
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Critical design issues in the targeted molecular imaging of cell surface receptors
Neil
Sim
and
David
Parker
*
Department of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. E-mail: david.parker@dur.ac.uk
First published on 25th February 2015
Abstract
The imaging of cell-surface receptors can be achieved using several methods, including single photon emission tomography (SPECT) positron emission tomography (PET), optical imaging and magnetic resonance imaging (MRI). The application of targeted MRI contrast agents is particularly well-suited to this task, provided that the agents reach the desired site efficiently and selectively. In addition, they should bind reversibly to the cell-surface receptor and give rise to a large change in cellular relaxation rate, in competition with binding to the natural substrate. Such approaches offer promise in the molecular imaging of neurotransmission in the brain, using conjugates that selectively target dopamine or glutamate receptor sub-types. Strategies based on the use of competitive antagonist vectors offer particular scope, as such conjugates are generally not taken into the target cell following cell surface receptor binding, in contrast to the use of MRI contrast agents based on agonists that tend to be internalised quickly or are designed to target intracellular sites.
Key learning points1. To target a cell-surface receptor, both cell specificity and receptor selectivity required.2. Address systems that are not internalized to reduce the risk of toxicity. 3. Use a competitive antagonist as a targeting vector, not an agonist, to avoid internalisation. 4. Avoid non-specific binding: consider competitive binding of the ligand moiety and at the metal centre. 5. For MRI, use a Gd probe in which relaxivity changes are not quenched by slow exchange kinetics and in which probe binding is reversible. |
1. Introduction
Molecular imaging has been defined as the non-invasive visualisation of biochemical events at the molecular level within cells, tissues or whole-bodied specimens.1 It is one of the most rapidly expanding areas of scientific and medical research. Over the past twenty years, efforts have been made to identify specific molecular events in vivo, leading to the design and development of targeted probes or contrast agents, to provide or enhance image contrast. Labelled antibodies, peptides, proteins and small molecules have each been employed in this manner, but it is often difficult to obtain sufficient signal intensity change, due to the relatively low probe-target to background ratio. Indeed, it is often the case that the target of interest is present in relatively low concentrations. Such a situation may arise for a cell-surface receptor that is expressed selectively in a particular cell type, or over-expressed in certain disease states. Nevertheless, these issues can be overcome by improving the contrast agent's pharmacokinetics and affinity, coupled with the use of rapid and sensitive, high-resolution imaging techniques.The selection of a particular molecular imaging modality is highly dependent upon the biochemical process to be examined and the nature of the information needed. The key design issues that need to be considered include: the spatial resolution required; the sensitivity needed i.e. the likely concentration of the target; whether dynamic information is to be obtained; whether whole body or regional imaging is required; the temporal resolution and the depth of penetration. By carefully considering each of these issues, it is possible to select the best imaging technique to use, (Fig. 1 and Table 1)
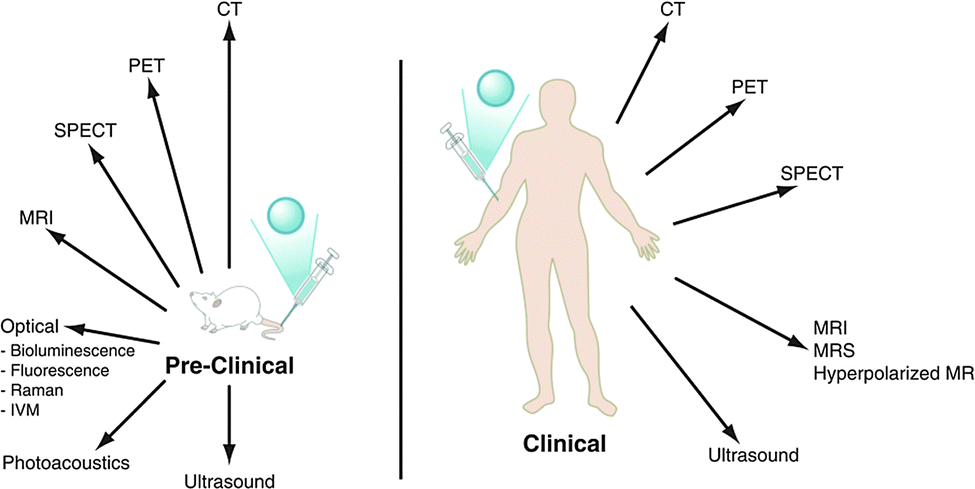 | ||
| Fig. 1 The major pre-clinical and clinical imaging modalities: CT = X-ray computed tomography; PET = positron emission tomography; SPECT = single photon emission computed tomography; MRI/MRS = magnetic resonance imaging/spectroscopy, (adapted from the original1). | ||
| Modality | Temporal resolution | Spatial resolution | Depth penetration | Sensitivity/M |
|---|---|---|---|---|
| a ND: not determined. | ||||
| X-ray CT | min | 0.5–1 mm | Unlimited | ND |
| MRI | min | 0.25 to 1 mm | Limited by magnet bore | 10−3–10−5 |
| PET | s–min | 5–10 mm | Unlimited | 10−11–10−12 |
| SPECT | min | 8–10 mm | Unlimited | 10−10–10−11 |
| Ultra-sound | s–min | 1–2 mm | A few cm | ND |
| Optical fluorescence imaging | s–min | 2–3 mm | <1 cm | 10−9–10−12 |
| Optical bioluminescence imaging | s–min | 3–5 mm | 1–2 cm | 10−15–10−17 |
| Photoacoustic imaging | s–min | 10 μm–1 mm | 6 mm–3 cm | ND |
This aim of this tutorial review article is to assess critically the progress made in the design and study of molecular imaging agents that target cell surface receptors. The review focuses on molecular MRI contrast agents based on Gd(III) conjugates, rather than conjugates of metal oxide or semiconductor particles. Whilst it is appreciated that the pharmacokinetics of a molecular imaging agent will largely determine the ratio of specific/non-specific binding in vivo and that the key issues of the toxicity, administration and delivery of the agent must be assessed in defining the potential for clinical translation, this review addresses the key chemical principles that must be considered first, in order to design an effective molecular imaging agent for future evaluation. Particular emphasis is given to imaging agents that target cell-surface receptors, comparing internalised versus non-internalised systems. Their merits are assessed with respect to those agents targeting a receptor found within the cell, and those that are rapidly internalized.
2. Receptor-targeted Gd(III)-MR contrast agents
There are numerous examples of extracellular Gd(III) contrast agents that have been designed to report upon local changes in pH, metal ion concentration and enzyme activity.2–4 Details of examples have also been described of contrast agents targeted towards abundant blood pool proteins, such as human serum albumin (HSA) or fibrin.5 However, this review will examine the contrast agents that have been designed to change their relaxivity-defined as the increment of the water proton relaxation rate per unit complex concentration-following association with cell-surface receptors, comparing where appropriate with systems involving an intracellular receptor.The fact that there are only a few reports of these contrast agents can be ascribed to the low sensitivity of the MRI method. Following administration of a Gd(III) complex, it has been suggested that there needs to be a 5 μM local Gd(III) concentration in order to observe a 5% change in the water relaxation rate, R1, for a contrast agent which possesses a relaxivity of 5 mM−1 s−1.6 This situation corresponds to about 107 Gd ions per cell, setting an approximate limit to the receptor density required in order to measure an observable change on contrast agent binding. Such thinking makes it difficult to imagine how to image the majority of cell surface receptors using MRI, as they are believed to be present at concentrations in the 10 to 100 nanomolar range. However, receptor density is likely to be heterogeneous, with high local concentrations and for a high affinity targeted gadolinium complex, the rate of dissociation from the receptor site will be slow and may be of the order of 1 s−1, if the forward rate of association is 107 s−1, i.e. when Kd = 0.1 μM. This means that the timescale of the imaging experiment (one or two seconds) is of the same order as the lifetime of the bound complex, enhancing the intrinsic sensitivity of the experiment.7 Furthermore, if the relaxivity of the receptor bound complex increases substantially compared to the unbound complex, the intrinsic sensitivity of the experiment is also increased. Such relaxivity enhancement is usually limited to the low field range of 0.4 to 1.5 Tesla (T), where rotational dynamics plays the major role in determining relaxivity for a Gd(III) contrast agent, when water exchange is fast (i.e. ∼107 s−1).4,5 Happily, this coincides with the magnetic field strengths of the older more common human imaging systems that operate at 1.5 T. The relaxivity enhancement reaches a maximum typically between 1 and 1.5 T, but drops quite rapidly beyond 1.5 T since rotational dynamics are no longer dominant. However, image resolution is greater at these higher magnetic fields (B) and so is sensitivity, as signal intensity in a magnetic resonance experiment increases as B3/2.
