be seen that the application of pressure to a sample of [10]CPP⊃C60 leads to the appearance of new Raman bands which are identical to those for C60˙− and [10]CPP˙+. Interestingly, the group also report a similar effect with [9]CPP and C60, (but not with [11]CPP and C60) even though Yamago could not discern any interaction between [9]CPP and C60 by NMR in solution (cf.Fig. 25).
 |
| | Fig. 96 785 nm Raman spectra of 1:1 CHCl3 solution mixtures of [10]CPP and C60 and in solid state after the application of a 6 GPa pressure. The spectra of the neutral C60 and [10]CPP together with those of the C60 anion and of the CPP radical cation are also shown. Reproduced with permission from ref. 109. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Majima and Fujitsuka reported a study on the optoelectronic properties of CPPs, specifically concerned with triplet-excited CPPs.110 They carried out phosphorescence measurements on [n]CPPs (n = 8 to 12) and the spectra they obtained are shown in Fig. 97. A clear correlation between CPP ring size and the emission maximum is seen, with shorter wavelengths being observed for larger CPP ring sizes; implicit in this is that the energy of the T1 state increases with an increase in phenylene units, the opposite of the trend seen in linear oligophenylenes. Flash photolysis experiments permitted the triplet lifetimes (τT) to be determined and these were found to be essentially invariant with CPP ring size, at around 60 μs.
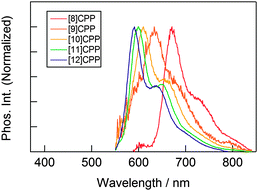 |
| | Fig. 97 Normalized phosphorescence spectra of [n]CPP (n = 8–12) in EEET solvent at 77 K. Excited at 350 nm. Phosphorescence spectra were measured by applying gate between 50 μs and 1 ms after excitation. Reproduced with permission from ref. 110. Copyright 2014 American Chemical Society. | |
Additional experiments permitted the determination of rate constants for various deactivation processes, which were different for each CPP under study. On this basis, schematic representations of the fates of the excited state(s) for [8]CPP and [12]CPP were presented, and these are reproduced in Fig. 98. A key finding of this study was that for the smaller CPPs, the energies of the T1 states were even lower than those of oligophenylenes, suggesting applications for the smaller CPPs as low band gap materials.
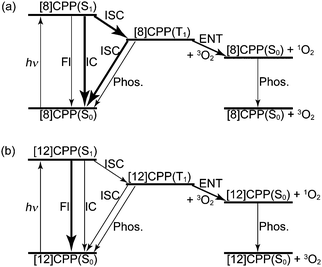 |
| | Fig. 98 Schematic energy diagrams of deactivation pathways of (a) [8]CPP and (b) [12]CPP. “Fl”, “IC”, “ISC”, “ENT” and “Phos” are fluorescence, internal conversion, intersystem crossing, energy transfer, and phosphorescence, respectively. Major deactivation pathways are indicated by bold arrows. Reproduced with permission from ref. 110. Copyright 2014 American Chemical Society. | |
The day after Majima and Fujitsuka's above-mentioned study was published, two papers on inclusion complexes of nanobelts were published. Isobe returned to the (12,8)-[4]CC2,8⊃C60 system he had previously studied extensively, this time probing its photoinduced electron transfer processes. In common with Majima and Fujitsuka, the Isobe group employed flash photolysis to gather data on the fate of the excited state formed upon photoirradiation of (12,8)-[4]CC2,8⊃C60.111 Electron paramagnetic resonance spectroscopy was also used, which confirmed the formation of (12,8)-[4]CC2,8˙+ and C60˙−, as well as a signal corresponding to a triplet charge-separated state. The key findings of Isobe's work are summarised in Fig. 99.
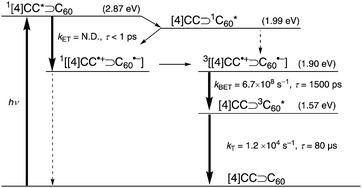 |
| | Fig. 99 Energetics of photodynamic processes in (P)-(12,8)-[4]CC2,8⊃C60. The bold arrows show the pathway confirmed by the present study, and the broken arrows show potential minor pathways. Reproduced with permission from ref. 111. Copyright 2014 American Chemical Society. | |
The other paper published on the same day was from López Navarrete, Baonza, Casado and co-workers, who built on their earlier work on the effects of high pressure on CPP fullerene inclusion complexes.112 They systematically studied the association of C70 with [9]- to [12]CPP. In the cases of [10]CPP⊃C70 and [11]CPP⊃C70, spectra were acquired at several different pressures, allowing trends to be discerned in the relationship between the applied pressure and the frequency of certain bands in the Raman IR spectrum (Fig. 100). Further Raman studies provided evidence for some reorientations of the host–guest complex under certain conditions. For example, [10]CPP⊃C70 ordinarily adopts a conformation where the CPP encircles the equatorial region of the fullerene (cf.Fig. 64–66), but under high pressure, it instead adopts a conformation more akin to a “standing” conformation of C70 in the host, associated with a deformation of the CPP away from a circular conformation, a process which appears to be irreversible. On the other hand, [11]CPP⊃C70, which ordinarily adopts a “standing” conformation of C70, is comparatively resistant to the effects of pressure on its conformation, but instead reorients under conditions of high temperature, with the CPP moving to encircle the equatorial region of C70.
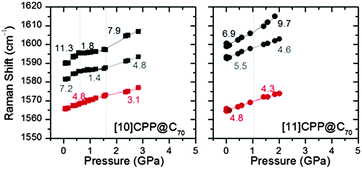 |
| | Fig. 100 Evolution of the Raman frequencies of the G modes of the [10]CPP⊃C70 and [11]CPP⊃C70 samples as a function of pressure. Inserted values correspond to the pressure coefficients of each linear trend expressed in units of cm−1 GPa−1. Black and grey dots correspond to the G1 and G2 bands of the [n]CPPs, respectively, while red dots correspond to the 1565 cm−1 band of C70. Reproduced with permission from ref. 112. Copyright 2014 Royal Society of Chemistry. | |
In the context of the studies above, mention should be made of a study by Zhao, published some few days later, which concerned a theoretical treatment of a fullerene inclusion complex, [6]CPPA⊃C70, although the host in this instance was a cycloparaphenyleneacetylene, i.e. not a CPP derivative.113
A second study in as many months from Fujitsuka, Majima and co-workers concerned the properties of CPP radical cations and anions.114 In order to observe the radicals spectroscopically, they were generated using irradiation with ionising radiation (i.e. γ-rays): a matrix of butyl chloride containing the CPP of interest as a solute was irradiated at 77 K, resulting in the formation of butyl chloride radical cations, which in turn abstracted an electron from the CPP solute molecules. Conversely, generation of CPP radical anions necessitated a matrix of 2-methyltetrahydrofuran, which is known to form radical anions of solute by an analogous process. When absorption spectra were acquired for the radicals so generated, a counter-intuitive trend was observed – for both the cations and anions, the absorption in the near infrared moved to lower energies as CPP ring size increased. This is the opposite of the by now well-established trend for the neutral CPPs, namely that the HOMO–LUMO transition energy increases with increasing CPP ring size. In an attempt to rationalise this finding, the group undertook computational modelling. Fig. 101 shows the calculated orbital energy levels for [12]CPP, its radical cation and radical anion; the main transitions responsible for the near-IR absorptions are shown as red arrows, whereas the main transitions responsible for the UV/vis absorptions are shown as blue arrows. The key to rationalising the trend for lower-energy near-IR absorption with increasing ring size is to recognise which orbitals are involved. Thus, in the radical cation, it is the transition from HOMO − 1/HOMO − 2 to the HOMO which is key, which correspond to HOMO − 1/HOMO − 2 and the HOMO in the corresponding neutral CPP. As can be seen in Fig. 102, as CPP ring size increases, the HOMO → LUMO gap increases, but the HOMO − 1/HOMO − 2 → HOMO gap decreases. A similar argument explains the trend for the radical anions: as CPP ring size increases, the HOMO → LUMO gap increases, but the LUMO → LUMO + 1/LUMO + 2 gap decreases.
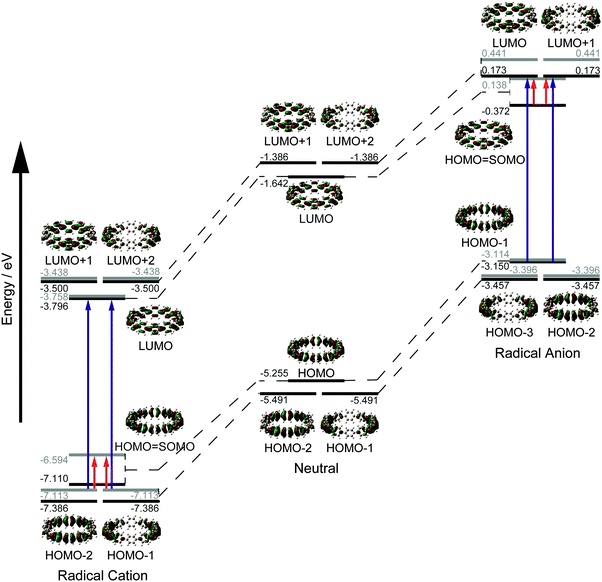 |
| | Fig. 101 Energy levels and MO patterns of radical cation, neutral, and radical anion states of [12]CPP calculated at the (U)B3LYP/6-31G(d) level assuming D6d symmetry as a representative. Numbers indicate energy levels in eV units. For the radical cation and radical anion, MO levels for α and β electrons are indicated by black and grey, respectively. Main transitions, which provide near-IR and UV absorption bands, are indicated by red and blue arrows, respectively. Reproduced with permission from ref. 114. Copyright 2014 American Chemical Society. | |
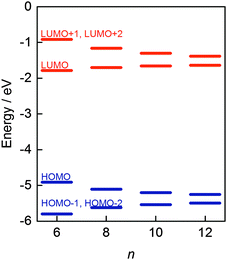 |
| | Fig. 102 Molecular orbitals for [n]CPPs, calculated at the B3LYP/6-31G(d) level of theory. Reproduced with permission from ref. 114. Copyright 2014 American Chemical Society. | |
Irle and Reddy reported an extensive study on vibronic effects on the photophysical properties of CPPs.115 Amongst other findings, their work rationalises the fairly constant absorption maximum seen at λ ≈ 340 nm (regardless of CPP ring size) on the basis of Jahn–Teller and pseudo-Jahn–Teller effects in the S2 and S3 states (which are doubly degenerate). They found that vibronic coupling between the S1 state and the higher excited singlet states increases with CPP ring size and as such, the ostensibly forbidden S0 → S1 transition becomes increasingly allowed as CPP ring size goes up. Their work also leads to a proposition which needs to be tested experimentally: that the absence of “visible” fluorescence for [6]CPP is somewhat of a misnomer, as a fluorescence emission is actually predicted in the near-infrared region, such an extremely large Stokes shift being accounted for by the large structural changes upon photoexcitation of [6]CPP and comparatively weak excited state vibronic coupling compared to larger CPPs (Fig. 103).
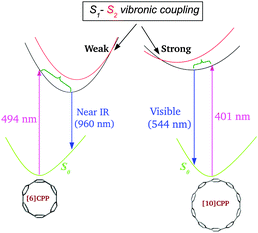 |
| | Fig. 103 Molecular orbitals for [n]CPPs, calculated at the B3LYP/6-31G(d) level of theory. Reproduced with permission from ref. 115. Copyright 2014 American Chemical Society. | |
Wegner published a review on CPPs with specific focus on substituted CPPs,116 which was followed by a report from Yamago and co-workers on La@C82⊂[11]CPP.117 Whereas Itami and Sinohara had already published on Gd@C82⊂[11]CPP in the context of purification of endohedral metallofullerenes (Fig. 82), this report from Yamago concerns the characterisation of charge transfer phenomena observed in La@C82⊂[11]CPP. Of particular interest are the comparisons Yamago has been able to draw between La@C82⊂[11]CPP and fullerene-SWCNT “peapods”. As shown in Fig. 104a, it known that endohedral metallofullerenes have appreciable dipoles due to the localisation of the metal atom near the edge of the fullerene cage; this in turn is due to partial charge transfer from the metal to the fullerene. Furthermore, it is known that in metallofullerene-SWCNT peapods, the dipoles of the “peas” align as shown in Fig. 104b, due to “pea–pea interactions”.
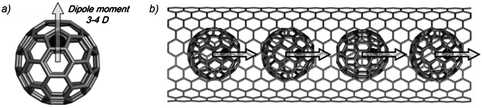 |
| | Fig. 104 (a) Dipole moment of an endohedral metallofullerene. (b) Alignment of dipoles in an endohedral metallofullerene-SWCNT “peapod” complex. Adapted with permission from ref. 117. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Upon successful synthesis of La@C82⊂[11]CPP, a Job's plot confirmed the 1:1 stoichiometry. A shift in the 1H-NMR resonance of [11]CPP upon addition of La⊂C82 (cf.Fig. 25) was clearly observed; conversely, [10]CPP did not interact with La⊂C82 as shown by NMR, despite previous reports of encapsulation of La⊂C82 in (10,10)-SWCNTs.118 Extensive electrochemical characterisation of La@C82⊂[11]CPP was carried out, both by cyclic voltammetry and differential pulse voltammetry. For comparison, C60⊂[10]CPP was also subjected to the same characterisation – it was found that for both C60 and [10]CPP, redox potentials were unaffected by complexation, but that for La@C82⊂[11]CPP, significant changes in the potentials occurred. In ortho-dichlorobenzene appreciable shifts to more negative potentials were observed for La@C82⊂[11]CPP compared to La⊂C82, both by CV and DPV (Fig. 105a). In a more polar solvent, nitrobenzene (Fig. 105b), the same cathodic shift was observed.
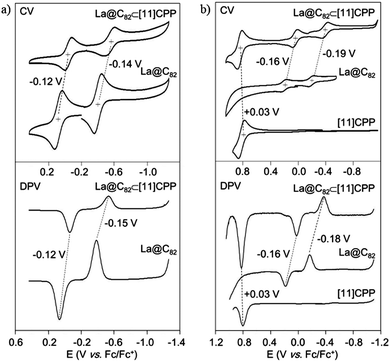 |
| | Fig. 105 Electrochemical behaviour of La@C82⊂[11]CPP, La⊂C82, and [11]CPP. Cyclic voltammograms and differential pulse voltammograms of La@C82⊂[11]CPP, La⊂C82, and [11]CPP are shown in (a) ortho-dichlorobenzene and (b) nitrobenzene with Bu4NPF6. Reproduced with permission from ref. 117. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
The Yamago group carried out computational modelling of La@C82⊂[11]CPP, locating two minima, which are depicted as “A” and “B” in Fig. 106. In conformer A, the metal atom is found in close proximity to the CPP nanobelt and the dipole of La⊂C82 is only 2° away from being perpendicular to the CPP tube axis. In contrast, conformer B involves the dipole of La⊂C82 being oriented almost parallel to the CPP tube axis. Whereas conformer B more closely mimics the arrangement seen in La⊂C82-SWCNT peapods (Fig. 104b), it is in fact conformer A which was computed to be lower in energy. The group obtained an X-ray crystal structure for La@C82⊂[11]CPP (Fig. 107), which shows that in the solid state La@C82⊂[11]CPP adopts a conformation which is not particularly close to either of the two computed minima, having the dipole at 66° to the CPP tube axis. Yamago notes that La@C82⊂[11]CPP lacks the “pea–pea” interactions that operate in La@C82⊂SWCNT and hence there is less impetus for the dipole(s) of La⊂C82 to align with the tube axis.
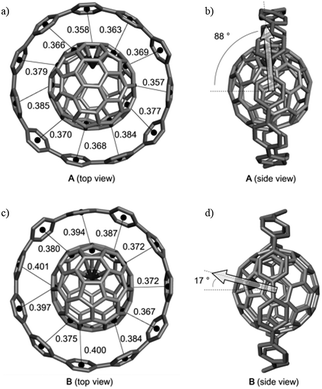 |
| | Fig. 106 Optimized structures of La@C82⊂[11]CPP conformer A, (a) top view and (b) side view, and conformer B, (c) top view and (d) side view, obtained through DFT calculations at the M06-2X/6-31G(d)-SDD level of theory. The numbers in (a) and (c) are the distances (nm) between the centroid of a phenylene unit of [11]CPP and the nearest centroid of a hexagon or pentagon of La⊂C82. Arrows in (b) and (d) are the dipole moments of La⊂C82, and the numbers in (b) and (d) indicate the tilt angles of the dipole moments from the CPP tube axis. Reproduced with permission from ref. 117. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
 |
| | Fig. 107 ORTEP diagram of La@C82⊂[11]CPP, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Solvent and disorder have been omitted for clarity. | |
Tretiak and co-workers have disclosed their theoretical studies on the optoelectronic properties of CPPs.119 The group employed time-dependent DFT and excited state dynamics simulations, determining that rapid (>100 fs) self-trapping of the lowest excitonic state due to electron–phonon coupling gives rise to localised excitation (albeit only for the larger CPPs). Fig. 108a depicts the group's calculated ground and excited state geometries for [6]-, [9]- and [12]CPP, and Fig. 108b depicts the calculated transition densities. For [12]CPP, Fig. 108c depicts the transition dipoles for the various states. The spatial localisation of the excitons renders the Condon approximation invalid and results in breaking of the optical selection rules.