2.1 Intracellular receptor-targeted contrast agents
First, it is necessary to assess the prior work that has been undertaken where the target site is found mainly within the cell, for which issues of cell permeability and retention require very careful consideration. Cancer cells are known to over-express a variety of receptors that are present on or in healthy cells. The higher concentration of these receptors allows targeted-contrast agents to overcome the inherent low sensitivity of MRI. The progesterone receptor (PR) is a useful marker for cancer progression and is frequently used to predict patient prognosis. Historically, progesterone receptor levels are determined from in vitro immunohistochemistry assays of patient samples gained by invasive biopsies. However, Meade and co-workers have described the development of progesterone receptor-targeted MRI contrast agents, which could potentially report the degree of cancer progression.Their initial complex, [Gd.L1.0] (Scheme 1), induced a 40% enhancement in the water proton longitudinal relaxation rate, R1, upon incubation with progesterone receptor positive cells, notwithstanding a relatively weak affinity for the progesterone receptor (IC50 = 1.91 μM).8 In order to increase relaxation rate enhancement, three further complexes were synthesised (Scheme 1), in an effort to maximise intracellular accumulation and progesterone receptor affinity. The effect of charge on the complex lipophilicity and cell permeability was assessed, along with the effect of the length of the spacer unit separating the progesterone motif and Gd(III) chelate.9
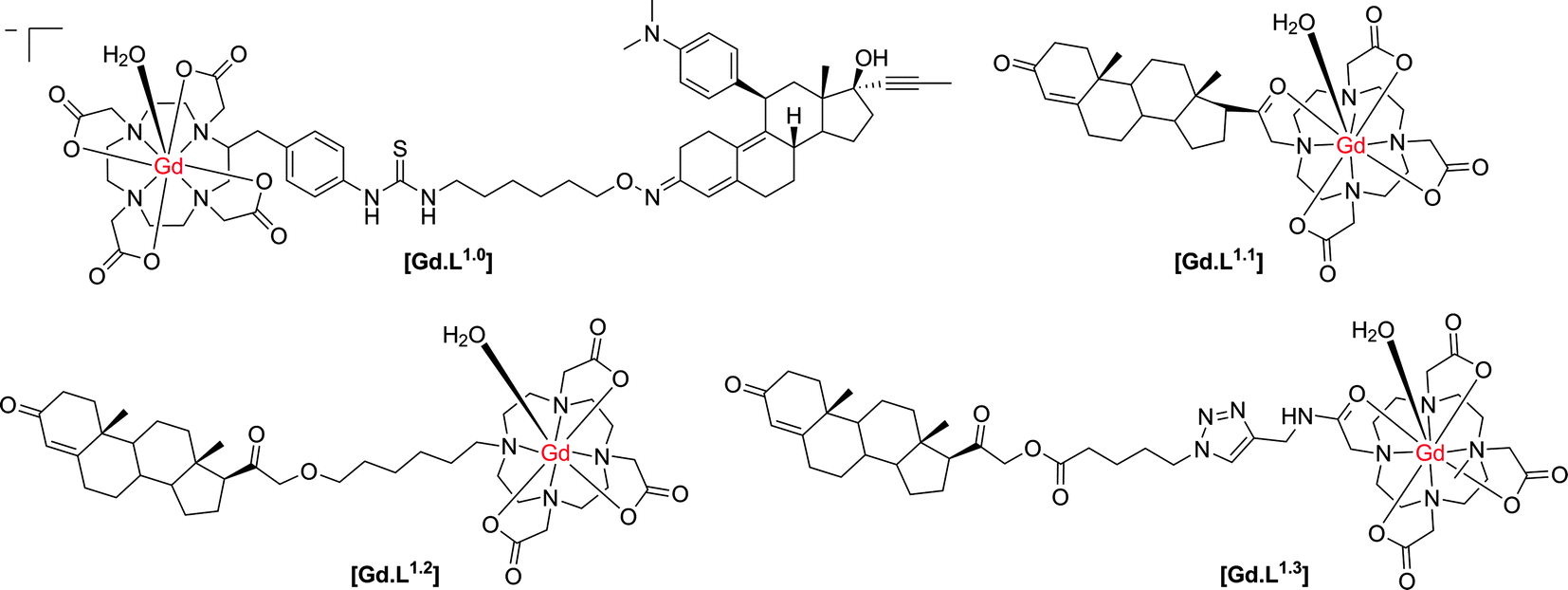 | ||
| Scheme 1 Structures of progesterone receptor-targeted contrast agents.8–10 | ||
In order to increase intracellular accumulation, it was reasoned that a complex with a more positive log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value would assist in enhancing cell permeability. Therefore, the neutral complexes [Gd.L1.1] and [Gd.L1.2] were selected as the lead complexes. The length and nature of the spacer group also had an effect on the affinity for the progesterone receptor. Surprisingly, [Gd.L1.1] with no spacer exhibited the largest affinity for the progesterone receptor, suggesting the possibility of a favourable interaction between the hydrophilic Gd(III) chelate and the receptor binding site. However, each complex exhibited an approximately 100-fold increase in progesterone receptor affinity over [Gd.L1.0], with IC50 values in the nanomolar range. Of the four complexes, [Gd.L1.2] showed the best charge and spacer properties and was selected for further evaluation.9
P value would assist in enhancing cell permeability. Therefore, the neutral complexes [Gd.L1.1] and [Gd.L1.2] were selected as the lead complexes. The length and nature of the spacer group also had an effect on the affinity for the progesterone receptor. Surprisingly, [Gd.L1.1] with no spacer exhibited the largest affinity for the progesterone receptor, suggesting the possibility of a favourable interaction between the hydrophilic Gd(III) chelate and the receptor binding site. However, each complex exhibited an approximately 100-fold increase in progesterone receptor affinity over [Gd.L1.0], with IC50 values in the nanomolar range. Of the four complexes, [Gd.L1.2] showed the best charge and spacer properties and was selected for further evaluation.9
The complex, [Gd.L1.2], was internalised in both progesterone receptor positive and negative cells, with the progesterone receptor positive cells exhibiting twice the accumulation of the complex over a 24 hour period, as compared to the progesterone negative cells. Furthermore, the non-targeted contrast agent, [Gd.DO3A], exhibited a negligible accumulation in both progesterone positive and negative cells, over the same time period. Such behaviour suggested that [Gd.L1.2] is retained longer in the progesterone receptor positive cells, by virtue of its interaction with the intracellular receptor. The non-specific uptake of [Gd.L1.2] into progesterone receptor negative cells was attributed to the higher hydrophobicity and cell permeability of the complex, with respect to [Gd.DO3A].10 Retention of [Gd.L1.2] in the uterus, ovaries and mammary glands over a 24 h period demonstrated the ability of the contrast agent to target progesterone receptor rich tissue. Importantly, [Gd.L1.2] did not accumulate in the abdominal tissue or fat. Finally, [Gd.L1.2] was shown to enhance the contrast of progesterone receptor positive tumours in the presence of progesterone receptor negative tumours in vivo. Irrespective of the mode of administration (subcutaneous vs. intra-peritoneal), a significant difference in the T1-weighted MR images between the two tumours was evident, 2 to 6 h post injection (Fig. 2).10
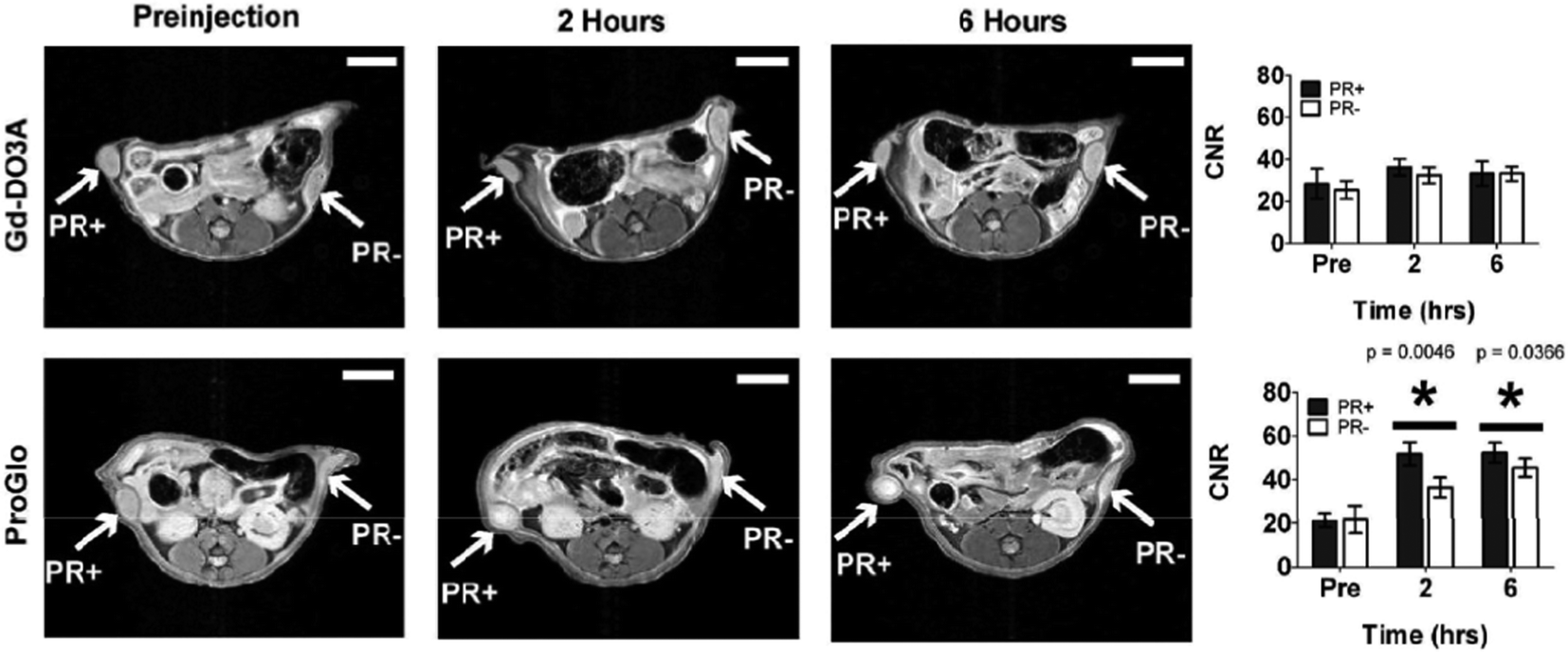 | ||
| Fig. 2 T 1-weighted MR images and contrast-to-noise plots (7 T) of mice bearing a tumour xenograft intraperitoneally injected with [Gd.DO3A] or [Gd.L1.2]. A significant contrast enhancement is observed in both progesterone receptor positive and negative tumours over a 6 h period, when injected with [Gd.L1.2].10 | ||
Despite the relative successes of [Gd.L1.2], it should be noted that this contrast agent is based on an N-alkyl–DO3A system. Such systems are likely to bind to protein non-specifically11via coordination at the Gd centre. Indeed, approximately 30% of [Gd.L1.2] was found to reside in the membrane and was therefore unavailable to interact with the progesterone receptor. Alternatively, the complex may bind reversibly to endogenous anions (e.g. bicarbonate), leading to displacement of the coordinated water molecule. This effect will tend to reduce the relaxivity gain observed upon receptor binding. Furthermore, the impact of non-specific protein binding on cellular uptake was not assessed, which may be a reason for their relatively poor specificity of cellular uptake (only 2:1). Finally, the poor water solubility and intracellular uptake of [Gd.L1.2] resulted in significant acute toxicity, possibly due to gadolinium retention that could be associated with premature dissociation of the Gd ion from its ligand.
In an attempt to reduce the toxicity associated with [Gd.L1.2], the lipophilicity of the complex was adjusted through modification of the linker unit.12 As expected, no change in progesterone receptor affinity was observed. However, an increase in cellular uptake of [Gd.L1.3] was achieved in progesterone positive tumours; 90% of the complex was found to localise within the cell, apparently in the cytoplasm, where one form of the progesterone receptor is located. Contrast-to-noise ratios in T1-weighted MR measurements of [Gd.L1.3] were comparable to those for [Gd.L1.2], but the increased water solubility resulted in an increase in cell viability.
Notwithstanding these improvements, [Gd.L1.3] is based on a mono-amide–DO3A system. Such complexes with amide carbonyl coordination giving a 5-ring chelate, have inherently slower water exchange rates (kex ∼ 106 s−1) and so the relaxivity gain at fields between 1.5 to 7 T, associated with modulation of the effective rotational correlation time, is quenched. Furthermore, the receptor-targeting moiety is attached through an ester linkage, which is very likely to be susceptible to hydrolysis by extracellular esterases. A chemical linkage that is less prone to metabolic transformation might give increased cellular retention in progesterone positive tumours, leading to an enhancement in signal contrast.
2.2 Cell surface receptor targeted contrast agents
In 1995, a seminal paper13 described how Gd-DTPA chelates conjugated to polylysine, loaded onto DNA containing transferrin, were able to label cells expressing the cell-surface transferrin receptor. A large enhancement in the T1-weighted MR image contrast ensued, compared to untreated cells. Furthermore, cells treated simultaneously with the Gd-DTPA loaded DNA-transferrin conjugate and also iron-loaded transferrin resulted in a negligible enhancement in T1-weighted MR contrast, due to competition for the receptor site. This demonstrated that the internalisation of the contrast agent was mediated by the cell surface receptor.13Folate is an essential vitamin for cells and binds with high affinity to the folate receptor present on the cell surface. Upon binding of folate, the folate receptor–ligand conjugate undergoes receptor-mediated endocytosis leading to folate internalization. Following release of the folate, the receptor returns to the cell surface ready for the next ligand to bind.
In systems targeting folate receptors, most reported work has used a derivative of folate itself. Such imaging agents are then likely to be quickly internalized, increasing the local concentration of the imaging agent in the desired cell type. There are several examples in the literature of polymeric, high molecular weight contrast agents that target the folate receptor, as this receptor can be expressed in high concentration, e.g. in certain ovarian cell lines.14–16 One example of a low molecular weight complex was reported by Kalber and co-workers,17 who synthesised a contrast agent by conjugation of folic acid to a [mono-amide-GdDO3A] chelate, separated by an ethylene glycol spacer (Fig. 3). Inductively coupled mass spectrometry (ICP-MS) measurements showed that the complex [Gd.L1.4] was internalised readily by folate receptor positive (FR+) cells after 1 hour, with more than double the accumulation than in folate receptor negative (FR−) cells. Although tumour perfusion studies were apparently not undertaken, the presence of free folic acid in the cell growth medium as a competitor resulted in a reduction in uptake of [Gd.L1.4] into FR+ cells, consistent with receptor-mediated internalisation. This study however, was carried out in vitro but was not seemingly undertaken in vivo, where it has more relevance.