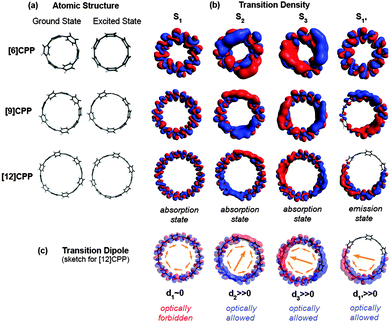 |
| | Fig. 108 (a) Structure of [6]-, [9]-, and [12]CPP molecules demonstrating conformational differences in the ground and excited states. (b) Orbital distribution of transition density for S1–S3 and S1′ transitions in [6]-, [9]-, and [12]CPPs. (c) Schematic of transition dipole shown for S1–S3 and S1′ transitions in [12]CPP. Red and blue colours correspond to negative and positive values of the transition density, respectively. Adapted with permission from ref. 119. Copyright 2014 American Chemical Society. | |
Isobe's most recent report on CPPs and related nanohoops describes the encapsulation of fullerenes in his previously reported (P)-(12,8)-[4]CA2,8 (Fig. 58).120 The main focus of the Isobe group's report is to compare these new (P)-(12,8)-[4]CA2,8⊃C60 and (P)-(12,8)-[4]CA2,8⊃C70 inclusion complexes (Fig. 109) with their previously reported (P)-(12,8)-[4]CC2,8⊃C60 and (P)-(12,8)-[4]CC2,8⊃C70 (Fig. 55 and 56). The key thermodynamic parameters for formation of the host–guest complexes were determined and it was found that the association constants (Ka) and ΔG values were very similar for all four supramolecular entities. However, the relative contributions of enthalpy and entropy to the Gibbs free energy were different for the [4]CA2,8 complexes compared to the [4]CC2,8 complexes. The [4]CA systems comprise a lengthened tube with respect to the [4]CC systems, as shown in Fig. 109. Values are reported for the length index (tf) in each case, a parameter defined by Isobe as part of a broader system of nomenclature for nanobelts and nanotubes.121 For the lengthened-tube [4]CA systems, the enthaplic term was more highly favourable for complexation, but this was offset by a less favourable (or indeed unfavourable) entropic term. To rationalise these findings, the Isobe group propose a model where the binding in (P)-(12,8)-[4]CA2,8⊃fullerenes consists not only of more extensive π–π interactions than for the [4]CC system, but also significant CH–π interactions between the hexyl sidechains on the nanohoop and the fullerene (which entail an entropic penalty). The authors present NMR data that support the notion of free rotation of the fullerene journal in the [4]CA bore (cf.Fig. 56), as well as providing a Hirshfeld analysis of the crystal structure of (P)-(12,8)-[4]CC2,8⊃C60 (cf.Fig. 91) and computational data on the conformations of the alkyl side chains.
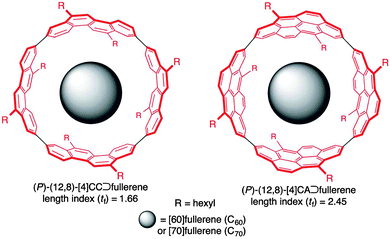 |
| | Fig. 109 (P)-(12,8)-[4]CA2,8⊃fullerenes, as reported by Isobe. Adapted with permission from ref. 120. Copyright 2015 Royal Society of Chemistry. | |
Itami, Segawa and co-workers followed their 2012 report65 of a “carbon nanocage” Y-branching unit (131, Fig. 48) by extending this approach to the synthesis of Y-branching units of different sizes.122 The group give a full account of the synthetic strategy towards their previously reported Y-branching unit 131, which they designate a [6.6.6] carbon nanocage. Thus, they propose a more general nomenclature for such nanocages, whereby for a [p,q,r] nanocage, the parameters denote the number of arylene units between the two nodal (or “polar”) arenes. The synthetic methodology previously reported for the [6.6.6] nanocage 131 required some slight modification in order to effect the synthesis of the two new nanocages this report describes, namely a [5,5,5] and a [4,4,4] nanocage. As shown in Scheme 58, their synthesis commenced from 1,3,5-tribromobenzene 271, which underwent three cycles of lithium–halogen exchange, addition to 2-ring ketone building block 272 and MOM protection of the resultant tertiary alcohol. This iterative approach ultimately gave tripodal fragment 278, possessing three halides for further elaboration. Exhaustive borylation gave the complementary tris(pinacolborane) fragment 279 (which required purification by GPC), which in turn could be elaborated by three-fold Suzuki–Miyaura coupling to give elongated tripodal fragment 280.
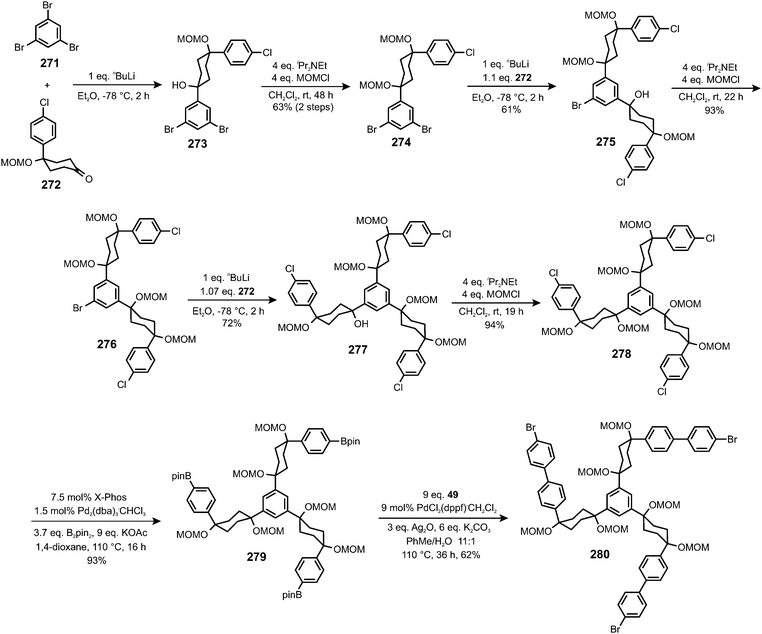 |
| | Scheme 58 Synthesis of pseudo-hemispherical building blocks for nanocage construction. | |
For construction of the fully cyclised nanocage precursors, more than one approach was shown to be applicable. For the smallest example, the union of two tripodal fragments could be achieved either by three-fold reductive nickel-mediated coupling (i.e.278 + 278 → 281, Scheme 59), or by three-fold Suzuki–Miyaura coupling (i.e.278 + 279 → 281). Both approaches proceeded in similarly modest yields, indicative of the strain inherent even in the non-aromatic precursor 281. For the larger nanocage, union of 280 (the homologue for 278) with 279 was also achieved by three-fold Suzuki–Miyaura coupling. Finally, both the precursors 281 and 283 underwent aromatisation using Itami's standard conditions, giving [4.4.4]nanocage 282 and [5.5.5]nanocage 284 respectively. The smaller of these, 282, was also characterised by X-ray crystallography (Fig. 110). Its structure in the solid state revealed an almost-spherical inner void; the minor distortion away from sphericality might be rationalised on the basis of the incorporation of molecules of solvent into the structure (not shown).
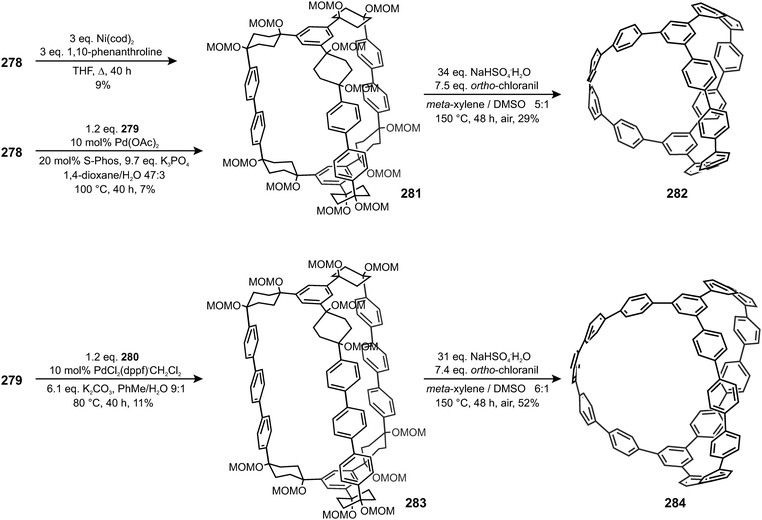 |
| | Scheme 59 Synthesis of carbon nanocage Y-branching units 282 and 284. | |
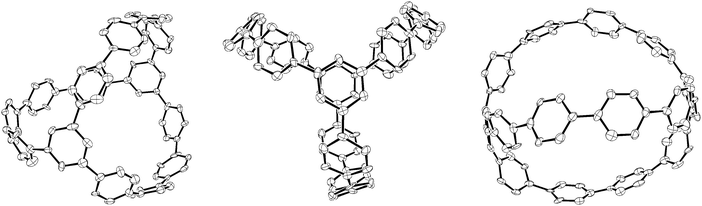 |
| | Fig. 110 ORTEP diagram showing three views of 282, showing ellipsoids at 30% probability. Solvent and hydrogen atoms have been omitted for clarity. | |
All three nanocages ([4.4.4]nanocage 282, [5.5.5]nanocage 284 and [6.6.6]nanocage 131) were comprehensively characterised by a variety of techniques, in order to discern the relationship(s) between nanocage size and various parameters. It was determined by cyclic voltammetry that the larger the nanocage, the more positive the value for Eox. Regarding the photophysical properties of the nanocages, the absorption and fluorescence spectra for 131, 282 and 284 are shown in Fig. 111 and it can be seen that the absorption maxima are increasingly red-shifted as nanocage size increases, in contrast to the essentially constant absorption maxima of CPPs themselves. Quantum yields were also observed to increase with increasing nanocage size. Extensive computational modelling was carried out and the strain energy per carbon atom was calculated for the three nanocages, as well as for [6]- to [16]CPP. These data are depicted in Fig. 112, and it can clearly be seen that the data for the nanocages adhere to the trendline that can be drawn for the CPP data (i.e. the smaller the diameter, the greater the strain energy per carbon). Orbital energy levels were calculated for the three nanocages (Fig. 113a) and the smallest cage was found to have the highest HOMO level, in keeping with the CV data. Itami and co-workers rationalise the increase in HOMO–LUMO gap with increasing nanocage diameter on the basis of orbital interactions between the three [n]paraphenylene bridges and the two nodal phenyl ring bridgeheads (Fig. 113b and c) – this is a wholly different argument to the one put forward to explain the same trend in CPPs themselves.
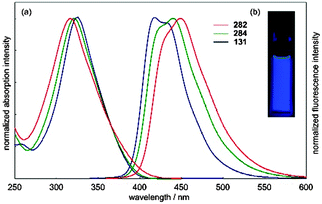 |
| | Fig. 111 (a) UV-vis absorption (solid line) and fluorescence (broken line) spectra of 131, 282 and 284 in chloroform. (b) Fluorescence of chloroform solution of 282. Adapted with permission from ref. 122. Copyright 2014 American Chemical Society. | |
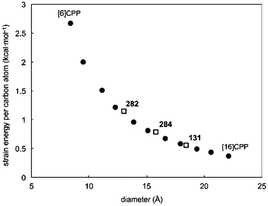 |
| | Fig. 112 Strain energies of carbon nanocages 131, 282 and 284 (square) and [6]–[16]CPP (filled circle) per carbon atom versus diameters (B3LYP/6-31G(d)). Adapted with permission from ref. 122. Copyright 2014 American Chemical Society. | |
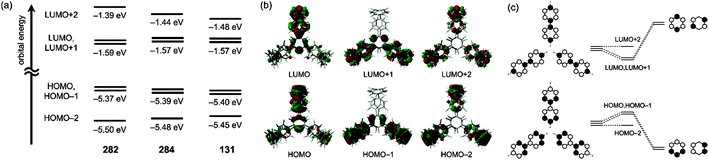 |
| | Fig. 113 (a) Energy diagrams of the frontier MOs of 131, 282 and 284. (b) Pictorial representations of the frontier MOs of 282 calculated at the B3LYP/6-31G(d) level of theory. (c) Components of frontier MOs of 131, 282 and 284. Adapted with permission from ref. 122. Copyright 2014 American Chemical Society. | |
Shortly after the above disclosure on nanocages, the Itami group published on the synthesis and characterisation of “cyclophenylenethienylenes” (CPTs), cyclic structures comprising alternating para-phenylene and 2,5-thienylene units.123 The synthetic approach in shown in Scheme 60. A double addition of a metallated silylacetylene 285 into cyclohexa-1,4-dione followed by MOM protection and desilylation gave L-shaped bis(acetylene) building block 288. This underwent “shotgun” cyclooligomerisation by means of a Glaser coupling giving products from dimer 289 through to hexamer 293. In order to be able to carry out this cyclooligomerisation on a sufficiently large scale, the Itami group were obliged to employ a biphasic reaction medium for this transformation. (Note that yields shown for 289–293 in Scheme 60 are the best yields obtained for each oligomer under a variety of reaction conditions). Although dimer 289 was the major product, this was not elaborated further, since [2]CPT (analogous with[4]CPP) was considered too strained a target to be synthetically viable. On the other hand, 290–293 each underwent multiple thiophene formations by addition of sodium sulfide to afford cyclo-cyclohexylidenetheinylenes (“CCyTs”) 294–297. The structure of [3]CCyT 294 was confirmed by X-ray crystallography (Fig. 114). Aromatisation of the CCyTs was carried out under Itami's standard conditions (with no addition of ortho-chloranil needed, in contrast to the nanocages in Scheme 59). The reaction was not successful for [3]CCyT 294, likely due to the predicted high strain energy of [3]CPT, but the larger CCyTs furnished the desired [4]- to [6]CPT as hoped. The structure of [4]CPT was also secured crystallographically (Fig. 115).
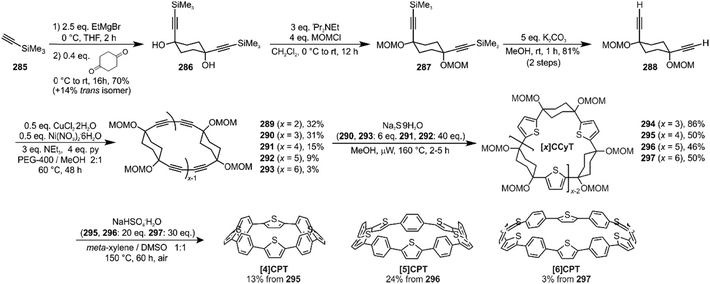 |
| | Scheme 60 Synthesis of cyclophenylenethienylenes (CPTs). | |
 |
| | Fig. 114 ORTEP diagram showing 294, showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Solvent has been omitted for clarity. | |
 |
| | Fig. 115 ORTEP diagram showing three views of [4]CPT (one of six molecules in the unit cell), showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Solvent has been omitted for clarity. | |
The X-ray structure of [4]CPT is very revealing – in the solid state, all four thiophene rings are oriented so that their sulfur atoms are pointing towards the interior of the macrocycle, so giving [4]CPT a cone shape overall, of C4v symmetry. Significantly, DFT calculations on a single molecule of [4]CPT in the geometry exhibited in the solid state predict a large dipole moment of 2.7 Debye. The NMR spectra for the CPTs are very simple, indicative of high symmetry and free rotation of both phenylene and thienylene groups on the NMR timescale. The absorption and emission spectra of the CPTs are shown in Fig. 116. It can be seen that the absorption maxima are increasingly red-shifted with increasing ring size. This is in contrast to CPPs (invariant absorption maxima), but in keeping with the observations for Itami's nanocages (Fig. 111). The main trend in the fluorescence of the CPTs (emission maxima increasingly blue-shifted with increasing ring size) is also the same as for the nanocages. Itami and co-workers have also carried out extensive computational studies on the CPTs, and conclude that the CPTs may truly be considered to be hybrid CPP–cyclothiophene structures, as in some regards their properties are closer to those of CPPs, yet in others they more closely mimic the cyclothiophenes (which do not possess a radial π system).
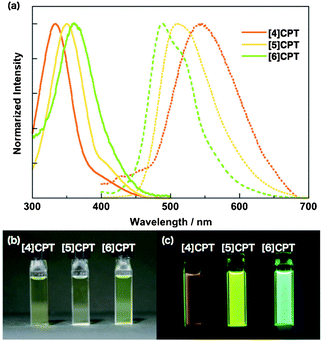 |
| | Fig. 116 (a) UV-vis absorption (broken line) and fluorescence spectra (solid line) of [4]CPT, [5]CPT, and [6]CPT in cyclohexane (absorption and fluorescence spectra were normalized). (b) Pictures of [4]CPT, [5]CPT, and [6]CPT in cyclohexane under (b) ambient light and (c) UV irradiation at 365 nm. Reproduced with permission from ref. 123. Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Yamago's final publication of 2014 returned to the dicationic [8]CPP2+ on which both his group87 and Jasti's group81 had published previously. A motivation for this current work was to attempt to resolve discrepancies between the spectroscopic data reported previously by the two groups – Yamago notes that the spectra reported by his group for [8]CPP2+ and [8]CPP˙+ are the same as those reported by Jasti for [8]CPP˙+ and ([8]CPP)2˙+, respectively. In this latest work, Yamago, Uchiyama and co-workers carried out magnetic circular dichroism spectroscopy on [8]CPP2+(SbF6−)2, as shown in Fig. 117.124 The MCD spectrum for [8]CPP2+ exhibits a derivative-shaped signal in the near-IR region, which the authors assign as the Faraday A term, which is negative. A smaller signal in the visible region is also assigned as having a negative Faraday A term. The authors assign these two terms as being due to transitions from the doubly degenerate HOMO to the LUMO, and from the doubly degenerate HOMO to the LUMO + 1, respectively. The frontier molecular orbitals for [8]CPP2+ were modelled using TDDFT and compared to those of all-cis-[32]annulene2+ (Fig. 118).
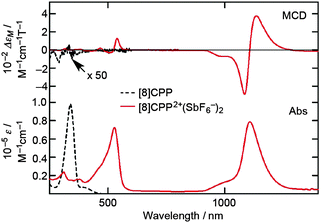 |
| | Fig. 117 Magnetic circular dichroism (top) and electronic absorption (bottom) spectra of [8]CPP2+(SbF6−)2 (red solid line) and [8]CPP (black broken line) in CH2Cl2 at room temperature under Ar. Reproduced with permission from ref. 124. Copyright 2015 American Chemical Society. | |
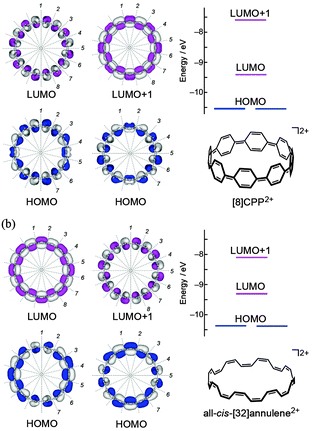 |
| | Fig. 118 Frontier molecular orbitals and energy levels of the optimized structures of [8]CPP2+ (a) and all-cis-[32]annulene dication (b). Calculations were performed at the B3LYP/6-31+G(d) level. Arbitrary nodal lines are drawn on the iso-surface plots. Reproduced with permission from ref. 124. Copyright 2015 American Chemical Society. | |
Yamago, Uchiyama and co-workers state that the MCD spectra for [8]CPP2+ (Fig. 117) and [8]CPP˙+ (not shown) that they have acquired are supportive of their assignments of the identity of these species and, therefore, are inconsistent with Jasti's reported assignments.81 Also of note, as shown in Fig. 118, the pattern of nodes and the orbital degeneracy for [8]CPP2+ are identical to those of (all-cis-[32]annulene)2+. The authors state that [8]CPP2+ possesses in-plane aromaticity (i.e. the CPP ring is aromatic “over all”) by virtue of its π electron count (conforming to the Hückel 4n + 2 rule, where n = 7). By extension, the dianion [8]CPP2− would also possess in-plane aromaticity (n = 8). This characteristic may be generalised further – for a CPP of any size, both the dication and dianion would conform to the Hückel rule and possess in-plane aromaticity. In support of this proposal, the authors present calculated nucleus-independent chemical shift values for neutral, dicationic and dianionic [5]- to [10]CPPs, which clearly show the anisotropic effects due to the in-plane aromaticity of the dicationic and dianionic forms. The authors suggest that the unexpected stability of [8]CPP2+ may be rationalised on the basis of this aromaticity; the work also suggests that although CPP dianions have not been reported to date, they ought to be stable entities.