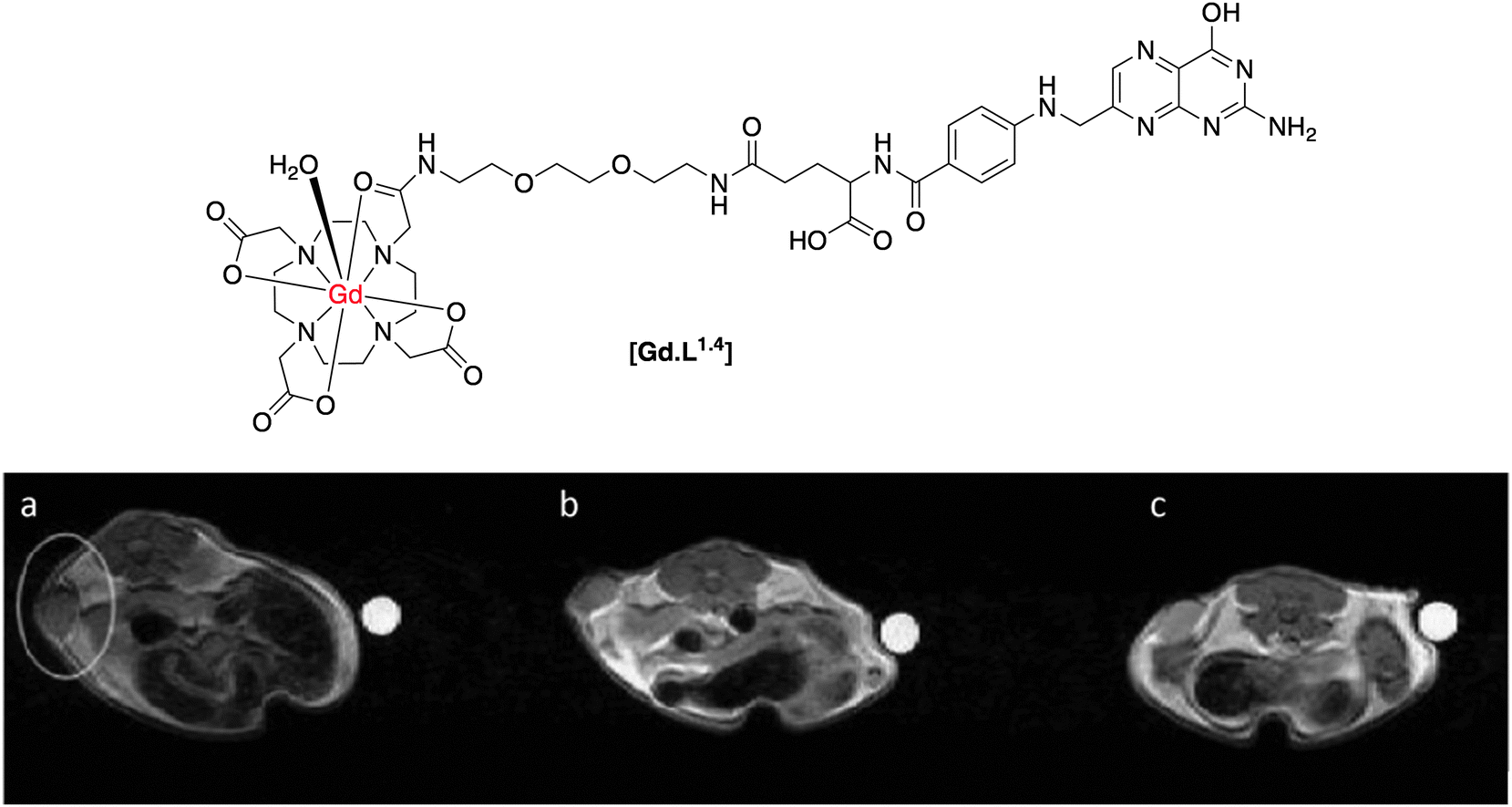 | ||
| Fig. 3 Top: structure of folate receptor-targeted contrast agent, [Gd.L1.4]; below: T1-weighted MR images of a folate receptor positive tumour xenograft: (a) pre-administration; (b) 2 h post and (c) 14 h post-administration of [Gd.L1.4].17 | ||
Despite confirmation of cellular labelling by ICP-MS measurements, there was no increase in cellular relaxation rate in vitro, after incubation of [Gd.L1.4]. On the other hand, when [Gd.L1.4] was administered to nude mice bearing a folate receptor-positive tumour xenograft, up to a 50% enhancement in R1 was observed over a 14 hour period, resulting in increased T1-weighted MR contrast (Fig. 3). In comparison, no increase in R1 was reported for tumour xenografts bearing folate receptor-negative cells, nor when the non-targeted agent, [Gd.DOTA]−, was used. In this case, it was suggested that the non-targeted [Gd.DOTA]− was rapidly washed out of the tumour, prior to the 2 h observation point.
The contrasting results obtained in vitro and in vivo results are worthy of consideration. Inherently, there are differences in delivery and transport in the two sets of studies; in the former case, the cells are in direct contact with the complex solution for 2, 4 and 14 h, whereas in the latter these times are the observation points of a dynamic flowing system. The relative degree of internalization and tumour wash-out are probably most significant. Indeed, the authors suggest a significant degree of cellular variation in folate receptor expression as one reason for the contrasting behaviour. This plausible assumption can be assessed by immunofluorescence staining experiments, although it is difficult to determine the real mechanism by which R1 increases in vivo. The intracellular fate of the complex is key. Once the complex is bound to the folate receptor, internalisation is likely to be fast via receptor-mediated endocytosis, resulting in the contrast agent residing inside an endosome. If it escapes from this compartment, it could bind non-specifically to intracellular proteins, modulating τR, and increasing R1. However, the expected relaxivity gain in vitro could be quenched if the contrast agent is trapped in a sub-cellular vesicle/endosome after internalisation, limiting the rate of water exchange on/off Gd, kex.18 Whatever the reasons for the discrepancies, the design of the complex could also limit the expected relaxivity gain. Mono-amide Gd(III) 5-ring chelates possess rather slow water exchange rates (around 106 s−1 at 298 K), quenching the relaxation enhancement that can arise from non-covalent binding to the more slowly rotating macromolecule.
A further example of a folate receptor-targeted contrast agent emanated from work in Durham in 2001.19 In a similar approach, folic acid was conjugated to a Gd(III) chelate. However, the ligand used to encapsulate the Gd(III) ion was a tetra-carboxy-substituted DOTA, giving the complex overall negative charge (Scheme 2).
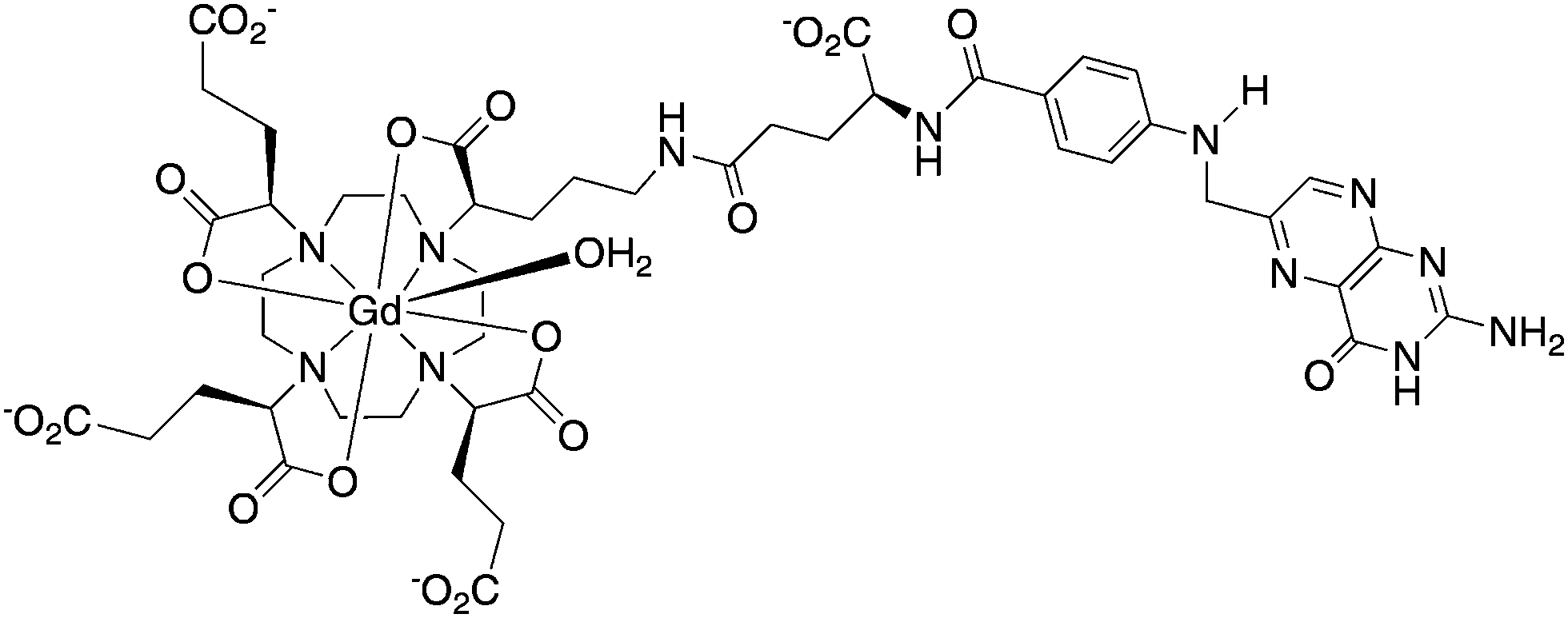 | ||
| Scheme 2 Structure of an anionic γ-folate-[Gd-gDOTA] conjugate.19 | ||
In work carried out in vitro only, this complex was found to exhibit an 80% increase in R1 in the presence of folate binding protein (FBP), but a negligible increase in the presence of the model protein serum albumin (pH 7, 60 MHz, 310 K). The complex had a fast water exchange rate (kex ∼ 107 s−1). Coupled with the overall negative charge that hinders non-specific binding, e.g. to serum albumin, the large R1 enhancement in the presence of FBP can be rationalised.
The formyl peptide receptors (FPRs) are a class of G-protein-coupled receptors present on the surface of neutrophils. Their main function is to mediate migration of neutrophils to a site of injury causing an inflammatory response. Long and co-workers20 synthesized an FPR-1-targeted MR contrast agent based on the hexapeptide, cFLFLFK, known to be an antagonist for this receptor. Conjugation of the peptide fragment to the Gd(III) chelate through the N-terminal lysine residue did not disrupt the affinity of the antagonist towards the receptor, with the apparent binding affinity only slightly decreasing (Kd = 4.5 vs. 2 nM for [Gd.L1.5] and native peptide, respectively).
The complex [Gd.L1.5] was intravenously administered to mice (1 mmol kg−1), which had been pre-injected with an inflammatory agent for 24 h. Upon administration, a significant increase in T1-weighted MR contrast ensued and was visible for up to 80 min. post injection at the site of inflammation (Fig. 4). The non-targeted complex, [Gd.DOTA]−, was used as a control. It gave rise to an initial increase in T1-weighted contrast after 1 minute, but was undetectable 80 min. post injection (Fig. 4). It is a more hydrophilic agent of lower molecular volume than [Gd.L1.5] that is expected to clear more quickly from inflamed tissue, and therefore has limited utility in comparative studies. The authors attribute the difference in time-dependent, T1-weighted contrast to specific targeting and binding of [Gd.L1.5] to the neutrophils' FPR-1 receptor, prolonging the contrast agent's lifetime at the site of inflammation.