The last report relevant to CPP chemistry from 2014 came from Stępień and co-workers.125 They describe the synthesis of discrete nanotube end-caps, whose structures include cyclooligo(hetero)arene nanobelts with radial π systems. As shown in Scheme 61, the Stępień group have adopted a templated approach, whereby three 2,7-dibromocarbazoles are tethered at nitrogen to a central mesitylene unit. In one step, strain-free precursor 298 can be induced to undergo a three-fold nickel mediated reductive coupling to give nanotube end-cap 300, which is computed to have a strain energy of 578 kJ mol−1. In a similar fashion, more extensively annulated precursor 299 affords nanotube end-cap 301; this is computed to have an greater strain energy of 603 kJ mol−1 (or even higher, depending on the computational methodology employed)! Final products 300 and 301 were oxidatively labile to the extent that their purification required column chromatography in an inert atmosphere using degassed solvents.
 |
| | Scheme 61 Synthesis of nanotube end-caps by three-fold reductive coupling. In the structures of the products 300 and 301, formal double bonds are omitted for clarity. | |
Stpień's approach to radial π systems is potentially highly significant for several reasons. It is conceptually distinct from the previously described approached whereby the key transformations take place over several steps. The approaches of Jasti and Itami have two distinct phases: firstly, cyclisation of a non-aromatised precursor, then secondly an aromatisation step which also incorporates strain energy in the nanohoop. On the other hand, the approaches of Yamago and Isobe do not require an aromatisation step, but nevertheless also have two distinct stages: firstly cyclisation (to an aromatic but non-strained intermediate), then secondly a step that incorporates the strain energy. In contrast, Stpień's approach consists of one step only: cyclisation and strain buildup are achieved simultaneously from a precursor in which all rings are already aromatic. The great brevity of the approach (one step to nanohoop 300 from a precursor 298, that is itself one step from commercial materials), as well as its ability to assemble nanohoops with such tremendous strain energies, renders this a powerful strategy that will undoubtedly find further application in the field of CPPs and related nanohoops. Both 300 and 301 incorporate [6]CPP as a structural subunit and the authors themselves speculate on the plausibility of accessing CPPs and derivatives using this strategy in conjunction with a removable tethering unit.
The NMR spectra for 300 and 301 are indicative of C3v symmetry in solution. Solid state structures were obtained by X-ray crystallography for both nanotube end-caps, and are shown in Fig. 119 and 120 – 300 and 301 are rigid bowl-shaped molecules with some structural similarities to SWCNT end-caps. The absorption spectra of 300 and 301 were significantly different from those observed for the CPPs, with 300 exhibiting several absorption maxima (the strongest being at 266 and 363 nm), whereas 301 exhibited a single absorption maximum at ≈320 nm. Both molecules were fluorescent with emission in the visible region (in contrast to [6]CPP).
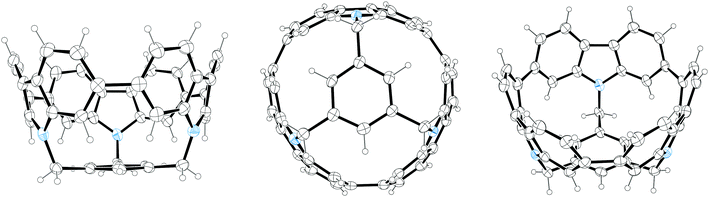 |
| | Fig. 119 ORTEP diagram showing three views of 300 (one of three molecules in the unit cell), showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Solvent has been omitted for clarity. | |
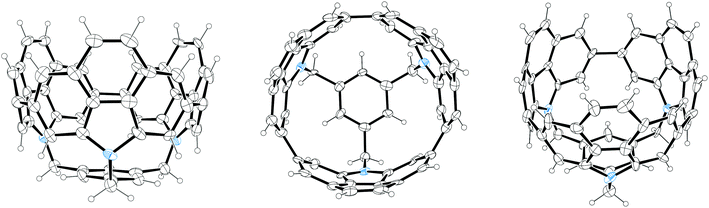 |
| | Fig. 120 ORTEP diagram showing three views of 301 (one of two molecules in the unit cell), showing ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Solvent has been omitted for clarity. | |
It was determined computationally that for all orbitals from HOMO − 3 to LUMO for 300, the amplitudes on the central mesitylene unit are near zero (Fig. 121). As regards the mechanism for the three-fold reductive coupling that gives rise to 300 and 301, both computational data and experimental observations support the oxidative insertion and ligand metathesis of three nickel atoms (giving an intermediate containing three aryl–Ni–aryl motifs), prior to any of the reductive eliminations occurring. One straightforward reason why this is the only plausible sequence of events is that if a reductive elimination occurred prior to formation of all three aryl–Ni–aryl motifs, the formation of a direct aryl–aryl bond would result in the remaining aryl bromides being pulled too far apart for full cyclisation to occur.
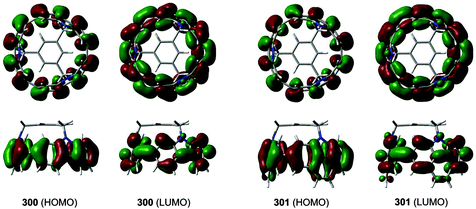 |
| | Fig. 121 Calculated HOMO and LUMO for 300 and 301. Reproduced with permission from ref. 125. Copyright 2015 American Chemical Society. | |
2015
Thus far, two reports on CPPs have appeared (simultaneously) in 2015. Chen, Swan and co-workers have further studied the Raman spectra of [6], [8], [10] and [12]CPP, employing group theory arguments to assign the observed Raman peaks.126 Itami, Segawa and co-workers have published only the third ever report (see also Scheme 24 and Fig. 57) on organometallic chemistry of CPPs, describing the formation and reactivity of η6-arene metal tricarbonyl complexes of CPPs, with a variety of metals and CPP ring sizes.127 As shown in Scheme 62, reaction of [9]CPP with one equivalent of chromium hexacarbonyl led to formation of η6-arene tricarbonylchromium complex 302 in 35% after purification. Synthesis of 302 was hampered by the ease with which the metal fragment was decomplexed from the arene upon exposure to light. Thus, the reaction and all subsequent isolation and purification procedures were carried out in the dark. At the outset, it was not obvious whether the metal would coordinate to the convex or the concave face of the CPP. The structure of 302 was solved crystallographically (Fig. 122) and it was seen that in the solid state, the metal is coordinated to the convex face (i.e. outside the CPP ring).
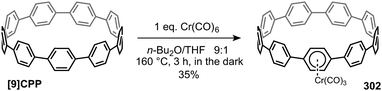 |
| | Scheme 62 Synthesis of CPP–metal tricarbonyl complex 302. | |
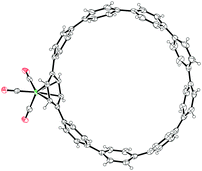 |
| | Fig. 122 ORTEP diagram showing the structure of 302 with ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Solvent has been omitted for clarity. | |
The 1H-NMR spectrum of 302 is reproduced in Fig. 123. The four protons on the phenylene unit complexed to Cr resonate as a singlet at δ = 5.46 ppm, significantly upfield of the normal aromatic region, as is typical for such η6-arene complexes. The equivalence of these four protons confirms the η6 mode of coordination in solution. Computational modelling of 302 allowed the estimation of the barrier to interconversion between the possible convex and concave rotamers of 302. This was calculated to be 51 kJ mol−1, which is not much higher than for the rotation of a phenylene unit in [9]CPP itself (44 kJ mol−1). Crucially, though, the concave rotamer of 302 was found to be higher in energy than its convex equivalent by 32 kJ mol−1. The orbitals of 302 were computed and unsurprisingly, the coordination of the metal was found to perturb the orbitals significantly with respect to those in uncomplexed [9]CPP (Fig. 124). As a consequence, the absorption spectrum of 302 is somewhat different from that of [9]CPP, having a maximum at 334 nm (slightly blue-shifted with respect to [9]CPP), but also having a lower intensity absorption at 440 nm.
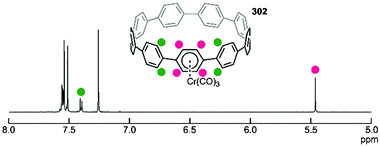 |
| | Fig. 123
1H-NMR spectrum of 302 (600 MHz, CDCl3). Adapted with permission from ref. 127. Copyright 2015 American Chemical Society. | |
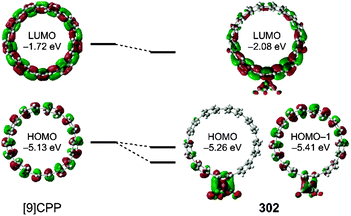 |
| | Fig. 124 Frontier molecular orbitals of [9]CPP (left) and 302 (right). Adapted with permission from ref. 127. Copyright 2015 American Chemical Society. | |
Itami, Segawa and co-workers then extended their efforts towards metal complexation with other CPPs and metals. All possible combinations of [9]CPP and [12]CPP with group six hexacarbonyls (chromium, molybdenum, tungsten) were tried – in each instance, formation of the desired product was observed by 1H-NMR and by mass spectrometry, but attempts at isolation failed due to the instability of the complexes upon exposure to light and/or air, with 302 being the only isolated CPP–metal complex reported in the present work.