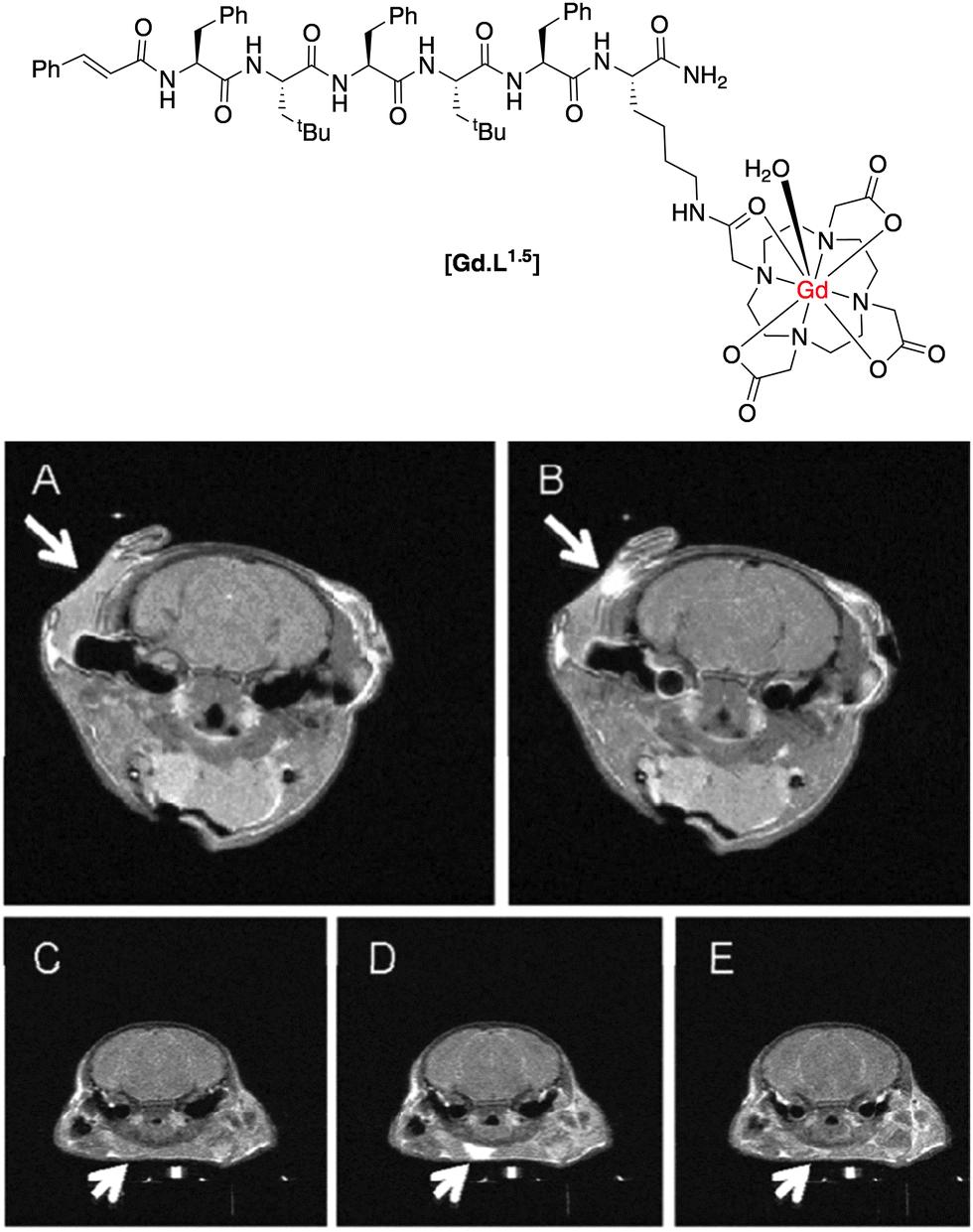 | ||
| Fig. 4 T 1-weighted MR images of a mouse brain: (A) pre-injection; (B) 80 min. post-injection of [Gd.L1.5]; in comparison, (C) pre-injection; (D) 1 min. post; (E) 80 min. post-injection of the control [Gd.DOTA]−.20 | ||
Of the examples of receptor-targeted MR contrast agents in the literature, there are very few that are targeted towards cell surface receptors. Dopamine is an abundant neurotransmitter in the central nervous system and mediates its effects through metabotropic dopamine receptors. The dopaminergic system is related to many neurological and psychological disorders. The D2/D3 dopamine receptors are the primary sites targeted by anti-Parkinsonism and anti-psychotic drugs.
Cohen and co-workers21,22 have synthesised a small series of D2/D3 receptor-targeted MR contrast agents based on an N-alkyl–DO3A system conjugated to modified dopamine receptor ligands (Scheme 3). In an inhibition assay, complex [Gd.L1.6] was shown to possess a large decrease in binding affinity for the dopamine D2-receptor, relative to the radiolabeled ligand, with an IC50 for [Gd.L1.6] of approximately 10 μM. The decrease in affinity reflects the large modification made to the spiperone ligand, inhibiting receptor binding. In an attempt to increase receptor affinity, further complexes were synthesised, reducing the number of structural modifications to the spiperone ligand and varying the length and nature of spacer (Table 2). An increase in D2 receptor binding affinity was found for [Gd.L1.7–1.8], demonstrating the need for the entire ligand moiety to be present in order to maintain affinity.
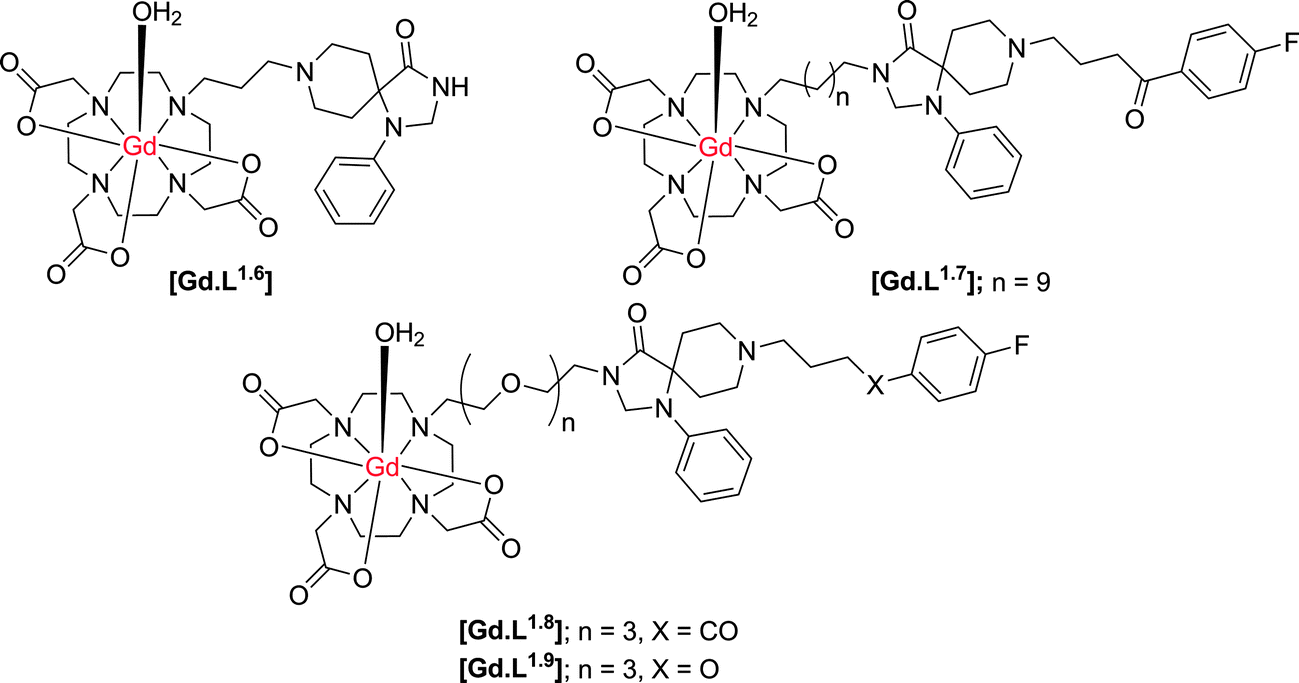 | ||
| Scheme 3 Structures of potential dopamine receptor-targeted MR contrast agents based on heptadentate ligands for the Gd ion (indicative hydration states at Gd are suggested, but were not determined). | ||
:1, 298 K) of dopamine receptor-targeted contrast agents21,22
| [Gd.L1.6] | [Gd.L1.7] | [Gd.L1.8] | [Gd.L1.9] | |
|---|---|---|---|---|
| IC50/M | 1 × 10−5 | 5 × 10−9 | 1 × 10−8 | 1 × 10−6 |
| r 1/mM−1 s−1 | 5.9 | 5.5 | 7.8 | ND |
Notwithstanding the demonstration of the ability of these conjugates to bind at the dopamine D2 receptor, there is a significant amount of work missing to fully characterise [Gd.L1.6–1.9] as dopamine-receptor targeted contrast agents. Firstly, there are no in vitro studies that demonstrate a change in relaxivity of these complexes upon binding to the dopamine receptor. The degree of relaxivity enhancement upon receptor binding would be limited due to the choice of the Gd(III) chelate, since N-alkyl–DO3A systems are likely to bind non-specifically to endogenous protein or anions11 leading to displacement of the coordinated water molecule, and a reduction of the relaxivity gain. Furthermore, specifically in [Gd.L1.8–1.9], the ligand moiety is positioned at a considerable distance from the Gd(III) chelate. Although the presence of this longer linker should tend not to inhibit the affinity of the complex for the receptor based on steric arguments, it could limit the relaxivity gain upon receptor binding. The expected increase in R1 is modulated by the rotational correlation time, τR. Therefore, the long spacer in these systems will induce independent motion of the slowly tumbling macromolecule and the Gd(III) chelate, resulting in only a small relaxivity enhancement on receptor binding.
3. Molecular imaging of glutamate receptors
Metabotropic glutamate receptors (mGluRs) are part of the glutamate neurotransmitter system. They are G-protein coupled receptors with a large extracellular N-terminus in which the orthosteric glutamate binding site is located. Eight subtypes have been characterised that are divided into three subgroups, depending on their distribution, amino acid sequence homology and the types of G-protein to which they are coupled. Group I consists of the mGluR subtypes 1 and 5, and their activation leads to calcium release from intracellular stores into the cytosol and subsequently to the activation of a variety of intracellular signalling molecules. The other two groups II (mGluR2,3) and III (mGluR4,6,7,8) are coupled to Gi/o-proteins and their activation results in a decreased intracellular formation of the secondary messenger, cyclic adenosine monophosphate (cAMP). Both group II and III receptors are mainly found on pre-synaptic terminals, whereas mGluR1 and mGluR5 are mostly localised on postsynaptic neurons (i.e. dendritic spines). Notably, the mGluR5 and mGluR3 receptors are expressed and fully functional on astrocytes.The mGluR5 receptor itself has several physiological roles in normal brain function. These receptors are widely distributed throughout the brain, reflecting functions such as emotion and motivational processes as well as differing aspects of cognitive function. The extensive roles of the mGluR5 makes it a key target for therapeutic intervention and a number of small molecules have been described that modulate the function of mGluR5.23–25 Numerous applications up to advanced clinical trials have been described.26 Examples for possible therapeutic purposes of mGluR5-modulators, include their use as antidepressant-/anxiolytic drugs (e.g. in the case of otherwise treatment resistant depression) as revealed in preclinical models.27 Other examples of possible therapeutic uses of functional mGluR5-modulators include treatment of schizophrenia28 and neurodegenerative disorders, such as Parkinson's disease,29 and the application of mGluR5-antagonists as treatment strategies for autism.30 There have been several molecular imaging approaches used to aid the visualisation of glutamate receptors. Recent examples have been reported relating to the use of PET/SPECT, optical and magnetic resonance methods for imaging.
3.1 PET and SPECT radiotracers for glutamate receptors
Both the PET and SPECT techniques possess very high sensitivity (10−11 – 10−12 M) and can be used to image whole bodied live subjects.1 There have been several reports concerning the synthesis and application of PET and SPECT tracers for glutamate receptors.31 The majority of these tracers are derived from well-known ligands (agonists) of glutamate receptors, with the incorporation of either an 18F/11C radiolabel for PET tracers and 123I/111In labels for SPECT. The introduction of an 11C label into a structure and the replacement of 1H by 19F offers a minimal perturbation to the structure, and hence is most likely not to change the behaviour of the imaging agent with respect to the parent ligand. Of the examples in the literature, the most well-characterised PET tracer for mGluR5 receptors is [18F]-SP203 (Scheme 4).32 This tracer was synthesised in a high radiochemical yield via nucleophilic substitution of the bromomethyl analogue with [18F]-KF,33 resulting in a ligand with an extremely high affinity (IC50 = 0.036 nM) and selectivity for mGluR5. Rapid uptake into the brain of rhesus monkeys was observed, with the radioactivity residing in mGluR5-rich regions. Although a moderate amount of activity accumulated in the skull during the in vivo experiments with monkeys,34 this was not observed in a pilot study with seven healthy human subjects.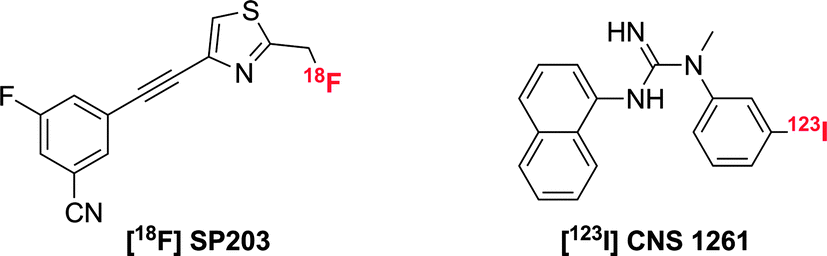 | ||
| Scheme 4 Structures of PET and SPECT agents for glutamate receptor imaging. | ||
The N-methyl-D-aspartate receptor (or NMDAR) is an ionotropic glutamate receptor that is involved in controlling synaptic plasticity and memory function. Uniquely, it requires co-activation by glutamate and either glycine or D-serine. For radiotracers synthesised to target NMDA receptors, the SPECT ligand, [123I]-CNS 1261 (Scheme 4) has shown the most potential.35 In human experiments, this tracer showed high distribution volumes in regions consistent with high NMDA receptor concentrations. Specific binding to the NMDA receptor was confirmed in a competition experiment with ketamine, but due to high degree of non-specific binding, the ability to detect small changes in receptor availability was limited.
Whilst there has been some success in the development of radiotracers for PET and SPECT imaging of glutamate receptors, there are many limitations associated with these methods. First, the intrinsic resolution of these techniques is low, (1–10 mm), which limits the ability to observe small changes at a receptor site. Furthermore, these methods require the efficient chemical synthesis of suitable radiolabelled tracers. Although the installation of a radiolabelled nucleus is probably a lot less perturbing to the biochemical activity of the ligand with respect to conjugation of a fluorophore or Gd-chelate to the ligand, this approach usually requires a cyclotron nearby to generate the short-lived radionuclide. The isotope must be introduced into the substrate in a high yielding and fast radiolabelling reaction.
3.2 Optical imaging of glutamate receptors
Similar to PET and SPECT, optical imaging is endowed with excellent sensitivity (10−9–10−12 M) and can be used to image bio-molecules, which are present in lower concentrations, such as the ionotropic glutamate receptors. Fluorescently labelled ligands offer advantages over radiolabelled analogues, as they are generally safer and easier to handle. However, the major limitation of optical imaging is the poor tissue penetration of photons in the range of 400–900 nm; penetration depths of no more than a few mm only are possible, due to co-absorption and light scattering.Chambers and co-workers have developed a four-component molecular probe for visualisation of AMPA receptors.36 The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA) is a non-NMDA-type ionotropic transmembrane receptor for glutamate. It mediates fast synaptic transmission in the central nervous system. The design of the probe (Fig. 5) was based on a polyamine ligand (known to bind in the AMPA channel), conjugated to a cyanine dye fluorescent reporter, via a photo-cleavable linker. The molecule also bears an electrophilic acrylamide moiety, which can undergo reaction with a nucleophilic partner, such as an amino acid side chain present on the AMPA receptor, to anchor the probe to the receptor (Fig. 5). The probe was found to block functional AMPA receptors in electrophysiology experiments. Removal of the polyamine block restored the receptor to its native state, after irradiation with 380 nm light.
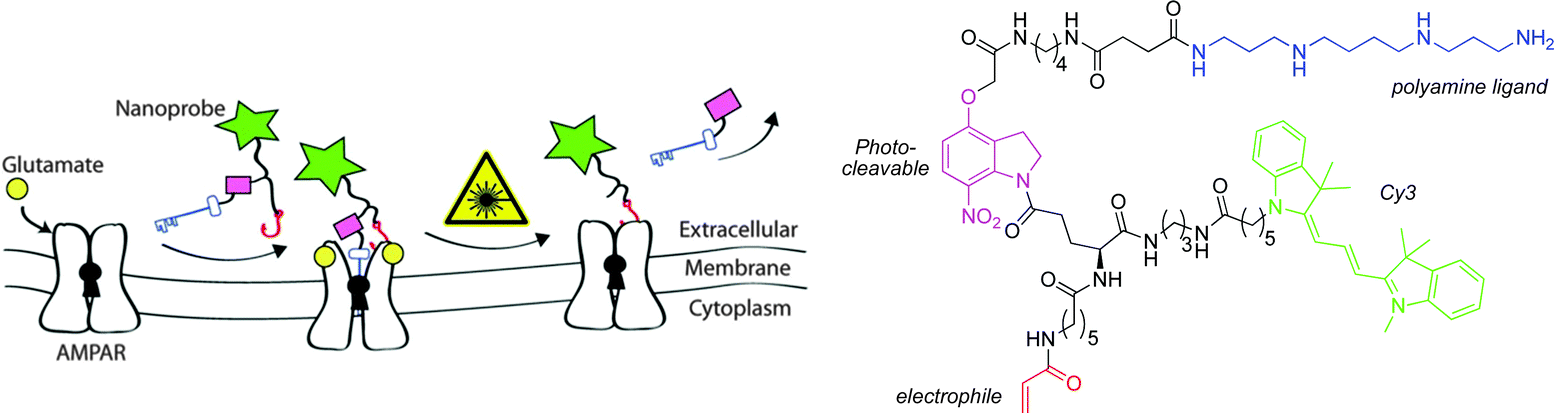 | ||
| Fig. 5 Schematic representation of the putative mechanism of action of the Cy3 probe.36 | ||
When cells were treated with the Cy3 probe for 2 mins and then continuously perfused with fresh buffer, fluorescence was observed around the synaptic junction, consistent with the expected distribution of AMPA receptors (Fig. 6). The fluorescence intensity was time-dependent, decreasing rapidly over a seven minute period (Fig. 6). In comparison, incubation of the Cy3 reporter lacking the targeting ligand gave rise to no cellular localisation in microscopy studies, suggesting that the acrylamide moiety does not react promiscuously with any nucleophile.36
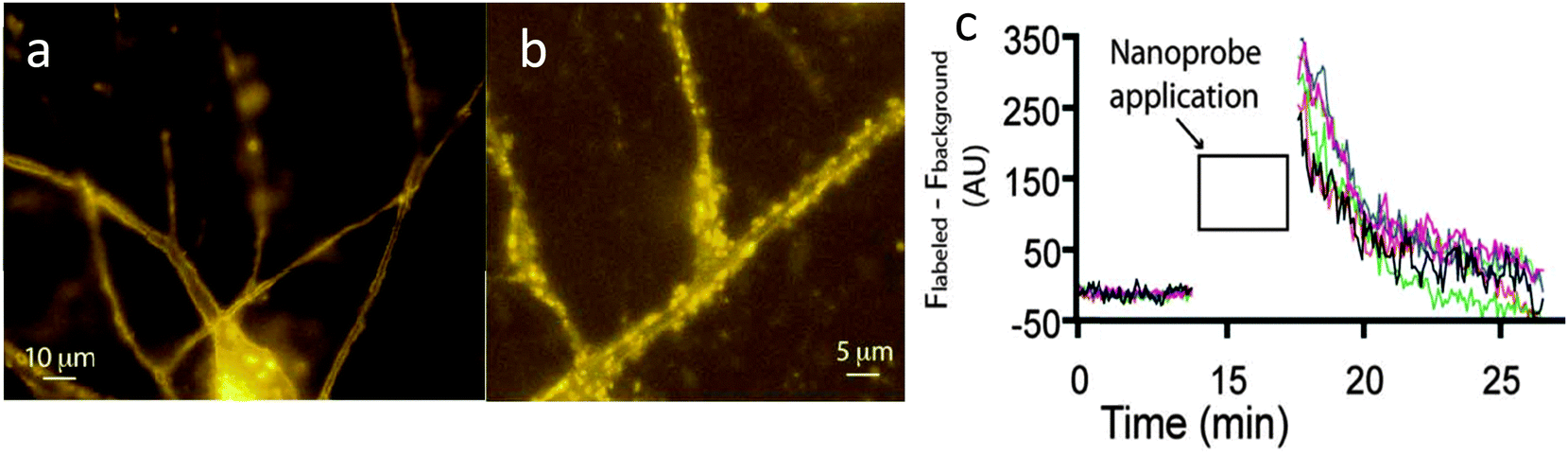 | ||
| Fig. 6 (a) Fluorescent images of neurons 30 seconds after incubation of Cy3 probe. (b) Magnified region after 5 minutes of washing with buffer. (c) Quantitative analysis showing a decrease in fluorescence intensity from the neuronal cells over a short period of time.36 | ||
Despite the visualisation of AMPA receptors, there are some limitations to this work. It is not clear whether or not imaging is performed after photo-dissociation of the polyamine ligand, and whether the Cy3 unit directly bound to the AMPA receptor is being visualised. The fact that the fluorescence decreases over time suggests that the positively charged polyamine is also labelling the plasma membrane non-specifically, and can then be washed out over time. It is unclear as to whether the probe is selective for labelling AMPA receptors, as all iGluRs possess a polyamine-binding site.
Another example exploiting the use of fluorescently labelled channel blockers is from Strømgaard and co-workers,37 who systematically replaced the aromatic head-group of polyamine iGluR channel blockers with fluorescent labels of varying size. It was found that as the size of the fluorophore increased, the compounds affinity for the AMPA and NMDA receptor decreased. Furthermore, the size of the fluorophore had an impact on the selectivity of the probes toward different receptors but, in general, a higher affinity for the AMPA receptor was observed.
The most promising BODIPY probe (Fig. 7) was used in cellular staining experiments of NMDA receptors transfected with the red fluorescent protein, mCherry. Upon incubation of the BODIPY probe, a considerable degree of non-specific binding was observed due to interaction of the positively charged polyamine with the plasma membrane. However, following a continuous five minute wash period, the non-specific staining was removed to reveal punctate fluorescence, overlapping with mCherry emission along the dendrites (Fig. 7). In contrast, cells treated with a similar compound but with a much weaker affinity for the NMDA receptor showed no cellular localisation, after the 5 min. wash period.37
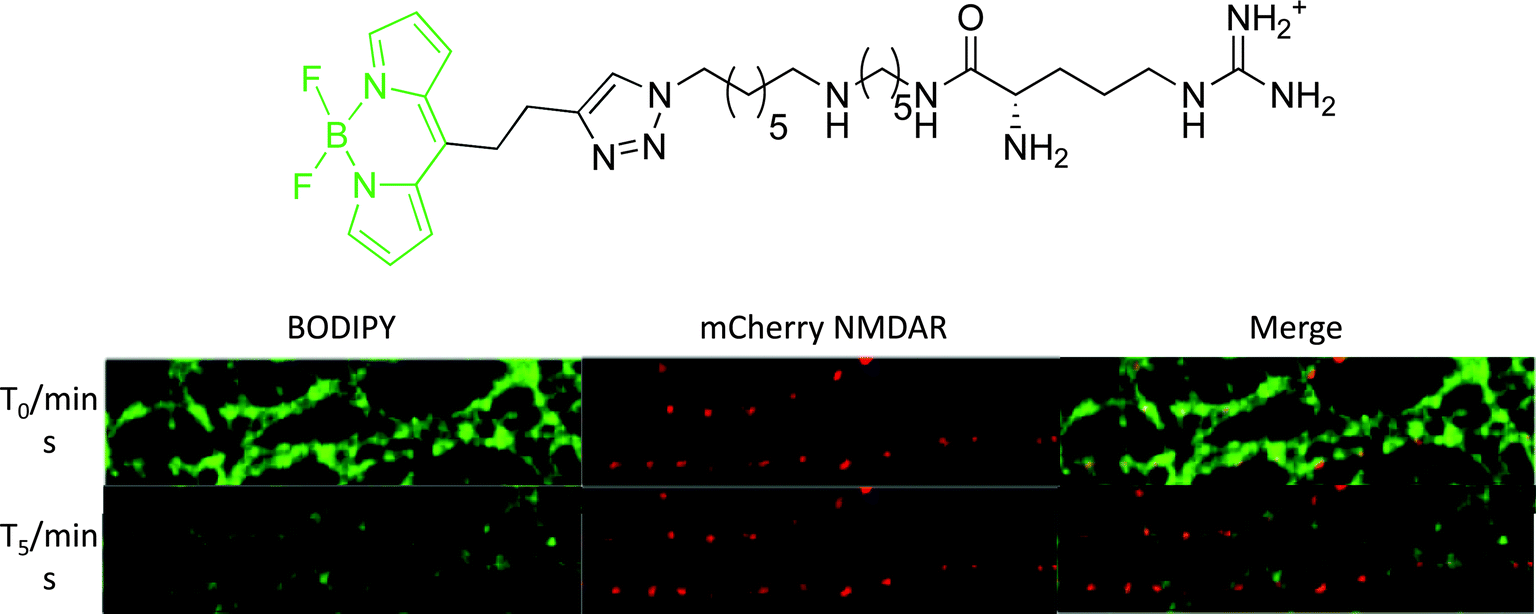 | ||
| Fig. 7 Above: structure of iGluR-targeted BODIPY probe; below: confocal microscopy images of neurons expressing mCherry-tagged NMDA receptors (red fluorescence). Cells were treated for 2 min. with 50 nM of BODIPY probe (green fluorescence) and then imaged at beginning of washing stage (T0) and following five minutes continuous washing (T5).37 | ||
Recently, Perrio and co-workers have synthesised three series of NMDA receptor NR2B subunit specific optical imaging probes by conjugation of fluorescein to derivatives of the non-competitive antagonist, ifenprodil.38,39 The point of attachment and length of spacer unit connecting the fluorophore to the ligand moiety was found to strongly dictate probe affinity towards the NMDA receptor. By measuring Ca2+ influx transients in a competitive assay, optimal affinity was demonstrated when conjugation occurred through modification of the benzylic hydroxyl group of ifenprodril. Generally, a longer spacer increased affinity for the NR2B subunit, although IC50 values were approximately 6-fold higher as compared to ifenprodil. Other pharmacological properties, such as sub-unit selectivity and neuroprotective effects were not compromised by the conjugation of the fluorophore.