Coordination of a metal to an arene in an η6 fashion significantly alters the reactivity of the arene in several regards, including lowering the pKa of the attached protons. The authors were able to exploit this modulation of reactivity to prepare several monofunctionalised CPPs, as shown in Scheme 63. Due to the instability of the CPP–metal complexes, these were not isolated. Rather, a one-pot, four-step protocol was implemented, comprising complexation, lithiation, addition of an electrophile and decomplexation. By this approach trimethylsilyl–[x]CPPs (x = 9, 12) 305 and 306 were prepared. The structure of 305 was confirmed by X-ray crystallography (Fig. 125). Other derivatives of the smaller CPP were prepared in an analogous fashion, namely borylated [9]CPP 307 and [9]CPP carboxylate ester 308. While the headline yields were moderate, the overall mass balance was high in each case, with most of the remaining material being recovered as unreacted starting material. The transformations shown in Scheme 63 are significant since they represent the first examples of the selective post-synthetic modification of CPPs. Although monofunctionalised CPPs have been prepared previously (e.g.261, Scheme 55), in that case the functional handle was incorporated during the multistep procedure prior to formation of the CPP ring. Also significant is the fact that the current work allows for clean monofunctionalisation of CPPs, whereas the authors state that traditional SEAr reactions on CPPs lead to complex, inseparable mixtures of isomeric multiply-functionalised CPPs, as exemplified by a reported unsuccessful bromination.
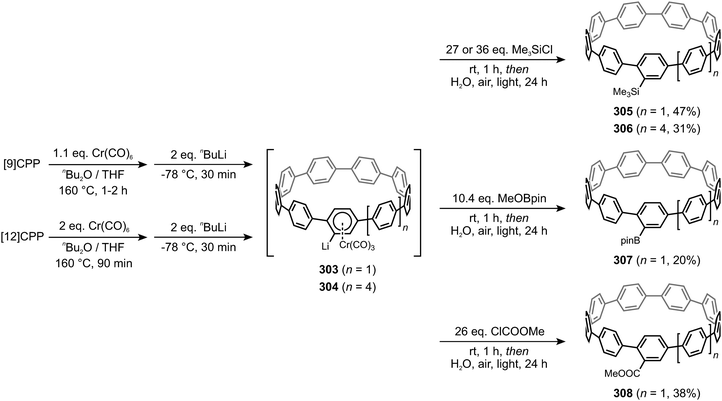 |
| | Scheme 63 Synthesis of monofunctionalised CPPs via the intermediacy of CPP–metal tricarbonyl complexes. | |
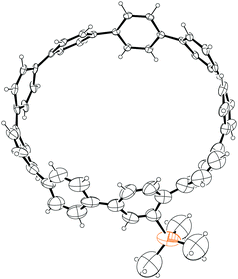 |
| | Fig. 125 ORTEP diagram showing the structure of 305 with ellipsoids at 30% probability. H atoms are shown as spheres of arbitrary radius. Solvent has been omitted for clarity. | |
Summary of synthetic approaches
As the field of CPP chemistry has evolved, several distinct strategies have evolved for the preparation of such nanohoops. These strategies are summarised in Scheme 64.
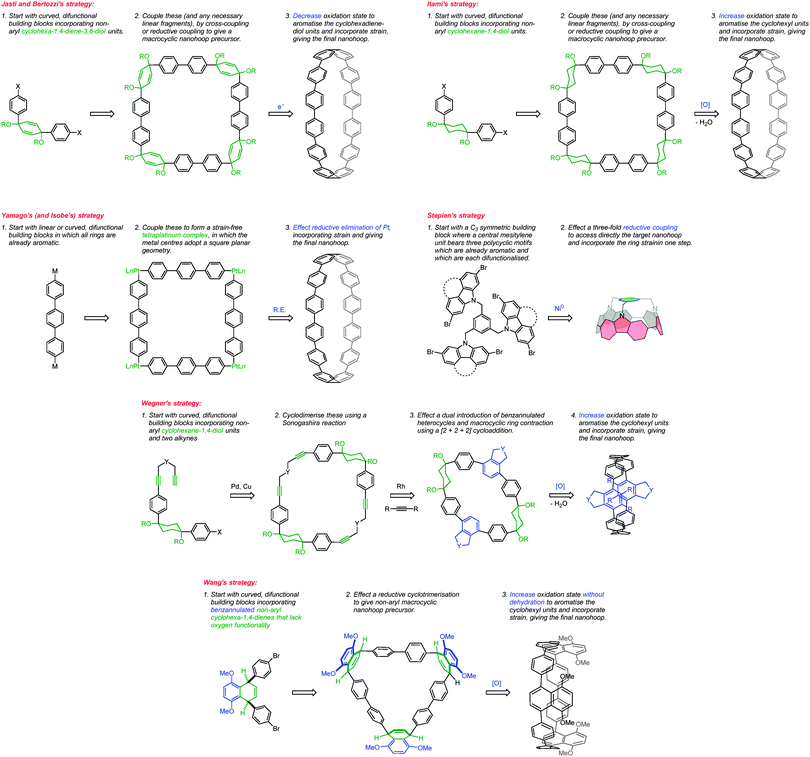 |
| | Scheme 64 Summary of different strategies for the synthesis of CPPs and related nanohoops. | |
Concluding remarks
Less than seven years have elapsed since the first report of the synthesis of CPPs, yet in this short time, a great deal of work has been reported that has had a transformative effect. From a synthetic perspective, a wide variety of CPP ring sizes are now accessible in a selective manner, thanks to methodology which has been extensively optimised. The brevity and high yielding nature of the various established routes to CPPs make access to them for diverse applications a realistic proposition. Indeed, at the time of writing, [9]-, [12]- and [15]CPP are commercially available from Kanto Chemical Corporation and from Tokyo Chemical Industries. Furthermore, the published synthetic methodology is sufficiently flexible that future reports on more novel CPP variants will undoubtedly appear, either making use of the known methods, or indeed reporting wholly novel approaches to the key radial π system. On the theoretical side, great insight has been gained into the origins of the structural and spectroscopic properties of the CPPs and related nanohoops, any many of their counter-intuitive properties have been rationalised. Applications of CPPs are beginning to emerge, foremost of which is their use for template “bottom-up” SWCNT synthesis. The use of CPP inclusion complexes to effect purification of endohedral metallofullerenes is significant and further reports on the supramolecular chemistry of CPPs will undoubtedly appear. A comparatively underexplored area so far is that of sensing applications for CPPs, but Itami's report on the pH-dependent fluorescence of [14,4]CPPy hints at the possibilities. Also, many authors have alluded to the characteristics of various CPP derivatives which would make them appropriate materials for organic electronics, but to date they remain an untapped resource in this regard. In summary, there is every reason to believe that activity in this field will continue to increase in future and hitherto unforeseen applications can certainly be expected.
Note added in proof
Since the preparation of this manuscript, the following relevant research has been published: Kim, Osuka and co-workers have reported the synthesis of cyclic 2,12-porphyrinylene nanohoops, which they term “[n]CPs”; they describe the preparation of Ni(II)-[n]CPs where n = 3 to 5.128 Zhao and co-workers have published a theoretical study on the nature of noncovalent interactions in [n]CPP⊃C70, where n = 10 to 12.129 Itami and co-workers have described a one-shot K-region-selective annulative π-extension reaction suitable for the synthesis of discrete nanographenes, and with possible applicability to CPPs.130 Jasti has published a personal account of his group's research, with particular emphasis on the smaller CPPs.131 Itami and co-workers have reported the isolation and characterisation of Li+@C60⊂[10]CPP.132 Isobe, Kono and co-workers have published further theoretical studies on carbonaceous molecular bearings, with particular emphasis on association thermodynamics and dual-mode rolling dynamics.133 Itami and co-workers reported a concise, high-yielding palladium-free synthesis of [10]CPP.134
References
- V. C. Parekh and P. C. Guha, J. Indian Chem. Soc., 1934, 11, 95 CAS.
- D. T. M. Wong and C. S. Marvel, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 1637 CrossRef CAS.
- O. M. Yaghi, H. Li and T. L. Groy, Z. Kristallogr. - New Cryst. Struct., 1997, 212, 453 CAS.
- R. Friederich, M. Nieger and F. Vögtle, Chem. Ber., 1993, 126, 1723 CrossRef CAS.
- J. Franke and F. Vögtle, Tetrahedron Lett., 1984, 25, 3445 CrossRef CAS.
- Y. Miyahara, T. Inazu and T. Yoshino, Tetrahedron Lett., 1983, 24, 5277 CrossRef CAS.
- J. E. McMurry, G. J. Haley, J. R. Matz, J. C. Clardy and J. Mitchell, J. Am. Chem. Soc., 1986, 108, 515 CrossRef CAS PubMed.
- S. Kammermeier, P. G. Jones and R. Herges, Angew. Chem., Int. Ed., 1996, 35, 2669 CrossRef CAS.
- M. Machón, S. Reich, J. Maultzsch, H. Okudera, A. Simon, R. Herges and C. Thomsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 155402 CrossRef.