When neuronal cells expressing Ds-Red labelled NMDA receptors were treated with fluorescein-ifenprodil, a punctate green fluorescence was observed from the fluorescein moiety (Fig. 8). This fluorescence image corresponded well with the red fluorescence from the genetically labelled NMDA receptors, demonstrating co-localisation of fluorescein-ifenprodil at the NMDA receptor (Fig. 8). Furthermore, when cells were pre-treated with ifenprodil and then loaded with fluorescein-ifenprodil, no fluorescence was observed, suggesting fluorescein-ifenprodil localises and binds in the same position on NMDA receptors as the native ligand (Fig. 8). Despite the increased sensitivity and resolution associated with optical imaging methods, this technique is limited by light scattering and absorbance in soft tissue, and has a low effective imaging depth of the order of 10 mm or so.
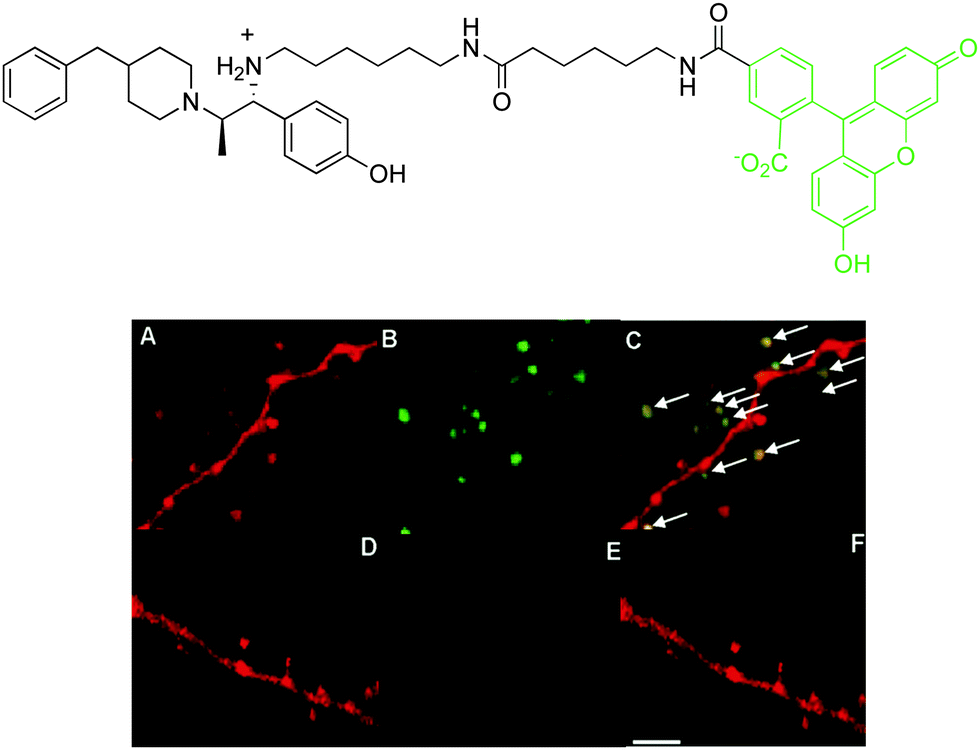 | ||
| Fig. 8 Above: structure of fluorescein-ifenprodil; below: confocal microscopy images of Ds-Red labelled NMDA receptors (red fluorescence): (A) Ds-Red emission; (B) visualisation of fluorescein emission after incubation of fluorescein-ifenprodil (10 μM, 15 min.); (C) merged image showing co-localisation. Images (D)–(F) are as for (A)–(C) but with a pre-incubation of ifenprodil (10 μM, 15 min.).39 Ds-Red is a red fluorescent protein. | ||
3.3 Gd(III)-MRI contrast agents for glutamate receptors
For brain imaging, more detailed spatio-temporal information is required about the chemical processes associated with neurotransmission and their relationship to cognitive behaviour, learning and disease status and progression. The utility of MRI is improved by the use of contrast agents, especially those that enhance specificity and sensitivity by targeting certain regions of interest, or that allow the monitoring of changes in chemical composition as a function of neural activity. This rationale has driven the recent development of agents that selectively target the various glutamate receptors that are found in high abundance on the cell surface of astrocytes.7,40,41 These responsive contrast agents must bind selectively and reversibly to the receptor, and not be internalized, so that changes in local contrast agent levels can be modulated following the extracellular glutamate bursts that periodically occur following a stimulus.The release of glutamate from a pre-synaptic cell takes place over a period of a few milliseconds and should lead to the displacement of a contrast agent that is already bound to the cell surface receptor. MRI has been hypothesised to be able to monitor the restoration to equilibrium that occurs over a period of a second or two.7 As the glutamate levels drop and the contrast agent competes for binding to the receptor, glutamate is displaced, leading to local signal intensity enhancement once more. Such a perturbational approach requires that the contrast agent is bound reversibly and with relatively high affinity to the target site, with minimal non-specific binding. For these reasons, conjugates of gadolinium contrast agents with established competitive antagonists for glutamate receptors have been examined. When bound to the receptor site, the rotational dynamics and second sphere of hydration around the gadolinium moiety are perturbed, leading to relaxivity enhancement and increased image intensity.
The targeting of metabotropic glutamate (mGluR5) receptors by new MRI contrast agents has been reported.40 The contrast agents were derived from a [Gd.DOTA]− complex with fast water exchange kinetics, linked via a glutarate arm to various selective mGluR5 antagonists (Scheme 5).
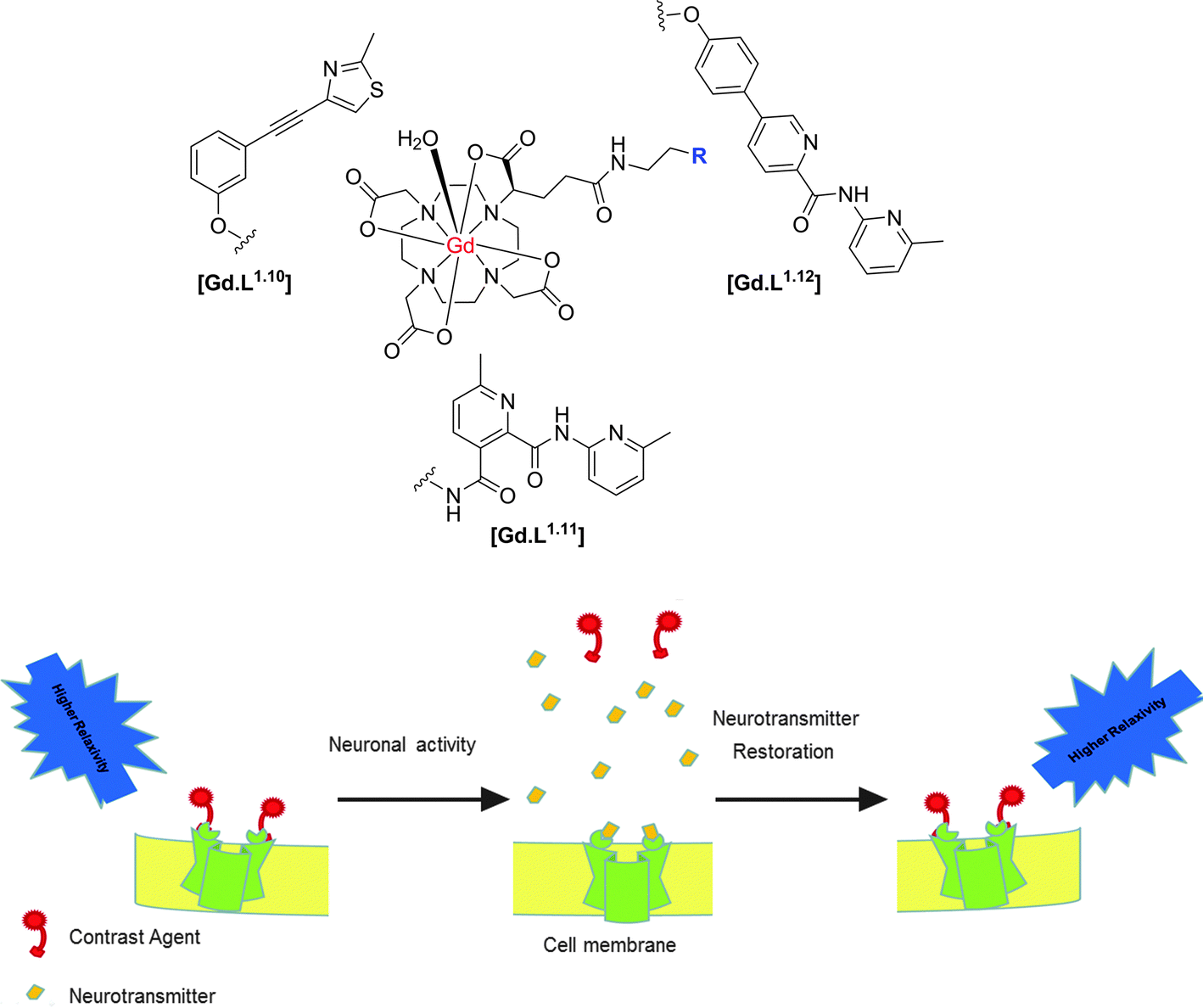 | ||
| Scheme 5 Structures of selected mGluR5 receptor-targeted contrast agents and putative mode of action, illustrating the relaxivity change as a function of reversible binding of the Gd(III) contrast agent to the cell surface receptor.40,41 | ||
Since appending a Gd(III) complex to an antagonist represents a substantial modification to the chemical structure of the antagonist, the antagonistic effect of the modified contrast agents was investigated. Based on changes in intracellular Ca2+ levels, only three of the eight complexes examined, [Gd.L1.10], [Gd.L1.11] and [Gd.L1.12] maintained a significant antagonistic effect.7
A complete study of the concentration-dependence on the cellular relaxation rate (R1,cell) enhancement of primary rat astrocytes expressing mGluR5 receptors was undertaken, by recording T1-weighted MR images at 3 Tesla. The complex derived from the most potent antagonist, gave rise to the largest enhancement in R1,cell (35% at 200 μM). However, this complex did not exhibit any antagonist effect and [Gd.L1.10] (R1,cell = 33% at 200 μM) was identified as a lead compound from the alkyne-based series of contrast agents. Of the dipyridyl amide systems, [Gd.L1.12] exhibited the largest enhancement in R1,cell (30% at 200 μM) and possessed a significant antagonist effect (Fig. 9). Such behaviour was attributed to the antagonist moiety being able to adopt a planar conformation, essential for mGluR5 affinity.7
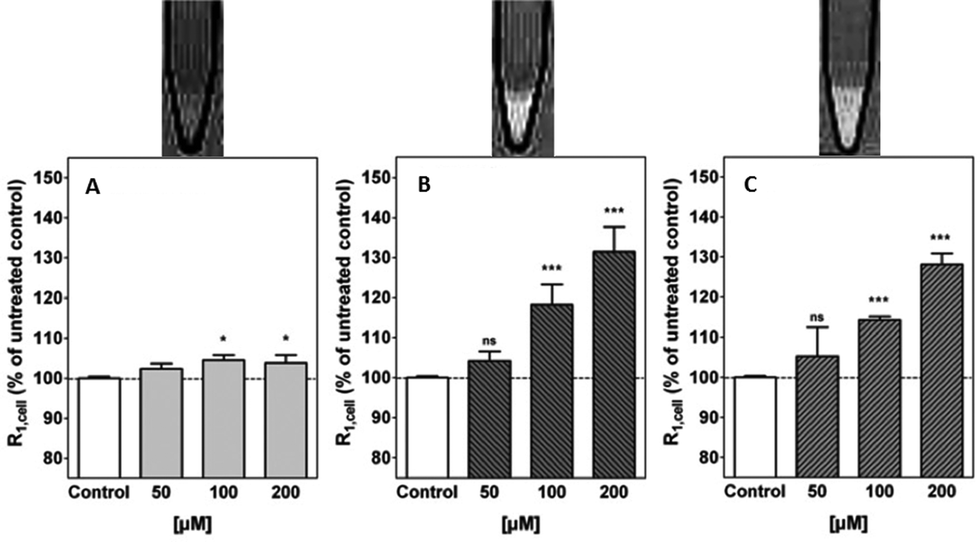 | ||
| Fig. 9 Above: representative T1-weighted MR images (3 T) of 1 × 107 cells; below: cellular relaxation rates, R1,cell, after treatment of primary rat astrocytes with (A) [Gd.DOTA], (B) [Gd.L1.10] and (C) [Gd.L1.12] (50 to 200 μM, 45 min.).7 | ||
Following MR experiments, the cells treated with [Gd.L1.10] and [Gd.L1.12] were subject to ICP-MS measurements to determine the total Gd(III) content. Between 6 to 60 × 107 Gd(III) ions per cell were present, which is well above the detection limit of modern MRI experiments. However, the cellular relaxivities were found to differ substantially (2.9 mM−1 s−1 and 7.4 mM−1 s−1, for [Gd.L1.10] and [Gd.L1.12], respectively), suggesting a different mechanism for R1,cell enhancement. The enhancement due to [Gd.L1.10] can be assigned to the huge increase in cell-associated Gd(III), whereas [Gd.L1.12] induces a comparable enhancement with only one third Gd(III) present. Such behaviour suggests that [Gd.L1.12] induces R1,cell enhancement due to its higher affinity for the mGluR5 receptor.7