- M. N. Jagadeesh, A. Makur and J. Chandrasekhar, J. Mol. Model., 2000, 6, 226 CrossRef CAS.
- G. J. Bodwell, Nat. Chem., 2014, 6, 383 CrossRef CAS PubMed.
- E. Kayahara, V. K. Patel and S. Yamago, J. Am. Chem. Soc., 2014, 136, 2284 CrossRef CAS PubMed.
- P. J. Evans, E. R. Darzi and R. Jasti, Nat. Chem., 2014, 6, 404 CrossRef CAS PubMed.
- T. Tahara and Y. Tobe, Chem. Rev., 2006, 106, 5274 CrossRef PubMed.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646 CrossRef CAS PubMed.
- F. Grein, J. Phys. Chem. A, 2002, 106, 3823 CrossRef CAS.
- A. Goller and U. W. Grummt, Chem. Phys. Lett., 2000, 321, 399 CrossRef CAS.
- R. Herges, Chem. Rev., 2006, 106, 4820 CrossRef CAS PubMed.
- N. I. Nijegorodov, W. S. Downey and M. B. Danailov, Spectrochim. Acta, Part A, 2000, 56, 783 CrossRef CAS.
- B. D. Steinberg and L. T. Scott, Angew. Chem., Int. Ed., 2009, 48, 5400 CrossRef CAS PubMed.
- H. Takaba, H. Omachi, Y. Yamamoto, J. Bouffard and K. Itami, Angew. Chem., Int. Ed., 2009, 48, 6112 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- B. M. Wong, J. Phys. Chem. C, 2009, 113, 21921 CAS.
- S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757 CrossRef CAS PubMed.
- A. Yahav-Levi, I. Goldberg and A. Vigalok, J. Am. Chem. Soc., 2006, 128, 8710 CrossRef CAS PubMed.
- D. Sundholm, S. Taubert and F. Pichierri, Phys. Chem. Chem. Phys., 2010, 12, 2751 RSC.
- Y. Segawa, H. Omachi and K. Itami, Org. Lett., 2010, 12, 2262 CrossRef CAS PubMed.
- T. Kawase, H. R. Darabi and M. Oda, Angew. Chem., Int. Ed., 1996, 35, 2664 CrossRef CAS.
- M. A. Ali and M. S. Krishnan, Mol. Phys., 2009, 107, 2149 CrossRef CAS.
- R. Jasti and C. R. Bertozzi, Chem. Phys. Lett., 2010, 494, 1 CrossRef CAS PubMed.
- S. Taubert, D. Sundholm and F. Pichierri, J. Org. Chem., 2010, 75, 5867 CrossRef CAS PubMed.
- S. M. Bachrach and D. Stück, J. Org. Chem., 2010, 75, 6595 CrossRef CAS PubMed.
- H. Omachi, S. Matsuura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 10202 CrossRef CAS PubMed.
- Y. Segawa, S. Miyamoto, H. Omachi, S. Matsuura, P. Šenel, T. Sasamori, N. Tokitoh and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 3244 CrossRef CAS PubMed.
- H. Omachi, Y. Segawa and K. Itami, Org. Lett., 2011, 13, 2480 CrossRef CAS PubMed.
- Y. Segawa, P. Šenel, S. Matsuura, H. Omachi and K. Itami, Chem. Lett., 2011, 40, 423 CrossRef CAS.
- T. Iwamoto, Y. Watanabe, Y. Sakamoto, T. Suzuki and S. Yamago, J. Am. Chem. Soc., 2011, 133, 8354 CrossRef CAS PubMed.
- C. Eaborn, K. J. Odell and A. Pidcock, J. Chem. Soc., Dalton Trans., 1978, 357 RSC.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed., 2011, 50, 8342 CrossRef CAS PubMed.
- T. J. Sisto, M. R. Golder, E. S. Hirst and R. Jasti, J. Am. Chem. Soc., 2011, 133, 15800 CrossRef CAS PubMed.
- S. Hitosugi, W. Nakanishi, T. Yamasaki and H. Isobe, Nat. Commun., 2011, 2, 492 CrossRef.
- B. M. Wong and J. W. Lee, J. Phys. Chem. Lett., 2011, 2, 2702 CrossRef CAS PubMed.
- E. S. Hirst, F. Wang and R. Jasti, Org. Lett., 2011, 13, 6220 CrossRef CAS PubMed.
- J. Xia and R. Jasti, Angew. Chem., Int. Ed., 2012, 51, 2474 CrossRef CAS PubMed.
- A. Yagi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2012, 134, 2962 CrossRef CAS PubMed.
- T. Sisto and R. Jasti, Synlett, 2012, 483 CAS.
- H. B. Li, A. J. Page, S. Irle and K. Morokuma, ChemPhysChem, 2012, 13, 1479 CrossRef CAS PubMed.
- E. H. Fort and L. T. Scott, J. Mater. Chem., 2011, 21, 1373 RSC.
- E. H. Fort, P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 16006 CrossRef CAS PubMed.
- K. Itami, Pure Appl. Chem., 2012, 84, 907 CrossRef CAS.
- S. Hitosugi, W. Nakanishi and H. Isobe, Chem. – Asian J., 2012, 7, 1550 CrossRef CAS PubMed.
- S. Muhammad, T. Minami, H. Fukui, K. Yoneda, S. Minamide, R. Kishi, Y. Shigeta and M. Nakano, Int. J. Quantum Chem., 2013, 113, 592 CrossRef CAS.
- K. Matsui, Y. Segawa and K. Itami, Org. Lett., 2012, 14, 1888 CrossRef CAS PubMed.
- Y. Segawa, A. Fukazawa, S. Matsuura, H. Omachi, S. Yamaguchi, S. Irle and K. Itami, Org. Biomol. Chem., 2012, 10, 5979 CAS.
- S. Fomine, M. G. Zolotukhin and P. Guadarrama, J. Mol. Model., 2012, 18, 4025 CrossRef CAS PubMed.
- Y. Ishii, Y. Nakanishi, H. Omachi, S. Matsuura, K. Matsui, H. Shinohara, Y. Segawa and K. Itami, Chem. Sci., 2012, 3, 2340 RSC.
- H. Omachi, Y. Segawa and K. Itami, Acc. Chem. Res., 2012, 45, 1378 CrossRef CAS PubMed.
- E. Kayahara, Y. Sakamoto, T. Suzuki and S. Yamago, Org. Lett., 2012, 14, 3284 CrossRef CAS PubMed.
-
(a) Y. Suzaki and K. Osakada, Organometallics, 2003, 22, 2193 CrossRef CAS;
(b) Y. Suzaki, T. Yagyu and K. Osakada, J. Organomet. Chem., 2007, 692, 326 CrossRef CAS.
- T. J. Sisto, X. Tian and R. Jasti, J. Org. Chem., 2012, 77, 5857 CrossRef CAS PubMed.
- J. Xia, J. W. Bacon and R. Jasti, Chem. Sci., 2012, 3, 3018 RSC.
- S. Hitosugi, T. Yamasaki and H. Isobe, J. Am. Chem. Soc., 2012, 134, 12442 CrossRef CAS PubMed.
- E. R. Darzi, T. J. Sisto and R. Jasti, J. Org. Chem., 2012, 77, 6624 CrossRef CAS PubMed.
- H.-B. Li, A. J. Page, S. Irle and K. Morokuma, J. Am. Chem. Soc., 2012, 134, 15887 CrossRef CAS PubMed.
- K. Matsui, Y. Segawa, T. Namikawa, K. Kamada and K. Itami, Chem. Sci., 2013, 4, 84 RSC.
- M. Fujitsuka, D. W. Cho, T. Iwamoto, S. Yamago and T. Majima, Phys. Chem. Chem. Phys., 2012, 14, 14585 RSC.
- T. Nishihara, Y. Segawa, K. Itami and Y. Kanemitsu, J. Phys. Chem. Lett., 2012, 3, 3125 CrossRef CAS PubMed.
- C. Camacho, T. A. Niehaus, K. Itami and S. Irle, Chem. Sci., 2013, 4, 187 RSC.
- E. S. Hirst and R. Jasti, J. Org. Chem., 2012, 77, 10473 CrossRef CAS PubMed.
- J. Xia, M. R. Golder, M. E. Foster, B. M. Wong and R. Jasti, J. Am. Chem. Soc., 2012, 134, 19709 CrossRef CAS PubMed.
- T. Nishiuchi, X. Feng, V. Enkelmann, M. Wagner and K. Müllen, Chem. – Eur. J., 2012, 18, 16621 CrossRef CAS PubMed.
- H. Isobe, S. Hitosugi, T. Yamasaki and R. Iizuka, Chem. Sci., 2013, 4, 1293 RSC.
- M. Fujitsuka, T. Iwamoto, E. Kayahara, S. Yamago and T. Majima, ChemPhysChem, 2013, 14, 1570 CrossRef CAS PubMed.
- P. J. Evans and R. Jasti, Top. Curr. Chem., 2012 DOI:10.1007/128_2012_415.
- A. V. Zabula, A. S. Filatov, J. Xia, R. Jasti and M. A. Petrukhina, Angew. Chem., Int. Ed., 2013, 52, 5033 CrossRef CAS PubMed.
- T. Matsuno, S. Kamata, S. Hitosugi and H. Isobe, Chem. Sci., 2013, 4, 3179 RSC.
- S. Hitosugi, Y. Nakamura, T. Matsuno, W. Nakanishi and H. Isobe, Tetrahedron Lett., 2012, 53, 1180 CrossRef CAS.