The enhancements in relaxation rates were tentatively assigned to an increase in the τR value of the complex upon receptor binding. This behaviour is reflected in the ability of [Gd.L1.12] to bind reversibly to cells possessing mGluR5 in the presence of glutamate, when studied by fluorescence spectroscopy.40 Internalisation of the contrast agents was ruled out by performing control experiments, where competition assays in the presence of unmodified antagonists resulted in no significant increase in relaxation rate. Furthermore, derivatives of the lead compounds were synthesised that included a remote biotin tag. These derivatives were shown to only label the mGluR5 receptors located on the surface of primary rat astrocytes using confocal and TIRF microscopy, after co-incubation with the fluorescent avidin-Alexafluor®-488 conjugate (Fig. 10).41
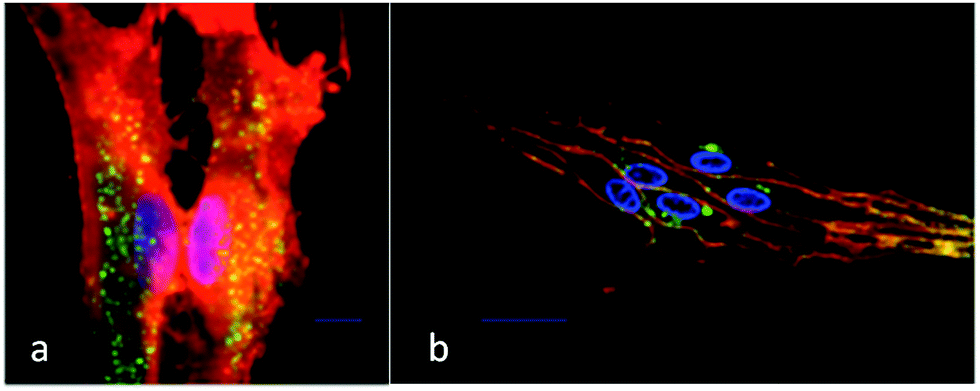 | ||
| Fig. 10 (a) Total internal reflectance fluorescence (TIRF) and (b) confocal microscopy images of primary rat astrocytes after incubation with a biotin derivative of [Gd.L1.12] showing predominant cell surface localisation. Green emission (avidin-AlexaFluor®-488) indicates the labelled mGluR5 receptors; the orange emission shows total plasma membrane staining and blue emission (Hoechst dye) highlights the nucleus.33 | ||
Similar work has been reported for NMDA receptors, in vitro and in cellulo. The synthesis and behaviour of competitive NMDAR antagonist conjugates has been reported, based on 3,4-diamino-3-cyclobutene-1,2-diones developed by Kinney.42 The NMDA receptor is a tetrameric complex, most commonly built up from alternating GluN1 and GluN2 sub-units. These receptor-targeted moieties are competitive antagonists that can bind to the GluN2 subunit where the glutamate binding site is located.43 In initial work,40 immunofluorescence staining experiments were undertaken and established the presence of the GluN2B subunit. The targeting moieties based on the competitive antagonist sub-unit were conjugated to modified [Gd.DOTA]− complexes that possess a single fast-exchanging water molecule, (Scheme 6).
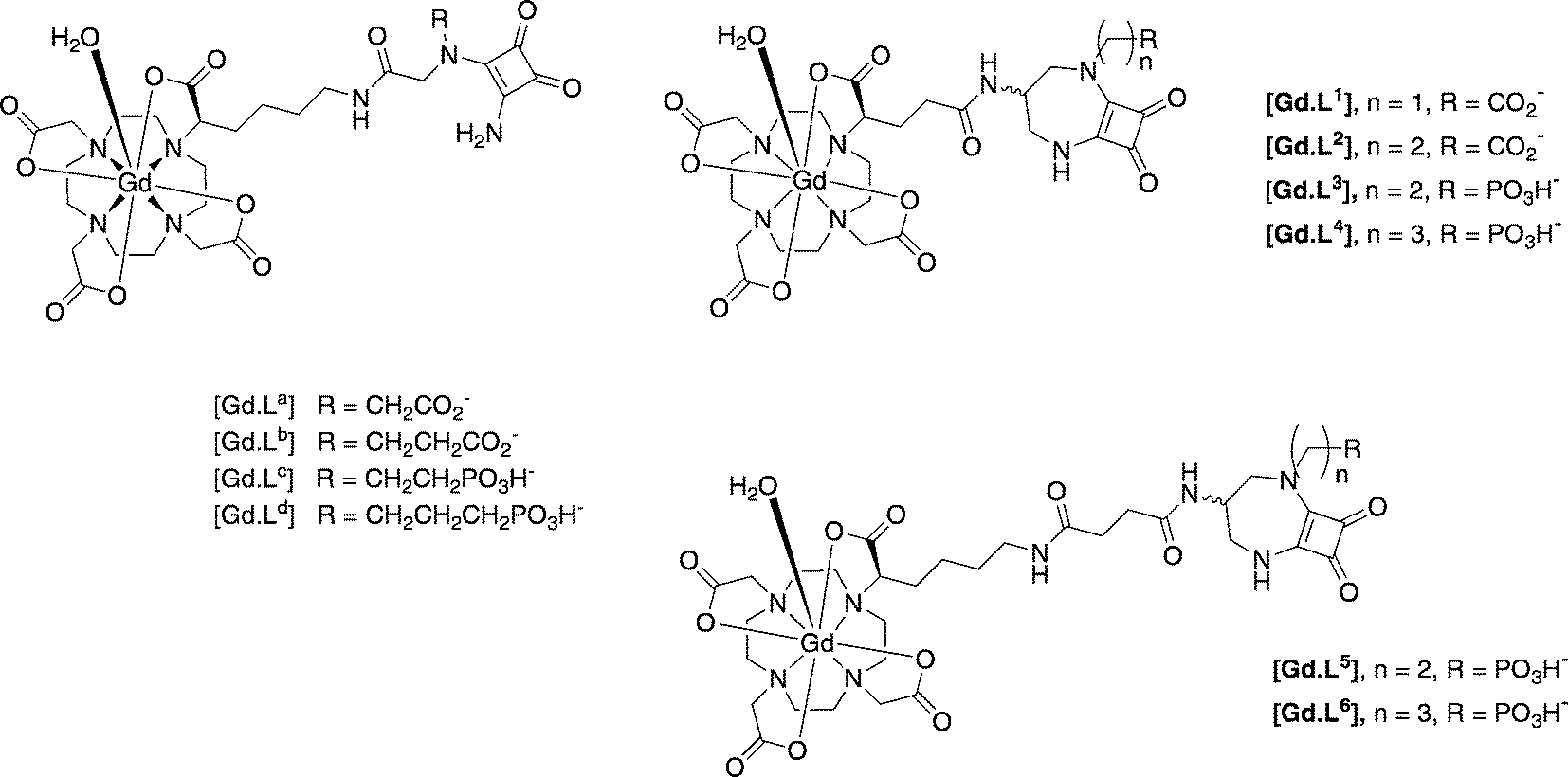 | ||
| Scheme 6 MRI contrast agents targeted to the NMDA receptor on neurons.44,45 | ||
As water exchange is fast at Gd, the relaxivity in the low to mid-field range (1 to 3 T), is dominated by rotational dynamics and is sensitive to receptor binding.5,6 Initial studies in cell suspensions of an NSC-34 cell line showed relaxation enhancements of up to 75% on cell surface binding, versus control cells lacking NMDA receptors or versus cells treated with non-targeted [Gd.DOTA]−.44 The conjugates were non-toxic, consistent with evidence from optical microscopy studies that revealed the conjugate clearly binding to the cell surface receptors, with no evidence for cell internalization. Furthermore, the confocal microscopy studies with the biotin-labelled derivative established both the specificity and the reversibility of probe binding, in the absence and presence of added glutamate.44,45 Such an approach augurs well for future studies in vivo, and further strengthens the case to use competitive antagonists as targeting vectors for the molecular imaging of cell-surface receptors. However, with anionic complexes of this molecular volume the blood brain barrier is not likely to be crossed. More immediate applications are best directed to address basic neuroscience investigations, as administration to the brain requires direct injection into the brain parenchyma. Nevertheless, applications beyond basic neuroscience can be envisaged, for example under disease-based circumstances when the blood–brain barrier is damaged or its permeability artificially enhanced.
4. Conclusions and future challenges
The design of a conjugate that can serve as an MR contrast agent for a cell-surface receptor must consider several issues at the outset.The selection of the cell-surface receptor to target must be undertaken judiciously. It should be present in sufficiently high abundance (i.e. a high receptor density) that cellular differentiation of contrast agent binding can occur, by minimising the degree of non-specific binding to other cell types. For several receptor types, e.g. folate, fast internalisation of the contrast agent is very likely to occur after receptor binding, if the contrast agent design is based on the natural substrate, or agonist. Such systems – like the putative ‘intracellular receptor targeted' contrast agents for progesterone receptors – raise concerns for future use in humans, as the internalised conjugate is likely to be eventually trapped in an endosome or lysosomal compartment, enhancing the chances of the long term retention of the toxic gadolinium ion. Mechanisms that increase the probability of the long-term retention of gadolinium are best avoided, as the debilitating and potentially fatal disease nephrogenic systemic fibrosis has been demonstrated to be associated with Gd retention in humans with impaired renal clearance. In these cases the contrast agent is circulating in the body for a much longer period of time. In particular, the use of Gd complexes and conjugates of DTPA-diamide ligands are of concern, as these complexes are inherently less stable with respect to acid-promoted dissociation of gadolinium,48 and have been shown to be more sensitive to metabolic degradation in cells, compared to macrocyclic analogues.49
Due to the intrinsic insensitivity of MRI, it is often necessary to establish or confirm the localisation profile of a Gd(III) complex through direct visualisation using an alternative imaging technique. Dual modality imaging agents are desirable for this purpose, but care must be taken in the design of the final complex. The structure of the parent complex must be minimally perturbed, such that changes to the overall complex charge and hydrophobicity are minimized; such changes may radically affect pharmacokinetics and pharmacodynamics. Simply appending a secondary reporter group can drastically alter the localisation profile of a receptor-targeted Gd complex, as was observed in the case of certain luminescent analogues of a mGluR5-targeted imaging agent, wherein probe localisation was determined by an added lipophilic chromophore (that probably encouraged non-specific uptake by macropinocytosis50) rather than by the receptor-targeting moiety.41
By critically assessing recent progress in molecular imaging for MRI, the following four main conclusions can be drawn in designing a Gd based molecular imaging agent. First, the gadolinium contrast agent should contain an MR-active core, conjugated to a known ligand for the receptor, in which fast water exchange occurs at Gd, such that τR, and not the rate of water exchange at the metal centre, limits the relaxivity enhancement that occurs on receptor binding. Such design criteria will tend to maximise the change of relaxivity from the unbound to the bound state.