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Nat. Chem., 2013, 5, 572 CrossRef CAS PubMed.
- E. Kayahara, T. Iwamoto, T. Suzuki and S. Yamago, Chem. Lett., 2013, 42, 621 CrossRef CAS.
- S. Hitosugi, R. Iizuka, T. Yamasaki, R. Zhang, Y. Murata and H. Isobe, Org. Lett., 2013, 15, 3199 CrossRef CAS PubMed.
- M. R. Golder, B. M. Wong and R. Jasti, Chem. Sci., 2013, 4, 4285 RSC.
- H.-B. Li, A. J. Page, S. Irle and K. Morokuma, J. Phys. Chem. Lett., 2013, 4, 3176 CrossRef CAS.
- T. Iwamoto, Y. Watanabe, H. Takaya, T. Haino, N. Yasuda and S. Yamago, Chem. – Eur. J., 2013, 19, 14061 CrossRef CAS PubMed.
- H. Chen, M. R. Golder, F. Wang, R. Jasti and A. K. Swan, Carbon, 2014, 67, 203 CrossRef CAS.
- J. M. Batson and T. M. Swager, Synlett, 2013, 2545 CAS.
- E. Kayahara, T. Iwamoto, H. Takaya, T. Suzuki, M. Fujitsuka, T. Majima, N. Yasuda, N. Matsuyama, S. Seki and S. Yamago, Nat. Commun., 2013, 4, 2694 Search PubMed.
- E. Kayahara, T. Kouyama, T. Kato, H. Takaya, N. Yasuda and S. Yamago, Angew. Chem., Int. Ed., 2013, 52, 13722 CrossRef CAS PubMed.
- F. Sibbel, K. Matsui, Y. Segawa, A. Studer and K. Itami, Chem. Commun., 2014, 50, 954 RSC.
- A. Yagi, G. Venkataramana, Y. Segawa and K. Itami, Chem. Commun., 2014, 50, 957 RSC.
- P. Li, T. J. Sisto, E. R. Darzi and R. Jasti, Org. Lett., 2014, 16, 182 CrossRef CAS PubMed.
- M. Banerjee, R. Shukla and R. Rathore, J. Am. Chem. Soc., 2009, 131, 1780 CrossRef CAS PubMed.
- B. L. Merner, K. S. Unikela, L. N. Dawe, D. W. Thompson and G. J. Bodwell, Chem. Commun., 2013, 49, 5930 RSC.
- S. Hitosugi, A. Matsumoto, Y. Kaimori, R. Iizuka, K. Soai and H. Isobe, Org. Lett., 2014, 16, 645 CrossRef CAS PubMed.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS.
- S. Yamago, E. Kayahara and T. Iwamoto, Chem. Rec., 2014, 14, 84 CrossRef CAS PubMed.
- F. E. Golling, M. Quernheim, M. Wagner, T. Nishiuchi and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1525 CrossRef CAS PubMed.
- D. A. Hines, E. R. Darzi, R. Jasti and P. V. Kamat, J. Phys. Chem. A, 2014, 118, 1595 CrossRef CAS PubMed . Erratum 2015, 119, 428.
- Y. Nakanishi, H. Omachi, S. Matsuura, Y. Miyata, R. Kitaura, Y. Segawa, K. Itami and H. Shinohara, Angew. Chem., Int. Ed., 2014, 53, 3102 CrossRef CAS PubMed.
- A.-F. Tran-Van, E. Huxol, J. M. Basler, M. Neuburger, J.-J. Adjizian, C. P. Ewels and H. A. Wegner, Org. Lett., 2014, 16, 1594 CrossRef CAS PubMed.
- A.-F. Tran-Van, S. Götz, M. Neuburger and H. A. Wegner, Org. Lett., 2014, 16, 2410 CrossRef CAS PubMed.
- Y. Kameoka, T. Sato, T. Koyama, K. Tanaka and T. Kato, Chem. Phys. Lett., 2014, 598, 69 CrossRef CAS.
- T. Nishihara, Y. Segawa, K. Itami and Y. Kanemitsu, Chem. Sci., 2014, 5, 2293 RSC.
- Y. Ishii, S. Matsuura, Y. Segawa and K. Itami, Org. Lett., 2014, 16, 2174 CrossRef CAS PubMed.
- C. Huang, Y. Huang, N. G. Akhmedov, B. V. Popp, J. L. Petersen and K. K. Wang, Org. Lett., 2014, 16, 2672 CrossRef CAS PubMed.
- S. Sato, T. Yamasaki and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8374 CrossRef CAS PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2004, 60, 627 CrossRef PubMed.
- T. Iwamoto, E. Kayahara, N. Yasuda, T. Suzuki and S. Yamago, Angew. Chem., Int. Ed., 2014, 53, 6430 CrossRef CAS PubMed.
- M. Peña Alvarez, P. Mayorga Burrezo, M. Kertesz, T. Iwamoto, S. Yamago, J. Xia, R. Jasti, J. T. López Navarrete, M. Taravillo, V. G. Baonza and J. Casado, Angew. Chem., Int. Ed., 2014, 53, 7033 CrossRef PubMed.
- M. Fujitsuka, C. Lu, T. Iwamoto, E. Kayahara, S. Yamago and T. Majima, J. Phys. Chem. A, 2014, 118, 4527 CrossRef CAS PubMed.
- S. Hitosugi, K. Ohkubo, R. Iizuka, Y. Kawashima, K. Nakamura, S. Sato, H. Kono, S. Fukuzumi and H. Isobe, Org. Lett., 2014, 16, 3352 CrossRef CAS PubMed.
- M. Peña Alvarez, P. Mayorga Burrezo, T. Iwamoto, L. Qiu, M. Kertesz, M. Taravillo, V. G. Baonza, J. T. López Navarrete, S. Yamago and J. Casado, Faraday Discuss., 2014, 173, 157 Search PubMed.
- K. Yuan, Y.-J. Guo, T. Yang, J.-S. Dang, P. Zhao, Q.-Z. Lia and X. Zhao, J. Phys. Org. Chem., 2014, 27, 772 CrossRef CAS.
- M. Fujitsuka, S. Tojo, T. Iwamoto, E. Kayahara, S. Yamago and T. Majima, J. Phys. Chem. Lett., 2014, 5, 2302 CrossRef CAS PubMed.
- V. S. Reddy, C. Camacho, J. Xia, R. Jasti and S. Irle, J. Chem. Theory Comput., 2014, 10, 4025 CrossRef CAS.
- A.-F. Tran-Van and H. A. Wegner, Beilstein J. Nanotechnol., 2014, 5, 1320 CrossRef PubMed.
- T. Iwamoto, Z. Slanina, N. Mizorogi, J. Guo, T. Akasaka, S. Nagase, H. Takaya, N. Yasuda, T. Kato and S. Yamago, Chem. – Eur. J., 2014, 20, 14403 CrossRef CAS PubMed.
- K. Suenaga, T. Okazaki, K. Hirahara, S. Bandow, H. Kato, A. Taninaka, H. Shinohara and S. Iijima, Appl. Phys. A: Mater. Sci. Process., 2003, 76, 445 CrossRef CAS.
- L. Adamska, I. Nayyar, H. Chen, A. K. Swan, N. Oldani, S. Fernandez-Alberti, M. R. Golder, R. Jasti, S. K. Doorn and S. Tretiak, Nano Lett., 2014, 14, 6539 CrossRef CAS PubMed.
- T. Matsuno, S. Sato, R. Iizuka and H. Isobe, Chem. Sci., 2015, 6, 909 RSC.
- T. Matsuno, H. Naito, S. Hitosugi, S. Sato, M. Kotani and H. Isobe, Pure Appl. Chem., 2014, 86, 489–495 CrossRef CAS.
- K. Matsui, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2014, 136, 16452 CrossRef CAS PubMed.
- H. Ito, Y. Mitamura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 159 CrossRef CAS PubMed.
- N. Toriumi, A. Muranaka, E. Kayahara, S. Yamago and M. Uchiyama, J. Am. Chem. Soc., 2015, 137, 82 CrossRef CAS PubMed.
- D. Myśliwiec, M. Kondratowicz, T. Lis, P. J. Chmielewski and M. Stępień, J. Am. Chem. Soc., 2015, 137, 1643 CrossRef PubMed.
- H. Chen, M. R. Golder, F. Wang, S. K. Doorn, R. Jasti, S. Tretiak and A. K. Swan, J. Phys. Chem. C, 2015, 119, 2879 CAS.
- N. Kubota, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 1356 CrossRef CAS PubMed.
- H. Jiang, T. Tanaka, H. Mori, K. H. Park, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 2219 CrossRef CAS PubMed.
- K. Yuan, Y. Guo and X. Zhao, J. Phys. Chem. C, 2015 DOI:10.1021/jp5129657.
- K. Ozaki, K. Kawasumi, M. Shibata, H. Ito and K. Itami, Nat. Commun., 2015, 6, 6251 CrossRef CAS PubMed.
- M. Golder and R. Jasti, Acc. Chem. Res., 2015 DOI:10.1021/ar5004253.
- H. Ueno, T. Nishihara, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201500544.
- H. Isobe, K. Nakamura, S. Hitosugi, S. Sato, H. Tokoyama, H. Yamakado, K. Ohno and H. Kono, Chem. Sci., 2015 10.1039/C5SC00335K.
- Y. Segawa, T. Kuwabara, K. Matsui, S. Kawai and K. Itami, Tetrahedron, 2015 DOI:10.1016/j.tet.2015.02.066.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a