Second, the incorporation of the contrast agent moiety into the conjugate should cause little or no perturbation to the affinity and selectivity of receptor binding. Moreover, the use of coordinatively unsaturated Gd centres should be avoided (e.g. those based on heptadentate ligands), as this is likely to lead to competitive and non-selective protein binding at the metal centre, as well as displacement of the Gd-bound water.11
Third, the contrast agent should bind to the receptor in a reversible manner and, where appropriate, with comparable affinity to the endogenous ligand. Such a situation arises for glutamate receptors in neuronal cells, for example, as in the mGluR5 and NMDA receptor types.
Finally, the targeting moiety may be better selected based on known competitive antagonists, rather than mimic or copy the structure of established agonists. In work with targeted SPECT tumour imaging using 111In radiolabelled conjugates, those based on somatostatin receptor antagonists were shown to be superior to systems based on the natural agonist structure.46,47 In contrast to an agonist, a competitive antagonist usually does not trigger receptor internalisation, following cell surface receptor binding. Provided that sufficient signal contrast can be obtained, such behaviour is arguably preferable as a strategy for contrast agent design, as it reduces the risk of longer term retention of the contrast agent and may feature more strongly in future work. Of course, each agent must be assessed carefully for its toxicity profile, in cellulo and in vivo prior to any use in humans.
Acknowledgements
We thank EPSRC and the ERC for support, Dr Anurag Mishra for the inspiration and Drs Jorn Engelmann, Sven Gottschalk and Robert Pal for their expertise.References
- M. L. James and S. S. Gambhir, Physiol. Rev., 2012, 92, 897–965 CrossRef CAS PubMed.
- M. P. Lowe, D. Parker, O. Reany, S. Aime, M. Botta, G. Castellano, E. Gianolio and R. Pagliarin, J. Am. Chem. Soc., 2001, 123, 7601–7609 CrossRef CAS PubMed.
- E. L. Que and C. J. Chang, Chem. Soc. Rev., 2010, 39, 51–60 RSC.
- M. C. Heffern, L. M. Matosiuk and T. J. Meade, Chem. Rev., 2014, 114, 4496 CrossRef CAS PubMed.
- P. Caravan, Acc. Chem. Res., 2009, 42, 851–862 CrossRef CAS PubMed.
- P. Caravan and Z. Zhang, Eur. J. Inorg. Chem., 2012, 1916–1923 CrossRef CAS.
- S. Gottschalk, J. Engelmann, G. A. Rolla, M. Botta, D. Parker and A. Mishra, Org. Biomol. Chem., 2013, 11, 6131–6141 CAS.
- J. Lee, M. J. Zylka, D. J. Anderson, J. E. Burdette, T. K. Woodruff and T. J. Meade, J. Am. Chem. Soc., 2005, 127, 13164–13166 CrossRef CAS PubMed.
- J. Lee, J. E. Burdette, K. W. MacRenaris, D. Mustafi, T. K. Woodruff and T. J. Meade, Chem. Biol., 2007, 14, 824–834 CrossRef CAS PubMed.
- P. A. Sukerkar, K. W. MacRenaris, T. J. Meade and J. E. Burdette, Mol. Pharmaceutics, 2011, 8, 1390–1400 CrossRef CAS PubMed.
- S. Aime, E. Gianolio, E. Terreno, G. B. Giovenzana, R. Pagliarin, M. Sisti, G. Palmisano, M. Botta, M. P. Lowe and D. Parker, J. Biol. Inorg. Chem., 2000, 5, 488–497 CAS.
- P. A. Sukerkar, K. W. MacRenaris, T. R. Townsend, R. A. Ahmed, J. E. Burdette and T. J. Meade, Bioconjugate Chem., 2011, 22, 2304–2316 CrossRef CAS PubMed.
- J. Faiz Kayyem, R. M. Kumar, S. E. Fraser and T. J. Meade, Chem. Biol., 1995, 2, 615–620 CrossRef.
- S. D. Konda, M. Aref, S. Wang, M. Brechbiel and E. C. Wiener, Magn. Reson. Mater. Phys., Biol. Med., 2001, 12, 104–113 CrossRef CAS.
- O. Saborowski, G. H. Simon, H.-J. Raatschen, M. F. Wendland, Y. Fu, T. Henning, R. Baehner, C. Corot, M.-H. Chen and H. E. Daldrup-Link, Contrast Media Mol. Imaging, 2007, 2, 72–81 CrossRef CAS PubMed.
- Z. Sideratou, D. Tsiourvas, T. Theodossiou, M. Fardis and C. M. Paleos, Bioorg. Med. Chem. Lett., 2010, 20, 4177–4181 CrossRef CAS PubMed.
- T. L. Kalber, N. Kamaly, P.-W. So, J. A. Pugh, J. Bunch, C. W. McLeod, M. R. Jorgensen, A. D. Miller and J. D. Bell, Molecular Imaging and Biology, 2011, 13, 653–662 CrossRef PubMed.
- S. Aime, C. Cabella, S. Colombatto, S. Geninatti Crich, E. Gianolio and F. Maggioni, Magn. Reson. Imaging, 2002, 16, 394–406 CrossRef PubMed.
- D. Messeri, PhD thesis, Durham University, 2002, p. 138.
- G. J. Stasiuk, H. Smith, M. Wylezinska-Arridge, J. L. Tremoleda, W. Trigg, S. K. Luthra, V. M. Iveson, F. N. E. Gavins and N. J. Long, Chem. Commun., 2013, 49, 564–566 RSC.
- I. Zigelboim, D. Offen, E. Melamed, H. Panet, M. Rehavi and Y. Cohen, J. Inclusion Phenom. Macrocyclic Chem., 2007, 59, 323–329 CrossRef CAS.
- I. Zigelboim, A. Weissberg and Y. Cohen, J. Org. Chem., 2013, 78, 7001–7012 CrossRef CAS PubMed.
- S. S. Kulkarni and A. H. Newman, Bioorg. Med. Chem. Lett., 2007, 17, 2074–2079 CrossRef CAS PubMed.
- S. R. Stauffer, ACS Chem. Neurosci., 2011, 2, 450–470 CrossRef CAS PubMed.
- K. J. Gregory, M. J. Noetzel, J. M. Rook, P. N. Vinson, S. R. Stauffer, A. L. Rodriguez, K. A. Emmitte, Y. Zhou, A. C. Chun, A. S. Felts, B. A. Chauder, C. W. Lindsley, C. M. Niswender and P. J. Conn, Mol. Pharmacol., 2012, 82, 860–875 CrossRef CAS PubMed.
- K. J. Gregory, E. N. Dong, J. Meiler and P. J. Conn, Neuropharmacology, 2011, 60, 66–81 CrossRef CAS PubMed.
- L. Pomierny-Chamioło, E. Poleszak, A. Pilc and G. Nowak, Pharmacol. Rep., 2010, 62, 1186–1190 CrossRef.
- P. J. Conn, C. W. Lindsley and C. K. Jones, Trends Pharmacol. Sci., 2009, 30, 25–31 CrossRef CAS PubMed.
- K. A. Johnson, P. J. Conn and C. M. Niswender, CNS Neurol. Disord.: Drug Targets, 2009, 8, 475–491 CAS.
- G. Dölen, E. Osterweil, B. S. Rao, G. B. Smith, B. D. Auerbach, S. Chattarji and M. F. Bear, Neuron, 2007, 56, 955–962 CrossRef PubMed.
- V. J. Majo, J. Prabhakaran, J. J. Mann and J. S. D. Kumar, Drug Discovery Today, 2013, 18, 173–184 CrossRef CAS PubMed.
- A. K. Brown, Y. Kimura, S. S. Zoghbi, F. G. Siméon, J.-S. Liow, W. C. Kreisl, A. Taku, M. Fujita, V. W. Pike and R. B. Innis, J. Nucl. Med., 2008, 49, 2042–2048 CrossRef PubMed.
- F. G. Siméon, A. K. Brown, S. S. Zoghbi, V. M. Patterson, R. B. Innis and V. W. Pike, J. Med. Chem., 2007, 50, 3256–3266 CrossRef PubMed.
- Y. Kimura, F. Siméon, J. Hatazawa, P. D. Mozley, V. Pike, R. Innis and M. Fujita, Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 1943–1949 CrossRef PubMed.
- J. Stone, K. Erlandsson, E. Arstad, L. Squassante, V. Teneggi, R. Bressan, J. Krystal, P. Ell and L. Pilowsky, Psychopharmacology, 2008, 197, 401–408 CrossRef CAS PubMed.
- D. Vytla, R. E. Combs-Bachmann, A. M. Hussey, I. Hafez and J. J. Chambers, Org. Biomol. Chem., 2011, 9, 7151–7161 CAS.
- N. G. Nørager, C. B. Jensen, M. Rathje, J. Andersen, K. L. Madsen, A. S. Kristensen and K. Strømgaard, ACS Chem. Biol., 2013, 8, 2033 CrossRef PubMed.
- P. Marchand, J. Becerril-Ortega, L. Mony, C. Bouteiller, P. Paoletti, O. Nicole, L. Barré, A. Buisson and C. Perrio, Bioconjugate Chem., 2011, 23, 21–26 CrossRef PubMed.
- M. Dhilly, J. Becerril-Ortega, N. Colloc'h, E. T. MacKenzie, L. Barré, A. Buisson, O. Nicole and C. Perrio, ChemBioChem, 2013, 14, 759–769 CrossRef CAS PubMed.
- A. Mishra, S. Gottschalk, J. Engelmann and D. Parker, Chem. Sci., 2012, 3, 131–135 RSC.
- A. Mishra, R. Mishra, S. Gottschalk, R. Pal, N. Sim, J. Engelmann, M. Goldberg and D. Parker, ACS Chem. Neurosci., 2014, 5, 128–137 CrossRef CAS PubMed.
- W. A. Kinney, N. E. Lee, D. T. Garrison, E. J. Podlesny, J. T. Simmonds, D. Bramlett, R. R. Notvest, D. M. Kowal and R. P. Tasse, J. Med. Chem., 1992, 35, 4720–4726 CrossRef CAS.
- W. A. Kinney, M. Abou-Gharbia, D. T. Garrison, J. Schmid, D. M. Kowal, D. R. Bramlett, T. L. Miller, R. P. Tasse, M. M. Zaleska and J. A. Moyer, J. Med. Chem., 1998, 41, 236–246 CrossRef CAS PubMed.
- N. Sim, S. Gottschalk, R. Pal, J. Engelmann, D. Parker and A. Mishra, Chem. Sci., 2013, 4, 3148 RSC.
- N. Sim, R. Pal, D. Parker, J. Engelmann, A. Mishra and S. Gottschalk, Org. Biomol. Chem., 2014, 12, 9389–9404 CAS.
- M. Ginj, H. Zhang, B. Waser, R. Cescato, D. Wild, X. Wang, J. Erchegy, J. Rivier, H. R. Macke and J. C. Reubi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16436 CrossRef CAS PubMed.
- D. Wild, M. Fani, R. Fischer, L. Del Pozzo, F. Kaul, S. Krebs, R. Fischer, J. E. Rivier, J. C. Reubi, H. R. Maecke and W. A. Weber, J. Nucl. Med., 2014, 55, 1248 CrossRef CAS PubMed.
- J. M. Idee, N. Fretellier, C. Robic and C. Corot, Crit. Rev. Toxicol., 2014, 44, 895 CrossRef CAS PubMed.
- E. Di Gregorio, E. Gianolio, R. Stefania, G. Barutello, G. Diglio and S. Aime, Anal. Chem., 2013, 85, 5627 CrossRef CAS PubMed.
- E. J. New, A. Congreve and D. Parker, Chem. Sci., 2010, 1, 111 RSC.
| This journal is © The Royal Society of Chemistry 2015 |